
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 07 July 2023
Sec. Plant Bioinformatics
Volume 14 - 2023 | https://doi.org/10.3389/fpls.2023.1205051
This article is part of the Research Topic Economic Plant Genome and Database Construction and Research View all 10 articles
 Zhengying Cao1,2Linyi Yang3Yaxuan Xin1,2Wenbin Xu4Qishao Li1,2Haorong Zhang1,2Yuxiang Tu1,2Yu Song5*Peiyao Xin1,2*
Zhengying Cao1,2Linyi Yang3Yaxuan Xin1,2Wenbin Xu4Qishao Li1,2Haorong Zhang1,2Yuxiang Tu1,2Yu Song5*Peiyao Xin1,2*The genus Neocinnamomum is considered to be one of the most enigmatic groups in Lauraceae, mainly distributed in tropical and subtropical regions of Southeast Asia. The genus contains valuable oilseed and medicinal tree species. However, there are few studies on the genus Neocinnamomum at present, and its interspecific relationship is still unclear. In order to explore the genetic structure and evolutionary characteristics of the Neocinnamomum chloroplast genome and to resolve the species relationships within the genus, comparative genomic and phylogenetic analyses were performed on the whole chloroplast genome sequences of 51 samples representing seven Neocinnamomum taxa. The whole Neocinnamomum chloroplast genome size ranged from 150,753-150,956 bp, with a GC content of 38.8%-38.9%. A total of 128 genes were annotated within the Neocinnamomum chloroplast genome, including 84 protein coding genes, 8 rRNA genes, and 36 tRNA genes. Between 71-82 SSRs were detected, among which A/T base repeats were the most common. The chloroplast genome contained a total of 31 preferred codons. Three highly variable regions, trnN-GUU-ndhF, petA-psbJ, and ccsA-ndhD, were identified with Pi values > 0.004. Based on the whole chloroplast genome phylogenetic tree, the phylogenetic relationships among the seven Neocinnamomum taxa were determined. N. delavayi and N. fargesii were the most closely related species, and N. lecomtei was identified as the most basal taxon. In this study, the characteristics and sequence variation of the chloroplast genomes of seven Neocinnamomum taxa were revealed, and the genetic relationship among the species was clarified. The results of this study will provide a reference for subsequent molecular marker development and phylogenetic research of Neocinnamomum.
Neocinnamomum (Neocinnamomeae, Lauraceae) is a genus of evergreen shrubs and small trees distributed across south-central, western, and northwestern China, as well as Nepal, Myanmar, and Vietnam, among other areas (Flora of China, 1982). To date, there are 7 known species of Neocinnamomum worldwide. Five of these species (N. caudatum, N. fargesii, N. lecomtei, N. mekongense, and N. delavayi) are located in China, and are primarily distributed across Hainan, Guangxi, Yunnan, and Sichuan, as well as Tibet (Flora of China, 1982). In 2017, Xu et al. (2017) discovered a variant of N. caudatum (N. caudatum var. macrocarpum) in Baise, Guangxi, China. Outside of China, N. atjehense and N. parvifolium (revised as N. delavayi) are also recorded in the world flora (WFO, 2023). Neocinnamomum includes important oil-bearing tree species as well as valuable medicinal resources. In China, N. caudatum is known as “Baigui” and N. delavayi is known as “Sangujin” (Wu, 1983). The bark and leaves of these species have a long history of use in traditional Chinese medicine (TCM) for dispelling wind and cold, promoting blood circulation, and relieving blood stasis (Jiangsu New Medical College, 1977). In addition, the seeds of Neocinnamomum species are rich in fatty acids (Gan et al., 2018). For example, the seeds of N. caudatum contain approximately 54% oil (dry weight), and the seeds of N. delavayi contain approximately 57% oil. These oils have the potential to be developed and promoted as raw materials for biodiesel production (Wang et al., 2013), which is important for sustainable energy production. Genomic research on the genus Neocinnamomum currently lags far behind that of other oil plants. In addition, the degree of exploitation and utilization of wild Neocinnamomum resources is also extremely low.
The genus Neocinnamomum was originally established by Liu in 1934, and was distinguished from other genera of Lauraceae by the presence of four-locular anthers with collateral pollen sacs (Liu, 1934). However, this feature appears to only exist in N. delavayi and a few individuals of N. caudatum. Because of this, Kostermans (Kostermans, 1974) suggested that these anther characteristics were insufficient to distinguish the genus Neocinnamomum. Instead, he suggested that the presence of compound cymes; shallow, fleshy fruiting receptacles; persistently enlarged tepals; and dichotomous leaves were better distinguishing characteristics. Despite these traits, Kostermans did not deny the morphological similarity between Neocinnamomum and Cinnamomum. Traditional classification systems have long considered the two genera to be closely related. However, more recent molecular phylogenetic studies have called this relationship into question, obscuring the phylogenetic position of the genus Neocinnamomum within Lauraceae. The development of molecular systematics techniques has allowed researchers to utilize chloroplast gene fragments, whole chloroplast genomes, and nrDNA to study Neocinnamomum phylogenetics (Chanderbali et al., 2001; Rohwer and Rudolph, 2005; Wang et al., 2010; Song et al., 2020). In contrast to the traditional understanding that Neocinnamomum and Cinnamomum are closely related, these recent studies indicate that Neocinnamomum is a monophyletic group with Cassytha and Caryodaphnopsis as its closest relatives.
Although the monophyly of the genus Neocinnamomum has been confirmed, the relationships between species within the genus are less clear. Kostermans (1974) divided the genus Neocinnamomum into six species, four of which are easily distinguished. However, N. mekongense and N. delavayi are extremely similar morphologically, and can be distinguished only based on the presence of pubescent twigs. Recently, Wang et al. (2010) used three molecular fragments of psbA-trnH, the trnK cpDNA region, and ITS nrDNA to draw a phylogenetic tree of the genus Neocinnamomum and found that N. mekongense and N. delavayi were located on the same branch. Despite this, molecular fragment analysis has not satisfactorily resolved the relationship between the two species. Compare with the use of chloroplast (cp) gene fragments, the whole cp genome can provide much more robust variation information (Wariss et al., 2017). For example, Ren et al. (2019) constructed a maximum likelihood (ML) phylogenetic tree using the whole chloroplast genomes of N. lecomtei, N. mekongense, and N. delavayi. This highly-supported analysis indicated that N. lecomtei and N. mekongense were located on different branches. Unfortunately, only 5 samples from 3 species of Neocinnamomum were used for the study, limiting the power of the results.
Chloroplasts are the central hub for photosynthetic and certain other metabolic reactions in plants (Tao et al., 2017). Like mitochondria, cp are maternally inherited semi-autonomously and contain a relatively independent genetic system. In addition, cp have been central drivers of evolutionary processes (Neuhaus and Emes, 2000; Xing and Liu, 2008). As sequencing technology has developed, the publication of an ever-increasing number of plant cp genomes has given us a deeper understanding of the structure and variation of cp genomes. For the vast majority of angiosperms, the cp genome is characterized by a closed, double-stranded circular structure, and consists of two inverted repeat (IRs) regions, a large single-copy (LSC) region, and a small single-copy (SSC) region (Jansen et al., 2011). The structural stability of the cp genome is largely maintained by the conserved IR region, which is characterized by a base substitution rate of only 1/4 of that of the SC region (Drouin et al., 2008). The cp genome offers several advantages over the nuclear and mitochondrial genomes, including structural conservation, low molecular weight, simple structure, genetic stability, and moderate evolution rate (slower than the nuclear genome but higher than the mitochondrial genome) (Goremykin et al., 2003). Owing to these unique characteristics, cp genomes are widely used to explore the phylogeny and genetic relationship among plant clades (Yang et al., 2018; Cao et al., 2022; Lan et al., 2022; Wang et al., 2023; Xin et al., 2023). The cp genome is particularly advantageous for studies of species identification, molecular geography, and speciation processes (Xu et al., 2001).
To date, only a few Neocinnamomum genomes have been sequenced, and detailed cp genomic comparisons and phylogenetic analyses are lacking. In order to further clarify the phylogenetic relationships among species of the genus Neocinnamomum, and to obtain useful genetic resources, we sequenced and assembled the cp genomes of 50 samples representing 7 Neocinnamomum taxa. Included in the analysis was one unique taxa recently collected from Wenshan, Yunnan, China, which shares some traits with N. complanifructum (merged into N. lecomtei). Based on 51 cp genome sequences (including 50 sequencing sequences and one sequence from NCBI), we analyzed the cp genome structure, gene content, codon usage frequencies, simple sequence repeats (SSRs), highly variable regions, and IR expansion and contraction. Finally, we reconstructed the phylogenetic relationships of 7 Neocinnamomum taxa. These results will be of great significance for studies of the population genetics, species identification, and conservation biology of the genus Neocinnamomum.
Fifty samples of either fresh leaves or silica gel-dried material representing 7 Neocinnamomum species were collected, including 5 samples of N. delavayi, 6 samples of N. mekongense, 4 samples of N. fargesii, 2 samples of N. caudatum var. macrocarpum, 8 samples of N. lecomtei, 23 samples of N. caudatum, and 2 samples of N. sp (Supplementary Table S1). Plant samples were primarily collected in the Yunnan, Sichuan, Guangxi, and Hainan provinces of China, at an altitude of 1100-2300 m. Only fresh, tender, healthy leaves with no visible pest or disease damage were collected. Fresh samples were dried in self-sealed bags of silica gel prior to DNA extraction and sequencing. At the same time, the cp genome sequence data of N. caudatum (RL01) was downloaded from NCBI database for subsequent analysis.
Total genomic DNA was extracted from fresh and silica gel-dried leaves using a modified cetyltrimethylammonium bromide (CTAB) method (Doyle and Doyle, 1987). DNA integrity was evaluated using 1% agarose gel electrophoresis. DNA purity and concentration were evaluated by Nanodrop. Using qualified DNA samples, 150 bp double-end libraries were constructed and sequenced on the Illumina sequencing platform at Novogene Bioinformatics Technology Co. Ltd (Beijing, China). More than 4.0 Gb of reads was generated per sample.
After the sequencing data were filtered and screened, the cp genome was automatically assembled using the GetOrganelle v1.7.1 (Jin et al., 2020). Bandage v0.8.1 (Wick et al., 2015) was used to visualize the assembly, remove redundant contigs, and edit the sequences into loops. The N. mekongense whole cp genome sequence (GenBank accession number NC_039718) was used as a reference, automatically annotated using CPGAVAS2 (http://47.96.249.172:16019/analyzer/home) (Liu et al., 2012), and adjusted using Geneious v9.1.7 (Kearse et al., 2012). The circular cp genome diagram was created using the OGDRAW online tool (https://chlorobox.mpimp-golm.mpg.de/OGDraw.html) (Lohse et al., 2013). The final assembled and annotated cp genome sequences were uploaded to the NCBI database to obtain GenBank accession numbers.
Eleven individuals of Neocinnamomum plants were selected for analysis (one sample was randomly selected from each taxon, and RL01 N. caudatum, 7751 N. caudatum, 6068 N. delavayi and 7683 N. mekongense).The relative synonymous codon usage (RSCU) of 11 cp genomes was statistically analyzed using CodonW1.4.2 software (https://galaxy.pasteur.fr/?form=codonw) (Liu and Xiu, 2005). Frequent codon usage (i.e., codon usage preference) is indicated when RSCU >1, while RSCU = 1 indicates no usage preference.
The SSRs present in 11 cp genomes were detected by MISA (https://webblast.ipk-gatersleben.de/misa/) (Beier et al., 2017), using the following parameters: the threshold values for the number mononucleotide, dinucleotide, trinucleotide, tetranucleotide, pentanucleotide, and hexanucleotide repeats were set to 10, 5, 4, 3, 3, and 3, respectively (Wu et al., 2021).
By examining the genes bordering IR regions, we can analyze IR expansion and contraction within the cp genomes of Neocinnamomum species. We manually examined the IR junctions of 11 cp genomes in Geneious software. To identify differences among the 11 cp genomes of the Neocinnamomum taxa, a comparative analysis of the full-length cp genome sequences was performed using mVISTA (http://genome.lbl.gov/vista/mvista/submit.shtml) (Frazer et al., 2004), with the cp genome of N. delavayi (6068) used as a reference. MAFFT v7.455 software (Katoh and Standley, 2013) was used to align the 51 cp genomes. The nucleotide polymorphism (Pi) between genomes was calculated using DnaSP v6.12.03 (Rozas et al., 2017), with a window length of 600 and a step size of 200 (Shen et al., 2022). The high-variance regions were screened by combining the mVISTA and Pi results.
Phylogenetic relationships were reconstructed based on the 51 cp genomes, using Caryodaphnopsis henryi, C. tonkinensis and C. malipoensis from NCBI database as outgroup. Both the ML and Bayesian Inference (BI) methods were used to construct the phylogenetic tree. Each of the complete cp genomes were aligned using MAFFT v7.455 (Katoh and Standley, 2013), and then manually edited using BioEdit v 7.2.5 (Hall, 1999). IQ-TREE v1.6.7 (Nguyen et al., 2015) was used to construct the ML phylogenetic tree, and the “GTR+F+R4” model was used for nucleic acid substitution. The support of each branch was verified by bootstrapping, with 1,000 iterations. BI analyses were performed using the Mrbayes v3.2.6 module of the CIPRES website (http://www.phylo.org/) (Huelsenbeck and Ronquist, 2001). Briefly, the processed data was uploaded to the CIPRES platform and jModeltest v2.1.10 (Posada, 2008) was used to determine the best nucleotide substitution model for phylogenetic reconstruction. Then, the Markov chain Monte Carlo (MCMC) algorithm was run for 1,000,000 generations. The results were sampled every 100 generations, and the first 25% of the generated trees were discarded. The “TPM1uf+I+G” model (freqA=0.3027, freqC=0.1972, freqG=0.1894, freqT=0.3107, R(a)[AC]=1.0000, R(b)[AG]=2.8265, R(c)[AT]=0.3346, R(d)[CG]=0.3346, R(e)[CT]=2.8265, R(f)[GT]=1.000, p-inv=0.6960, gamma shape=1.0440) was used to construct the BI phylogenetic tree. The phylogenetic trees were visualized and adjusted using FigTree v1.4.3 (Dong et al., 2022b).
The structure of the Neocinnamomum cp genome is a typical circular double-stranded tetrad (Figure 1). The cp genome size of 51 Neocinnamomum plant samples ranged from 150,753 to 150,956 bp (Supplementary Table S2). The GC content was between 38.8%-38.9%, with N. lecomtei and N. mekongense having a higher GC content than the other studied taxa. We observed some variation in the length and GC content of the LSC, SSC, and IR regions of the cp genomes. The LSC region ranged in length between 91,850-92,006 bp, and had a GC content of 37.4%-37.5%. The SSC region ranged in length between 18,096-18,457 bp, and had a GC content of 33.2%-33.4%. The IR region ranged in length between 20,257-20,425 bp, and had a GC content of 44.5%-44.6%. Overall, the GC content of the IR region was significantly higher than in the SSC and LSC regions.
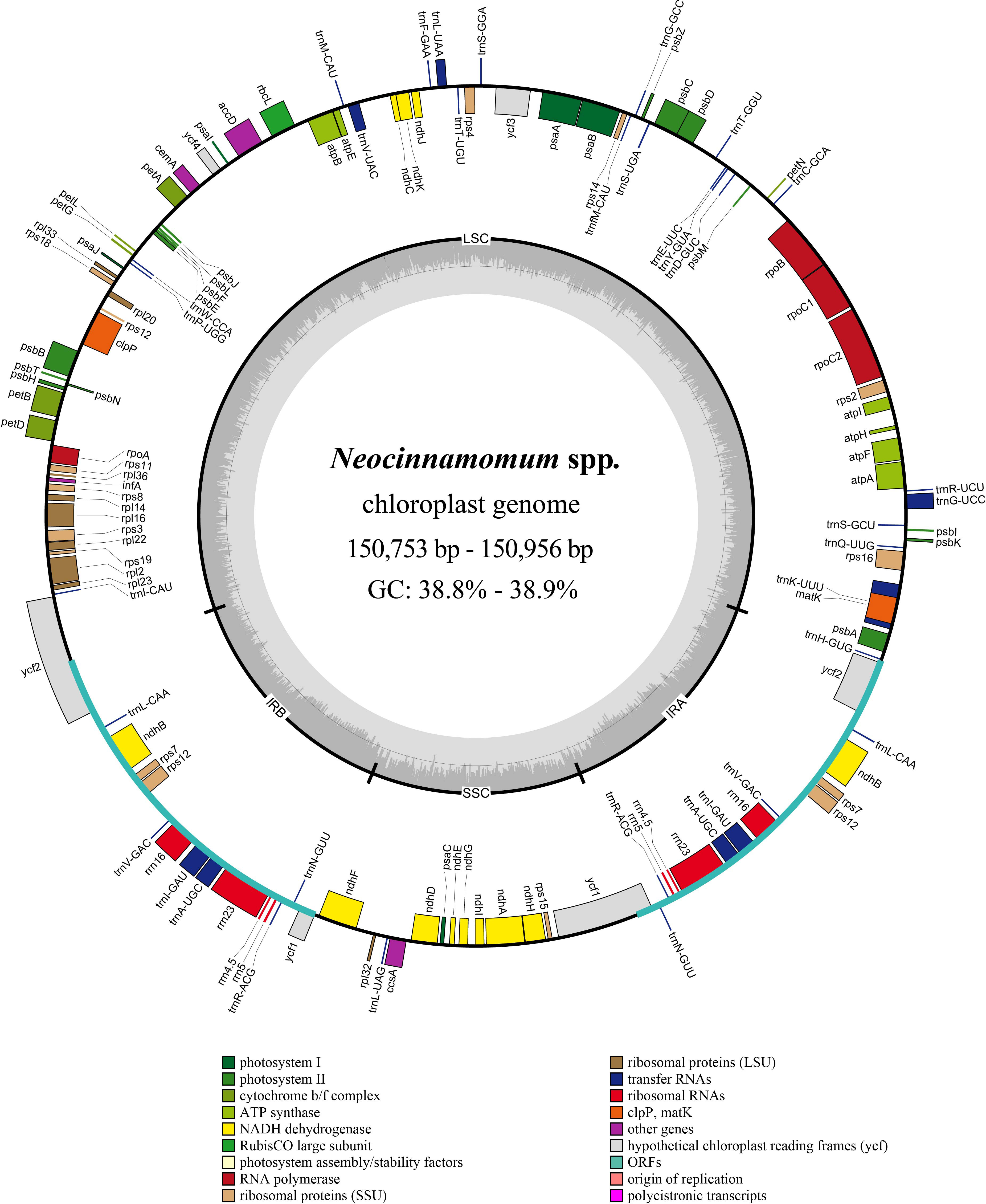
Figure 1 Circular map of the Neocinnamomum chloroplast genome. Genes belonging to different functional groups are color-coded. In the inner circle, the GC and AT content are denoted by the dark gray and light gray, respectively.
A total of 128 genes were annotated within the Neocinnamomum cp genome, including 84 protein coding genes, 8 rRNA genes, and 36 tRNA genes (Table 1). The coding genes were primarily composed of self-replication genes, photosynthesis genes, ycf genes, and “other” genes. Two copies each of 15 genes were located in the IR region, including 5 protein coding genes (ndhB, rps7, rps12, ycf1, and ycf2), 4 rRNA genes (rrn4.5, rrn5, rrn16, and rrn23) and 6 tRNA genes (trnA-UGC, trnI-CAU, trnL-CAA, trnN-GUU, trnR-ACG, and trnV-GAC). In addition, 15 genes (9 protein coding genes and 6 tRNA genes) contained one intron and 3 genes (rps12, clpP, ycf3) contained two introns.

Table 1 Genes present in the Neocinnamomum chloroplast genome.
Using MEGAX64 to determine the codon usage bias of the cp genomes, 64 codons, encoding 20 amino acids, were detected (Figure 2). Among these, three codons had RSCU = 1 and 31 codons had RSCU > 1, indicating a preference for these codons within the Neocinnamomum cp genome. Among 11 cp genomes of the 7 Neocinnamomum taxa, the AGA codon encoding arginine (R) was the most frequently used among the synonymous codons, with RSCU values ranging from 1.82-1.83. Least frequently used was the CGC codon encoding arginine (R), with RSCU values ranging from 0.41-0.42. Among the 20 amino acids, the most common were arginine (R), leucine (L), and serine (S), using 6 codons. The next most common were alanine (A), glycine (G), proline (P), threonine (T), and valine (V), using 4 codons. The least common were tryptophan (W) and methionine (M), using 1 codon. Except for methionine (M) and tryptophan (W), the preferred codons for all amino acids ended in A/U, indicating a preference for codons ending in these bases across the Neocinnamomum cp genome.
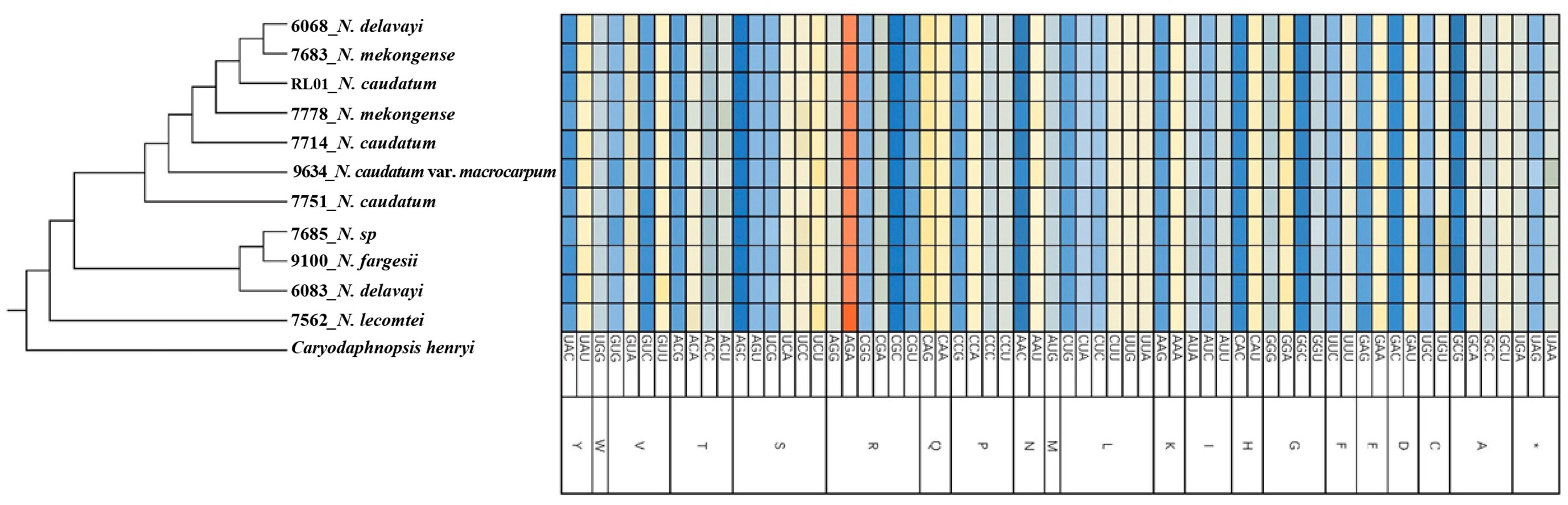
Figure 2 Relative synonymous codon usage (RSCU) of 20 amino acids and the stop codon for 11 chloroplast genomes. 20 uppercase letters such as A, C and D are abbreviations of 20 amino acids, and the symbol "*" is the stop codon.
SSRs are widely distributed in chloroplast genomes. We detected between 71-82 SSRs across each of 11 cp genomes representing 7 Neocinnamomum taxa, for a total of 828 SSRs. The SSRs ranged from single to hexanucleotide repeats, although not all six types of SSR were present in each taxa (Figure 3). Single nucleotide SSRs were the most common (52-63), and were mainly comprised of A/T repeats, with only 1-3 C/G repeats. There were 5-7 dinucleotide SSRs, all of which were AT/AT repeats. Only one trinucleotide SSR, an AAT/ATT repeat, was identified, with only one repeat per sample. There were 11-12 tetranucleotide SSRs, including four repeat types. However, the AAAC/GTTT repeat was present only in N. delavayi (6068), N. mekongense (7683), and N. caudatum (RL01). There were 0-2 pentanucleotide SSRs, including three repeat types (AATTC/AATTG, AAAAC/GTTTT, and ACGAT/ATCGT), which were present only in N. delavayi (6083) and N. caudatum (7714). The hexanucleotide AATTAG/AATTCT SSR was identified only in N. caudatum var. macrocarpum and N. caudatum (7751), with one repeat per sample.
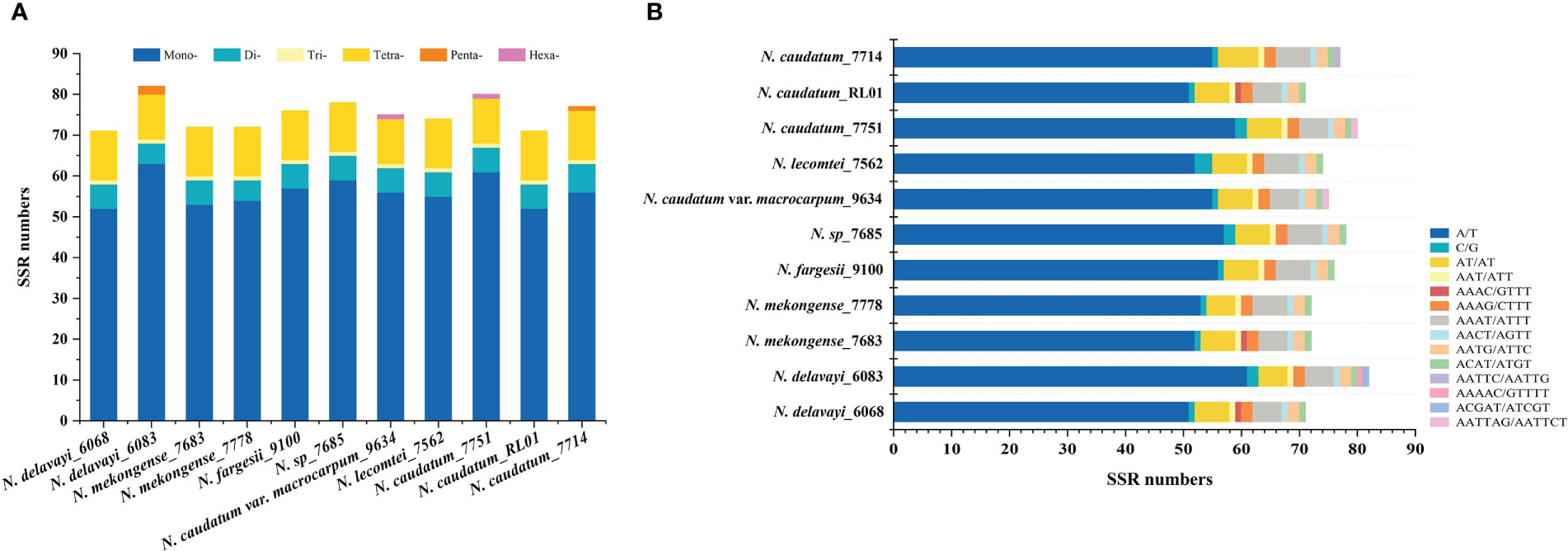
Figure 3 Comparison of SSRs across 11 Neocinnamomum cp genomes. (A) The type frequency of different SSR types. (B) The type frequency of SSR motifs in different repeat class types.
IR junctions analysis of 11 cp genomes representing 7 Neocinnamomum taxa indicated the presence of a contracted expansion of the tetrad structure SC/IR boundary (Figure 4). The LSC/IRb (JLB), IRb/SSC (JSB), SSC/IRa (JSA), and IRa/LSC (JLA) linkage boundaries were primarily associated with four genes: ycf1, ycf2, ndhF, and ndhH. The LSC/IRb (JLB) boundary and the SSC/IRa (JSA) boundary were located within the coding regions of ycf2 and ycf1, respectively. In three samples (7683, 6068, and RL01), the ycf2 gene expanded 3109 bp into the IRb region, which was 5 bp less than in other samples. The ycf1 gene is different in both N. delavayi and N. lecomtei compared to the other taxa, with an additional expansion of 9 bp and 162 bp into the IRa region, respectively. The IRb/SSC (JSB) boundary was located in the noncoding region between ycf1 and ndhF, with trnF of N. lecomtei closest to the boundary at 59 bp. The IRa/LSC (JLA) boundary was located in the noncoding region between ycf2 and ndhH, with trnH of three samples (7683, 6068, and PL01) farthest from the boundary at 34 bp. Interestingly, the IR boundaries of N. lecomtei differed significantly from the other taxa. We hypothesize that this taxa either has an increased rate of evolution or diverged earlier. The distribution of genes at the SC/IR boundary was similar for samples 7683, 6068, and PL01, but differed somewhat from other samples of their respective taxa. Collectively, although the SC/IR boundary of the Neocinnamomum cp genome was relatively conservative, it also exhibited significant diversity.
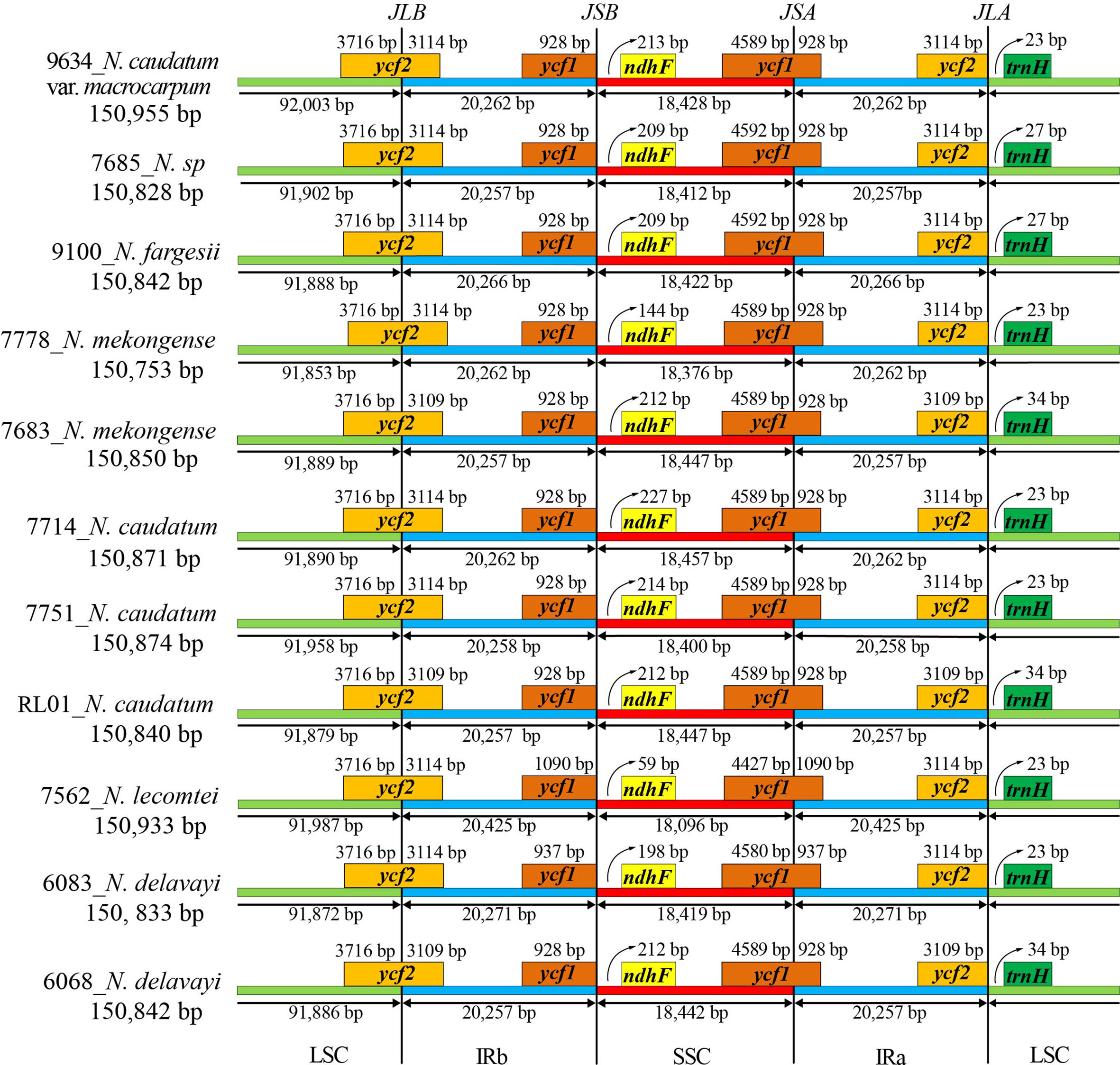
Figure 4 Expansion and contraction of the IR/SC boundary of the Neocinnamomum chloroplast genome.
Using mVISTA software, the N. delavayi (6068) cp genome was used as the reference and compared with the full-length cp genomes of N. delavayi (6083), N. mekongense (7683 and 7778), N. fargesii (9100), N. caudatum (7714, 7751 and RL01), N. caudatum var. macrocarpum (9634), N. sp (7685), and N. lecomtei (7562) (Figure 5). Overall, both gene composition and order was relatively conserved across all 7 Neocinnamomum taxa. The IR region was more conserved than the LSC and SSC regions. The coding region was more conserved than the non-coding region, and variants primarily occurred in the spacer regions of adjacent genes, such as trnN-GUU-ndhF, petA-psbJ, rbcL-accD, and psbE-petL.
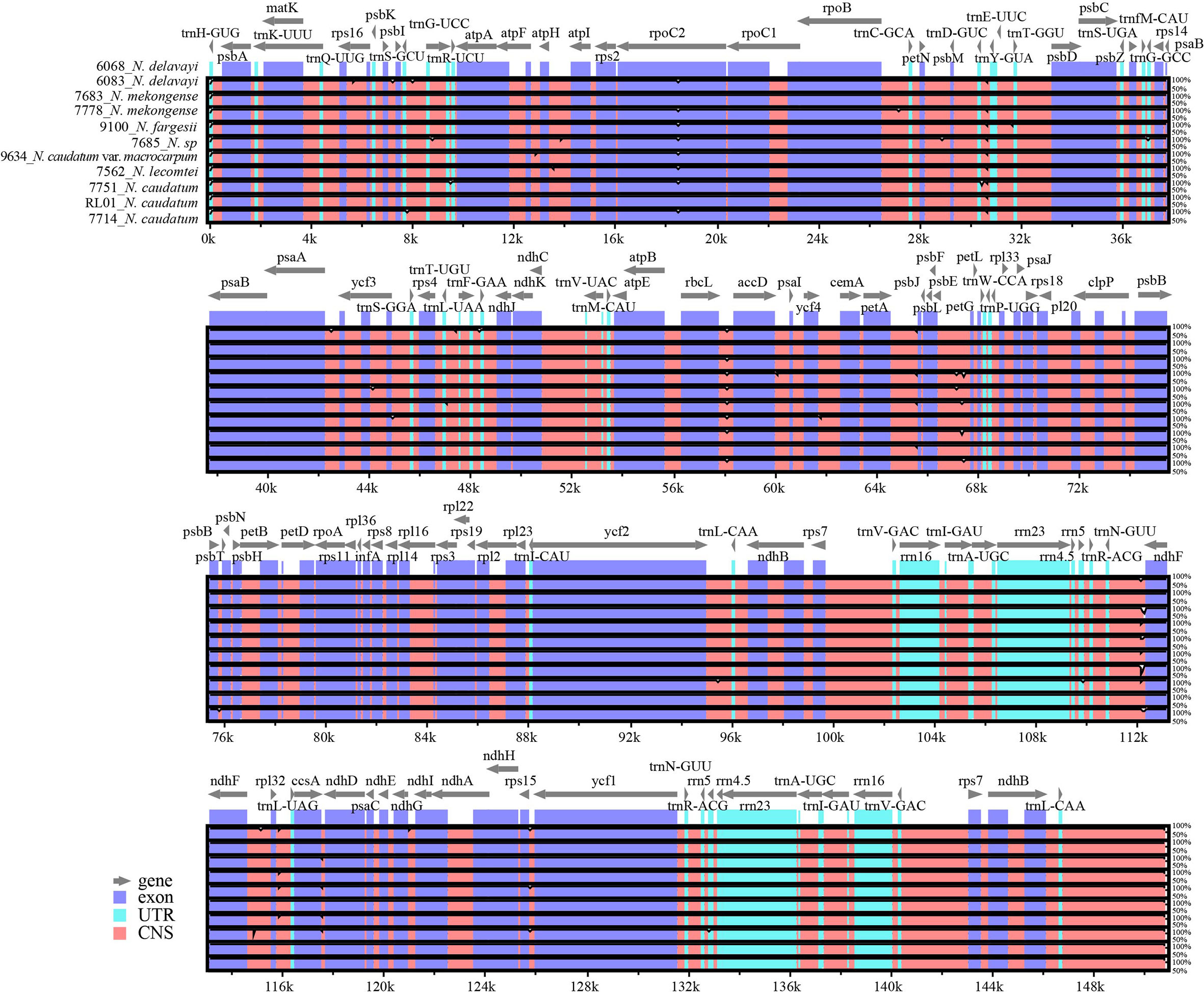
Figure 5 Complete chloroplast genome alignments for seven species of Neocinnamomum. mVISTA software was used to perform the alignment, with the N. delavayi (6068) chloroplast genome used as the reference sequence. The gray arrows indicate genes and their directions.
DnaSP software was used to compare the nucleotide variation values (Pi) between all genes and intergenic regions of the cp genomes. The Pi values of the 51 Neocinnamomum cp genomes varied from 0 to 0.01571, with a mean of 0.00098 (Figure 6). Although the Neocinnamomum cp genome was highly conserved, we identified three divergent hotspot regions (Pi > 0.004): trnN-GUU-ndhF, petA-psbJ, and ccsA-ndhD. Among them, petA-psbJ was located in the LSC region and had a Pi value of 0.00696. Both trnN-GUU-ndhF and ccsA-ndhD were located in the SSC region and had Pi values of 0.01571 and 0.00522, respectively.
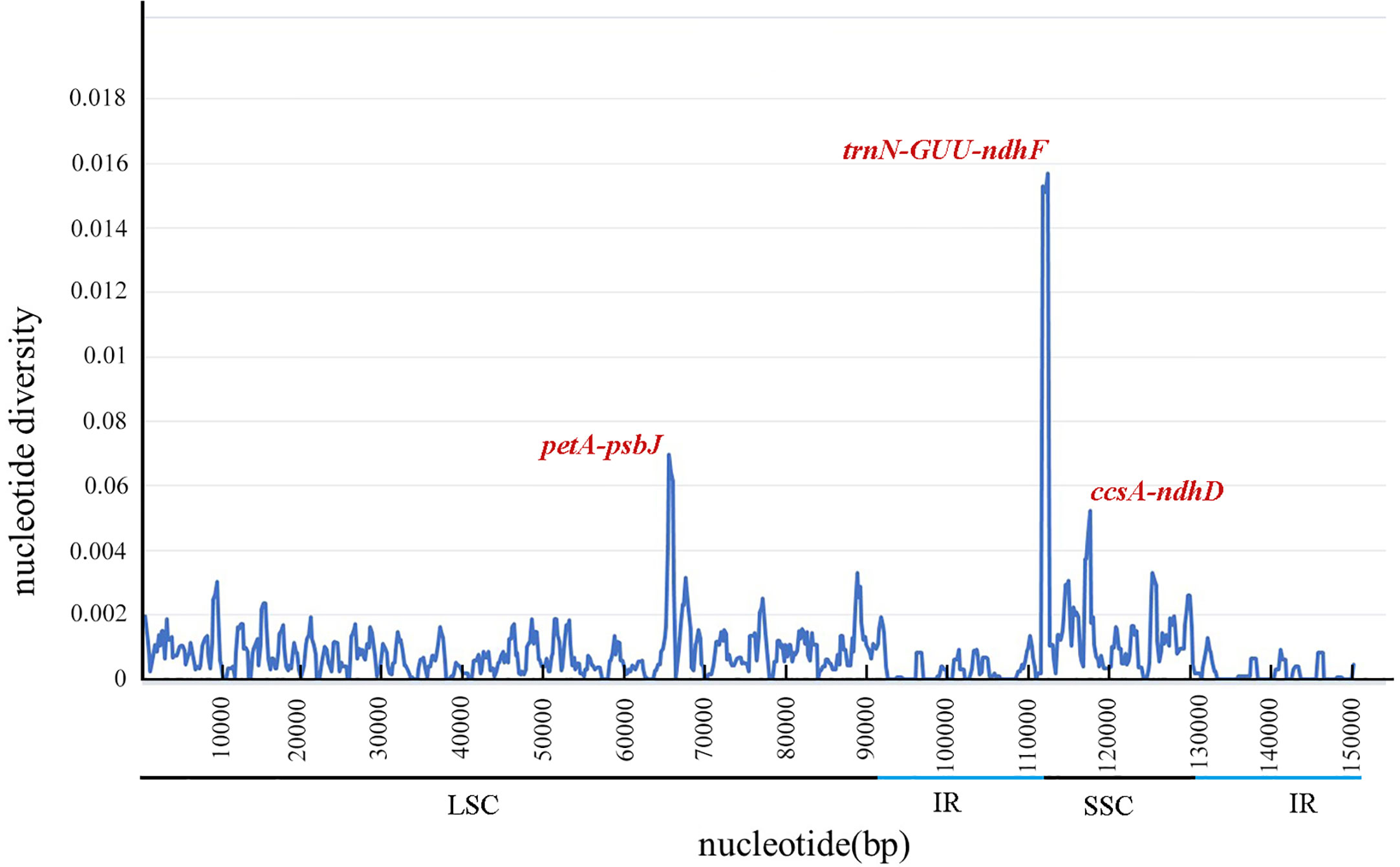
Figure 6 Nucleotide diversity (Pi) in matching areas of Neocinnamomum chloroplast genomes.
The phylogenetic relationships among 7 taxa of Neocinnamomum were reconstructed based on the cp genomes, with C. henryi, C. tonkinensis and C. malipoensis used as outgroup (Figure 7). In the ML phylogenetic tree, 40.91% of the branches had 100% support and 93.18% of the branches had ≥ 75% support. In the BI phylogenetic tree, only a small branch had a support rate of 0.5, and the other branches had a support of 1. The topology of the phylogenetic tree constructed based on both ML and BI methods was nearly identical. Overall, the 51 samples representing 7 Neocinnamomum taxa were divided into six branches. Clade I included only N. caudatum. Clade II included N. mekongense, as well as N. delavayi (6068) and N. caudatum (RL01). Clade III included only N. caudatum var. macrocarpum. Clade IV included only N. caudatum (7751). Clade V included N. delavayi, N. fargesii, and N. sp. Clade VI included only N. lecomtei, which was first differentiated as a basal taxon.
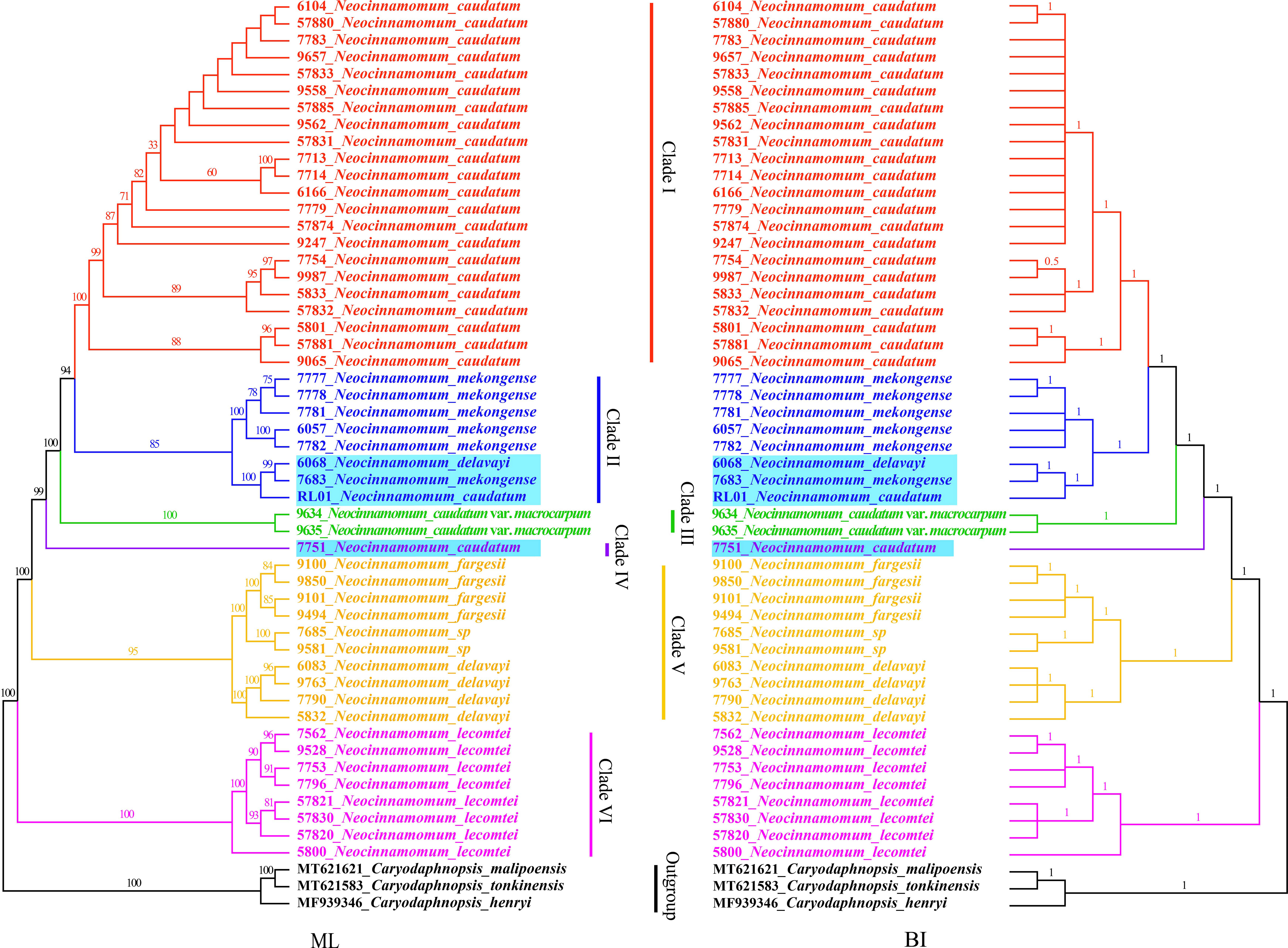
Figure 7 Phylogenetic tree constructed with Neocinnamomum chloroplast genomes.
The structure, size, and gene content of the cp genomes of the 7 Neocinnamomum taxa were relatively conserved. The 51 Neocinnamomum cp genomes exhibited a typical tetrad structure, with a length between 150,753 bp-150,956 bp and a GC content of 38.8%-38.9%. Notably, the Neocinnamomum chloroplast genome is smaller than that of most other Lauraceae genera (except for Cassytha). Song et al. (2017) found that the cp genomes of the core Lauraceae group ranged from 150,749bp to 152,739bp in length, whereas the cp genomes of the basal Lauraceae group ranged from 157,577bp to 158,530bp in length. Their analysis indicated that the core Lauraceae group lost trnI-CAU, rpl23, rpl2, and a fragment of ycf2, as well as the intergenic regions of these genes in IRb region. To a great extent, the loss of fragments in the IR region will lead to plastid contraction, which may account for the smaller Neocinnamomum cp genome. A total of 128 genes were annotated in the Neocinnamomum cp genome, including 84 protein-coding genes, 8 rRNA genes, and 36 tRNA genes. After excluding duplicates, a total of 113 unique genes were identified. These results are consistent with previous studies on Neocinnamomum cp genomes (Song et al., 2017; Tian, 2021). Overall, the cp genomes were very similar between all 7 taxa of Neocinnamomum, with no significant differences in either gene sequence or gene content.
SSR markers are widely employed in plant germplasm research because they are rich in polymorphism, strongly co-dominant, highly reproducible, stable, and reliable (Echt et al., 1996). We detected a total of 828 SSRs across 11 Neocinnamomum cp genomes, with 71-82 detected per sample. Single nucleotide repeats accounted for 74.64% of all SSRs, followed by tetranucleotide repeats (15.58%). These results differ from studies of the Lauraceae genera Litsea (Liu et al., 2021) and Ocotea (Trofimov et al., 2022), the cp genomes of which contain more dinucleotide repeats than tetranucleotide repeats. According to the principle of base complementarity, the 6 repeat types include 14 repeat motifs, among which A/T accounted for 72.71% of all SSRs, followed by AT/AT and AAAT/ATTT, which accounted for 7.85% and 7.25% of all SSRs, respectively. These results indicate that A/T are the preferred bases across the cp genomes of Neocinnamomum taxa. Studies suggest that energy consumption affects base preference. Because the nitrogen content of A/T bases is lower than that of G/C bases, A/T enrichment can make base mutations consume less energy (Niu et al., 2017). In addition to affecting the SSR types and codon bias, base bias can also affect the stability of the four partitions of the cp genome (Mukhopadhyay et al., 2007). To date, there are few published studies of SSR markers in the Neocinnamomum cp genome. The identification of SSRs in the Neocinnamomum cp genome can provide a reference for the development of molecular markers and the analysis of genetic variation within the genus Neocinnamomum.
During cp genome evolution, the boundaries of the IR region may undergo contraction and expansion. This process is central to cp genome evolution and is the primary driver of cp genome diversity among species (Park et al., 2018). Song et al. (2017) detected a double intact copy of the ycf2 gene, as well as one intact copy and one fragment of ycf1, in the plastids of basal Lauraceae group. However, only one intact copy and one fragment of both ycf2 and ycf1 were detected in the plastids of Neocinnamomum. Consistent with Song’s results, we identified one complete copy and one fragment of ycf1 and ycf2 located at the boundary between the IR region and the SC region, respectively. The ycf1 fragment at the IRb/SSC boundary and the ycf2 fragment at the IRa/LSC boundary were considered pseudogenes. Zhao et al. (2018) found that variation in the lengths of ycf1, ycf2, ψycf1, and ndhF-ψycf1 drives the contraction and expansion of the IR region in the Lindera cp genome. This is similar to Neocinnamomum, wherein the contraction and expansion of the IR region was driven by variation in the lengths ycf1, ycf2, ndhF-ψycf1, and trnH-ψycf2.
Although the cp genome is relatively conserved and exhibits a slow rate of evolution, it also contains several mutation sites such as trnH-psbA and trnQ-rps16, among other fragments (Nithaniyal and Parani, 2016; Linh et al., 2022). We performed a comparative analysis of the cp genomes of 7 Neocinnamomum taxa by combining the use of the DnaSP 6 software with the mVISTA online program. Three hypervariable regions, trnN-GUU-ndhF, petA-psbJ, and ccsA-ndhD, were identified. These hypervariable regions can be used as candidate molecular markers for the development of specific DNA barcodes to aid in the taxonomic identification of Neocinnamomum samples. Among these, the highly-variable petA-psbJ was previously identified as a variation hotspot in the Lauraceae species Litsea glutinosa, Persea americana, and Machilus chuanchienensis (Hinsinger et al., 2017; Song et al., 2016; Bai et al., 2022). The variability of the IR region was significantly lower than that of the SSC and LSC regions, and the variability of the coding regions was much lower than that of the noncoding regions, in the Neocinnamomum cp genome. This result is consistent with studies of cp genomes in other higher plants, including Hernandia nymphaeifolia (Li et al., 2020) and Saposhnikovia divaricata (Yi et al., 2022). These results also suggest that the noncoding regions evolve faster than the coding regions in the genus Neocinnamomum. Therefore, priority should be given to noncoding regions when screening DNA barcodes for Neocinnamomum species.
Neocinnamomum has been confirmed as a monophyletic by several studies. However, the phylogenetic relationships between species within the genus remain controversial. N. delavayi and N. mekongense are virtually indistinguishable in terms of morphological and molecular characteristics, and N. mekongense was once considered to be a variety of N. delavayi (Handel-Mazzetti, 1925). Wang et al. (2010), based on the phylogenetic study of psbA-trnH, trnK, and ITS, reported that N. delavayi and N. mekongense are closely related sister taxa. Li et al. (2016) used four species of Neocinnamomum in a phylogenetic study of Caryodaphnopsis based on RPB2, LEAFY, and ITS sequences, and the results supported the sister relationship between N. delavayi and N. mekongense. However, our phylogenetic analysis yielded different results. In our whole cp genome phylogenetic tree, N. delavayi was most closely related to and formed a sister group with N. fargesii and N. sp, while it was distant from N. mekongense. We speculate that the selection and combination of different gene fragments led to divergent phylogenetic trees. In addition, differences in biparental and maternal inheritance could have caused the divergent results of the ITS and cpDNA evolutionary trees (Wu, 2018). Of course, sampling bias may also lead to phylogenetic errors, and increasing the sampling size can effectively improve the overall phylogenetic accuracy (Murphy et al., 2001; Zwickl and Hillis, 2002; Dong et al., 2022a). Although the results are different, it has been recognized that the cp genome can well analyze the phylogeny of Lauraceae, and the whole cp genome is more effective than the fragment (Tian et al., 2021). N. sp, a special taxa recently collected from Wenshan, Yunnan, China, is morphologically similar to N. complanifructum, which has been merged into N. lecomtei. Our results indicated that N. sp is a sister taxon of N. fargesii. In addition, nesting was observed for 4 samples, including N. caudatum (RL01), N. caudatum (7751), N. delavayi (6068), and N. mekongense (7683), which may have been due to partial gene exchange between closely distributed species (Wu et al., 2022). The geographic distributions of N. delavayi, N. mekongense, and N. caudatum are similar, and often overlap, suggesting that hybridization may have occurred among these species. However, whether these four samples represent hybrids will require further morphological and molecular studies.
In this study, we sequenced and assembled 50 cp genomes representing 7 Neocinnamomum taxa, and obtained the cp genome data of N. caudatum (RL01) from NCBI. A total of 51 cp genomes were analyzed. Similar to most angiosperms, the Neocinnamomum cp genome exhibited a relatively conserved gene structure and gene content. Sequence variations were primarily concentrated in the LSC and SSC regions. Three hotspot regions and 71-82 SSRs were identified, which may be used as resources for DNA barcoding and molecular marker development for further genetic diversity analyses and molecular marker-assisted breeding. The whole cp genome phylogenetic tree revealed that the 51 samples representing 7 Neocinnamomum taxa were divided into six branches. Among them, N. lecomtei was the most primitive, basal taxon, and N. delavayi was found to be most closely related to N. fargesii. These results deepen our understanding of the Neocinnamomum cp genome and provide the basis for subsequent taxonomic identification, phylogenetic evolution, and population genetics studies of Neocinnamomum species.
The 50 new sequencing data presented in the study are deposited in the NCBI (https://www.ncbi.nlm.nih.gov/nuccore), accession numbers OR085909- OR085920, and OR095860- OR095897.
PX and YS designed the research study. YS and WX contributed materials. ZC annotated and analyzed the genomes, wrote the manuscript. LY isolated DNA, assembled the genomes. YX, QL, HZ and YT helped ZC to analyze the data. All authors contributed to the article and approved the submitted version.
This work was funded by Yunnan Science and Technology Talents and Platform Program (No. 202205AF150022), the National Natural Science Foundation of China (No. 32260060, 32060710), and the Special Program for Technology Bases and Talents of Guangxi (Grant No. 2022AC20002).
We thank TopEdit (www.topeditsci.com) for its linguistic assistance during the preparation of this manuscript. We also thank Xishuangbanna Tropical Botanical Garden, CAS. Genomic data processing and analyses were conducted at the High Performance Computing Cluster from the Institutional Center for Shared Technologies and Facilities of Xishuangbanna Tropical Botanical Garden.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1205051/full#supplementary-material
Bai, X., Peng, J., Yang, Y., Xiong, B. (2022). The complete chloroplast genome sequence of Machilus chuanchienensis (Lauraceae): genome structure and phylogenetic analysis. Genes (Basel) 13 (12), 2402. doi: 10.3390/genes13122402
Beier, S., Thiel, T., Münch, T., Scholz, U., Mascher, M. (2017). MISA-web: a web server for microsatellite prediction. Bioinformatics 33, 2583–2585. doi: 10.1093/bioinformatics/btx198
Cao, Z., Zhao, W., Xin, Y., Shen, W., Wang, F., Li, Q., et al. (2022). Characteristics of the complete chloroplast genome of Pourthiaea (Rosaceae) and its comparative analysis. Horticulturae 8 (12), 1144. doi: 10.3390/horticulturae8121144
Chanderbali, A. S., Werff, H., Renner, S. S. (2001). Phylogeny and historical biogeography of lauraceae: evidence from the chloroplast and nuclear genomes. Ann. Missouri Bot. Gard. 88, 104–134. doi: 10.2307/2666133
Dong, Z., Qu, S., Landrein, S., Yu, W. B., Xin, J., Zhao, W., et al. (2022a). Increasing taxa sampling provides new insights on the phylogenetic relationship between Eriobotrya and Rhaphiolepis. Front. Genet. 13. doi: 10.3389/fgene.2022.831206
Dong, Z., Zhang, R., Shi, M., Song, Y., Xin, Y., Li, F., et al. (2022b). The complete plastid genome of the endangered shrub Brassaiopsis angustifolia (Araliaceae): comparative genetic and phylogenetic analysis. PloS One 17 (6), e0269819. doi: 10.1371/journal.pone.0269819
Doyle, J. J., Doyle, J. L. (1987). A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 19 (1), 11–15.
Drouin, G., Daoud, H., Xia, J. (2008). Relative rates of synonymous substitutions in the mitochondrial, chloroplast and nuclear genomes of seed plants. Mol. Phylogenet. Evol. 49 (3), 827–831. doi: 10.1016/j.ympev.2008.09.009
Echt, C. S., May-Marquardt, P., Hseih, M., Zahorchak, R. (1996). Characterization of microsatellite markers in eastern white pine. Genome 39 (6), 1102–1108. doi: 10.1139/g96-138
Editorial Committee of Chinese Flora of China Academy of Sciences (1982). Flora of China Vol. 31 (Beijing: Science Press), 229.
Frazer, K. A., Pachter, L., Poliakov, A., Rubin, E. M., Dubchak, I. (2004). VISTA: computational tools for comparative genomics. Nucleic. Acids Res. 32, W273–W279. doi: 10.1093/nar/gkh458
Gan, Y., Song, Y., Chen, Y., Liu, H., Yang, D., Xu, Q., et al. (2018). Transcriptome analysis reveals a composite molecular map linked to unique seed oil profile of Neocinnamomum caudatum (Nees) merr. BMC Plant Biol. 18 (1), 303. doi: 10.1186/s12870-018-1525-9
Goremykin, V. V., Hirsch-Ernst, K. I., Wolfl, S., Hellwig, F. H. (2003). Analysis of the Amborella trichopoda chloroplast genome sequence suggests that Amborella is not a basal angiosperm. Mol. Biol. Evol. 20 (9), 1499–1505. doi: 10.1093/molbev/msg159
Hall, T. A. (1999). BioEdit: a user-friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nucleic. Acids Symp. Ser. 41, 95–98. doi: 10.1021/bk-1999-0734.ch008
Handel-Mazzetti, H. (1925). Cinnamomum delavayi lecomte var. mekongense. Anz Akad Wiss Wien Math-Naturwiss Kl 62, 218.
Hinsinger, D. D., Strijk, J. S. (2017). Toward phylogenomics of lauraceae: the complete chloroplast genome sequence of Litsea glutinosa (Lauraceae), an invasive tree species on Indian and pacific ocean islands. Plant Gene 9, 71–79. doi: 10.1016/j.plgene.2016.08.002
Huelsenbeck, J. P., Ronquist, F. (2001). MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17 (8), 754–755. doi: 10.1093/bioinformatics/17.8.754
Jansen, R. K., Saski, C., Lee, S. B., Hansen, A. K., Daniell, H. (2011). Complete plastid genome sequences of three rosids (Castanea, Prunus, Theobroma): evidence for at least two independent transfers of rpl22 to the nucleus. Mol. Biol. Evol. 28 (1), 835–847. doi: 10.1093/molbev/msq261
Jiangsu New Medical College (1977). Dictionary of traditional Chinese medicine (Shanghai: Shanghai People Press).
Jin, J. J., Yu, W. B., Yang, J. B., Song, Y., dePamphilis, C. W., Yi, T. S., et al. (2020). GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21 (1), 241. doi: 10.1186/s13059-020-02154-5
Katoh, K., Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30 (4), 772–780. doi: 10.1093/molbev/mst010
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28 (12), 1647–1649. doi: 10.1093/bioinformatics/bts199
Kostermans, A. J. G. H. (1974). A monograph of the genus Neocinnamomum liou ho. Reinwardtia 9, 85–96.
Lan, Z., Shi, Y., Yin, Q., Gao, R., Liu, C., Wang, W., et al. (2022). Comparative and phylogenetic analysis of complete chloroplast genomes from five Artemisia species. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.1049209
Li, J., Liu, Q., Zhang, J., Yang, Y., Zhang, S., Zhang, Y. (2020). Chloroplast genome of endangered mangrove plants Hernandia nymphaeifolia and its phylogenetic evolution. J. Northwest Forest. Univ. 35 (5), 54–61. doi: 10.3969/j.issn.1001-7461.2020.05.09
Li, L., Madriñán, S., Li, J. (2016). Phylogeny and biogeography of Caryodaphnopsis (Lauraceae) inferred from low-copy nuclear gene and ITS sequences. Taxon 65 (3), 433–443. doi: 10.12705/653.1
Linh, N. N., Hang, P. L. B., Hue, H. T. T., Ha, N. H., Hanh, H. H., Ton, N. D., et al. (2022). Species discrimination of novel chloroplast DNA barcodes and their application for identification of Panax (Aralioideae, araliaceae). PhytoKeys 188, 1–18. doi: 10.3897/phytokeys.188.75937
Liu, H. (1934). Lauracées de chine et d’Indochine: contribution à l’étude systématique et phytogéographique (Paris: Hermann et Cie).
Liu, C., Chen, H. H., Tang, L. Z., Khine, P. K., Han, L. H., Song, Y., et al. (2021). Plastid genome evolution of a monophyletic group in the subtribe lauriineae (Laureae, lauraceae). Plant Diversity 44 (4), 377–388. doi: 10.1016/j.pld.2021.11.009
Liu, C., Shi, L., Zhu, Y., Chen, H., Zhang, J., Lin, X., et al. (2012). CpGAVAS, an integrated web server for the annotation, visualization, analysis, and GenBank submission of completely sequenced chloroplast genome sequences. BMC Genomics 13, 715. doi: 10.1186/1471-2164-13-715
Liu, Q., Xue, Q. (2005). Comparative studies on codon usage pattern of chloroplasts and their host nuclear genes in four plant species. J. Genet. 84 (1), 55–62. doi: 10.1007/BF02715890
Lohse, M., Drechsel, O., Kahlau, S., Bock, R. (2013). OrganellarGenomeDRAW–a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic. Acids Res. 41, W575–W581. doi: 10.1093/nar/gkt289
Mukhopadhyay, P., Basak, S., Ghosh, T. C. (2007). Nature of selective constraints on synonymous codon usage of rice differs in GC-poor and GC-rich genes. Gene 400 (1-2), 71–81. doi: 10.1016/j.gene.2007.05.027
Murphy, W. J., Eizirik, E., Johnson, W. E., Zhang, Y. P., Ryder, O. A., O’Brien, S. J. (2001). Molecular phylogenetics and the origins of placental mammals. Nature 409 (6820), 614–618. doi: 10.1038/35054550
Neuhaus, H. E., Emes, M. J. (2000). Nonphotosynthetic metabolism in plastids. Annu. Rev. Plant Physiol. Plant Mol. Biol. 51 (51), 111–140. doi: 10.1146/annurev.arplant.51.1.111
Nguyen, L. T., Schmidt, H. A., von Haeseler, A., Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32 (1), 268–274. doi: 10.1093/molbev/msu300
Nithaniyal, S., Parani, M. (2016). Evaluation of chloroplast and nuclear DNA barcodes for species identification in Terminalia l. Biochem. Syst. Ecol. 68, 223–229. doi: 10.1016/j.bse.2016.08.001
Niu, Z., Xue, Q., Wang, H., Xie, X., Zhu, S., Liu, W., et al. (2017). Mutational biases and GC-biased gene conversion affect GC content in the plastomes of dendrobium genus. Int. J. Mol. Sci. 18 (11), 2307. doi: 10.3390/ijms18112307
Park, S., An, B., Park, S. (2018). Reconfiguration of the plastid genome in Lamprocapnos spectabilis: IR boundary shifting, inversion, and intraspecific variation. Sci. Rep. 8 (1), 13568. doi: 10.1038/s41598-018-31938-w
Posada, D. (2008). jModelTest: phylogenetic model averaging. Mol. Biol. Evol. 25, 1253–1256. doi: 10.1093/molbev/msn083
Ren, S., Song, Y., Zhao, M., Xu, W. (2019). The plastid genome sequence of Neocinnamomum delavayi (Lec.) liou. Mitochondrial. DNA B Resour 4 (2), 3711–3712. doi: 10.1080/23802359.2019.1679051
Rohwer, J. G., Rudolph, B. (2005). Jumping genera: the phylogenetic positions of Cassytha, Hypodaphnis, and Neocinnamomum (Lauraceae) based on different analyses of trnK intron sequences. Ann. Mo. Bot. Gard. 92, 153–178. doi: 10.3417/2014033
Rozas, J., Ferrer-Mata, A., Sánchez-DelBarrio, J. C., Guirao-Rico, S., Librado, P., Ramos-Onsins, S. E., et al. (2017). DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 34 (12), 3299–3302. doi: 10.1093/molbev/msx248
Shen, W., Dong, Z., Zhao, W., Ma, L., Wang, F., Li, W., et al. (2022). Complete chloroplast genome sequence of Rosa lucieae and its characteristics. Horticulturae 8 (9), 788. doi: 10.3390/horticulturae8090788
Song, Y., Yao, X., Tan, Y., Gan, Y., Corlett, R. T. (2016). Complete chloroplast genome sequence of the avocado: gene organization, comparative analysis, and phylogenetic relationships with other lauraceae. C. Can. J. For. Res. 46, 1293–1301. doi: 10.1139/cjfr-2016-0199
Song, Y., Yu, W., Tan, Y., Jin, J., Wang, B., Yang, J., et al. (2020). Plastid phylogenomics improve phylogenetic resolution in the lauraceae. J. Syst. Evol. 58 (4), 423–439. doi: 10.1111/jse.12536
Song, Y., Yu, W. B., Tan, Y., Liu, B., Yao, X., Jin, J., et al. (2017). Evolutionary comparisons of the chloroplast genome in lauraceae and insights into loss events in the magnoliids. Genome Biol. Evol. 9 (9), 2354–2364. doi: 10.1093/gbe/evx180
Tao, X., Ma, L., Nie, B., Wang, Y., Liu, Z. (2017). The draft and characterization of the complete chloroplast genome of Vicia sativa cv. langjian No.3. Pratacult. Sci. 34 (2), 321–330. doi: 10.11829/j.issn.1001-0629.2016-0054
Tian, Y. (2021). The complete chloroplast genomes of lauraceae species: comparative genomic and phylogenetic analyses (Nanjing University). doi: 10.27235/d.cnki.gnjiu.2021.001893
Tian, Y., Zhou, J., Zhang, Y., Wang, S., Wang, Y., Liu, H., et al. (2021). Research progress in plant molecular systematics of lauraceae. Biol. (Basel) 10 (5), 391. doi: 10.3390/biology10050391
Trofimov, D., Cadar, D., Schmidt-Chanasit, J., Rodrigues de Moraes, P. L., Rohwer, J. G. (2022). A comparative analysis of complete chloroplast genomes of seven Ocotea species (Lauraceae) confirms low sequence divergence within the Ocotea complex. Sci. Rep. 12 (1), 1120. doi: 10.1038/s41598-021-04635-4
Wang, Z., Li, J., Conran, J. G., Li, H. (2010). Phylogeny of the southeast Asian endemic genus Neocinnamomum h. liu (Lauraceae). Plant Syst. Evol. 290 (1), 173–184. doi: 10.1007/s00606-010-0359-1
Wang, Z., Yang, J., Tan, Y., Hu, G., Long, C. (2013). Comprehensive evaluation of woody oil-bearing plants in yunnan as sources for biodiesel. Plant Diversity Res. 35 (5), 630–640. doi: 10.7677/ynzwyj201312168
Wang, F., Zhao, W., Dong, Z., Ma, L., Li, W., Li, Z., et al. (2023). Analysis of the chloroplast genome characteristics of 6 species of Yucca. Bull. Bot. Res. 43 (1), 109–119. doi: 10.7525/j.issn.1673-5102.2023.01.012
Wariss, H. M., Yi, T., Wang, H., Zhang, R. (2017). The chloroplast genome of a rare and an endangered species Salweenia bouffordiana (Leguminosae) in China. Conserv. Genet. Resour. 10 (3), 405–407. doi: 10.1007/s12686-017-0836-8
WFO (2023) Neocinnamomum atjehense kosterm. Available at: http://www.worldfloraonline.org/taxon/wfo-0000381722 (Accessed February 23, 2023).
Wick, R. R., Schultz, M. B., Zobel, J., Holt, K. E. (2015). Bandage: interactive visualization of de novo genome assemblies. Bioinformatics 31 (20), 3350–3352. doi: 10.1093/bioinformatics/btv383
Wu, Y. (2018). The molecular phylogenetic analysis of mulberry based on the nucleus ITS and chloroplast DNA sequences (Southwest University).
Wu, L., Cui, Y., Wang, Q., Xu, Z., Wang, Y., Lin, Y., et al. (2021). Identification and phylogenetic analysis of five Crataegus species (Rosaceae) based on complete chloroplast genomes. Planta 254 (1), 14. doi: 10.1007/s00425-021-03667-4
Wu, Z. Y., Milne, R. I., Liu, J., Slik, F., Yu, Y., Luo, Y. H., et al. (2022). Phylogenomics and evolutionary history of Oreocnide (Urticaceae) shed light on recent geological and climatic events in SE Asia. Mol. Phylogenet. Evol. 175, 107555. doi: 10.1016/j.ympev.2022.107555
Xin, Y., Yu, W. B., Eiadthong, W., Cao, Z., Li, Q., Yang, Z., et al. (2023). Comparative analyses of 18 complete chloroplast genomes from eleven Mangifera species (Anacardiaceae): sequence characteristics and phylogenomics. Horticulturae 9 (1), 86. doi: 10.3390/horticulturae9010086
Xing, S. C., Liu, C. J. (2008). Progress in chloroplast genome analysis. Prog. Biochem. Biophys. 35, 21–28. doi: 10.3321/j.issn:1000-3282.2008.01.004
Xu, D. H., Abe, J., Kanazawa, A., Gai, J. Y., Shimamoto, Y. (2001). Identification of sequence variations by PCR-RFLP and its application to the evaluation of cpDNA diversity in wild and cultivated soybeans. Theor. Appl. Genet. 102 (5), 683–688. doi: 10.1007/s001220051697
Xu, W., Xia, B., Zhang, S., Chen, Z. (2017). A new variety of Neocinnamomum h. liou from guangxi, China. Guihaia 37 (7), 855–858. doi: 10.11931/guihaia.gxzw201610026
Yang, Z., Zhao, T., Ma, Q., Liang, L., Wang, G. (2018). Comparative genomics and phylogenetic analysis revealed the chloroplast genome variation and interspecific relationships of Corylus (Betulaceae) species. Front. Plant Sci. 9. doi: 10.3389/fpls.2018.00927
Yi, S., Lu, H., Wang, W., Wang, G., Xu, T., Li, M., et al. (2022). The chloroplast genome of wild Saposhnikovia divaricata: genomic features, comparative analysis, and phylogenetic relationships. Genes 13 (5), 931. doi: 10.3390/genes13050931
Zhao, M. L., Song, Y., Ni, J., Yao, X., Tan, Y. H., Xu, Z. F. (2018). Comparative chloroplast genomics and phylogenetics of nine Lindera species (Lauraceae). Sci. Rep. 8 (1), 8844. doi: 10.1038/s41598-018-27090-0
Keywords: Neocinnamomum, chloroplast genome, genome comparison, sequence characteristic, phylogenetic analysis
Citation: Cao Z, Yang L, Xin Y, Xu W, Li Q, Zhang H, Tu Y, Song Y and Xin P (2023) Comparative and phylogenetic analysis of complete chloroplast genomes from seven Neocinnamomum taxa (Lauraceae). Front. Plant Sci. 14:1205051. doi: 10.3389/fpls.2023.1205051
Received: 20 April 2023; Accepted: 29 May 2023;
Published: 07 July 2023.
Edited by:
Gao Jihai, Chengdu University of Traditional Chinese Medicine, ChinaReviewed by:
Huasheng Peng, China Academy of Chinese Medical Sciences, ChinaCopyright © 2023 Cao, Yang, Xin, Xu, Li, Zhang, Tu, Song and Xin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peiyao Xin, xpytgyx@163.com; Yu Song, songyu@gxnu.edu.cn
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.