 Xuanjiao Bai†
Xuanjiao Bai† Gang Wang
Gang Wang Jinping Han
Jinping Han
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci., 13 February 2023
Sec. Plant Bioinformatics
Volume 14 - 2023 | https://doi.org/10.3389/fpls.2023.1119041
This article is part of the Research TopicStructural Variation of the Chloroplast Genome and Related Bioinformatics ToolsView all 11 articles
Introduction: The Aristolochia, as an important genus comprised of over 400 species, has attracted much interest because of its unique chemical and pharmacological properties. However, the intrageneric taxonomy and species identification within Aristolochia have long been difficult because of the complexity of their morphological variations and lack of high-resolution molecular markers.
Methods: In this study, we sampled 11 species of Aristolochia collected from distinct habitats in China, and sequenced their complete chloroplast (cp) genomes.
Results: The 11 cp genomes of Aristolochia ranged in size from 159,375bp (A. tagala) to 160,626 bp (A. tubiflora), each containing a large single-copy (LSC) region (88,914-90,251 bp), a small single-copy (SSC) region (19,311-19,917 bp), and a pair of inverted repeats (IR) (25,175-25,698 bp). These cp genomes contained 130-131 genes each, including 85 protein-coding genes (CDS), 8 ribosomal RNA genes, and 37-38 transfer RNA genes. In addition, the four types of repeats (forward, palindromic, reverse, and complement repeats) were examined in Aristolochia species. A. littoralis had the highest number of repeats (168), while A. tagala had the lowest number (42). The total number of simple sequence repeats (SSRs) is at least 99 in A. kwangsiensis, and, at most, 161 in A. gigantea. Interestingly, we detected eleven highly mutational hotspot regions, including six gene regions (clpP, matK, ndhF, psbT, rps16, trnK-UUU) and five intergenic spacer regions (ccsA-ndhD, psbZ-trnG-GCC, rpl33-rps18, rps16-trnQ-UUG, trnS-GCU-trnG-UCC). The phylogenetic analysis based on the 72 protein-coding genes showed that 11 Aristolochia species were divided into two clades which strongly supported the generic segregates of the subgenus Aristolochia and Siphisia.
Discussion: This research will provide the basis for the classification, identification, and phylogeny of medicinal plants of Aristolochiaceae.
Aristolochia, a type genus of the family Aristolochiaceae, is widely distributed in tropical, subtropical, and temperate areas. Approximately 45 species are distributed in China, and 33 are endemic (Huang et al., 2003). Many species of Aristolochia possess a long history of medicinal value. For example, A. manshuriensis was commonly used as a traditional Chinese medicine to alleviate pathogenic fire. The dry mature fruits of A. contorta and A. debilis were called “Fructus Aristolochiae” and had been used to relieve cough and alleviate hemorrhoids. Else species such as A. fangchi, A. tagala, and A. kwangsiensis are widely used in folk medicine and are important medicinal plants. However, the outbreak of renal disease among the group of young women who followed the same slimming medicine containing A. fangchi sounds an alarm about the delayed toxic effects of Aristolochia species (Vanherweghem et al., 1993; Tomlinson et al., 2020). After decades of investigation, increasing research verified the aristolochic acid contained in the Aristolochia species was the main causative factor of nephropathy and may be the potential to cause cancer (Stefanovic et al., 2006; Jelaković et al., 2019). Hence, the Aristolochia species have been excluded from the Chinese pharmacopeia and banned to utilize for medicinal purposes in many countries (Kim and Lim, 2019). Yet the conflict between the medicinal value and potential nephrotoxicity and teratogenicity makes the illegal addition of Aristolochia in medicines and health products still rampant (Maggini et al., 2018; Ji et al., 2021). Recently, modern studies gradually discovered the new bioactivities of Aristolochia species such as insecticidal, anti-bacterial, anti-nociceptive, and anti-inflammatory effects (Kuo et al., 2011; Salome et al., 2020). Therefore, the strict supervision and accurate utilization of the Aristolochia species are important to implement the medicinal value.
Elucidating the relationships between species of genus Aristolochia is crucial for understanding and harnessing the medicinal properties of the different species. However, as a diverse genus with a large number of species distributed widely in geography, the circumscription and infrageneric classification of genus Aristolochia have been complicated and ambiguous. In the cladistic analysis based on morphological characters, many infrageneric taxa have been recognized by different authors (Ohi-Toma and Murata, 2016). For example, González et al. proposed that genus Aristolochia should be divided into three subgenera (Aristolochia, Pararistolochia and Siphisia), while Stevenson et al. indicated that the genus consisted of four genera in two subtribes Aristolochiinae and Isotrematinae (González and Rudall, 2003; Buchwalder et al., 2014; Ohi-Toma and Murata, 2016). Besides, in the Flora of China, it is also stated a controversy that some species of Aristolochia should be transferred to the genus Isotrema (Huang et al., 2003). Molecular markers are a reliable alternative that is independent of morphological feathers, enabling them to address the taxonomic challenges arising from the blurring morphological characters (Wu et al., 2020). Numerous molecular methods have been applied to Aristolochia and have advanced the understanding of the relationships of the genus Aristolochia (Wanke et al., 2006; González et al., 2014; Zhao et al., 2021). The phylogenetic trees produced with three gene sequences rbcL, phyA and matk of Aristolochia supported that Aristolochia was composed of two lineages corresponding to Aristolochiinae and Isotrematinae, respectively (Ohi-Toma et al., 2006). Based on the combined analysis using two plastid genic spacers (rps16-trnK and petB-petD) and two nuclear genes (phyA and ITS2), the phylogeny construction results confirmed that genus Aristolochia was divided into two well-supported clades representing subtribe Aristolochiinae and Isotrematinae, and Zhu et al. suggested Aristolochia subgenus Siphisia should be treated as an independent genus Isotrema (Zhu et al., 2019a). However, the results of different studies are not completely consistent, and the taxonomic systems of Aristolochia are still controversial (Wanke et al., 2006; Buchwalder et al., 2014; Ohi-Toma and Murata, 2016). With the new values and species of Aristolochia gradually published, effective methods to resolve the phylogenetic relationships and assess the previous classification of Aristolochia species are urgently needed (Yang et al., 2018; Luo et al., 2020).
With the rapid development of next-generation DNA sequencing (NGS) technologies, obtaining a complete plastome sequence has become a laboratory routine (Shi et al., 2019). The complete chloroplast (cp) genomes, as the important organelle DNA in plants, are characterized by a large size, containing richer variant site information to be an attractive tool for phylogenetic studies of plants (Niu et al., 2018). Compared with the phylogenetic analysis based on the limited phylogenetic information provided by short fragments of nuclear and cp DNA, the cp genome has significant advantages in phylogenetic resolution, particularly at low taxonomic levels (Parks et al., 2009; Wilkinson et al., 2017; Wu et al., 2021). For example, plastid genome data provided strong support for the sister relationship of sect. Macroceras and sect. Diphyllon of the genus Epimedium (Guo et al., 2022). In recent years, several cp genomes of Aristolochia have been reported (Kim and Lim, 2019; Zhao et al., 2021). The molecular structure and phylogenetic analyses of cp genomes of Aristolochia debilis and Aristolochia contorta revealed a close phylogenetic relationship with Piperaceae, Laurales, and Magnoliales (Zhou et al., 2017). Nevertheless, the compared analysis of multiple Aristolochia chloroplasts is still deficient, which is unable to comprehensively illustrate the intricate phylogenetic relationships and systematic evolution of Aristolochia.
In this study, we reported eleven complete Aristolochia cp genomes including five of subgenera Siphisia (A. fulvicoma, A. hainanensis, A. griffithii, A. kwangsiensis and A. dabieshanensis), three in subgenera Aristolochia (A. tagala, A. debilis, A. tubiflora) and another three species (A. gigantea, A. littoralis, A. neolongifolia) with unclear subgenera information. The comparative genomic analyses were conducted to explore the features and structural differentiation of the sequences. Analysis of simple sequence repeats (SSRs) could screen out potential molecular polymorphic markers for analyzing the genetic diversity and structure of Aristolochia populations in the future. Highly variable regions would provide candidate DNA barcodes for further studying Aristolochia species identification. Phylogenetic analysis performed by constructing phylogenetic trees enabled to reveal the interspecific relationship of Aristolochia species. This study enriched the valuable complete cp genome resources of Aristolochia and will contribute to further research on the identification and phylogenetic relationships within the species of the genus Aristolochia.
Eleven species of Aristolochia were newly collected from the Hainan, Yunnan, Xizang, Guizhou, and Hubei Provinces of China (Supplementary Table 1). Thereinto, referring to the Flora of China (http://www.iplant.cn/), five species (A. fulvicoma, A. hainanensis, A. griffithii, A. kwangsiensis and A. dabieshanensis) were divided into subgenus Siphisia, and other three species of A. tagala, A. debilis and A. tubiflora were recorded in subgenus Aristolochia. Besides, three species without taxonomic information on subgenus, A. gigantea, A. littoralis, and A. neolongifolia, were collected to explore the phylogeny. The 11 individuals were frozen at -80°C and the total genomic DNA was isolated from fresh leaves using the Plant Genomic DNA Kit (TIANGEN, Beijing, China) by the manufacturer’s instructions. DNA integrity was examined by electrophoresis in 1% (w/v) agarose gel and their concentration was measured using a NanoDrop 2000C spectrophotometer (Thermo Scientific; Waltham, MA, USA).
The quantified DNA was used to construct shotgun libraries with insert sizes of 300~500bp and a paired-end library was constructed by TruSeq™ Nano DNA Sample Prep Kit (Illumina, San Diego, CA, USA). Then paired-end sequencing was performed to obtain 150 bp sequences at both ends of each read according to the manufacturer’s manual for the Illumina NovaSeq platform (Illumina, San Diego, CA, USA). Low-quality regions in the original data were trimmed using the software Trimmomatic (Bolger et al., 2014). Then the clean cp reads were screened and compared with the Aristolochia sequences published at the National Centre for Biological Information. SOAPdenovo 2 was used to splice the extracted reads into several contigs (Luo et al., 2012). The assembled contigs were connected to cp genome sequences by using the NOVOPlasty (Dierckxsens et al., 2017), and gaps were filled by the GapCloser module in SOAP package. Lastly, the genes, introns and boundaries of coding regions were compared with reference sequences, A. debilis (NC036153), and assembled into complete cp genomes. Genome annotation was performed referring to the complete cp genomes of Aristolochia and corrected manually. All of the annotated genomes were deposited in GenBank with the accession numbers listed in Supplementary Table 1.
Chloroplast circular maps were drawn in OGDRAW v1.3.1 (http://ogdraw.mpimp-golm.mpg.de/) according to the adjusted genome annotation. The GC content was analyzed using MEGA (Tamura et al., 2013). The SSRs were identified by MISA software (Beier et al., 2017) with the thresholds of 10, 5, 4, 3, 3, and 3 repeat units for mono-, di-, tri-, tetra-, penta-, and hexanucleotides, respectively. To identify the long repeat motifs, REPuter (Kurtz et al., 2001) was used to locate direct, reverse, complementary and palindromic sequences, with a minimum repeat size of 30bp and Hamming distance of 3. Statistical analysis was accomplished by GraphPad Prism (GraphPad Sofware, La Jolla, CA, USA).
The whole-genome alignment for the 11 Aristolochia cp genomes was performed and plotted using mVISTA software (Dubchak and Ryaboy, 2006). Comparison of boundaries of the large single-copy (LSC), small single-copy (SSC) and two inverted repeats (IR) regions was analyzed using IRscope (Amiryousefi et al., 2018). The nucleotide diversity (Pi) of shared genes and intergenic spacers was calculated using DnaSP (Librado and Rozas, 2009). The cp genomes of the 11 Aristolochia species together with those Aristolochia species available in NCBI, which were A. bracteolata (MT130705), A. tagala (NC041455), A. debilis (NC036153), A. delavayi (MW413320), A. kaempferi (NC041452), A. mollissima (NC041457), A. kunmingensis (NC041451), A. moupinensis (NC041454), A. kwangsiensis (NC052833) and A. macrophylla (NC041453), were used for phylogenetic analyses. The cp genomes of Asarum pulchellum (MZ440306) and Piper kadsura (NC027941) were included as the outgroup to root the tree. Considering the better-supported trees yielded by protein-coding data sets, a total of 72 protein-coding genes which were shared by these species were extracted to perform ML analysis using PhyloSuite software (Zhang et al., 2020; Guo et al., 2022). The maximum-likelihood (ML) analysis was performed based on the generated data using IQ-TREE with 1000 bootstrap replicates (Nguyen et al., 2015).
The complete cp genomes of 11 Aristolochia species were all typical quadripartite structures with the total length from 159,375 bp (A. tagala) to 160,626 bp (A. tubiflora) (Figure 1; Table 1). The consisted LSC region (88914-90251 bp) and SSC region (19311-19917 bp) were separated by two inverted repeat (IR) regions (50350-51396 bp) (Table 1). The total number of unique genes annotated is from 130 to 131, comprising 85 protein-coding genes (CDS), 37-38 tRNA and 8 rRNA genes (Table 1). GC contents of the plastomes of 11 Aristolochia species ranged slightly from 38.3% to 38.8%, and the GC contents of the four regions were not balanced. The IR regions had the highest GC content (43.4-43.6%), followed by the LSC regions (36.6-37.2%) and the SSC regions (32.8-33.8%) (Supplementary Table 2). The cumulative length of CDS ranged from 77,466 (A. littoralis) to 79,074 bp (A. gigantea) and the GC contents were 38.9% to 39.2% (Table 1; Supplementary Table 2). Moreover, the GC% content of the first position was higher compared to those of the second and third positions (Supplementary Table 2).
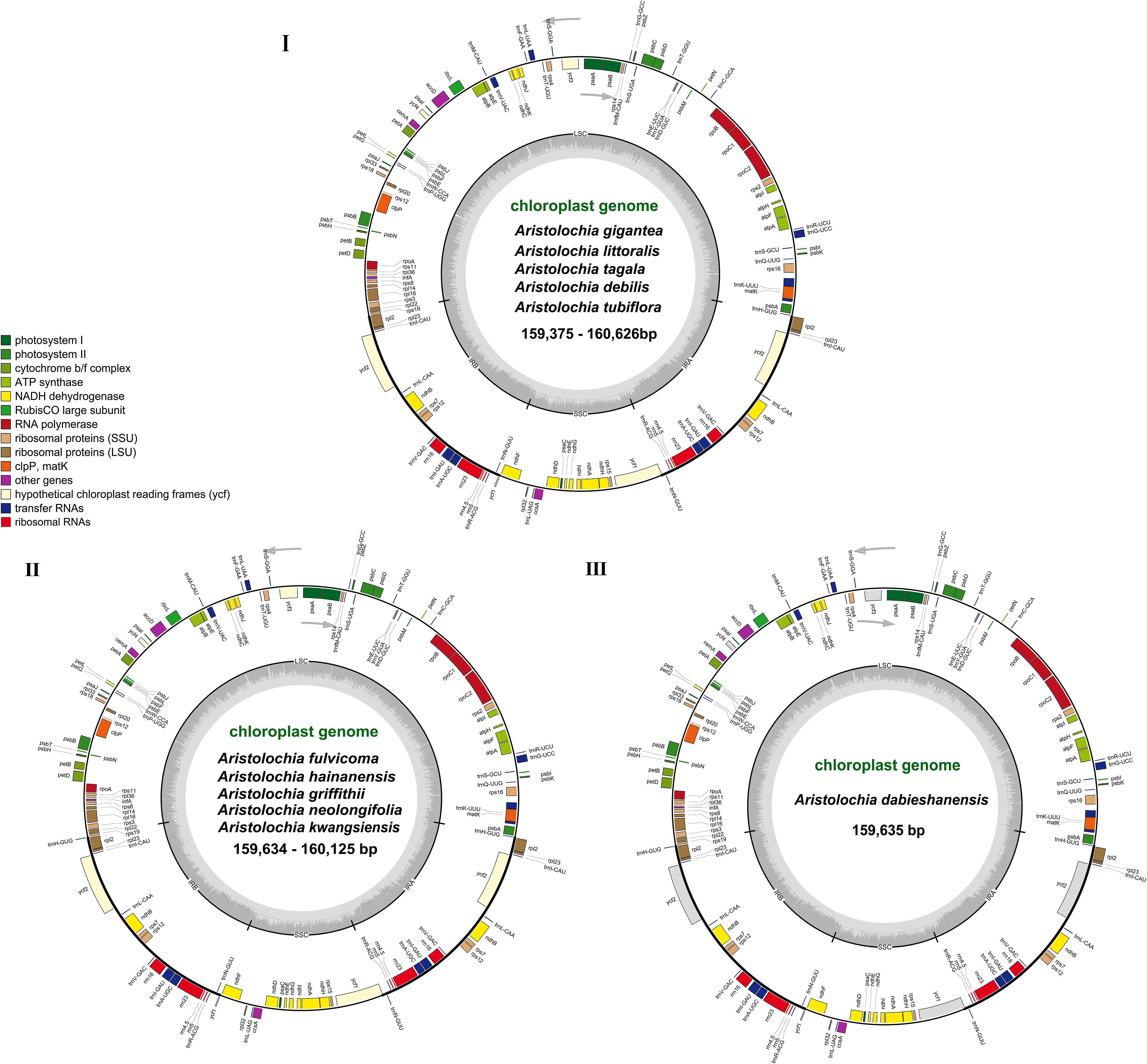
Figure 1 Gene maps of the complete cp genome of 11 species of Aristolochia. Three types of cp genome of (I) A. gigantea, A. littoralis, A. tagala, A. debilis and A. tubiflora. (II) A. fulvicoma, A. hainanensis, A. griffithii, A. neolongifolia and A. kwangsiensis. (III) A. dabieshanensis. Genes on the inside of the circle are transcribed clockwise, while those outside are transcribed counter clockwise. The darker gray in the inner circle corresponds to the GC content, whereas the lighter gray corresponds to AT content.
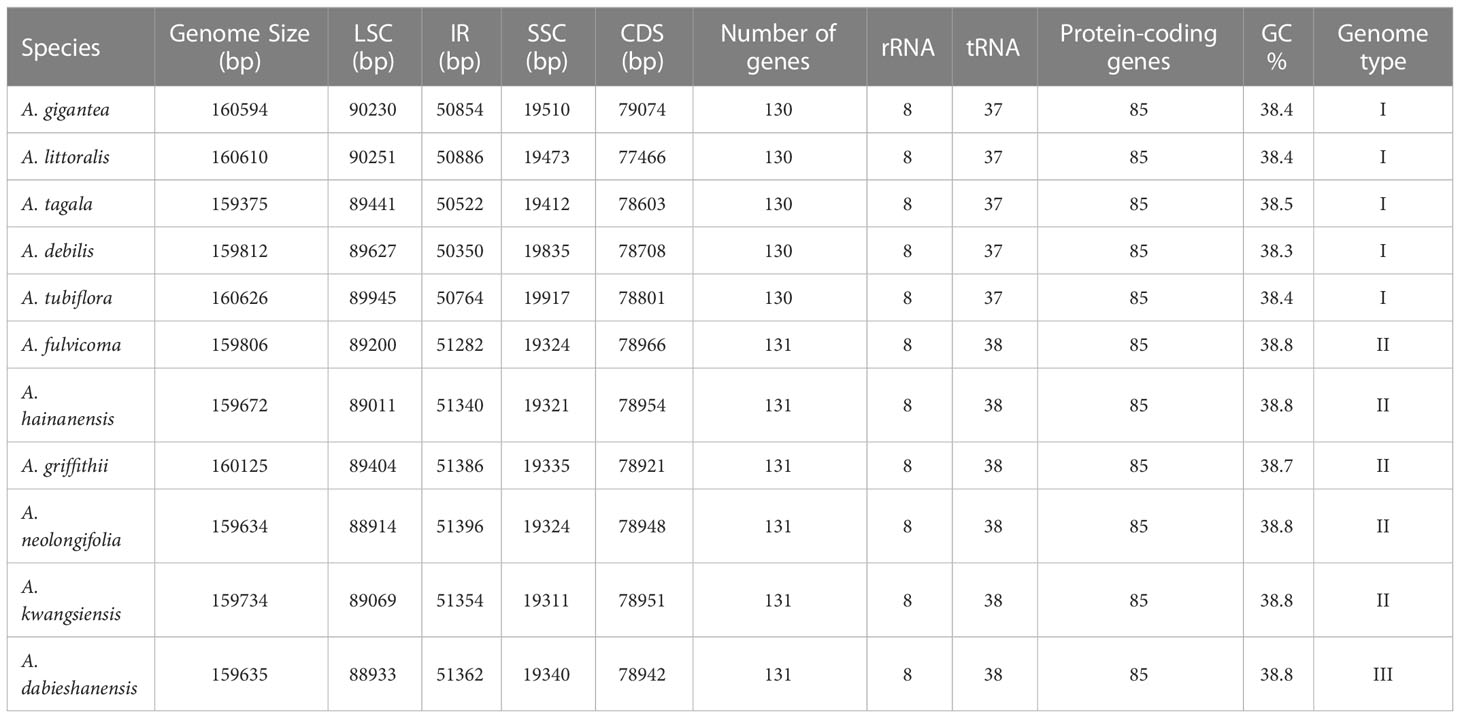
Table 1 Genome structure of complete chloroplast (cp) genomes of Aristolochia species.
A total of 817 repeats were identified in 11 Aristolochia species, including 288 reverse repeats, 260 palindromic repeats, 175 complement repeats, and 94 direct repeats (Supplementary Table 3). For each Aristolochia species, the number of repeat sequences varied greatly. A. littoralis had the largest number of repeats (168), while A. tagala had the smallest number of repeats (42). Four types of repeating motifs were detected in all 11 species (Figure 2A; Supplementary Table 3). The length of these repeats was mainly concentrated in 30-49 bp. Repeats with a length of ≥50bp only existed in A. gigantea and A. littoralis (Figure 2B; Supplementary Table 4).
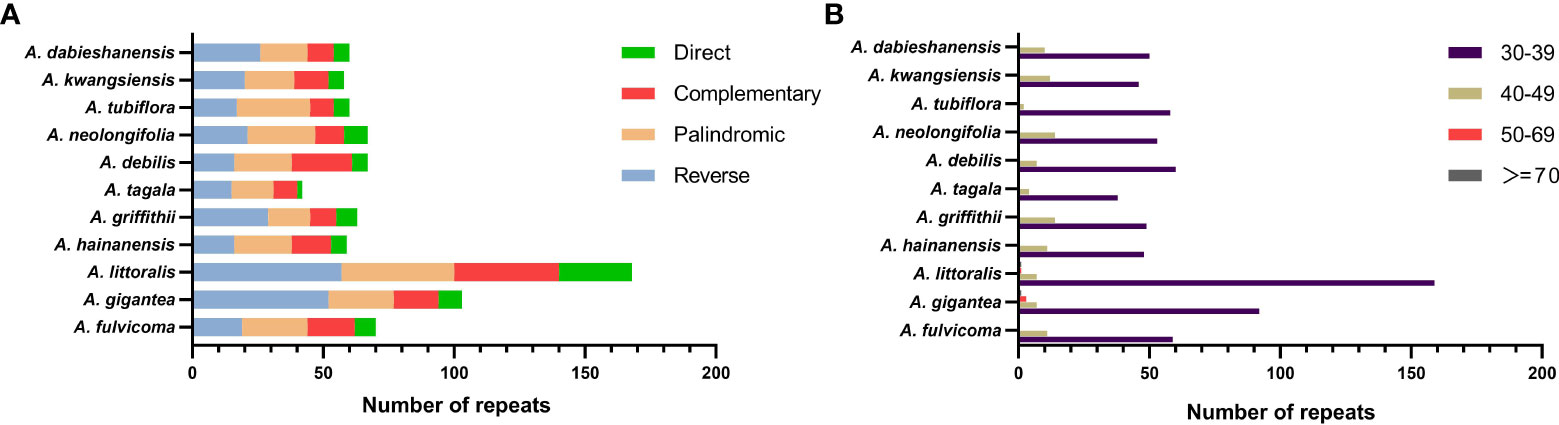
Figure 2 Long repeat sequences in the chloroplast genome of 11 species of Aristolochia. (A) Frequency of four types of repeats; (B) Length of repeat sequences.
Six kinds of SSRs were screened in the cp genomes of 11 Aristolochia species. The number of SSRs identified in 11 Aristolochia plastomes ranged from 99 in A. kwangsiensis to 161 in A. gigantea (Supplementary Table 5). In these SSRs, mono-nucleotide repeats were the largest in number, which accounted for the percent of 59.57%-72.61% in all types of SSRs (Figure 3A; Supplementary Table 5). The base composition of the repeating motifs had a certain base preference, mainly the repeating motifs rich in A-T (Supplementary Table 5). Eleven species all contained six kinds of repeat except for A. kwangsiensis and A. dabieshanensis which were without Hexa (Figure 2A; Supplementary Table 5). Regarding the SSRs distribution, these SSRs were mainly found in the LSC regions (Figure 3B; Supplementary Table 6).
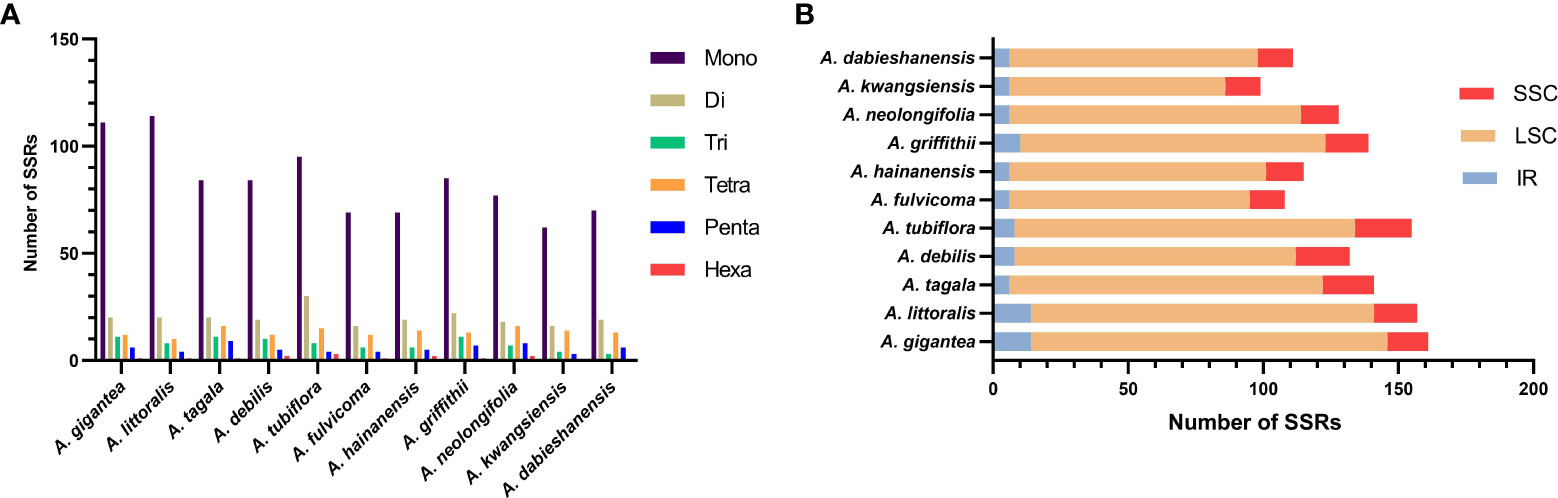
Figure 3 Amounts and distribution of SSRs in the chloroplast genome of 11 species of Aristolochia. (A) Amounts of the SSRs; (B) Distribution of SSRs.
Divergence hotspots are important for discovering DNA markers and barcodes in species identification (Kong et al., 2021). In this study, the cp genomes of 11 species of Aristolochia were compared using mVISTA with the A. debilis genome as the reference genome. Overall, the comparative genomic analysis revealed that the 11 Aristolochia cp genomes were relatively conserved. Most variations are discovered in the conserved noncoding sequences, and only a few in coding genes, such as accD, ndhF and ycf1 (Figure 4). The results indicated that the coding-gene sequences were more conserved than the noncoding sequences. Moreover, the nucleic acid variation analyses showed the intergenic spacers had more polymorphisms (average Pi=0. 04049) than the gene regions (average Pi=0.01546) (Figure 5). The highly variable regions comprised the genes regions: clpP, matK, ndhF, psbT, rps16, trnK-UUU (Pi>0.035). Among the six highly variable regions, five regions clpP, matK, psbT, rps16, and trnK-UUU were located in the LSC, and ndhF was located in the SSC. The intergenic spacer regions with high variations were screened as follows: ccsA-ndhD, psbZ-trnG-GCC, rpl33-rps18, rps16-trnQ-UUG, trnS-GCU-trnG-UCC (Pi>0.060). Among the five highly variable regions, four regions, rpl33-rps18, rps16-trnQ-UUG, psbZ-trnG-GCC, and trnS-GCU-trnG-UCC were located in the LSC, and ccsA-ndhD were located in the SSC. It was confirmed that the variations in the LSC and SSC regions were remarkably higher than those in the IR regions of cp.
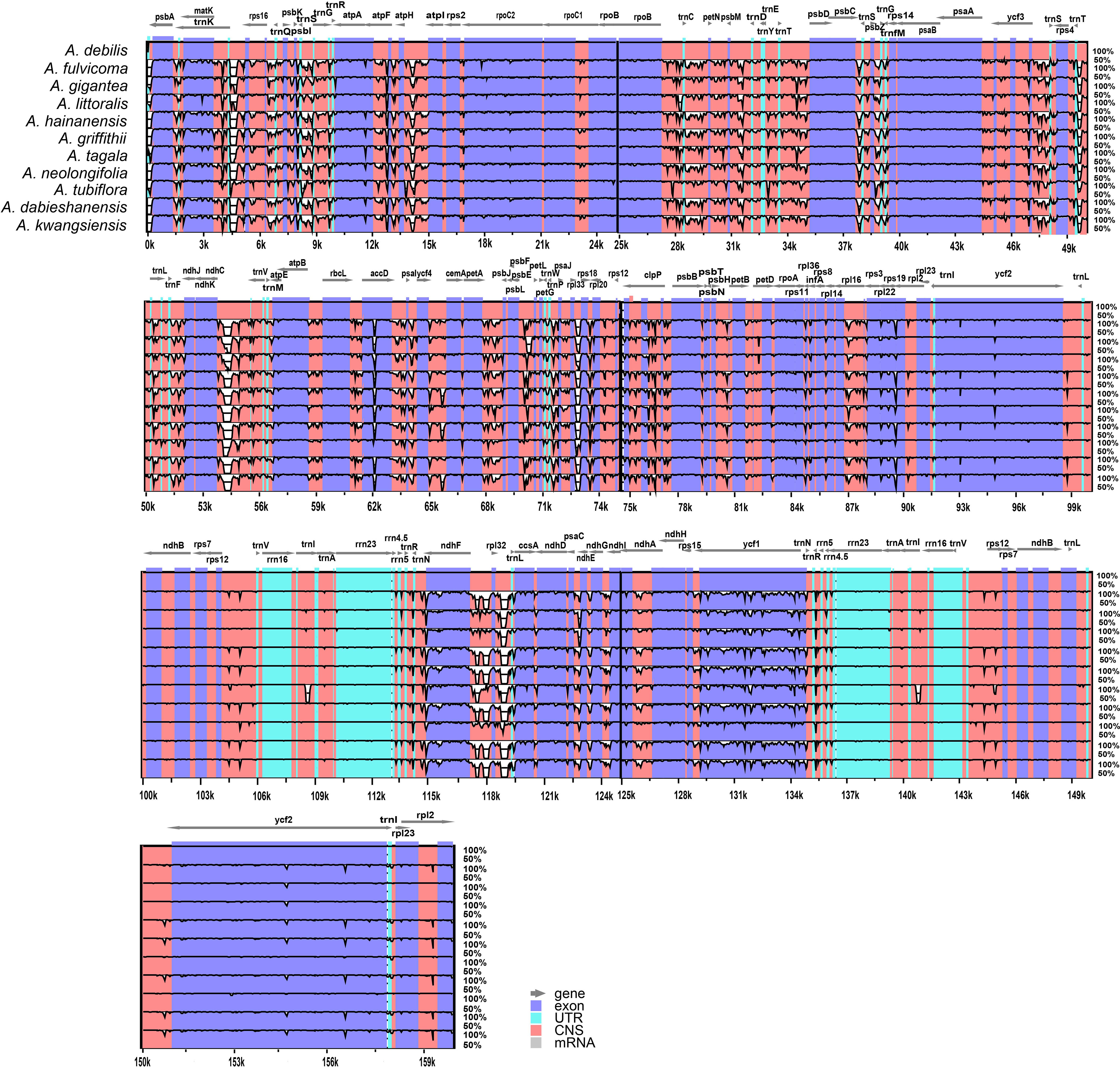
Figure 4 Sequence identity plot comparing the four chloroplast genomes of species of Aristolochia with Aristolochia debilis as a reference using mVISTA. Gray arrows and thick black lines above the alignment indicate genes with their orientation. Purple bars represent exons, blue bars represent UTRs, and red bars represent noncoding sequences (CNS). Y-scale represents the percent identity ranging from 50% to 100%.
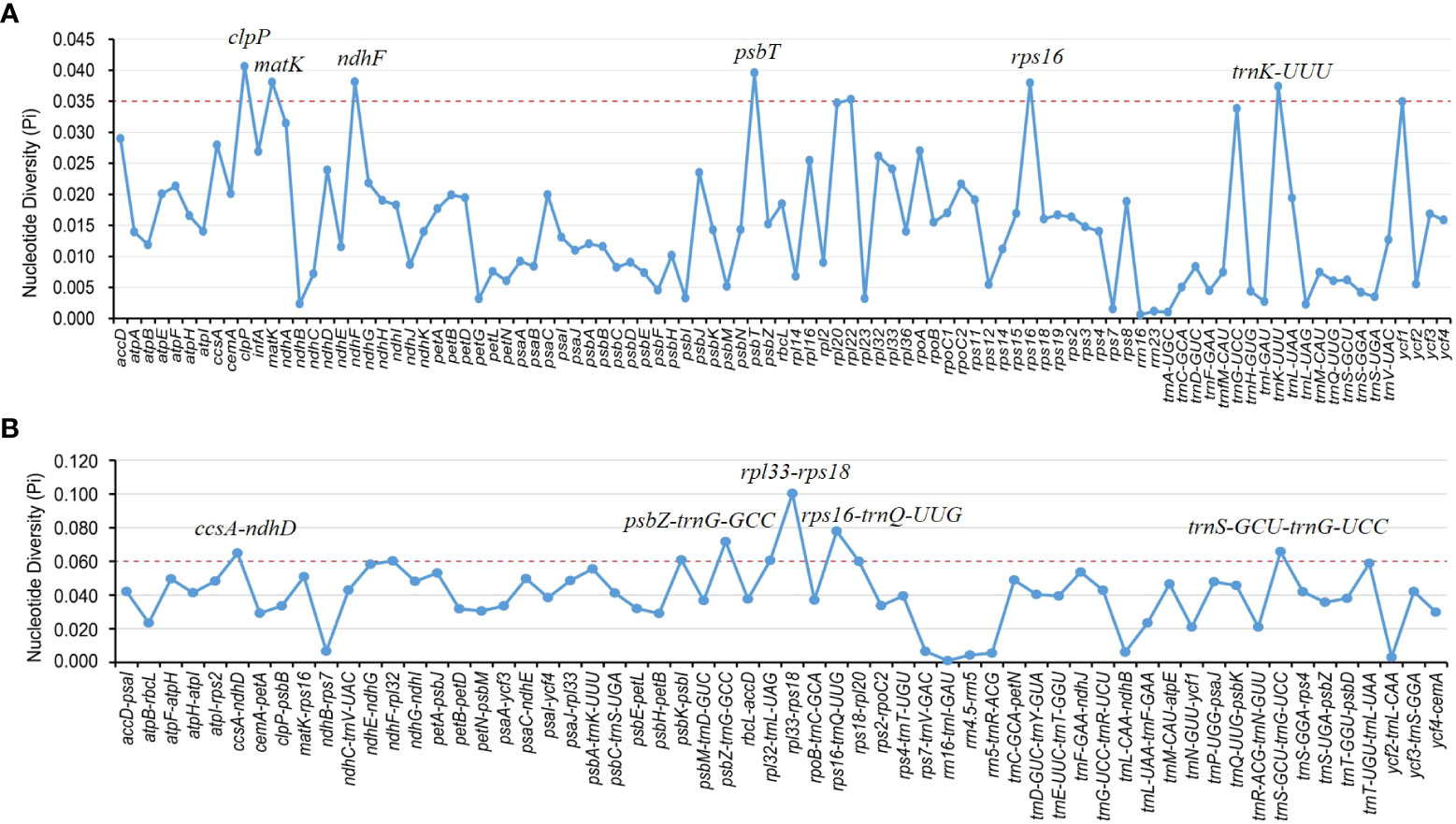
Figure 5 Nucleotide diversity (Pi) of shared various regions in 11 Aristolochia species chloroplast genomes. (A) Pi values in the genes regions. (B) Pi values in the intergenic spacers regions.
Chloroplast genomes play an important role in phylogenetic studies, and it is necessary to solve complex evolutionary relationships (Zhang et al., 2011). In our study, to obtain a more accurate analysis of the Aristolochia phylogeny, available Aristolochia genomes downloaded from NCBI were also included to construct the phylogenetic tree. A total of eighteen Aristolochia species were contained, and Asarum pulchellum and Piper kadsura served as the outgroup (Figure 6). Phylogenetic analyses using the ML method and sequences of 72 CDS strongly supported the identification of two clades among Aristolochia species, and they corresponded to subgenus Aristolochia (Clade A) and subgenus Siphisia (Clade B), as classified in Flora of China (Huang et al., 2003). Within the subgenus Aristolochia, A. gigantea and A. littoralis formed a monophyletic cluster, which was a sister to the other five Aristolochia species (A. bracteolate, A. tagala, A. delavayi, A. tubiflora and A. debilis). In subgenus Siphisia, A. macrophylla diverged first. Then A. griffithii showed a sister relationship with remaining Siphisia species. The monophyletic cluster comprising A. fulvicoma, A. kwangsiensis and A. hainanensis was a sister to the cluster composed of A. kunmingensis, A. neolongifolia and A.moupinensis, and both were sister to the other three species (A. kaempferi, A. mollissima and A. dabieshanensis).
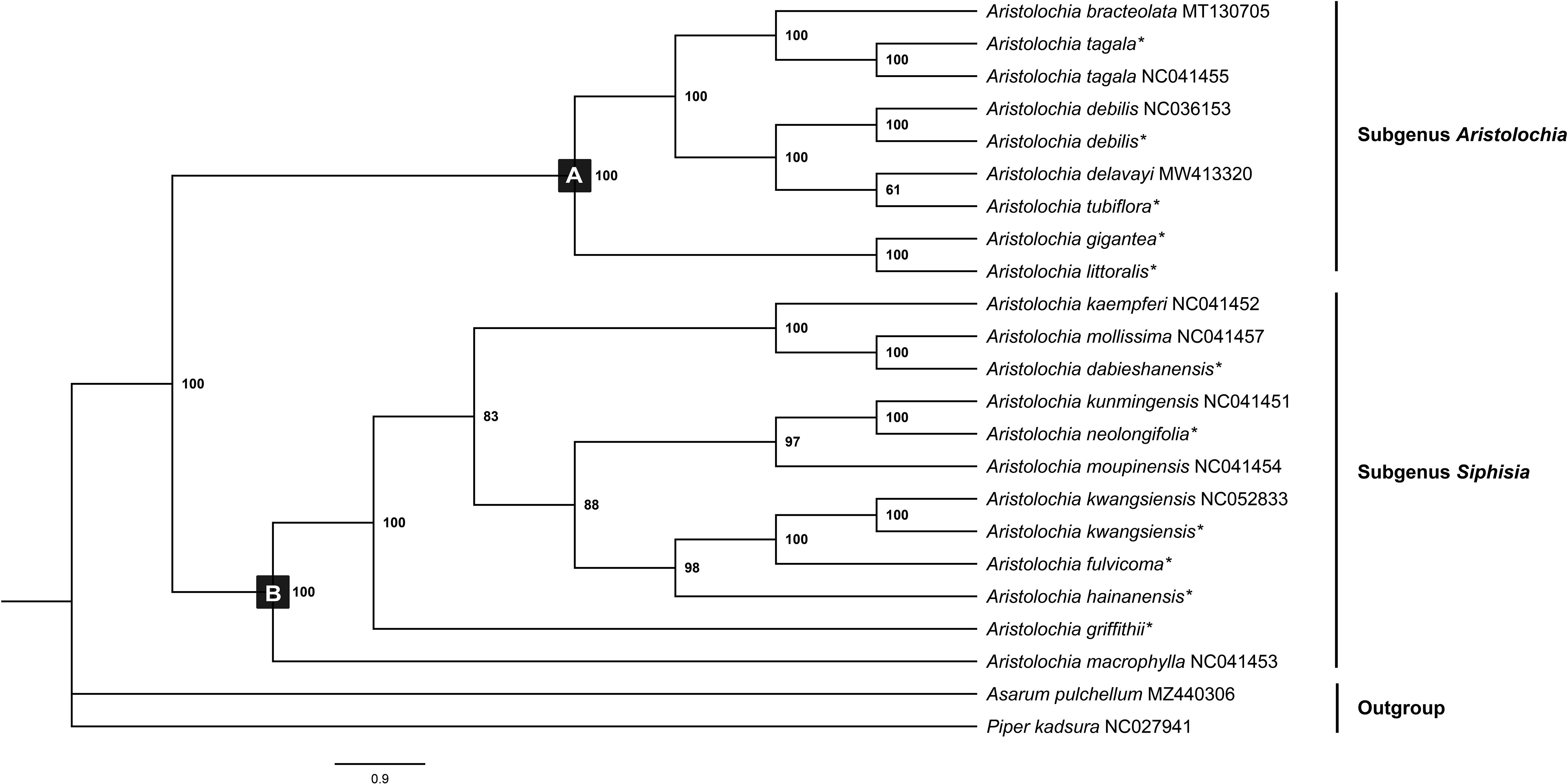
Figure 6 Phylogenetic tree inferred from the CDS of the 72 protein-coding genes of Aristolochia species using Maximum Likelihood (ML) method. The numbers near by nodes are values for bootstrap support. Species with newly sequenced chloroplast genomes are marked with asterisks.
The boundaries of IR region are hot spots for gene duplications or deletions (Yue et al., 2008). In this study, the expansion and contraction of the IR region in 11 Aristolochia cp genomes were analyzed. Results showed that all Aristolochia plastomes have the SSC/IRb boundary within the pseudogene (ψ) ycf1 gene and the SSC/IRa border within the ycf1 gene except the A. tagala which between the ycf1 and gene trnN (Figure 7). However, there were some differences in the IR/LSC border area, and three types of plastomes were characterized by IR/LSC boundary variation (Figures 1, 7). A. debilis, A. tubiflora, A. tagala, A. gigantea and A. littorali were grouped together in Clade A and classified as Type I, because the LSC-IRb border of cp genomes was located within the genic spacer of rps19-rpl2 as well as the LSC-IRa border was located within the gene trnH. Type II and III corresponded to Clade B which contained one more repeat of trnH-GUG in the IRb region and the LSC-IRb border was located within the gene rps19 (A. fulvicoma, A. hainanensis, A. griffithii, A. neolongifolia and A. kwangsiensis) or the genic spacer of rps19-trnH (A. dabieshanensis). Besides, the IRa regions of Type II and III had slightly expanded, resulting in trnH being located in the IR region, and the IRa/LSC border was located in the genic spacer of trnH-psbA. Compared with the type I, the IR region of the remaining two types of plastomes expanded approximately 0.4-1.0 k (Table 1; Figure 7).
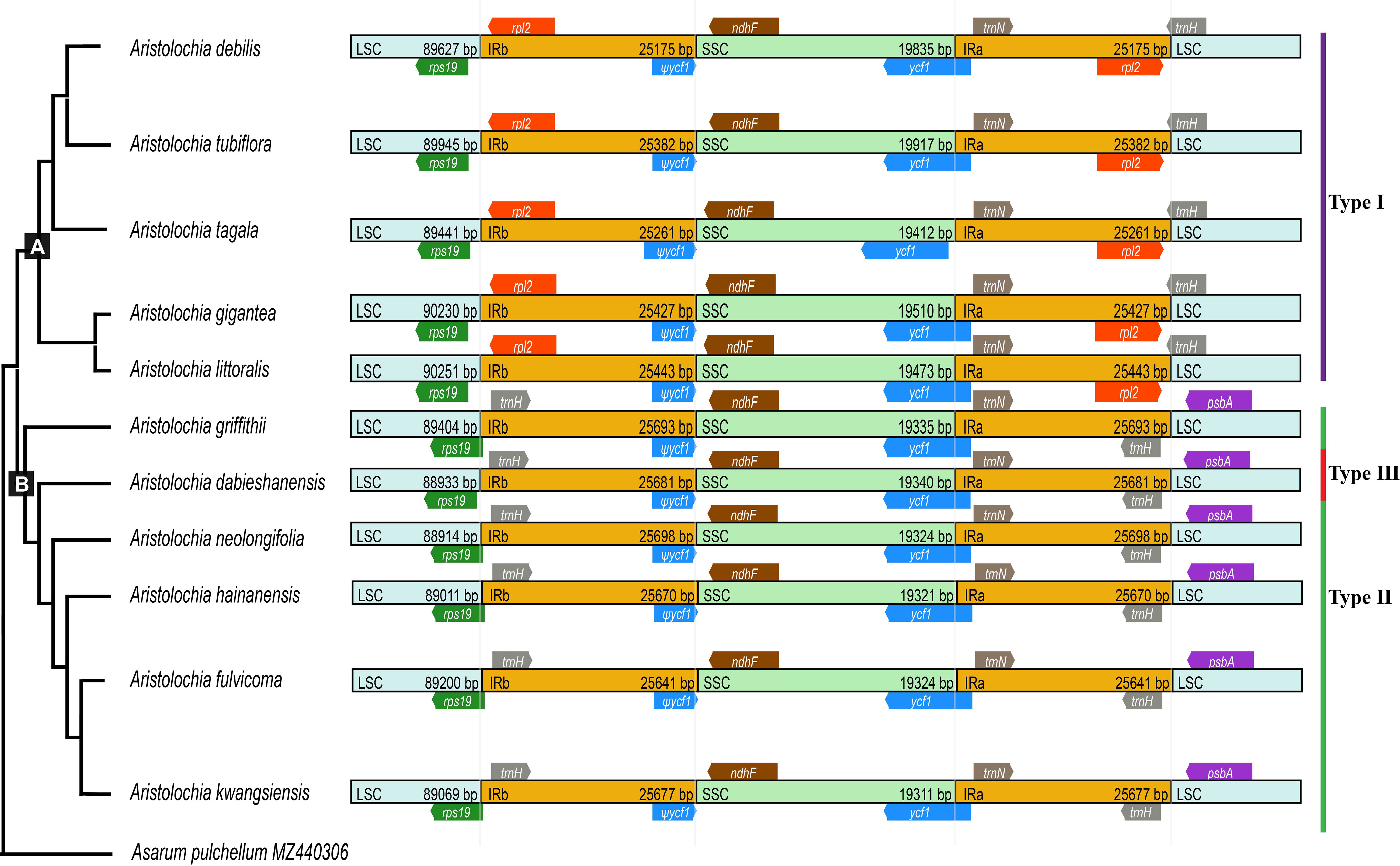
Figure 7 Comparison of boundaries of the LSC, SSC, and IR regions among 11 Aristolochia plastomes.
In this study, we reported eleven sequenced complete cp genomes of Aristolochia (Figure 1; Table 1). Our comparative analyses indicated that the overall eleven cp genomes showed a highly conserved feature in terms of structures. The GC content of eleven Aristolochia plants ranged from 38.3% to 38.8%, which was the same as that previously reported Aristolochia plastome (Zhou et al., 2017; Li et al., 2019). Besides, the IR regions had the highest GC content among the four regions of these Aristolochia species, which was consistent with most other angiosperms (Wu et al., 2020). SSRs, also known as cp microsatellites, were short tandem repeat sequences consisting of 1-6 bp nucleotides as repeating units. In all types of SSRs in this study, A or T repeats accounted for the majority, and mono-nucleotides were the predominant type. The richness of A/T in cp genomes can be explained by the easier strand separation for increasing the slipped-strand mispairing as compared to GC/CG and other tracts (George et al., 2015). Widely distributed SSRs in cp genomes provide the available molecular markers for the species of interest or closely related species (Varshney et al., 2005; Vu et al., 2020). In orchids, SSR markers were developed for recognizing valuable plants, investigating intraspecific genetic variation and reconstructing phylogeographic patterns (Tsa et al., 2014). The SSRs detected in the Aristolochia species were of great significance for the phylogenetic research and classification of Aristolochia plants. Additionally, four types of long repeat sequences were all identified in 11 Aristolochia species including direct, reverse, complement and palindromic. Most of the repeats were reversed and palindromic. These long repeat sequences were not only abundant in mutations but also very important in phylogenetic analyses (Wu et al., 2021). All the identified repeats in this study may be useful for the population genetics studies of these 11 species in the future.
Although the cp genomes among 11 Aristolochia species have a highly conserved feature, there were some small changes presented among these species on the boundary between the IR and LSC regions. Plastid genomes have been divided into three types according to the boundary of LSC/IR, which has a certain relationship corresponding to the clades of two subgenera Aristolochia and Siphisia. By comparing the length variation of IR, LSC and SSC regions among these cp genomes, we also found that the IR region of the plastomes of subgenus Siphisia expanded approximately 0.4-1.0 k to the LCS region compared to the subgenus Aristolochia (Figure 7). The expansions and contractions of the boundaries of the IR regions are considered to be the main reason for the size change of cp (Zhang et al., 2016). Besides, the deletion of one copy of trnH-GUG gene was observed in subgenus Aristolochia species, which resulted in the total of 37 tRNA genes in the species of subgenus Aristolochia and 38 in Siphisia (Table 1). A previous study also reported the loss of the trnH-GUG genes was one of the major differences between the plastomes of the two subgenera Siphisia and Aristolochia (Li et al., 2019). These sequence variations might be the result of boundary contraction and expansion between the LSC/IR regions in plants (Wang et al., 2022). Plastid genomes have the characteristics of high conservation and slow evolutionary rate, thus the special characteristics presented in their structure are often phylogenetically informative (Pascual-Diaz et al., 2021). In general, broad sampling and more evidence from the genomes will be necessary for the further understanding of the interspecies relationships of Aristolochia.
Species of Aristolochia are controversially officinal and strictly forbidden in the present. The identification of Aristolochia species is important to supervise the abuse and protect customer safety. Morphological evidence is a conventional method for plant classification and identification. However, morphological traits are easily affected by the natural environment and artificial treatment, which hardly meet the requirements of detection in practical application. DNA studies can achieve the accurate authentication of similar species within a genus based on reliable molecular evidence. Numerous DNA regions, such as the nuclear genes ITS2, and cp genes matK, rbcL, trnH-psbA and trnL-trnF, have been applied to the identification of Aristolochia species (Li et al., 2014; Dechbumroong et al., 2018). However, multiple primers were required to achieve the authentication of different Aristolochia species, and the existence of long sequence deletions or poly-A/T sequences also resulted in the difficulty of sequencing analysis (Wu et al., 2015). In this study, the results of mVISTA analysis suggested that the hypervariable intergenic regions were mostly distributed in the non-coding regions, and rarely in coding genes. Moreover, our comparative results have shown that the used cp markers appeared to be relatively low in nucleotide diversity, which may be insufficient to distinguish the species within genus Aristolochia. Thus, to achieve better species resolution, future molecular markers can focus on the more variable regions of the cp genomes, such as clpP, psbT, rps16, ycf1 and rpl33-rps18 (Chang et al., 2021).
With increasing taxon samples of Aristolochia species, our phylogenetic analyses of cp genome sequences have substantially improved the phylogenetic resolution and provided robust inference of the intraspecific relationships. In the current study, phylogenetic trees of the genus Aristolochia were constructed based on CDS sequences from a total of 18 Aristolochia species, including eleven species we sequenced and other seven downloaded from NCBI. Regarding the division of genus Aristolochia, our phylogenetic analyses have confirmed the division of two clades representing the species of subgenus Aristolochia and Siphisia, respectively. This cp phylogeny concurs well with previously published phylogenetic trees based on several nuclear/plastid regions (Zhu et al., 2019a). Compared with the phylogenetic results, it is further confirmed that the species clustered in subgenus Siphisia also could be corresponded with the Isotrema species, which is consistent with the classification based on the morphological characteristics, number of chromosomes and molecular data (Huang et al., 2003; Ohi-Toma et al., 2006; Zhu et al., 2019b). Our result provided stronger support that the subgenus Siphisia was clustered as an independent clade, and may contribute to the reinstatement of Isotrema as a new generic delimitation of Aristolochia subgenus Siphisia. In general, the phylogenetic tree conducted in this study demonstrated that the cp genomes can be used as essential evidence to resolve the intergeneric and interspecies relationships within genus Aristolochia.
In this study, the complete cp genomes of eleven species of genus Aristolochia were sequenced and compared. All of these cp genomes were obvious quadripartite structures and comparatively conserved on the length, GC content and gene content. The high variations were mostly found in LCS and SSC regions, and variable regions could serve as potential markers for species identification. Phylogenetic results indicated that the genus Aristolochia was composed of two main clades, corresponding to the division of subgenus Siphisia and subgenus Aristolochia. Moreover, combined with the analyses of IR/LSC boundaries, a whole duplication of trnH-GUG gene was observed in subgenus Siphisia, and it may be associated with the expansion of its IR region. In conclusion, this study provides an important foundation for species identification and valuable insight into the phylogenetic relationships of the Aristolochia.
The data presented in the study are deposited in the NCBI repository (https://www.ncbi.nlm.nih.gov/), and the accession numbers were OP895634, OP925753, OP950686-OP950694.
JH, XB, GW, YR and YS conceived and designed the study. XB, GW collected and analyzed the data. XB, YR and YS wrote the manuscript. All authors have directly contributed to this manuscript. All authors contributed to the article and approved the submitted version.
This study is supported by the National Key Research and Development Program of China (2019YFC1604701) and CAMS Innovation Fund for Medical Sciences, China (CIFMS, 2021-I2M-1-071).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1119041/full#supplementary-material
Amiryousefi, A., Hyvonen, J., Poczai, P. (2018). IRscope: an online program to visualize the junction sites of chloroplast genomes. Bioinformatics 34 (17), 3030–3031. doi: 10.1093/bioinformatics/bty220
Beier, S., Thiel, T., Muench, T., Scholz, U., Mascher, M. (2017). MISA-web: a web server for microsatellite prediction. Bioinformatics 33 (16), 2583–2585. doi: 10.1093/bioinformatics/btx198
Bolger, A. M., Lohse, M., Usadel, B. (2014). Trimmomatic: a flexible trimmer for illumina sequence data. Bioinf. (Oxford England) 30 (15), 2114–2120. doi: 10.1093/bioinformatics/btu170
Buchwalder, K., Samain, M.-S., Sankowsky, G., Neinhuis, C., Wanke, S. (2014). Nomenclatural updates of aristolochia subgenus pararistolochia (Aristolochiaceae). Aust. Systematic Bot. 27 (1), 48–55. doi: 10.1071/sb13042
Chang, H., Zhang, L., Xie, H., Liu, J., Xi, Z., Xu, X. (2021). The conservation of chloroplast genome structure and improved resolution of infrafamilial relationships of crassulaceae. Front. Plant Sci. 12. doi: 10.3389/fpls.2021.631884
Dechbumroong, P., Aumnouypol, S., Denduangboripant, J., Sukrong, S. (2018). DNA Barcoding of aristolochia plants and development of species-specific multiplex PCR to aid HPTLC in ascertainment of aristolochia herbal materials. PLoS One 13 (8), e0202625. doi: 10.1371/journal.pone.0202625
Dierckxsens, N., Mardulyn, P., Smits, G. (2017). NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45 (4), e18. doi: 10.1093/nar/gkw955
Dubchak, I., Ryaboy, D. V. (2006). VISTA family of computational tools for comparative analysis of DNA sequences and whole genomes. Methods Mol. Biol. (Clifton N.J.) 338, 69–89.
George, B., Bhatt, B. S., Awasthi, M., George, B., Singh, A. K. (2015). Comparative analysis of microsatellites in chloroplast genomes of lower and higher plants. Curr. Genet. 61 (4), 665–677. doi: 10.1007/s00294-015-0495-9
González, F., Rudall, P. J. (2003). Structure and development of the ovule and seed in aristolochiaceae, with particular reference to saruma. Plant Systematics Evol. 241 (3-4), 223–244. doi: 10.1007/s00606-003-0050-x
González, F., Wagner, S. T., Salomo, K., Symmank, L., Samain, M.-S., Isnard, S., et al. (2014). Present trans-pacific disjunct distribution of aristolochia subgenus isotrema (Aristolochiaceae) was shaped by dispersal, vicariance and extinction. J. Biogeography 41 (2), 380–391. doi: 10.1111/jbi.12198
Guo, M., Pang, X., Xu, Y., Jiang, W., Liao, B., Yu, J., et al. (2022). Plastid genome data provide new insights into the phylogeny and evolution of the genus epimedium. J. Adv. Res. 36, 175–185. doi: 10.1016/j.jare.2021.06.020
Jelaković, B., Dika, Ž., Arlt, V. M., Stiborova, M., Pavlović, N. M., Nikolić, J., et al. (2019). Balkan Endemic nephropathy and the causative role of aristolochic acid. Semin. Nephrol. 39 (3), 284–296. doi: 10.1016/j.semnephrol.2019.02.007
Ji, H., Hu, J., Zhang, G., Song, J., Zhou, X., Guo, D. (2021). Aristolochic acid nephropathy: A scientometric analysis of literature published from 1971 to 2019. Med. (Baltimore) 100 (27), e26510. doi: 10.1097/MD.0000000000026510
Kim, K., Lim, C. E. (2019). The complete chloroplast genome sequence of aristolochia manshuriensis kom. (Aristolochiaceae). Mitochondrial DNA B Resour 4 (2), 3515–3516. doi: 10.1080/23802359.2019.1675484
Kong, B. L.-H., Park, H.-S., Lau, T.-W. D., Lin, Z., Yang, T.-J., Shaw, P.-C. (2021). Comparative analysis and phylogenetic investigation of Hong Kong ilex chloroplast genomes. Sci. Rep. 11 (1), 5153. doi: 10.1038/s41598-021-84705-9
Kuo, P.-C., Li, Y.-C., Wu, T.-S. (2011). Chemical constituents and pharmacology of the aristolochia species. J. Traditional Complementary Med. 2 (4), 249–266.
Kurtz, S., Choudhuri, J. V., Ohlebusch, E., Schleiermacher, C., Stoye, J., Giegerich, R. (2001). REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29 (22), 4633–4642. doi: 10.1093/nar/29.22.4633
Li, M., Au, K.-Y., Lam, H., Cheng, L., But, P. P.-H., Shaw, P.-C. (2014). Molecular identification and cytotoxicity study of herbal medicinal materials that are confused by aristolochia herbs. Food Chem. 147, 332–339. doi: 10.1016/j.foodchem.2013.09.146
Li, X., Zuo, Y., Zhu, X., Liao, S., Ma, J. (2019). Complete chloroplast genomes and comparative analysis of sequences evolution among seven aristolochia (Aristolochiaceae) medicinal species. Int. J. Mol. Sci. 20 (5), 1–27. doi: 10.3390/ijms20051045
Librado, P., Rozas, J. (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25 (11), 1451–1452. doi: 10.1093/bioinformatics/btp187
Luo, R. B., Liu, B. H., Xie, Y. L., Li, Z. Y., Huang, W. H., Yuan, J. Y., et al. (2012). SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1, 1–6. doi: 10.1186/2047-217x-1-18
Luo, Y. J., Ni, S. D., Jiang, Q., Huang, B. G., Liu, Y., Huang, Y. S. (2020). Aristolochia yachangensis, a new species of aristolochiaceae from limestone areas in guangxi, China. PhytoKeys 153, 49–61. doi: 10.3897/phytokeys.153.52796
Maggini, V., Menniti-Ippolito, F., Firenzuoli, F. (2018). Aristolochia, a nephrotoxic herb, still surfs on the web, 15 years later. Intern. Emerg. Med. 13 (5), 811–813. doi: 10.1007/s11739-018-1813-2
Nguyen, L. T., Schmidt, H. A., von Haeseler, A., Minh, B. Q. (2015). IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32 (1), 268–274. doi: 10.1093/molbev/msu300
Niu, Y. T., Jabbour, F., Barrett, R. L., Ye, J. F., Zhang, Z. Z., Lu, K. Q., et al. (2018). Combining complete chloroplast genome sequences with target loci data and morphology to resolve species limits in triplostegia (Caprifoliaceae). Mol. Phylogenet Evol. 129, 15–26. doi: 10.1016/j.ympev.2018.07.013
Ohi-Toma, T., Murata, J. (2016). Nomenclature of isotrema, siphisia, and endodeca, and their related infrageneric taxa of aristolochia (Aristolochiaceae). Taxon 65 (1), 152–157. doi: 10.12705/651.11
Ohi-Toma, T., Sugawara, T., Murata, H., Wanke, S., Neinhuis, C., Murata, J. (2006). Molecular phylogeny of aristolochia sensu lato (Aristolochiaceae) based on sequences of rbcL, matK,and phyA genes, with special reference to differentiation of chromosome numbers. Systematic Bot. 31 ((3), 481–492.
Parks, M., Cronn, R., Liston, A. (2009). Increasing phylogenetic resolution at low taxonomic levels using massively parallel sequencing of chloroplast genomes. BMC Biol. 7, 84. doi: 10.1186/1741-7007-7-84
Pascual-Diaz, J. P., Garcia, S., Vitales, D. (2021). Plastome diversity and phylogenomic relationships in asteraceae. Plants (Basel) 10 (12), 1–16. doi: 10.3390/plants10122699
Salome, D. D. C., Cordeiro, N. M., Valerio, T. S., Santos, D. A., Alves, P. B., Alviano, C. S., et al. (2020). Aristolochia trilobata: Identification of the anti-inflammatory and antinociceptive effects. Biomedicines 8 (5), 1–21. doi: 10.3390/biomedicines8050111
Shi, L., Chen, H., Jiang, M., Wang, L., Wu, X., Huang, L., et al. (2019). CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 47 (W1), W65–W73. doi: 10.1093/nar/gkz345
Stefanovic, V., Toncheva, D., Atanasova, S., Polenakovic, M. (2006). Etiology of Balkan endemic nephropathy and associated urothelial cancer. Am. J. Nephrol. 26 (1), 1–11. doi: 10.1159/000090705
Tamura, K., Stecher, G., Peterson, D., Filipski, A., Kumar, S. (2013). MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30 (12), 2725–2729. doi: 10.1093/molbev/mst197
Tomlinson, T., Fernandes, A., XXXP.Grollman, P. (2020). Aristolochia herbs and iatrogenic disease: The case of portland's powders. Yale J. OF Biol. AND Med. 93, 355–363.
Tsa, C.-C., Wu, P.-Y., Kuo, C.-C., Huang, M.-C., Yu, S.-K., Hsu, T.-W., et al. (2014). Analysis of microsatellites in the vulnerable orchid gastrodia flavilabella: the development of microsatellite markers, and cross-species amplification in gastrodia. Botanical Stud. 55, 72.
Vanherweghem, J., Depierreux, M., Tieiemans, C., Abramowicz, D., Dratwa, M., Jadoul, J., et al. (1993). Rapidly progressive interstitial renal fibrosis in young women: association with slimming regimen including Chinese herbs. Lancet 341 (8842), 387–391.
Varshney, R. K., Graner, A., Sorrells, M. E. (2005). Genic microsatellite markers in plants: features and applications. Trends Biotechnol. 23 (1), 48–55. doi: 10.1016/j.tibtech.2004.11.005
Vu, H. T., Tran, N., Nguyen, T. D., Vu, Q. L., Bui, M. H., Le, M. T., et al. (2020). Complete chloroplast genome of paphiopedilum delenatii and phylogenetic relationships among orchidaceae. Plants (Basel) 9 (1), 1–27. doi: 10.3390/plants9010061
Wang, Y., Wen, F., Hong, X., Li, Z., Mi, Y., Zhao, B. (2022). Comparative chloroplast genome analyses of paraboea (Gesneriaceae): Insights into adaptive evolution and phylogenetic analysis. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.1019831
Wanke, S., Gonza´lez, F., Neinhuis, C. (2006). Systematics of pipevines: combining morphological and fast-evolving molecular characters to investigate the relationships within subfamliy aristolochioideae (Aristolochiaceae). Int. J. Plant Sci. 167 (6), 1215–1227.
Wilkinson, M. J., Szabo, C., Ford, C. S., Yarom, Y., Croxford, A. E., Camp, A., et al. (2017). Replacing Sanger with next generation sequencing to improve coverage and quality of reference DNA barcodes for plants. Sci. Rep. 7, 46040. doi: 10.1038/srep46040
Wu, L., Nie, L., Wang, Q., Xu, Z., Wang, Y., He, C., et al. (2021). Comparative and phylogenetic analyses of the chloroplast genomes of species of paeoniaceae. Sci. Rep. 11 (1), 1–10, 14643. doi: 10.1038/s41598-021-94137-0
Wu, L., Nie, L., Xu, Z., Li, P., Wang, Y., He, C., et al. (2020). Comparative and phylogenetic analysis of the complete chloroplast genomes of three paeonia section moutan species (Paeoniaceae). Front. Genet. 11. doi: 10.3389/fgene.2020.00980
Wu, L., Sun, W., Wang, B., Zhao, H., Li, Y., Cai, S., et al. (2015). An integrated system for identifying the hidden assassins in traditional medicines containing aristolochic acids. Sci. Rep. 5 (1), 1–10. doi: 10.1038/srep11318
Yang, B., Ding, H. B., Zhou, S. S., Zhu, X., Li, R., Maw, M. B., et al. (2018). Aristolochia sinoburmanica (Aristolochiaceae), a new species from north Myanmar. PhytoKeys 94), 13–22. doi: 10.3897/phytokeys.94.21557
Yue, F., Cui, L., dePamphilis, C. W., Moret, B. M., Tang, J. (2008). Gene rearrangement analysis and ancestral order inference from chloroplast genomes with inverted repeat. BMC Genomics 9 Suppl 1, S25. doi: 10.1186/1471-2164-9-S1-S25
Zhang, D., Gao, F., Jakovlic, I., Zou, H., Zhang, J., Li, W. X., et al. (2020). PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour 20 (1), 348–355. doi: 10.1111/1755-0998.13096
Zhang, Y., Du, L., Liu, A., Chen, J., Wu, L., Hu, W., et al. (2016). The complete chloroplast genome sequences of five epimedium species: Lights into phylogenetic and taxonomic analyses. Front. Plant Sci. 7. doi: 10.3389/fpls.2016.00306
Zhang, Y. J., Ma, P. F., Li, D. Z. (2011). High-throughput sequencing of six bamboo chloroplast genomes: phylogenetic implications for temperate woody bamboos (Poaceae: Bambusoideae). PloS One 6 (5), e20596. doi: 10.1371/journal.pone.0020596
Zhao, J., Yue, X. L., He, Z. R., Zhou, X. M. (2021). The complete chloroplast genome of endangered species aristolochia delavayi franch. (Aristolochiaceae) in southwestern China. Mitochondrial DNA B Resour 6 (8), 2339–2341. doi: 10.1080/23802359.2021.1931506
Zhou, J., Chen, X., Cui, Y., Sun, W., Li, Y., Wang, Y., et al. (2017). Molecular structure and phylogenetic analyses of complete chloroplast genomes of two aristolochia medicinal species. Int. J. Mol. Sci. 18 (9), 1–15. doi: 10.3390/ijms18091839
Zhu, X.-X., Li, X.-Q., Liao, S., Du, C., Wang, Y., Wang, Z.-H., et al. (2019a). Reinstatement of isotrema, a new generic delimitation of aristolochia subgen. siphisia (Aristolochiaceae). Phytotaxa 401 (1), 1–23. doi: 10.11646/phytotaxa.401.1.1
Keywords: Aristolochia, taxonomy, phylogenetic relationship, comparative analysis, chloroplast genome
Citation: Bai X, Wang G, Ren Y, Su Y and Han J (2023) Insights into taxonomy and phylogenetic relationships of eleven Aristolochia species based on chloroplast genome. Front. Plant Sci. 14:1119041. doi: 10.3389/fpls.2023.1119041
Received: 08 December 2022; Accepted: 20 January 2023;
Published: 13 February 2023.
Edited by:
Weijun Kong, Capital Medical University, ChinaReviewed by:
Zhichao Xu, Northeast Forestry University, ChinaCopyright © 2023 Bai, Wang, Ren, Su and Han. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jinping Han, aGFwcHlteXJhMjAwN0AxNjMuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.