
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 15 September 2022
Sec. Plant Pathogen Interactions
Volume 13 - 2022 | https://doi.org/10.3389/fpls.2022.991077
 Peng Yan1†
Peng Yan1† Jiawen Yu1†Xinmei Fang1Shuying Li1Shan Han1Tiantian Lin1Yinggao Liu1
Jiawen Yu1†Xinmei Fang1Shuying Li1Shan Han1Tiantian Lin1Yinggao Liu1 Chunlin Yang1Fang He1Tianhui Zhu1*
Chunlin Yang1Fang He1Tianhui Zhu1* Shujiang Li1,2*
Shujiang Li1,2*Arthrinium phaeospermum is the main pathogen that causes Bambusa pervariabilis × Dendrocalamopsis grandis blight. It secretes the cutinase transcription factor ApCtf1β, which has been shown to play an important role in B. pervariabilis × D. grandis virulence. However, knowledge about the interaction target genes of ApCtf1β in B. pervariabilis × D. grandis remains limited. A cDNA library for the yeast two-hybrid system was constructed from B. pervariabilis × D. grandis shoots after 168 h treatment with A. phaeospermum. The library was identified as 1.20 × 107 cfu, with an average insert >1,000 bp in size and a 100% positive rate, providing a database for the subsequent molecular study of the interaction between A. phaeospermum and B. pervariabilis × D. grandis. The yeast two-hybrid (Y2H), bimolecular fluorescence complementation (BiFC), and glutathione-S-transferase (GST) pull-down assays were used to screen for and identify two ApCtf1β interacting target proteins, BDUbc and BDSKL1, providing a reliable theoretical basis to study the molecular mechanism underlying B. pervariabilis × D. grandis resistance in response to A. phaeospermum, which would, in turn, establish a platform to develop new strategies for the sustainable and effective control of the blight diseases of forest trees.
Hybrid bamboo (Bambusa pervariabilis × Dendrocalamopsis grandis) is widely used as greenery, paper making and bamboo weaving to produce bamboo shoots and as an ornamental. It has also been introduced in the Yangtze River basin to build ecological barriers (Zhao et al., 2004; Li et al., 2020). An infestation of A. phaeospermum has recently caused extensive dieback of hybrid bamboos (Zhu et al., 2009). Our previous studies were mainly focused on investigating the physiology of disease development and biological characteristics of pathogenic fungi, determining the virulence of pathogenic toxin proteins, isolating and screening antagonistic fungi, as well as the hybrid bamboo blight fungus genome and the transcriptome, proteome, and metabolome of hybrid bamboo induced by pathogen infestation and toxin proteins (Li and Zhu, 2012; Li et al., 2013a,b). However, the molecular mechanism by which Bambusa pervariabilis × Dendrocalamopsis grandis responds to A. phaeospermum is not yet understood.
Pathogenic fungi can cause a wide range of plant diseases. Their hyphae can directly penetrate the plant’s epidermal cells or form special infestation structures (e.g., adherent spores) that invade the plant to extract nutrients. Plants have evolved an elaborate, cell-based immune system with innate and induced defenses to escape pathogenic fungi (Dangl and Jones, 2001; Ausubel, 2005; Jones and Dangl, 2006; Dodds and Rathjen, 2010). The earliest known mechanism of plant-pathogen interactions was the gene-for-gene hypothesis proposed by Flor in the mid-twentieth century (Flor, 1942). Following advances in research and accumulated experimental evidence, the zigzag model is now widely recognized and accepted by the academic community (Jones and Dangl, 2006). The study of the molecular mechanism by which pathogenic fungi infest hosts is centered on the functional regulatory network of pathogenic key genes. The key genes related to the development of the pathogenic fungus infesting structure and pathogenicity are explored using gene knockout and backfill overexpression technologies and phenotype analysis of mutants. The cloning and functional study of key pathogenicity genes interacting within the plant immune system can help resolve the host molecular resistance mechanism (Zhang et al., 2015). Therefore, it is essential to screen for and identify target proteins involved in fungi-host interactions and clarify the interaction mechanism between target protein function and pathogenic effector genes to resolve the molecular mechanisms underlying fungal pathogenicity and plant disease resistance. The yeast two-hybrid (Y2H) assay has been widely used to determine protein–protein interactions in organisms (Chen et al., 2012). Furthermore, constructing cDNA libraries is a popular methodology in molecular biology. By combining cDNA libraries and yeast two-hybrid technology, we can study the interactions between known proteins and target proteins more conveniently, which is important for studying function-specific proteins. Currently, the yeast two-hybrid system has been used to screen for target proteins that interact with pathogenic fungi on host plants such as rice (Oryza sativa L.; Singh et al., 2014), Arabidopsis (Arabidopsis thaliana; Napoli et al., 2021), potato (Solanum tuberosum L
The cuticle is a biopolyester on the surface of higher plants, whose main roles include controlling the water exchange between plants and the environment, regulating plant body temperature, preventing mechanical damage, and defending against pathogens. The cuticle is the first barrier against pathogenic infestation. The cuticle also involved in signal production and transmission during plant development and plant-pathogen interactions. The study of cutinase originated from the search for the mechanism of plant pathogens, where pathogenic fungus secrete cutinase to break down the cuticle when invading the plant epidermis (Li et al., 2002; Wang et al., 2016), invade the interior, and eventually affect plant growth. Transcriptional activation of cutinase genes in both Fusarium solani and Aspergillus nidulans is mediated by the cutinase transcription factor CTF1 (Li and Kolattukudy, 1997; Li et al., 2002). In Fusarium solani f. sp. pisi (Rogers et al., 1994), Curvularia lunata (Liu et al., 2016), and other plant pathogens, deletion of the gene encoding cutinase reduces virulence or eliminates pathogenicity to the host plant (Davies et al., 2000; Li et al., 2003; Zhang et al., 2005; Skamnioti and Gurr, 2007; Marino et al., 2012; Liu et al., 2016; Wang et al., 2016). However, in other studies, cutinase knockout mutants did not lose pathogenicity (Chasan, 1992; Stahl and Schäfer, 1992; Sweigard et al., 1992).To date, several studies have demonstrated that cutinases are also associated with the triggering of host-derived signals, fungal spore attachment, and carbon acquisition during saprophytic growth (Köller, 1991; Deising et al., 1992; Kolattukudy et al., 1995; Skamnioti and Gurr, 2007; Cheung et al., 2009; Järvinen et al., 2009; Auyong, 2015). Cutinases are essential for spore adhesion in Uromyces viciae-fabae and Colletotrichum graminicola (Deising et al., 1992; Pascholati et al., 1992) and may also play a role in inducing defense responses (Parker and Köller, 1998; Zhang et al., 2014). For example, in Sclerotinia sclerotiorum, SsCut acts as a PAMP to induce defense responses in several host plants. Degradation of the plant epidermis by cutinases secreted by fungal pathogens may lead to the release of DAMPs (De Lorenzo et al., 2011).
In our previous study, the cutinase transcription factor ApCtf1β was confirmed to be the key virulence factor of A. phaeospermum in the stress response of B. pervariabilis × D. grandis (Fang et al., 2021). However, few studies have investigated the screening and functions of cutinase transcription factors in host plants, especially in B. pervariabilis × D. grandis, thus severely limiting our understanding of the pathogenesis of A. phaeospermum and the molecular mechanism of the response of bamboo to its infection. In the present study, we constructed a yeast library of hybrid bamboo infested with A. phaeospermum and screened the ApCtf1β reciprocal protein using a yeast two-hybrid assay. Verification was via GST pull-down and fluorescent bimolecular complementation assays. The findings of this study will provide a reliable theoretical basis for the study of the A. phaeospermum pathogenic pathway and the molecular mechanism underlying B. pervariabilis × D. grandis resistance in response to pathogenic infection, establishing a platform for developing new strategies for sustainable and effective control of wilting blight diseases in forest trees.
Arthrinium phaeospermum was isolated from diseased B. pervariabilis × D. grandis using the tissue isolation method. The accession number for A. phaeospermum whole genome information in the GenBank database is QYRS0000000.1. The isolate was stored at the China Forestry Culture Collection Center (reference number: cfcc 86,860; http://www.cfcc-caf.org.cn/, Accessed on April 6, 2007).
One-year-old B. pervariabilis × D. grandis (bamboo) seedlings purchased from Shuyang Qichen Bamboo Seedling Co., Ltd. (Suqian, China) and Nicotiana benthamiana (tobacco) seedlings purchased from Huayueyang Biotech Co., Ltd. (Beijing, China) were planted in the greenhouse of Sichuan Agricultural University, Chengdu, Sichuan, China at a temperature of 25°C and humidity of 60%. The accession number for dual-seq of B. pervariabilis × D. grandis infected by A.phaeospermum in the NCBI database is SAMN19312317.
Arthrinium phaeospermum was inoculated onto PDA (containing 200 g of potato, 20 g of glucose, and 20 g of agar per liter) and incubated at 25°C for 5 days. Small wounds were created at the shoot nodes of B. pervariabilis × D. grandis via needle pricking, inoculated with a 5-mm fungal cake, and covered with film to retain moisture. Total RNA was extracted using TRIzol (Beijing TransGen Biotech Co., Ltd., China) after 168 h of A. phaeospermum treatment. Residual DNA was removed using DNase I digestion enzyme, and total RNA concentration and purity were determined using a spectrophotometer. RNA integrity was detected via 1% agarose gel electrophoresis—a 28S:18S ratio of 2:1 indicated suitable RNA integrity, and an A260/A280 ratio between 1.8 and 2.0 indicated suitable RNA purity. mRNA was isolated and purified using the Oligotex mRNA Midi Kit(QIAGEN-CN, Shanghai, China). The quality and concentration of mRNA were detected using 1% agarose gel and a spectrophotometer (NANODROP, ThermoFisher Scientific-CN, Shanghai, China), respectively.
mRNA obtained from the previous step was reverse-transcribed to obtain the first strand of cDNA using the CloneMiner II cDNA library construction kit according to the manufacturer’s instructions. Next, first-strand cDNA was used as the PCR template to synthesize the second strand of cDNA. The cDNA was ligated with three frames of attB recombinant connectors (one copy of each of the three connectors), separated, and collected. Approximately 500–4,000 bp of double-stranded cDNA was collected and ligated into the vector via the BP recombination reaction. The recombination products were electrotransformed into Escherichia coli DH10B receptor cells, and the cDNA library was obtained by culturing E. coli in a shaking incubator. After 1-h incubation, 10 μl of the transformed E. coli stock solution was diluted 100 times, and 50 μl of the diluted suspension was aspirated and coated with LB medium (50 mg/L Amp) to identify library capacity. Twenty-four single clones were randomly selected from the colonies grown on the medium and identified via colony PCR. Their insert fragment length and recombination rate were analyzed. The remaining bacterial broth was mixed with sterile glycerol to a final concentration of 20% and stored in liquid nitrogen at −80°C to be used as the primary library broth. Library volume identification was calculated as follows: cfu/ml = clones on medium/50 μl × dilution × 1 × 103 μl. Total library CFU = cfu/ml × total volume of library broth (ml).
The validated primary library was added to the corresponding resistant LB medium and incubated overnight at 30°C in a shaker. The plasmids were extracted, their OD measured, and their presence detected via electrophoresis at the end of the culture period. The extracted plasmid was diluted to 300 ng/μl and mixed with the pGADT7-DEST vector for LR recombination reaction. The reaction product was electrotransformed into E. coli DH10B receptor cells, and the transformed product was inoculated into a new 15-ml centrifuge tube and incubated for 1 h in a shaker at 37°C and 250 r/min. The culture was coated with medium, and 24 single clones were randomly picked from the resulting colonies. The clones were identified via colony PCR and analyzed for insert length and recombination rate. Sterile glycerol was added to the remaining bacterial broth to a final concentration of 20%, and the mixture was stored at −80°C in liquid nitrogen to be used as the secondary library broth.
The Y187 yeast strain was transformed with 5 μg of secondary library plasmid, and 100 μl of dilutions (at 1:10, 1:100, 1:1,000, and 1:10,000) were spread on 150 mm SD/−Leu mediums. The medium were inverted and incubated at 30°C until clones appeared (at 3–6 days). Transformants were collected to determine the titer of the library, and 24 clones were picked for PCR identification using the T7SP primer and 3′AD primer. Electrophoresis was performed to determine the size of inserted cDNA fragments.
According to the manufacturer’s instructions, the total RNA of A. phaeospermum was extracted using the TransZol Up RNA Extraction Kit (TransGen). The extracted products were tested for purity and integrity via agarose electrophoresis and microspectrophotometry. According to the manufacturer’s instructions, RNA that met the target requirements was reverse-transcribed to synthesize cDNA for backup using the All-in-One First-Strand cDNA Synthesis Super Mix for PCR (TransGen a). The homologous primers ApCtf1β-F/ApCtf1β-R (Supplementary Table S1) were designed based on the ApCtf1β1 and ApCtf1β2 genes (MK789640 and MK789641) in the NCBI database. PCR amplification was performed using A. phaeospermum cDNA as the template. The PCR products were detected via 1% agarose gel electrophoresis, recovered, and ligated to the cloning vector pEASY-Blunt Zero (TransGen) to obtain the recombinant vector. The ligated products were transformed into receptor cells DH5α. Positive transformants were picked following verification via colony PCR and sent to Tsingke for sequencing to confirm that the target genes were not mutated.
The homologous recombinant primers of ApCtf1β were designed (Supplementary Table S1). The cloned plasmid successfully sequenced in the previous step was used as a template for the PCR reaction, and the PCR products were recovered. The pGBKT7 vector was recovered via overnight digestion at 37°C using the restriction endonuclease XmaI. The recovered ApCtf1β fragment was ligated with the linearized pGBKT7 vector at 50°C for 15 min using the Trelief™ SoSoo Cloning Kit (Tsingke) with homologous recombinase and transferred into E. coli receptor DH5α for colony PCR identification. Plasmids were extracted from the positive clones using magnetic beads (Shan et al., 2011) and sent to Tsingke for sequencing to confirm that the target genes were not mutated.
The yeast receptor state Y2HGold was prepared for the decoy vector toxicity assay, self-activation assay, and subsequent library screening experiments using the standard PEG/LiAc method (Chen et al., 2009). Plasmids containing the decoy vector (pGBKT7-ApCtf1β), negative control (pGBKT7), positive control (pGBKT7-53 + pGADT7-T), and negative control (pGBKT7-Lam + pGADT7-T) were also transferred into Y2H Gold receptor cells using the PEG/LiAc method. Reactions were constructed according to Table 1 and cultured at 30°C for 5–7 days. Growth was observed to determine the presence of self-activation and toxic effects.
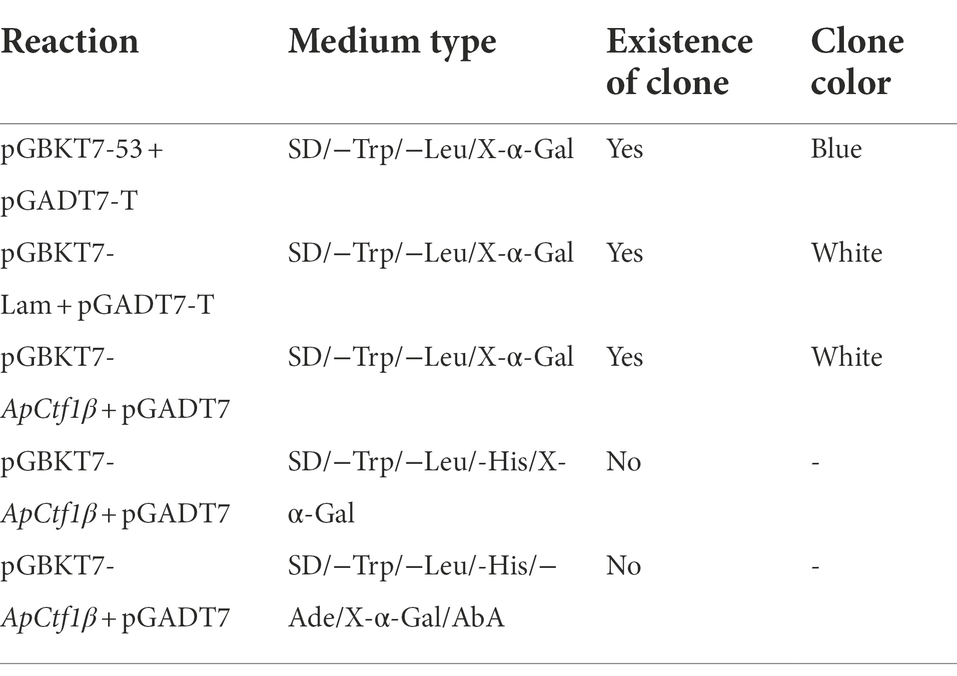
Table 1. Self-activation and toxicity assay reaction constructs.
The extracted pGBKT7-ApCtf1β plasmid was transferred into the yeast receptor cells Y2HGold. Yeast monoclonal clones that grew normally in SD/−Trp medium were selected for verification via PCR. Single colonies of Y2HGold yeast strain transformed with the recombinant plasmid were picked and inoculated in 50 ml of SD/−Trp liquid medium. Y2H Gold yeast solution (5 ml) transformed with the bait gene and 1 ml of B. pervariabilis × D. grandis cDNA library (Y187) were combined in a 2 L conical flask. A 45 ml volume of 2 × YPDA was added, and the mixture was incubated at 30°C and 40 r/min for 24 h. The bacteria were centrifuged at 1,000 × g for 10 min in a 50 ml centrifuge tube, collected, and resuspended in 10 ml of 0.5× YPDA solution (Kana concentration at 50 μg/ml). Next, the bacteria were coated with SD/−Trp/−Leu/X-α-Gal/AbA (DDO/X/A) and SD/−Trp/−Leu/-His/X-α-Gal/AbA (TDO/X/A) separately and cultured on/AbA (TDO/X/A)-deficient medium. After approximately 3–5 days, the blue yeast monoclones were selected onto SD/−Trp/−Leu/-His/−Ade/X-α-Gal/AbA (QDO/X/A) plates for further screening. Positive clones growing on QDO/X/A were sequenced and verified one-to-one.
Plasmid DNA was extracted from the blue colonies growing on the QDO/X/A medium using the yeast plasmid DNA extraction kit. The extracted plasmid DNA was transferred into E. coli DH5α receptor cells and coated with ampicillin medium to screen the bacteria transferred into pGADT7. The plasmid sequences were compared with data from the NCBI database, and the gene information represented by the plasmid sequences was clarified. The clones most likely related to the function of ApCtf1β were selected and validated based on the gene information. To exclude false positives and obtain proteins with definite interactions with ApCtf1β, candidate proteins screened from the library were constructed on the pGADT7 vector and then co-transferred into yeast cells coated with QDO/X/A medium and with the decoy plasmid. We observed whether and to what extent the yeast colonies turned blue to verify the interaction further.
Specific primers were designed based on the sequences of BDUbc, BDNADP-ME, BDWGA-3, BDSKL1, and BDGolS2 genes in the NCBI database (SAMN19312317; Supplementary Table S1). PCR amplification was performed using B. pervariabilis × D. grandis cDNA as a template. The PCR products were detected via 1% agarose gel electrophoresis, and the PCR products were recovered and ligated to the cloning vector pEASY-Blunt Zero to obtain the recombinant plasmids. The ligated products were transformed into receptor cells DH5α. The positive transformants were picked after correct verification via colony PCR and sent to Tsingke. We designed pSPYNE (R)173-ApCtf1β, pSPYCE (M)-BDUbc, pSPYCE (M)-BDNADP-ME, pSPYCE (M)-BDWGA-3, pSPYCE (M)-BDSKL1, and pSPYCE (M)-BDGolS2 homologous recombination primers (Supplementary Table S1). PCR reactions were performed using the cloned plasmids successfully sequenced in the previous step as templates, and the PCR products were recovered. The pSPYNE (R)173 and pSPYCE (M) vectors were recovered following overnight digestion with the restriction endonuclease BamHI at 37°C. The recovered ApCtf1β fragment was ligated with linearized pSPYNE (R)173 using homologous recombinase. BDUbc, BDNADP-ME, BDWGA-3, BDSKL1, and BDGolS2 fragments were ligated with the linearized pSPYCE (M) vector at 50°C for 15 min, transferred into E. coli receptor cells DH5α, and subjected to colony PCR for identification. The plasmids were extracted from the positive clones and sent to Tsingke for sequencing. The sequences were compared with the correct ones to confirm that the target genes were not mutated.
Nicotiana benthamiana plants grown at 25°C and incubated for approximately 30 days were selected for Agrobacterium infiltration experiments. The constructed BiFC recombinant vector was transformed into Agrobacterium tumefaciens GV3101 (pSoup-19) using the freeze–thaw method (Weigel and Glazebrook, 2006). The monoclonal Agrobacterium containing the target vector was picked into 5 ml of LB medium containing the corresponding antibiotics and cultured until the logarithmic phase of Agrobacterium growth was reached (OD600 = 0.5–0.6). Bacteria were collected following centrifugation at 5,000 rpm for 10 min at 25°C, resuspended in an immersion solution (containing 10 mM MgCl2, 10 mM MES, and 150 μM acetosyringone; pH = 5.6) to an OD600 of 1.0, and left to rest at room temperature for 2–3 h. Equal volumes of two bacteriophages containing different plasmids were mixed. A small opening was made on the back of a N. benthamiana leaf using a 1-mm needle. The bacteriophage was aspirated using the needle and injected into the leaf at the wound site. The water-stained areas of the tobacco leaves were marked. After the infiltrated plants were incubated at approximately 21°C for 2 days, observations were made under laser confocal microscope (FV3000, Olympus, Tokyo, Japan), 488 nM excitation and 509 nM emission filters were used. The genes bZIP63 and bZIP1, which have been shown to interact under laboratory conditions, were used as positive controls. The experimental setup is shown in Table 2.
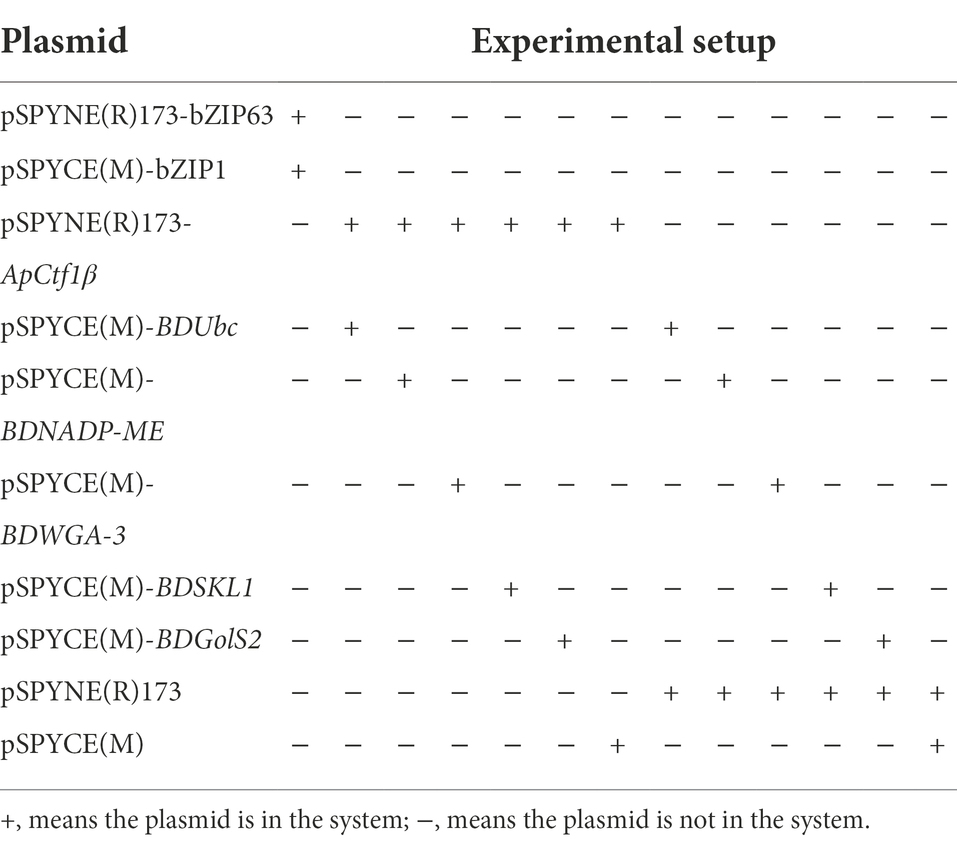
Table 2. BiFC experimental setup.
The homologous recombinant primers for PGEX-6P-1-ApCtf1β, pET28a-BDUbc, and pET28a-BDSKL1 were designed (Supplementary Table S1), and the PCR reaction was performed using the cloned plasmids successfully sequenced in the previous step as templates. The PGEX-6P-1 and pET28a vectors were recovered following overnight digestion with the restriction endonuclease BamHI at 37°C. The recovered ApCtf1β fragment was ligated with the linearized PGEX-6P-1 vector, while BDUbc and BDSKL1 fragments were ligated with the linearized vector pET28a at 50°C for 15 min. The plasmids were transferred into E. coli receptor DH5α for colony PCR identification. The positive clones were extracted from the plasmids, sent to Tsingke for sequencing, and compared with the correct sequences to confirm that no mutations were found in the target gene.
Primary nuclear expression and purification of GST-ApCtf1β, His-BDUbc, and His-BDSKL1 fusion proteins were performed according to the method described by Xu (2020). The eluate containing purified proteins was concentrated and dialyzed, and 10-μl samples were subjected to SDS-PAGE.
The GST pull-down experiment was performed according to the method described by Xu. The Glutathione-agarose resin was first treated with GST-tagged protein mixed with glutathione-agarose resin homogenate for the control group and GST-ApCtf1β fusion protein mixed with glutathione-agarose resin homogenate for the experimental group. Next, His-BDUbc or His-BDSKL1 fusion proteins were added to the control and experimental groups, respectively. The supernatants were collected following overnight incubation with agitation. Finally, the supernatant was separated by SDS-PAGE, and the protein bands were transferred onto PVDF membranes via the wet transfer method for immunoblotting.
Blast1 was used for sequence homology comparisons, and ExPASy’s ProtParam tool2 was used for the physicochemical property (isoelectric point, molecular weight) analysis of proteins, Predictprotein3 was used for secondary structure prediction of the protein encoded by the gene, and SWISS-MODEL4 was used for tertiary structure prediction of the protein encoded by this gene. MEGA 7 was used for amino acid sequence homology and phylogenetic analysis, and a neighbor-joining evolutionary tree was constructed with 1,000 replicates and under all other default settings.
Bambusa pervariabilis × D. grandis shoots inoculated with A. phaeospermum for 0, 8, 24, 72, 120, 168, and 240 h were used as starting material. RNA was extracted and reverse transcribed to cDNA. Real-time fluorescent quantitative primers qBDUbc-F/R and qBDSKL1-F/R were designed with reference to the BDUbc and BDSKL1 sequences (Supplementary Table S1). Three software programs, geNorm, Normfinder, and BestKeeper, were used to statistically analyze the expression stability of candidate reference genes (Li et al., 2021) in the B. pervariabilis × D. grandis after infestation by A. phaeospermum (Primers were shown in Supplementary Table S1; Vandesompele et al., 2002; Silver et al., 2006; Xie et al., 2012). To determine the optimal number of reference genes, the GeNorm program was used to determine whether increasing the number of reference genes could improve the stability of the normalization factor by calculating the pairwise variation values Vn/Vn + 1 of different candidate genes using the rank order normalization factors NFn and NFn + 1 based on the stability of different gene expression (Vandesompele et al., 2002). TransScript Green One-Step qRT-PCR SuperMix (TransGen) was used for qRT-PCR, with three reactions for each treatment group. The mean values were calculated, and the data were analyzed using the 2-ΔΔCt method.
mRNA analysis showed a homogeneous diffuse elastic distribution of the bands in the range of 500–2,000 bp, demonstrating that the quality of the mRNA bands of the validated cDNA library was well maintained without chemical degradation (Figure 1). Double-stranded (ds) cDNA bands showed a diffuse homogeneous longitudinal distribution of the structure over a wide length range, indicating that the structural integrity of the cDNA bands was satisfactory and the quality performance was high (Supplementary Figure S1). A total of 1,400 monoclonal clones were counted on medium, and the total library capacity was 1.12 × 107 CFU (Figure 1A). The 24 monoclonal clones randomly selected for colony PCR were successfully amplified, and the average length of the amplified fragments was greater than 1,000 bp, indicating that the cDNA insertion rate in the primary library was 100% (Figure 1B). A total of 1,500 monoclonal clones were counted on medium, the secondary library titer was calculated to be 3.00 × 106 cfu/ml (Figure 1C), and the total library capacity was 1.20 × 107 CFU. The 24 monoclonal clones randomly selected for colony PCR were successfully amplified, and the average length of the amplified fragments was greater than 1,000 bp, indicating that the insertion rate of cDNA in the secondary library was 100% (Figure 1D). The results showed that the constructed library capacity was large enough to meet the library requirements for screening the reciprocal proteins. A total of 800 monoclones were counted on the medium showed that the yeast working solution titer was 8.00 × 107 cfu/ml (Figure 1E). The 24 monoclones randomly selected for colony PCR were successfully amplified with a 100% positive rate (Figure 1F). This yeast library can be used for downstream yeast two-hybrid screening.
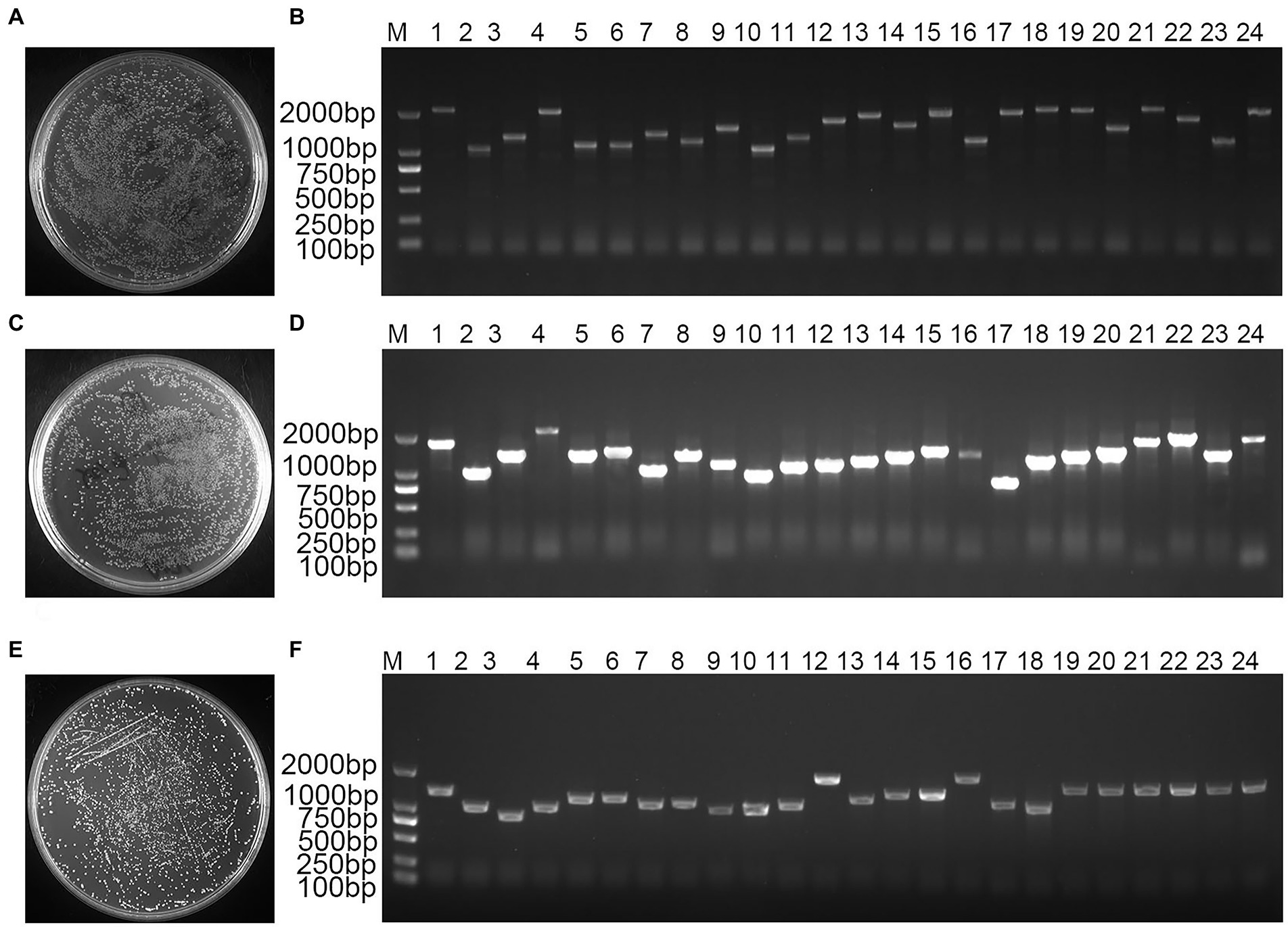
Figure 1. Bambusa pervariabilis × Dendrocalamopsis grandi cDNA library quality assay. (A) Primary library titer assay at 100-fold dilution; (B) PCR detection of primary library insert cDNA fragments, 1–24: 24 randomly selected single clones from the primary library; (C) secondary library titer assay at 100-fold dilution; (D) PCR for secondary library insert cDNA fragment, 1–24: 24 randomly selected monoclonal clones from secondary library; (E) Yeast working solution library titer assay at 100-fold dilution; and (F) PCR for yeast working solution insert library fragment, 1–24: 24 randomly selected monoclonal clones from yeast working solution; M: DL2000 DNA Marker.
The ApCtf1β gene sequence was amplified from the A. phaeospermum cDNA and was 1,236 bp in length. pGBKT7-ApCtf1β was obtained by homologous recombination of the ApCtf1β fragment with pGBKT7. The plasmid was sequenced, verified positive and without base mutations (Supplementary Figure S2).
The result showed that the positive control strain grew on the QDO/X/A medium and was blue, indicating that it could activate the AbA and X-α-Gal reporter genes; therefore, the positive control experiment was successful (Supplementary Figures S3 A,B). The negative control strain grew well on the DDO medium but failed to grow on the QDO/X/A medium, indicating that it could not activate the AbA and X-α-Gal reporter genes; therefore, the negative control experiment was successful (Supplementary Figures S3 C,D). The decoy pGBKT7-ApCtf1β and pGADT7 grew on DDO/X medium and were blue, indicating that the pGBKT7-ApCtf1β plasmid was successfully transferred into the yeast strain. The normal size of the transformed yeast cells indicated that the decoy plasmid was not toxic. pGBKT7-ApCtf1β grew blue on TDO/X but not on QDO/X/A, indicating that pGBKT7-ApCtf1β had slight self-activation but can be inhibited under QDO/X/A screening conditions. It can thus be used for subsequent screening experiments that include QDO/X/A screening (Supplementary Figures S3 E–G).
The results showed that yeast broth produced approximately 144 positive clones on the QDO/X/A medium (Supplementary Figures S4 A,B). The blue clones on the QDO/X/A screening plate were picked and transferred to new QDO/X/A screening plates for further screening. A total of 122 clones were obtained growing blue (Supplementary Figures S4 C,D), and PCR products were sequenced. All sequencing results are shown in Supplementary Table S2. The sequencing results were combined to exclude duplicates, shifts, and clones with non-coding regions. Seven significant sequences were screened: BDUbc (ubiquitin-conjugating enzyme), BDNADP-ME(NADP-dependent malic enzyme), BDWGA-3(agglutinin isolectin 3), BDSKL1(shikimate kinase-like protein 1), BDBBTI(Bowman-Birk type bran trypsin inhibitor-like), BDnsLTP1(non-specific lipid transfer protein-like 1), and BDGolS2(galactinol synthase 2).
The results showed that yeast strains grew normally on the DDO medium but not on the QDO/X/A medium, indicating that none of these genes were self-activated (Figure 2A).The positive control can grow normally and turn blue on DDO/X/A, while the negative control cannot grow on DDO/X/A, indicating the success of the control experiment. pGBKT7-ApCtf1β + pGADT7-BDUbc, pGBKT7-ApCtf1β + pGADT7-BDNADP-ME, pGBKT7-ApCtf1β + pGADT7-BDWGA-3, pGBKT7-ApCtf1β + pGADT7-BDSKL1, and pGBKT7-ApCtf1β + pGADT7-BDGolS2 could grow blue clones on QDO/X/A medium. pGBKT7-ApCtf1β + pGADT7-BDBBTI and pGBKT7-ApCtf1β + pGADT7-BDnsLTP1 did not grow blue clones when transformed into QDO/X/A medium, proving that the reporter gene was not activated (Figure 2B). Therefore, ApCtf1β can interact with BDUbc, BDNADP-ME, BDWGA-3, BDSKL1, and BDGolS2; however, there is no interaction with BDBBTI and BDnsLTP1.

Figure 2. (A) Self-activation validation of positive candidate gene from gradient dilution spot medium; pGBKT7 + pGADT7-BDUbc, pGBKT7 + pGADT7-BDNADP-ME, pGBKT7 + pGADT7-BDWGA-3,pGBKT7 + pGADT7-BDSKL1,pGBKT7 + pGADT7-BDBBTI, pGBKT7 + pGADT7-BDnsLTP1, and pGBKT7 + pGADT7-BDGolS2 were the experimental groups; pGBKT7-53 + pGADT7-T was the positive control; pGBKT7-Lam + pGADT7-T was the negeative control; (B) Validation of positive candidate gene from gradient dilution spot medium. pGBKT7-ApCtf1β + pGADT7-BDUbc, pGBKT7-ApCtf1β + pGADT7-BDNADP-ME, pGBKT7-ApCtf1β + pGADT7-BDWGA-3, pGBKT7-ApCtf1β + pGADT7-BDSKL1, pGBKT7-ApCtf1β + pGADT7-BDBBTI, pGBKT7-ApCtf1β + pGADT7-BDnsLTP1, and pGBKT7-ApCtf1β + pGADT7-BDGolS2 were the experimental groups; pGBKT7-53 + pGADT7-T was the positive control; pGBKT7-Lam + pGADT7-T was the negeative control.
The ApCtf1β gene sequence was amplified from the A. phaeospermum cDNA with a full gene length of 1,236 bp. The BDUbc, BDNADP-ME, BDWGA-3, BDSKL1, and BDGolS2 gene sequences were amplified from B. pervariabilis × D. grandis cDNA with lengths of 591, 1,713, 624, 573, and 1,008 bp, respectively. PCR of the bacteriophage solution matched the theoretical values, and the sequencing was verified as positive and without base mutations (Supplementary Figure S5).The results showed that a strong fluorescent signal was detected using laser confocal microscopy in the positive control. In contrast, no fluorescent signal was obtained for the negative control, showing the success of the experimental setup. Compared to the negeative control, which had no fluorescent signal in the cells, the transient expressed pSPYNE(R)173-ApCtf1β + pSPYCE(M)-BDUbc and pSPYNE(R)173-ApCtf1β + pSPYCE(M)-BDSKL1 showed a strong fluorescent signal in N. benthamiana cells (Figure 3). These findings demonstrate that the decoy protein ApCtf1β interacts with the prey proteins BDUbc and BDSKL1 in N. benthamiana cells in the stomata but not with the three proteins BDNADP-ME, BDWGA-3, and BDGolS2. Therefore, two prey proteins, BDUbc and BDSKL1, were selected for in vitro validation.

Figure 3. Fluorescent laser confocal detection of Nicotiana benthamiana epidermal cells. (A) pSPYNE(R)173-bZIP63 + pSPYCE(M)-bZIP1; (B) pSPYNE(R)173-ApCtf1β + pSPYCE(M)-BDUbc; (C) pSPYNE(R)173-ApCtf1β + pSPYCE(M)-BDNADP-ME; (D) pSPYNE(R)173-ApCtf1β + pSPYCE(M)-BDWGA-3; (E) pSPYNE(R)173-ApCtf1β + pSPYCE(M)-BDSKL1; (F) pSPYNE(R)173-ApCtf1β + pSPYCE(M)-BDGolS2; (G) pSPYNE(R)173 + pSPYCE(M); (H) pSPYNE(R)173-ApCtf1β + pSPYCE(M); (I) pSPYNE(R)173 + pSPYCE(M)-BDUbc; (J) pSPYNE(R)173 + pSPYCE(M)-BDNADP-ME; (K) pSPYNE(R)173 + pSPYCE(M)-BDWGA-3; (L) pSPYNE(R)173 + pSPYCE(M)BDSKL1; (M) pSPYNE(R)173 + pSPYCE(M)-BDGolS2; (A) positive control; (B–F) experimental groups; (G–M) negative controls; UV, GFP fluorescence; DIC, bright field; and Merged, superimposition of GFP fluorescence and bright field.
The ApCtf1β gene sequence amplified from the cDNA of A. phaeospermum was 1,236 bp long. Amino acid sequence sorting showed that ApCtf1β had no signal peptide or transmembrane region and that there was a long continuous disordered sequence at the C-terminus. Therefore, ApCtf1β was intercepted at 1–217 aa for expression, i.e., 651 bp. The BDUbc and BDSKL1 sequences were amplified from the B. pervariabilis × D. grandis cDNA, producing fragments 591 and 573 bp long, respectively. Following amino acid sequence sorting, BDUbc and BDSKL1 had no signal peptide, transmembrane region, and low disorder. Therefore, they were expressed at full length, i.e., 197 and 191 aa, respectively. PCR using both bacterial broths produced fragments between 500 and 750 bp in length, consistent with theoretical values, and were sequenced and verified as positive without base mutations (Supplementary Figure S6). The GST-ApCtf1β fusion protein was expected to be 49 kDa in size, as expected by SDS-PAGE (Figure 4). The His-BDUbc and His-BDSKL1 fusion proteins were expected to be 21.31 and 20.89 kDa in size, respectively. The actual SDS-PAGE results showed a target band of the expected size (Figure 4). This result indicates that the GST-ApCtf1β, His-BDUbc, and His-BDSKL1 fusion proteins were successfully obtained and that the GST-pull down experiment can be used in further investigations. The results of the ApCtf1β and BDUbc protein in vitro interaction experiment are shown in Figure 5A. The hybridization results of the corresponding His-tagged antibody showed that the eluted control group did not contain the His-BDUbc fusion protein, indicating that the BDUbc protein and the GST-tagged protein did not interact and, therefore, could not be adsorbed on the GST column. The experimental group contained the His-BDUbc fusion protein, indicating that the BDUbc protein and GST-ApCtf1β fusion protein in ApCtf1β interactively bound to adsorb on the GST column. Similarly, the results of the in vitro interaction assay between the ApCtf1β and BDSKL1 proteins are shown in Figure 5B. The hybridization results of the his-tagged antibody showed that the eluted control group did not contain the His-BDSKL1 fusion protein and that the experimental group contained the His-BDSKL1 fusion protein, thus indicating that BDSKL1 protein can interact with ApCtf1β in GST-ApCtf1β fusion protein and adsorb to the GST column. The GST pull-down assay results showed that the ApCtf1β protein can interact with the BDUbc and BDSKL1 proteins in vitro.
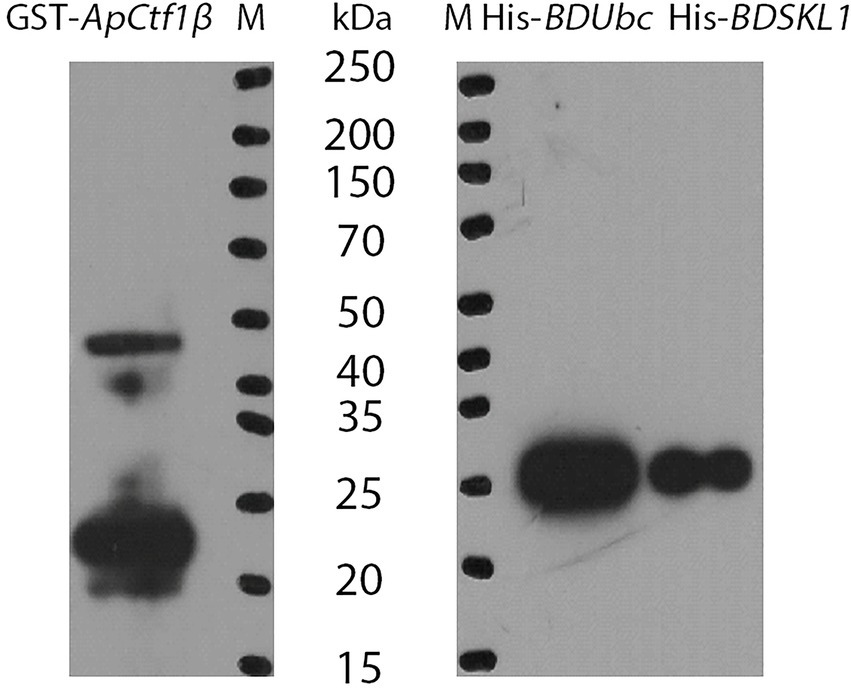
Figure 4. Immunoblot analysis of fusion proteins. M: Protein Marker.
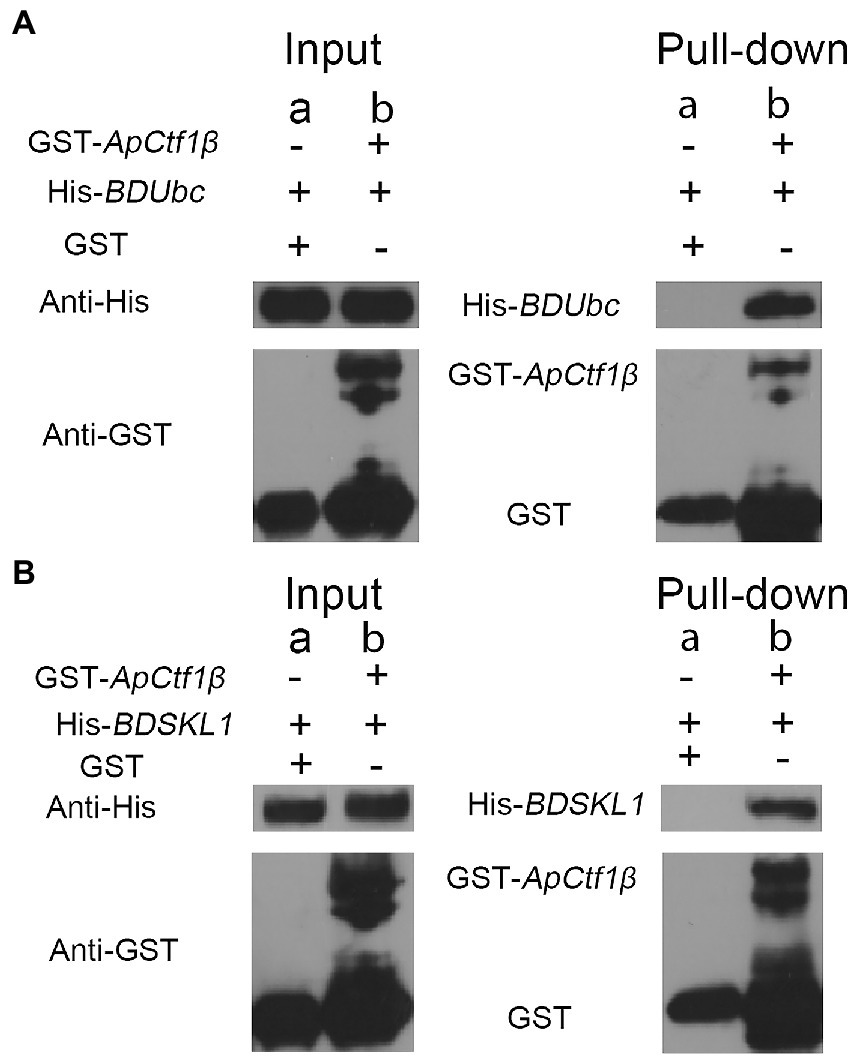
Figure 5. (A) GST pull-down assay of ApCtf1β interacting with the BDUbc protein in vitro; (B) GST pull-down assay of ApCtf1β interacting with the BDSKL1 protein in vitro; a: control group; b: experimental group.
Blast and multiple alignment analysis using ClustalW revealed that BDUbc and BDSKL1 have a high identity with other reported plant Ubcs and SKL1s in GenBank. BDUbc shares 97.45% identity with DlUbc (AGY80454.1), 93.88% identity with ZpUbc(KAG8054101.1), ObUbc(XP006645263.1), SbUbc (XP002459014.1), and OsUbc (XP015618841.1), respectively (Figure 6A). BDSKL1 shares 93.16, 92.63, 91.58, 88.42, and 88.59% identity with FnSKL1 (AIA26165.1), FsSKL1 from (AIA26167.1), PeSKL1 from (AIA26163.1), SiSKL1 from Setaria italica (AIA26163.1), and SbSKL1 from (XP_002442345.1), respectively (Figure 6B). The results from the database were compared using the NCBI online blast nucleic acid sequence analysis tool. Comparison analysis using MEGA 5.1 showed that the annotation of different families, genera, and model plants was selected as the phase protein gene sequence. The phylogenetic tree was constructed using the neighbor-joining algorithm, and the results showed that BDUbc was more closely related to Dendrocalamus latiflorus, while BDSKL1 was more closely related to the bamboo subfamily (Figures 6C,D). The basic physicochemical properties of the proteins encoded by the BDUbc and BDSKL1 genes were analyzed using ProtParam, The numbers of amino acid residues of BDUbc and BDSKL1 were 196 and 190, the instability coefficients were 41.68 and 34.93, the theoretical isoelectric points were 5.06 and 4.72, and the predicted molecular weights were 21.31 and 20.89 kDa, respectively. The amino acid sequences were analyzed for hydrophilicity using Protscale, and both proteins were presumed to be hydrophilic. Signal peptide and transmembrane domain prediction using online software SignalP-5.0 and TMHMM showed that both proteins have no signal peptide and no transmembrane structural domain. The secondary structure prediction of the amino acid sequence encoded by BDUbc and BDSKL1 through the SPOMA website showed that the BDUbc protein contained α-helix (47.96%), extended strand (11.22%), and random coil (36.73%). BDSKL1 contained α-helix (63.68%), extended strand (8.68%), extended strand (8.42%), β-turn (6.32%), and random coil (21.58%; Figures 7A,B). SWISS-MODEL was used to model the tertiary structural homology of the translated proteins of BDUbc and BDSKL1 (Figures 7C,D). The comparison results showed that the BDUbc global model quality assessment (GMQE) score was 0.67, and QMEANDisCo global score was 0.69 ± 0.06. The BDSKL1 global model quality assessment (GMQE) score was 0.80, and QMEANDisCo global score was 0.78 ± 0.06. Initially, the functions were determined to have similarities.
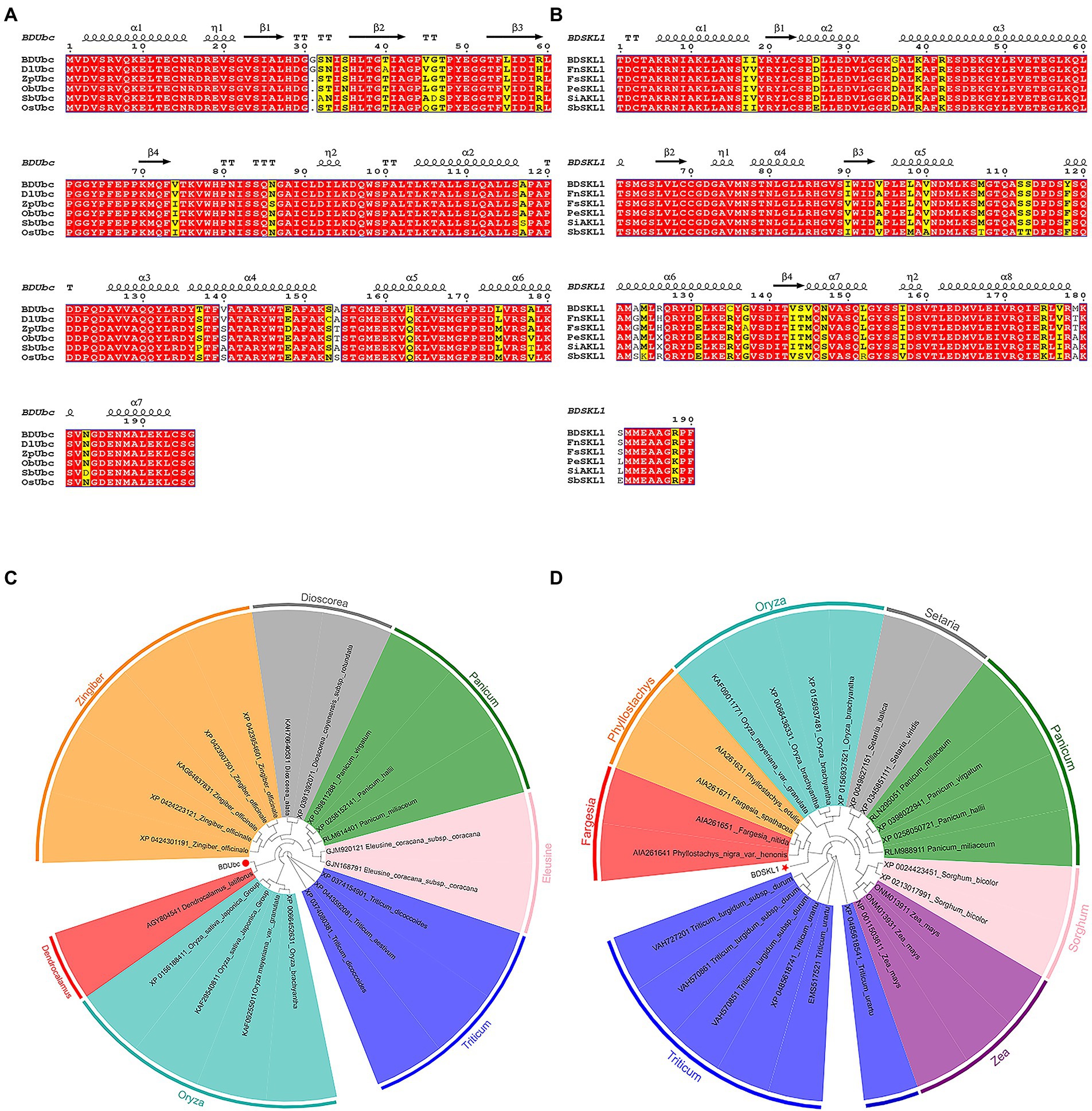
Figure 6. Multiple alignment of proteins and target gene phylogenetic tree based on neighbor-joining algorithm. (A) Multiple alignment of the obtained BDUbc protein with BDUbc proteins from other plants; (B) Multiple alignment of the obtained BDSKL1 protein with BDSKL1 proteins from other plants. Completely identical amino acid sequences are indicated using white words and red backgrounds; highly conserved amino acid sequences are represented by black words and yellow backgrounds; non-conserved amino acid sequences are represented by gray words and white backgrounds. α-Helices are displayed as large squiggles and η-helices as small squiggles; strict β-turns are shown as TT letters and β-strands as arrows. Strictly conserved amino acid residues are shown as. (C) BDUbc phylogenetic tree; (D) BDSKL1 phylogenetic tree. The numbers above/below the nodes indicated bootstrap number.
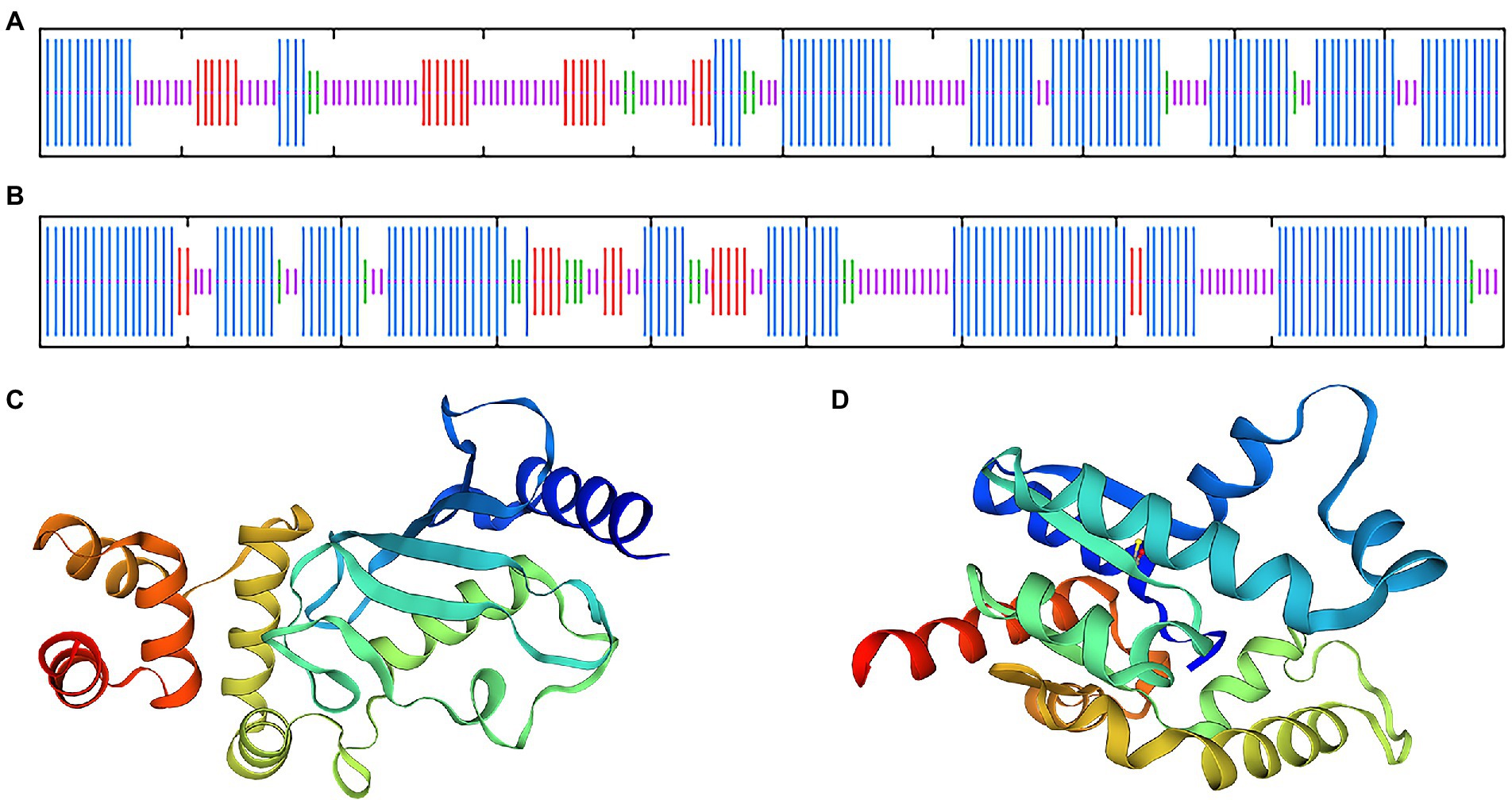
Figure 7. Transmembrane region, signal peptide, hydrophobicity, structure prediction of BDUbc and BDSKL1. (A,B) Secondary structure prediction of BDUbc and BDSKL1, α-helix, random coil, extended strand, and β-turn are represented with blue, pink, red and green regions; (C,D) tertiary structure prediction of BDUbc and BDSKL1.
The results were shown in the Supplementary Table S3 calculated that both GAPDH and Actin were the two most stable expression genes. The pairwise variation values of all candidate reference genes were less than the threshold value of 0.15, and the value of V2/3 was 0.045, which was smaller than 0.15, indicating that two reference genes were sufficient for accurate normalization of these samples sufficient (Supplementary Figure S8). Therefore, we chosed GAPDH and Actin as double reference to analyze the changes in expression. The results showed that the expression of BDUbc increased significantly at 8 h after infestation, then decreased slowly, with the highest expression at 72 h. The relative expression at 240 h was approximately twice as high as that at 0 h. The expression of BDSKL1 increased significantly at 8 h after infestation to reach the highest value, reached the lowest at 72 h, increased significantly at 120 h, and then decreased slowly. The relative expression at 240 h was lower than at 0 h (Figure 8).
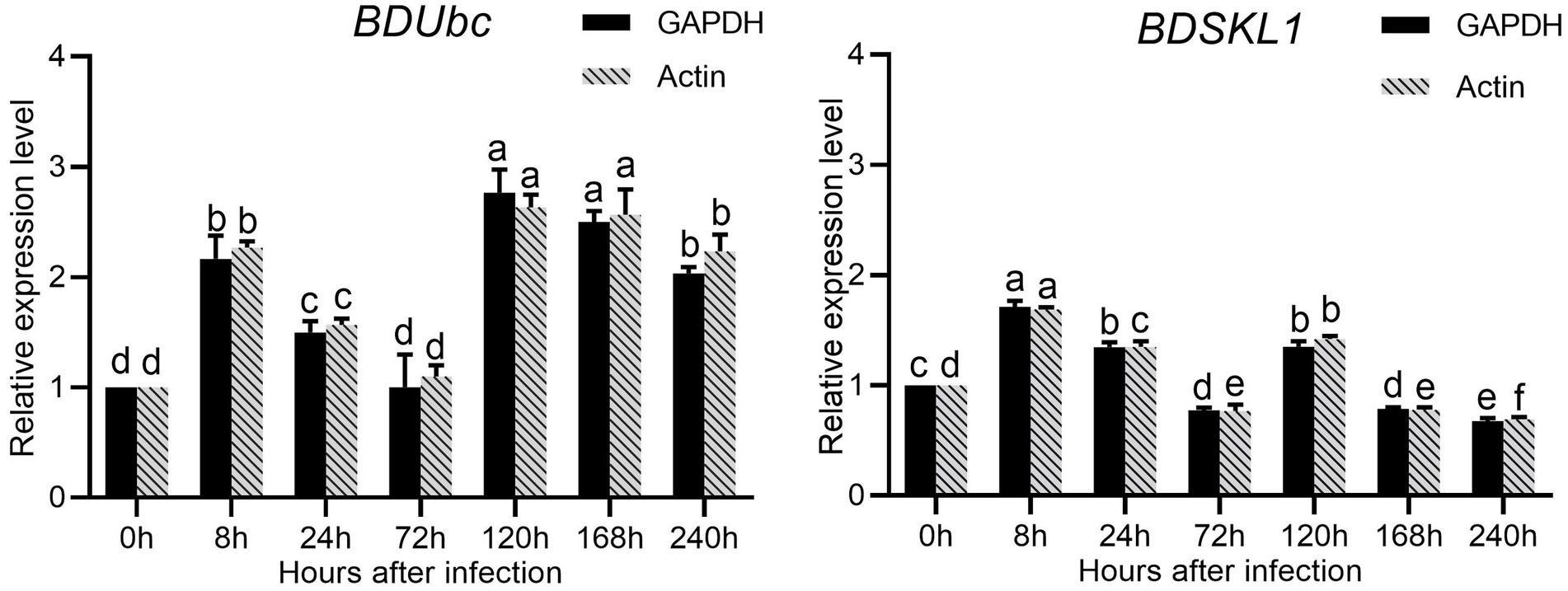
Figure 8. Relative expression levels of BDUbc and BDSKL1.There are significant differences in the relative expression of different lowercase letters (p < 0.05).
High-quality cDNA libraries are a strong guarantee for screening reciprocal proteins in yeast two-hybrid systems, and cDNA library quality depends mainly on RNA integrity and library capacity (Xu et al., 2021). Plants often exhibit high expression of disease-resistance-associated genes when infested with pathogens. These genes are expressed in relatively low abundance without pathogen stimulation and are potential targets for disease-causing genes in response to pathogen infestation. Therefore, yeast libraries were selected from bamboo hybrid shoots infested with A. phaeospermum spore suspension for 168 h to provide a more comprehensive picture of the expression of genes in yeast libraries. A study reported that the library capacity of the yeast two-hybrid library constructed using Phyllostachys pupescens was 1.2 × 106 CFU with a 99% recombination rate (Zheng and Ming-bing, 2012), while the library capacity of Dendrocalamus sinicus was 1.04 × 105 CFU with a 95% recombination rate (Wang et al., 2006). The average length of both libraries was approximately 1,000 bp. By contrast, the library capacity of the B. pervariabilis × D. grandi yeast secondary library was 1.20 × 107 CFU, with inserts averaging over 1,000 bp and a recombinant insertion rate of 100%, all of which were slightly higher than those of other bamboo subfamilies, and could satisfy the screening of intercalated proteins with low expression in abundance. The CDS sequence analysis of the two intercalated proteins showed that both were full-length CDS insertions into the secondary library. The above results indicate that the constructed B. pervariabilis × D. grandi library is of good quality and can be used to screen host target proteins for the keratinase transcription factor ApCtf1β of A. phaeospermum and to provide a database for the subsequent screening of the reciprocal target proteins of other related pathogenic genes of A. phaeospermum in B. pervariabilis × D. grandi and their mechanism.
Bambusa pervariabilis × D. grandi has a well-developed cuticle, which, in addition to its barrier function of water retention and cleansing, has a complex defense function of promoting overall plant development and regulating plant-pathogen interactions (Dickman, 1986; Barthlott and Neinhuis, 1997; Serrano et al., 2014). Arthrinium phaeospermum must degrade the cuticle to invade B. pervariabilis × D. grandi by secreting cuticle-degrading enzymes, including esterases, cutinases, and lipases, which catalyze the hydrolysis of ester bonds in lipoproteins, fats, and waxes to penetrate the outermost cuticle barrier. Cutinases are considered important enzymes, as they allow the fungus to penetrate the cuticle and infect the plant and function as an attachment to the plant epidermis, cuticle invasion, and signal production (Beys da silva et al., 2010; Ortiz-Urquiza and Keyhani, 2013). The cutinase transcription factor is a gene that was significantly up-regulated when in the growth environment of the host plant culture, as identified by our previous transcriptome sequencing analysis of A. phaeospermum under different culture conditions. Furthermore, the gene was found to be a key pathogenic gene for A. phaeospermum infestation of B. pervariabilis × D. grandi using knockdown and backfill tests. Therefore, ApCtf1β was used as a bait to screen for and validate two interacting proteins, BDUbc, belonging to the plant ubiquitin/protease pathway, and BDSKL1, belonging to the shikimate pathway.
The plant ubiquitin/proteasome system is the main pathway for intracellular protein degradation and plays a vital role in plant growth, development, morphogenesis, and disease resistance (Hershko, 2005). Recent studies have also shown that certain pathogenic microorganisms can mimic components of the host plant’s ubiquitin/protease system, thereby exploiting the system to benefit the pathogen (Dielen et al., 2011). The ubiquitin/proteasome pathway consists mainly of the ubiquitin-activating enzyme (E1), ubiquitin-conjugating enzyme (E2), ubiquitin-protein ligase (E3), proteasomes, and deubiquitinases (DUBs). The reaction process starts with activating the ubiquitin molecule by E1 using ATP. The activated ubiquitin molecule is attached to E1 by a thioester bond. E1 further binds to E2 and transfers the ubiquitin molecule to E2 via cross-esterification. E2 then binds to E3, mainly responsible for recruiting the substrate. Thus, the substrate is labeled with ubiquitin and later degraded by the proteasome into small polypeptides. Simultaneously, the ubiquitin molecule is re-cycled via the action of a deubiquitinating enzyme (Zeng et al., 2006; Stone, 2014). Ubiquitination plays an essential role in the plants’ biotic and abiotic stress responses (Zeng et al., 2006; Dielen et al., 2011; Marino et al., 2012; Stone, 2014). It has been known as early as 2006 that the ubiquitin/proteasome system may play an important role in plant-microbe interactions (Zeng et al., 2006), and much indirect evidence is available. Next, Dielen et al. (2011), published in Molecular Phytopathology, emphasize that the ubiquitin/proteasome system is involved in every step of the plant defense response. The ubiquitin/proteasome system is not only a defense weapon for the host plant but also a target for some pathogens to attack, thereby inhibiting the normal functioning of the system or using it for the benefit of the pathogen. Therefore, it is easy to propose a scenario in which the pathogen disrupts the ubiquitin/proteasome system of the host plant while infesting the plant, thereby disrupting the homeostasis of the host plant cell and facilitating pathogen infestation or a scenario in which the pathogen secretes effector proteins into the host plant cell, mimicking components of the host plant’s ubiquitin/proteasome system, thus exploiting the host’s ubiquitin/proteasome system for the benefit of the pathogen. The proteasome system is used to facilitate the successful infestation of the pathogen. There is experimental evidence that OsUBC26 expression in rice is induced by P. ramorum and JA treatment. Zhiguo et al. (2015) also showed that the expression of rice ubiquitin-conjugating enzymes could be induced by at least two phytohormones among IAA, 6-BA, GA, and ABA, further suggesting that the rice ubiquitin-conjugating enzyme E2 are involved in response to the infestation signaling pathway of rice blast and are likely to be associated with the JA signaling pathway. The ubiquitin-conjugating enzyme E2 mainly determines the length and topology of the ubiquitin chain, and ubiquitin chain-tagged protein molecules with different topologies will perform different functions. For example, ubiquitin chain-tagged protein molecules linked to K48 are mainly sent to the 26S proteasome for degradation, whereas ubiquitin chain-tagged protein molecules linked to K63 primarily function in signal transduction. Therefore, based on the qRT-PCR results, we can speculate that ubiquitin-binding enzyme genes induced at the early stage of A. phaeospermum infestation are likely to play a role in signaling the disease resistance defense response in B. pervariabilis × D. grandi. Conversely, ubiquitin-conjugating enzyme genes that were induced later are likely to play a role in initiating the defense response in B. pervariabilis × D. grandi, for example, by degrading certain defense-response-related repressor proteins through the ubiquitination pathway, thereby initiating the B. pervariabilis × D. grandi defense response. Conversely, the ubiquitin-conjugating enzyme genes may play a role in initiating defense responses in bamboo. For example, the ubiquitination pathway degrades several defense response-related repressor proteins, thereby initiating the disease-resistant defense response in B. pervariabilis × D. grandi.
The shikimate pathway functions at the critical interface between primary and secondary metabolism by directing carbon from the glycolytic and pentose phosphate pathways to synthesize a wide range of physiologically important aromatic compounds (Le Maréchal and Azerad, 1976). In plants, these include aromatic amino acids, phenylpropanoids, lignans, hormones, pigments, plant antitoxins, alkaloids, UV protectors, and electron carriers (Takeya et al., 1996). The major metabolites of the shikimate pathway are also considered to be blight point substrates for other secondary metabolic pathways. Shikimate kinase (SK) catalyzes the fifth reaction of the manganate pathway, which directs carbon from a central metabolic pool to a wide range of secondary metabolites involved in plant development and growth and stress responses. It is thought that plant SKs act as regulatory points in the mangiferous acid pathway, facilitating metabolic flow to specific secondary metabolite pools (Herrmann, 1995). The rapid induction of plant SK transcripts by fungal inducers (Görlach et al., 1995), significant sensitivity of plant SK activity to cellular ATP energy charge (Collins et al., 2017), and the differential expression of three rice SK genes at specific developmental stages and in response to biological stress have demonstrated that (Kasai et al., 2005). It was shown that AtSKL1 is a functionally distinct direct homologous cluster evolved from plant SK gene repeats. The Arabidopsis genome contains an SKL1 (AtSKL1; At3g26900) gene, and the ATP-binding site of AtSKL1 is highly conserved; however, AtSKL1 has a structural domain different from the ancestral manganate kinase and lacks the conserved SK manganate binding residues and catalytic residues. Therefore, AtSKL1 does not catalyze the SK reaction (Fucile et al., 2008). The main role of AtSK1 is to increase the carbon flux of specific metabolite pools in response to environmental stresses or tissue-specific developmental requirements. For example, tomato SK transcripts are induced approximately 17-fold by fungal excitons, potentially leading to a redirection of carbon flow to plant antitoxin biosynthesis. Moreover, the tomato SK induction pattern is not delayed and is a direct response to infestation with the pathogen (Görlach et al., 1995). Mutations in AtSKL1 in Arabidopsis lead to an albino phenotype, and loss-of-function mutations in SKL1 (e.g., SKL1-8) result in the loss of signals necessary to carry out the nuclear-encoded chloroplast developmental program. Previous studies have shown that a similar down-regulation of photosynthetic gene expression has been reported under different stress conditions (Bilgin et al., 2010). The qRT-PCR results showed that expression of the manganate kinase SKL1 gradually increased after dark spore rhodopsin infestation of B. pervariabilis × D. grandi, which may be due to the enhanced photosynthesis of the hybrid bamboo during the pre-invasion period of the pathogen. This occurrence directly induced the biosynthesis of antitoxin substances in pro × green bamboo through the SK mode, followed by a gradual decrease in expression to inhibit the photosynthetic capacity of B. pervariabilis × D. grandi in response to plant cells. Moreover, photo-oxidative stress results in induced genes, which are mainly divided into detoxification and stress-related genes, e.g., peroxidases, WRKY, and HSPs. These genes are induced for cellular defense and survival when the plant is under attack by pathogenic microorganisms and under uncomfortable environmental conditions.
In the present study, a cDNA library of B. pervariabilis × D. grandi infected by A. phaeospermum was constructed for the first time and screened for reciprocal target proteins of ApCtf1β in the host using the yeast two-hybrid technique. In vivo validation using the BiFC assay and in vitro validation using the GST pull-down assay yielded two interaction proteins, BDUbc and BDSKL1. Our findings have established the foundation for understanding the target protein functions and the resistance mechanism of B. pervariabilis × D. grandi in response to pathogenic stress. Moreover, they will provide a reliable theoretical basis to study the pathogenic pathway of A. phaeospermum and the molecular mechanism underlying the resistance of hybrid bamboo in response to pathogen infestation. This study has provided a basis for developing new strategies for the sustainable and effective control of forest tree blight wilting diseases.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/genbank/, QYRS0000000.1, MK789640, and MK789641.
PY and JY: conceptualization. PY: methodology, software, and writing–original draft preparation. SyL, TZ, and SH: validation. PY and XF: formal analysis. TL and JY: investigation. TL: resources. YL and CY: data curation. SjL and TL: writing–review and editing. PY and FH: visualization. SH: supervision. TZ: project administration. SjL: funding acquisition. All authors contributed to the article and approved the submitted version.
This research was funded by the National Natural Science Foundation of China, grant number 32171795.
We thank Charlesworth English editing for its linguistic assistance during the preparation of this manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.991077/full#supplementary-material
1. ^http://www.ncbi.nlm.nih.gov/blast/
2. ^http://www.expasy.org/tools/protparam.html
Ausubel, F. M. (2005). Are innate immune signaling pathways in plants and animals conserved? Nat. Immunol. 6, 973–979. doi: 10.1038/ni1253
Auyong, A. S. (2015). The role of Cutinase and its impact on pathogenicity of Colletotrichum truncatum. J. Plant Pathol. Microbiol. 6:3. doi: 10.4172/2157-7471.1000259
Barthlott, W., and Neinhuis, C. (1997). Purity of the sacred lotus, or escape from contamination in biological surfaces. Planta 202, 1–8. doi: 10.1007/s004250050096
Beys da silva, W. O., Santi, L., Corrêa, A. P. F., Silva, L. A. D., Bresciani, F. R., Schrank, A., et al. (2010). The entomopathogen Metarhizium anisopliae can modulate the secretion of lipolytic enzymes in response to different substrates including components of arthropod cuticle. Fungal Biol. 114, 911–916. doi: 10.1016/j.funbio.2010.08.007
Bilgin, D. D., Zavala, J. A., Zhu, J., Clough, S. J., Ort, D. R., and Delucia, E. H. (2010). Biotic stress globally downregulates photosynthesis genes. Plant Cell Environ. 33, 1597–1613. doi: 10.1111/j.1365-3040.2010.02167.x
Chen, J., Carter, M. B., Edwards, B. S., Cai, H., and Sklar, L. A. (2012). High throughput flow cytometry based yeast two-hybrid array approach for large-scale analysis of protein-protein interactions. Cytom. Part A 81A, 90–98. doi: 10.1002/cyto.a.21144
Chen, S. J., Lin, G. H., Hsiung, P.-A., and Hu, Y. H. (2009). “Parallel compiler,” in Hardware Software Co-Design of a Multimedia SOC Platform (Dordrecht: Springer).
Cheung, I. W. Y., Nakayama, S., Hsu, M. N. K., Samaranayaka, A. G. P., and Li-Chan, E. C. Y. (2009). Angiotensin-I converting enzyme inhibitory activity of Hydrolysates from oat (Avena sativa) proteins by in silico and in vitro analyses. J. Agric. Food Chem. 57, 9234–9242. doi: 10.1021/jf9018245
Collins, S. L., Ladwig, L. M., Petrie, M. D., Jones, S. K., Mulhouse, J. M., Thibault, J. R., et al. (2017). Press–pulse interactions: effects of warming, N deposition, altered winter precipitation, and fire on desert grassland community structure and dynamics. Glob. Chang. Biol. 23, 1095–1108. doi: 10.1111/gcb.13493
Dangl, J. L., and Jones, J. D. G. (2001). Plant pathogens and integrated defence responses to infection. Nature 411, 826–833. doi: 10.1038/35081161
Davies, K. A., De Lorono, I., Foster, S. J., Li, D., Johnstone, K., and Ashby, A. M. (2000). Evidence for a role of cutinase in pathogenicity of Pyrenopeziza brassicae on brassicas. Physiol. Mol. Plant Pathol. 57, 63–75. doi: 10.1006/pmpp.2000.0282
De Lorenzo, G., Brutus, A., Savatin, D. V., Sicilia, F., and Cervone, F. (2011). Engineering plant resistance by constructing chimeric receptors that recognize damage-associated molecular patterns (DAMPs). FEBS Lett. 585, 1521–1528. doi: 10.1016/j.febslet.2011.04.043
Deising, H., Nicholson, R. L., Haug, M., Howard, R. J., and Mendgen, K. (1992). Adhesion pad formation and the involvement of cutinase and esterases in the attachment of uredospores to the host cuticle. Plant Cell 4, 1101–1111. doi: 10.2307/3869478
Dickman, M. B. (1986). A rapid and sensitive plate assay for the detection of Cutinase produced by plant pathogenic fungi. Phytopathology 76:473. doi: 10.1094/Phyto-76-473
Dielen, A. S., Badaoui, S., Canderesse, T., and German, R. S. (2011). The ubiquitin/26S proteasome system in plant-pathogen interactions: a never-ending hide-and-seek game. Mol. Plant Pathol. 12:103. doi: 10.1111/j.1364-3703.2010.00673.x
Dodds, P. N., and Rathjen, J. P. (2010). Plant immunity: towards an integrated view of plant–pathogen interactions. Nat. Rev. Genet. 11, 539–548. doi: 10.1038/nrg2812
Fang, X., Yan, P., Guan, M., Han, S., Qiao, T., Lin, T., et al. (2021). Comparative transcriptomics and gene knockout reveal virulence factors of Arthrinium phaeospermum in Bambusa pervariabilis × Dendrocalamopsis grandis. J. Fungi 7:1001. doi: 10.3390/jof7121001
Fucile, G., Falconer, S., and Christendat, D. (2008). Evolutionary diversification of plant Shikimate kinase gene duplicates. PLoS Genet. 4:e1000292. doi: 10.1371/journal.pgen.1000292
Görlach, J., Raesecke, H. R., Rentsch, D., Regenass, M., Roy, P., Zala, M., et al. (1995). Temporally distinct accumulation of transcripts encoding enzymes of the prechorismate pathway in elicitor-treated, cultured tomato cells. Proc. Natl. Acad. Sci. 92, 3166–3170. doi: 10.1073/pnas.92.8.3166
Herrmann, K. M. (1995). The Shikimate pathway: early steps in the biosynthesis of aromatic compounds. Plant Cell 7, 907–919. doi: 10.2307/3870046
Hershko, A. (2005). The ubiquitin system for protein degradation and some of its roles in the control of the cell-division cycle (Nobel lecture). ChemInform 36:49. doi: 10.1002/chin.200549271
Järvinen, R., Silvestre, A. J. D., Holopainen, U., Kaimainen, M., Nyyssölä, A., Gil, A. M., et al. (2009). Suberin of potato (Solanum tuberosum Var. Nikola): comparison of the effect of cutinase CcCutl with chemical depolymerization. J. Agric. Food Chem. 57, 9016–9027. doi: 10.1021/jf9008907
Jones, J. D. G., and Dangl, J. L. (2006). The plant immune system. Nature 444, 323–329. doi: 10.1038/nature05286
Kasai, K., Kanno, T., Akita, M., Ikejiri-Kanno, Y., Wakasa, K., and Tozawa, Y. (2005). Identification of three shikimate kinase genes in rice: characterization of their differential expression during panicle development and of the enzymatic activities of the encoded proteins. Planta 222, 438–447. doi: 10.1007/s00425-005-1559-8
Kolattukudy, P. E., Li, D., Hwang, C.-S., and Flaishman, M. A. (1995). Host signals in fungal gene expression involved in penetration into the host. Can. J. Bot. 73, 1160–1168. doi: 10.1139/b95-373
Köller, W. (1991). Role of Cutinase in the penetration of apple leaves by Venturia inaequalis. Phytopathology 81:1375. doi: 10.1094/Phyto-81-1375
Le Maréchal, P., and Azerad, R. (1976). The shikimate pathway. Biochimie 58, 1123–1128. doi: 10.1016/S0300-9084(76)80090-9
Li, D., Ashby, A. M., and Johnstone, K. (2003). Molecular evidence that the extracellular Cutinase Pbc1 is required for pathogenicity of Pyrenopeziza brassicae on oilseed rape. Mol. Plant-Microbe Interact. 16, 545–552. doi: 10.1094/MPMI.2003.16.6.545
Li, L. S., Chen, L. N., Xia, T. Z., and Hanqi, Y. (2021). Selection of reference genes for quantitative real-time PCR analysis in Cephalostachyum pingbianense. Plant Physiol. J. 57, 225–234. doi: 10.13592/j.cnki.ppj.2020.0449
Li, D., and Kolattukudy, P. E. (1997). Cloning of Cutinase transcription factor 1, a Transactivating protein containing Cys6Zn2 binuclear cluster DNA-binding motif. J. Biol. Chem. 272, 12462–12467. doi: 10.1074/jbc.272.19.12462
Li, S. J., Liang, M., Zhu, T. H., and Yang, C. (2013b). Selection of an antagonistic bacterium against Arthrinium phaeospermum and its antibacterial protein analysis. J. Nanjing For. Univ. Sci. Educ. 37:6. doi: 10.3969/j.issn.1000-2006.2013.06.006
Li, D., Sirakova, T., Rogers, L., Ettinger, W. F., and Kolattukudy, P. E. (2002). Regulation of constitutively expressed and induced Cutinase genes by different zinc finger transcription factors in Fusarium solani f. sp. pisi (Nectria haematococca). J. Biol. Chem. 277, 7905–7912. doi: 10.1074/jbc.M108799200
Li, S., Tang, Y., Fang, X., Qiao, T., Han, S., and Zhu, T. (2020). Whole-genome sequence of Arthrinium phaeospermum, a globally distributed pathogenic fungus. Genomics 112, 919–929. doi: 10.1016/j.ygeno.2019.06.007
Li, S. J., and Zhu, T. H. (2012). Binding site of toxic protein from Arthrinium phaeospermum on plasmalemma of hybrid bamboo. J. Zhejiang Univ. Life Sci. 38, 355–361. doi: 10.3785/j.issn.1008-9209.2012.04.001
Li, S. J., Zhu, T. H., Zhu, H. M. Y., Liang, M., Qiao, T. M., Han, S., et al. (2013a). Purification of protein AP-toxin from Arthrinium phaeospermum causing blight in Bambusa pervariabilis × Dendrocalamopisis grandis and its metabolic effects on four bamboo varieties. Phytopathology 103, 135–145. doi: 10.1094/PHYTO-07-12-0164-R
Liu, T., Hou, J., Wang, Y., Jin, Y., Borth, W., Zhao, F., et al. (2016). Genome-wide identification, classification and expression analysis in fungal–plant interactions of cutinase gene family and functional analysis of a putative ClCUT7 in Curvularia lunata. Mol. Gen. Genomics. 291, 1105–1115. doi: 10.1007/s00438-016-1168-1
Marino, D., Peeters, N., and Rivas, S. (2012). Ubiquitination during Plant Immune Signaling. Plant Physiol. 160, 15–27. doi: 10.1104/pp.112.199281
McLellan, H., Armstrong, M. R., and Birch, P. R. J. (2021). Yeast two-hybrid screening for identification of protein-protein interactions in Solanum tuberosum. Methods Mol. Biol. 2354, 95–110. doi: 10.1007/978-1-0716-1609-3_4
Napoli, R. S., Allen, P. J., Parish, R. W., and Li, S. F. (2021). The Arabidopsis MYB5 transcription factor interacts with CASEIN KINASE2 BETA3 subunit in a yeast two-hybrid system. MicroPubl. Biol. 2021. doi: 10.17912/micropub.biology.000486
Ortiz-Urquiza, A., and Keyhani, N. (2013). Action on the surface: entomopathogenic Fungi versus the insect cuticle. Insects 4, 357–374. doi: 10.3390/insects4030357
Parker, D. M., and Köller, W. (1998). Cutinase and other Lipolytic Esterases protect bean leaves from infection by Rhizoctonia solani. Mol. Plant-Microbe Interact. 11, 514–522. doi: 10.1094/MPMI.1998.11.6.514
Pascholati, S. F., Yoshioka, H., Kunoh, H., and Nicholson, R. L. (1992). Preparation of the infection court by Erysiphe graminis f. sp. hordei: cutinase is a component of the conidial exudate. Physiol. Mol. Plant Pathol. 41, 53–59. doi: 10.1016/0885-5765(92)90048-Z
Rogers, L. M., Flaishman, M. A., and Kolattukudy, P. E. (1994). Cutinase gene disruption in Fusarium solani f sp pisi decreases its virulence on pea. Plant Cell 6, 935–945. doi: 10.1105/tpc.6.7.935
Serrano, M., Coluccia, F., Torres, M., Haridon, F., and Métraux, J.-P. (2014). The cuticle and plant defense to pathogens. Front. Plant Sci. 5:274. doi: 10.3389/fpls.2014.00274
Shan, Z., Zhou, Z. W., Wu, Q., and Chen, H. (2011). Rapid extraction of plasmid DNA from Escherichia coli using magnetic bead separation method. J. Sichuan Agric. Univ. 29, 230–234. doi: 10.3969/j.issn.1000-2650.2011.02.017
Silver, N., Best, S., Jiang, J., and Thein, S. L. (2006). Selection of housekeeping genes for gene expression studies in human reticulocytes using real-time PCR. BMC Mol. Biol. 7:33. doi: 10.1186/1471-2199-7-33
Singh, R., Dangol, S., and Jwa, N.-S. (2014). Yeast two-hybrid system for dissecting the Rice MAPK Interactome. Methods Mol. Biol. 1171, 195–216. doi: 10.1007/978-1-4939-0922-3_16
Skamnioti, P., and Gurr, S. J. (2007). Magnaporthe grisea Cutinase2 mediates Appressorium differentiation and host penetration and is required for full virulence. Plant Cell 19, 2674–2689. doi: 10.1105/tpc.107.051219
Song, M., Fang, S. Q., Li, X., Liu, W. B., Lin, C., and Miao, W. (2022). Identification and screening of proteins interacting with CsAtf1 transcription factor in Colletotrichum siamense. Chin. J. Trop. Crop. 43:7. doi: 10.3969/j.issn.1000-2561.2022.01.008
Stahl, D. J., and Schäfer, W. (1992). Cutinase is not required for fungal pathogenicity on pea. Plant Cell 4, 621–629. doi: 10.1105/tpc.4.6.621
Stone, S. L. (2014). The role of ubiquitin and the 26S proteasome in plant abiotic stress signaling. Front. Plant Sci. 5:135. doi: 10.3389/fpls.2014.00135
Sweigard, J. A., Chumley, F. G., and Valent, B. (1992). Disruption of a Maanaporthe arisea cutinase gene. Mol. Gen. Genet. MGG 232, 183–190. doi: 10.1007/BF00279995
Takeya, K., Qiao, Z. S., Hirobe, C., and Itokawa, H. (1996). Cytotoxic azadirachtin-type limonoids from Melia azedarach. Phytochemistry 42, 709–712. doi: 10.1016/0031-9422(96)00044-1
Vandesompele, J., Preter, K. D., Pattyn, F., Poppe, B., Roy, N. V., Paepe, A. D., et al. (2002). Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 3, RESEARCH0034–RESEARCH3411. doi: 10.1186/gb-2002-3-7-research0034
Wang, Y., Chen, J., Li, D. W., Zheng, L., and Huang, J. (2016). CglCUT1 gene required for cutinase activity and pathogenicity of Colletotrichum gloeosporioides causing anthracnose of Camellia oleifera. Eur. J. Plant Pathol. 147, 103–114. doi: 10.1007/s10658-016-0983-x
Wang, Y., Gong, X. H., and Li, X. (2006). Construction of cDNA library from leaves of Dendrocalamus sinicu. J. Bamboo Res. 4, 21–23.
Weigel, D., and Glazebrook, J. (2006). Transformation of agrobacterium using the freeze-thaw method. Cold Spring Harb Protoc 2006:pdb.prot4666. doi: 10.1101/pdb.prot4666
Xie, F., Xiao, P., Chen, D., Xu, L., and Zhang, B. (2012). miRDeepFinder: a miRNA analysis tool for deep sequencing of plant small RNAs. Plant Mol. Biol. 80, 75–84. doi: 10.1007/s11103-012-9885-2
Xu, C. Y. (2020). Pull-down and co-immunoprecipitation assays of interacting proteins in plants. Bull. Bot. 55, 62–68. doi: 10.11983/CBB19143
Xu, Y., Li, R., Song, J., Wang, D., Zhou, L., Li, J. J., et al. (2021). Construction of a sea-island cotton yeast two-hybrid library under the conditions of infection with Verticillium dahliae and treatment with hormones. Plant Prot. 47, 46–55. doi: 10.16688/j.zwbh.2019691
Zeng, L. R., VegaSánchez, M. E., Zhu, T., and Wang, G. L. (2006). Ubiquitination-mediated protein degradation and modification: an emerging theme in plant-microbe interactions. Cell Res. 16, 413–426. doi: 10.1038/sj.cr.7310053
Zhang, Z., Henderson, C., Perfect, E., Carver, T. L. W., Thomas, B. J., Skamnioti, P., et al. (2005). Of genes and genomes, needles and haystacks: Blumeria graminis and functionality. Mol. Plant Pathol. 6, 561–575. doi: 10.1111/j.1364-3703.2005.00303.x
Zhang, K., Liu, Y. Y., Peng, D. L., Wu, Z. D., Mao, L. M., Tang, D. B., et al. (2020). Screening and identification of interacting proteins of sweet potato WRKY transcription factor IbWRKY61. J. Agric. Biotechnol. 28, 2118–2129. doi: 10.3969/j.issn.1674-7968.2020.12.003
Zhang, X., Wang, J., Cao, K., Xu, C., and Cao, W. (2015). An expressed sequence tags analysis for leaves of Chinese milk vetch (Astragalus sinicus). Legum. Res. An Int. J. 38:1. doi: 10.5958/0976-0571.2015.00001.6
Zhang, H., Wu, Q., Cao, S., Zhao, T., Chen, L., Zhuang, P., et al. (2014). A novel protein elicitor (SsCut) from Sclerotinia sclerotiorum induces multiple defense responses in plants. Plant Mol. Biol. 86, 495–511. doi: 10.1007/s11103-014-0244-3
Zhao, M. R., Huang, J. D., Yao, J. P., Li, F. L., and Zhang, X. L. (2004). Introduction effect of Bambusa ervariabilis× Dendrocalamopsis daii in dry hot valley of Huaning. For. Investig. Plan 29, 24–26.
Zheng, L. N., and Ming-bing, Z. (2012). Construction and analysis of cDNA libraries of the rapidly-elongating internodes of Phyllostachys pupescens. J. Fujian For. Sci. Technol. 5. doi: 10.3969/j.issn.1002-7351.2012.03.05
Zhiguo, E., Zhang, Y., Li, T., Wang, L., and Zhao, H. (2015). Characterization of the ubiquitin-conjugating enzyme gene family in Rice and evaluation of expression profiles under abiotic stresses and hormone treatments. PLoS One 10:e0122621. doi: 10.1371/journal.pone.0122621
Keywords: Arthrinium phaeospermum, ApCtf1β, Y2H, BiFC, GST pull-down, qRT-PCR, interaction protein
Citation: Yan P, Yu J, Fang X, Li S, Han S, Lin T, Liu Y, Yang C, He F, Zhu T and Li S (2022) Identification of the interacting proteins of Bambusa pervariabilis × Dendrocalamopsis grandis in response to the transcription factor ApCtf1β in Arthrinium phaeospermum. Front. Plant Sci. 13:991077. doi: 10.3389/fpls.2022.991077
Edited by:
Pramod Prasad, ICAR-Indian Institute of Wheat and Barley Research, IndiaReviewed by:
Shailesh Pandey, Forest Research Institute (FRI), IndiaCopyright © 2022 Yan, Yu, Fang, Li, Han, Lin, Liu, Yang, He, Zhu and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tianhui Zhu, MTA2MjdAc2ljYXUuZWR1LmNu; Shujiang Li, MTQwODdAc2ljYXUuZWR1LmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.