 Yujiao Yang
Yujiao Yang Xiaolei Yu
Xiaolei Yu Pei Wei1
Pei Wei1- 1State Key Laboratory of Hybrid Rice, Laboratory of Plant Systematics and Evolutionary Biology, College of Life Sciences, Wuhan University, Wuhan, China
- 2Biology Experimental Teaching Center, School of Life Science, Wuhan University, Wuhan, China
Isoetes is a famous living fossil that plays a significant role in the evolutionary studies of the plant kingdom. To explore the adaptive evolution of the ancient genus Isoetes from China, we focused on Isoetes yunguiensis (Q.F. Wang and W.C. Taylor), I. shangrilaensis (X. Li, Y.Q. Huang, X.K. Dai & X. Liu), I. taiwanensis (DeVol), I. sinensis (T.C. Palmer), I. hypsophila_GHC (Handel-Mazzetti), and I. hypsophila_HZS in this study. We sequenced, assembled, and annotated six individuals’ chloroplast genomes and transcriptomes, and performed a series of analyses to investigate their chloroplast genome structures, RNA editing events, and adaptive evolution. The six chloroplast genomes of Isoetes exhibited a typical quadripartite structure with conserved genome sequence and structure. Comparative analyses of Isoetes species demonstrated that the gene organization, genome size, and GC contents of the chloroplast genome are highly conserved across the genus. Besides, our positive selection analyses suggested that one positively selected gene was statistically supported in Isoetes chloroplast genomes using the likelihood ratio test (LRT) based on branch-site models. Moreover, we detected positive selection signals using transcriptome data, suggesting that nuclear-encoded genes involved in the adaption of Isoetes species to the extreme environment of the Qinghai-Tibetan Plateau (QTP). In addition, we identified 291–579 RNA editing sites in the chloroplast genomes of six Isoetes based on transcriptome data, well above the average of angiosperms. RNA editing in protein-coding transcripts results from amino acid changes to increase their hydrophobicity and conservation in Isoetes, which may help proteins form functional three-dimensional structure. Overall, the results of this study provide comprehensive transcriptome and chloroplast genome resources and contribute to a better understanding of adaptive evolutionary and molecular biology in Isoetes.
Introduction
Lycopsids, the sister of the remaining vascular plants and an important bridge between non-vascular bryophytes and vascular plants, is a key group in evolution (Pryer et al., 2001), but it currently includes only three major lineages (Lycopodiaceae, Isoetaceae, and Selaginellaceae). These groups were widely distributed over the Carboniferous, being the dominant plants on earth (DiMichele et al., 2001). It was not until the end of the Carboniferous that these groups began to diminish due to dramatic changes in climate and environment (Liu et al., 2005). Isoetes is an ancient heterosporous lycopsids that occupies a unique position in plant evolution and there are ~200 extant species (Pigg, 1992). Phylogenetic analyses show that this genus is one of the earliest basal vascular plants, which can date back to the Devonian (Pigg, 2001; Pryer et al., 2001). The modern distribution of Isoetes is influenced by geographic variation (Liu et al., 2004), and they grow in a variety of habitats, including seasonal pools, intermittent streams, and high-altitude wetlands (Pfeiffer, 1922; Taylor and Hickey, 1992). To date, six Isoetes species have been reported in China: I. yunguiensis (Qing-Feng et al., 2002), I. hypsophila, I. shangrilaensis (Li et al., 2019), I. taiwanensis (DeVol, 1972), I. sinensis (Palmer, 1927), and I. orientalis (Hong et al., 2005). Species of the genus Isoetes are widely distributed in China, about 100–4,300 m above the sea level. However, it does not fit the hypothesis that the distribution of polyploids is more likely at high altitudes (Liu et al., 2004). I. hypsophila (2n = 22) inhabits altitudes above 4,000 m on the QTP, which is the youngest, largest, and highest plateau in the world, while I. sinensis (4n = 44) inhabits low-altitude environments in the Middle and Lower Yangtze Plain (MYP; Xing et al., 2002; Liu et al., 2004).
The QTP is characterized by low temperature, low oxygen, and strong radiation, which offers a unique extreme environment for studying adaptive evolution (Qiao et al., 2016a,b). Although the adaptive evolution of QTP has been studied previously in animals (Hao et al., 2019) and plants (Zhang et al., 2019; Guo et al., 2020), these studies are far from sufficient because different organisms adapt to high altitudes through multiple genetic routes (Hao et al., 2019). Notably, hitherto, no study of adaptive evolution has been conducted on Isoetes based on combined transcriptome and chloroplast genome analysis.
Chloroplasts are photosynthetic organelles that play an irreplaceable role in plant growth and development (Liu et al., 2012). Most chloroplast genomes have a circular structure ranging from 110 to 190 kb in size and consist of a relatively conserved quadripartite structure, including two Inverted Repeat (IR) regions, a Small Single Copy (SSC), and a Large Single Copy (LSC) region (Palmer, 1985; Green, 2011; Yu et al., 2019). Despite the chloroplast genome being relatively conservative in gene content, structure, and gene order (Shahzadi et al., 2020; Yu et al., 2021), numerous early evolutionary modifying mutational events frequently occur in the chloroplast genome, including inversions, contractions, substitutions, gene loss, duplications, and pseudogenes (Raubeson and Jansen, 1992a,b; Henriquez et al., 2020; Shahzadi et al., 2020). As the chloroplast is the center of photosynthesis, the study of the chloroplast genome is important for discovering the mechanisms of plant photosynthesis.
RNA editing is a post-transcriptional modification that changes nucleotide sequences of RNA by nucleotide insertions/deletions or transitions (Takenaka et al., 2013), this phenomenon occurs in different regions of the chloroplast genome such as protein-coding regions, introns, and tRNAs (Schallenberg-Rüdinger and Knoop, 2016; Stefan, 2016). Although RNA editing is not limited to protein-coding regions, it may play a fundamental role in these regions, potentially affecting species phenotype and evolution by maintaining the basic functions of genes. In protein-coding genes, it generally implies generating start/stop codons, restoring codons for amino acids, or removing internal stop codons (Schallenberg-Rüdinger and Knoop, 2016). In other regions, RNA editing performs different functions; for example, in tRNA, the processing of precursor RNA molecules may require editing, while in introns, editing events appear to be required for efficient splicing in some cases (Binder et al., 1994). RNA editing is considered to be an indirect repair mechanism for the correction of DNA mutations on the RNA level by converting specific cytidine to uridine (C-to-U) or uridine to cytidine (U-to-C; Chateigner-Boutin and Small, 2011; Ichinose and Sugita, 2016). In plants, the most common type of RNA editing in the chloroplast genome is C-to-U editing. In contrast, U-to-C RNA editing is present abundantly in hornworts (Kugita et al., 2003), and ferns (Wolf et al., 2004; Guo et al., 2015), but not in seed plants (Tillich et al., 2006). The Selaginella chloroplast genome has a particularly high number of C-to-U editing events, but no U-to-C editing (Hecht et al., 2011; Oldenkott et al., 2014). As a close relative of Selaginella, the research on the chloroplast genome of Isoetes is still limited to the phylogenetic analysis (Schafran et al., 2018; Wood et al., 2020; Pereira et al., 2021b). In addition, the research on RNA editing sites is limited to prediction using the PREPACT tool (Oldenkott et al., 2014), and lacks transcriptome data for verification.
In this study, we sequenced and compared the transcriptomes and chloroplast genomes of the six individuals (five species) which are distributed at an altitude between 100 and 4,300 m above sea level in China. Based on the generated dataset, we analyzed a total of six individuals’ chloroplast genomes and transcriptomes of Isoetes with the aim of (i) evaluating the structural features of the chloroplast genome, (ii) identifying RNA editing sites in chloroplast genomes of six individuals based on RNA-Seq data, and (iii) studying the genetic mechanism of its adaptation to high altitude.
Materials and methods
Sampling, DNA/RNA extraction, and sequencing
Plants were harvested from type localities whenever possible in China (Supplementary Table S1). Then, we collected the leaves of I. yunguiensis, I. shangrilaensis, I. taiwanensis, I. sinensis, and I. hypsophila, washed them with distilled water, fixed them in RNAlater solution (Takara, Dalian, China) immediately, and stored them in a −80°C freezer for DNA and RNA extraction. DNA quality and DNA concentration were measured on a NanoDrop 2000 (Supplementary Table S2).
We sequenced the transcriptomes and chloroplast genomes of the six individuals. Total genomic DNA was extracted using an extract Plant DNA kit (TIANGEN, China), while total RNA was isolated using the RNAiso Plus kit (TaKaRa, Dalian, China). Afterward, a paired-end library with an insert size of 350 bp was constructed using the Truseq Nano DNA HT Sample Prep Kit (Illumina, United States), and the RNA-sequencing library was generated using the VAHTS mRNA-seq v2 Library Prep Kit for Illumina® (Vazyme, NR601).
Chloroplast genome and transcriptome de novo assembly and annotation
Raw data were processed by removing linker sequences and removing low-quality reads at the Q20 cutoff, and subsequent analyses were based on these filtered high-quality sequences. The de novo assembly of the chloroplast genome was carried out using GetOrganelle with parameter settings as follows: ‘-R 15 -k 21,45,65,85,105 -F embplant_pt’ (Jin et al., 2020). Then, the wrong bases of the organelle genomes were corrected using BWA (Li and Durbin, 2010) and Pilon with default parameters. The chloroplast genome annotations were performed in GeSeq (Tillich et al., 2017). The tRNA genes were further verified using the tRNAscan-SE program (Schattner et al., 2005). We used Geneious (Kearse et al., 2012) to validate the annotated six chloroplast genomes by comparison with reference chloroplast genomes of Isoetes nuttallii, and Isoetes cangae (NCBI accession numbers: NC_038073, MG019394). Finally, the resulting chloroplast genome maps were drawn with Chloroplot (Zheng et al., 2020).
We assembled the transcriptomes by de novo assembly of high-quality RNA-Seq data using Trinity (Grabherr et al., 2011), followed by splicing of Trinity-obtained contigs into transcripts, after which only the longest transcripts in each cluster were selected as unigenes for subsequent analysis.
Comparative genome analysis
Comparative genomics and visualization of six Isoetes chloroplast genomes were performed using mVISTA software (Frazer et al., 2004) with annotations of I. taiwanensis as a reference. The genes on the IR, SSC, and LSC boundaries were visualized using the tool IRscope (Amiryousefi et al., 2018) based on the annotation information.
Identification of RNA editing sites
Geneious (Kearse et al., 2012) was used to map the RNA reads from each individual to their chloroplast genomes. Variants with <5× read depth and <2.5% of RNA reads mapped to a given fragment were excluded to address possible sequencing errors. RNA editing efficiency was calculated by dividing the edited reads by the total mapped reads.
Phylogenetic analysis
To determine the phylogenetic relationships between Isoetes species, the CDS of chloroplast genome sequences and the transcriptome data were used to construct trees. For the phylogenetic tree constructed from chloroplast genome sequences, 72 protein-coding genes shared by 34 species were extracted. In addition, for the genes in the IR region, we only extracted one copy of them. The 28 completed chloroplast genome sequences were downloaded from the NCBI Organelle Genome Resource database. GenBank information for all of the chloroplast genomes used for the present phylogenetic analyses is found in Supplementary Table S3. For the phylogenetic tree constructed from transcriptome data, we first aligned the amino acid sequences of each single-copy gene orthologous using muscle with default parameters (Edgar, 2004). The alignment file of each orthologous gene was then concatenated into a super gene alignment, which was further trimmed to remove poorly aligned regions using trimal (Capella-Gutiérrez et al., 2009). Among them, the genome data of the two species Selaginella moellendorffii (Banks et al., 2011) and Isoetes taiwanensis (Wickell et al., 2021) have been published, so the genome protein file was used in the construction of the tree. Selaginella moellendorffii was set as the outgroup.
Maximum likelihood (ML) analysis was performed using the RAxML v 8.0.5 software package (Stamatakis, 2014) with 1,000 non-parametric bootstrap replicates. Bayesian Inference (BI) phylogenies were inferred using MrBayes 3.2.6(Ronquist et al., 2012) under JC + I + G model (2 parallel runs, 2,000,000 generations), in which the initial 25% of sampled data are discarded as burn-in.
Orthologous gene identification and positive selection analysis
We used the branch-site model in the PAML (Yang, 2007) CODEML program to identify positively selected genes in the six Isoetes chloroplast genomes collected from China. The null model refers to the assumption that all branches evolve at the same rate, and the alternative model differs from the null model by allowing the foreground branches to evolve at different rates. In addition, the likelihood ratio test (LRT; Nielsen and Yang, 1998) was used to evaluate the statistical significance of each pair of nested models. We set I. hypsophila collected from high altitudes as the foreground branch in the branch-site model and others as the background branch.
The Reciprocal best hit (RBH) algorithm is the most commonly used algorithm based on the Basic Local Alignment Search Tool (BLAST; Altschul et al., 1997), which defines orthologous genes as sequences of a pair of genes from two genomes that are the best hits to each other (Moreno-Hagelsieb and Latimer, 2008). Next, we identified positively selected genes in the six transcriptomes based on orthologous genes by using the CODEML program’s branch-site model, where the foreground branch was set identically to the chloroplast genome. Only orthologous genes with p < 0.05 were considered positively selected genes; otherwise, orthologous genes were considered non-positively selected genes. KEGG enrichment analysis was performed using the OmicShare tools. These genes were annotated using eggNOG-mapper (Huerta-Cepas et al., 2017) in the eggNOG database.
Results
Transcriptome features and orthologous genes
Illumina pair-end sequencing produced 613,197,232 raw reads for six individuals, and 608,937,022 clean reads were obtained after removing low-quality reads and ambiguous nucleotides (Supplementary Table S4). Q30 and Q20 of clean reads were above 93% and 97%, respectively, which indicated that these data could be used in subsequent analysis. Based on Trinity assembly, extraction of the longest transcript yielded a total of 457,357 unigenes, and 2,798 single-copy orthologous genes were identified using RBH.
Chloroplast genome features
The complete chloroplast genomes of the six Isoetes species ranged from 145,479 bp (I. shangrilaensis) to 146,380 bp (I. hypsophila_GHC), with 38%–38.1% GC content, which were composed of four regions, including LSC (91,740–91,880 bp) and SSC (27,218–27,272 bp) region separated by two IRs (13,207–13,691 bp; Figure 1; Table 1). A total of 135 genes were annotated from Isoetes chloroplast genome: 84 protein-coding genes, 8 rRNA genes, 36 tRNA genes, and 7 pseudogenes (Table 2). Among these genes, 24 were duplicated in the IR regions: 6 protein-coding genes, 10 tRNA, and 8 rRNA genes. There were 18 genes with introns, of which 16 had one intron, and two genes (clpP, ycf3) had two introns. Internal stop codons were observed in the CDS of 16 genes in Isoetes, except for I. hypsophila which rps3 without internal stop codons. In addition, except for rpoC1 and rpoC2, all genes contained a single internal stop codon. The rpoC2 gene revealed three internal stop codons and observed two premature stop codons in rpoC1.
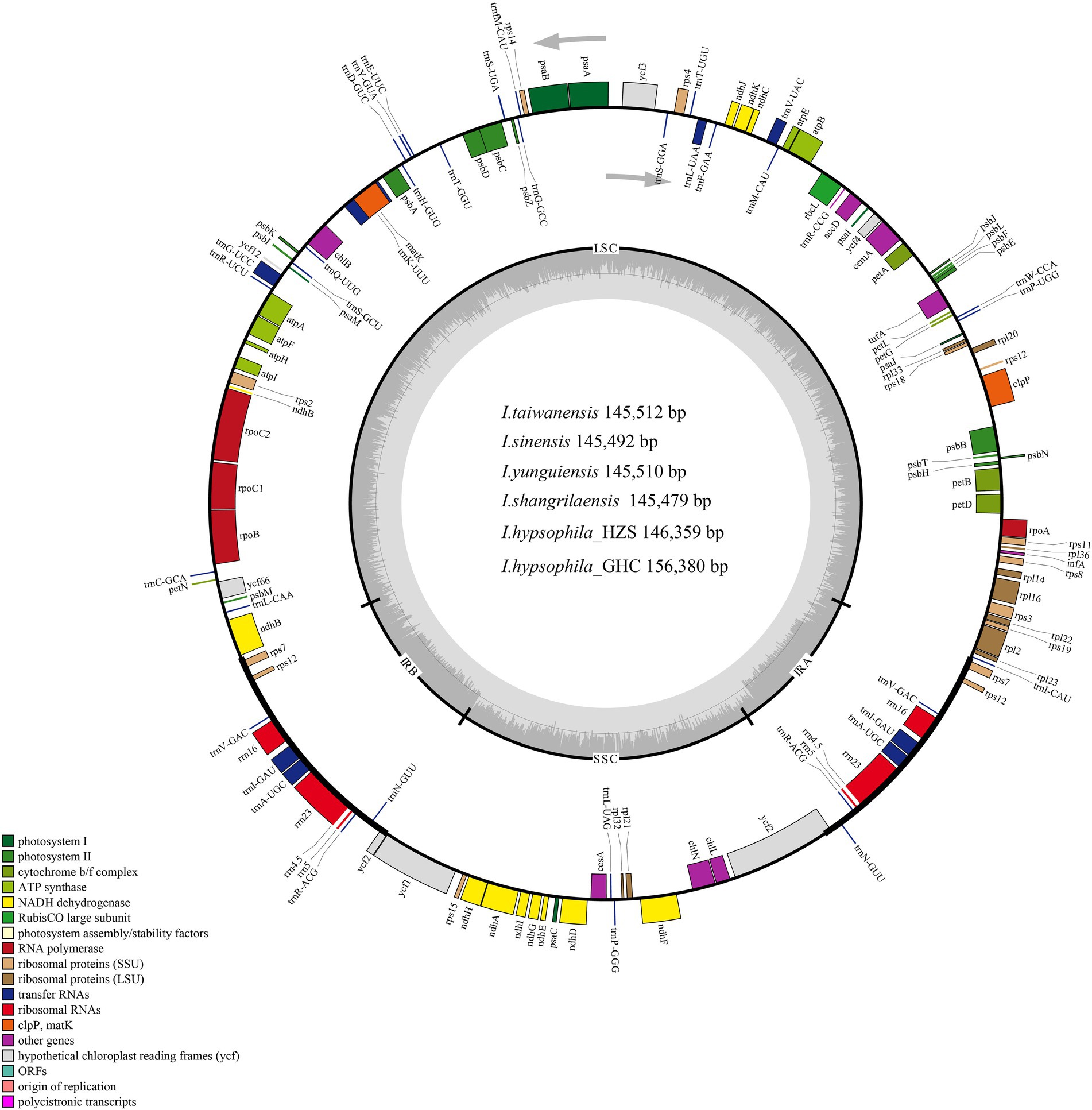
Figure 1. Chloroplast genome maps of Isoetes species. Genes belonging to different functional groups are color-coded. The darker grey in the inner circle shows the GC content, while the lighter grey shows the AT content.
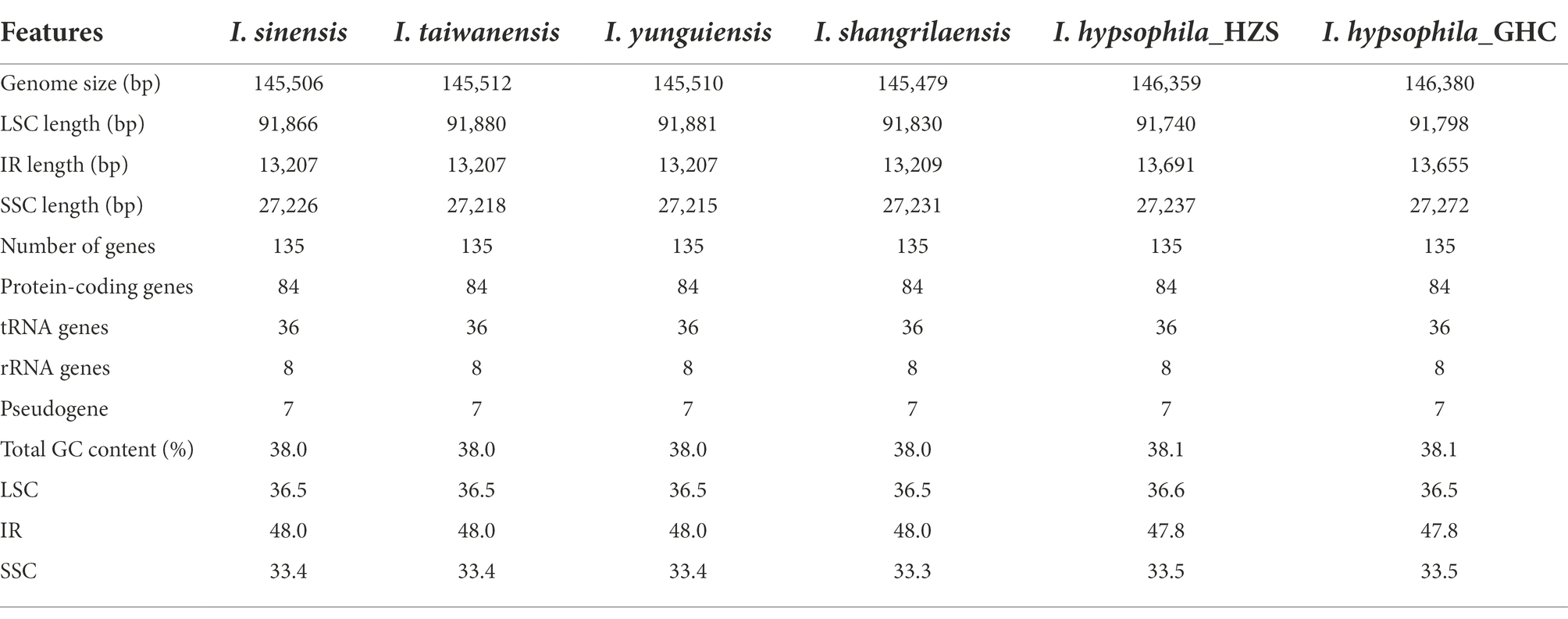
Table 1. The basic characteristics of the chloroplast genomes of six Isoetes individuals.

Table 2. Gene annotation of the Isoetes chloroplast genomes.
Comparative genome analysis
To investigate the extent of divergence in the sequences of the chloroplast genomes of the genus Isoetes, the six Isoetes chloroplast genome sequences were aligned by using the mVISTA, with the I. taiwanensis annotation as a reference. The results of sequence alignment revealed intragenus sequence differences in the chloroplast genome, and the results showed that the highly differentiated regions were mainly located in intergenic regions, such as trnR-trnN, and there were also variant regions in the coding regions such as rpoC2, rpoB, ycf1, and ycf2 (Figure 2). Overall, the high degree of gene order conservation was detected in the six chloroplast genomes, indicating evolutionary conservation at the genome scale.
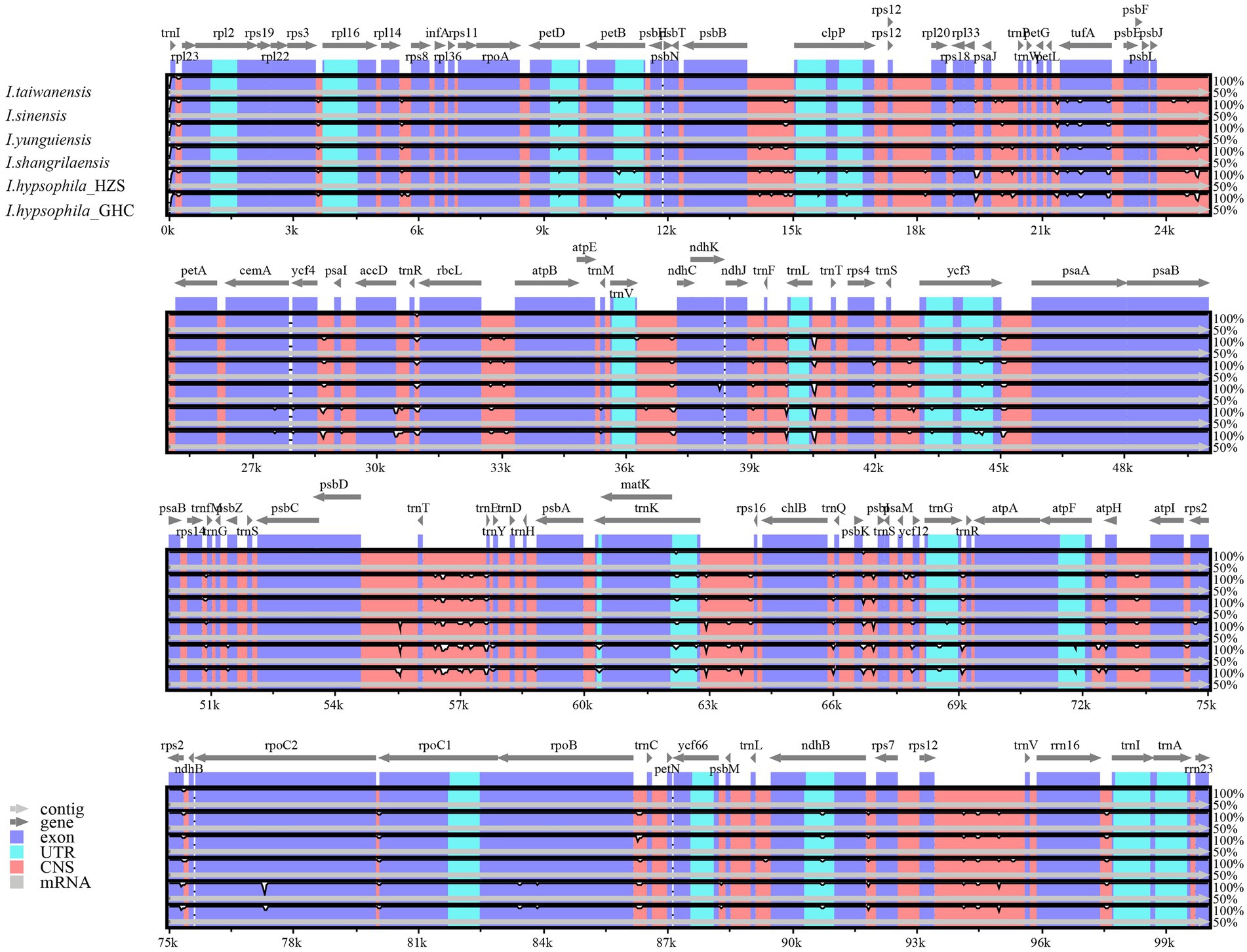
Figure 2. Comparison of the borders of LSC, SSC, and IR regions among six Isoetes chloroplast genomes.
A comprehensive comparison of the IR-SSC and IR-LSC boundaries of the chloroplast genomes of the six Isoetes individuals is presented in Figure 3. The genes ndhB, rps7, ycf2, and rpl23 are located at the junction of the LSC/IRa, IRa/SSC, SSC/Irb, and IRb/LSC borders, respectively. The ycf2 gene is located at the junction of the IRb/SSC and the border has moved toward the SSC region because there are 30 bp sequences situated at SSC region and the trnI gene is located in the LSC, 37–60 bp from the IRa/LSC boundary. Overall, the chloroplast genomic structure of the six Isoetes individual is concordant, while differences in the lengths of four regions lead to six genome sizes ranging from 145,479 to 146,380 bp.
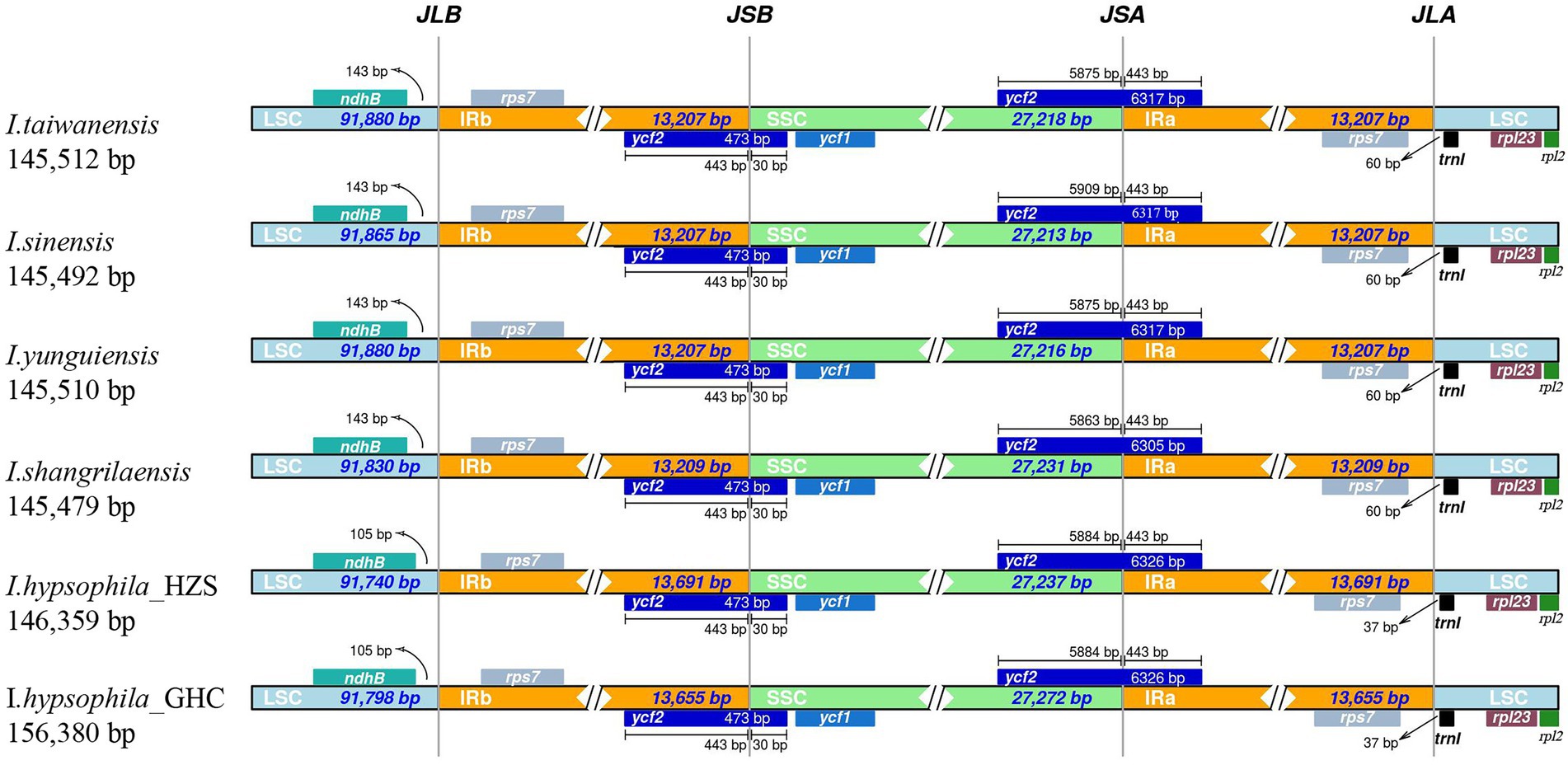
Figure 3. Global alignment of chloroplast genomes of six Isoetes, with the I. taiwanensis genome as the reference. Gray arrows indicate the direction of gene transcription. Red blocks indicate conserved non-coding sequences (CNS), and blue blocks indicate conserved genes. The y-axis represents the percent identity within 50%–100%.
RNA editing analysis
To identify RNA editing sites, all the transcriptome reads were mapped to the chloroplast genomes using Geneious software. The type, position, and editing efficiency of the editing sites are presented in Supplementary Table S5. The RNA editing analyses revealed the presence of 291 (I. hypsophila_GHC) to 579 (I. taiwanensis) RNA editing sites in six individuals. All editing types appearing in internal stop codons are U-to-C editing, which results in codon changes from stop codons (UAA, UGA) to glutamine (CAA) or arginine (CGA).
We found that the majority (nearly 78%) of edits in coding regions resulted in non-synonymous amino acid changes in six Isoetes individuals, and <5% of edits were synonymous rather than coding regions (UTRs and introns) and tRNAs have also found some RNA editing sites (Figure 4A). In addition, we investigated the effect of RNA editing on the hydrophobicity of the encoded amino acid, with the vast majority of non-synonymous RNA editing converting codons for hydrophilic amino acids to codons for hydrophobicity (Figure 4B).
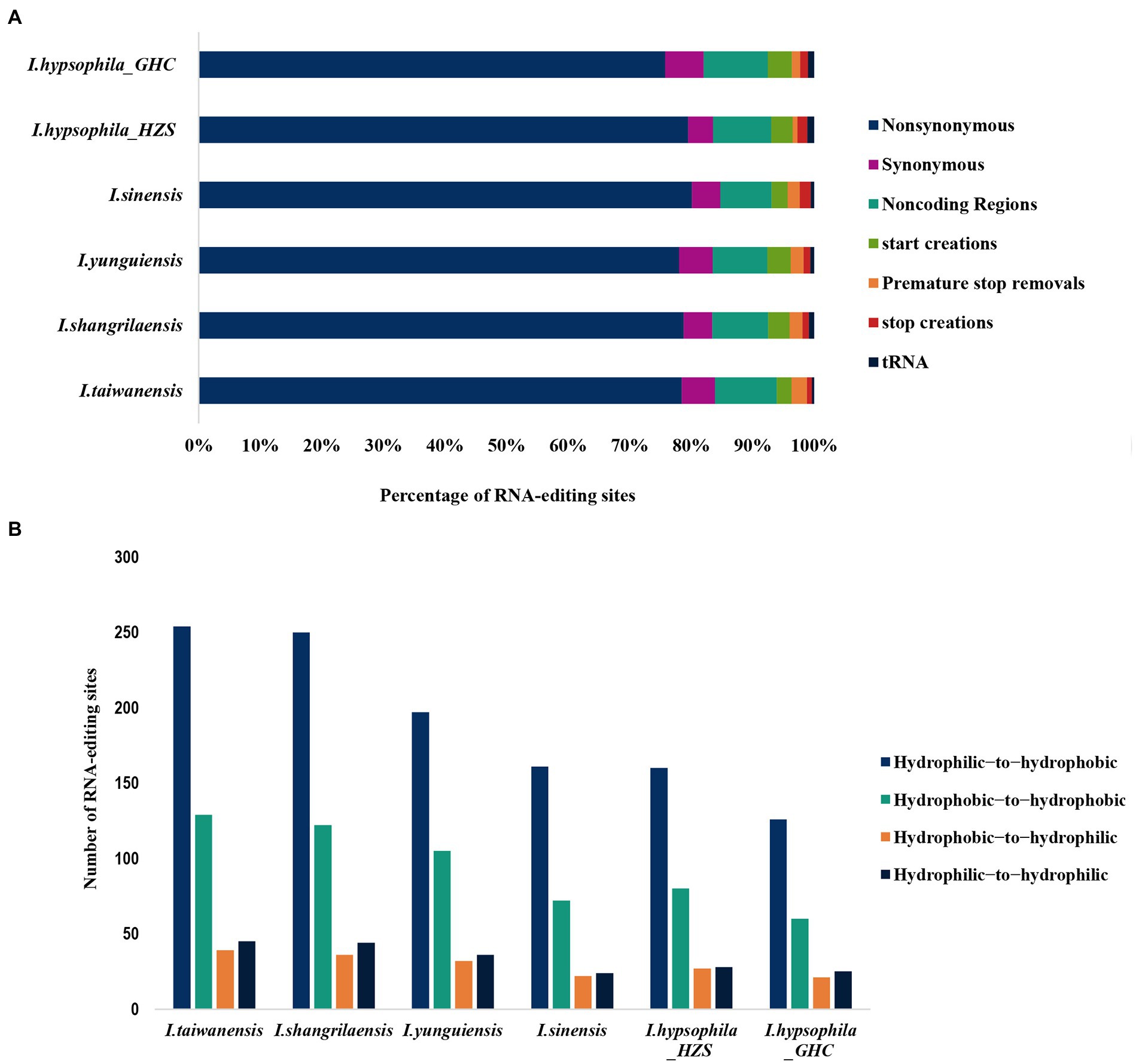
Figure 4. (A) Percentage of specific sequence modifications produced by RNA editing in sampled individuals. Histograms represent 100% of RNA edits detected. (B) Comparison of edit sites that lead to a change in hydrophobicity/hydrophilicity of the resulting amino acid via non-synonymous RNA editing.
Phylogenetic relationships and positive selection analysis
In order to reconstruct a phylogeny for further selection analyses, two phylogenetic trees were constructed using transcriptome data and the CDS from the chloroplast genomes, respectively. Although the long evolutionary history of Isoetes, with its split from the closest extant relatives in the Devonian, is a confounding factor in establishing phylogenetic relationships in this genus, our chloroplast-based phylogeny analysis may contribute to understanding the diversification of Isoetes and provide a highly robust framework for investigating the evolutionary history of the genus. The backbone of the phylogenetic reconstruction and most of the clades agree with previous studies by Larsén and Rydin (2016) and Pereira et al. (2017). For species from China, we found all phylogenetic trees exhibited similar clustering, which showed two different evolutionary branches. The resulting phylogenetic trees demonstrated that alpine species I. hypsophila were located on one branch, whereas other species were located on another branch (Figure 5).
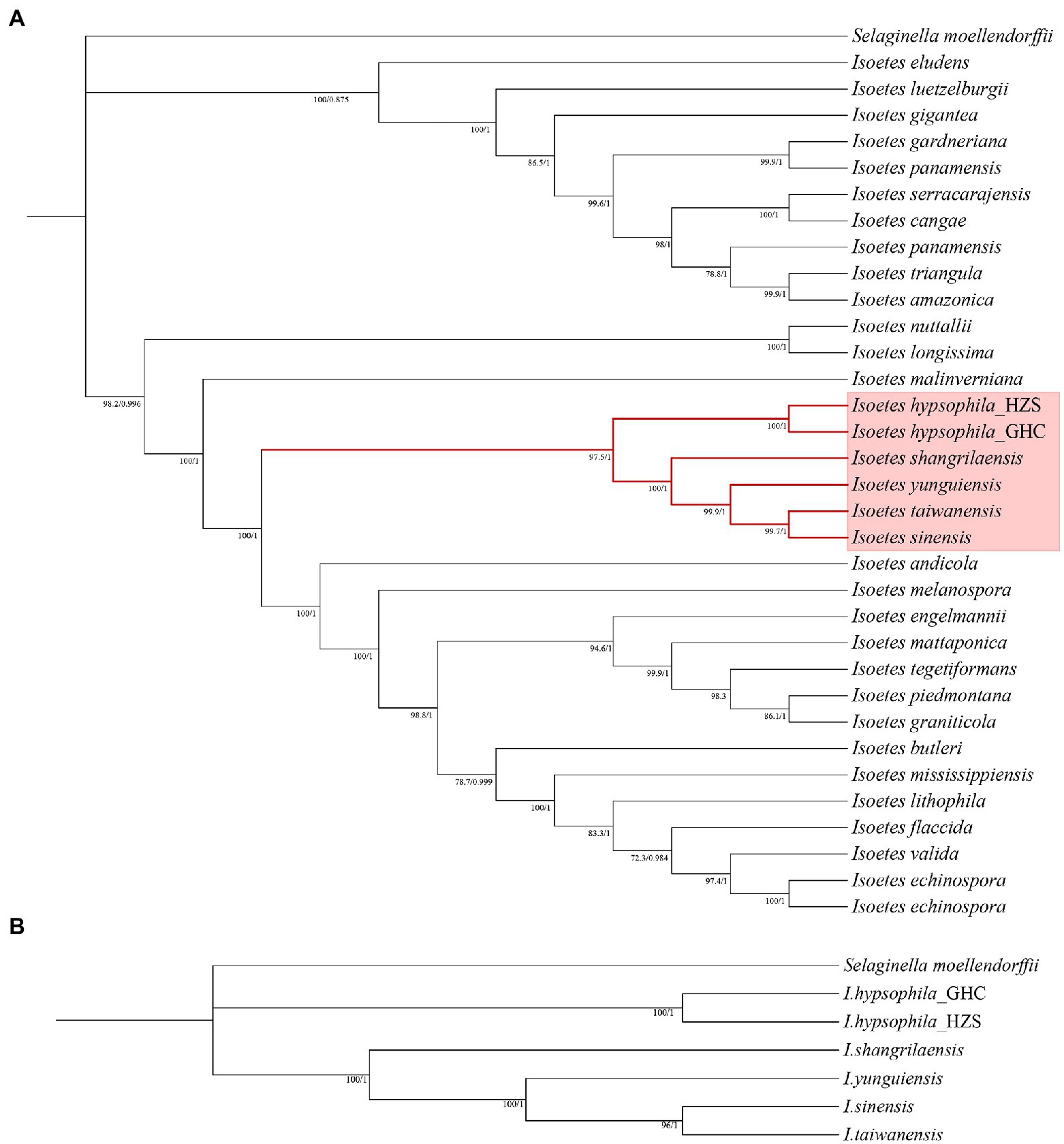
Figure 5. (A) Phylogenetic tree constructed using protein-coding regions of the chloroplast genomes. (B) Phylogenetic tree constructed using transcriptome data.
For the chloroplast genome, a total number of 86 common genes were involved in the positive selection analysis, of which 16 genes with internal stop codons were corrected back to normal coding sequences before analysis (Supplementary Table S6). The branch-site model detected only one gene (rps3) containing sites that had been subject to positive selection (Table 3). In addition, a total of 2,798 single-copy orthologs were identified in transcriptome data among the six individuals. Of these genes, 46 positively selected genes (PSGs) were annotated and enriched on the KEGG pathway (Supplementary Table S7). The top 20 clusters of the KEGG functional analyses with their representative enriched pathway are shown in Figure 6. The pathway “carbon fixation in photosynthetic organisms” was enriched, in which three PSGs encode fructose-1,6-bisphosphatase (FBP), ribulose-phosphate 3-epimerase (RPE), and malate dehydrogenase (MDH).

Table 3. Positively selected genes and sites detected in the chloroplast genomes of Isoetes species.
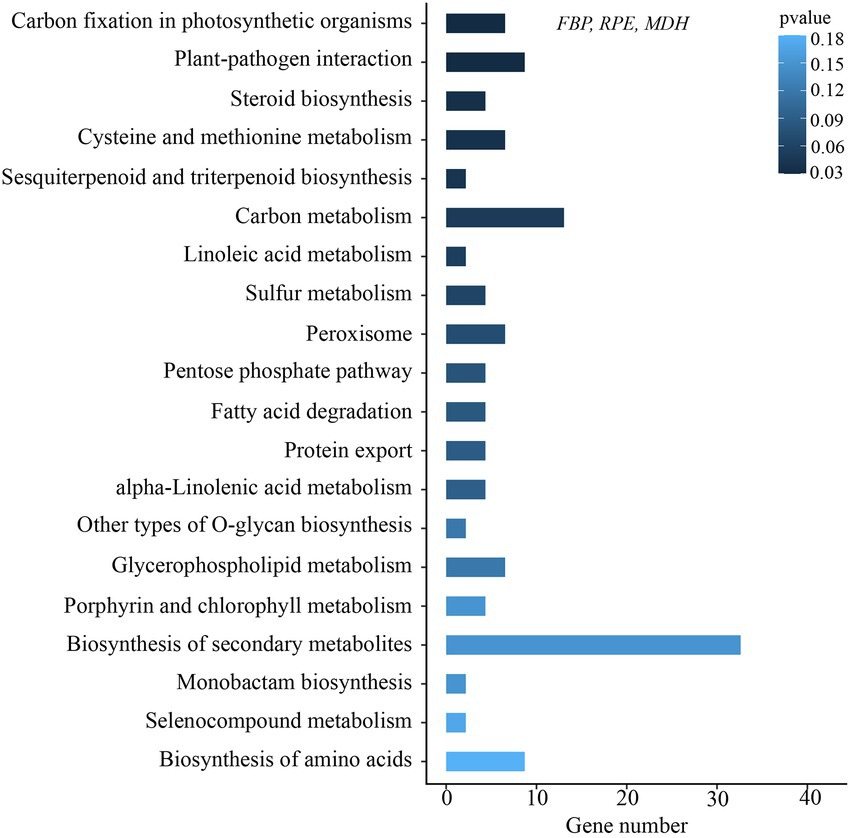
Figure 6. Distribution of KEGG classifications of PSGs in I. hypsophila.
Discussion
Chloroplast genome features
The whole chloroplast genome sequences newly obtained herein for six individuals are not only very similar in size (145,479–146,380 bp; Table 1), but also in overall structure, gene order, and content (Figure 2). These findings are consistent with a previous study on Isoetes, which showed that the chloroplast sequences and gene arrangements were conserved (Pereira et al., 2021a), Although the sequence is conserved, there are still some features worth discussing. Both large-scale studies (Shaw et al., 2014) and specific case studies (Ye et al., 2018; Alwadani et al., 2019) have demonstrated higher differences in non-coding regions, some of the non-coding sequences we detected had hypervariable regions, such as psbB-clpP, psbJ-petA, trnK-rps16, rpoB-trnC, and rps12-trnV. Furthermore, we found greater differences in chloroplast genome sequences between the high-altitude species I. hypsophila and other Isoetes species, such as rpoC2 and rpoB, suggesting that altitude may drive genetic differentiation, as evidenced by other studies (Liu et al., 2020). These newly discovered regions can be used for subsequent species identification and provide additional phylogenetic information.
In terms of the GC content of the six Isoetes, the total GC content of the complete chloroplast genome is ~38%, similar to the previously published (Pereira et al., 2021a). In general, the effect of GC content on chloroplast genome stability is more pronounced because GC base pairs and AT base pairs are thermodynamically different in stability (Yang et al., 2021). Among the LSC, SSC, and IR regions, the IR regions have the highest GC content, followed by the LSC and SSC regions. The IR region has the highest GC content among the four regions, probably because of the high G/C content of rRNAs in this region.
RNA editing sites
In plants, RNA editing plays an irreplaceable role in growth and development. RNA editing has been observed in the chloroplasts of extant descendants of early land plants other than liverworts and mosses. In angiosperm chloroplasts, RNA editing is mostly restricted to a C-to-U conversion, and the conversion occurs at about 30 different positions (Yu et al., 2020), whereas the range of variation in RNA-editing sites is even more remarkable in hornworts and fern chloroplasts. It is rare in the moss Physcomitrella patens, which holds only 11 C-to-U edit sites (Rüdinger et al., 2009), and is completely absent from the liverwort Marchantia polymorpha (Rüdinger et al., 2008), whereas it is most abundant in hornworts and fern, with over 300 different positions (Stern et al., 2010). The reduced number of PPR genes and absence of RNA editing in marchantiid liverworts are most probably secondary losses, as the organellar RNA editing and plant-specific extensions of PPR genes were also found in jungermanniid liverworts (Rüdinger et al., 2008; Zhang et al., 2020). Our analyses revealed obvious differences in the number of RNA editing sites in the chloroplast genomes of the genus Isoetes, that is, the range of number from 291 in I. hypsophila_GHC to 579 in I. taiwanensis (Supplementary Table S5). Interestingly, we found that there were large differences in the number of RNA editing sites of the same species in different populations, including 291 RNA editing sites in I. hypsophila_GHC, and 354 RNA editing sites in I. hypsophila_HZS. This difference also exists in the RNA editing of the mitochondria of the three ecotypes of Arabidopsis thaliana (Zehrmann et al., 2008). This situation may suggest that the environment has a greater influence on the RNA editing site, and subsequent studies can focus on this aspect. The pentatricopeptide repeat (PPR) is a family of RNA binding proteins involved in specific RNA processing events such as RNA editing, translation initiation, and transcript processing (Ichinose and Sugita, 2018). Previous studies have shown that chloroplast RNA editing abundance is positively correlated with the PPR gene family (Rudinger et al., 2012; Xu et al., 2018). Taken together, these species with a large number of RNA editing sites are early landing plants. Through RNA editing, PPR proteins can act as “repair” factors, alleviating DNA damage caused by increased UV exposure during adaptive landing (Zhang et al., 2020).
A pseudogene is rendered non-functional through the introduction of stop codons predominantly in the chloroplast genome. Conversely, in mosses and ferns, genes that contain internal stop codons can still make proteins function properly because U-to-C RNA editing can convert translation internal stop codons (such as the UAA termination signal) into CAA triplet encoding the amino acid glycine. In addition, there are some special cases, such as in Selaginellaceae, because the absence of U-to-C RNA editing cannot eliminate the internal stop codons, so some genes become nonfunctional pseudogenes (Gerke et al., 2020). However, Isoetes, which is the closest relative of Selaginellaceae, can convert internal stop codons to functional amino acids. This indicates that these two species that diverged from a common Lycopsida ancestor may evolve different mechanisms to achieve the same ends.
For each species, roughly 78% of the RNA-editing events did not involve a non-synonymous amino acid change in start or stop codons (Figure 4A). Furthermore, we found that the proportion of Isoetes RNA editing sites in non-coding regions and tRNAs are similar (Figure 4A), while we observed that RNA editing greatly increased the proportion of hydrophobic amino acids (Figure 4B), and the hydrophobicity has long been considered as one of the primary drivers of protein folding and protein function (Moelbert et al., 2004; Li et al., 2016). Therefore, we speculate that the increase in the hydrophobicity due to a large number of RNA editing may facilitate the translation of mRNA into polypeptides with folded structures at the appropriate locations, which are often necessary for proteins to form functional three-dimensional (3D) structures (Yura and Go, 2008).
Adaptive evolution
With the recent development of genome technology, the investigation of genome-wide molecular mechanisms of high-altitude adaptation has attracted great attention in the last few years (Chen et al., 2019; Hao et al., 2019; Zhang et al., 2021). Even though whole nuclear genome sequencing allows investigation of the impact of selection events at the genome-wide level, it is expensive and not easily available. On the contrary, transcriptome sequencing has been described as a powerful method for genome-wide analysis of high-altitude adaptation and is cheaper and easier available than the nuclear genome. Additionally, QTP is the highest plateau in the world, with an extreme environment of hypoxia, low temperature, and strong solar radiation (Mao et al., 2021). Solar radiation is one of the main stresses faced by alpine plants, and chloroplasts, as the site for photosynthesis, may have acquired adaptive strategies to strong solar radiation (Yoshida et al., 2019). Thus, both transcriptome and chloroplast genomes represent a great system to study the footprint of alpine plants in adaptation to QTP. Nevertheless, previous studies have either focused on transcriptome or chloroplast genome to study altitude adaptation, while the feasibility of integrating transcriptome and chloroplast genome to uncover the adaptive mechanisms to QTP remains less explored. Thus, in this study, we integrated the chloroplast genome and transcriptome data to explore the molecular mechanism of the adaptation of alpine plants to the high altitude of QTP, and we expect our research can provide a reference for future genome-wide studies on the adaptive evolution of alpine plants on the QTP.
Alpine species on the QTP have to evolve to have a high ability to adapt to extremely harsh environments (Thompson et al., 2000; Norsang et al., 2011). Intense UV radiation is a major environmental stressors for plants, and recent studies have revealed candidate genes for plateau adaptability, mainly associated with UV radiation in a variety of plants (Mao et al., 2021). We anticipated that some genes in the chloroplast genome of I. hypsophila might have undergone adaptive evolution to adapt to the alpine environment, although overall genome size, structure, and gene number have changed slightly. Using I. hypsophila as the foreground, the branch-site model detected rps3 as the possible positively selected gene (PSG). The rps3 (Ribosomal protein S3) gene encodes ribosomal small subunit protein 3, which belongs to the ribosomal protein S3P family and is a part of the ribosomal 40s subunit (Korovesi et al., 2018), and plays important roles in repairing damaged DNA and apoptosis (Dong et al., 2017; Kim et al., 2018). The branch-site model detected two positively selected sites (96S,123Y) of rps3 in I. hypsophila, which were probably involved in the protection of I. hypsophila from strong UV radiation, drought, and other stressful environments in higher altitudes.
In addition, our study revealed many nuclear-encoded genes involved in high-altitude adaption and the genes may play an important role in the adaption to the high-altitude environment of QTP. Through the positive selection analysis of transcriptome data, a total of 46 positive selection genes were enriched in the KEGG pathway, and the “carbon fixation in photosynthetic organisms” pathway was significantly enriched (Figure 6), of which all three detected PSGs encode proteins with functions related to photosynthesis (NADP-MDH, RPE, and FBP). In plants, the NADP-MDH is the key enzyme controlling the malate valve, which plays a role in the export of reducing equivalents in photosynthesizing chloroplasts (Vaseghi et al., 2018). The RPE is an enzyme in the chloroplast-localized oxidized pentose phosphate pathway that is essential for both the Calvin cycle and the reverse pentose phosphate pathway (Liu et al., 2015). The FBPase is a rate-limiting enzyme in the carbohydrate metabolism and the Calvin cycle, which plays a pivotal role in carbohydrate biosynthesis (Lee et al., 2008). A previous study reported that the increased cell growth rate and enhanced photosynthetic activity could be achieved by increasing the levels of FBP aldolase in Anabaena (Ma et al., 2007). Given that high elevations are characterized by high levels of UV radiation, which pose a serious challenge to plant photosynthesis and may lead to positive selection of the related genes (Li et al., 2020; Moutinho et al., 2020), it is possible that these three PSGs were driven by natural selection in high elevation environments and may contribute to high-altitude adaptation in Isoetes species.
Data availability statement
The data presented in the study are deposited in the NCBI repository, accession number: SRR18531157, SRR18531155, SRR18531156, SRR18531154, SRR18531159, SRR18531158, OM283821, OM283822, OM283818, OM283820, OM283819, and OM283817.
Author contributions
XinL and XiaL designed the study. YY and XY assembled, annotated, and analyzed the plastomes. PW and YY detected RNA editing sites. YY drafted the manuscript. CL and ZC revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by a grant from the National Natural Science Foundation of China (31170203).
Acknowledgments
We sincerely thank Xiaokang Dai, Weiwei Lu, and Shengnan Wang, for their critical assistance on sample collection.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.924559/full#supplementary-material
References
Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
Alwadani, K. G., Janes, J. K., and Andrew, R. L. (2019). Chloroplast genome analysis of box-ironbark Eucalyptus. Mol. Phylogenet. Evol. 136, 76–86. doi: 10.1016/j.ympev.2019.04.001
Amiryousefi, A., Hyvonen, J., and Poczai, P. (2018). IRscope: an online program to visualize the junction sites of chloroplast genomes. Bioinformatics 34, 3030–3031. doi: 10.1093/bioinformatics/bty220
Banks, J. A., Nishiyama, T., Hasebe, M., Bowman, J. L., Gribskov, M., DePamphilis, C., et al. (2011). The Selaginella genome identifies genetic changes associated with the evolution of vascular plants. Science 332, 960–963. doi: 10.1126/science.1203810
Binder, S., Marchfelder, A., and Brennicke, A. (1994). RNA editing of tRNAPhe and tRNACys in mitochondria of Oenothera berteriana is initiated in precursor molecules. Mol. Gen. Genet. MGG 244, 67–74. doi: 10.1007/BF00280188
Capella-Gutiérrez, S., Silla-Martínez, J. M., and Gabaldón, T. (2009). trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973. doi: 10.1093/bioinformatics/btp348
Chateigner-Boutin, A. L., and Small, I. (2011). Organellar RNA editing. Wiley Interdiscip. Rev. RNA 2, 493–506. doi: 10.1002/wrna.72
Chen, J.-H., Huang, Y., Brachi, B., Yun, Q.-Z., Zhang, W., Lu, W., et al. (2019). Genome-wide analysis of cushion willow provides insights into alpine plant divergence in a biodiversity hotspot. Nat. Commun. 10, 1–12. doi: 10.1038/s41467-019-13128-y
DiMichele, W. A., Pfefferkorn, H. W., and Gastaldo, R. A. (2001). Response of late carboniferous and early Permian plant communities to climate change. Annu. Rev. Earth Planet. Sci. 29, 461–487. doi: 10.1146/annurev.earth.29.1.461
Dong, J., Aitken, C. E., Thakur, A., Shin, B.-S., Lorsch, J. R., and Hinnebusch, A. G. (2017). Rps3/uS3 promotes mRNA binding at the 40S ribosome entry channel and stabilizes preinitiation complexes at start codons. Proc. Natl. Acad. Sci. 114, E2126–E2135. doi: 10.1073/pnas.1620569114
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Frazer, K. A., Pachter, L., Poliakov, A., Rubin, E. M., and Dubchak, I. (2004). VISTA: computational tools for comparative genomics. Nucleic Acids Res 32, W273–W279. doi: 10.1093/nar/gkh458
Gerke, P., Szövényi, P., Neubauer, A., Lenz, H., Gutmann, B., McDowell, R., et al. (2020). Towards a plant model for enigmatic U-to-C RNA editing: the organelle genomes, transcriptomes, editomes and candidate RNA editing factors in the hornwort Anthoceros agrestis. New Phytol. 225, 1974–1992. doi: 10.1111/nph.16297
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652. doi: 10.1038/nbt.1883
Green, B. R. (2011). Chloroplast genomes of photosynthetic eukaryotes. Plant J. 66, 34–44. doi: 10.1111/j.1365-313X.2011.04541.x
Guo, W. H., Grewe, F., and Mower, J. P. (2015). Variable frequency of plastid RNA editing among ferns and repeated loss of Uridine-to-Cytidine editing from vascular plants. PLoS One 10:e0117075. doi: 10.1371/journal.pone.0117075
Guo, W., Xin, M., Wang, Z., Yao, Y., Hu, Z., Song, W., et al. (2020). Origin and adaptation to high altitude of Tibetan semi-wild wheat. Nat. Commun. 11, 5085. doi: 10.1038/s41467-020-18738-5
Hao, Y., Xiong, Y., Cheng, Y., Song, G., Jia, C., Qu, Y., et al. (2019). Comparative transcriptomics of 3 high-altitude passerine birds and their low-altitude relatives. Proc. Natl. Acad. Sci. 116, 11851–11856. doi: 10.1073/pnas.1819657116
Hecht, J., Grewe, F., and Knoop, V. (2011). Extreme RNA editing in coding islands and abundant microsatellites in repeat sequences of Selaginella moellendorffii mitochondria: the root of frequent plant mtDNA recombination in early tracheophytes. Genome Biol. Evol. 3, 344–358. doi: 10.1093/gbe/evr027
Henriquez, C. L., Abdullah,, Ahmed, I., Carlsen, M. M., Zuluaga, A., Croat, T. B., et al. (2020). Evolutionary dynamics of chloroplast genomes in subfamily Aroideae (Araceae). Genomics 112, 2349–2360. doi: 10.1016/j.ygeno.2020.01.006
Hong, L., Qing-Feng, W., and Taylor, W. C. (2005). Isoetes orientalis (Isoetaceae), a new hexaploid quillwort from China. Novon 15, 164–167.
Huerta-Cepas, J., Forslund, K., Coelho, L. P., Szklarczyk, D., Jensen, L. J., von Mering, C., et al. (2017). Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. Mol. Biol. Evol. 34, 2115–2122. doi: 10.1093/molbev/msx148
Ichinose, M., and Sugita, M. (2016). RNA editing and its molecular mechanism in plant organelles. Genes (Basel) 8, 5. doi: 10.3390/genes8010005
Ichinose, M., and Sugita, M. (2018). The DYW domains of Pentatricopeptide repeat RNA editing factors contribute to discriminate target and non-target editing sites. Plant Cell Physiol. 59, 1652–1659. doi: 10.1093/pcp/pcy086
Jin, J. J., Yu, W. B., Yang, J. B., Song, Y., dePamphilis, C. W., Yi, T. S., et al. (2020). GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21, 241. doi: 10.1186/s13059-020-02154-5
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Kim, W., Youn, H., Lee, S., Kim, E., Kim, D., Sub Lee, J., et al. (2018). RNF138-mediated ubiquitination of rpS3 is required for resistance of glioblastoma cells to radiation-induced apoptosis. Exp. Mol. Med. 50:e434. doi: 10.1038/emm.2017.247
Korovesi, A. G., Ntertilis, M., and Kouvelis, V. N. (2018). Mt-rps3 is an ancient gene which provides insight into the evolution of fungal mitochondrial genomes. Mol. Phylogenet. Evol. 127, 74–86. doi: 10.1016/j.ympev.2018.04.037
Kugita, M., Yamamoto, Y., Fujikawa, T., Matsumoto, T., and Yoshinaga, K. (2003). RNA editing in hornwort chloroplasts makes more than half the genes functional. Nucleic Acids Res. 31, 2417–2423. doi: 10.1093/nar/gkg327
Larsén, E., and Rydin, C. (2016). Disentangling the phylogeny of Isoetes (Isoetales), using nuclear and plastid data. Int. J. Plant Sci. 177, 157–174. doi: 10.1086/684179
Lee, S. K., Jeon, J. S., Boernke, F., Voll, L., Cho, J. I., Goh, C. H., et al. (2008). Loss of cytosolic fructose-1, 6-bisphosphatase limits photosynthetic sucrose synthesis and causes severe growth retardations in rice (Oryza sativa). Plant Cell Environ. 31, 1851–1863. doi: 10.1111/j.1365-3040.2008.01890.x
Li, H., and Durbin, R. (2010). Fast and accurate long-read alignment with burrows–wheeler transform. Bioinformatics 26, 589–595. doi: 10.1093/bioinformatics/btp698
Li, X., Huang, Y., Dai, X., and Liu, X. (2019). Isoetes shangrilaensis, a new species of Isoetes from Hengduan mountain region of Shangri-la. Yunnan. Phytotaxa 397, 65–73. doi: 10.11646/phytotaxa.397.1.6
Li, W., Li, K., Huang, Y., Shi, C., Hu, W.-S., Zhang, Y., et al. (2020). SMRT sequencing of the Oryza rufipogon genome reveals the genomic basis of rice adaptation. Commun. Biol. 3, 1–11. doi: 10.1038/s42003-020-0890-8
Li, Z. C., Liu, Z. Q., Zhong, W. Q., Huang, M. H., Wu, N., Xie, Y., et al. (2016). Large-scale identification of human protein function using topological features of interaction network. Sci. Rep. 6, 1–11. doi: 10.1038/srep37179
Liu, X., Gituru, W. R., and Wang, Q. F. (2004). Distribution of basic diploid and polyploid species of Isoetes in East Asia. J. Biogeogr. 31, 1239–1250. doi: 10.1111/j.1365-2699.2004.01115.x
Liu, M., Li, X., Liu, Y., Shi, Y., and Ma, X. (2015). Analysis of differentially expressed genes under UV-B radiation in the desert plant Reaumuria soongorica. Gene 574, 265–272. doi: 10.1016/j.gene.2015.08.026
Liu, X., Ma, Y., Wan, Y., Li, Z., and Ma, H. (2020). Genetic diversity of Phyllanthus emblica from two different climate type areas. Front. Plant Sci. 11:580812. doi: 10.3389/fpls.2020.580812
Liu, J., Qi, Z.-C., Zhao, Y.-P., Fu, C.-X., and Xiang, Q.-Y. J. (2012). Complete cpDNA genome sequence of Smilax China and phylogenetic placement of Liliales–influences of gene partitions and taxon sampling. Mol. Phylogenet. Evol. 64, 545–562. doi: 10.1016/j.ympev.2012.05.010
Liu, X., Wang, J. Y., and Wang, Q. F. (2005). Current status and conservation strategies for Isoetes in China: a case study for the conservation of threatened aquatic plants. Oryx 39, 335–338. doi: 10.1017/S0030605305000712
Ma, W. M., Wei, L. Z., Wang, Q. X., Shi, D. J., and Chen, H. B. (2007). Increased activity of the non-regulated enzymes fructose-1,6-bisphosphate aldolase and triosephosphate isomerase in Anabaena sp strain PCC 7120 increases photosynthetic yield. J. Appl. Phycol. 19, 207–213. doi: 10.1007/s10811-006-9125-8
Mao, K. S., Wang, Y., and Liu, J. Q. (2021). Evolutionary origin of species diversity on the Qinghai–Tibet plateau. J. Syst. Evol. 59, 1142–1158. doi: 10.1111/jse.12809
Moelbert, S., Emberly, E., and Tang, C. (2004). Correlation between sequence hydrophobicity and surface-exposure pattern of database proteins. Protein Sci. 13, 752–762. doi: 10.1110/ps.03431704
Moreno-Hagelsieb, G., and Latimer, K. (2008). Choosing BLAST options for better detection of orthologs as reciprocal best hits. Bioinformatics 24, 319–324. doi: 10.1093/bioinformatics/btm585
Moutinho, A. F., Bataillon, T., and Dutheil, J. Y. (2020). Variation of the adaptive substitution rate between species and within genomes. Evol. Ecol. 34, 315–338. doi: 10.1007/s10682-019-10026-z
Nielsen, R., and Yang, Z. (1998). Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics 148, 929–936. doi: 10.1093/genetics/148.3.929
Norsang, G., Kocbach, L., Stamnes, J., Tsoja, W., and Pincuo, N. (2011). Spatial distribution and temporal variation of solar UV radiation over the Tibetan plateau. Appl. Phys. Res. 3, 37. doi: 10.5539/apr.v3n1p37
Oldenkott, B., Yamaguchi, K., Tsuji-Tsukinoki, S., Knie, N., and Knoop, V. (2014). Chloroplast RNA editing going extreme: more than 3400 events of C-to-U editing in the chloroplast transcriptome of the lycophyte Selaginella uncinata. RNA 20, 1499–1506. doi: 10.1261/rna.045575.114
Palmer, J. D. (1985). Comparative organization of chloroplast genomes. Annu. Rev. Genet. 19, 325–354. doi: 10.1146/annurev.ge.19.120185.001545
Pereira, J. B., Giulietti, A. M., Pires, E. S., Laux, M., Watanabe, M. T., Oliveira, R. R., et al. (2021a). Chloroplast genomes of key species shed light on the evolution of the ancient genus Isoetes. J. Syst. Evol. 59, 429–441. doi: 10.1111/jse.12693
Pereira, J. B. S., Giulietti, A. M., Prado, J., Vasconcelos, S., Watanabe, M. T. C., Pinange, D. S. B., et al. (2021b). Plastome-based phylogenomics elucidate relationships in rare Isoetes species groups from the Neotropics. Mol. Phylogenet. Evol. 161, 107–177. doi: 10.1016/j.ympev.2021.107177
Pereira, J., Labiak, P. H., Stützel, T., and Schulz, C. (2017). Origin and biogeography of the ancient genus Isoëtes with focus on the Neotropics. Bot. J. Linn. Soc. 185, 253–271. doi: 10.1093/botlinnean/box057
Pfeiffer, N. E. (1922). Monograph of the Isoetaceae. Ann. Mo. Bot. Gard. 9, 79–233. doi: 10.2307/2990000
Pigg, K. B. (1992). Evolution of Isoetalean Lycopsids. Ann. Mo. Bot. Gard. 79, 589–612. doi: 10.2307/2399754
Pigg, K. B. (2001). Isoetalean lycopsid evolution: from the Devonian to the present. Am. Fern J. 91, 99–114. doi: 10.1640/0002-8444(2001)091[0099:Ileftd]2.0.Co;2
Pryer, K. M., Schneider, H., Smith, A. R., Cranfill, R., Wolf, P. G., Hunt, J. S., et al. (2001). Horsetails and ferns are a monophyletic group and the closest living relatives to seed plants. Nature 409, 618–622. doi: 10.1038/35054555
Qiao, Q., Huang, Y. Y., Qi, J., Qu, M. Z., Jiang, C., Lin, P. C., et al. (2016a). The genome and transcriptome of Trichormus sp NMC-1: insights into adaptation to extreme environments on the Qinghai-Tibet plateau. Sci. Rep. 6, 1–10. doi: 10.1038/srep29404
Qiao, Q., Wang, Q., Han, X., Guan, Y. L., Sun, H., Zhong, Y., et al. (2016b). Transcriptome sequencing of Crucihimalaya himalaica (Brassicaceae) reveals how Arabidopsis close relative adapt to the Qinghai-Tibet plateau. Sci. Rep. 6, 1–8. doi: 10.1038/srep21729
Qing-Feng, W., Xing, L., Taylor, W. C., and Zhao-Rong, H. (2002). Isoetes yunguiensis (Isoetaceae), a new basic diploid quillwort from China. Novon 12, 587–591. doi: 10.2307/3393143
Raubeson, L. A., and Jansen, R. K. (1992a). Chloroplast DNA evidence on the ancient evolutionary split in vascular land plants. Science 255, 1697–1699. doi: 10.1126/science.255.5052.1697
Raubeson, L. A., and Jansen, R. K. (1992b). A rare chloroplast-DNA structural mutation is shared by all conifers. Biochem. Syst. Ecol. 20, 17–24. doi: 10.1016/0305-1978(92)90067-N
Ronquist, F., Teslenko, M., Van Der Mark, P., Ayres, D. L., Darling, A., Höhna, S., et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Rüdinger, M., Funk, H. T., Rensing, S. A., Maier, U. G., and Knoop, V. (2009). RNA editing: only eleven sites are present in the Physcomitrella patens mitochondrial transcriptome and a universal nomenclature proposal. Mol. Gen. Genomics. 281, 473–481. doi: 10.1007/s00438-009-0424-z
Rüdinger, M., Polsakiewicz, M., and Knoop, V. (2008). Organellar RNA editing and plant-specific extensions of pentatricopeptide repeat proteins in jungermanniid but not in marchantiid liverworts. Mol. Biol. Evol. 25, 1405–1414. doi: 10.1093/molbev/msn084
Rudinger, M., Volkmar, U., Lenz, H., Groth-Malonek, M., and Knoop, V. (2012). Nuclear DYW-type PPR gene families diversify with increasing RNA editing frequencies in liverwort and Moss mitochondria. J. Mol. Evol. 74, 37–51. doi: 10.1007/s00239-012-9486-3
Schafran, P. W., Zimmer, E. A., Taylor, W. C., and Musselman, L. J. (2018). A whole chloroplast genome phylogeny of diploid species of Isoetes (Isoetaceae, Lycopodiophyta) in the southeastern United States. Castanea 83, 224–235. doi: 10.2179/17-132
Schallenberg-Rüdinger, M., and Knoop, V. (2016). “Coevolution of organelle RNA editing and nuclear specificity factors in early land plants,” in Advances in Botanical Research (Elsevier), 37–93.
Schattner, P., Brooks, A. N., and Lowe, T. M. (2005). The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 33, W686–W689. doi: 10.1093/nar/gki366
Shahzadi, I., Abdullah,, Mehmood, F., Ali, Z., Ahmed, I., and Mirza, B. (2020). Chloroplast genome sequences of Artemisia maritima and Artemisia absinthium: comparative analyses, mutational hotspots in genus Artemisia and phylogeny in family Asteraceae. Genomics 112, 1454–1463. doi: 10.1016/j.ygeno.2019.08.016
Shaw, J., Shafer, H. L., Leonard, O. R., Kovach, M. J., Schorr, M., and Morris, A. B. (2014). Chloroplast DNA sequence utility for the lowest phylogenetic and phylogeographic inferences in angiosperms: the tortoise and the hare IV. Am. J. Bot. 101, 1987–2004. doi: 10.3732/ajb.1400398
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Stefan, R. (2016). Genomes and Evolution of Charophytes, Bryophytes, Lycophytes and Ferns Academic Press.
Stern, D. B., Goldschmidt-Clermont, M., and Hanson, M. R. (2010). Chloroplast RNA metabolism. Annu. Rev. Plant Biol. 61, 125–155. doi: 10.1146/annurev-arplant-042809-112242
Takenaka, M., Zehrmann, A., Verbitskiy, D., Hartel, B., and Brennicke, A. (2013). RNA editing in plants and its evolution. Annu. Rev. Genet. 47, 335–352. doi: 10.1146/annurev-genet-111212-133519
Taylor, W. C., and Hickey, R. J. (1992). Habitat, evolution, and speciation in Isoetes. Ann. Mo. Bot. Gard. 79, 613–622. doi: 10.2307/2399755
Thompson, L. G., Yao, T., Mosley-Thompson, E., Davis, M. E., Henderson, K. A., and Lin, P. (2000). A high-resolution millennial record of the south asian monsoon from himalayan ice cores. Science 289, 1916–1919. doi: 10.1126/science.289.5486.1916
Tillich, M., Lehwark, P., Morton, B. R., and Maier, U. G. (2006). The evolution of chloroplast RNA editing. Mol. Biol. Evol. 23, 1912–1921. doi: 10.1093/molbev/msl054
Tillich, M., Lehwark, P., Pellizzer, T., Ulbricht-Jones, E. S., Fischer, A., Bock, R., et al. (2017). GeSeq–versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45, W6–W11. doi: 10.1093/nar/gkx391
Vaseghi, M. J., Chibani, K., Telman, W., Liebthal, M. F., Gerken, M., Schnitzer, H., et al. (2018). The chloroplast 2-cysteine peroxiredoxin functions as thioredoxin oxidase in redox regulation of chloroplast metabolism. Elife 7:e38194. doi: 10.7554/eLife.38194
Wickell, D., Kuo, L.-Y., Yang, H.-P., Dhabalia Ashok, A., Irisarri, I., Dadras, A., et al. (2021). Underwater CAM photosynthesis elucidated by Isoetes genome. Nat. Commun. 12, 1–13. doi: 10.1038/s41467-021-26644-7
Wolf, P. G., Rowe, C. A., and Hasebe, M. (2004). High levels of RNA editing in a vascular plant chloroplast genome: analysis of transcripts from the fern Adiantum capillus-veneris. Gene 339, 89–97. doi: 10.1016/j.gene.2004.06.018
Wood, D., Besnard, G., Beerling, D. J., Osborne, C. P., and Christin, P.-A. (2020). Phylogenomics indicates the “living fossil” Isoetes diversified in the Cenozoic. PLoS One 15:e0227525. doi: 10.1371/journal.pone.0227525
Xing, L., Yong, W., Qing-Feng, W., and You-Hao, G. (2002). Chromosome numbers of the Chinese Isoetes and their taxonomical significance. J. Syst. Evol. 40, 351–356.
Xu, Z., Xin, T., Bartels, D., Li, Y., Gu, W., Yao, H., et al. (2018). Genome analysis of the ancient Tracheophyte Selaginella tamariscina reveals evolutionary features relevant to the Acquisition of Desiccation Tolerance. Mol. Plant 11, 983–994. doi: 10.1016/j.molp.2018.05.003
Yang, Z. (2007). PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591. doi: 10.1093/molbev/msm088
Yang, X., Zhou, T., Su, X., Wang, G., Zhang, X., Guo, Q., et al. (2021). Structural characterization and comparative analysis of the chloroplast genome of Ginkgo biloba and other gymnosperms. J. For. Res. 32, 765–778. doi: 10.1007/s11676-019-01088-4
Ye, W. Q., Yap, Z. Y., Li, P., Comes, H. P., and Qiu, Y. X. (2018). Plastome organization, genome-based phylogeny and evolution of plastid genes in Podophylloideae (Berberidaceae). Mol. Phylogenet. Evol. 127, 978–987. doi: 10.1016/j.ympev.2018.07.001
Yoshida, K., Yokochi, Y., and Hisabori, T. (2019). New light on chloroplast redox regulation: molecular mechanism of protein thiol oxidation. Front. Plant Sci. 10:1534. doi: 10.3389/fpls.2019.01534
Yu, X., Jiang, W., Tan, W., Zhang, X., and Tian, X. (2020). Deciphering the organelle genomes and transcriptomes of a common ornamental plant Ligustrum quihoui reveals multiple fragments of transposable elements in the mitogenome. Int. J. Biol. Macromol. 165, 1988–1999. doi: 10.1016/j.ijbiomac.2020.10.075
Yu, X., Tan, W., Zhang, H., Jiang, W., Gao, H., Wang, W., et al. (2019). Characterization of the complete mitochondrial genome of Harpalus sinicus and its implications for phylogenetic analyses. Genes (Basel) 10, 724. doi: 10.3390/genes10090724
Yu, X. L., Wang, W. X., Yang, H. X., Zhang, X. Y., Wang, D., and Tian, X. X. (2021). Transcriptome and comparative chloroplast genome analysis of Vincetoxicum versicolor: insights Into molecular evolution and phylogenetic implication. Front. Genet. 12:602528. doi: 10.3389/fgene.2021.602528
Yura, K., and Go, M. (2008). Correlation between amino acid residues converted by RNA editing and functional residues in protein three-dimensional structures in plant organelles. BMC Plant Biol. 8, 79–11. doi: 10.1186/1471-2229-8-79
Zehrmann, A., van der Merwe, J. A., Verbitskiy, D., Brennicke, A., and Takenaka, M. (2008). Seven large variations in the extent of RNA editing in plant mitochondria between three ecotypes of Arabidopsis thaliana. Mitochondrion 8, 319–327. doi: 10.1016/j.mito.2008.07.003
Zhang, Z. H., Chang, X., Su, D. Y., Yao, R., Liu, X. D., Zhu, H., et al. (2021). Comprehensive transcriptome analyses of twoOocystisalgae provide insights into the adaptation to Qinghai–Tibet plateau. J. Syst. Evol. 59, 1209–1219. doi: 10.1111/jse.12589
Zhang, J., Fu, X.-X., Li, R.-Q., Zhao, X., Liu, Y., Li, M.-H., et al. (2020). The hornwort genome and early land plant evolution. Nat. Plants 6, 107–118. doi: 10.1038/s41477-019-0588-4
Zhang, T. C., Qiao, Q., Novikova, P. Y., Wang, Q., Yue, J. P., Guan, Y. L., et al. (2019). Genome of Crucihimalaya himalaica, a close relative of Arabidopsis, shows ecological adaptation to high altitude. Proc. Natl. Acad. Sci. U. S. A. 116, 7137–7146. doi: 10.1073/pnas.1817580116
Keywords: Isoetes, chloroplast genome, transcriptome, adaptive evolution, RNA editing
Citation: Yang Y, Yu X, Wei P, Liu C, Chen Z, Li X and Liu X (2022) Comparative chloroplast genome and transcriptome analysis on the ancient genus Isoetes from China. Front. Plant Sci. 13:924559. doi: 10.3389/fpls.2022.924559
Edited by:
Xiaohua Jin, Institute of Botany (CAS), ChinaReviewed by:
Gang Yao, South China Agricultural University, ChinaXiaoguo Xiang, Nanchang University, China
Copyright © 2022 Yang, Yu, Wei, Liu, Chen, Li and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoyan Li, eGlhb3lhbmxpeHlAc2luYS5jb20=; Xing Liu, eGluZ2xpdUB3aHUuZWR1LmNu