 Yan-Yan Guo
Yan-Yan Guo Jia-Xing Yang
Jia-Xing Yang Hong-Kun Li
Hong-Kun Li- College of Plant Protection, Henan Agricultural University, Zhengzhou, China
The size of the chloroplast genome (plastome) of autotrophic angiosperms is generally conserved. However, the chloroplast genomes of some lineages are greatly expanded, which may render assembling these genomes from short read sequencing data more challenging. Here, we present the sequencing, assembly, and annotation of the chloroplast genomes of Cypripedium tibeticum and Cypripedium subtropicum. We de novo assembled the chloroplast genomes of the two species with a combination of short-read Illumina data and long-read PacBio data. The plastomes of the two species are characterized by expanded genome size, proliferated AT-rich repeat sequences, low GC content and gene density, as well as low substitution rates of the coding genes. The plastomes of C. tibeticum (197,815 bp) and C. subtropicum (212,668 bp) are substantially larger than those of the three species sequenced in previous studies. The plastome of C. subtropicum is the longest one of Orchidaceae to date. Despite the increase in genome size, the gene order and gene number of the plastomes are conserved, with the exception of an ∼75 kb large inversion in the large single copy (LSC) region shared by the two species. The most striking is the record-setting low GC content in C. subtropicum (28.2%). Moreover, the plastome expansion of the two species is strongly correlated with the proliferation of AT-biased non-coding regions: the non-coding content of C. subtropicum is in excess of 57%. The genus provides a typical example of plastome expansion induced by the expansion of non-coding regions. Considering the pros and cons of different sequencing technologies, we recommend hybrid assembly based on long and short reads applied to the sequencing of plastomes with AT-biased base composition.
Introduction
The average chloroplast genome (plastome) size of land plants is 151 kb, with most species ranging from 130–170 kb in length, and the average GC content is 36.3% (NCBI database, 4,281 land plant plastomes, March 17, 2020) (Supplementary Table 1). However, previous studies documented that the plastome size of some lineages was extremely enlarged (Chumley et al., 2006; Kim et al., 2015; Blazier et al., 2016; Weng et al., 2017; Lim et al., 2018; Gruzdev et al., 2019; Li H. et al., 2020). The largest chloroplast genome of angiosperm is Pelargonium transvaalense (242, 575 bp), with the inverted repeat (IR) region of the species expanding to 87,724 bp (Weng et al., 2017). Of more than 4,000 land plant plastomes from NCBI, 82 sequences examined to date had chloroplast genome sizes over 170 kb, and seven of these species had chloroplast genome sizes over 200 kb (Supplementary Table 1). The expansion of the plastomes of these species is mainly caused by gene duplications in the IR regions (Chumley et al., 2006; Weng et al., 2017; Sinn et al., 2018; Li H. et al., 2020) or the expansion of repeat sequences in non-coding regions (Dugas et al., 2015; Li H. et al., 2020).
Chloroplast genome sequences have been widely used in studies of phylogeny, evolution, and population genetics of angiosperms (Tonti-Filippini et al., 2017). The accurate assembly and annotation of plastome sequences are the foundation of these studies. At present, most of the studies used Illumina short reads for chloroplast genome assembly. However, the short-reads method occasionally does not perform well owing to biased coverage depth, which may lead to fragmented genome assemblies (Ferrarini et al., 2013; Sinn et al., 2018). Recently, a few studies used long reads (Ferrarini et al., 2013; Wu et al., 2014; Chen et al., 2015; Xiang et al., 2016; Cauzsantos et al., 2017; Lee et al., 2020; Zhang et al., 2020) or combined short and long reads for chloroplast genome assembly (Ruhlman et al., 2017; Lin et al., 2018; Wang et al., 2018; Yan et al., 2019; Li H. et al., 2020). Ruhlman et al. (2017) combined long and short read data to investigate repeat sequences in Monsonia emarginata (Geraniaceae). Wang et al. (2018) compared short-read (Illumina) data only assembly, long-read (Oxford nanopore) data only assembly, and hybrid assembly involving short- and long-read data to test the accuracy of chloroplast genome assembly. They suggested that hybrid assembly provides highly accurate and complete chloroplast genome assembly.
Cypripedium is a genus of Orchidaceae, mainly distributed in the temperate regions of the Northern Hemisphere, with high ornamental and economic values. The genus is a good example to study the evolution of orchids. Owing to the large genome size (average 1C = 31.3 pg) (Leitch et al., 2009), molecular evolution studies of the genus are relatively rare. Cypripedium is one of the genera with chloroplast genome expansion (>170 kb) (Supplementary Table 1). At present, three chloroplast genomes of the genus have bene reported: Cypripedium japonicum (Kim et al., 2015), C. formosanum (Lin et al., 2015), and C. calceolus (Zhang et al., 2019). The sequenced chloroplast genomes showed that the genome sizes of the three species are larger than those of most other species of angiosperms, with relatively low GC contents (33.9–34.5%), and that the expansion of the genome size correlated with the expansion of the LSC region. Lin et al. (2015) found an ∼62 kb inversion in C. formosanum. However, the inversion was not observed in C. japonicum, which is the sister species of C. formosanum. In addition, Kim et al. (2015) found AT-rich regions in C. japonicum, and owing to the difficulty of sequencing poly(A), poly(T), or poly(AT) regions, they proposed a method for improving the success rates of these AT-rich regions, but the method was limited. Considering that the extents of chloroplast genome expansion and genome structure variation at the genus level are unknown, Cypripedium provides an ideal example to study the chloroplast genome evolution of angiosperm with expanded plastome size.
In this study, we selected two species of Cypripedium with clear morphological and habitat differentiation to investigate the chloroplast genome variation in the genus. Cypripedium tibeticum is widely distributed in southwest China, and Cypripedium subtropicum is restricted to southeast Yunnan Province and northern Vietnam. We sequenced, assembled, and annotated the complete chloroplast genomes of C. tibeticum and C. subtropicum using a combination of Illumina and PacBio sequencing platforms; then, we compared the two genomes with the previously reported chloroplast genomes of the genus in terms of genome size, gene number, genome structure, GC content, and substitution rates of the coding genes; finally, we discussed the expansion mechanism of Cypripedium and investigated the repeat sequences of the genus.
Materials and Methods
Taxon Sampling and Library Construction
We sampled fresh leaves of C. tibeticum and C. subtropicum from Huanglong, Sichuan and Malipo, Yunnan, China. Total genomic DNA was isolated using the CTAB method (Doyle and Doyle, 1987). The extracted total genomic DNA was used for library construction with 350 bp and 20 kb insert sizes and then sequenced on MGI2000 (MGI, Shenzhen, China) and PacBio RS-II platforms for the short and long reads, respectively.
Sequence Assembly and Annotation
The potential chloroplast genome reads were filtered in reference to the three chloroplast genomes of the genus reported in previous studies. Short reads were extracted with a script in NOVOPlasty 3.8.1 (Dierckxsens et al., 2017), and long reads were extracted with BLASR (Chaisson and Tesler, 2012). The hybrid assembly of the chloroplast genomes was performed in SPAdes 3.14.0 based on the filtered reads, with careful error correction and different K-mers (21, 33, 55, and 77) (Bankevich et al., 2012). Then, we used coding genes as seed sequences to test the assembly with NOVOPlasty 3.8.1 (Dierckxsens et al., 2017). The two methods generated almost identical results, except for the AT-biased repeat regions, and the short read only method failed to obtain the assemblies of these regions. The assembled sequences were annotated in Geneious Prime 2020 (Biomatters Ltd., Auckland, New Zealand), coupled with manual correction. The three plastome sequences downloaded from GenBank were reannotated for the following comparison. In addition, we found a paper reporting the chloroplast genome of C. tibeticum (GenBank accession No. MN561380) with samples collected from Qinling Mountains of China (Li J. et al., 2020), but the sequence was not yet accessible at the time of the analysis (June 2, 2020), so we were not able to perform further comparisons.
Linear plastome maps were generated with OGDRAW (Greiner et al., 2019). The boundaries of the IR and SC regions were defined by Repeat Finder embedded in Geneious Prime 2020. We calculated GC content in Geneious Prime 2020. Then, we visualized the genome rearrangement of the genus using the progressiveMauve algorithm (Darling et al., 2010) with IRa removed.
Nucleotide Substitution Rate Analyses
We used the CODEML program in PAML v. 4.9 (model = 0) (Yang, 2007) to calculate the average non-synonymous substitution rate (dN) and synonymous substitution rate (dS) for 79 protein coding genes by the F3X4 codon model. Gapped regions were excluded for rate estimation (cleandata = 1). The input tree [C. subtropicum, (C. formosanum, C. japonicum), and (C. calceolus, C. tibeticum)]simplified from Li et al. (2011) was used for the following analyses. In matK, only three sequences could be used for substitution rate estimation, and C. formosanum, (C. calceolus, C. tibeticum) was used as input tree. In addition, we counted the numbers of indels in the intergenic spacer regions and introns in the plastomes of Cypripedium using DnaSP v6.12.03 (Rozas et al., 2017) with unalignable regions removed using GBlocks (Talavera and Castresana, 2007) with the default settings.
Repeat Sequence Analysis
Simple sequence repeats (SSRs) (≥10 bp) were detected via MISA (Beier et al., 2017), and the minimum thresholds for mono-, di-, tri-, tetra-, penta-, and hexa-nucleotides were set to 10, 5, 4, 3, 3, and 3, respectively. In addition, tandem repeats were identified with Tandem Repeats Finder v4.09 (Benson, 1999) with default parameters, the identity of repeats was set at 90%, and overlapped repeats were removed manually. Dispersed repeats (≥30 bp) and palindromic repeats (≥20 bp) were identified with Vmatch1.
Results
Plastomes of Cypripedium tibeticum and Cypripedium subtropicum
We obtained the full chloroplast genome sequences of 197,815 bp for C. tibeticum and 212,668 bp for C. subtropicum (GenBank accession Nos. MT937101 and MT937100, respectively). The plastid genomes of the two species showed typical quadripartite structure, with two identical copies of the IR region separated by an LSC region and a small single copy (SSC) region (Figures 1, 2). The LSC regions of the two species expanded to 117,193 and 129,998 bp, respectively, the IR regions of the two species (27,764 and 27,628 bp, respectively) were similar to those of the previously sequenced species, and the SSC regions of the two species (25,094 and 27,414 bp, respectively) were slightly larger than those of the other three species (Table 1). The gene number of the genus was conserved and consisted of 131–132 genes, including 85–86 protein coding genes (seven duplicated in the IR region), 38 tRNAs (eight duplicated in the IR region), and eight rRNAs (four duplicated in the IR region). A total of 15 genes contained one intron, including six tRNA genes (trnG-UCC, trnK-UUU, trnL-UAA, trnV-UAC, trnA-UGC, and trnI-GAU) and nine protein coding genes (rps16, rpl2, rpl16, rpoC1, petB, petD, atpF, ndhA, and ndhB), while the other three protein coding genes (clpP, rps12, and ycf3) contained two introns (Supplementary Table 2). We found pseudogenization of the matK gene owing to a single-base deletion-induced frameshift in C. subtropicum. Corresponding to the expansion of the genome size and the relatively conserved gene number, the gene density of the genus ranged from 0.62 to 0.75. The GC contents of the total genomes were 30.5% for C. tibeticum and 28.2% for C. subtropicum (Figure 2), and the GC contents of the two genomes were uneven. The GC contents of the IR regions (42.5 and 42.6%) were higher, whereas the LSC regions (26.5 and 23.7%) and SSC regions (22.4 and 20.6%) had lower GC content (Table 1).
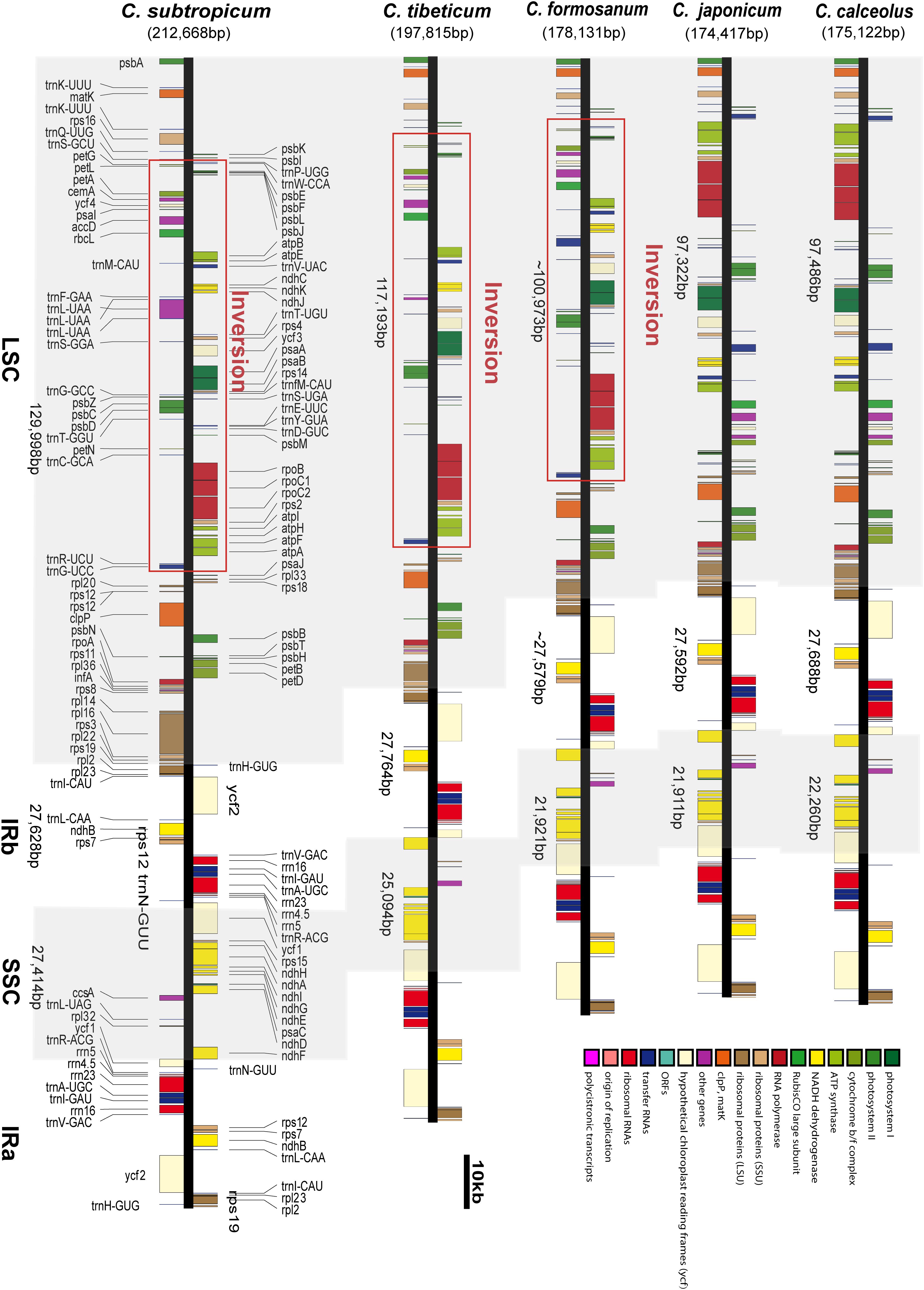
Figure 1. The chloroplast genome structures of five species of Cypripedium. The red lines indicate the inversion spanning from trnG-UCC to trnP-UGG in the LSC region.
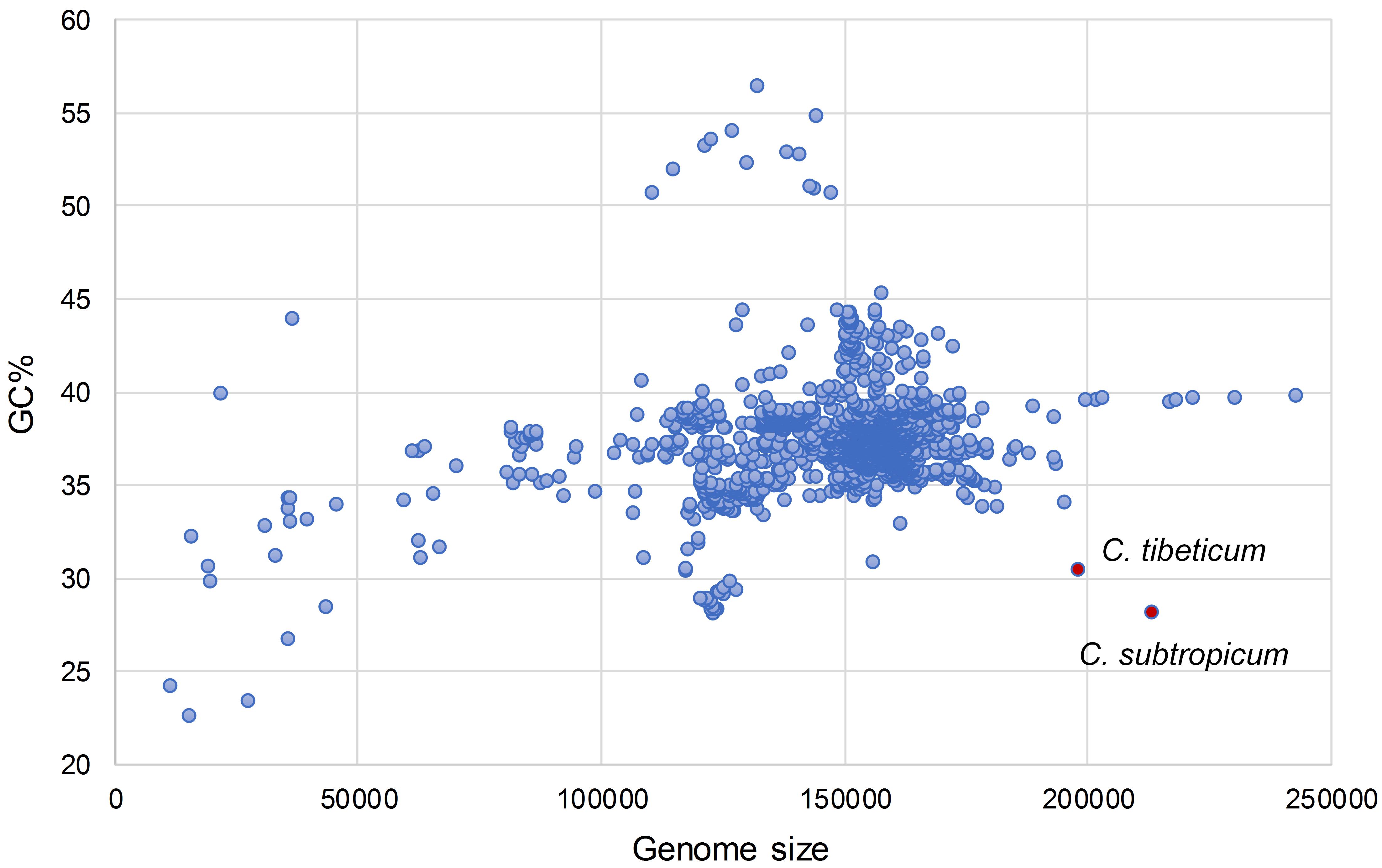
Figure 2. The plastomes of C. tibeticum and C. subtropicum exhibit atypical size and GC content.
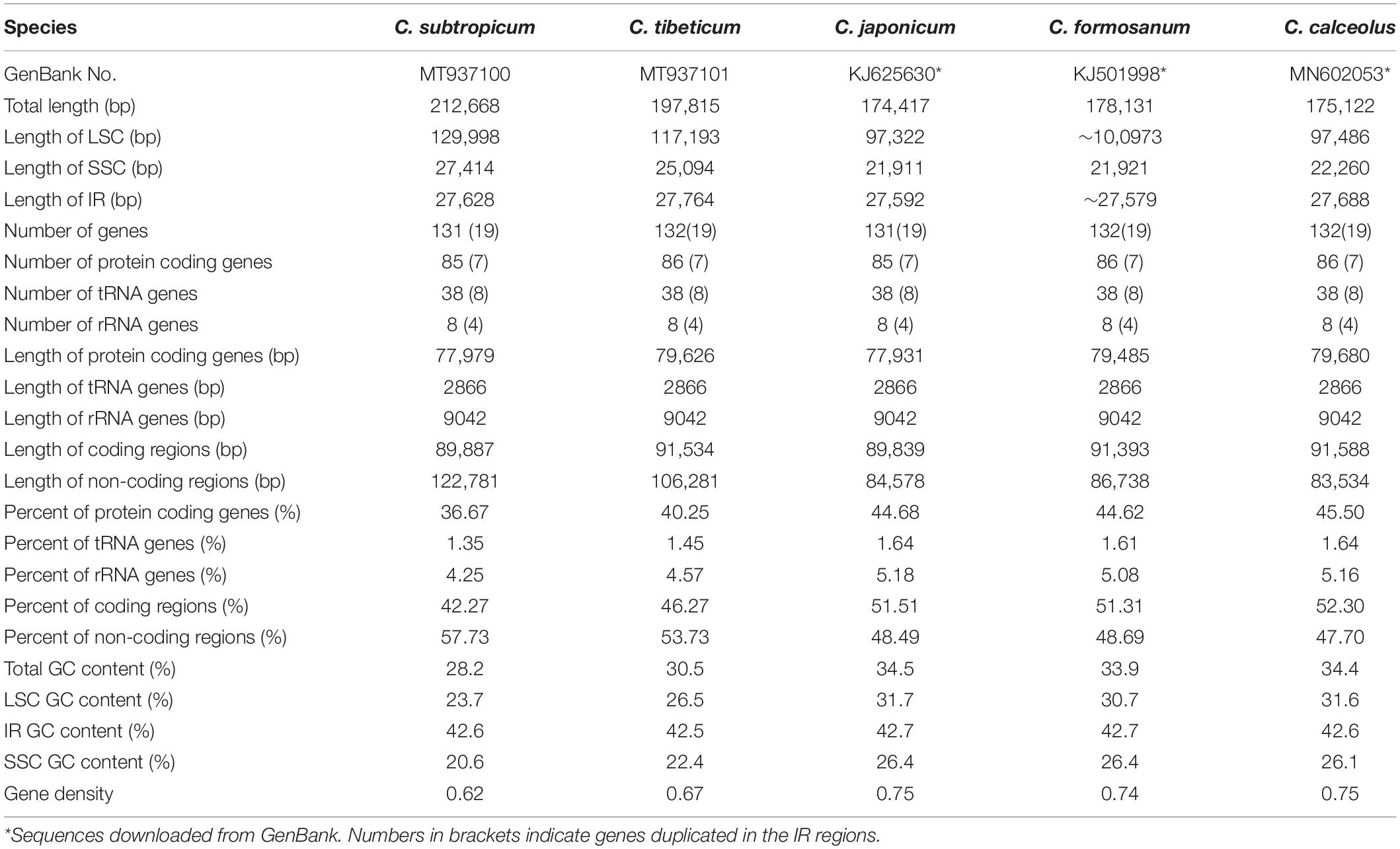
Table 1. General characteristics of the plastomes of the five Cypripedium species included in this study.
Approximately 36.67–45.50% of the genome encoded proteins, 1.35–1.64% encodes tRNA, and 4.25–5.18% encodes ribosomal RNA (Table 1). The length of the coding regions of the five species was approximately 90 kb, whereas the length of non-coding regions ranged from 84 kb in C. calceolus to 123 kb in C. subtropicum (Table 1). Correspondingly, the non-coding regions of some genes were extremely expanded in the two species; for example, the intron of rpl16 expanded to 7.5 kb in C. subtropicum, the intron of trnK-UUU expanded to 7.7 kb in C. subtropicum and to 6 kb in C. tibeticum, the intergenic region between psbA and trnK-UUU expanded to 4.3 kb in C. subtropicum, the intergenic region between rbcL and atpB expanded to 4.9 kb in C. tibeticum, the intergenic region between trnL-UAG and ccsA expanded to 2.5 kb in C. tibeticum and to 3.4 kb in C. subtropicum (Supplementary Table 3). However, some of the non-coding regions were conserved in length, such as the introns of ndhB and rpl2, which had no length variation (Supplementary Table 3).
The LSC/IR boundary and the IR/SSC boundary were stable in the genus (Figure 3). The LSC/IRb boundary in all five species was located on rpl22, while one end of SSC was located on ycf1, and the other end of SSC was located on the truncated copy of ycf1 (Figure 3). The gene order of the genus was conserved, apart from the ∼75 kb inversion spanning from trnG-UCC to trnP-UGG in the LSC region (Figure 1 and Supplementary Figure 2). In addition, the intergeneric regions adjacent to the long inversions also had high AT contents compared to the other two species without long inversions.
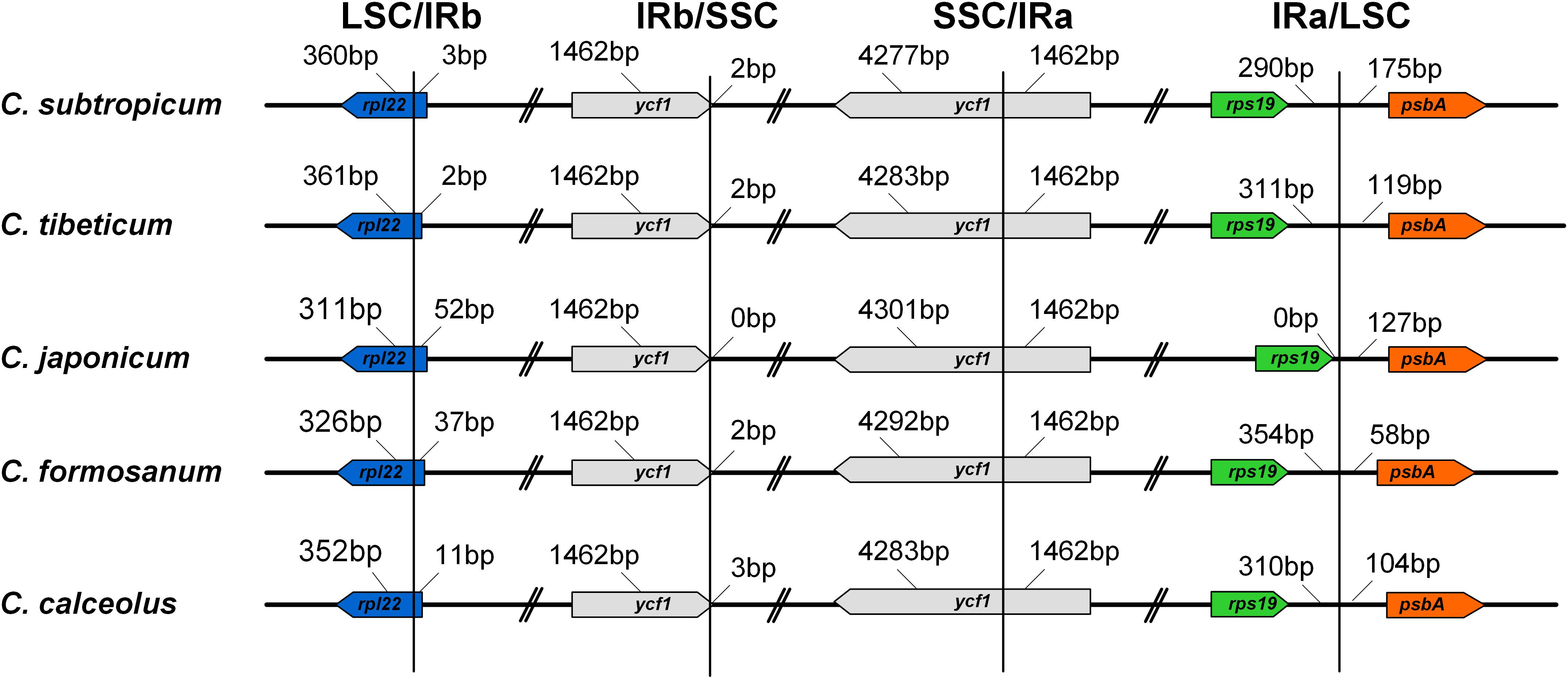
Figure 3. Comparison of the border positions of LSC, SSC, and IR regions among the five Cypripedium chloroplast genomes.
Repeat Sequences in Cypripedium
The plastomes of Cypripedium ranged from 252 repeats (5,362 bp) in C. japonicum to 930 repeats (30,453 bp) in C. subtropicum, and these repeat sequences represented 3.07% in C. japonicum to 14.32% in C. subtropicum of the chloroplast genome length, the newly sequenced two species had increased numbers and sizes of repeat sequences (Table 2). The total length of SSR, tandem repeats, and dispersed repeats revealed 3.05-fold, 2.56-fold, and 15.88-fold variations, respectively (Table 2). The repeat sequences in the genus were dominated by SSR (182–535), followed by tandem repeat (54–120) or dispersed repeat (12–191), whereas palindromic repeats were the fewest (three to 84) (≥30 bp) (Table 2). C. subtropicum (84) and C. tibeticum (54) had more palindromic repeats than the other three species (three to four). All the five species shared a 48-bp (TATAGTGTGGTAGAAAGAGCTATATATAGCTCTTTCTAC CACACTATA) palindromic repeat located in the intergeneric region between psbM and petN, and most of the other palindromic repeats were species-specific. The longest repeats reached 156 bp in C. tibeticum (a 78-bp motif repeated twice), 180 bp in C. subtropicum (a 44-bp motif repeated 4.1 times). Unexpectedly, when the repeat length was set to 20 bp, the palindromic repeat number increased substantially (149–5,481) (Table 2), with most of the palindromic repeats having lengths between 20–25 bp (Supplementary Table 5). These repeats were strongly AT-biased, and tandem A/T, AT/TA, AAT/ATT, AAAT/ATTT, and AATAT/ATATT were the five dominant motif types in the SSR (Figure 4). Most of these repeat sequences were located in the non-coding regions of LSC and SSC regions and rarely appeared in the IR region (Figure 4).
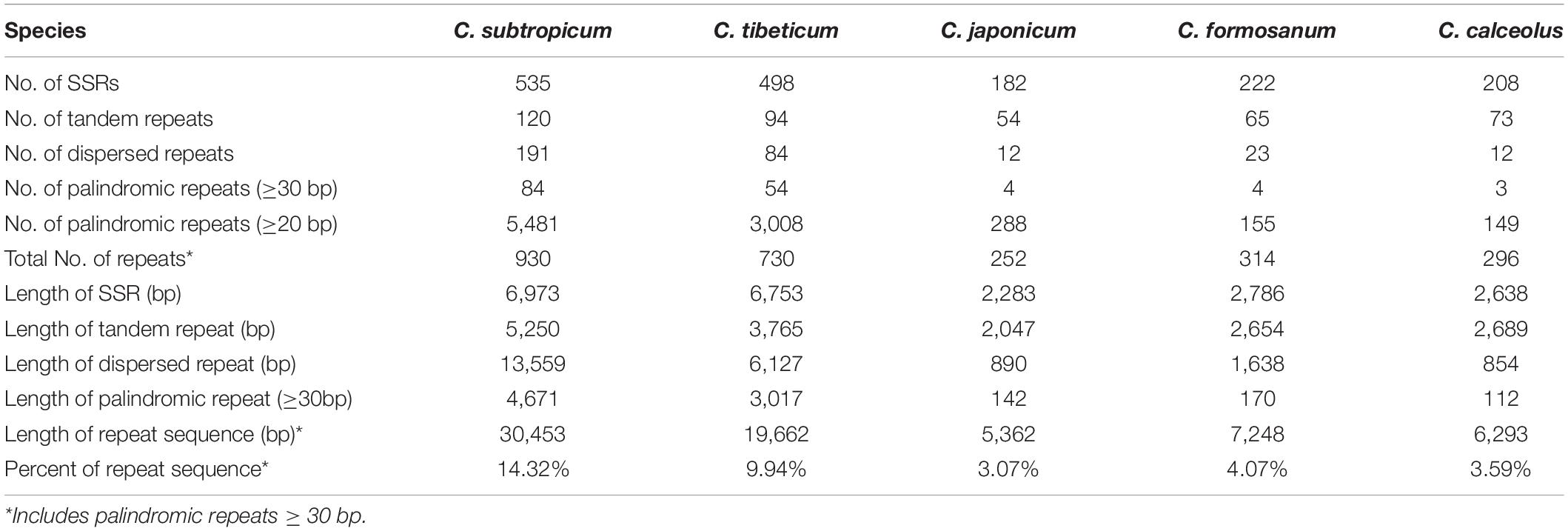
Table 2. Summary of repeat sequences in the plastomes of five Cypripedium species.
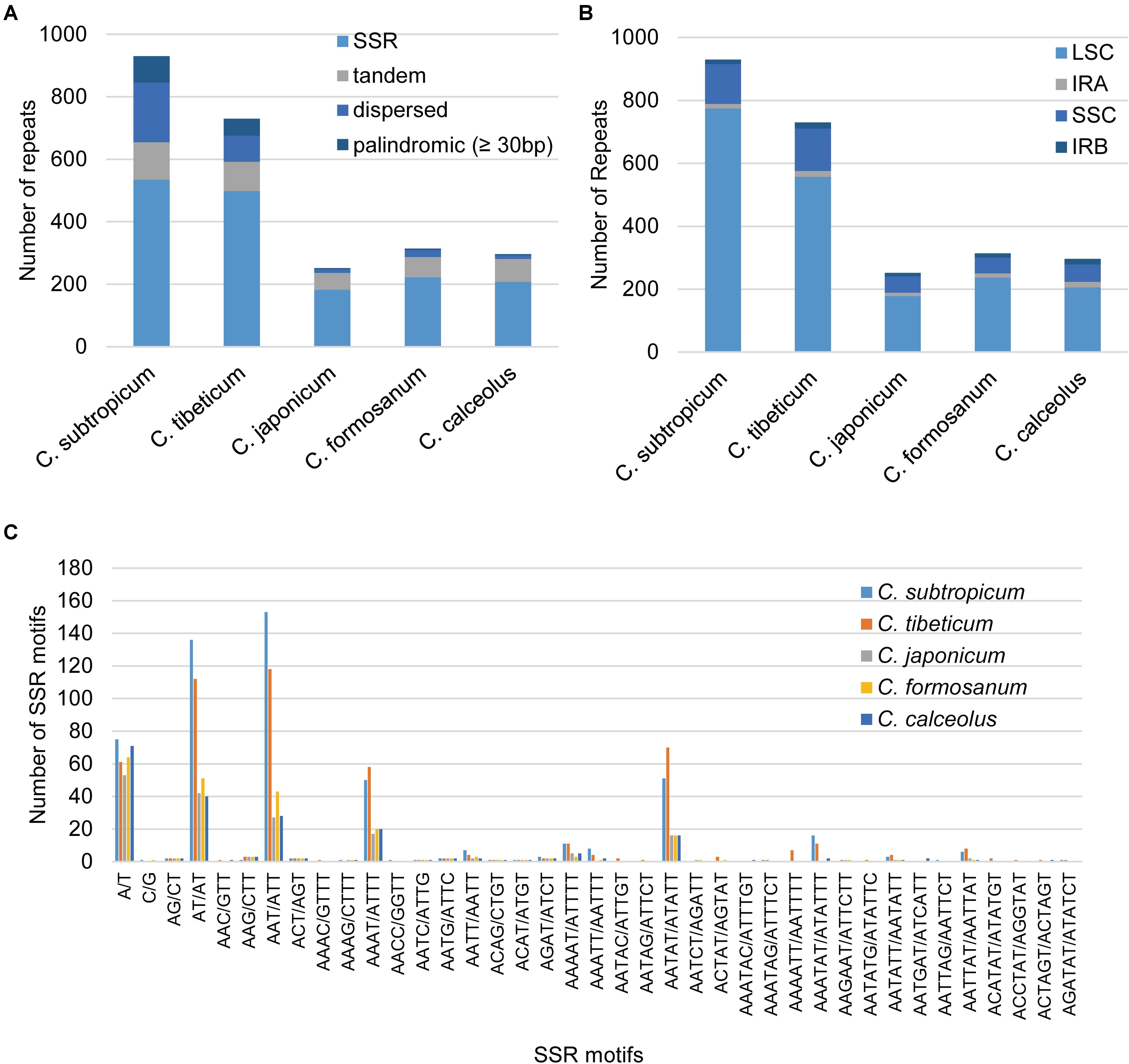
Figure 4. Statistics of the repeat elements in the five Cypripedium chloroplast genomes. (A) Number and type of repeats; (B) the number of repeat sequences in different regions (palindromic repeats excluded); (C) number and type of SSR.
Nucleotide Substitution Rate Analyses
Mean synonymous and non-synonymous divergence was extremely low (dN = 0.0069, dS = 0.0287), and varied among genes (Supplementary Table 6). The most rapidly evolving genes in the genus was rpl33, while the sequences of some protein coding genes were identical in the five species, such as atpH, petG, petN, psaI, psbF, rpl23, rps12, and rps14 (Supplementary Table 6 and Figure 5). Most of the expanded AT-rich regions were unalignable, and the expanded IGS in LSC and SSC regions had more indels than the non-coding regions without expansion (Supplementary Table 7).
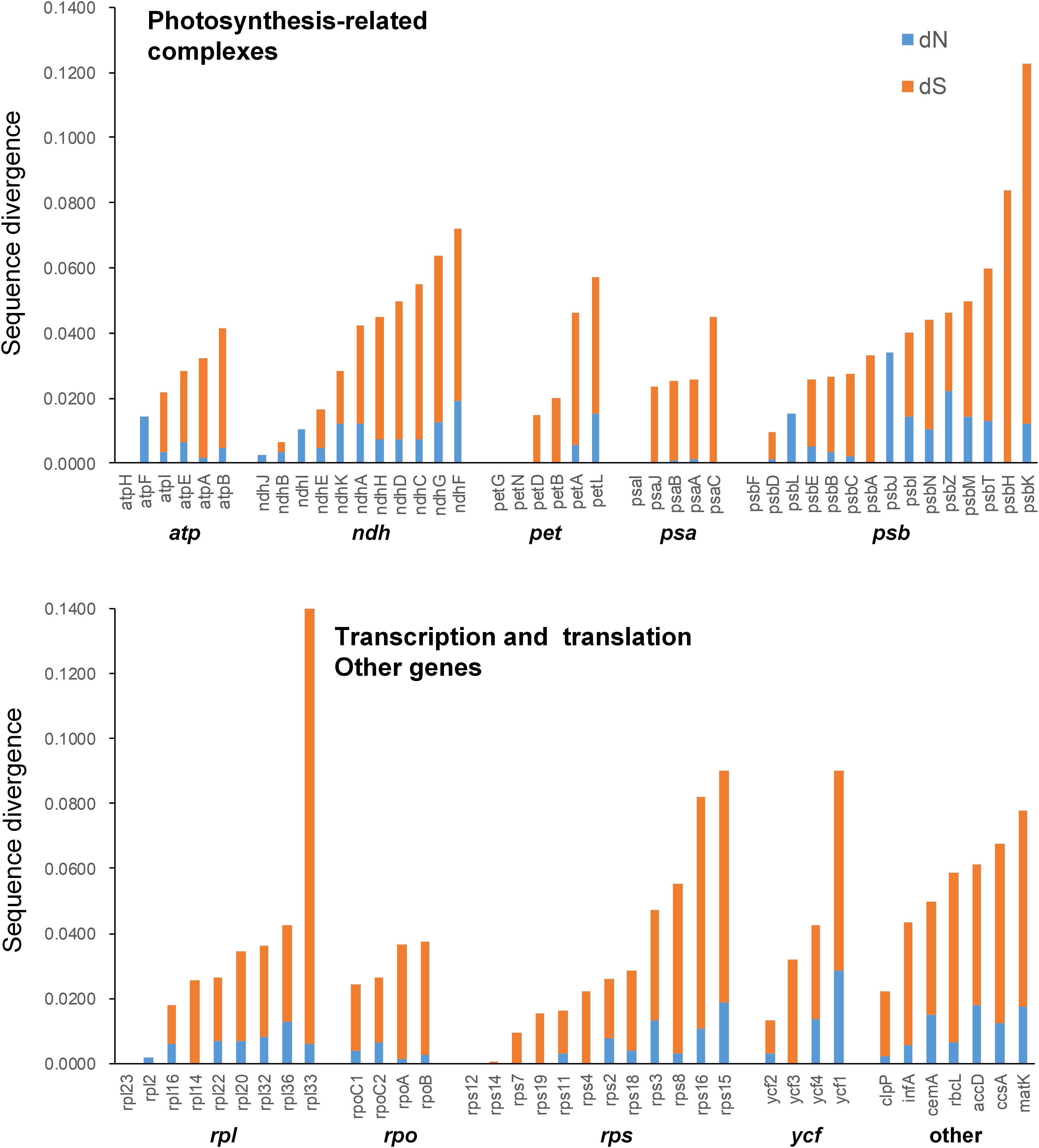
Figure 5. Non-synonymous substitution rate (dN), synonymous substitution rate (dS), and dN/dS for each gene.
Discussion
The Plastome Characters of Cypripedium tibeticum and Cypripedium subtropicum
To date, the chloroplast genome of C. subtropicum (212,668 bp) is the largest of Orchidaceae and the sixth largest of sequenced land plants. Following the expansion of the plastomes, the gene density of the genus decreased to 0.62 in C. subtropicum (Table 1), while the gene density of most angiosperms is over 0.80 (the average plastome size of land plants is 151 kb, ∼130 genes, and the gene density is approximately 0.86). The mean synonymous and non-synonymous divergence was low (dN = 0.0069, dS = 0.0287) (Supplementary Table 6). The divergence rate of coding genes is even lower than the coniferous forest tree genus Picea, the mean synonymous and non-synonymous of Picea was 0.017 ± 0.024 and 0.040 ± 0.031 (Sullivan et al., 2017). The low substitution rates might explain the unresolved relationships among sections in Cypripedium (Li et al., 2011).
Interestingly, the overall GC contents of C. subtropicum (28.2%) and C. tibeticum (30.5%) plastomes are much lower than those of the three species sequenced in previous studies, and the GC content of C. subtropicum is the lowest in the sequenced autotrophic species to date (Figure 2 and Table 1). The GC content of the genus is lower than the average GC content of land plant plastomes (37.6%) (Supplementary Table 1). The high AT content is induced by repetitive sequences composed of poly(A), poly(T), or poly(AT) regions in non-coding regions in the single copy region, especially in the LSC region (Kim et al., 2015). Tandem AAT/ATT is the most abundant repeat in newly sequenced species, whereas A/T mononucleotide is the most abundant repeat in the three species sequenced in previous studies (Figure 4). In addition, we found 26 records of the NCBI plastome database with GC content below 30%, and these lower GC content species were restricted to parasitic/mycoheterotrophic plants, mosses, and liverworts (Supplementary Table 5), such as the holoparasitic Balanophora reflexa (11.6%) and B. laxiflora (12.2%) (Su et al., 2019), the parasitic Pilostyles hamiltoni (22.7%) (Bellot and Renner, 2016), and the mycoheterotrophic Gastrodia elata (26.7%) (Ma & Jin, unpublished data, MF163256.1). In Cypripedium, the GC contents of the coding regions are similar to those of other species, whereas the non-coding regions have relatively lower GC contents, including intergenic spacer regions and introns, and some of the intergenic regions with GC contents lower than 10%, e.g., trnL-ccsA (1.3% in C. subtropicum to 7.2% in C. japonicum) and psbA-trnK (2.5% in C. subtropicum).
Although the gene order of the two species is conserved, the two species sequenced in this study share an ∼75 kb inversion in the LSC region; the inversion was also found in C. formosanum (Lin et al., 2015), and the gene order and orientation in the long inversion were completely conserved (Figure 1). Apart from the long inversion, the chloroplast genome structure of the genus is conserved. According to the section delimitation of the genus (Chen et al., 2013), the three species with long inversions are ascribed to different sections: C. tibeticum belongs to section Cypripedium, C. subtropicum belongs to section Subtropica, C. formosanum belongs to section Flabellinervia, which suggests that the long inversion occurred independently in the genus. Large inversions were also found in other lineages, a 47 kb inversion (petN to clpP) in Paphiopedilum fairrieanum (unpublished data), a 30 kb inversion (trnG-GCC to trnE-UUC) in Hevea brasiliensis (Tangphatsornruang et al., 2011), an 8 kb inversion (ycf4 to atpE) in Annona cherimola (Blazier et al., 2016), a 24 kb inversion (trnQ-UUG to trnT-GGU) in Viscum minimum (Petersen et al., 2015), a 42 kb inversion (clpP to trnC-GCA) in Passiflora edulis (Cauzsantos et al., 2017), and a length portion of LSC (trnH-GUG to trnT-GGU) in Asarum (Sinn et al., 2018). The mechanism of these long inversions is mostly unclear, but Sinn et al. (2018) found that the inversion in Asarum is flanked by long AT-rich regions, and they proposed that intramolecular recombination induced a long inversion in the genus. The AT-biased repeat sequences were also found in the flanking regions of the three Cypripedium species with the long inversion, but there are some other regions of the plastome with AT-rich repeat sequences; thus, the relationship between the inversion and the AT-rich sequences remains uncertain.
Furthermore, despite the increases in the chloroplast genome size of the two sequenced species, the gene number (131–132 genes) of the genus is rather conserved, and the genus encodes all the coding genes commonly found in the chloroplast genomes (Table 1 and Supplementary Table 2). The one gene variation in the two newly sequenced species was due to the pseudogenization of matK in C. subtropicum. The pseudogenization of matK was also observed in C. japonicum (Guo et al., 2012; Kim et al., 2015). Interestingly, the pseudogenization of matK occurred independently, and both pseudogenization events were frameshift mutations induced by non-triplet nucleotide deletions following a 10-bp mononucleotide (T) repeat 294 bp from the start codon, a 10-bp deletion and a 1-bp deletion (Supplementary Figure 1). The non-triplet indels of matK have been reported in previous studies (Kores et al., 2000; Freudenstein and Senyo, 2008; Kocyan et al., 2008; Logacheva et al., 2011), and 82% of the pseudogene entries in GenBank are from Orchidaceae (Barthet et al., 2015). MatK is a rapidly evolving chloroplast gene that encodes maturase in the plastome and is related to the splicing of the group IIA introns of seven genes (atpF, rpl2, rps12, trnV-UAC, trnI-GAU, trnA-UGC, and trnK-UUU) in land plants (Zoschke et al., 2010). Some angiosperm lineages present coevolution between matK and the seven group IIA introns, such as the parasitic Cuscuta (McNeal et al., 2009) and the mycoheterotroph Rhizanthella gardneri (Delannoy et al., 2011). However, there is no parallel loss of matK and the seven group IIA introns in Cypripedium: matK in C. subtropicum and C. japonicum are pseudogenes, while the seven group IIA introns are all retained in the two species. We infer that the matK gene in the genus might be at the transition from a functional gene to a pseudogene, and that there are other mechanisms regulating the splicing of the group IIA introns.
The Expansion Mechanism of Cypripedium
The plastome expansion of Cypripedium is strongly correlated with the proliferation of AT-biased non-coding regions. The five species with larger chloroplast genomes all belong to Pelargonium of Geraniaceae (Supplementary Table 1). However, the plastomes of the two genera expanded in different ways. The plastomes of Pelargonium incorporated a large portion of what was once the SSC region into the IR region, which induced massive gene duplications (Chumley et al., 2006; Weng et al., 2017). In Cypripedium, the lengths of the IR region (27,764 and 27,628 bp) and the SSC region (27,764 and 27,628 bp) are conserved, but the LSC regions of the two species expanded to 117,193 and 129,998 bp respectively, which are 20–30 kb larger than the LSC regions of the three species sequenced in previous studies (Figure 1 and Table 1; Kim et al., 2015; Lin et al., 2015; Zhang et al., 2019). The coding regions of the sequenced species were approximately 90 kb, and the non-coding regions varied in different species. The non-coding region of C. subtropicum expanded to 122,781 bp (approximately 57.73% of the chloroplast genome), whereas the non-coding region of C. japonicum was 83,534 bp (approximately 47.7% of the chloroplast genome) (Table 1). Given that coding regions of the genus are conserved, we infer that the plastome expansion in the genus is strongly correlated with the proliferation of non-coding regions, especially the non-coding regions in LSC regions, and the two species are typical examples of plastome expansion without gene duplication. These AT-rich repeat sequences led to the plastome expansion of Cypripedium. Dispersed repeats contributed most to plastome expansion, followed by SSR and then tandem repeats (Table 2). The expanded regions appeared as unalignable insertions, where the number of indels are correlated with the length of the non-coding regions (Supplementary Table 7). Furthermore, other studies have proposed that repeat sequences lead to plastome expansion; however, the expansion of these species is also associated with gene duplications caused by boundary shifts in IR regions, e.g., watercress (Yan et al., 2019) and Rhododendron delavayi (Li H. et al., 2020). Interestingly, short palindromic repeats (20–25 bp) are prevalent in the plastomes of C. subtropicum and C. tibeticum (Supplementary Table 5). Smith (2020) indicated that panlindromic repeats are mutational hotspots, and contribute to plastome expansion in chlamydomonadalean Chlorosarcinopsis eremi. In addition to this study, the proliferation of non-coding regions was mainly documented in algae (Muñoz-Gómez et al., 2017; Gaouda et al., 2018; Smith, 2018), and the non-coding DNA of Haematococcus lacustris comprises over 90% of the plastome (Smith, 2018). In addition, we also found high AT regions in Paphiopedilum (trnS-trnG, trnE-trnT, and trnP-psaJ) (unpublished data). AT-rich regions were also found in some other genera, e.g., Asarum (Sinn et al., 2018). However, the AT-rich regions in most cases do not contribute to plastome size expansion.
Finally, there is coverage bias related to the GC content in the short-read sequencing technologies (Browne et al., 2020), which means that the AT-rich regions exhibit under-coverage in the high-throughput dataset compared to the GC neutral regions. In this study, we failed to obtain parts of the plastomes of the two species through a short-read dataset, especially the AT-rich repetitive regions. We are also unable to circularize the plastomes of some Paphiopedilum species due to the lengthy AT-rich regions (unpublished data). Sinn et al. (2018) obtained fragmented plastomes in Asarum owing to the lengthy AT-biased regions. Moreover, Zhang et al. (2020) found that two repeat fragments were missing in the short-read assembly compared to the long-read assembly. In contrast, most studies indicated that the coverage depth of long-read sequencing is relatively even (Ferrarini et al., 2013) and could yield highly-accurate plastome assemblies (Wang et al., 2018). Considering the limitation of short-read sequencing and the fact that most of the plastomes in GenBank are obtained based on short-read data, the species with AT-biased plastomes might be underrepresented. Considering the pros and cons of different sequencing technologies, we recommend that the research of plastomes with AT-biased base composition and lengthy repetitive sequence use hybrid assembly, which will take advantage of the high throughput of second-generation sequencing and the longer read length of third-generation sequencing and reduce the coverage biases introduced by DNA sequencing methods, especially the species containing long repetitive elements, and the combination of the two sequencing platforms will greatly simplify the assembly.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
Y-YG conceived and designed the study. Y-YG, J-XY, H-SZ, and H-KL analyzed the data. Y-YG wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by grants from the National Natural Science Foundation of China (No. U1804117) and Key Scientific Research Projects of Henan Province (No. 17A180023).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Xi-Bin Guo and Ting-Hong Zhou for help with sample collection.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2021.609729/full#supplementary-material
Supplementary Figure 1 | Alignment of the matK gene of the genus. The red arrows indicate the non-triplet nucleotide deletions.
Supplementary Figure 2 | Synteny alignment of chloroplast genomes of Cypripedium. Locally collinear blocks of the sequences are color-coded and connected by lines.
Supplementary Table 1 | List of land plants with sequenced plastomes available from GenBank. The species with expanded chloroplast genomes (>170 kb) are marked in red.
Supplementary Table 2 | List of genes identified in the chloroplast genomes of Cypripedium.
Supplementary Table 3 | Lengths of intergenic spacer regions and introns in the plastomes of Cypripedium.
Supplementary Table 4 | Species with GC content lower than 30% from GenBank database.
Supplementary Table 5 | The number of palindromic repeats (≥20 bp) in the chloroplast genomes of Cypripedium.
Supplementary Table 6 | Non-synonymous substitution rate (dN), synonymous substitution rate (dS), and dN/dS for each gene.
Supplementary Table 7 | Numbers of indels in intergenic spacer regions and introns in the plastomes of Cypripedium.
Footnotes
References
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Barthet, M. M., Moukarzel, K., Smith, K. N., Patel, J., and Hilu, K. W. (2015). Alternative translation initiation codons for the plastid maturase MatK: unraveling the pseudogene misconception in the Orchidaceae. BMC Evol. Biol. 15:210. doi: 10.1186/s12862-015-0491-1
Beier, S., Thiel, T., Münch, T., Scholz, U., and Mascher, M. (2017). MISA-web: a web server for microsatellite prediction. Bioinformatics 33, 2583–2585. doi: 10.1093/bioinformatics/btx198
Bellot, S., and Renner, S. S. (2016). The plastomes of two species in the endoparasite genus Pilostyles (Apodanthaceae) each retain just five or six possibly functional genes. Genome Biol. Evol. 8, 189–201. doi: 10.1093/gbe/evv251
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. doi: 10.1093/nar/27.2.573
Blazier, J. C., Ruhlman, T. A., Weng, M.-L., Rehman, S. K., Sabir, J. S., and Jansen, R. K. (2016). Divergence of RNA polymerase α subunits in angiosperm plastid genomes is mediated by genomic rearrangement. Sci. Rep. 6:24595. doi: 10.1038/srep24595
Browne, P. D., Nielsen, T. K., Kot, W., Aggerholm, A., Gilbert, M. T. P., Puetz, L., et al. (2020). GC bias affects genomic and metagenomic reconstructions, underrepresenting GC-poor organisms. GigaScience 9:giaa008. doi: 10.1093/gigascience/giaa008
Cauzsantos, L. A., Munhoz, C. D. F., Rodde, N., Cauet, S., Santos, A. A., Penha, H. A., et al. (2017). The chloroplast genome of Passiflora edulis (Passifloraceae) assembled from long sequence reads: structural organization and phylogenomic studies in Malpighiales. Front. Plant Sci. 8:334. doi: 10.3389/fpls.2017.00334
Chaisson, M. J., and Tesler, G. (2012). Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): application and theory. BMC Bioinform. 13:238. doi: 10.1186/1471-2105-13-238
Chen, S.-C., Liu, Z., Chen, L., and Li, L. (2013). The Genus Cypripedium in China. Beijing: Science Press.
Chen, X., Li, Q., Li, Y., Qian, J., and Han, J. (2015). Chloroplast genome of Aconitum barbatum var. puberulum (Ranunculaceae) derived from CCS reads using the PacBio RS platform. Front. Plant Sci. 6:42. doi: 10.3389/fpls.2015.00042
Chumley, T. W., Palmer, J. D., Mower, J. P., Fourcade, H. M., Calie, P. J., Boore, J. L., et al. (2006). The complete chloroplast genome sequence of Pelargonium x hortorum: organization and evolution of the largest and most highly rearranged chloroplast genome of land plants. Mol. Biol. Evol. 23, 2175–2190. doi: 10.1093/molbev/msl089
Darling, A. E., Mau, B., and Perna, N. T. (2010). progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5:e0011147. doi: 10.1371/journal.pone.0011147
Delannoy, E., Fujii, S., Colas Des Francs-Small, C., Brundrett, M., and Small, I. (2011). Rampant gene loss in the underground orchid Rhizanthella gardneri highlights evolutionary constraints on plastid genomes. Mol. Biol. Evol. 28, 2077–2086. doi: 10.1093/molbev/msr028
Dierckxsens, N., Mardulyn, P., and Smits, G. (2017). NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45:e18. doi: 10.1093/nar/gkw955
Doyle, J., and Doyle, J. (1987). A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 19, 11–15.
Dugas, D. V., Hernandez, D., Koenen, E. J., Schwarz, E., Straub, S., Hughes, C. E., et al. (2015). Mimosoid legume plastome evolution: IR expansion, tandem repeat expansions, and accelerated rate of evolution in clpP. Sci. Rep. 5:16958. doi: 10.1038/srep16958
Ferrarini, M., Moretto, M., Ward, J. A., Šurbanovski, N., Stevanović, V., Giongo, L., et al. (2013). An evaluation of the PacBio RS platform for sequencing and de novo assembly of a chloroplast genome. BMC Genom. 14:670. doi: 10.1186/1471-2164-14-670
Freudenstein, J. V., and Senyo, D. M. (2008). Relationships and evolution of matK in a group of leafless orchids (Corallorhiza and Corallorhizinae; Orchidaceae : Epidendroideae). Am. J. Bot. 95, 498–505. doi: 10.3732/ajb.95.4.498
Gaouda, H., Hamaji, T., Yamamoto, K., Kawai-Toyooka, H., Suzuki, M., Noguchi, H., et al. (2018). Exploring the limits and causes of plastid genome expansion in volvocine green algae. Genome Biol. Evol. 10, 2248–2254. doi: 10.1093/gbe/evy175
Greiner, S., Lehwark, P., and Bock, R. (2019). OrganellarGenomeDRAW (OGDRAW) version 1.3. 1: expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 47, W59–W64. doi: 10.1093/nar/gkz238
Gruzdev, E. V., Kadnikov, V. V., Beletsky, A. V., Kochieva, E. Z., Mardanov, A. V., Skryabin, K. G., et al. (2019). Plastid genomes of carnivorous plants Drosera rotundifolia and Nepenthes × ventrata reveal evolutionary patterns resembling those observed in parasitic plants. Int. J. Mol. Sci. 20:4107. doi: 10.3390/ijms20174107
Guo, Y.-Y., Luo, Y.-B., Liu, Z.-J., and Wang, X.-Q. (2012). Evolution and biogeography of the slipper orchids: eocene vicariance of the conduplicate genera in the Old and New World tropics. PLoS One 7:e38788. doi: 10.1371/journal.pone.0038788
Kim, J. S., Kim, H. T., and Kim, J.-H. (2015). The largest plastid genome of monocots: a novel genome type containing AT residue repeats in the slipper orchid Cypripedium japonicum. Plant Mol. Biol. Rep. 33, 1210–1220. doi: 10.1007/s11105-014-0833-y
Kocyan, A., De Vogel, E. F., Conti, E., and Gravendeel, B. (2008). Molecular phylogeny of Aerides (Orchidaceae) based on one nuclear and two plastid markers: a step forward in understanding the evolution of the Aeridinae. Mol. Phylogenet. Evol. 48, 422–443. doi: 10.1016/j.ympev.2008.02.017
Kores, P. J., Weston, P. H., Molvray, M., and Chase, M. W. (2000). “Phylogenetic relationships within the Diurideae (Orchidaceae): inferences from plastid matK DNA sequences,” in Monocots: Systematics and Evolution, eds K. L. Wilson and D. A. Morrison (Collingwood: CSIRO publishing), 449–456.
Lee, C., Ruhlman, T. A., and Jansen, R. K. (2020). Unprecedented intraindividual structural heteroplasmy in Eleocharis (Cyperaceae, Poales) plastomes. Genome Biol. Evol. 12, 641–655. doi: 10.1093/gbe/evaa076
Leitch, I. J., Kahandawala, I., Suda, J., Hanson, L., Ingrouille, M. J., Chase, M. W., et al. (2009). Genome size diversity in orchids: consequences and evolution. Ann. Bot. 104, 469–481. doi: 10.1093/aob/mcp003
Li, H., Guo, Q., Li, Q., and Yang, L. (2020). Long-reads reveal that Rhododendron delavayi plastid genome contains extensive repeat sequences, and recombination exists among plastid genomes of photosynthetic Ericaceae. PeerJ 8:e9048. doi: 10.7717/peerj.9048
Li, J., Xu, B., Yang, Q., and Liu, Z.-L. (2020). The complete chloroplast genome sequence of Cypripedium tibeticum King Ex Rolfe (Orchidaceae). Mitochondrial DNA B 5, 150–151. doi: 10.1080/23802359.2019.1698357
Li, J.-H., Liu, Z.-J., Salazar, G. A., Bernhardt, P., Perner, H., Tomohisa, Y., et al. (2011). Molecular phylogeny of Cypripedium (Orchidaceae: Cypripedioideae) inferred from multiple nuclear and chloroplast regions. Mol. Phylogenet. Evol. 61, 308–320. doi: 10.1016/j.ympev.2011.06.006
Lim, C. E., Lee, S.-C., So, S., Han, S.-M., Choi, J.-E., and Lee, B.-Y. (2018). The complete chloroplast genome sequence of Asarum sieboldii Miq. (Aristolochiaceae), a medicinal plant in Korea. Mitochondrial DNA B 3, 118–119. doi: 10.1080/23802359.2018.1424577
Lin, C.-S., Chen, J. J., Huang, Y.-T., Chan, M.-T., Daniell, H., Chang, W.-J., et al. (2015). The location and translocation of ndh genes of chloroplast origin in the Orchidaceae family. Sci. Rep. 5:9040. doi: 10.1038/srep09040
Lin, M., Qi, X., Chen, J., Sun, L., Zhong, Y., Fang, J., et al. (2018). The complete chloroplast genome sequence of Actinidia arguta using the PacBio RS II platform. PLoS One 13:e0197393. doi: 10.1371/journal.pone.0197393
Logacheva, M. D., Schelkunov, M. I., and Penin, A. A. (2011). Sequencing and analysis of plastid genome in mycoheterotrophic orchid Neottia nidus-avis. Genome Biol. Evol. 3, 1296–1303. doi: 10.1093/gbe/evr102
McNeal, J. R., Kuehl, J. V., Boore, J. L., Leebens-Mack, J., and Depamphilis, C. W. (2009). Parallel loss of plastid introns and their maturase in the genus Cuscuta. PLoS One 4:e5982. doi: 10.1371/journal.pone.0005982
Muñoz-Gómez, S. A., Mejía-Franco, F. G., Durnin, K., Colp, M., Grisdale, C. J., Archibald, J. M., et al. (2017). The new red algal subphylum Proteorhodophytina comprises the largest and most divergent plastid genomes known. Curr. Biol. 27, 1677–1684. doi: 10.1016/j.cub.2017.04.054
Petersen, G., Cuenca, A., and Seberg, O. (2015). Plastome evolution in hemiparasitic mistletoes. Genome Biol. Evol. 7, 2520–2532. doi: 10.1093/gbe/evv165
Rozas, J., Ferrer-Mata, A., Sánchez-Delbarrio, J. C., Guirao-Rico, S., Librado, P., Ramos-Onsins, S. E., et al. (2017). DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 34, 3299–3302. doi: 10.1093/molbev/msx248
Ruhlman, T. A., Zhang, J., Blazier, J. C., Sabir, J. S. M., and Jansen, R. K. (2017). Recombination-dependent replication and gene conversion homogenize repeat sequences and diversify plastid genome structure. Am. J. Bot. 104, 559–572. doi: 10.3732/ajb.1600453
Sinn, B. T., Sedmak, D. D., Kelly, L. M., and Freudenstein, J. V. (2018). Total duplication of the small single copy region in the angiosperm plastome: rearrangement and inverted repeat instability in Asarum. Am. J. Bot. 105, 71–84. doi: 10.1002/ajb2.1001
Smith, D. R. (2018). Haematococcus lacustris: the makings of a giant-sized chloroplast genome. Aob Plants 10:hy058. doi: 10.1093/aobpla/ply058
Smith, D. R. (2020). Can green algal plastid genome size be explained by DNA repair mechanisms? Genome Biol. Evol. 12, 3797–3802. doi: 10.1093/gbe/evaa012
Su, H.-J., Barkman, T. J., Hao, W., Jones, S. S., Naumann, J., Skippington, E., et al. (2019). Novel genetic code and record-setting AT-richness in the highly reduced plastid genome of the holoparasitic plant Balanophora. Proc. Natl. Acad. Sci. U.S.A. 116, 934–943. doi: 10.1073/pnas.1816822116
Sullivan, A. R., Schiffthaler, B., Thompson, S. L., Street, N. R., and Wang, X.-R. (2017). Interspecific plastome recombination reflects ancient reticulate evolution in Picea (Pinaceae). Mol. Biol. Evol. 34, 1689–1701. doi: 10.1093/molbev/msx111
Talavera, G., and Castresana, J. (2007). Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 56, 564–577. doi: 10.1080/10635150701472164
Tangphatsornruang, S., Uthaipaisanwong, P., Sangsrakru, D., Chanprasert, J., Yoocha, T., Jomchai, N., et al. (2011). Characterization of the complete chloroplast genome of Hevea brasiliensis reveals genome rearrangement, RNA editing sites and phylogenetic relationships. Gene 475, 104–112. doi: 10.1016/j.gene.2011.01.002
Tonti-Filippini, J., Nevill, P. G., Dixon, K., and Small, I. (2017). What can we do with 1000 plastid genomes? Plant J. 90, 808–818. doi: 10.1111/tpj.13491
Wang, W., Schalamun, M., Morales-Suarez, A., Kainer, D., Schwessinger, B., and Lanfear, R. (2018). Assembly of chloroplast genomes with long- and short-read data: a comparison of approaches using Eucalyptus pauciflora as a test case. BMC Genom. 19:977. doi: 10.1186/s12864-018-5348-8
Weng, M. L., Ruhlman, T. A., and Jansen, R. K. (2017). Expansion of inverted repeat does not decrease substitution rates in Pelargonium plastid genomes. New Phytol. 214, 842–851. doi: 10.1111/nph.14375
Wu, Z., Gui, S., Quan, Z., Pan, L., Wang, S., Ke, W., et al. (2014). A precise chloroplast genome of Nelumbo nucifera (Nelumbonaceae) evaluated with Sanger, Illumina MiSeq, and PacBio RS II sequencing platforms: insight into the plastid evolution of basal eudicots. BMC Plant Biol. 14:289. doi: 10.1186/s12870-014-0289-0
Xiang, B., Li, X., Qian, J., Wang, L., Ma, L., Tian, X., et al. (2016). The complete chloroplast genome sequence of the medicinal plant Swertia mussotii using the PacBio RS II platform. Molecules 21:1029. doi: 10.3390/molecules21081029
Yan, C., Du, J., Gao, L., Li, Y., and Hou, X. (2019). The complete chloroplast genome sequence of watercress (Nasturtium officinale R. Br.): genome organization, adaptive evolution and phylogenetic relationships in Cardamineae. Gene 699, 24–36. doi: 10.1016/j.gene.2019.02.075
Yang, Z. (2007). PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591. doi: 10.1093/molbev/msm088
Zhang, L.-J., Ding, R., Meng, W.-W., Hu, H.-L., Chen, X.-H., and Wu, H.-H. (2019). The complete chloroplast genome sequence of the threatened Cypripedium calceolus (Orchidaceae). Mitochondrial DNA B 4, 4220–4222. doi: 10.1080/23802359.2019.1693933
Zhang, Y., An, D., Li, C., Zhao, Z., and Wang, W. (2020). The complete chloroplast genome of greater duckweed (Spirodela polyrhiza 7498) using PacBio long reads: insights into the chloroplast evolution and transcription regulation. BMC Genom. 21:76. doi: 10.1186/s12864-020-6499-y
Keywords: plastome expansion, repeat sequence, hybrid assembly, AT-biased base composition, long-read sequencing, palindromic repeat, inversion
Citation: Guo Y-Y, Yang J-X, Li H-K and Zhao H-S (2021) Chloroplast Genomes of Two Species of Cypripedium: Expanded Genome Size and Proliferation of AT-Biased Repeat Sequences. Front. Plant Sci. 12:609729. doi: 10.3389/fpls.2021.609729
Received: 24 September 2020; Accepted: 20 January 2021;
Published: 09 February 2021.
Edited by:
Gerald Matthias Schneeweiss, University of Vienna, AustriaReviewed by:
Maria D. Logacheva, Lomonosov Moscow State University, RussiaEric de Camargo Smidt, Federal University of Paraná, Brazil
Copyright © 2021 Guo, Yang, Li and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yan-Yan Guo, Z3VveXlAaGVuYXUuZWR1LmNu