 Jingxue Wang
Jingxue Wang Sanjay K. Singh
Sanjay K. Singh Chunfang Du3
Chunfang Du3 Sitakanta Pattanaik
Sitakanta Pattanaik Ling Yuan
Ling Yuan- 1College of Life Sciences, Shanxi University, Taiyuan, China
- 2Department of Plant and Soil Sciences, University of Kentucky, Lexington, KY, USA
- 3Cotton Research Institute of Shanxi Academy of Agricultural Sciences, Yuncheng, China
Rapeseed (Brassica napus) is an important oil seed crop, providing more than 13% of the world’s supply of edible oils. An in-depth knowledge of the gene network involved in biosynthesis and accumulation of seed oil is critical for the improvement of B. napus. Using available genomic and transcriptomic resources, we identified 1,750 acyl-lipid metabolism (ALM) genes that are distributed over 19 chromosomes in the B. napus genome. B. rapa and B. oleracea, two diploid progenitors of B. napus, contributed almost equally to the ALM genes. Genome collinearity analysis demonstrated that the majority of the ALM genes have arisen due to genome duplication or segmental duplication events. In addition, we profiled the expression patterns of the ALM genes in four different developmental stages. Furthermore, we developed two B. napus near isogenic lines (NILs). The high oil NIL, YC13-559, accumulates significantly higher (∼10%) seed oil compared to the other, YC13-554. Comparative gene expression analysis revealed upregulation of lipid biosynthesis-related regulatory genes in YC13-559, including SHOOTMERISTEMLESS, LEAFY COTYLEDON 1 (LEC1), LEC2, FUSCA3, ABSCISIC ACID INSENSITIVE 3 (ABI3), ABI4, ABI5, and WRINKLED1, as well as structural genes, such as ACETYL-CoA CARBOXYLASE, ACYL-CoA DIACYLGLYCEROL ACYLTRANSFERASE, and LONG-CHAIN ACYL-CoA SYNTHETASES. We observed that several genes related to the phytohormones, gibberellins, jasmonate, and indole acetic acid, were differentially expressed in the NILs. Our findings provide a broad account of the numbers, distribution, and expression profiles of acyl-lipid metabolism genes, as well as gene networks that potentially control oil accumulation in B. napus seeds. The upregulation of key regulatory and structural genes related to lipid biosynthesis likely plays a major role for the increased seed oil in YC13-559.
Introduction
In higher plants, cell proliferation, maturation, and desiccation are three key stages of seed development. Seed maturation requires accumulation of storage reserves of carbohydrates, storage proteins, and triacylglycerides (TAG) (Huang, 1992; Goldberg et al., 1994). TAG is high in energy-density, and vital in lipid homeostasis, cellular energy balance, and plant growth and maintenance (Durrett et al., 2008). Seed oils are utilized in human food, raw materials for non-food uses, and high-energy biofuels (Kumar and Sharma, 2015). Brassica napus is one of the most important oilseed crops, providing approximately 13% of the world’s supply of vegetable oils (Hajduch et al., 2006). In China, annual B. napus production reaches 14 million tons, of which human consumption accounts for more than 6 million tons, approximately 20% of the total edible vegetable oils (Shen and Fu, 2011). B. napus seeds contain oil, carbohydrates, and proteins as major storage reserves. The mature seeds contain approximately 45% (w/w) storage oil and 25% (w/w) proteins. The three major seed proteins are cruciferin (40–50%), napin (20%) and a hydrophobic polypeptide associated with the proteinaceous membrane surrounding the storage oil bodies (20%). In addition, about 10% of the storage reserve in B. napus seeds are soluble sugars (Murphy et al., 1989).
Over the past few decades, significant progress has been made in understanding the lipid biosynthesis using the model plant, Arabidopsis, which possesses more than 700 lipid related genes (Beisson et al., 2003). Most of the genes encoding key enzymes in lipid biosynthesis in plants have been isolated and characterized. Previous studies have revealed the importance of fatty acid (FA) metabolism in plant morphology, growth, pollen and seed development, defense, and stress responses (Mou et al., 2000; Kachroo et al., 2003, 2004; Zheng et al., 2005; Zhang et al., 2008; Dong et al., 2009). In most seeds, glycolysis in plastids supplies carbon for FA synthesis; however, green seeds can also use light to generate NADPH and ATP, thus enabling the bypass of glycolysis and an increased metabolic flux (Goffman et al., 2005; Hay and Schwender, 2011). Assembly of TAG occurs in the endoplasmic reticulum (ER) in association with the lipid droplets (LDs), or oil bodies. The plastid FA synthetic pathway determines the acyl chain length and the level of saturation in seed oils. ACETYL-CoA CARBOXYLASE (ACCase) is the first committed enzyme that controls the flux of carbon into FAs. In the plastid, acyl carrier protein (ACP)-linked acyl chains (acyl-ACP) are synthesized by ACCase, FATTY ACID SYNTHASE (FAS), and KETOACYL ACP SYNTHETASE I and II (KASI and II). The saturated acyl-ACP can either be hydrolyzed by the FATTY ACYL-ACP THIOESTERASES B (FATB), or desaturated by acyl-ACP desaturases, and further hydrolyzed by the FATTY ACYL-ACP THIOESTERASES A (FATA) (Li-Beisson et al., 2010). TAG are formed through three sequential acyl-CoA-dependent acylation of the glycerol backbone, catalyzed by enzymes such as ACYL-CoA GLYCEROL-3-ACYLTRANSFERASE (GPAT), ACYL-CoA PHOSPHATIDIC ACID ACYLTRANSFERASE (LPAAT), and ACYL-CoA DIACYLGLYCEROL ACYLTRANSFERASE (DGAT) (Li-Beisson et al., 2010). Although significant progress has been made in isolating structural genes encoding key enzymes, and elucidating their roles in lipid biosynthesis, the regulation of these genes is not well understood. During the past decade, several families of transcription factors (TF) have been reported to be able to enhance seed oil contents. Functionally characterized TFs include SHOOTMERISTEMLESS (STM), LEAFY COTYLEDON1 (LEC1), LEC2, ABSCISIC ACID INSENSITIVE3 (ABI3), FUSCA3 (FUS3) and WRINKLED1 (WRI1) (Mu et al., 2008; Li et al., 2015; Roscoe et al., 2015). STM, a member of the class-I KNOX HOMEODOMAIN-containing proteins, is known to regulate shoot apical meristem architecture in plants. In Arabidopsis, altered expression of STM affects in vitro organogenesis and somatic embryogenesis. Moreover, STM affects somatic embryogenesis by modulating the expression of LEC1 (Elhiti et al., 2012). LEC1 belongs to the NFY-B type CCAT-box binding TF family; whereas LEC2, ABI3, and FUS3 are plant specific B3-domain TFs. WRI1, a member of the APETALA2/ETHYLENE RESPONSE ELEMENT BINDING PROTEIN (AP2/EREBP) TF family, is a conserved master regulator that controls the genes encoding at least 15 enzymes in FA synthetic and glycolytic pathways, including PYRUVATE DEHYDROGENASE and ACCase (Bates et al., 2013; Ma et al., 2013). ABI3, FUS3, and WRI1 work downstream of LEC1 and LEC2. In addition, WRI1 has been shown to be a direct target of LEC2. Arabidopsis wri1 mutants accumulate 80% less seed oil (Focks and Benning, 1998). Overexpression of WRI1 enhances the oil contents in several crop plants, including potato (Hofvander et al., 2016), B. napus (Wu et al., 2014; Li et al., 2015), Camelina sativa (An and Suh, 2015), and maize (Shen et al., 2010). Recently, the mediator subunit 15 (MED15) has been implicated FA biosynthesis. The mediator is a highly conserved multi-protein complex and an important component of RNA polymerase II-mediated transcription machinery in eukaryotes. Arabidopsis MED15 overexpression increases FA content in seedlings and mature seeds whereas MED15 silencing results in reduced FA accumulation. Moreover, MED15 interacts with WRI1 and has been shown to be associated with the promoters of WRI1 target genes (Kim et al., 2016). TFs often mediate the connections between phytohormones and biological processes, such as lipid biosynthesis. Many of these lipid-biosynthesis regulators act downstream of multiple phytohormone pathways (Söderman et al., 2000; Cernac and Benning, 2004; Santos-Mendoza et al., 2008).
Brassica napus (genome AACC, 2n = 38) arises from hybridization between the diploids B. rapa (Asian cabbage, genome AA, 2n = 20) and B. oleracea (Mediterranean cabbage, genome CC, 2n = 18) (Allender and King, 2010). Over the past decade, a growing number of genomic resources for B. napus have become available, most noticeably the whole genome sequence of B. napus cultivar Darmor-bzh, a European winter oilseed (Hilliker et al., 2008; Chalhoub et al., 2014; Wang et al., 2015). Efforts have been made to either identify genomic loci controlling oil contents, or alter the quality and quantity of seed oil of B. napus (Katavic et al., 2014; Wu et al., 2014; Elahi et al., 2015, 2016; Li et al., 2015; Liu S. et al., 2016). Niu et al. (2009) have analyzed the FA biosynthesis-related genes using serial analysis of gene expression (SAGE), and concluded that 17–21 days after flowering (DAF) are crucial for the transition of young seeds to sink tissues (Niu et al., 2009). Transcriptome analysis of developing seeds from four different plant species, Ricinus communis, B. napus, Euonymus alatus and Tropaeolum majus, which differ in their oil storage tissues, as well as TAG structures and contents, has revealed both conserved and distinct species-specific expression patterns for the genes involved in lipid biosynthesis (Troncoso-Ponce et al., 2011). Recently, a comparative mapping of Arabidopsis lipid-related orthologous genes in B. napus has also been reported (Subramaniam et al., 2011).
As a close relative to Arabidopsis, B. napus is considered an ideal crop plant for translating information from the model species (Snowdon and Friedt, 2004). However, frequent duplication and rearrangement events in the B. napus genome preclude the establishment of a simple one-to-one relationship between B. napus and Arabidopsis (Parkin et al., 2005). Although the biochemical pathway for de novo lipid biosynthesis is well understood, much less is known about how qualitative and quantitative lipid productions are controlled during seed development of oil seed crops, such as B. napus. Advancements in high throughput technologies have resulted in surge of various -omic data addressing different aspects of plant growth and development. Microarray and next-generation sequencing (NGS) technologies have revolutionized our ability to monitor and analyze the global transcriptomic changes of plants in response to developmental and environmental cues. Genome-wide transcriptomic analyses of genetic variants can lead to identification of a set of genes that contribute to phenotypic changes. An insightful approach to understand the multi-step TAG biosynthetic pathway, and its complex regulatory network, is to study plant near isogenic lines (NILs) with near identical genetic background but different levels of TAG accumulation. Transcriptomic analysis of such NILs can potentially yield distinctive gene expression profiles that allow the identification of critical genes involved in TAG biosynthesis and candidate target genes for further characterization. For this purpose, we have developed a pair of B. napus NILs that vary in seed oil content by ∼10%. We identified differentially expressed genes (DEGs) of the two NILs. Our results suggest that changes in expression of the genes related to phyotohormones and lipid-regulating TF likely resulted in increased expression of FA and TAG pathway genes in the high oil-accumulating NIL, YC13-559.
Materials and Methods
Plant Materials
The parental donor of the NILs was a high-oil B. napus variety, YN171, developed by the Nanjing Agricultural Research Institute, China. Breeding in Shanxi province to select regional agronomic performance, such as growth, yield, and oil contents, has developed several B. napus lines. The NILs were developed by conventional method of repeated back-crossing (BC) of YN171 with locally domesticated recipient for up to five generations. BC individuals were then selfed and a number of lines selected from each crossing stream. YC13-554 and -559 are two individual NILs having genetic similarity coefficient of 0.94 as determined by SSR analysis. FA analysis also showed that YC13-559 accumulated significantly higher oil (∼10%) than YC13-554 (Liu Z. et al., 2016). This pair of NILs were selected for three consecutive years of field trial for agronomic characteristics and oil accumulation.
Near isogenic lines, YC13-554 and YC13-559, were grown in experimental fields in Yuncheng city, Shanxi, China, in the 2014 growing season. Following artificial pollination, siliques in the middle of main florescence were date-tagged. Immature seeds were separated from siliques at 19 days after pollination (DAP) as described previously (Niu et al., 2009), frozen immediately in liquid nitrogen, and stored at -80°C until total RNA isolation.
RNA Isolation and Illumina Sequencing
Total RNA were isolated from 2 g of immature seeds collected at 19 DAP using the RNeasy Plant Mini Kit (Qiagen, Chatsworth, CA, USA) following manufacturer’s instructions. RNA quantity was determined using a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA). Quality of RNA samples were determined using an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA). RNA samples with RNA integrity number (RIN) above 8 were used for library preparation. cDNA libraries were prepared using the TruSeq RNA Sample Prep Kit (Illumina, San Diego, CA, USA) according to the manufacturer’s protocol. The libraries were then pooled together and sequenced on an Illumina HiSeq 2000 (2X 101bp).
Gene Expression Quantification
Raw Illumina sequence reads were processed using the prinseq-lite-0.20.4 (Schmieder and Edwards, 2011) for removal of low-quality reads (Singh et al., 2015). Subsequently, pre-processed reads were assessed for quality control with FastQC (version 0.11.3; Babraham Bioinformatics, Cambridge, UK). Read mapping was performed by Bowtie2 (Langmead and Salzberg, 2012) using reference sequence downloaded from the B. napus genome database (Chalhoub et al., 2014). Differential gene expression analysis was carried out using the DESeq2 Bioconductor package in R (Love et al., 2014). DEGs were identified using two criteria: (a) log2 fold-change ≥ 1 and (b) an adjusted p-value less than 0.05.
Gene Ontology Analysis
Corresponding Arabidopsis thaliana orthologs for all DEGs were determined by the reciprocal best hits method. A list of Arabidopsis orthologs was uploaded to BiNGO plug-in (Maere et al., 2005) of Cytoscape (Shannon et al., 2003) for gene ontology analysis. Enrichment analysis was based on a hypergeometric test. P-values were adjusted using Benjamini–Hochberg’s FDR; only FDR < 0.05 was considered significant. For pathway analysis, a MapMan mapping file was specifically generated for B. napus genes by the Mercator tool, which bins all genes according to hierarchical ontologies after searching a variety of databases. MapMan v.3.5.1 (Thimm et al., 2004) was used to visualize the DEGs. In this study, the MCScanX tool kit (Wang et al., 2012a) with default parameter was used to identify genome collinearity. The Protein ANalysis THrough Evolutionary Relationships (PANTHER) Classification System and analysis tools were used to categorize DEGs by protein class (Mi et al., 2016).
Identification and Expression Analysis of Acyl-lipid Metabolism Genes of B. napus
In order to identify genes related to acyl-lipid metabolism, B. napus gene sequences were analyzed against a list of acyl-lipid metabolism genes obtained from the “Arabidopsis Acyl-Lipid Metabolism” website (ARALIP)1. Locally installed MultiLoc2 tool was used for prediction of sub-cellular localization of proteins (Blum et al., 2009).
To analyze the temporal expression of acyl-lipid metabolism genes in sequential stages of seed filling of B. napus, RNA-seq data of seeds at four developmental stages (2, 4, 6, and 8 weeks after pollination), and microarray data of eight developmental stages (10, 15, 20, 25, 30, 35, 40, and 45 days), were obtained from the sequence read archive database (accession number SRP069360) and Gene Expression Omnibus (accession number GSE43918), respectively (Leinonen et al., 2011; Barrett et al., 2013). Unsupervised hierarchical clustering was performed using hclust function, and clusters were then extracted using the cuttree function in R base package2. The heat-map corresponding to the hierarchical clustering was generated with the heatmap.2 function of the R packages gplots3.
Quantitative Real-Time PCR
RNA isolated from immature seeds of YC13-554 and YC13-559 were reverse-transcribed using the Superscript III Reverse Transcriptase (Invitrogen, USA), following the manufacturer’s instructions. Quantitative real-time PCR (qRT-PCR) was performed as described previously (Pattanaik et al., 2010). All PCR reactions were performed in triplicate and repeated two times. The comparative cycle threshold (Ct) method (bulletin no. 2; Applied Biosystems)4 was used to measure transcript levels. B. napus actin (GenBank accession number AF111812) was used as a reference gene (Niu et al., 2009). The relative expression of each gene is presented as the ratio of that in YC13-559 and YC13-554. The normalized expression in YC13-559 was divided by the normalized expression in YC13-554 and log2 transformed. The data represent the mean values ± SD of three replicates. Primer sequences for the genes have been listed in Supplementary Table S5.
Results and Discussion
The Near Isogenic Lines YC13-559 and YC13-554 Differ Significantly in Oil Content
Near isogenic lines, with near identical genetic background but varying mainly in the alleles responsible for a trait of interest, are useful in identification of target genes and pathways. Starting in 2009, we developed two B. napus NILs, YC13-554 and YC13-559, through repeated backcrossing of a high-yielding production line, YN171 with the recipient line. The two NILs share a genetic similarity coefficient of 0.94 based on SSR marker analysis. Agronomic trait analysis of the NILs showed several major differences, including 1000-seed weight and seed oil content. While plant height, primary branch height, length of main inflorescence, and number of pods per main inflorescence, were indistinguishable, YC13-559 accumulated significantly higher (∼10%) seed oil than YC13-554. The 1000-seed weight, which negatively correlates with seed oil content (Tang et al., 1997), is 30% less in YC13-559 compared to YC13-554. High oil accumulation is inversely correlated with protein content in seeds. As no significant seed size difference between the two NILs was observed, we speculate that a decrease of proteins or other metabolites led to the reduced seed weight. The results of three consecutive years of field trial (2012–2014) demonstrated that oil accumulation was higher in YC13-559 compared to YC13-554. It is important to note that the data generated in this study were based on field trial results. In field production, even a small percentage increase in seed oil translates into significant crop values (Lardizabal et al., 2008). The two NILs are ideal materials for the investigation of genetic and gene regulatory variations that contribute to TAG accumulation.
Analysis of Acyl-lipid-Metabolism Gene Expression during B. napus Seed Development Reveals Genetic Complexity and Critical Timing for TAG Biosynthesis
The amphidiploid B. napus shows significantly greater genome-level divergence compared to its close relative, Arabidopsis. We sought to clarify the number and distribution of acyl-lipid-metabolism (ALM) genes in B. napus. Based on homologies to 771 Arabidopsis ALM genes, we identified a total of 1750 lipid-related genes in B. napus (Supplementary Table S1). B. rapa and B. oleracea, two diploid parents of B. napus, contributed almost equally to the ALM genes; 881 and 863 genes were derived from the AA and CC genomes, respectively (Supplementary Table S1). B. napus ALM genes were found to be present in over 19 chromosomes, with chromosomes A03 and C03 harboring the largest numbers of ALM genes (126 and 122, respectively), while chromosomes A06 and C06 contain the least 70 and 68, respectively. Figure 1A summarizes the distribution of these genes into 12 different functional categories. The most abundant ALM genes were found to function in FA elongation, followed by phospholipid signaling and TAG biosynthesis. The maximum numbers of ALM proteins (∼37%) were predicted to be distributed in the cytoplasm, followed by organelles involved in ALM, such as the ER (∼18%) and chloroplasts (∼12%) (Supplementary Table S1). Whole-genome, segmental, and tandem gene duplications are common mechanisms for the generation of evolutionary novel functions (Wang et al., 2012b). In this study, we used the MCScanX toolkit (Wang et al., 2012a), with default parameters to detect collinear genomic regions and classify duplicated ALM genes according to their most likely mode of generation. The ALM genes, distributed on all nineteen chromosome pairs, were subjected to substantial duplication events (Supplementary Table S1). Among the duplicated genes, 1499 (85.6%) were predicted to have originated from whole genome duplication or segmental duplication events (Supplementary Table S1). The significantly higher numbers of ALM genes found in B. napus compared to Arabidopsis, are most likely due to frequent duplication events.
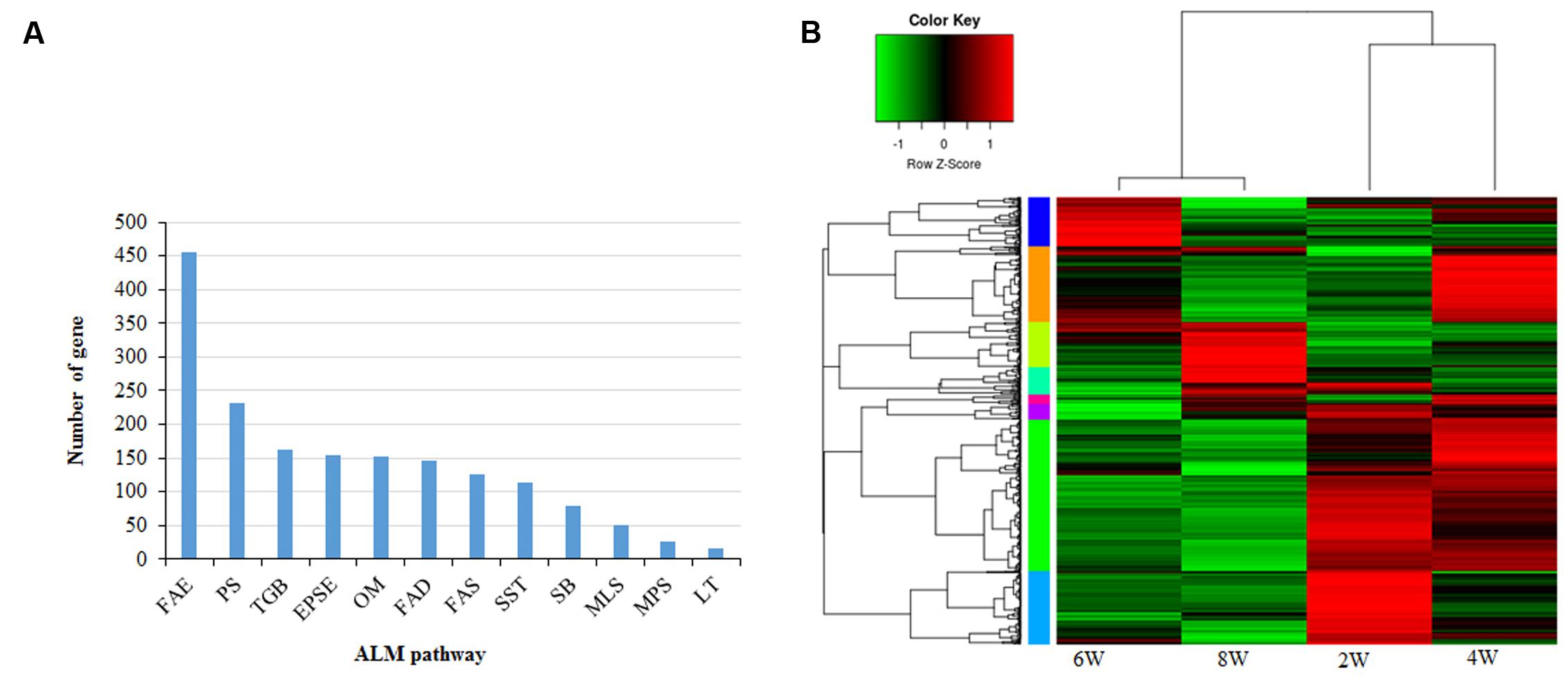
FIGURE 1. Acyl-lipid-metabolism (ALM) genes in Brassica napus (A) Identification and distribution of ALM genes in B. napus into twelve different functional categories and number of genes in each category is shown (B) ALM genes were categorized into 8 distinct clusters based on their expression pattern. Heat map shows the relative expression of all groups. Clusters are color-coded by row sidebars: light orange (cluster 1), green–yellow (cluster 2), green (cluster 3), cyan (cluster 4), aqua (cluster 5); blue (cluster 6); purple (cluster 7) and pink (cluster 8). FAE, Fatty Acid Elongation; PS, Phospholipid Signaling, TGB, Triacylglycerol Biosynthesis; EPSE, Eukaryotic Phospholipid Synthesis and Editing; OM, Oxylipin Metabolism; FAD, Fatty Acid Degradation; FAS, Fatty Acid Synthesis; SST, Suberin Synthesis Transport; SB, Sphingolipid Biosynthesis; MLS, Mitochondrial Lipopolysaccharide Synthesis; MPS, Mitochondrial Phospholipid Synthesis; LT, Lipid Trafficking; 2W, 2 weeks; 4W, 4 weeks; 6W, 6 weeks and 8W, 8 weeks.
Developing seeds accumulate TAGs either in embryonic tissues (e.g., B. napus and soybean), or in the endosperm (e.g., castor bean). Analyzing the specific gene expression patterns during seed development will aid in understanding the mechanism of seed oil accumulation. We examined the temporal changes in ALM gene expression during B. napus seed filling by analyzing RNA-seq data. Expression profiles of ALM genes during different stages of seed development were studied using publicly available RNA-seq datasets deposited in sequence read archive (SRA) database (accession number SRP069360). These datasets contain the expression data for four seed developmental stages, i.e., 2, 4, 6, and 8 weeks after pollination (WAP). To avoid background noise, ALM genes having expression levels equal to, or more than 1, read per kilobase per million (RPKM) in at least two samples, were selected for expression analysis (Topa and Honkela, 2016) (Supplementary Table S2). A total of 1205 genes thus generated from four developmental stages were grouped into 8 different clusters based on their expression patterns (Figure 1B; Supplementary Figure S1). Genes in cluster 1 (n = 203) and 3 (n = 408), which constitute ∼50% of all analyzed genes, were most highly expressed during week 4 of seed development (Supplementary Figure S1). Members of these two clusters are mainly involved in FA synthesis, FA elongation, and phospholipid signaling. As seeds aged, cluster 2 (n = 123) gene expression increased continuously, while cluster 5 (n = 198) gene expression decreased. Cluster 2 was overrepresented by genes involved in TAG biosynthesis, whereas cluster 5 genes are mostly related to FA elongation, phospholipid signaling, and TAG degradation. The expression of genes related to FA-elongation, TAG synthesis, and degradation peaked in week 4 (Supplementary Figure S2). Between weeks 6 and 8, the expression of most lipid related genes were gradually reduced. Previous studies on B. napus seed filling (Hajduch et al., 2006; Jolivet et al., 2011) show that seed weight, FA accumulation, and protein production increase continuously during the first 5 weeks after flowering. Seed weight and FA synthesis remain unchanged between weeks 4 and 5, while protein production continues. Our results are in good agreement with the reported characteristics of B. napus seed filling, showing the timing of ALM gene expression is slightly ahead of lipid accumulation.
To further verify our data, we analyzed a microarray dataset, obtained from Gene Expression Omnibus (accession number GSE43918), that includes B. napus tissues from week 2–6 of seed development (Huang et al., 2013). A total of 597 probes that mapped uniquely to the ALM genes, were included in this analysis (Supplementary Table S3). As shown in Supplementary Figure S3A, B. napus ALM genes formed three clusters, corresponding to week 2 (10 and 15 days), week 3–4 (20, 25, and 30 days), and week 5–6 (35, 40, and 45 days), of seed filling. Based on expression patterns during seed development, all probes were further classified into 16 different clusters. Similar to RNA-seq data, and a previous report (Niu et al., 2009), we observed that approximately 50% of analyzed probes were expressed the highest at 3–4 weeks (Supplementary Figure S3B). Seed LDs comprise a TAG core surrounded by a monolayer of phospholipids embedded with proteins such as caleosin and oleosin. Consistent with a report by Jolivet et al. (2011), we found that the expression of caleosin- and oleosin-family genes increased as seeds aged. Several TF genes that are known to be involved in FA biosynthesis and TAG accumulation, including WRI1, FUS3, and ABI3, have their highest expression in 3–4 weeks. Collectively, both RNA-seq and microarray analysis revealed that weeks 3–4 during seed development is critical for TAG biosynthesis in B. napus as expression of genes encoding key enzymes and transcriptional regulators peak at this stage.
RNA-seq Identifies Differentially Expressed Genes in the NILs
Illumina transcriptome sequencing was performed for libraries prepared from the two NILs. After the quality control, RNA-seq generated approximately 23.5 and 22.6 million pair-end reads (100-bp in size), as well as 7.9 and 7.5 million singletons for YC13-554 and YC13-559, respectively (Table 1). On an average, 86% reads were mapped to the reference genome using the software, Bowtie2 (Langmead and Salzberg, 2012). Based on RPKM, we found close overlap between the two NILs. When 0.1 RPKM was set as the threshold, 60,896 common genes were found to express in both NILs. When the threshold was increased to 1 RPKM, 46,797 common genes were found (Figure 2A). We identified 1234 candidate DEGs (twofold change) between the two NILs (Figure 2B, Supplementary Table S4). In YC13-559, 635 genes were upregulated, while 599 genes were downregulated compared to YC13-554. The upregulated and downregulated DEGs were classified into different class of proteins using PANTHER. More than 35% of DEGs belonged to enzyme class of proteins. Nucleic acid binding proteins and transporters were two other abundant class of proteins in DEGs (Figure 2C).
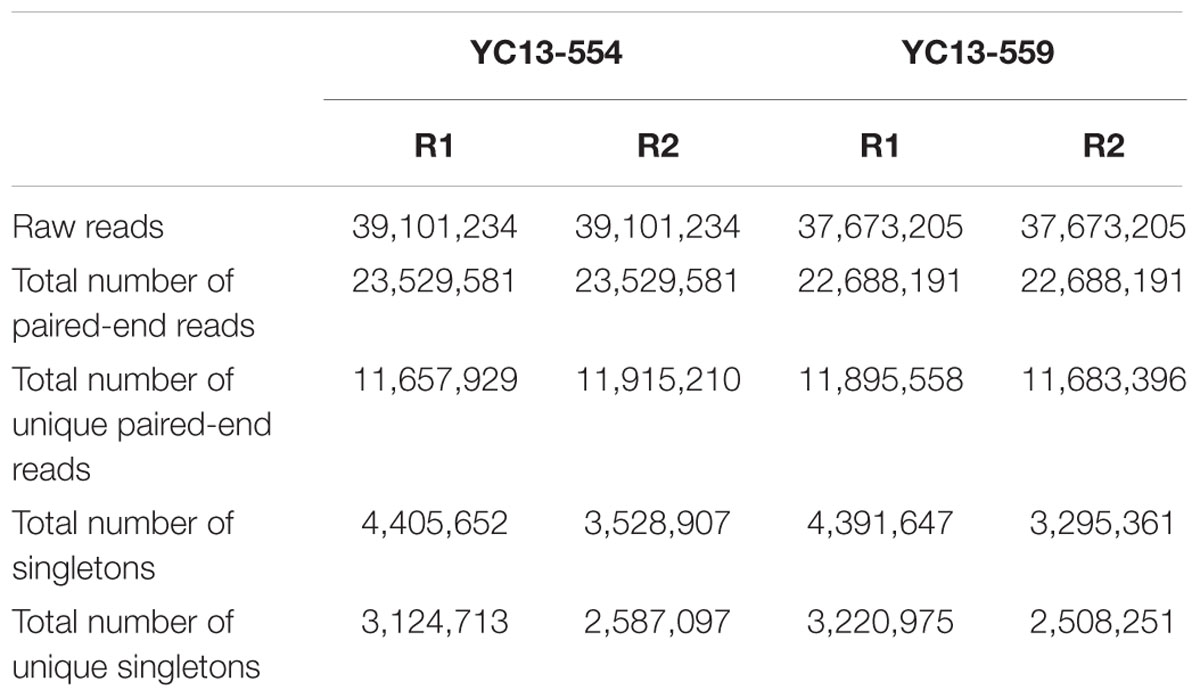
TABLE 1. Summary of transcriptome data.
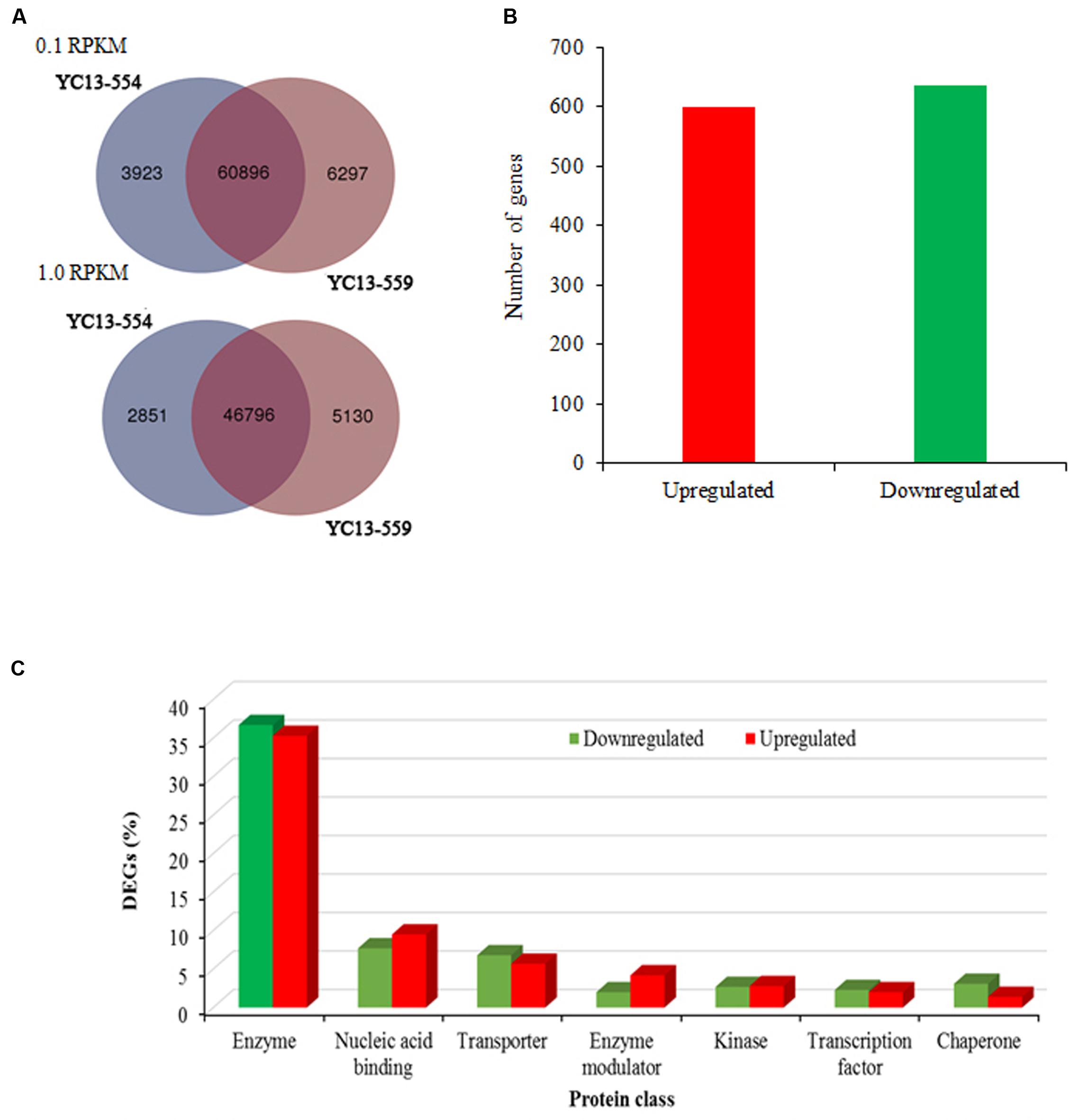
FIGURE 2. Differential expression and classification of transcripts. (A) Venn diagrams showing overlap of expressed genes having at least 0.1 reads per kilobase of transcript per million mapped reads (RPKM) and 1.0 RPKM expression value in near- isogenic lines YC13-554 and YC13-559. (B) Number of upregulated and downregulated genes. (C) Protein classification of the DEGs (n = 1234 genes; 635 upregulated and 599 downregulated) using PANTHER (Protein Analysis Through Evolutionary Relationships). Protein classes encompassing at least 2% of the upregulated or downregulated genes are shown.
Quantitative RT-PCR of Selected ALM Genes Validates the RNA-seq Results
We identified a number of FA biosynthesis- and accumulation-related genes whose expression changed significantly in YC13-559 compared to YC13-554 and chose a set of 10 genes (7 upregulated and 3 downregulated) from that list to validate their expression by qRT-PCR (Figure 3; Supplementary Table S5). In addition to the 3 downregulated genes, 7 ALM genes, including pyruvate kinase (PK), long-chain acyl-CoA synthetase1, 6, and 7 (LACS1, 6, and 7), alpha/beta-Hydrolases superfamily protein (alpha/beta hydrolase), DGAT1, and 3-ketoacyl-CoA synthase 16 (KCS16), were upregulated. The qRT-PCR results were in agreement with the RNA-seq data, confirming the quality and accuracy of our RNA-seq experiment.
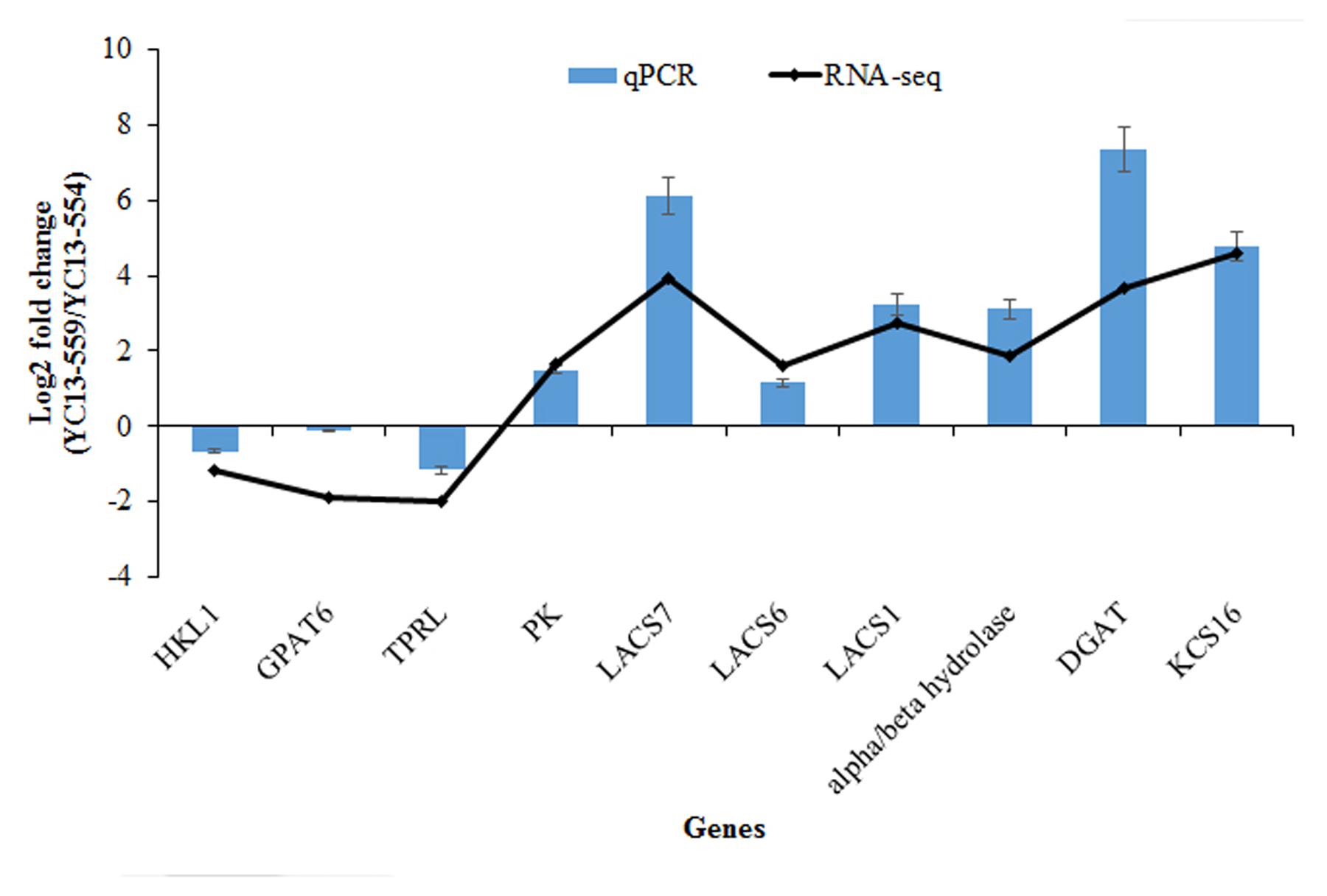
FIGURE 3. Validation of RNA-seq results using quantitative real-time polymerase chain reaction (qRT-PCR). The relative abundance of each gene is presented as the ratio of YC13-559 and YC13-554 lines. B. napus actin was used as an internal control for normalization. The normalized expression (2-ΔCT) in YC13-559 sample was divided by the normalized expression in YC13-554 and log2 transformed. The data represent the mean values ± SD of three replicates. HKL1, hexokinase-like 1; GPAT6, glycerol-3-phosphate acyltransferase 6; TPRL, tetratricopeptide repeat (TPR)-like superfamily; protein PK, pyruvate kinase family protein; LACS7, long-chain acyl-CoA synthetase 7; LACS6, long-chain acyl-CoA synthetase 6; LACS1, long-chain acyl-CoA synthetase 1; alpha/beta-Hydrolases, alpha/beta-Hydrolases superfamily protein; DGAT1, acyl-CoA:diacylglycerol acyltransferase 1; KCS16, 3-ketoacyl-CoA synthase 16.
Gene Ontology and MapMan Functionally Classify the Differentially Expressed Genes
Gene Ontology (GO) annotation is a major tool for gene enrichment analysis of genome-scale experiments. The enrichment analysis of the transcriptomic data from the two NILs, according to three major GO terms, namely the biological process, cellular component and molecular function, and, is given in Figure 4. The GO term, “biological process,” includes the majority of genes involved in metabolic process (GO: 0008152), cellular process (GO: 0009987), and response to stress (GO: 0006950) (Figure 4A). Lipid metabolic process (GO: 0006629) and carbohydrate metabolic process (GO: 0005975) were two most affected metabolic processes. Lipid metabolism is closely connected to that of carbohydrates which are regularly metabolized to form acetyl-CoA, a precursor for FA biosynthesis. The metabolism process was overrepresented by children terms, including lipid biosynthesis (GO: 0006629), monocarboxylic acid biosynthesis (GO: 0072330), and oxylipin metabolism (GO: 0031407). Several genes related to JA and oxylipin metabolism, such as LIPOXYGENASE 2 (LOX2), LOX3, JASMONIC ACID CARBOXYL METHYLTRANSFERASE (JMT), and JASMONATE RESISTANT 1 (JAR1), were found to be differentially expressed in our transcriptome data. JA and FA synthesis are well connected as the acyl-CoA pool generated from FA biosynthetic pathway is used for JA synthesis (Fu et al., 2015). The DEGs in the category “cellular components” were predicted to localize mostly in cytoplasm or plasma- and cell-membranes (Figure 4B). Notably, cell organelles, such as ER and plastid, which are known sites of FA and TAG biosynthesis, are also enriched in our dataset. Within the category “molecular function,” the most abundant GO term is catalytic activity (GO: 0003824), followed by protein binding (GO: 0005515) (Figure 4C). The majority of catalytic activities are associated with hydrolases and transferases.
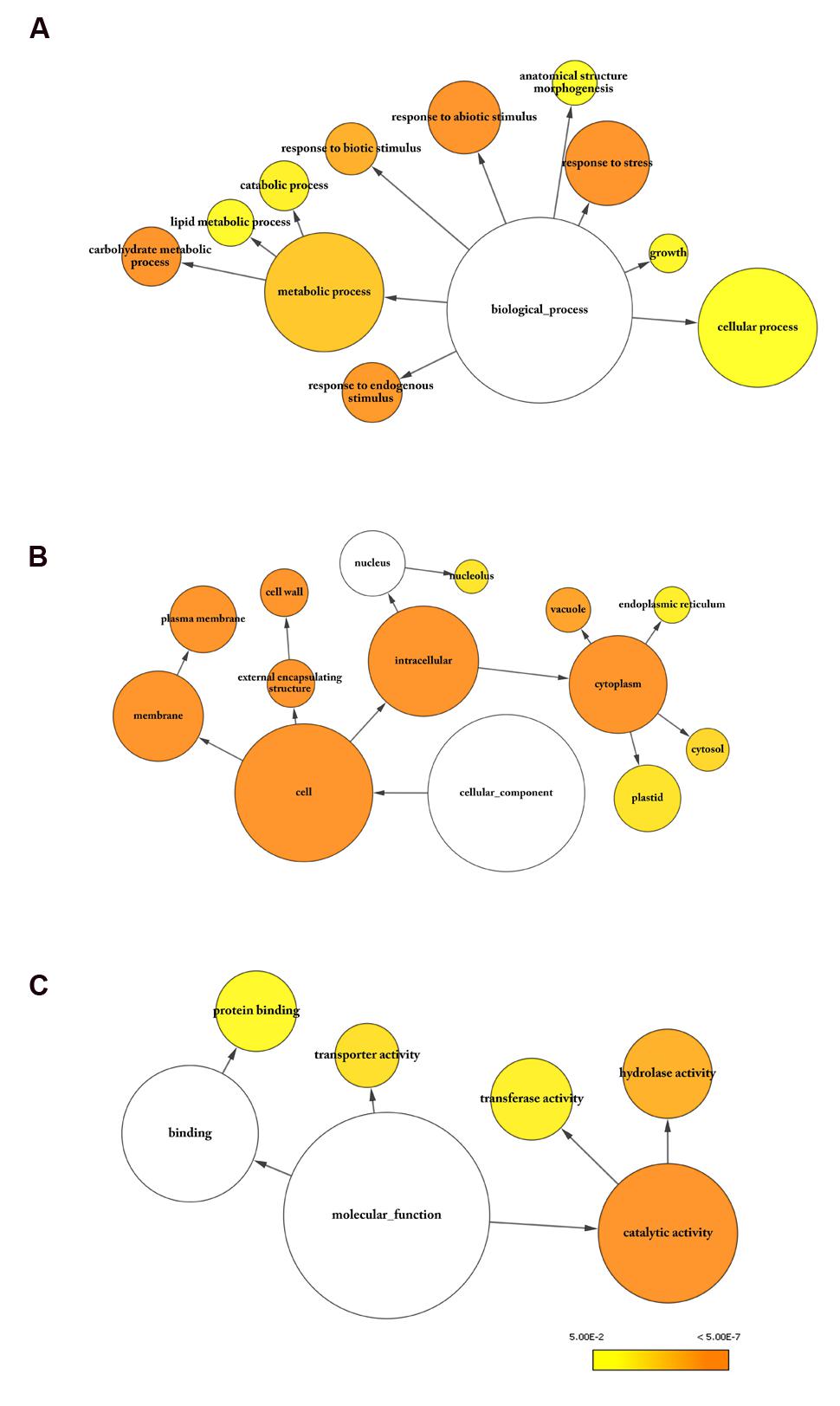
FIGURE 4. Gene Ontology (GO) analysis of DEGs. GO analysis was carried out using BinGO (Maere et al., 2005). Enriched categories for (A) biological process, (B) cellular component, and (C) molecular function are shown. Goslim_Plants categories with significant enrichment in the dataset were highlighted in color. The size of the node is proportional to the number of molecules within the group, and the color of the node represents the significance of enrichment. Benjamini and Hochberg p-value legend is indicated below.
As a complementary approach to GO term enrichment analysis, we explored the putative functions of the DEGs using MapMan (Thimm et al., 2004), which allows the visualization of varietal specific changes in different metabolic processes (Figure 5). Each gene in MapMan is initially organized in bins rather than as pathways, allowing the genes to be assigned into a pathway even when their functions are only tentatively predicted. Figure 5 shows the mapping of the DEGs to overall metabolism, cellular responses, and regulation. Consistent with the GO analysis, the DEGs were enriched in similar functional categories and pathways by MapMan. Although a significant fraction of DEGs could not be assigned to specific functional categories, a large number of genes were assigned to lipid metabolism, cell wall, protein, signaling, and transport. Notably, many genes were linked to stress, consistent with the involvement of lipid related genes in response to both biotic (Shah, 2005) and abiotic stress (Golldack et al., 2014; Higashi et al., 2015; Hou et al., 2016). An additional bin comprises genes involved in biosynthesis of secondary metabolites, such as flavonoids and phenylpropanoids. Another significantly affected category involves genes related to phytohormone biosynthesis and signaling. A total of 41 genes related to JA, ethylene, abscisic acid, gibberellins (GAs), auxin, and brassinosteroid were found to be differentially expressed. Previously, altered FA productions have been reported in Arabidopsis mutants deficient in JA and auxin metabolism (Niu et al., 2009). GAs play crucial roles in many aspects of plant life cycles, from seed germination and development to reproduction. GA signal perception and transduction are controlled by the GA receptor GIBBERELLIN INSENSITIVE DWARF (GID) and DELLA repressor proteins (Daviere and Achard, 2013). In many plant species, GA biosynthesis increases in the early stages of seed development, continues to seed maturity, and begins to decrease at the onset of dormancy. GA3 is also involved in the regulation of FA biosynthesis in Arabidopsis. Loss of DELLA function or exogenous application of GA3 affect the expression of key regulatory genes, including LEC1, LEC2, FUS3, ABI3 and WRI1, as well as their targets in Arabidopsis (Chen et al., 2012). A cytochrome p450, ent-kaurenoic acid oxidase (KAO), catalyzes the three-step oxidation of ent-kaurenoic acid to GA12, the precursor of all GAs, thereby determining the endogenous GA levels in plants (Regnault et al., 2014). In our data set, the expression of a KAO1 homolog was significantly higher in YC13-559 compared to YC13-554. The higher expression of KAO1 likely resulted in the increase of endogenous GA level, and was probably, at least in part, responsible for increased expression of the regulatory and structural genes in YC13-559 FA biosynthetic pathway. Inactivation of phytohormones is a mechanism to maintain hormone homeostasis in plants. In Arabidopsis, two members of the SABATH family methyltransferases, GIBBERELLIN METHYLTRANSFERASE (GAMT) 1 and GAMT2, mediate the transfer of methyl groups to GA, resulting in the biologically inactive methyl esters. Overexpression of GAMT1 or GAMT2 in Arabidopsis, petunia, and tobacco lead to GA deficiency and the resulting transgenic plants are dwarf with reduced fertility (Varbanova et al., 2007). In our data set, the expression of a GAMT homolog was higher in YC13-559 compared to YC13-554. It appears that the biosynthesis and catabolism of GA, involving activation and deactivation of the GA, are affected more significantly in YC13-559 than in YC13-554.
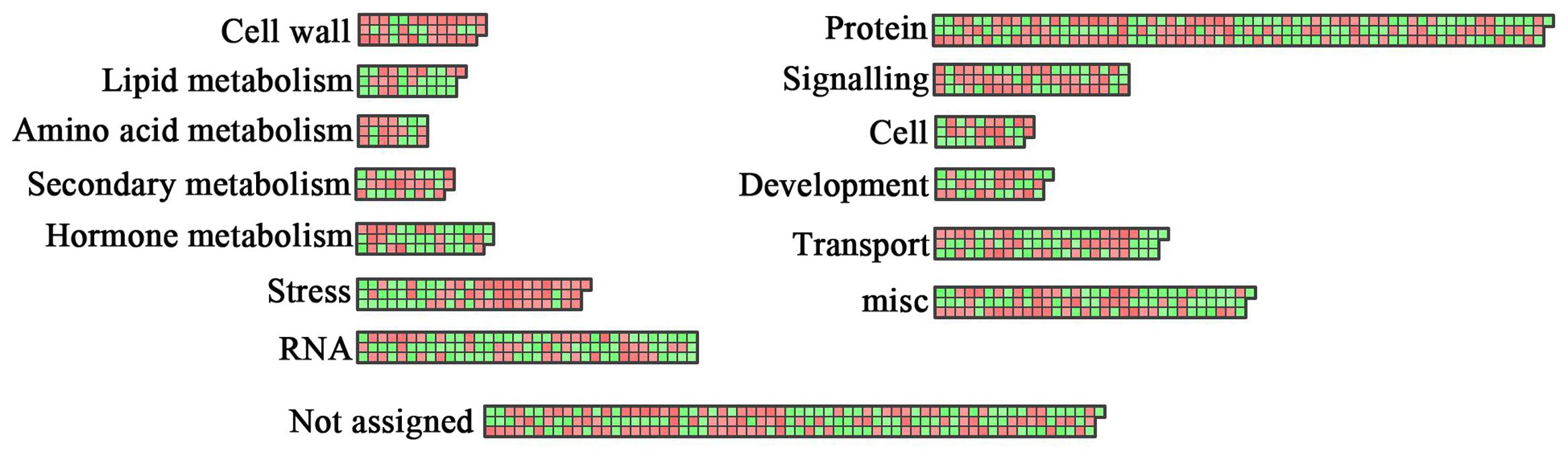
FIGURE 5. Functional analysis of DEGs using MapMan. Overview of the distribution of DEGs in different functional groups as identified by MapMan (Thimm et al., 2004) analysis. Red boxes correspond to up-regulated genes and green boxes to down-regulated genes.
Jasmonic acid is essential for a number of biological processes in plants, including growth, development, and response to wounding. Long-chain FAs serve as the precursors of JA biosynthesis in plants. CORONATINE INSENSITIVE 1 (COI1) plays a key role in JA signal perception and transduction. The Arabidopsis loss-of-function mutant, coi1, exhibits altered FA biosynthesis and composition in seeds (Niu et al., 2009). On the other hand, mutations in key FA biosynthetic genes have been shown to affect JA signaling. Mutation in the Arabidopsis ssi2 locus, that encodes the plastidic stearoyl–ACP desaturase, compromises JA responses (Kachroo et al., 2003; Nandi et al., 2003), suggesting the existence of a feedback regulatory loop between the JA signaling and FA biosynthesis pathways. A number of genes encoding enzymes for JA biosynthesis and conversion of JA to methyl jasmonate (MeJA) were differentially expressed in our data set. Increased expression of genes encoding LOX2, that oxidizes ś-linolenic acid, and JMT, that converses JA to MeJA, were observed in YC13-559 relative to YC13-554.
The category ‘RNA’ includes the genes related to RNA metabolism and TFs (Supplementary Table S6). A total of 57 TFs in this category can be further classified into 28 families that are overrepresented by MYB, WRKY, and homeobox TFs. FA biosynthesis is a rigorous process in which transcriptional regulation is no doubt vital; however, to date only very few TFs have been identified to be responsible for FA biosynthesis and storage. We thus analyzed the co-expression of these TFs and the FA biosynthetic genes. A total of 1262 genes (1205 ALM and 57 TF genes), were categorized into 7 clusters (Supplementary Figure S4A). Clusters 3 and 4 were the largest clusters containing approximately 50% of the identified TFs. The members of cluster 3 were found to have highest expression at early stage (2-week) of seed filling, while the members of cluster 4 maximally expressed during week 4 (Supplementary Figure S4B). Cluster 3 and 4 are rich in MYB, AP2/ERF and Dof TFs. Some members of these TF families are known to be involved in direct or indirect regulation of ALM genes. For example, overexpression of GmDof4 or GmDof11 from Glycine max enhance lipid content in transgenic Arabidopsis (Wang et al., 2007).
Key Regulatory Genes in FA and TAG Biosynthetic Pathways are Upregulated in YC13-559
Expression of the structural genes in FA and TAG biosynthetic pathways are regulated by a number of TFs, including STM, LEC1, LEC2, ABI3, FUS3, and WRI1 (Figure 6A). Recently, ABI4, an AP2/ERF TF, and ABI5, a bZIP TF, have also been implicated in TAG biosynthesis in Arabidopsis (Yang et al., 2011; Kong et al., 2013). Accumulating evidence has begun to uncover the gene regulatory network underlying lipid biosynthesis in plants. LEC1 and LEC2 likely act downstream of STM, but upstream of ABI3 and FUS3. ABI3 and FUS3 are known to regulate each other (Figure 6A) (Baud et al., 2007; Mu et al., 2008; Santos-Mendoza et al., 2008; Maeo et al., 2009). Expression of STM, LEC1, LEC2, ABI3, and FUS3, were moderately higher in YC13-559 compared to YC13-554. The B. napus genome contains two LEC1 and four LEC2 homologs of Arabidopsis. Expression of two LEC1 and three LEC2 homologs were 1.7–2.5 fold higher in YC13-559 relative to YC13-554. B. napus contains four FUS3 homologs of Arabidopsis and expression of one was higher in YC13-559 compared to YC13-554. In Arabidopsis, WRI1 is directly controlled by LEC1 (Mu et al., 2008) and LEC2 (Baud et al., 2007). We therefore analyzed the expression of the genes encoding WRI1 in the two NILs. B. napus has 3 Arabidopsis WRI1 homologs. Expression of all three homologs were 1.5–1.8 fold higher in YC13-559 compared to YC13-554. WRI1 regulates expression of the late step genes in the glycolic and FA biosynthetic pathways. Enhanced expression of WRI1 homologs in YC13-559 correlated with expression of the genes involved in seed oil biosynthesis, such as PK, PDHC, and LACS, known targets of WRI1 (Pouvreau et al., 2011). ABI4 and ABI5 repress lipid breakdown by increasing expression of DGAT1, a rate-limiting gene in TAG biosynthesis (Yang et al., 2011; Kong et al., 2013). The expression of ABI4 and ABI5 were 5- and 3-fold higher, respectively, in YC13-559 relative to YC13-554 (Figure 6B).
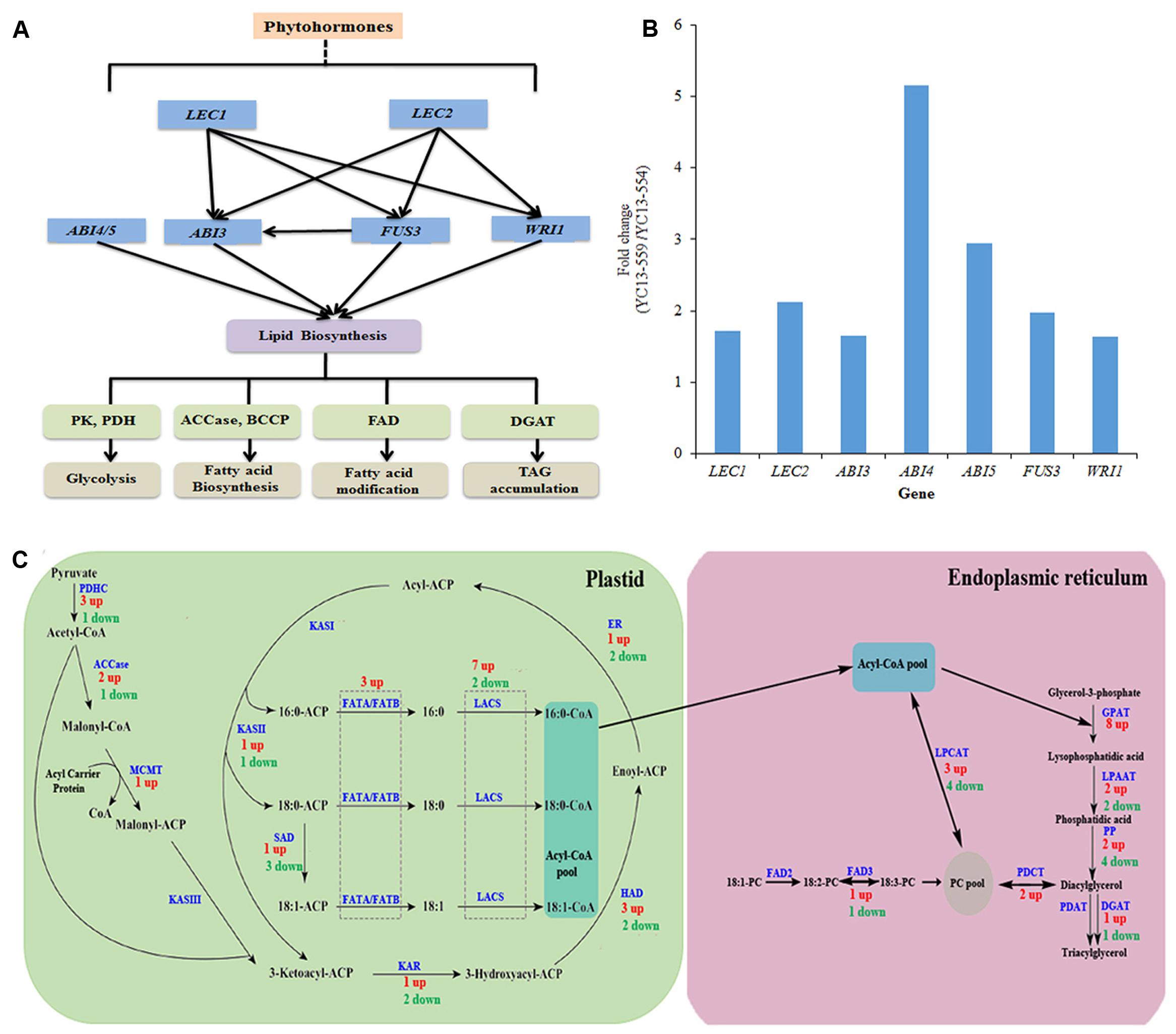
FIGURE 6. Overview of gene network involved in seed oil biosynthesis in B. napus (A). A simplified scheme of transcription factors (TF) network involved in regulation seed oil biosynthesis B. napus (B) Relative expression levels of different TF genes in YC13-559. TFs whose expression changed more than 1.5-fold in YC13-559 relative to YC13-554 are shown. The RPKM values for multiple copies of same TF were summed. (C) Overview of de novo fatty acid (FA) and triacylglycerol (TAG) biosynthesis pathways. Genes whose expression changed more than 1.5-fold in YC13-559 relative to YC13-554 were included in this study. Genes were mapped onto de novo FA and TAG biosynthesis pathway. Numbers refers to the numbers of genes, and “up” refers to up-regulation in YC13-559 line compare to YC13-554 line, and “down” to down-regulation. Same enzyme catalyze several steps are highlighted with dotted rectangle. Lipid substrates are abbreviated: 16:0, palmitic acid; 18:0, stearic acid. PDHC, pyruvate dehydrogenase complex; ACCase, acetyl-CoA carboxylase; MCMT, malonyl-CoA : ACP malonyltransferase; ACP, acyl carrier protein; KASI/II/III, ketoacyl-ACP synthase I/II/III; KAR, ketoacyl-ACP reductase; HAD, hydroxyacyl-ACP dehydrase; ER, enoyl-ACP reductase; FATA/B, fatty acyl-ACP thioesterase A/B; SAD, stearoyl-ACP desaturase; LACS, long-chain acyl-CoA synthetase; GPAT, glycerol-3-phosphate acyltransferase; LPAAT, 1-acylglycerol-3-phosphate acyltransferase, PP, phosphatidate phosphatase; PDCT, Phosphatidylcholine:diacylglycerol cholinephosphotransferase; PDAT, phospholipid :diacylglycerol acyltransferase; DGAT, acyl-CoA : diacylglycerol acyltransferase, LPCAT, lysophospholipid acyltransferase; FAD2/3, fatty acid desaturase.
A Number of Key Structural Genes in the FA and TAG Biosynthetic Pathways Are Upregulated in YC13-559
Biosynthesis of TAG involves three major steps in two sub-cellular compartments (Figure 6C). The first step is de novo FA synthesis in the plastid, mainly by the FA synthase complex (FAS) using acetyl-CoA as starting substrate. Subsequently, TAG is derived from FA and glycerol backbone in the ER. Finally, TAG is stored, by association with LDs, in the cytoplasm (Li-Beisson et al., 2010; Bates et al., 2013). PK catalyzes the conversion of phosphoenol pyruvate (PEP) to pyruvate, which is translocated to the plastid via the BILE ACID: SODIUM SYMPORTER FAMILY PROTEIN 2 (BASS2) transporter, and acts as an initial precursor for FA biosynthesis (Furumoto et al., 2011). Expression of PK and BASS2 were higher (1.6–10 fold) in YC13-559 compared to YC13-554, suggesting a potential increase of initial substrate for FA biosynthesis in YC13-559. The conversion of pyruvate to FAs involves at least 14 enzymes and/or protein complexes (Figure 6C). As in Arabidopsis, most of the individual enzymes are encoded by multiple homologous genes in B. napus. The pyruvate dehydrogenase complex (PDHC), catalyzing the oxidative decarboxylation of pyruvate to form acetyl-CoA, consists of three subunits, E1, E2, and E3. We found that 3 PDHC genes were upregulated (1.5-15 fold) while one was downregulated in YC13-559. ACCase converts acetyl-CoA into malonyl-CoA in the first committed step of FA synthesis. In YC13-559, two of the 3 identified ACCase genes were upregulated (1.5–1.9 fold) and one was downregulated. FAs are elongated in a series of condensation reactions with malonyl-ACP. This process is catalyzed by the FAS that comprises five components, namely MALONYL-CoA-ACP TRANSACYLASE (MCMT), 3-OXOACYL:ACP-SYNTHASE (KAS) I, KASII and KASIII, 3-OXOACYL-ACP REDUCTASE (KAR), HYDROXYACYL-ACP DEHYDRATASE (HAD) and ENOYL-ACP REDUCTASE (EAR). All of these genes, except KASI and KASII, were highly expressed in YC13-559 compared to YC13-554. FAs synthesized in the plastids are transported to the ER for TAG biosynthesis. LACS catalyze the esterification of FAs to acyl-CoAs, a key activation step that is essential for the utilization of FAs by most lipid metabolic enzymes. LACS are thus important enzymes for supplying the acyl-CoA pool for phospholipids and TAG biosynthesis (Shockey et al., 2002). Recent findings suggest that LACS are involved in lipid trafficking between ER and plastids in Arabidopsis (Jessen et al., 2015). Expression of seven LACS genes were 1.5–15 fold higher in YC13-559 compared to YC13-554, suggesting an increased acyl-CoA pool and trafficking rate for YC13-559.
Glycerol-3-phosphate (G-3-P) is an initial substrate for TAG biosynthesis (Figure 6C). GLYCEROL3-PHOSPHATE ACYLTRANSFERASE (GPAT) utilizes G-3-P and acyl-CoA as substrates to form lysophosphatidic acid (Lyso-PA). Expression of eight GPAT genes were significantly higher (1.5–15 fold) in YC13-559, possibly contributing to higher TAG accumulation. Acylation of Lyso-PA is catalyzed by 1-ACYL-sn-GLYCEROL-3-PHOSPHATE ACYLTRANSFERASE (LPAAT), resulting in phosphatidic acid (PA) formation. Next, PA is dephosphorylated by PHOSPHATIDATE PHOSPHATASE (PP) to form diacylglycerol (DAG). Two LPAAT genes were upregulated and two were downregulated in YC13-559. Four PP genes were downregulated while two were upregulated in YC13-559. In addition, two genes encoding PHOSPHATIDYLCHOLINE:DIACYLGLYCEROL CHOLINEPHOSPHOTRANSFERASE (PDCT), that catalyzes the transfer of phosphocholine head-groups from PC to DAG (Lu et al., 2009), were 2–4.5 fold higher in YC13-559. The final conversion of DAG into TAG is catalyzed by two enzymes: DIACYLGLYCEROL O-ACYLTRANSFERASE (DGAT) and PHOSPHOLIPID DIACYLGLYCEROL ACYLTRANSFERASE1 (PDAT). In B. napus, DGATs are encoded by four homologous genes of Arabidopsis DGAT1 and DGAT2. In Arabidopsis, DGAT2 seems to be more efficient in DAG to TAG conversion than DGAT1 (Zhou et al., 2013). The expression of DGAT2 in YC13-559 was approximately 6-fold higher compared to that of YC13-554. No significant difference in the expression of PDAT was observed in the two NILs. TAGs are stored in LDs which consist of caleosins and oleosins. In our data set, the expression of four out of ten caleosin genes were higher in YC13-559 relative to YC13-554. We did not observe increase in oleosin expression in the two NILs.
Key Structural Genes in the Glucosinolate Biosynthetic Pathway Are Downregulated in YC13-559
Glucosinolates (GLS) are a group of sulfur-containing specialized metabolites in Brassica seeds that negatively affect nutritional quality. GLS and their breakdown products play roles in plant defense against pest and pathogens (Troncoso-Ponce et al., 2011). A previous study showed that overexpression of STM in B. napus downregulates key genes in the GLS pathway, and reduces GLS accumulation (Elhiti et al., 2012). In our data set, the expression of STM was higher in YC13-559 compared to YC13-554. We therefore analyzed the expression of key GLS pathway genes, including CYTOCHROMEP45 CYP79B2 (CYP79B2), METHYLTHIOALKYLMALATE SYNTHASE1 (MAM1), CYTOCHROMEP45 CYP83B1 (CYP83B1), and SULPHOTRANSFERASE5a (ST5a), in the two NILs. The transcript levels of CYP79B2, CYP83B1 and ST5a were lower in YC13-559 relative to YC13-554, suggesting that higher expression of STM possibly affected the GLS pathway in YC13-559. Our future efforts will include profiling of GLS accumulation in the NILs.
Glucosinolate metabolism is tightly connected to phytohormones. Auxins, in particular, indole-3-acetic acid (IAA), are essential for plant growth. Indolic GLS are derived from tryptophan (Trp) and linked to IAA biosynthesis (Bak and Feyereisen, 2001). The enzymes, CYP83B1 and CYP83B2, are important for indolic GLS and IAA biosynthesis. CYP83B2 converts Trp to indo-3-acetaldoxime (IAOx), a precursor of IAA and indole-GLS. CYP83B1 catalyzes the conversion of IAOx to S-alkyl thiohydroximates to regulate the flux of IAOx between IAA and indolic GLS. In Arabidopsis, loss-of-function of CYP83B1 results in auxin overproduction phenotypes (Bak and Feyereisen, 2001). In our data set, the transcript levels of both CYP83B1 and CYP83B2 were lower in YC13-559 compared to YC13-554, indicating possible upregulation of auxin biosynthesis. To support our assumption, we analyzed the expression of auxin metabolism genes in the two NILs. Transcript level of NITRILASE, which is involved in IAA production, was higher in YC13-559 relative to YC13-554. Amino acid-conjugated IAA are biologically inactive, perhaps to help maintain IAA homeostasis in plants. Several IAA-amido synthetases catalyze the conjugation of amino acids to IAA (Bak and Feyereisen, 2001). In addition, IAA conjugates can be hydrolyzed by amidohydrolases, such as IAR3 and ILL2 (Rampey et al., 2004). In YC13-559, we found upregulation of IAA-amido synthetase and downregulation of IAR3 (Supplementary Table S4). Our results suggest that upregulation of amido-synthatase, and downregulation of amido hydrolase, possibly play roles in maintaining optimal auxin levels during seed development. Previously, Taipalensuu et al. (1997) reported that the myrosinase-binding protein, MYROSINASE-BINDING PROTEIN 2 (MIB2), is involved in the conversion of metabolized GLS to other defense compounds (Taipalensuu et al., 1997). We found that the expression of MIB2 was more than fourfold higher in YC13-559 compared to YC13-554.
Conclusion
Comparative transcriptome analysis of a pair of B. napus NILs, YC13-554 and YC13-559 which differ significantly in seed oil accumulation revealed 1,234 candidate DEGs between the two lines that are mostly involved in lipid and carbohydrate metabolism. In addition, a number of genes associated with signal transduction, transport, and biosynthesis of phytohormones, such as JA, GA, and IAA, were found to be differentially expressed. Identification of DEGs related to phytohormones in the NILs is of particular interest. Phytohormones impose drastic phenotypes, often through interactions with signal pathway proteins that form complexes with TFs. Therefore, small changes in phytohormone metabolism can affect a large number of transcriptional cascades that regulate various biological processes. The differential expression of several lipid-related TF genes, such as STM, LEC1, ABI3, WRI1, FUS3, in the NILs, are potentially influenced by these phytohormones. The upregulation of these TFs in YC13-559 probably led to the increased expression of key pathway genes, including PK, ACCase, LACS, FATA/FATB, and DGAT, resulting in higher seed oil content in YC13-559. In addition, the differential expression of key TF genes is likely responsible for the downregulation of GLS pathway genes in YC13-559. It is highly possible that genetic variations affecting other pathways, e.g., that of carbohydrate metabolism, also contribute to the distinct lipid phenotypes of the NILs. Our comparative transcriptomic analysis of the B. napus NILs to elucidate DEGs that potentially affect seed oil accumulation provides a broader insight into gene networks involved in lipid biosynthesis and metabolism.
Author Contributions
JW and CD designed research; CL, JF, JW, and CD performed experiments; SS, JW, SP and LY analyzed data; SS, LY, SP, and JW wrote the paper.
Funding
This project is supported by Shanxi Provincial Science and Technology Research Grant (20140311010-4) to JW and by the National Science Foundation under Cooperative Agreement No. 1355438, to LY.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
We thank Que Kong, of Michigan State University, and Kathy Shen, of University of Kentucky, for helpful comments and critical review of the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2016.01498
Footnotes
- ^ http://aralip.plantbiology.msu.edu
- ^ https://www.R-project.org
- ^ https://cran.r-project.org/web/packages/gplots/index.html
- ^ http://www.appliedbiosystems.com
References
Allender, C. J., and King, G. J. (2010). Origins of the amphiploid species Brassica napus L. investigated by chloroplast and nuclear molecular markers. BMC Plant Biol. 10, 54–54. doi: 10.1186/1471-2229-10-54
An, D., and Suh, M. C. (2015). Overexpression of Arabidopsis WRI1 enhanced seed mass and storage oil content in Camelina sativa. Plant Biotechnol. Rep. 9, 137–148. doi: 10.1007/s11816-015-0351-x
Bak, S., and Feyereisen, R. (2001). The involvement of two P450 enzymes, CYP83B1 and CYP83A1, in auxin homeostasis and glucosinolate biosynthesis. Plant Physiol. 127, 108–118. doi: 10.1104/pp.127.1.108
Barrett, T., Wilhite, S. E., Ledoux, P., Evangelista, C., Kim, I. F., Tomashevsky, M., et al. (2013). NCBI GEO: archive for functional genomics data sets—update. Nucleic Acids Res. 41, D991–D995. doi: 10.1093/nar/gks1193
Bates, P. D., Stymne, S., and Ohlrogge, J. (2013). Biochemical pathways in seed oil synthesis. Curr. Opin. Plant Biol. 16, 358–364. doi: 10.1016/j.pbi.2013.02.015
Baud, S., Mendoza, M. S., To, A., Harscoet, E., Lepiniec, L., and Dubreucq, B. (2007). WRINKLED1 specifies the regulatory action of LEAFY COTYLEDON2 towards fatty acid metabolism during seed maturation in Arabidopsis. Plant J. 50, 825–838. doi: 10.1111/j.1365-313X.2007.03092.x
Beisson, F., Koo, A. J. K., Ruuska, S., Schwender, J., Pollard, M., Thelen, J. J., et al. (2003). Arabidopsis genes involved in acyl lipid metabolism. A 2003 census of the candidates, a study of the distribution of expressed sequence tags in organs, and a web-based database. Plant Physiol. 132, 681–697. doi: 10.1104/pp.103.022988
Blum, T., Briesemeister, S., and Kohlbacher, O. (2009). MultiLoc2: integrating phylogeny and gene ontology terms improves subcellular protein localization prediction. BMC Bioinformatics 10:274. doi: 10.1186/1471-2105-10-274
Cernac, A., and Benning, C. (2004). WRINKLED1 encodes an AP2/EREB domain protein involved in the control of storage compound biosynthesis in Arabidopsis. Plant J. 40, 575–585. doi: 10.1111/j.1365-313X.2004.02235.x
Chalhoub, B., Denoeud, F., Liu, S., Parkin, I. A. P., Tang, H., Wang, X., et al. (2014). Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 345, 950–953. doi: 10.1126/science.1253435
Chen, M., Du, X. U. E., Zhu, Y., Wang, Z., Hua, S., Li, Z., et al. (2012). Seed fatty acid reducer acts downstream of gibberellin signalling pathway to lower seed fatty acid storage in Arabidopsis. Plant Cell Environ. 35, 2155–2169. doi: 10.1111/j.1365-3040.2012.02546.x
Daviere, J. M., and Achard, P. (2013). Gibberellin signaling in plants. Development 140, 1147–1151. doi: 10.1242/dev.087650
Dong, C.-H., Zolman, B. K., Bartel, B., Lee, B.-H., Stevenson, B., Agarwal, M., et al. (2009). Disruption of Arabidopsis CHY1 Reveals an Important Role of Metabolic Status in Plant Cold Stress Signaling. Mol. Plant 2, 59–72. doi: 10.1093/mp/ssn063
Durrett, T. P., Benning, C., and Ohlrogge, J. (2008). Plant triacylglycerols as feedstocks for the production of biofuels. Plant J. 54, 593–607. doi: 10.1111/j.1365-313X.2008.03442.x
Elahi, N., Duncan, R. W., and Stasolla, C. (2015). Decreased seed oil production in FUSCA3 Brassica napus mutant plants. Plant Physiol, Biochem. 96, 222–230. doi: 10.1016/j.plaphy.2015.08.002
Elahi, N., Duncan, R. W., and Stasolla, C. (2016). Modification of oil and glucosinolate content in canola seeds with altered expression of Brassica napus LEAFY COTYLEDON1. Plant Physiol. Biochem. 100, 52–63. doi: 10.1016/j.plaphy.2015.12.022
Elhiti, M., Yang, C., Chan, A., Durnin, D. C., Belmonte, M. F., Ayele, B. T., et al. (2012). Altered seed oil and glucosinolate levels in transgenic plants overexpressing the Brassica napus SHOOTMERISTEMLESS gene. J. Exp. Bot. 63, 4447–4461. doi: 10.1093/jxb/ers125
Focks, N., and Benning, C. (1998). wrinkled1: a novel, low-seed-oil mutant of Arabidopsis with a deficiency in the seed-specific regulation of carbohydrate metabolism. Plant Physiol. 118, 91–101. doi: 10.1104/pp.118.1.91
Fu, W., Shen, Y., Hao, J., Wu, J., Ke, L., Wu, C., et al. (2015). Acyl-CoA N-acyltransferase influences fertility by regulating lipid metabolism and jasmonic acid biogenesis in cotton. Sci. Rep. 5:11790. doi: 10.1038/srep11790
Furumoto, T., Yamaguchi, T., Ohshima-Ichie, Y., Nakamura, M., Tsuchida-Iwata, Y., Shimamura, M., et al. (2011). A plastidial sodium-dependent pyruvate transporter. Nature 476, 472–475. doi: 10.1038/nature10250
Goffman, F. D., Alonso, A. P., Schwender, J., Shachar-Hill, Y., and Ohlrogge, J. B. (2005). Light enables a very high efficiency of carbon storage in developing embryos of rapeseed. Plant Physiol. 138, 2269–2279. doi: 10.1104/pp.105.063628
Goldberg, R. B., de Paiva, G., and Yadegari, R. (1994). Plant embryogenesis: zygote to seed. Science 266, 605–614. doi: 10.1126/science.266.5185.605
Golldack, D., Li, C., Mohan, H., and Probst, N. (2014). Tolerance to drought and salt stress in plants: unraveling the signaling networks. Front. Plant Sci. 5:151. doi: 10.3389/fpls.2014.00151
Hajduch, M., Casteel, J. E., Hurrelmeyer, K. E., Song, Z., Agrawal, G. K., and Thelen, J. J. (2006). Proteomic analysis of seed filling in Brassica napus. Developmental characterization of metabolic isozymes using high-resolution two-dimensional gel electrophoresis. Plant Physiol. 141, 32–46. doi: 10.1104/pp.105.075390
Hay, J., and Schwender, J. (2011). Computational analysis of storage synthesis in developing Brassica napus L. (oilseed rape) embryos: flux variability analysis in relation to 13C metabolic flux analysis. Plant J. 67, 513–525. doi: 10.1111/j.1365-313X.2011.04611.x
Higashi, Y., Okazaki, Y., Myouga, F., Shinozaki, K., and Saito, K. (2015). Landscape of the lipidome and transcriptome under heat stress in Arabidopsis thaliana. Sci. Rep. 5:10533. doi: 10.1038/srep10533
Hilliker, A., Xiang, D., Datla, R., Li, F., Cutler, A., Malik, M. R., et al. (2008). Development of a Brassica seed cDNA microarray. Genome 51, 236–242. doi: 10.1139/G07-115
Hofvander, P., Ischebeck, T., Turesson, H., Kushwaha, S. K., Feussner, I., Carlsson, A. S., et al. (2016). Potato tuber expression of Arabidopsis WRINKLED1 increase triacylglycerol and membrane lipids while affecting central carbohydrate metabolism. Plant Biotechnol. J. 14, 1883–1898. doi: 10.1111/pbi.12550
Hou, Q., Ufer, G., and Bartels, D. (2016). Lipid signalling in plant responses to abiotic stress. Plant Cell Environ. 39, 1029–1048. doi: 10.1111/pce.12666
Huang, A. H. C. (1992). Oil bodies and oleosins in seeds. Annu. Rev. Plant Physiol. Plant Mol. Biol. 43, 177–200. doi: 10.1146/annurev.pp.43.060192.001141
Huang, D., Koh, C., Feurtado, J. A., Tsang, E. W., and Cutler, A. J. (2013). MicroRNAs and their putative targets in Brassica napus seed maturation. BMC Genomics 14, 1–25. doi: 10.1186/1471-2164-14-140
Jessen, D., Roth, C., Wiermer, M., and Fulda, M. (2015). Two activities of long-chain acyl-coenzyme a synthetase are involved in lipid trafficking between the endoplasmic reticulum and the plastid in Arabidopsis. Plant Physiol. 167, 351–366. doi: 10.1104/pp.114.250365
Jolivet, P., Boulard, C., Bellamy, A., Valot, B., d’Andréa, S., Zivy, M., et al. (2011). Oil body proteins sequentially accumulate throughout seed development in Brassica napus. J. Plant Physiol. 168, 2015–2020. doi: 10.1016/j.jplph.2011.06.007
Kachroo, A., Lapchyk, L., Fukushige, H., Hildebrand, D., Klessig, D., and Kachroo, P. (2003). Plastidial fatty acid signaling modulates salicylic acid– and jasmonic acid–mediated defense pathways in the Arabidopsis ssi2 mutant. Plant Cell 15, 2952–2965. doi: 10.1105/tpc.017301
Kachroo, A., Venugopal, S. C., Lapchyk, L., Falcone, D., Hildebrand, D., and Kachroo, P. (2004). Oleic acid levels regulated by glycerolipid metabolism modulate defense gene expression in Arabidopsis. Proc. Natl. Acad. Sci. U.S.A. 101, 5152–5157. doi: 10.1073/pnas.0401315101
Katavic, V., Shi, L., Yu, Y., Zhao, L., Haughn, G. W., and Kunst, L. (2014). Investigation of the contribution of oil biosynthetic enzymes to seed oil content in Brassica napus and Arabidopsis thaliana. Can. J. Plant Sci. 94, 1109–1112. doi: 10.4141/cjps2013-161
Kim, M. J., Jang, I. C., and Chua, N. H. (2016). MED15 subunit mediates activation of downstream lipid-related genes by Arabidopsis WRINKLED1. Plant Physiol. 171, 1951–1964. doi: 10.1104/pp.16.00664
Kong, Y., Chen, S., Yang, Y., and An, C. (2013). ABA-insensitive (ABI) 4 and ABI5 synergistically regulate DGAT1 expression in Arabidopsis seedlings under stress. FEBS Lett. 587, 3076–3082. doi: 10.1016/j.febslet.2013.07.045
Kumar, M., and Sharma, M. P. (2015). Assessment of potential of oils for biodiesel production. Renew. Sustain. Energy Rev. 44, 814–823. doi: 10.1016/j.rser.2015.01.013
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Lardizabal, K., Effertz, R., Levering, C., Mai, J., Pedroso, M. C., Jury, T., et al. (2008). Expression of Umbelopsis ramanniana DGAT2A in seed increases oil in soybean. Plant Physiol. 148, 89–96. doi: 10.1104/pp.108.123042
Leinonen, R., Sugawara, H., Shumway, M., and on behalf of the International Nucleotide Sequence Database Collaboration. (2011). The sequence read archive. Nucleic Acids Res. 39(Database issue), D19-D21. doi: 10.1093/nar/gkq1019
Li, Q., Shao, J., Tang, S., Shen, Q., Wang, T., Chen, W., et al. (2015). Wrinkled1 accelerates flowering and regulates lipid homeostasis between oil accumulation and membrane lipid anabolism in Brassica napus. Front. Plant Sci. 6:1015. doi: 10.3389/fpls.2015.01015
Li-Beisson, Y., Shorrosh, B., Beisson, F., Andersson, M. X., Arondel, V., Bates, P. D., et al. (2010). Acyl-lipid metabolism. Arabidopsis Book 8:e0133. doi: 10.1199/tab.0133
Liu, S., Fan, C., Li, J., Cai, G., Yang, Q., Wu, J., et al. (2016). A genome-wide association study reveals novel elite allelic variations in seed oil content of Brassica napus. Theor. Appl. Genet. 129, 1203–1215. doi: 10.1007/s00122-016-2697-z
Liu, Z., Xian, S., Wang, Y., Fan, Wang, J., and Du, C. (2016). Construction and identification of near-isogenic lines of high (low) oil content in Brassica napus L. Acta Agric. Boreali Sin. 31.
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15:550. doi: 10.1186/s13059-014-0550-8
Lu, C., Xin, Z., Ren, Z., Miquel, M., and Browse, J. (2009). An enzyme regulating triacylglycerol composition is encoded by the ROD1 gene of Arabidopsis. Proc. Natl. Acad. Sci. U.S.A. 106, 18837–18842. doi: 10.1073/pnas.0908848106
Ma, W., Kong, Q., Arondel, V., Kilaru, A., Bates, P. D., Thrower, N. A., et al. (2013). WRINKLED1, a ubiquitous regulator in oil accumulating tissues from Arabidopsis embryos to oil palm mesocarp. PLoS ONE 8:e68887. doi: 10.1371/journal.pone.0068887
Maeo, K., Tokuda, T., Ayame, A., Mitsui, N., Kawai, T., and Tsukagoshi, H. (2009). An AP2-type transcription factor, WRINKLED1, of Arabidopsis thaliana binds to the AW-box sequence conserved among proximal upstream regions of genes involved in fatty acid synthesis. Plant J. 60, 476–487. doi: 10.1111/j.1365-313X.2009.03967.x
Maere, S., Heymans, K., and Kuiper, M. (2005). BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 21, 3448–3449. doi: 10.1093/bioinformatics/bti551
Mi, H., Poudel, S., Muruganujan, A., Casagrande, J. T., and Thomas, P. D. (2016). PANTHER version 10: expanded protein families and functions, and analysis tools. Nucleic Acids Res. 44, D336–342. doi: 10.1093/nar/gkv1194
Mou, Z., He, Y., Dai, Y., Liu, X., and Li, J. (2000). Deficiency in fatty acid synthase leads to premature cell death and dramatic alterations in plant morphology. Plant Cell 12, 405–417. doi: 10.1105/tpc.12.3.405
Mu, J., Tan, H., Zheng, Q., Fu, F., Liang, Y., Zhang, J., et al. (2008). LEAFY COTYLEDON1 is a key regulator of fatty acid biosynthesis in Arabidopsis. Plant Physiol. 148, 1042–1054. doi: 10.1104/pp.108.126342
Murphy, D. J., Cummins, I., and Kang, A. S. (1989). Synthesis of the major oil-body membrane protein in developing rapeseed (Brassica napus) embryos. Integration with storage-lipid and storage-protein synthesis and implications for the mechanism of oil-body formation. Biochem. J. 258, 285–293. doi: 10.1042/bj2580285
Nandi, A., Krothapalli, K., Buseman, C. M., Li, M., Welti, R., Enyedi, A., et al. (2003). Arabidopsis sfd mutants affect plastidic lipid composition and suppress dwarfing, cell death, and the enhanced disease resistance phenotypes resulting from the deficiency of a fatty acid desaturase. Plant Cell 15, 2383–2398. doi: 10.1105/tpc.015529
Niu, Y., Wu, G.-Z., Ye, R., Lin, W.-H., Shi, Q.-M., Xue, L.-J., et al. (2009). Global analysis of gene expression profiles in Brassica napus developing seeds reveals a conserved lipid metabolism regulation with Arabidopsis thaliana. Mol. Plant 2, 1107–1122. doi: 10.1093/mp/ssp042
Parkin, I. A., Gulden, S. M., Sharpe, A. G., Lukens, L., Trick, M., Osborn, T. C., et al. (2005). Segmental structure of the Brassica napus genome based on comparative analysis with Arabidopsis thaliana. Genetics 171, 765–781. doi: 10.1534/genetics.105.042093
Pattanaik, S., Kong, Q., Zaitlin, D., Werkman, J. R., Xie, C. H., Patra, B., et al. (2010). Isolation and functional characterization of a floral tissue-specific R2R3 MYB regulator from tobacco. Planta 231, 1061–1076. doi: 10.1007/s00425-010-1108-y
Pouvreau, B., Baud, S., Vernoud, V., Morin, V., Py, C., and Gendrot, G. (2011). Duplicate maize Wrinkled1 transcription factors activate target genes involved in seed oil biosynthesis. Plant Physiol. 156, 674–686. doi: 10.1104/pp.111.173641
Rampey, R. A., LeClere, S., Kowalczyk, M., Ljung, K., Sandberg, G., and Bartel, B. (2004). A family of auxin-conjugate hydrolases that contributes to free indole-3-acetic acid levels during Arabidopsis germination. Plant Physiol. 135, 978–988. doi: 10.1104/pp.104.039677
Regnault, T., Davière, J.-M., Heintz, D., Lange, T., and Achard, P. (2014). The gibberellin biosynthetic genes AtKAO1 and AtKAO2 have overlapping roles throughout Arabidopsis development. Plant J. 80, 462–474. doi: 10.1111/tpj.12648
Roscoe, T. T., Guilleminot, J., Bessoule, J. J., Berger, F., and Devic, M. (2015). Complementation of seed maturation phenotypes by ectopic expression of ABSCISIC ACID INSENSITIVE3, FUSCA3 and LEAFY COTYLEDON2 in Arabidopsis. Plant Cell Physiol. 56, 1215–1228. doi: 10.1093/pcp/pcv049
Santos-Mendoza, M., Dubreucq, B., Baud, S., Parcy, F., Caboche, M., and Lepiniec, L. (2008). Deciphering gene regulatory networks that control seed development and maturation in Arabidopsis. Plant J. 54, 608–620. doi: 10.1111/j.1365-313X.2008.03461.x
Schmieder, R., and Edwards, R. (2011). Quality control and preprocessing of metagenomic datasets. Bioinformatics 27, 863–864. doi: 10.1093/bioinformatics/btr026
Shah, J. (2005). Lipids, lipases, and lipid-modifying enzymes in plant disease resistance. Annu. Rev. Phytopathol. 43, 229–260. doi: 10.1146/annurev.phyto.43.040204.135951
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. doi: 10.1101/gr.1239303
Shen, B., Allen, W. B., Zheng, P., Li, C., Glassman, K., Ranch, J., et al. (2010). Expression of ZmLEC1 and ZmWRI1 increases seed oil production in maize. Plant Physiol. 153, 980–987. doi: 10.1104/pp.110.157537
Shen, J.-X., and Fu, T.-D. (2011). Rapeseed production, improvement and edible oil supply in China. J. Agric. Sci. Technol. 1, 1–18.
Shockey, J. M., Fulda, M. S., and Browse, J. A. (2002). Arabidopsis contains nine long-chain acyl-coenzyme a synthetase genes that participate in fatty acid and glycerolipid metabolism. Plant Physiol. 129, 1710–1722. doi: 10.1104/pp.003269
Singh, S. K., Wu, Y., Ghosh, J. S., Pattanaik, S., Fisher, C., Wang, Y., et al. (2015). RNA-sequencing reveals global transcriptomic changes in Nicotiana tabacum responding to topping and treatment of axillary-shoot control chemicals. Sci. Rep. 5:18148. doi: 10.1038/srep18148
Snowdon, R. J., and Friedt, W. (2004). Molecular markers in Brassica oilseed breeding: current status and future possibilities. Plant Breed. 123, 1–8. doi: 10.1111/j.1439-0523.2003.00968.x
Söderman, E. M., Brocard, I. M., Lynch, T. J., and Finkelstein, R. R. (2000). Regulation and function of the Arabidopsis ABA-insensitive4 gene in seed and abscisic acid response signaling networks. Plant Physiol. 124, 1752–1765. doi: 10.1104/pp.124.4.1752
Subramaniam, S., Fahy, E., Gupta, S., Sud, M., Byrnes, R. W., Cotter, D., et al. (2011). Bioinformatics and systems biology of the lipidome. Chem. Rev. 111, 6452–6490. doi: 10.1021/cr200295k
Taipalensuu, J., Eriksson, S., and Rask, L. (1997). The myrosinase-binding protein from Brassica napus seeds possesses lectin activity and has a highly similar vegetatively expressed wound-inducible counterpart. Eur. J. Biochem. 250, 680–688. doi: 10.1111/j.1432-1033.1997.00680.x
Tang, Z. L., Li, J. N., Zhang, X. K., Chen, L., and Wang, R. (1997). Genetic variation of yellow-seeded rapeseed lines (Brassica napus L.) from different genetic sources. Plant Breed. 116, 471–474. doi: 10.1111/j.1439-0523.1997.tb01033.x
Thimm, O., Blasing, O., Gibon, Y., Nagel, A., Meyer, S., Kruger, P., et al. (2004). MAPMAN: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 37, 914–939. doi: 10.1111/j.1365-313X.2004.02016.x
Topa, H., and Honkela, A. (2016). Analysis of differential splicing suggests different modes of short-term splicing regulation. Bioinformatics 32, i147–i155. doi: 10.1093/bioinformatics/btw283
Troncoso-Ponce, M. A., Kilaru, A., Cao, X., Durrett, T. P., Fan, J., Jensen, J. K., et al. (2011). Comparative deep transcriptional profiling of four developing oilseeds. Plant J. 68, 1014–1027. doi: 10.1111/j.1365-313X.2011.04751.x
Varbanova, M., Yamaguchi, S., Yang, Y., McKelvey, K., Hanada, A., Borochov, R., et al. (2007). Methylation of Gibberellins by Arabidopsis GAMT1 and GAMT2. Plant Cell 19, 32–45. doi: 10.1105/tpc.106.044602
Wang, H.-W., Zhang, B., Hao, Y.-J., Huang, J., Tian, A.-G., Liao, Y., et al. (2007). The soybean Dof-type transcription factor genes, GmDof4 and GmDof11, enhance lipid content in the seeds of transgenic Arabidopsis plants. Plant J. 52, 716–729. doi: 10.1111/j.1365-313X.2007.03268.x
Wang, X., Wu, J., Liang, J., Cheng, F., and Wang, X. (2015). Brassica database (BRAD) version 2.0: integrating and mining Brassicaceae species genomic resources. Database 2015: bav093. doi: 10.1093/database/bav093
Wang, Y., Tang, H., Debarry, J. D., Tan, X., Li, J., Wang, X., et al. (2012a). MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49. doi: 10.1093/nar/gkr1293.
Wang, Y., Wang, X., and Paterson, A. H. (2012b). Genome and gene duplications and gene expression divergence: a view from plants. Ann. N. Y. Acad. Sci. 1256, 1–14. doi: 10.1111/j.1749-6632.2011.06384.x
Wu, X.-L., Liu, Z.-H., Hu, Z.-H., and Huang, R.-Z. (2014). BnWRI1 coordinates fatty acid biosynthesis and photosynthesis pathways during oil accumulation in rapeseed. J. Integr. Plant Biol. 56, 582–593. doi: 10.1111/jipb.12158
Yang, Y., Yu, X., Song, L., and An, C. (2011). ABI4 activates DGAT1 expression in Arabidopsis seedlings during nitrogen deficiency. Plant Physiol. 156, 873–883. doi: 10.1104/pp.111.175950
Zhang, D.-S., Liang, W.-Q., Yuan, Z., Li, N., Shi, J., Wang, J., et al. (2008). Tapetum degeneration retardation is critical for aliphatic metabolism and gene regulation during rice pollen development. Mol. Plant 1, 599–610. doi: 10.1093/mp/ssn028
Zheng, H., Rowland, O., and Kunst, L. (2005). Disruptions of the Arabidopsis enoyl-CoA reductase gene reveal an essential role for very-long-chain fatty acid synthesis in cell expansion during plant morphogenesis. Plant Cell 17, 1467–1481. doi: 10.1105/tpc.104.030155
Keywords: Brassica napus, lipid biosynthesis, near isogenic lines, seed oil, RNA-sequencing, gene expression, acyl-lipid metabolism
Citation: Wang J, Singh SK, Du C, Li C, Fan J, Pattanaik S and Yuan L (2016) Comparative Transcriptomic Analysis of Two Brassica napus Near-Isogenic Lines Reveals a Network of Genes That Influences Seed Oil Accumulation. Front. Plant Sci. 7:1498. doi: 10.3389/fpls.2016.01498
Received: 15 July 2016; Accepted: 20 September 2016;
Published: 29 September 2016.
Edited by:
Niranjan Baisakh, Louisiana State University, USAReviewed by:
Gunvant Baliram Patil, University of Missouri, USAManoj Majee, National Institute of Plant Genome Research, India
Copyright © 2016 Wang, Singh, Du, Li, Fan, Pattanaik and Yuan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jingxue Wang, amluZ3h1ZXdAc3h1LmVkdS5jbg== Ling Yuan, bHl1YW4zQHVreS5lZHU=
†These authors have contributed equally to this work.