 Yinping Song
Yinping Song Hao Jia
Hao Jia Qing Ma1
Qing Ma1 Lulu Zhang
Lulu Zhang Youhua Wang
Youhua Wang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol. , 17 October 2024
Sec. Vascular Physiology
Volume 15 - 2024 | https://doi.org/10.3389/fphys.2024.1461519
Pulmonary hypertension is a progressive disease of the pulmonary arteries that begins with increased pulmonary artery pressure, driven by progressive remodeling of the small pulmonary arteries, and ultimately leads to right heart failure and death. Vascular remodeling is the main pathological feature of pulmonary hypertension, but treatments for pulmonary hypertension are lacking. Determining the process of vascular proliferation and dysfunction may be a way to decipher the pathogenesis of pulmonary hypertension. In this review, we summarize the important pathways of pulmonary hypertension pathogenesis. We show how these processes are integrated and emphasize the benign role of aerobic exercise, which, as an adjunctive therapy, may be able to modify vascular remodeling in pulmonary hypertension.
Pulmonary hypertension (PH) is a severe pulmonary vascular disease characterized by excessive vascular cell proliferation, increased extracellular matrix deposition, and accumulation of inflammatory cells within the walls of the pulmonary vasculature, which contribute to increased pulmonary vascular resistance (Pullamsetti et al., 2017). During the pathologic progression of PH, there is a progressive narrowing of the pulmonary lumen and an increase in pulmonary artery pressure, which ultimately leads to right ventricular failure and death (Humbert et al., 2019). The Seventh World Symposium on Pulmonary Hypertension has held in Barcelona. It is endorsed by several patients’ organizations, the European Respiratory Society, the International Society for Heart and Lung Transplantation, the Société de Pneumologie de Langue Française and the European Reference Network for Rare Lung Diseases (ERN-LUNG). In recent years, the European Commission has launched programmes for a higher level of research and care in the field of rare diseases. Symbolically, the 2024 Symposium started for the first time with the patients’ perspective, showing the respect and importance given to patients’ priorities. The translational aspects of modern pulmonary vascular research have been highlighted by the development of novel treatment approaches directly targeting the basic drivers of pulmonary vascular remodeling through activin signaling inhibition (Humbert et al., 2023). Similarly, refined treatment approaches for PH highlighted the complementarity of the different treatment modalities targeting different mechanisms of the disease.
In previous studies, PH is classified according to the level of pulmonary artery pressure; it was considered mild to moderate when the pulmonary artery pressure is between 25 and 45 mmHg, and severe PH when it exceeds 45–50 mmHg. The Sixth World Symposium on PH recommended that the diagnosis be made by setting the minimum mean pulmonary artery pressure from 25 mmHg to 20 mmHg (Simonneau et al., 2019). Growing evidence that even a mean pulmonary artery pressure slightly above this threshold is associated with an increased risk of death (Maron et al., 2016; Assad et al., 2017; Douschan et al., 2018), irrespective of the underlying causative factor. This provides more opportunities and options for treating patients in the early stages of the disease.
The previous of clinical treatments for PH remain vasodilators (Goldenberg et al., 2019). Currently, drug development for pulmonary hypertension is undergoing a dramatic shift, with the advent of novel drugs such as tyrosine kinase inhibitors and activin receptor ligand traps (including sotatercept, which was approved by the FDA in March 2024 for the treatment of PH in adults). Sotatercept is a fusion protein that traps activins and growth differentiation factors involved in pulmonary arterial hypertension. In patients with pulmonary arterial hypertension who were receiving stable background therapy, sotatercept resulted in a greater improvement in exercise capacity as assessed by the 6-minute walk test (Hoeper et al., 2023). But, sotatercept-like drugs might be detrimental effects in the heart.
The 3-year survival rate for patients with PH is 68%–70% (Hurdman et al., 2012; Farber et al., 2015). Current therapies mainly target pulmonary vasoconstriction and do not effectively intervene or reverse pulmonary arterial vascular remodeling. Therefore, there is an urgent need for new explorations to directly address targeting pulmonary artery remodeling in this PH pathology.
In addition to the extensive research on medication, little is known about the impact of lifestyle on PH. Currently, complex exercise rehabilitation interventions are being developed for patients with pulmonary hypertension: the Supervised Pulmonary Hypertension Exercise Rehabilitation (SPHERe) trial is also underway (Ennis et al., 2023). Notably, aerobic exercise is effective against skeletal and cardiac dysfunction in PH (Vieira et al., 2020). Exercise rehabilitation training as an adjunct to disease-specific treatments is often effective and safe for patients with PH (Dong and Li, 2022). It can improve exercise capacity and endurance, skeletal and respiratory muscle performance, cardiorespiratory function and quality of life in patients with PH. Therefore, the aim of this review is to investigate the pathways at the level of pathomechanisms and to illustrate the efficacy and safety of performing aerobic exercise in order to provide a theoretical basis for the prevention and treatment of PH.
Vascular dysfunction and vascular remodeling are thought to be central mechanisms in the development of high pulmonary arterial pressure, which is chronically elevated by dysregulation of functional vascular signaling pathways driven by relevant triggers. This process leads to high afterload and failure of the right ventricle. The process of pulmonary vascular remodeling involves all the layers of the membrane structure of the vascular wall; the intima consists of endothelial cells, the middle layer consists mainly of smooth muscle cells (SMCs), and the tunica consists of fibroblasts. It is complicated by the presence of cellular heterogeneity within the compartments of the pulmonary artery wall.
Most studies have concluded that the pulmonary artery endothelium is the initial site of PH disease (Stenmark et al., 2012; Kuebler et al., 2017). Pulmonary vasoconstriction occurs early in PH pathology, in turn, vasoconstriction is closely related to endothelial disfunction. The extent to which the endothelium is compromised in PH is not fully understood. In one study, based on cross-sectional analysis of the pulmonary arteries, the normal intima accounted for approximately 10% of the total thickness. Idiopathic PH, which is typical of severe PH, shows marked thickening and disorganization of the intima; in severe PH disease, the intima of the pulmonary arteries in PH shows an increase in thickness of about three times compared to normal pulmonary arteries (Stacher et al., 2012). The thickened intima leads to increased pulmonary vascular resistance. Using the volume density parameter of alveolar septa as a reference, PH is associated with a doubling of the intima compared to more normal pulmonary arteries (Stacher et al., 2012). The types of intimal thickening are diverse. They can be briefly categorized according to the predominance of collagen and mucin, fibroblasts or endothelial cells. Endothelial-like cells proliferate abnormally in a disorganized and chaotic manner and are prone to develop lesions. Studies have shown that high expression of angiogenic markers including vascular endothelial growth factor (VEGF), VEGF receptor and hypoxia inducible factor (HIF) are detected in these diseased endothelial tissues (Bryant et al., 2012).
Hypoxia is also a major causative agent of PH, and hypoxia leads to endothelial cell dysfunction through activation of receptor-γ cofactor-1α by endothelial peroxisome proliferators, increased formation of reactive oxygen species (ROS), mitochondrial dysfunction, nuclear factor κB (NF-κB) activation, and subsequent secretion of interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α) (Ye et al., 2016). Endothelial dysfunction leads to decreased production of vasodilators such as nitric oxide (NO) and prostacyclin and increased production of vasoconstrictors such as endothelin-1 (ET-1). ET-1 is a potent vasoconstrictor, and its prolonged overexpression not only affects vascular tone but also induces vascular remodeling. Therefore, it plays an important role in the pathogenesis of PH. In addition, ET-1 is involved in lung microvascular remodeling (Stam et al., 2018). There is substantial evidence that the endothelium may play a key role as an inflammatory cell signaling hub (Pugliese et al., 2015). As a signaling hub, the endothelium maintains feed-forward interactions between resident fibroblasts and macrophages. In addition, prolonged hypoxia disabled the FIS1 deSUMOylation by diminishing the availability of SENP1 in mitochondria via inducing miR (microRNA)-138 and consequently resulted in mitochondrial dysfunction and metabolic reprogramming in pulmonary endothelium (Zhou et al., 2023). The specific mechanism(s) of the initiating stimulus(s) or injury(s) leading to abnormal endothelial cell proliferation are not fully understood, but may include hypoxia, shear stress, inflammation, or response to drugs or toxins in the context of genetic susceptibility.
PH is a progressive disease that is typically characterized by an abnormal proliferation of SMCs in the pulmonary artery mesentery, thus leading to progressive apoptosis of the pulmonary arteries. Numerous studies have shown that hypoxia causes pulmonary vasoconstriction and vascular remodeling (Wilkins et al., 2015; Huang et al., 2022). Prolonged exposure to hypoxia leads to hypoxic PH. Due to the superimposed release of hypoxic and growth factors, the pulmonary arteries undergo an excessive muscularization process, with mesenchymal smooth muscle cell remodeling as the main remodeling. Pulmonary artery smooth muscle cells (PASMCs) are the predominant cell type in the inner layer of the pulmonary arteries and contain contractile proteins that are regulated by calcium and control vascular tone (Lyle et al., 2017). When dysregulated, SMCs contract abnormally, which in turn leads to sustained vasoconstriction, ultimately triggering vascular smooth muscle remodeling, a disease hallmark of PH.
PASMCs are a cell type without terminal differentiation and can maintain significant plasticity (Gomez and Owens, 2012). Under hypoxia, overproliferation and anti-apoptosis of PASMCs in small pulmonary arteries can cause pulmonary artery remodeling to occur. SMCs have been shown to transmit inflammatory signals in lung tissue through their secretion of pro-inflammatory cytokines (Dreymueller et al., 2014). In addition, the proliferative and quasi-synthetic phenotypic switch observed in PASMCs under hypoxia is mediated by hypoxia-inducible factor-1(HIF-1) driven expression of miR-9-1 and miR-9-3 (Shan et al., 2014). HIF-2α is a homologue of HIF-1α, chronic hypoxia enhances HIF-2α stability, which causes increased arginase expression and dysregulates normal vascular NO homeostasis, suggesting that HIF-2α contributes to the development of hypoxic pulmonary vascular remodeling by upregulating these vasoconstrictors through the endothelium. HIF-1-dependent upregulation of miR-210 leads to anti-apoptosis in PASMCs by targeting the transcription factor E2F3 (Gou et al., 2012). Hypoxia-induced muscularization of non-muscularized pulmonary artery vessels involves pre-existing smooth muscle cell progenitors that undergo dedifferentiation, migration to distal vessels, proliferation and redifferentiation (Sheikh et al., 2014). Thus, multiple mechanisms act synergistically and ultimately contribute to the pro-proliferative, pro-growth, and anti-apoptotic phenotypes of PASMCs during the pathologic process of PH.
As hypoxia activates ROS signaling, it stimulates the production of α-smooth muscle actin (α-SMA), a classical marker of activated fibroblasts (Barman et al., 2014). The work of Chai et al. demonstrated that hypoxia induces the proliferation of pulmonary artery adventitia fibroblast (PAAF), migration and vascular remodeling, as observed in the pulmonary artery wall of hypoxic rats in vivo, where hypoxia induced medial and lateral thickening as well as excessive fibrin and collagen deposition (Chai et al., 2018).
Fibroblasts provide mechanical strength to tissues by producing extracellular matrix and providing matrix support. In addition, fibroblasts are able to respond to various stimuli such as vasodilation or hypoxia (Stenmark et al., 2013). In response to these stimuli, pulmonary artery endothelial fibroblasts exhibit a distinct pro-inflammatory phenotype characterized by an increase in chemokines, cytokines and adhesion molecules (Kitamura et al., 2011). In addition, interactions between fibroblasts and leukocytes at sites of chronic inflammation appear to promote the continued survival and retention of leukocytes, which in turn leads to a delay or disappearance of the inflammatory lesion (Buckley, 2011).
Thus, pulmonary artery endothelial cells (PAECs), PASMCs, and pulmonary artery adventitia fibroblasts (PAAFs) are able to interact cooperatively with each other in response to stimuli induced during the pathologic process of PH, such as altered fluid shear stress, stretch, and hypoxia. Pulmonary artery constriction, regulated by contraction and relaxation of PASMCs, can be modulated by paracrine factors released by PAECs. ECs release of the endothelium-derived constricting factors (EDCF), ET-1, from PAEC causes pulmonary vasoconstriction by activating ET receptors (e.g., ETA) and thromboxane/prostaglandin endoperoxide (TP) receptors, respectively, on the membrane of PASMC. Release of the endothelium-derived relaxing factors (EDRF), NO and prostacyclin (PGI2), and the endothelium-derived hyperpolarizing factors (EDHF) from PAEC (Makino et al., 2011). Serotonin synthesized by PAECs is transferred through these gap junctions to PASMCs where it activates TGF-β1 signaling, which in turn induces a more differentiated phenotype. Since TGF-β1 is an important regulator of fibrosis, this is an important way in which PAECs and PASMCs respond to PH (Gairhe et al., 2012).
In the pathological progression of PH, chronic hypoxia, vasoconstriction, endothelial dysfunction, mitochondrial abnormalities, and inflammation are among the many factors that can activate the HIF signaling pathway, which in turn triggers alterations in the intima-media, endothelium, and epima-media of the pulmonary vasculature, inflammatory cells, and cardiomyocytes, leading ultimately to pulmonary vascular remodeling and right ventricular failure.
Hypoxic environments are widely used as a major stimulus for proliferative vasculopathy in PH models prepared in small animals; hypoxic environments also trigger reversible pulmonary vascular remodeling when humans arrive to reach high altitudes (Sydykov et al., 2021). Cellular sensing of oxygen is multilayered and tissue-specific, but the hypoxia-inducible transcription factors HIF-1α and HIF-2α are key regulators of hypoxic adaptation in pulmonary vascular cells (Semenza, 2012).
It has been shown that HIF-1α expression is upregulated in the pulmonary arteries of patients with PH (Labrousse-Arias et al., 2015). The cellular sources of increased HIF-1α expression in lung tissues of patients with PH are pulmonary artery endothelial cells (Bryant et al., 2016) and pulmonary artery SMCs (Kurosawa et al., 2019; Marsboom et al., 2012). The proliferation and quasi-synthetic phenotypic switch observed in PASMCs under hypoxia is mediated by HIF-1 drive (Shan et al., 2014).
Clinical results have shown that the degree of perivascular inflammation in PH patients correlates with pulmonary hemodynamics and vascular remodeling (Stacher et al., 2012). In lung biopsies from PH patients, almost all inflammatory cell lineages were found in the vicinity of remodeled pulmonary vessels, such as macrophages, mast cells, T lymphocytes, B lymphocytes, dendritic cells, and neutrophils (Savai et al., 2012).
Inflammatory mechanisms play an important role in PH pathology. Autoantibodies (e.g., antinuclear antibodies), and elevated levels of pro-inflammatory cytokines Interleukin-1 (IL-1) and IL-6 were detected in some patients with PH. Histologic findings in the lungs have also shown an inflammatory infiltrate (macrophages and lymphocytes) at the lesion in patients with severe PH (Yaku et al., 2022). Interleukin-1 β (IL-1β) is a key inflammatory cytokine released in response to inflammasome activation and is an important mediator of the inflammatory response. Elevated serum levels of IL-1β in patients with PH are associated with worsening PH (Pickrell and Youle, 2015). IL-1β may be derived in part from neutrophils and T cells infiltrated in diseased pulmonary vessels, a key component of chronic hypoxia-induced inflammation in PH mice, i.e., it contains Pyrin domain 3 (NLRP3) of the Nod-like receptor family and contains the positive staining for apoptosis-associated speckled protein (ASC) of the caspase recruitment domain proved this (Cero et al., 2015). Elevations in lung IL-1β, IL-1βR and MyD88 preceded pulmonary hypertension in hypoxic mice. Knockdown of IL-1βR or a molecular adaptor of mouse myeloid differentiation primary response protein 88 prevented hypoxia-induced PH (Parpaleix et al., 2016). In PASMCs, prostacyclin modulates vasodilatation and has antiproliferative effects. This vasodilatory effect is mediated through the second messenger cyclic adenosine monophosphate (cAMP). IL-1β attenuates the conversion of ATP to cAMP in PASMCs by down-regulating adenylate cyclase. In addition, IL-1β can regulate PASMC growth through the IL-1R1/MyD88 pathway (Yang et al., 2014).
As can be seen, inflammatory factors are clearly drivers and contributors to the pathologic process of PH. some of these inflammatory factors show a strong correlation with the severity of PH disease (notably interleukins IL-1, IL-6 (Hu et al., 2020). Not only is inflammation part of the pathology of PH, but inflammation may indeed drive several key pulmonary vasculopathic features of the disease.
ROS, including O2- and H2O2, play important roles as mediators and signaling molecules capable of activating multiple pathways involved in the control of pulmonary vascular tone, cell proliferation and apoptosis, inflammation, and fibrosis. Supraphysiologic doses of H2O2, in turn, are highly correlated with pathophysiologic responses leading to vasoconstriction and PH.
In the pulmonary vasculature, Nox isozymes can be activated by a number of stimuli (e.g., G-protein-coupled receptor agonists, angiotensin II, thrombin, endothelin, 5-hydroxytryptophan, thromboxane A2 (Karpińska et al., 2017). Nox-derived ROS are able to activate NF-κβ, activate MAPK, and cause aberrant cell proliferation, as well as potassium channel regulation in response to changes in oxygen concentration (Dias-Freitas et al., 2016; Brendel et al., 2020). Both Nox1 and Nox4 induce proliferation in PASMCs (Mittal et al., 2007). Nox4 expression in PASMCs can be induced by several stimuli, including hypoxia, shear stress, and endoplasmic reticulum stress (Bedard and Krause, 2007; Lambeth et al., 2007). The effect of Nox4 on the expression of smooth muscle differentiation markers is required, and there is a correlation between the reduction of Nox4 expression and the loss of smooth muscle differentiation markers, such as smooth α-actin and myosin heavy chain, over multiple passages in PASMCs (Clempus et al., 2007; Sturrock et al., 2006), a process in which Nox4 is involved through the induction of TGF-β1 (Sturrock et al., 2006). In PASMCs TGF-β activation of Nox4 leads to intracellular ROS production (Waghray et al., 2005). Under hypoxic conditions Nox4 increases ROS production, stimulates proliferation of PASMCs and inhibits apoptosis in lung fibroblasts (Li et al., 2008).
Endoplasmic reticulum (ER) stress has been a hot topic in the study of disease mechanisms. Research surrounding ER stress and PH has consequently been initiated, with numerous findings suggesting that the unfolded protein response (UPR) plays an important role in the onset and progression of PH pathology. Nowadays, inhibition of ER stress is considered as a new potential intervention in the clinical treatment of PH. Several studies have demonstrated that treatments that reduce ER stress through the use of chemical chaperones can reverse or treat PH in animal models (Wu et al., 2016; Dromparis et al., 2013; Martin and Pabelick, 2014).
It has been shown that vascular remodeling is closely related to ER stress, especially to the abnormal proliferation of PASMCs (Koyama et al., 2014). It has been suggested that proliferation of PASMCs and resistance to apoptosis are critical for vascular remodeling in PH (Masson et al., 2022; Perros et al., 2008; Wang et al., 2014). ER stress is a fundamental cellular response that promotes the proliferation of PASMCs and enhances inflammatory responses (Chen et al., 2018). Therefore, it has been suggested that abnormal proliferation of PASMCs is the most important cause of pulmonary vascular remodeling in PH (Santos-Ribeiro et al., 2016; Yu et al., 2017). Studies have shown that autophagy is involved in the pathogenesis and development of MCT-induced PH (Long et al., 2013; Feng et al., 2021). And eIF2α can activate ER autophagy after ER stress. Meanwhile, eIF2α plays a key role in regulating cell proliferation and hypertrophy, and is involved in regulating the proliferation and migration of vascular smooth muscle (Jiang et al., 2016; Liu et al., 2013). All of the above suggest that ER stress is involved in the abnormal proliferation of PASMCs in PH. Inflammatory cell recruitment and persistence of inflammation in PH are two key components of pathological vascular remodeling (Hassoun et al., 2009). Activation of UPR and production of pro-inflammatory biomolecules were due to ET-1 signaling in rat PASMCs (Yeager et al., 2012). The occurrence of ER stress promotes the proliferation and inflammatory state of PASMCs, which contributes to PH pathology. The persistent inflammation produced by PASMCs is associated with vascular remodeling, and inhibiting this process may be a potential approach for treating PH. Although the function of ER stress in PASMCs has been extensively studied, the mechanism remains incompletely understood. In conclusion, inhibition of the aforementioned proliferation of pulmonary artery smooth muscle cells due to ER stress is also a potential therapeutic option for PH.
Patients with PH may even develop thrombotic arteriopathy, which indicates a prolonged activation of platelets and endothelial cells, with the continuous involvement of coagulation factors. This endothelial activation is associated with endothelial cell proliferation and plexiform lesion formation (Guignabert and Dorfmüller, 2017).
Patients with PH tend to exhibit thrombocytopenia, and those with moderate to severe disease have a poorer prognosis, suggesting that platelets appear to play a role in disease progression. Impaired platelet function in patients with PH leading to their chronic activation and degranulation, increased expression of thrombopoietin in the pulmonary arteries, and an increase in the mean platelet volume suggest that platelet activation, aggregation, and depletion may be increased in the pulmonary circulation (Vrigkou et al., 2019; Zheng et al., 2015). Platelet depletion also stimulates platelet production in the bone marrow, resulting in more platelets and potentially stronger thrombogenic effects. Activated endothelial cells may promote thrombosis through von Willebrand factor (vWF)-mediated platelet activation, factor X activation and tissue factor (TF) production (Nogueira-Ferreira et al., 2014). In rodent models of PH, the activity of this protein is positively correlated with pathological hemodynamic changes and vascular remodeling. The pathological role of TF is not only related to its prothrombotic effects, but also to its direct proliferative and migratory effects. In addition, decreased histone trimethylation and increased histone acetylation of the vWF promoter in the PH endothelium facilitated the binding of NF-κB2 to the vWF promoter and drove the transcription of vWF. epigenetic regulation of the vWF promoter contributes to the creation of a localized environment that is conducive to in situ thrombosis in pulmonary arteries. It reveals a direct link between inflammatory pathways and platelet adhesion in the pulmonary vascular wall, suggesting a possible role of in situ thrombosis in the development or progression of PH (Manz et al., 2022).
Microparticles derived from platelets and the endothelium are an increasingly well-recognized signal in PH (Zheng et al., 2015). The activation of platelets, apart from releasing granules, increases the surface expression of various adhesion molecules and receptor (e.g., selectin P, gp IIIa/IIb) as well as the production of thromboxane A2 (TxA2), which in turn activates other platelets and promotes vasoconstriction and local thrombosis (Kazimierczyk and Kamiński, 2018). Evidence of platelet function abnormalities and dysregulation of the coagulation cascade have been found in PH patients (Bazan and Fares, 2018). Coagulation processes are involved in most of the major pathophysiological pathways of PH, either directly (e.g., thrombus formation and thrombotic arteriopathy) or indirectly (through the production and release of vasoactive substances) (Kazimierczyk and Kamiński, 2018). Therefore, platelets are activated and function abnormally, platelet aggregation is diminished, there is catabolism, there are defects in the initiation of the coagulation process and propagation of clots, and there is diminished thrombin formation in patients with PH. The procoagulant activity is impaired, which may be due to the sustained and prolonged activation of the procoagulant process (Vrigkou et al., 2020). PH patients have higher levels of procoagulant microparticles (MPs) in the pulmonary vasculature, which are fragments of circulating cell membranes released by activated and/or apoptotic cells that stimulate thrombosis by providing coagulation factor activation (Nadaud et al., 2013). As a result, patients with PH have reduced levels of PGI2 and increased levels of TxA2. Upon activation, platelets release TxA2 and platelet-derived growth factor (PDGF), among others, which induce activation of nearby platelets and contribute to platelet amplification and aggregation. Meanwhile, PGI2, which is mainly produced by inactivated endothelium, has vasodilatory and antiplatelet aggregation effects. Thus, this imbalance appears to be the result of impaired endothelial function and/or enhanced platelet activation in the pulmonary vasculature, with important implications for disease progression.
Considerable progress has been made in the genomics of pulmonary arterial hypertension since the 6th World Symposium on Pulmonary Hypertension, with the identification of 17 genes, as well as common variants that confer a modest increase in PH risk. Gene and variant curation by an expert panel now provides a robust framework for knowing which genes to test and how to interpret variants in clinical practice. Researchers recommend that genetic testing be offered to specific subgroups of symptomatic patients with PH, and to children with certain types of group 3 PH. Testing of asymptomatic family members and the use of genetics in reproductive decision-making require the involvement of genetics experts (Austin et al., 2024). In 2020, investigators for the United States Pulmonary Hypertension Scientific Registry (USPHSR) provided data from the first US PH patient registry to include genetic information. Genetic testing identified pathogenic or suspected pathogenic variants in 67 of 499 (13%) USPHSR participants (Badlam et al., 2021).
Pathogenic mutations in BMPR2 are the most common cause of heritable pulmonary arterial hypertension (HPAH) in both adults and children (Welch and Chung, 2020). The loss of BMPR2 promotes endothelial dysfunction, endothelial to mesenchymal transition and pulmonary arterial smooth muscle cell hyperproliferation (Theilmann et al., 2020; Hiepen et al., 2019; Hopper et al., 2016). Genes with causal mutations identified in children with PH differs from the prevalence of causal gene mutations identified in adults with PH. For example, mutations in TBX4, a transcription factor in the T-box gene family that modulates lung development, are found more commonly in children with PH (Zhu et al., 2018).
Clinical observations, molecular discoveries, and laboratory technology have made genetic counseling and testing possible for patients diagnosed with PH, especially group 1 PAH. Recent studies in scRNA-seq have significantly deepened our understanding of PH by revealing cellular heterogeneity and uncovering key molecular pathways involved in disease progression. Dysregulated endothelial cell subpopulations with proliferative and angi ogenic phenotypes contribute to aberrant vascular remodeling, as exemplified by the role of CD74 in endothelial dysfunction—its knockdown regulates endothelial cell proliferation and barrier integrity, suggesting a potential therapeutic target (Rodor et al., 2022). Additionally, scRNA-seq analyses have identified significant changes in immune cells: macrophages interact with other disease-related cells via the PI3K/Akt pathway (Miao et al., 2022); T cells and natural killer cells drive heightened inflammation through upregulation of CCL5 (Kumar et al., 2024); and neutrophil subsets expressing high levels of MMP9 correlate with increased mortality in idiopathic pulmonary arterial hypertension patients (Zhang et al., 2023). These insights highlight the complex interplay between endothelial dysfunction and immune-mediated inflammation in PH, offering new potential therapeutic targets, particularly in immune cell regulation and endothelial function. Furthermore, to advance gene- and pathway-specific care and targeted therapies, gene-specific registries will be essential to support patients and their families and to lay the foundation for genetically informed clinical trials.
In conclusion, the causes of pulmonary arterial hypertension include increased vascular resistance, etc., of which hypoxia is the most important factor in the formation of pulmonary arterial hypertension, under the effect of hypoxia, inflammation, ROS, ER stress, platelet activation, thrombosis and genetics together lead to pathological changes in pulmonary arterial vascular configuration (Figure 1).
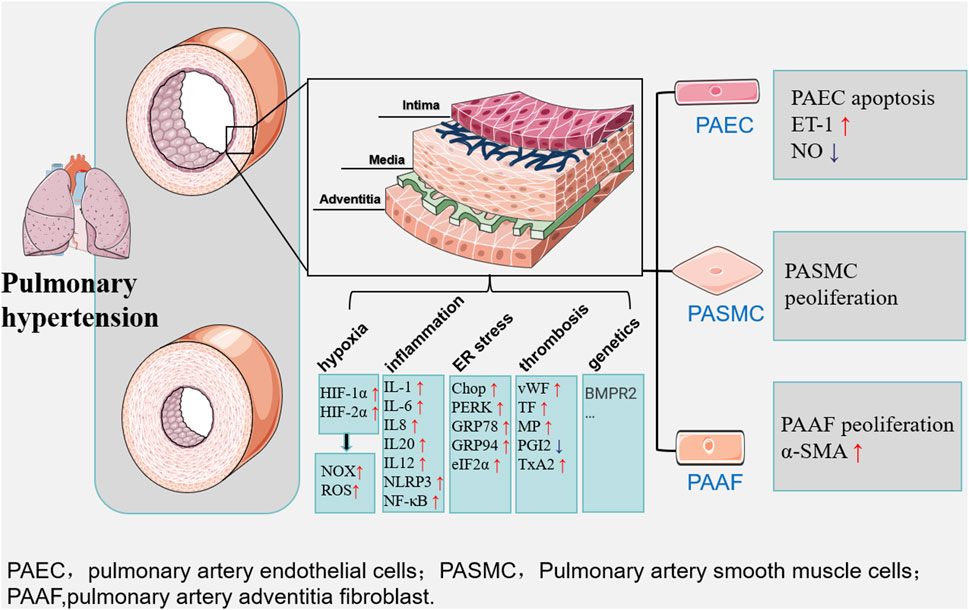
Figure 1. Multiple factors such as hypoxia, inflammation and ER stress lead to long-term maladaptation and dysfunction of the pulmonary artery vasculature, ultimately leading to the development of pulmonary hypertension. The main manifestations are changes in the structure and function of vascular endothelial cells, smooth muscle cells, and extracellular matrix.
Aerobic exercise has emerged as adjuvant therapy for many diseases, and the beneficial relationship between aerobic exercise and lung disease has gradually become closer.
Based on the existing literature, it seems that researchers prefer to discuss the benefits of aerobic exercise on each layer of the vessel separately rather than as a whole, and these beneficial effects are mainly concentrated in the pulmonary artery intima and pulmonary artery media.
Aerobic exercise training opposes endothelial dysfunction via enhanced endothelial NO synthase and increased NO production and bioactivity, which improves NO-dependent vasodilation of large conduit vessels. As such, exercise training improves vascular endothelial function (Fiuza-Luces et al., 2018). Exercise training studies also demonstrate that exercise training is associated with an increase in endothelial function (Ramos et al., 2015). Long-term aerobic exercise training also induces shear stress-mediated arterial remodeling that results in larger conduit and peripheral artery sizes. Aerobic exercise also reduces the wall thickness of conduit arteries (Hellsten and Nyberg, 2015). These vascular structural adaptations markedly increase the luminal reserve of the vessel and reduce the probability of a flow-limiting stenosis (Fiuza-Luces et al., 2018).
Aerobic exercise and inflammation: Aerobic exercise can facilitate lung regeneration with mild inflammatory effect, and proper aerobic exercise is beneficial for lung damage repair and regeneration, which should be considered as an ideal supportive therapy for patients with different respiratory diseases (Wu et al., 2022).
Aerobic exercise and ROS: Exercise improves endothelial function and arterial stiffness by reducing inflammatory and oxidative damage signaling in vascular tissue together with an increase in antioxidant enzymes and nitric oxide availability, globally promoting functional performance (El et al., 2022). In the case of drug resistance, effective aerobic exercise could reduce ROS, activate SOD, inhibit HIF-1 and acetaldehyde dehydrogenase 1 (ALDH1), and cause a reduction in cancer stem cells to sensitise cells to drug again and ultimately inhibit the malignant proliferation of tumours. Therefore, in the treatment of lung adenocarcinoma, the inhibitory effect of aerobic exercise on oxidative stress can be used as an effective adjunct measure in the treatment of lung adenocarcinoma (Yang et al., 2022).
Aerobic exercise and ER stress: Study showed 8-week exercise and choline intervention also inhibited the protein expression of myocardial MFN2, PERK/eIF2α/ATF4, and NLRP3/caspase-1/IL-1β signaling pathways, thereby effectively reducing mitochondrial fusion, endoplasmic reticulum stress, and inflammation. Aerobic exercise increases can improve cardiac function in cardiovascular disease (CVD) rats (Ma et al., 2021).
Aerobic exercise and thrombosis: Exercise is considered a double-edged sword since, on one hand, acute exercise can be a direct cause of a thrombotic event, and on the other hand, exercise training is a potent intervention for lowering the risk of cardiovascular events. Thus, for patients at risk, the safer recommendation would accordingly be to initiate exercise programs at low to moderate intensities. Studies combining platelet reactivity assay, clinical measures of hemostatic markers, and the novel functional measure of clot microstructure will provide a new level of detailed prediction of the susceptibility to harmful arterial blood clots (Olsen et al., 2021).
Therefore, although many studies have examined the effects of regular physical exercise on the cardiovascular and respiratory systems, the specific effects of physical exercise on the pathological mechanisms of pulmonary hypertension have not been fully studied, especially the lack of exercise and the mechanism of pulmonary artery vascular hyperplasia. Therefore, future research on aerobic exercise and vascular hyperplasia should be increased.
Patients with PH have reduced exercise capacity and quality of life. Despite initial concerns that exercise training might exacerbate symptoms in such patients, several studies have reported that exercise rehabilitation training improves function and quality of life (Figure 2).
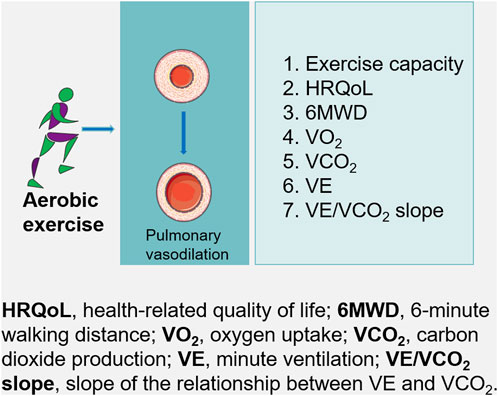
Figure 2. For patients with pulmonary hypertension, aerobic exercise may lead to substantial improvements in exercise capacity, which may improves pulmonary hypertension.
European Respiratory Society published a statement on exercise training in PH reporting improvements in exercise capacity, muscular function, quality of life and potentially right ventricular function (Grünig et al., 2019). However, it was noted that further studies were needed to consolidate these findings and the impact of exercise training on disease risk profiles and to establish optimal training methodology. European Cardiac and Respiratory Societies Guidelines for the diagnosis and treatment of PH recommend supervised exercise training for people with PAH under medical therapy (Class of recommendation: 1; level of evidence A) (Humbert et al., 2022). With the broadening of international acceptance of exercise training in PH, albeit based on a limited body of evidence. In patients with PH, supervised exercise-based rehabilitation may result in a large increase in exercise capacity. Changes in exercise capacity remain heterogeneous and cannot be explained by subgroup analysis. It is likely that exercise-based rehabilitation increases health-related quality of life (HRQoL) and is probably not associated with an increased risk of a serious adverse events. Exercise training may result in a large reduction in mean pulmonary arterial pressure (Morris et al., 2023).
The beneficial effects of exercise rehabilitation training were mainly demonstrated by improvements in peak oxygen uptake (pVO2), 6-minute walk distance (6MWD), hemodynamics, cardiorespiratory fitness, and exercise capacity in patients with different types of PH.
PH patients receiving PH medication and inoperable patients were randomly assigned to training and control groups. The training regimen consisted of low-load intermittent cycle ergometer training, walking, low weight (500–1,000 g) dumbbell training for a single muscle group, and respiratory training for at least 1.5 h per day. Results showed a significant increase in pVO2/kg in the training group after 15 weeks (mean increase of 24.3% relative to baseline). The training group also showed significant improvements in cardiac index, mean pulmonary artery pressure, pulmonary vascular resistance, 6MWD, quality of life scores and exercise tolerance at rest and during exercise (Ehlken et al., 2016). It was also shown that 8 weeks of exercise significantly improved a predictor of death in PH: cardiorespiratory fitness (Santos-Lozano et al., 2019). The efficacy of low-dose exercise and breathing training as an add-on treatment for severe chronic PH. Their effects are a powerful complement to the beneficial results of new medical treatments.
Cardiopulmonary exercise testing (CPET) is a comprehensive methodology well studied in PAH with roles in diagnosis, treatment response, and prognosis. Submaximal and maximal exercise data is a valuable tool in detecting abnormal hemodynamics associated with exercise-induced and resting pulmonary hypertension as well as right ventricular dysfunction. Pulsatile pulmonary vascular pressure-flow relationships in PH allow for the assessment of RV hydraulic load. 12 weeks of aerobic training can significantly improve right ventricular systolic blood pressure in patients with PH (Atef and Abdeen, 2021). The increased granularity of CPET may help further risk stratify patients to inform prognosis and better individualize treatment decisions (Sherman and Saggar, 2023). Chronic obstructive pulmonary disease or diffuse interstitial lung disease, CPET helps to understand the nature of exertional limitation. Different studies have shown that the presence of PH significantly reduces peak oxygen consumption and oxygen pulse volume in these chronic lung diseases, confirming that the typical ventilatory limitation of the underlying respiratory disease is associated with cardiovascular restriction (Blanco et al., 2020).
Decreased exercise capacity is a common feature of PH. The 6-min walk test (6MWT) is often the most common method incorporated into the clinical assessment of the disease (Babu et al., 2017). The test can be used to assess the efficacy of interventions and to provide prognostic information (Holland et al., 2014). In the past, the PM6M was used to assess the efficacy of treatment for PH, and it is now used as one of the basic elements in the multiparametric assessment of risk of death (Boucly et al., 2014). For example, in PH due to chronic thromboembolic disease, which can be resolved surgically, PM6M is routinely performed before and after pulmonary endarterectomy as a tool to assess disease severity, functional capacity and prognosis (Richter et al., 2014).
In conclusion, for patients with PH, supervised exercise rehabilitation may lead to substantial improvements in exercise capacity, which in turn improves PH.
YS: Funding acquisition, Writing–original draft, Writing–review and editing. HJ: Formal Analysis, Funding acquisition, Writing–review and editing. QM: Funding acquisition, Writing–original draft. LZ: Writing–review and editing, Investigation. XL: Writing–review and editing, Investigation. YW: Supervision, Writing–review and editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This publication was supported by grants from Natural Science Basic Research Program of Shaanxi Province (Grant nos. 2023-JC-QN-0925), Philosophy and Social Science Research Specialization of Shaanxi Province (Grant nos. 2024QN309), Xi’an Fanyi University Regular Programs of Research (Grant nos. 22B17), Philosophy and Social Science Research Specialization of Shaanxi Province (Grant nos. 2023ZD0645), Xi’an Fanyi University's Key Projects on Teaching Reform (Grant nos. J23A01).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Assad T. R., Maron B. A., Robbins I. M., Xu M., Huang S., Harrell F. E., et al. (2017). Prognostic effect and longitudinal hemodynamic assessment of borderline pulmonary hypertension. JAMA Cardiol. 2 (12), 1361–1368. doi:10.1001/jamacardio.2017.3882
Atef H., Abdeen H. (2021). Effect of exercise on sleep and cardiopulmonary parameters in patients with pulmonary artery hypertension. Sleep. Breath. 25 (4), 1953–1960. doi:10.1007/s11325-020-02286-9
Austin E. D., Aldred M. A., Alotaibi M., Gräf S., Nichols W. C., Trembath R. C., et al. (2024). Genetics and precision genomics approaches to pulmonary hypertension. Eur. Respir. J. 29, 2401370. doi:10.1183/13993003.01370-2024
Babu A. S., Arena R., Morris N. R. (2017). Evidence on exercise training in pulmonary hypertension. Adv. Exp. Med. Biol. 1000, 153–172. doi:10.1007/978-981-10-4304-8_10
Badlam J. B., Badesch D. B., Austin E. D., Benza R. L., Chung W. K., Farber H. W., et al. (2021). United States pulmonary hypertension scientific registry: baseline characteristics. Chest 159 (1), 311–327. doi:10.1016/j.chest.2020.07.088
Barman S. A., Chen F., Su Y., Dimitropoulou C., Wang Y., Catravas J. D., et al. (2014). NADPH oxidase 4 is expressed in pulmonary artery adventitia and contributes to hypertensive vascular remodeling. Arterioscler. Thromb. Vasc. Biol. 34 (8), 1704–1715. doi:10.1161/ATVBAHA.114.303848
Bazan I. S., Fares W. H. (2018). Hypercoagulability in pulmonary hypertension. Clin. Chest Med. 39, 595–603. doi:10.1016/j.ccm.2018.04.005
Bedard K., Krause K. H. (2007). The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol. Rev. 87 (1), 245–313. doi:10.1152/physrev.00044.2005
Blanco I., Valeiro B., Torres-Castro R., Barberán-García A., Torralba Y., Moisés J., et al. (2020). Effects of pulmonary hypertension on exercise capacity in patients with chronic obstructive pulmonary disease. Arch. Bronconeumol (Engl Ed) 56 (8), 499–505. doi:10.1016/j.arbres.2019.10.015
Boucly A., Weatherald J., Savale L., Jaïs X., Cottin V., Prevot G., et al. (2014). Risk assessment, prognosis and guideline implementation in pulmonary arterial hypertension. Eur. Respir. J. 50 (2), 1700889. doi:10.1183/13993003.00889-2017
Brendel H., Shahid A., Hofmann A., Mittag J., Bornstein S. R., Morawietz H., et al. (2020). NADPH oxidase 4 mediates the protective effects of physical activity against obesity-induced vascular dysfunction. Cardiovasc Res. 116 (10), 1767–1778. doi:10.1093/cvr/cvz322
Bryant A. J., Carrick R. P., McConaha M. E., Jones B. R., Shay S. D., Moore C. S., et al. (2016). Endothelial HIF signaling regulates pulmonary fibrosis-associated pulmonary hypertension. Am. J. Physiol. Lung Cell Mol. Physiol. 310 (3), L249–L262. doi:10.1152/ajplung.00258.2015
Buckley C. D. (2011). Why does chronic inflammation persist: an unexpected role for fibroblasts. Immunol. Lett. 138 (1), 12–14. doi:10.1016/j.imlet.2011.02.010
Cero F. T., Hillestad V., Sjaastad I., Yndestad A., Aukrust P., Ranheim T., et al. (2015). Absence of the inflammasome adaptor ASC reduces hypoxia-induced pulmonary hypertension in mice. Am. J. Physiol. Lung Cell Mol. Physiol. 309 (4), L378–L387. doi:10.1152/ajplung.00342.2014
Chai X., Sun D., Han Q., Yi L., Wu Y., Liu X. (2018). Hypoxia induces pulmonary arterial fibroblast proliferation., migration., differentiation and vascular remodeling via the PI3K/Akt/p70S6K signaling pathway. Int. J. Mol. Med. 41 (5), 2461–2472. doi:10.3892/ijmm.2018.3462
Chen R., Zhong W., Shao C., Liu P., Wang C., Wang Z., et al. (2018). Docosahexaenoic acid inhibits monocrotaline-induced pulmonary hypertension via attenuating endoplasmic reticulum stress and inflammation. Am. J. Physiol. Lung Cell Mol. Physiol. 314 (2), L243-L255–L255. doi:10.1152/ajplung.00046.2017
Clempus R. E., Sorescu D., Dikalova A. E., Pounkova L., Jo P., Sorescu G. P., et al. (2007). Nox4 is required for maintenance of the differentiated vascular smooth muscle cell phenotype. Arterioscler. Thromb. Vasc. Biol. 27 (1), 42–48. doi:10.1161/01.ATV.0000251500.94478.18
Dias-Freitas F., Metelo-Coimbra C., Roncon-Albuquerque R. Jr. (2016). Molecular mechanisms underlying hyperoxia acute lung injury. Respir. Med. 119, 23–28. doi:10.1016/j.rmed.2016.08.010
Dong C., Li Y. (2022). Exercise rehabilitation training in patients with pulmonary hypertension: a review. Heart Lung Circ. 31 (10), 1341–1348. doi:10.1016/j.hlc.2022.06.660
Douschan P., Kovacs G., Avian A., Foris V., Gruber F., Olschewski A., et al. (2018). Mild elevation of pulmonary arterial pressure as a predictor of mortality. Am. J. Respir. Crit. Care Med. 197 (4), 509–516. doi:10.1164/rccm.201706-1215OC
Dreymueller D., Martin C., Schumacher J., Groth E., Boehm J. K., Reiss L. K., et al. (2014). Smooth muscle cells relay acute pulmonary inflammation via distinct ADAM17/ErbB axes. J. Immunol. 192 (2), 722–731. doi:10.4049/jimmunol.1302496
Dromparis P., Paulin R., Stenson T. H., Haromy A., Sutendra G., Michelakis E. D. (2013). Attenuating endoplasmic reticulum stress as a novel therapeutic strategy in pulmonary hypertension. Circulation 127 (1), 115–125. doi:10.1161/CIRCULATIONAHA.112.133413
Ehlken N., Lichtblau M., Klose H., Weidenhammer J., Fischer C., Nechwatal R., et al. (2016). Exercise training improves peak oxygen consumption and haemodynamics in patients with severe pulmonary arterial hypertension and inoperable chronic thrombo-embolic pulmonary hypertension: a prospective., randomized., controlled trial. Eur. Heart J. 37 (1), 35–44. doi:10.1093/eurheartj/ehv337
El A. M., Álvarez-Bustos A., Sosa P., Angulo J., Rodríguez-Mañas L. (2022). Effect of physical activity/exercise on oxidative stress and inflammation in muscle and vascular aging. Int. J. Mol. Sci. 23 (15), 8713. doi:10.3390/ijms23158713
Ennis S., Sandhu H. K., Bruce J., Seers K., Pincus T., Underwood M., et al. (2023). Development of a complex exercise rehabilitation intervention for people with pulmonary hypertension: the supervised pulmonary hypertension exercise rehabilitation (SPHERe) trial. BMJ Open 13 (8), e066053. doi:10.1136/bmjopen-2022-066053
Farber H. W., Miller D. P., Poms A. D., Badesch D. B., Frost A. E., Muros-Le Rouzic E., et al. (2015). Five-Year outcomes of patients enrolled in the REVEAL Registry. Chest 148 (4), 1043–1054. doi:10.1378/chest.15-0300
Feng W., Wang J., Yan X., Zhang Q., Chai L., Wang Q., et al. (2021). ERK/Drp1-dependent mitochondrial fission contributes to HMGB1-induced autophagy in pulmonary arterial hypertension. Cell Prolif. 54 (6), e13048. doi:10.1111/cpr.13048
Fiuza-Luces C., Santos-Lozano A., Joyner M., Carrera-Bastos P., Picazo O., Zugaza J.L., et al. (2018). Exercise benefits in cardiovascular disease: beyond attenuation of traditional risk factors. Nat. Rev. Cardiol. 15 (2), 731–743. doi:10.1038/s41569-0180065-1
Gairhe S., Bauer N. N., Gebb S. A., McMurtry I. F. (2012). Serotonin passes through myoendothelial gap junctions to promote pulmonary arterial smooth muscle cell differentiation. Am. J. Physiol. Lung Cell Mol. Physiol. 303 (9), L767–L777. doi:10.1152/ajplung.00183.2012
Goldenberg N. M., Rabinovitch M., Steinberg B. E. (2019). Inflammatory basis of pulmonary arterial hypertension: implications for perioperative and critical care medicine. Anesthesiology 131 (4), 898–907. doi:10.1097/ALN.0000000000002740
Gomez D., Owens G. K. (2012). Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovasc Res. 95 (2), 156–164. doi:10.1093/cvr/cvs115
Gou D., Ramchandran R., Peng X., Yao L., Kang K., Sarkar J., et al. (2012). miR-210 has an antiapoptotic effect in pulmonary artery smooth muscle cells during hypoxia. Am. J. Physiol. Lung Cell Mol. Physiol. 303 (8), L682–L691. doi:10.1152/ajplung.00344.2011
Grünig E., Eichstaed C., Barberà J. A., Benjamin N., Blanco I., Bossone E., et al. (2019). ERS statement on exercise training and rehabilitation in patients with severe chronic pulmonary hypertension. Eur. Respir. J. 53 (2), 1800332. doi:10.1183/13993003.00332-2018
Guignabert C., Dorfmüller P. (2017). Pathology and pathobiology of pulmonary hypertension. Semin. Respir. Crit. Care Med. 38 (5), 571–584. doi:10.1055/s-0037-1606214
Hassoun P. M., Mouthon L., Barberà J. A., Eddahibi S., Flores S. C., Grimminger F., et al. (2009). Inflammation., growth factors., and pulmonary vascular remodeling. J. Am. Coll. Cardiol. 54 (1 Suppl. l), S10–S19. doi:10.1016/j.jacc.2009.04.006
Hellsten Y., Nyberg M. (2015). Cardiovascular adaptations to exercise training. Compr. Physiol. 6 (1), 1–32. doi:10.1002/cphy.c140080
Hiepen C., Jatzlau J., Hildebrandt S., Kampfrath B., Goktas M., Murgai A., et al. (2019). BMPR2 acts as a gatekeeper to protect endothelial cells from increased TGFβ responses and altered cell mechanics. PLoS Biol. 17 (12), e3000557. doi:10.1371/journal.pbio.3000557
Hoeper M. M., Badesch D. B., Ghofrani H. A., Gibbs J. S. R., Gomberg-Maitland M., McLaughlin V. V., et al. (2023). Phase 3 trial of sotatercept for treatment of pulmonary arterial hypertension. N. Engl. J. Med. 388 (16), 1478–1490. doi:10.1056/NEJMoa2213558
Holland A. E., Spruit M. A., Troosters T., Puhan M. A., Pepin V., Saey D., et al. (2014). An official European Respiratory Society/American Thoracic Society technical standard: field walking tests in chronic respiratory disease. Eur. Respir. J. 44 (6), 1428–1446. doi:10.1183/09031936.00150314
Hopper R. K., Moonen J. R., Diebold I., Cao A., Rhodes C. J., Tojais N. F., et al. (2016). In pulmonary arterial hypertension, reduced BMPR2 promotes endothelial-to-mesenchymal transition via HMGA1 and its target slug. Circulation 133 (18), 1783–1794. doi:10.1161/CIRCULATIONAHA.115.020617
Hu Y., Chi L., Kuebler W. M., Goldenberg N. M. (2020). Perivascular inflammation in pulmonary arterial hypertension. Cells 9 (11), 2338. doi:10.3390/cells9112338
Huang X., Akgün E. E., Mehmood K., Zhang H., Tang Z., Li Y. (2022). Mechanism of hypoxia-mediated smooth muscle cell proliferation leading to vascular remodeling. Biomed. Res. Int. 24, 3959845. doi:10.1155/2022/3959845
Humbert M., Guignabert C., Bonnet S., Dorfmüller P., Klinger J. R., Nicolls M. R., et al. (2019). Pathology and pathobiology of pulmonary hypertension: state of the art and research perspectives. Eur. Respir. J. 53 (1), 1801887. doi:10.1183/13993003.01887-2018
Humbert M., Kovacs G., Hoeper M. M., Badagliacca R., Berger R. M., Brida M., et al. (2022). 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur. Respir. J. 60 (6), 2200879. doi:10.1183/13993003.00879-2022
Hurdman J., Condliffe R., Elliot C. A., Davies C., Hill C., Wild J. M., et al. (2012). ASPIRE registry: assessing the Spectrum of Pulmonary hypertension Identified at a REferral centre. Eur. Respir. J. 39 (4), 945–955. doi:10.1183/09031936.00078411
Jiang L., Zang D., Yi S., Li X., Yang C., Dong X., et al. (2016). A microRNA-mediated decrease in eukaryotic initiation factor 2α promotes cell survival during PS-341 treatment. Sci. Rep. 6, 21565. doi:10.1038/srep21565
Karpińska O., Baranowska-Kuczko M., Kloza M., Ambroz Ewicz E., Kozłowski T., Kasacka I., et al. (2017). Activation of CB1 receptors by 2-arachidonoylglycerol attenuates vasoconstriction induced by U46619 and angiotensin II in human and rat pulmonary arteries. Am. J. Physiol. Regul. Integr. Comp. Physiol. 312 (6), R883-R893–R893. doi:10.1152/ajpregu.00324.2016
Kazimierczyk R., Kamiński K. (2018). The role of platelets in the development and progression of pulmonary arterial hypertension. Adv. Med. Sci. 63 (2), 312–316. doi:10.1016/j.advms.2018.04.013
Kitamura H., Cambier S., Somanath S., Barker T., Minagawa S., Markovics J., et al. (2011). Mouse and human lung fibroblasts regulate dendritic cell trafficking, airway inflammation, and fibrosis through integrin αvβ8-mediated activation of TGF-β. J. Clin. Invest 121 (7), 2863–2875. doi:10.1172/JCI45589
Koyama M., Furuhashi M., Ishimura S., Mita T., Fuseya T., Okazaki Y., et al. (2014). Reduction of endoplasmic reticulum stress by 4-phenylbutyric acid prevents the development of hypoxia-induced pulmonary arterial hypertension. Am. J. Physiol. Heart Circ. Physiol. 306 (9), H1314–H1323. doi:10.1152/ajpheart.00869.2013
Kuebler W. M., Nicolls M. R., Olschewski A., Abe K., Rabinovitch M., Stewart D., et al. (2017). A pro-con debate: current controversies in PAH pathogenesis at the American Thoracic Society International Conference in 2017. Am. J. Physiol. Lung Cell Mol. Physiol. 315 (4), L502-L516–L516. doi:10.1152/ajplung.00150.2018
Kumar S., Mickael C., Kumar R., Prasad R. R., Campbell N. V., Zhang H., et al. (2024). Single cell transcriptomic analyses reveal diverse and dynamic changes of distinct populations of lung interstitial macrophages in hypoxia-induced pulmonary hypertension. Front. Immunol. 15, 1372959. doi:10.3389/fimmu.2024.1372959
Kurosawa R., Satoh K., Kikuchi N., Kikuchi H., Saigusa D., Al-Mamun M. E., et al. (2019). Identification of celastramycin as a novel therapeutic agent for pulmonary arterial hypertension. Circ. Res. 125 (3), 309–327. doi:10.1161/CIRCRESAHA.119.315229
Labrousse-Arias D., Castillo-González R., Rogers N. M., Torres-Capelli M., Barreira B., Aragonés J., et al. (2015). HIF-2α-mediated induction of pulmonary thrombospondin-1 contributes to hypoxia-driven vascular remodelling and vasoconstriction. Cardiovasc Res. 109 (1), 115–130. doi:10.1093/cvr/cvv243
Lambeth J. D., Kawahara T., Diebold B. (2007). Regulation of Nox and Duox enzymatic activity and expression. Free Radic. Biol. Med. 43 (3), 319–331. doi:10.1016/j.freeradbiomed.2007.03.028
Li S., Tabar S. S., Malec V., Eul B. G., Klepetko W., Weissmann N., et al. (2008). NOX4 regulates ROS levels under normoxic and hypoxic conditions., triggers proliferation., and inhibits apoptosis in pulmonary artery adventitial fibroblasts. Antioxid. Redox Signal 10 (10), 1687–1698. doi:10.1089/ars.2008.2035
Liu X., Bennett R. L., Cheng X., Byrne M., Reinhard M. K., May W. S. Jr. (2013). PKR regulates proliferation., differentiation., and survival of murine hematopoietic stem/progenitor cells. Blood 121 (17), 3364–3374. doi:10.1182/blood-2012-09-456400
Long L., Yang X., Southwood M., Lu J., Marciniak S. J., Dunmore B. J., et al. (2013). Chloroquine prevents progression of experimental pulmonary hypertension via inhibition of autophagy and lysosomal bone morphogenetic protein type II receptor degradation. Circ. Res. 112 (8), 1159–1170. doi:10.1161/CIRCRESAHA.111.300483
Lyle M. A., Davis J. P., Brozovich F. V. (2017). Regulation of pulmonary vascular smooth muscle contractility in pulmonary arterial hypertension: implications for therapy. Front. Physiol. 8, 614. doi:10.3389/fphys.2017.00614
Ma M., Chen W., Hua Y., Jia H., Song Y., Wang Y. (2021). Aerobic exercise ameliorates cardiac hypertrophy by regulating mitochondrial quality control and endoplasmic reticulum stress through M2 AChR. J. Cell Physiol. 236 (9), 6581–6596. doi:10.1002/jcp.30342
Makino A., Firth A. L., Yuan J. X. (2011). Endothelial and smooth muscle cell ion channels in pulmonary vasoconstriction and vascular remodeling. Compr. Physiol. 1 (3), 1555–1602. doi:10.1002/cphy.c100023
Manz X. D., Szulcek R., Pan X., Symersky P., Dickhoff C., Majolée J., et al. (2022). Epigenetic Modification of the von Willebrand Factor Promoter Drives Platelet Aggregation on the Pulmonary Endothelium in Chronic Thromboembolic Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 205 (7), 806–818. doi:10.1164/rccm.202109-2075OC
Maron B. A., Hess E., Maddox T. M., Opotowsky A. R., Tedford R. J., Lahm T., et al. (2016). Association of borderline pulmonary hypertension with mortality and hospitalization in a large patient cohort: insights from the veterans affairs clinical assessment, reporting, and tracking program. Circulation 133 (13), 1240–1248. doi:10.1161/CIRCULATIONAHA.115.020207
Marsboom G., Toth P. T., Ryan J. J., Hong Z., Wu X., Fang Y. H., et al. (2012). Dynamin-related protein 1-mediated mitochondrial mitotic fission permits hyperproliferation of vascular smooth muscle cells and offers a novel therapeutic target in pulmonary hypertension. Circ. Res. 110 (11), 1484–1497. doi:10.1161/CIRCRESAHA.111.263848
Martin Y. N., Pabelick C. M. (2014). Sex differences in the pulmonary circulation: implications for pulmonary hypertension. Am. J. Physiol. Heart Circ. Physiol. 306 (9), H1253–H1264. doi:10.1152/ajpheart.00857.2013
Masson B., Le Ribeuz H., Sabourin J., Laubry L., Woodhouse E., Foster R., et al. (2022). Orai1 inhibitors as potential treatments for pulmonary arterial hypertension. Circ. Res. 131 (9), e102–e119. doi:10.1161/CIRCRESAHA.122.321041
Miao R., Dong X., Gong J., Li Y., Guo X., Wang J., et al. (2022). Examining the development of chronic thromboembolic pulmonary hypertension at the single-cell level. Hypertension 79 (3), 562–574. doi:10.1161/HYPERTENSIONAHA.121.18105
Mittal M., Roth M., König P., Hofmann S., Dony E., Goyal P., et al. (2007). Hypoxia-dependent regulation of nonphagocytic NADPH oxidase subunit NOX4 in the pulmonary vasculature. Circ. Res. 101 (3), 258–267. doi:10.1161/CIRCRESAHA.107.148015
Morris N. R., Kermeen F. D., Jones A. W., Lee J. Y., Holland A. E. (2023). Exercise-based rehabilitation programmes for pulmonary hypertension. Cochrane Database Syst. Rev. 3 (3), CD011285. doi:10.1002/14651858.CD011285.pub2
Nadaud S., Poirier O., Girerd B., Blanc C., Montani D., Eyries M., et al. (2013). Small platelet microparticle levels are increased in pulmonary arterial hypertension. Eur. J. Clin. Invest 43, 64–71. doi:10.1111/eci.12018
Nogueira-Ferreira R., Ferreira R., Henriques-Coelho T. (2014). Cellular interplay in pulmonary arterial hypertension: implications for new therapies. Biochim. Biophys. Acta 1843 (5), 885–893. doi:10.1016/j.bbamcr.2014.01.030
Olsen L. N., Fischer M., Evans P. A., Gliemann L., Hellsten Y. (2021). Does exercise influence the susceptibility to arterial thrombosis? An integrative perspective. Front. Physiol. 12, 636027. doi:10.3389/fphys.2021.636027
Parpaleix A., Amsellem V., Houssaini A., Abid S., Breau M., Marcos E., et al. (2016). Role of interleukin-1 receptor 1/MyD88 signalling in the development and progression of pulmonary hypertension. Eur. Respir. J. 48 (2), 470–483. doi:10.1183/13993003.01448-2015
Perros F., Montani D., Dorfmüller P., Durand-Gasselin I., Tcherakian C., Le Pavec J., et al. (2008). Platelet-derived growth factor expression and function in idiopathic pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 178 (1), 81–88. doi:10.1164/rccm.200707-1037OC
Pickrell A. M., Youle R. J. (2015). The roles of PINK1., parkin., and mitochondrial fidelity in Parkinson's disease. Neuron 85 (2), 257–273. doi:10.1016/j.neuron.2014.12.007
Pugliese S. C., Poth J. M., Fini M. A., Olschewski A., El Kasmi K. C., Stenmark K. R. (2015). The role of inflammation in hypoxic pulmonary hypertension: from cellular mechanisms to clinical phenotypes. Am. J. Physiol. Lung Cell Mol. Physiol. 308 (3), L229–L252. doi:10.1152/ajplung.00238.2014
Pullamsetti S. S., Savai R., Seeger W., Goncharova E. A. (2017). Translational advances in the field of pulmonary hypertension. From cancer biology to new pulmonary arterial hypertension therapeutics. Targeting cell growth and proliferation signaling hubs. Am. J. Respir. Crit. Care Med. 195 (4), 425–437. doi:10.1164/rccm.201606-1226PP
Ramos J. S., Dalleck L. C., Tjonna A. E., Beetham K. S., Coombes J. S. (2015). The impact of high-intensity interval training versus moderate-intensity continuous training on vascular function: a systematic review and meta-analysis. Sports Med. 45 (5), 679–692. doi:10.1007/s40279-015-0321-z
Richter M. J., Milger K., Tello K., Stille P., Seeger W., Mayer E., et al. (2014). Heart rate response during 6-minute walking testing predicts outcome in operable chronic thromboembolic pulmonary hypertension. BMC Pulm. Med. 16 (1), 96. doi:10.1186/s12890-016-0260-y
Rodor J., Chen S. H., Scanlon J. P., Monteiro J. P., Caudrillier A., Sweta S., et al. (2022). Single-cell RNA sequencing profiling of mouse endothelial cells in response to pulmonary arterial hypertension. Cardiovasc Res. 118 (11), 2519–2534. doi:10.1093/cvr/cvab296
Santos-Lozano A., Fiuza-Luces C., Fernández-Moreno D., Llavero F., Arenas J., López J. A., et al. (2019). Exercise benefits in pulmonary hypertension. J. Am. Coll. Cardiol. 73 (22), 2906–2907. doi:10.1016/j.jacc.2019.03.489
Santos-Ribeiro D., Mendes-Ferreira P., Maia-Rocha C., Adão R., Leite-Moreira A. F., Brás-Silva C. (2016). Pulmonary arterial hypertension: basic knowledge for clinicians. Arch. Cardiovasc Dis. 109 (10), 550–561. doi:10.1016/j.acvd.2016.03.004
Savai R., Pullamsetti S. S., Kolbe J., Bieniek E., Voswinckel R., Fink L., et al. (2012). Immune and inflammatory cell involvement in the pathology of idiopathic pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 186 (9), 897–908. doi:10.1164/rccm.201202-0335OC
Semenza G. L. (2012). Hypoxia-inducible factors in physiology and medicine. Cell 148 (3), 399–408. doi:10.1016/j.cell.2012.01.021
Shan F., Li J., Huang Q. Y. (2014). HIF-1 alpha-induced up-regulation of miR-9 contributes to phenotypic modulation in pulmonary artery smooth muscle cells during hypoxia. J. Cell Physiol. 229 (10), 1511–1520. doi:10.1002/jcp.24593
Sheikh A. Q., Lighthouse J. K., Greif D. M. (2014). Recapitulation of developing artery muscularization in pulmonary hypertension. Cell Rep. 6 (5), 809–817. doi:10.1016/j.celrep.2014.01.042
Sherman A. E., Saggar R. (2023). Cardiopulmonary exercise testing in pulmonary arterial hypertension. Heart Fail Clin. 19 (1), 35–43. doi:10.1016/j.hfc.2022.08.015
Simonneau G., Montani D., Celermajer D. S., Denton C. P., Gatzoulis M. A., Krowka M., et al. (2019). Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 53 (1), 1801913. doi:10.1183/13993003.01913-2018
Stacher E., Graham B. B., Hunt J. M., Gandjeva A., Groshong S. D., McLaughlin V. V., et al. (2012). Modern age pathology of pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 186 (3), 261–272. doi:10.1164/rccm.201201-0164OC
Stam K., van Duin R. W., Uitterdijk A., Krabbendam-Peters I., Sorop O., Danser A. H. J., et al. (2018). Pulmonary microvascular remodeling in chronic thrombo-embolic pulmonary hypertension. Am. J. Physiol. Lung Cell Mol. Physiol. 315 (6), L951-L964–L964. doi:10.1152/ajplung.00043.2018
Stenmark K. R., Frid M. G., Yeager M., Li M., Riddle S., McKinsey T., et al. (2012). Targeting the adventitial microenvironment in pulmonary hypertension: a potential approach to therapy that considers epigenetic change. Pulm. Circ. 2012 Jan-Mar 2 (1), 3–14. doi:10.4103/2045-8932.94817
Stenmark K. R., Yeager M. E., El Kasmi K. C., Nozik-Grayck E., Gerasimovskaya E. V., Li M., et al. (2013). The adventitia: essential regulator of vascular wall structure and function. Annu. Rev. Physiol. 75, 23–47. doi:10.1146/annurev-physiol-030212-183802
Sturrock A., Cahill B., Norman K., Huecksteadt T. P., Hill K., Sanders K., et al. (2006). Transforming growth factor-beta1 induces Nox4 NAD(P)H oxidase and reactive oxygen species-dependent proliferation in human pulmonary artery smooth muscle cells. Am. J. Physiol. Lung Cell Mol. Physiol. 290 (4), L661-L673–L673. doi:10.1152/ajplung.00269.2005
Sydykov A., Mamazhakypov A., Maripov A., Kosanovic D., Weissmann N., Ghofrani H. A., et al. (2021). Pulmonary hypertension in acute and chronic high altitude maladaptation disorders. Int. J. Environ. Res. Public Health 18 (4), 1692. doi:10.3390/ijerph18041692
Theilmann A. L., Hawke L. G., Hilton L. R., Whitford M. K. M., Cole D. V., Mackeil J. L., et al. (2020). Endothelial BMPR2 loss drives a proliferative response to BMP (bone morphogenetic protein) 9 via prolonged canonical signaling. Arterioscler. Thromb. Vasc. Biol. 40 (11), 2605–2618. doi:10.1161/ATVBAHA.119.313357
Vieira J. S., Cunha T. F., Paixão N. A., Dourado P. M., Carrascoza L. S., Bacurau A. V. N., et al. (2020). Exercise intolerance establishment in pulmonary hypertension: preventive effect of aerobic exercise training. Life Sci. 261, 118298. doi:10.1016/j.lfs.2020.118298
Vrigkou E., Tsangaris I., Bonovas S., Kopterides P., Kyriakou E., Konstantonis D., et al. (2019). Platelet and coagulation disorders in newly diagnosed patients with pulmonary arterial hypertension. Platelets 30 (5), 646–651. doi:10.1080/09537104.2018.1499890
Vrigkou E., Tsantes A. E., Kopterides P., Orfanos S. E., Armaganidis A., Maratou E., et al. (2020). Coagulation profiles of pulmonary arterial hypertension patients, assessed by non-conventional hemostatic tests and markers of platelet activation and endothelial dysfunction. Diagn. (Basel) 10 (10), 758. doi:10.3390/diagnostics10100758
Waghray M., Cui Z., Horowitz J. C., Subramanian I. M., Martinez F. J., Toews G. B., et al. (2005). Hydrogen peroxide is a diffusible paracrine signal for the induction of epithelial cell death by activated myofibroblasts. FASEB J. 19 (7), 854–856. doi:10.1096/fj.04-2882fje
Wang G., Liu X., Meng L., Liu S., Wang L., Li J., et al. (2014). Up-regulated lipocalin-2 in pulmonary hypertension involving in pulmonary artery SMC resistance to apoptosis. Int. J. Biol. Sci. 10 (7), 798–806. doi:10.7150/ijbs.9470
Welch C. L., Chung W. K. (2020). Genetics and other omics in pediatric pulmonary arterial hypertension. Chest 157 (5), 1287–1295. doi:10.1016/j.chest.2020.01.013
Wilkins M. R., Ghofrani H. A., Weissmann N., Aldashev A., Zhao L. (2015). Pathophysiology and treatment of high-altitude pulmonary vascular disease. Circulation 131 (6), 582–590. doi:10.1161/CIRCULATIONAHA.114.006977
Wu Y., Adi D., Long M., Wang J., Liu F., Gai M. T., et al. (2016). 4-Phenylbutyric acid induces protection against pulmonary arterial hypertension in rats. PLoS One 11 (6), e0157538. doi:10.1371/journal.pone.0157538
Wu Z., Zhang Z., Zhou Z., Rao S., Li S. (2022). Effect of aerobic exercise on lung regeneration and inflammation in mice. Immunobiology 227 (6), 152296. doi:10.1016/j.imbio.2022.152296
Yaku A., Inagaki T., Asano R., Okazawa M., Mori H., Sato A., et al. (2022). Regnase-1 prevents pulmonary arterial hypertension through mRNA degradation of interleukin-6 and platelet-derived growth factor in alveolar macrophages. Circulation 146 (13), 1006–1022. doi:10.1161/CIRCULATIONAHA.122.059435
Yang H., Liu Y., Kong J. (2022). Effect of aerobic exercise on acquired gefitinib resistance in lung adenocarcinoma. Transl. Oncol. 14 (11), 101204. doi:10.1016/j.tranon.2021.101204
Yang P. S., Kim D. H., Lee Y. J., Lee S. E., Kang W. J., Chang H. J., et al. (2014). Glycyrrhizin., inhibitor of high mobility group box-1., attenuates monocrotaline-induced pulmonary hypertension and vascular remodeling in rats. Respir. Res. 15, 148. doi:10.1186/s12931-014-0148-4
Ye J. X., Wang S. S., Ge M., Wang D. J. (2016). Suppression of endothelial PGC-1α is associated with hypoxia-induced endothelial dysfunction and provides a new therapeutic target in pulmonary arterial hypertension. Am. J. Physiol. Lung Cell Mol. Physiol. 310 (11), L1233–L1242. doi:10.1152/ajplung.00356.2015
Yeager M. E., Belchenko D. D., Nguyen C. M., Colvin K. L., Ivy D. D., Stenmark K. R. (2012). Endothelin-1., the unfolded protein response., and persistent inflammation: role of pulmonary artery smooth muscle cells. Am. J. Respir. Cell Mol. Biol. 46 (1), 14–22. doi:10.1165/rcmb.2010-0506OC
Yu L., Tu Y., Jia X., Fang K., Liu L., Wan L., et al. (2017). Resveratrol protects against pulmonary arterial hypertension in rats via activation of silent information regulator 1. Cell Physiol. Biochem. 42 (1), 55–67. doi:10.1159/000477115
Zhang R., Zhang J., Zhang Y. L., Gong S. G., Zhao Q. H., Wang X. J., et al. (2023). Single-cell transcriptome analysis of peripheral neutrophils from patients with idiopathic pulmonary arterial hypertension. Hypertension 80 (8), 1784–1794. doi:10.1161/HYPERTENSIONAHA.123.21142
Zheng Y. G., Yang T., Xiong C. M., He J. G., Liu Z. H., Gu Q., et al. (2015). Platelet distribution width and mean platelet volume in idiopathic pulmonary arterial hypertension. Heart Lung Circ. 24, 566–572. doi:10.1016/j.hlc.2014.11.025
Zhou X., Jiang Y., Wang Y., Fan L., Zhu Y., Chen Y., et al. (2023). Endothelial FIS1 DeSUMOylation protects against hypoxic pulmonary hypertension. Circ. Res. 133 (6), 508–531. doi:10.1161/CIRCRESAHA.122.321200
Keywords: pulmonary hypertension, aerobic exercise, ER stress, ROS, thrombosis, 6-min walk distance
Citation: Song Y, Jia H, Ma Q, Zhang L, Lai X and Wang Y (2024) The causes of pulmonary hypertension and the benefits of aerobic exercise for pulmonary hypertension from an integrated perspective. Front. Physiol. 15:1461519. doi: 10.3389/fphys.2024.1461519
Received: 08 July 2024; Accepted: 26 September 2024;
Published: 17 October 2024.
Edited by:
Michael S. Wolin, New York Medical College, United StatesReviewed by:
Ran Wang, Anhui Medical University, ChinaCopyright © 2024 Song, Jia, Ma, Zhang, Lai and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yinping Song, c3lwQHhhZnkuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.