 Dathe Z. Benissan-Messan
Dathe Z. Benissan-Messan Hua Zhu
Hua Zhu Weina Zhong1
Weina Zhong1 Tao Tan
Tao Tan Jianjie Ma
Jianjie Ma Peter H. U. Lee
Peter H. U. Lee
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol. , 06 November 2020
Sec. Striated Muscle Physiology
Volume 11 - 2020 | https://doi.org/10.3389/fphys.2020.583393
This article is part of the Research Topic Calcium Homeostasis in Skeletal Muscle Function, Plasticity and Disease View all 14 articles
Since its identification in 2009, multiple studies have indicated the importance of MG53 in muscle physiology. The protein is produced in striated muscles but has physiologic implications reaching beyond the confines of striated muscles. Roles in muscle regeneration, calcium homeostasis, excitation-contraction coupling, myogenesis, and the mitochondria highlight the protein’s wide-reaching impact. Numerous therapeutic applications could potentially emerge from these physiologic roles. This review summarizes the current literature regarding the role of MG53 in the skeletal muscle. Therapeutic applications are discussed.
More than 70 TRIM family proteins exist. They are characterized by the presence of a tripartite motif composed of a RING finger, B-box motifs, and a coiled coil region at their N-terminus. Among them is a 53 kD protein named Mitsugumin 53 (MG53) (Cai et al., 2009a; McNeil, 2009). MG53, also known as TRIM72, was cloned in 2009 using an immunoproteomic library in an attempt to identify proteins involved in myogenesis, calcium signaling, and striated muscle integrity (Weisleder et al., 2008; Tan et al., 2016). Since its identification, multiple physiologic functions have been attributed to this protein, which have made it an attractive candidate in translational medicine as a potential therapeutic agent. The protein is primarily expressed in striated muscles. This review of literature details the current understanding of the role of MG53 in skeletal muscle. Therapeutic applications of the protein relative to skeletal muscle diseases are discussed.
Skeletal muscles are made of bundles of contractile units called myofibers. Myofibers are made of myofibrils which are in turn composed of longitudinal thick and thin filaments (Ciciliot and Schiaffino, 2010). Myofibers form fascicles and bundles of fascicles form muscle tissue (Mukund and Subramaniam, 2020). Myofibers arise during development from the fusion of mononucleate precursor cells known as myoblasts and satellites cells which are stem cells that are activated during regeneration following muscle injury to proliferate and differentiate into myofibers (Blanco-Bose et al., 2001). Skeletal muscles are highly vascularized and innervated (Mukund and Subramaniam, 2020).
Contraction of muscle fibers occur following conduction of impulses through motor neurons via neuromuscular junctions through excitation-contraction (EC) coupling (Takeshima et al., 1994; Mukund and Subramaniam, 2020). Acetylcholine receptors in the plasma membrane of skeletal muscle cells are activated by the release of acetylcholine from motor neurons and leads to membrane depolarization (Lee, 2010; Ahn et al., 2016). The membrane potential induced through depolarization is propagated to the interior of muscle cells via transverse tubules (T-tubules) and leads to the activation of dihydropyridine receptors (DHPR), which in turn activate ryanodine receptor 1 (RyR1), a calcium channel on the sarcoplasmic reticulum membrane (Ahn et al., 2016). RyR1 is responsible for the release of calcium ions into the cytosol (Takeshima et al., 1994). Calcium ions in the cytosol further activate more RyR1 to release more calcium into the cytosol, a phenomenon referred to as calcium-ion induced calcium release (Takeshima et al., 1994). Entry of extracellular calcium into the cell through store-operated calcium entry (SOCE) via Orai1 or through the transient receptor potential canonical (TRPC) channels also contribute to the calcium supply during skeletal muscle contraction (Ahn et al., 2016). Following contraction, the sarcoplasmic endoplasmic reticulum calcium ion ATPase 1a (SERCA1a) uptakes cytosolic calcium and leads to a return to a resting level and a replenishment of the calcium content of the sarcoplasmic reticulum (Ahn et al., 2016).
In addition to being a contractile machine, the skeletal muscle is considered an endocrine organ and secretes several myokines that modulate tissue function (Iizuka et al., 2014; Karstoft and Pedersen, 2016; Giudice and Taylor, 2017; Hoffmann and Weigert, 2017; Bian et al., 2019). Striated muscles are constantly subjected to stress and an active injury-repair mechanism is necessary for their optimum function. Repeated use, stress, diseases, and other injuries would otherwise impair muscle function and movement. The repair of the plasma membrane following injury requires the translocation of intracellular vesicles to the site of injury (Cai et al., 2009a,b). These vesicles fuse with the plasma membrane in a calcium dependent manner (Cai et al., 2009a). Dysferlin and caveolae are essential components of the membrane repair machinery and participate in vesicle fusion to the plasma membrane (Bansal et al., 2003; Glover and Brown, 2007).
MG53 is composed of a TRIM domain at the N-terminus, a PRY domain (associated with SPRY), and a SPRY domain (sequence repeat in the dual-specificity kinase sp1A and ryanodine receptor) at the C-terminus (Cai et al., 2009a; Ahn et al., 2016). Generally, TRIM containing proteins control important cellular processes in intracellular signaling, innate immunity, transcription, autophagy, and carcinogenesis (Hatakeyama, 2017). Genetic analysis of the TRIM domain reveals that it is made up of a Really Interesting New Gene (RING) domain with ubiquitin E3 ligase activity, a B-box with a zinc-binding domain, and coiled-coil moieties (Ozato et al., 2008; Cai et al., 2009a; Tan et al., 2016). The RING finger domain is a zinc finger motif containing a Cys3HisCys4 amino acid motif which binds two zinc cations (Zhang et al., 2017). The E3 ubiquitin ligase activity of the RING domain catalyzes ubiquitin conjugation which targets proteins for degradation (Hatakeyama, 2017). The B-box domains are zinc binding as well and have been shown to play roles in the innate immune system defense (Ozato et al., 2008). The coiled-coil domain mediates interactions among TRIM proteins which leads to protein assembly into high molecular mass complexes (Ozato et al., 2008; Zhang et al., 2017). The PRY domain is involved in protein binding and has been suggested to be an interface for the protein-protein interaction of MG53 with other proteins (Ahn et al., 2016). At the C-terminus, MG53 has a SPRY domain, which is responsible for the binding of target proteins (Zhang et al., 2017). MG53 is primarily produced in cardiac and skeletal muscles (Cai et al., 2009a; Hu and Xiao, 2018). RNA analysis has revealed expression in the lung and kidney (Cai et al., 2009a; Jia et al., 2014; Duann et al., 2015). The protein was also noted to be expressed by macrophages (Sermersheim et al., 2020). Recombinant human MG53 (rhMG53) has been purified and is available for therapeutic applications (Weisleder et al., 2012; Jia et al., 2014; Duann et al., 2015; Li et al., 2015; Liu J. et al., 2015; Zhu et al., 2015; Yao et al., 2016; Adesanya et al., 2019; Bian et al., 2019; Chandler et al., 2019; Guan et al., 2019a,b; Liu et al., 2020; Sermersheim et al., 2020).
MG53 binds to Orai1 via its PRY-SPRY region and co-localizes with Orai1 to the plasma membranes of skeletal muscles to enhance extracellular calcium entry via by SOCE, while reducing intracellular calcium release through RyR1, and increasing the expression of TRPC3, TRPC4, and calmodulin 1(CaM1) (Figure 1A; Ahn et al., 2016). MG53 also attenuates SERCA1a activity, leading to a rise in cytosolic calcium (Lee et al., 2012). The TRIM and PRY domains of MG53 constitute the binding region to SERCA1a and the interaction of MG53 and SERCA1a reduces the activity of SERCA1a, which takes up calcium from the cytosol to the sarcoplasmic reticulum (Lee et al., 2012; Ahn et al., 2016). Together, this leads to a transient increase in cytosolic calcium, which results in efficient skeletal muscle contractions (Ahn et al., 2016).
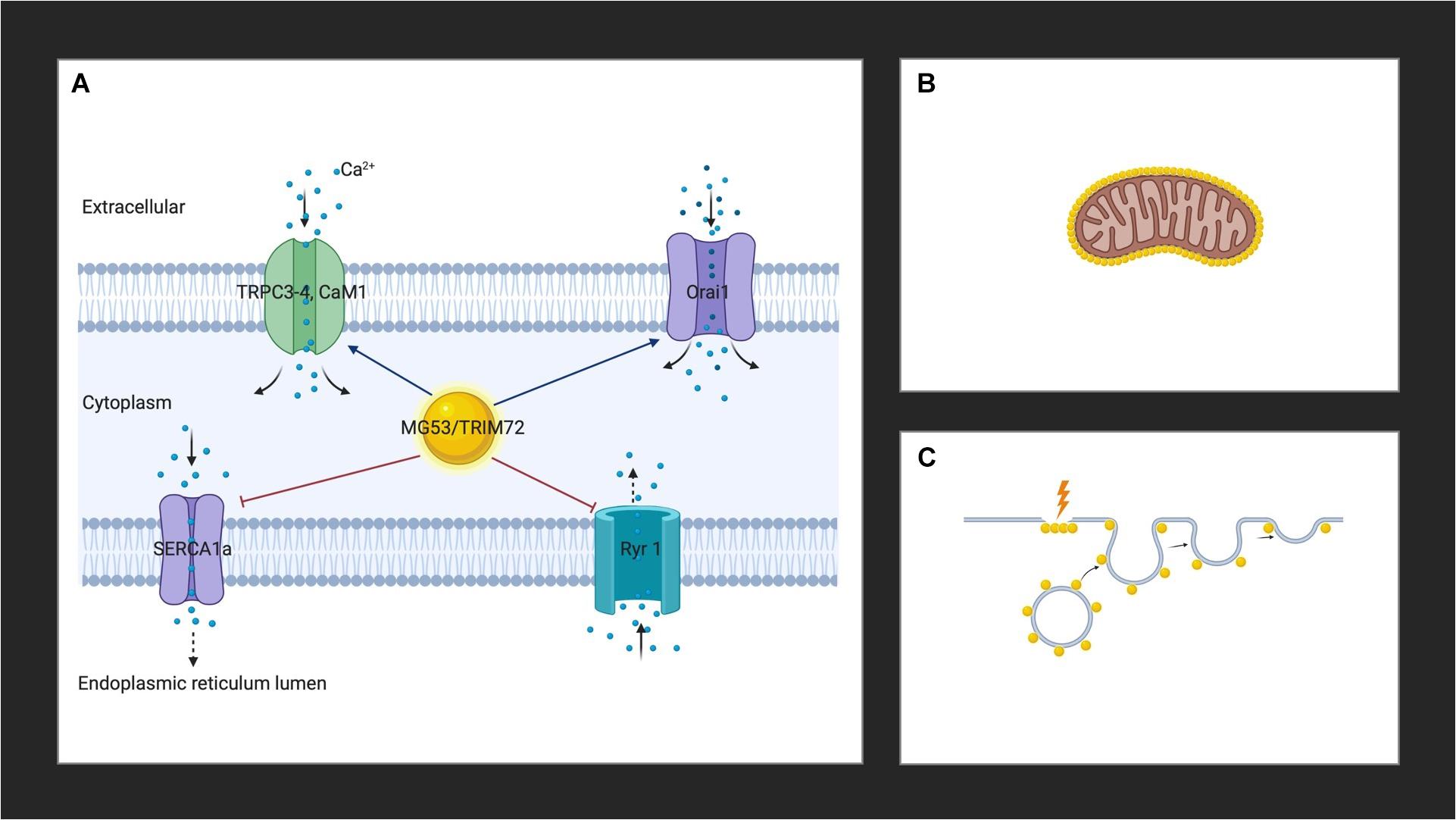
Figure 1. (A) MG53 controls cytosolic calcium through interactions with Orai1, RyR1, TRPC3, TRPC4, CaM1, and SERCA. (B) MG53 aggregates around the mitochondria of skeletal muscle in the presence of reactive oxygen species. (C) MG53 promotes plasma membrane repair by facilitating vesicle translocation following injury. Golden spheres represent MG53. Created with BioRender.com.
Studies in high fat diet (HFD)-induced metabolic syndrome mouse models have demonstrated an aggregation of MG53 around the mitochondria of skeletal muscle in the presence of reactive oxygen species (ROS) which suggests a role for the protein as a mitochondrial guardian (Figure 1B; Ma et al., 2015). Since HFD feeding induces an elevation in mitochondrial activity as a result of increased production of ROS, MG53 is thought to play a protective role (Ma et al., 2015). More recently, MG53 overexpression was also suggested to promote mitochondrial autophagy to help remove damaged mitochondria to relieve CKD-induced muscle atrophy in skeletal muscle cells by upregulating autophagy and beclin 1 regulator 1 (Ambra1), a key regulator involved in autophagosome formation (Lijie et al., 2019). These studies herald the opening of new avenues regarding the widening reach of the activity of MG53. Further studies will undoubtedly elucidate the role of the protein in the mitochondria.
The role of MG53 in the plasma membrane was one of the first discovered physiological functions of the protein. It is now well established that MG53 facilitates vesicle translocation to the plasma membrane where it leads to repair following injury (Figure 1C; Cai et al., 2009a). Cholesterol exposure in the plasma membrane following skeletal muscle injury is thought to mediate the translocation of MG53 upon membrane damage (Han, 2011; Zhu et al., 2011). The protein appears as a monomer in the reduced environment of the cytoplasm of an intact cell, and acute muscle plasma membrane disruption leads to exposure of the cell interior to the external oxidized environment (Cai et al., 2009a). A change in the oxidation state of MG53 ensues, which initiates oligomerization (Cai et al., 2009a; McNeil, 2009). Targeted mutagenesis revealed that MG53 oligomer formation requires a conserved cystidine residue (C242) and mutations in C242 do not result in the formation of oligomers (Matsuda et al., 2012). Oligomerization of MG53 through oxidation of the thiol group of cysteine at 242 and a leucine zipper motif between the two coiled-coil domains induces the nucleation of intracellular vesicles coated with MG53, the trafficking of vesicles to the injury sites, and the resealing of the injured membranes (Hwang et al., 2011; Ahn et al., 2016). At the membrane, MG53 concentrates at the site of injury in damaged adult muscle fibers (Cai et al., 2009a; Weisleder et al., 2012; Lek et al., 2013). The protein binds to phosphatidylserine in the plasma membrane to mediate vesicle accumulation at the site of injury (Cai et al., 2009a; Tan et al., 2016). Polymerase I and transcript release factor (PTRF; also known as cavin-1) recognize cholesterol at the injury site and act as a docking protein to tether MG53 together with its associated vesicles to the damage site (Zhu et al., 2011). Once associated with intracellular vesicles and the inner leaflet of the sarcolemma of striated muscle, fusion is mediated through membrane proteins such as caveolin-3 (Cav-3) (Weisleder et al., 2009). While the exact interaction of MG53 with Cav-3 to control intracellular vesicle trafficking and membrane remodeling in muscle is unknown, the binding of Cav-3 to MG53 is known to be essential for vesicle fusion (Cai et al., 2009b). Together with dysferlin, PTRF, and non-muscle myosin type IIA, MG53 constitute the membrane repair system (Cai et al., 2009c; Zhu et al., 2011; Lin et al., 2012). The ensuing vesicle fusion to the plasma membrane requires the presence of calcium and results in the formation of a membrane patch which seals the site of injury (Cai et al., 2009b; Weisleder et al., 2009; Lek et al., 2013). A recently proposed novel model of membrane repair including MG53 involves the formation of cap and shoulder proteins (Demonbreun and McNally, 2016; Demonbreun et al., 2016). Following membrane disruption, annexins proteins (A1, A2, A5, and A6) bind phospholipids in the presence of calcium ions to form a “cap” at the site of damage (Demonbreun et al., 2016). Repair “shoulder” proteins, such as dysferlin, Epsin 15 homology domain-containing proteins (EHD1, EHD2), MG53, and BIN1 then localizes adjacent to the repair cap to form a membrane repair complex system and seals the injured site (Demonbreun et al., 2016).
Mammalian skeletal muscles are made of multinucleated myofibers formed during development by fusion of mononucleated muscle progenitors (Ciciliot and Schiaffino, 2010). Myogenic differentiation is a tightly regulated process in which mononucleated myoblasts expressing myogenic marker proteins [MyoD, Myogenin, Myosin Heavy Chain (MyHC), and others] proliferate and fuse to form multinucleated myotubes. Matured myotubes convert into contractile myofibers (Okino et al., 2020). The formation of new myofibers or myofiber segments following necrosis is called muscle regeneration (Ciciliot and Schiaffino, 2010). Mature skeletal muscle tissues possess the ability to regenerate from the interaction of satellite cells and their surrounding microenvironment (Mukund and Subramaniam, 2020). Satellite cells are characterized by the expression of transcription factors PAX3 and PAX7 and are located between the basal lamina and the sarcolemma (Relaix et al., 2006; Buckingham, 2007; von Maltzahn et al., 2013). Muscle regeneration following injury is characterized by an inflammatory reaction dominated by the invasion of macrophages, followed by the activation, migration, and proliferation of satellite cells, and their differentiation and fusion into muscle fibers (Ciciliot and Schiaffino, 2010). The maturation of these newly formed myofibers and the remodeling of the regenerated muscle follows (Ciciliot and Schiaffino, 2010). Local apoptosis seen in atrophic muscle can also lead to an increase in the number of satellite cells (Ciciliot and Schiaffino, 2010).
Quiescent satellite cells are marked by the expression of PAX7 as well as several other molecular markers, and is notable for the absence of Myogenic Regulatory Factor (MRF), Myogenic Differentiation1 (MYOD1) and Myogenin (MYOG) (Rocheteau et al., 2015; Mukund and Subramaniam, 2020). Skeletal muscle-specific TGFb family member, myostatin, suppresses satellite cell activation (McCroskery et al., 2003). Sprouty1 (SPRY1), a tyrosine inhibitor kinase, is necessary for maintenance and re-entry of PAX7 satellite cells into quiescence (Shea et al., 2010). Satellite cells are activated in response to injury. Activated satellite cells are characterized by PAX7 and the expression of MRFs [MYOD1, MYOG, and Myogenic Factor (MYF)5] (Mukund and Subramaniam, 2020). PAX7 is absolutely required for normal function of satellite cells in regenerative myogenesis in both neonatal and adult skeletal cells (von Maltzahn et al., 2013). Deficiency in PAX7 results in cell-cycle arrest and precocious differentiation (von Maltzahn et al., 2013). The migration to an injury site and proliferation of satellite cells is governed by chemo-attractants released from the extracellular matrix or inflammatory cells (Mukund and Subramaniam, 2020).
Insulin-like growth factors, IGF-I and IGF-II, are essential for skeletal muscle development, hypertrophy, and regeneration. IGF-I plays a key role in the differentiation of satellite cells and the development, hypertrophy, and regeneration of skeletal muscles (Zhang et al., 2017). Likewise, satellite cells lose their muscle differentiation activity when treated with anti-IGF-II antibody or antisense IGF-II, whereas IGF-I overexpression in mice leads to hypertrophy and hyperplasia, leading to increased muscle mass and force generation (Jung and Ko, 2010). IGF-I and IGF-II transduce cellular signaling through the IGF-I receptor (IGFR) that subsequently recruits insulin receptor substrate-1 (IRS-1) (Yi et al., 2013). The recruited IRS-1 can activate the phosphoinositide 3 kinase (PI(3)K)/Akt and Ras/Raf/MEK/ERK pathways. The Ras/Raf/MEK/ERK pathway controls muscle fiber type whereas the PI(3)K/Akt pathway induces muscle differentiation and hypertrophy (Jung and Ko, 2010). The activated Akt targets a mammalian target of rapamycin (mTOR), glycogen synthase kinase 3β (GSK3β), and Forkhead box O (FOXO) (Jung and Ko, 2010). mTOR activity is not involved in muscle differentiation (Jung and Ko, 2010). Phosphorylated GSK3β and FOXO stimulate muscle hypertrophy and suppress muscle atrophy. IRS-1 overexpression inhibits myogenic differentiation through continuous FOXO inhibition and causes repression of MyHC during differentiation (Okino et al., 2020). Additionally, IRS-1 and insulin signaling is reduced during myogenesis, and myoblasts with higher IRS-1 levels are eliminated during differentiation (Bian et al., 2019; Okino et al., 2020).
Intracellularly, evidence suggests that MG53 is a negative regulator of IGF-induced myogenesis (Jung and Ko, 2010; Lee et al., 2010). The MG53 promoter contains E-boxes [which are short DNA motifs within myogenic cis-regulatory element (CREs)] and a Myocyte Enhancer Factor (MEF)-binding site for the myogenic transcription factors MyoD and MEF (Jung and Ko, 2010; Soleimani et al., 2018). MG53 is gradually expressed during myogenesis because the transcription of MG53 requires MyoD and Akt (Jung and Ko, 2010; Hong et al., 2016). MG53 then acts as an E3 ligase, targeting the insulin receptor and IRS-1 for ubiquitin-dependent degradation (Song et al., 2013; Yi et al., 2013). This negatively regulates skeletal myogenesis by reducing IGF-I-initiated IRS-1 tyrosine phosphorylation and Akt phosphorylation without affecting IGF-I-elicited IGFR tyrosine phosphorylation and ERK1/2 phosphorylation (Yi et al., 2013). Myogenesis and IGF-I-induced IRS-1 tyrosine phosphorylation were prevented in C2C12 myoblasts by MG53 overexpression but enhanced by MG53 knockdown (Jung and Ko, 2010). The RING domain of MG53 with E3 ligase activity is necessary for the inhibition of IGF-I-elictied IRS-1 phosphorylation (Yi et al., 2013). MG53-induced ubiquitination and degradation of another kinase, focal adhesion kinase (FAK), is also thought to inhibit myogenesis (Nguyen et al., 2014). FAK regulates heterochromatin remodeling to modulate myogenin expression during skeletal myogenesis and regulate the expression of genes involved in membrane fusion, including caveolae (Nguyen et al., 2014). Its inhibition by MG53 is also thought to reduce myogenesis (Nguyen et al., 2014).
Meanwhile, exogenous MG53 has been noted to facilitate the differentiation of C2C12 skeletal myoblasts to myotubes by enhancing vesicle trafficking and membrane fusion (Cai et al., 2009a; Bian et al., 2019). A recent study published by Bian et al. provided evidence that increased levels of MG53 in circulation do not negatively impact myogenesis (Bian et al., 2019). Transgenic mice with sustained elevation of MG53 in circulation (tPA-MG53) have a longer lifespan compared to their wild type counterparts due to enhanced repair capacity after injury and enhanced muscle regeneration (Bian et al., 2019). The authors also did not observe any changes in IRS-1 activity in the skeletal muscles as a result of MG53 overexpression. MG53 was noted to restore satellite cell proliferation in MG53 knockout (mg53–/–) mice (Bian et al., 2019). This evidence led to the hypothesis that MG53 may improve muscle regeneration by modulating the activity of satellite cells (Bian et al., 2019). This resulting dichotomous functions between the intracellular mechanism of MG53 and the effect of extracellular MG53 application remains to be elucidated.
MG53 is an endogenous protein and therefore therapies based on its use are less likely to elicit an immune system reaction. The recombinant human form of the protein (rhMG53) is readily available for therapeutic application and studies have shown limited toxicity (Weisleder et al., 2012; Alloush and Weisleder, 2013; Jia et al., 2014; Duann et al., 2015; Liu J. et al., 2015; Tan et al., 2016). Here, we present a few studies highlighting the potential therapeutic applications for MG53.
When applied intravenously, rhMG53 can potentially treat multiple pathologies in rodent and large animal disease models (Weisleder et al., 2012; Jia et al., 2014; Duann et al., 2015; Liu J. et al., 2015; Zhu et al., 2015). The myokine has been shown to play a protective role in skeletal muscles, kidneys, lungs, brains, skin, hearts, and corneas (He et al., 2012; Weisleder et al., 2012; Jia et al., 2014; Duann et al., 2015; Li et al., 2015; Yao et al., 2016; Adesanya et al., 2019; Chandler et al., 2019). Exogenously administered rhMG53 travels in the circulation and concentrates at the healing edge of injury sites and contributes to regeneration following injury (Weisleder et al., 2012; Bian et al., 2019). Muscle cells and tissues treated with rhMG53 show resistance to mechanical, chemical, and ultraviolet damage (Weisleder et al., 2012). Intravenous injection of the protein protects skeletal muscles against acute and chronic insults (Weisleder et al., 2012; Zhu et al., 2015; Bian et al., 2019). Weisleder et al. (2012) noted in mouse models that 300 nM rhMG53 was sufficient to protect muscle cells against mechanical damage and prevent muscle injury.
The importance of the protein in membrane repair has been highlighted by multiple studies. MG53-/- mice with targeted protein deletion develop progressive muscle pathology with age characterized by defects in skeletal muscle contractility after injury and defective membrane repair (Cai et al., 2009a). Morphologically, a pathologic increase in the number of central nuclei, a measure of defective muscle repair, and decreased diameter of muscle fibers, were observed in isolated skeletal muscles of older mg53-/- mice when compared to younger counterparts and wild type littermates (Cai et al., 2009a). Following cardiotoxin-induced muscle injury, genetically enhanced tPA-MG53 mice overexpressing MG53 displayed enhanced regeneration when compared to wild type and mg53-/- mice (Bian et al., 2019). The regenerative ability of MG53 is thought to be mediated via satellite cell proliferation (Bian et al., 2019).
Muscular dystrophies are an inherited group of disorders characterized by progressive muscle weakness and atrophy and linked to a loss of function of dystrophin and its associated proteins (Glover and Brown, 2007). Muscular dystrophies can be caused by genetic mutations in over thirty different genes, many of which encode for proteins essential for the integrity of muscle cell structure and membranes (He et al., 2012). Mutations in the dystrophin gene in Duchenne muscular dystrophy (DMD) and Becker muscular dystrophy (BMD), or sarcoglycan genes in limb-girdle muscular dystrophies (LGMDs) lead to muscle sarcolemma destabilization and fragility (He et al., 2012). DMD is an X-linked inherited muscle wasting disorder with limited treatment options characterized by compromised muscle structure and decreased muscle function (Burkin and Wuebbles, 2012; Weisleder et al., 2012). Dysferlinopathies are a heterogenous group of progressive muscular dystrophies characterized by mutations in the DYSF gene, leading to reduced or null expression of the dysferlin protein and resulting in varied phenotypes (Flix et al., 2013). Dysferlin is a type II muscle surface membrane, expressed in the sarcolemma, in intracellular vesicles, and in T-tubules (Glover and Brown, 2007; Han, 2011; Flix et al., 2013). Although dysferlin is important in membrane repair, dysferlin itself does not participate in the recruitment of intracellular vesicles for membrane repair and dysferlin -/- muscle retains accumulation of vesicles near membrane damage sites. Dysferlin is known to interact with several proteins including AHNAK, MG53, and Cav3 (Flix et al., 2013). MG53 and AHNAK interact directly with dysferlin (Flix et al., 2013). Like dysferlin, many mutations of Cav3 have been linked to muscular dystrophy (Cai et al., 2009c). Additionally, Cav3 has also been shown to be essential for MG53-mediated vesicle translocation (Cai et al., 2009c).
Cav3 binds to MG53, which in turn interacts with dysferlin (Flix et al., 2013). In the absence of dystrophin, there is weakening of the attachment of the muscle cells to the surrounding basal lamina, which results in reduced force transmission, loss of muscle integrity, altered cell signaling, and weakened sarcolemma that is easily damaged during muscle contraction (Burkin and Wuebbles, 2012). MG53 interacts with dysferlin to facilitate vesicle trafficking to sites of membrane damage (Cai et al., 2009c). Systemic delivery and muscle specific overexpression of the human MG53 gene by recombinant adeno-associated virus (AAV) vectors enhanced membrane repair, ameliorated pathology, and improved muscle and heart functions in δ-sarcoglycan (δ-sarcoglycan) deficient TO-s hamsters (He et al., 2012). Overexpression of MG53 increased dysferlin levels and facilitated its trafficking to the muscle membrane through the participation of Cav3 (He et al., 2012). In addition, MG53 protected muscle cells by activating cell survival kinases, such as Akt, extracellular signal-regulated kinases (ERK1/2), and glycogen synthase kinase-3β (GSK-3β), and inhibiting proapoptotic protein like Bax (He et al., 2012). Even though MG53 delivery would not constitute a cure for dysferlonopathies, a palliation of pathology could be obtained through its delivery in some sarcoglyconopathies.
Maintenance of plasma membrane integrity is vital for cell survival. The constant stress imposed on the skeletal muscle system over time requires a regular system of maintenance to ensure that it continuously fulfills its mechanical and endocrine functions. Aging, cachexia, sarcopenia and disease states such as cancers are conditions associated with compromised muscle integrity and muscle atrophy where MG53 could play a therapeutic role. An increase in MG53 expression level and other plasma membrane repair proteins has already been observed in sedentary ad libitum aging in animal studies by Hord et al. (2016). The authors postulated that the increased requirement for phospholipid membrane repair in the aging skeletal muscle necessitated the increased presence of these proteins. The study of the function of MG53 in these diseases states and conditions could further shed light on the restoration of muscle integrity and function. Given its protective roles, MG53 is an ideal candidate therapeutic agent for maintaining skeletal muscle health.
Skeletal muscles play an important role in glucose control. They are responsible for 70–90% of insulin-stimulated glucose metabolism (Song et al., 2013; Tan et al., 2016). Song et al. reported increased MG53 expression in animal models and human patients with diabetes (Song et al., 2013). They also showed that an upregulation of MG53 occurred before the onset of diabetes (Song et al., 2013). They proposed that an upregulation of MG53 causes insulin insensitivity through the degradation of IRS-1. These observations have not been substantiated by other investigators, and many others failed to observe an upregulation of MG53 in diabetic animals and humans (Ma et al., 2013, 2015, 2017; Xu et al., 2013; Yi et al., 2013; Yuan et al., 2013; Zabielski et al., 2016). Rather, serum samples from high fat diet induced diabetic mice (Ma et al., 2015) and db/db mice (Wang et al., 2020) showed reduced levels of MG53. It is also well-known that a loss of IRS-1 does not directly result in diabetes, owing to a robust compensatory mechanism among the different IRS subtypes (Tamemoto et al., 1994; Terauchi et al., 1997; Laustsen et al., 2002). A recent proteomic study specifically examined the interactions of IRS-1 in skeletal muscle from normal individuals, obese insulin-resistant non-diabetic control subjects, and patients with type 2 diabetes before and after insulin infusion, and failed to identify an enhancement of MG53 (Caruso et al., 2014). Thus, the mounting evidence do not support the proposed role of MG53 in the regulation of IRS-1 in diabetes.
Liu F. et al. (2015) proposed a different mechanism of MG53 mediated diabetes in 2015. The researchers generated transgenic mice by using the alpha myosin heavy chain (α-MHC) promoter to drive expression of MG53 and found that overexpression of MG53 was powerful enough to induce whole body insulin resistance and compromised glucose uptake (Liu F. et al., 2015). MG53 was posited to enter the nucleus and serve as a transcription factor to activate the peroxisome proliferation-activated receptor alpha (PPAR-α) which in turn mediated excessive lipid uptake induced cytotoxicity. Our group successfully generated a MG53 transgenic mouse line to achieve sustained elevation of circulating MG53 by fusing a tPA secretory peptide at the N-terminus of the MG53 protein (tPA-MG53) (Yao et al., 2016; Bian et al., 2019). tPA-MG53 mice live a healthy lifespan with enhanced tissue repair and regeneration capacity (Yao et al., 2016; Bian et al., 2019). To test the role of elevated circulating MG53 in the development of diabetes, we crossed tPA-MG53 mice with db/db mice and found that tPA-MG53/db/db mice develop diabetes at a rate similar to its db/db littermates, suggesting overexpression of MG53 has negligible effects on the development of diabetes (Wang et al., 2020). A separate transgenic mouse model with overexpression of MG53 was generated by Ham and Mahoney (2013). While they used the same α-MHC promoter to drive overexpression of MG53 as Liu F. et al. (2015), they observed that IRS-1 protein levels were higher in transgenic animals when compared to that of their wild type littermates (Ham and Mahoney, 2013). This observation suggests that a feedback mechanism compensates for the downregulation of IRS-1 by MG53. Other studies with mg53-/-, tPA-MG53, and their wild type littermates also failed to show alteration of PPAR-α (Bian et al., 2019).
Wu et al. (2019) recently implicated MG53 in the development of diabetes. They showed that the binding affinity between MG53 and insulin receptor (IR) is over threefold higher than that of insulin and insulin receptor (Kd: MG53 and IR is 8 nM vs. insulin and IR is 28 nM) (Wu et al., 2019). Thus, MG53 serves as a novel agonist of IR and a competitive antagonist of insulin. If this hypothesis holds true, not only would the authors have successfully identified the receptor for MG53 but they would also have shown that targeting endogenous MG53 could treat diabetes. This would also imply that the high levels of circulating MG53 present in tPA-MG53 mice could block the insulin pathway and greatly facilitate the development of diabetes. However, as mentioned above, tPA-MG53 mice live a healthy lifespan and when crossed with db/db mice, tPA-MG53/db/db mice are indistinguishable from db/db littermates in terms of growth rate, glucose tolerance test, and insulin tolerance test (Bian et al., 2019; Wang et al., 2020). These studies highlight the deep controversies surrounding the protein that will need to be resolved by future studies.
Much is still unknown regarding the physiologic impact of MG53 in skeletal muscle despite the presence of numerous studies involving the protein. MG53 undoubtedly plays an important role in several arenas of physiology. It is involved in calcium homeostasis, muscle contraction, and regeneration. Its role as an agent in plasma membrane repair is uncontested. Emerging roles in mitochondrial protection further highlight the fact that not all is known about the protein. The therapeutic applications of the protein in treating skeletal muscle pathologies are numerous and include the use of the protein as an agent of repair, in providing a treatment for certain types of dystrophies, and in modulating muscle regeneration.
DB-M, JM, and PL designed the manuscript. DB, HZ, WZ, TT, JM, and PL wrote, read, edited, and approved the final version of manuscript. All authors contributed to the article and approved the submitted version.
This work was partially supported by a postdoctoral Ruth L. Kirschstein National Research Service Award to DB-M (2T32AI106704-06) from ARTIST, a K08-GM126315 grant to PL, and R01-AG056919, R01-AR061385, R01-AR070752, R01-DK106394, and R01-HL138570, as well as by DOD grant W81XWH-18-1-0787 to JM.
JM was the founder of TRIM-edicine, Inc., a university spin-off biotechnology company that is developing recombinant MG53 protein as a therapeutic reagent for regenerative medicine. Patents on the use of MG53 are held by Rutgers University and The Ohio State University.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Adesanya, T. M. A., Russell, M., Park, K. H., Zhou, X., Sermersheim, M. A., Gumpper, K., et al. (2019). MG 53 protein protects aortic valve interstitial cells from membrane injury and fibrocalcific remodeling. J. Am. Heart Assoc. 8:e009960. doi: 10.1161/jaha.118.009960
Ahn, M. K., Lee, K. J., Cai, C., Huang, M., Cho, C. H., Ma, J., et al. (2016). Mitsugumin 53 regulates extracellular Ca(2+) entry and intracellular Ca(2+) release via Orai1 and RyR1 in skeletal muscle. Sci. Rep. 6:36909. doi: 10.1038/srep36909
Alloush, J., and Weisleder, N. (2013). TRIM proteins in therapeutic membrane repair of muscular dystrophy. JAMA Neurol. 70, 928–931. doi: 10.1001/jamaneurol.2013.469
Bansal, D., Miyake, K., Vogel, S. S., Groh, S., Chen, C. C., Williamson, R., et al. (2003). Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature 423, 168–172. doi: 10.1038/nature01573
Bian, Z., Wang, Q., Zhou, X., Tan, T., Park, K. H., Kramer, H. F., et al. (2019). Sustained elevation of MG53 in the bloodstream increases tissue regenerative capacity without compromising metabolic function. Nat. Commun. 10:4659. doi: 10.1038/s41467-019-12483-0
Blanco-Bose, W. E., Yao, C. C., Kramer, R. H., and Blau, H. M. (2001). Purification of mouse primary myoblasts based on alpha 7 integrin expression. Exp. Cell Res. 265, 212–220. doi: 10.1006/excr.2001.5191
Buckingham, M. (2007). Skeletal muscle progenitor cells and the role of Pax genes. C. R. Biol. 330, 530–533. doi: 10.1016/j.crvi.2007.03.015
Burkin, D. J., and Wuebbles, R. D. (2012). A molecular bandage for diseased muscle. Sci. Transl. Med. 4:139fs119. doi: 10.1126/scitranslmed.3004082
Cai, C., Masumiya, H., Weisleder, N., Matsuda, N., Nishi, M., Hwang, M., et al. (2009a). MG53 nucleates assembly of cell membrane repair machinery. Nat. Cell Biol. 11, 56–64. doi: 10.1038/ncb1812
Cai, C., Masumiya, H., Weisleder, N., Pan, Z., Nishi, M., Komazaki, S., et al. (2009b). MG53 regulates membrane budding and exocytosis in muscle cells. J. Biol. Chem. 284, 3314–3322. doi: 10.1074/jbc.M808866200
Cai, C., Weisleder, N., Ko, J. K., Komazaki, S., Sunada, Y., Nishi, M., et al. (2009c). Membrane repair defects in muscular dystrophy are linked to altered interaction between MG53, caveolin-3, and dysferlin. J. Biol. Chem. 284, 15894–15902. doi: 10.1074/jbc.M109.009589
Caruso, M., Ma, D., Msallaty, Z., Lewis, M., Seyoum, B., Al-janabi, W., et al. (2014). Increased interaction with insulin receptor substrate 1, a novel abnormality in insulin resistance and type 2 diabetes. Diabetes 63, 1933–1947. doi: 10.2337/db13-1872
Chandler, H. L., Tan, T., Yang, C., Gemensky-Metzler, A. J., Wehrman, R. F., Jiang, Q., et al. (2019). MG53 promotes corneal wound healing and mitigates fibrotic remodeling in rodents. Commun. Biol. 2:71. doi: 10.1038/s42003-019-0316-7
Ciciliot, S., and Schiaffino, S. (2010). Regeneration of mammalian skeletal muscle: basic mechanisms and clinical implications. Curr. Pharmaceutical Design 16, 906–914. doi: 10.2174/138161210790883453
Demonbreun, A. R., and McNally, E. M. (2016). Plasma membrane repair in health and disease. Curr. Top. Membr. 77, 67–96. doi: 10.1016/bs.ctm.2015.10.006
Demonbreun, A. R., Quattrocelli, M., Barefield, D. Y., Allen, M. V., Swanson, K. E., and McNally, E. M. (2016). An actin-dependent annexin complex mediates plasma membrane repair in muscle. J. Cell Biol. 213, 705–718. doi: 10.1083/jcb.201512022
Duann, P., Li, H., Lin, P., Tan, T., Wang, Z., Chen, K., et al. (2015). MG53-mediated cell membrane repair protects against acute kidney injury. Sci. Transl. Med. 7:279ra236. doi: 10.1126/scitranslmed.3010755
Flix, B., De La Torre, C., Castillo, J., Casal, C., Illa, I., and Gallardo, E. (2013). Dysferlin interacts with calsequestrin-1, myomesin-2 and dynein in human skeletal muscle. Int. J. Biochem. Cell Biol. 45, 1927–1938. doi: 10.1016/j.biocel.2013.06.007
Giudice, J., and Taylor, J. M. (2017). Muscle as a paracrine and endocrine organ. Curr. Opin. Pharmacol. 34, 49–55. doi: 10.1016/j.coph.2017.05.005
Glover, L., and Brown, R. H. Jr. (2007). Dysferlin in membrane trafficking and patch repair. Traffic 8, 785–794. doi: 10.1111/j.1600-0854.2007.00573.x
Guan, F., Huang, T., Wang, X., Xing, Q., Gumpper, K., Li, P., et al. (2019a). The TRIM protein Mitsugumin 53 enhances survival and therapeutic efficacy of stem cells in murine traumatic brain injury. Stem Cell Res. Ther. 10:352. doi: 10.1186/s13287-019-1433-4
Guan, F., Zhou, X., Li, P., Wang, Y., Liu, M., Li, F., et al. (2019b). MG53 attenuates lipopolysaccharide-induced neurotoxicity and neuroinflammation via inhibiting TLR4/NF-kappaB pathway in vitro and in vivo. Prog. Neuropsychopharmacol. Biol. Psychiatry 95:109684. doi: 10.1016/j.pnpbp.2019.109684
Ham, Y. M., and Mahoney, S. J. (2013). Compensation of the AKT signaling by ERK signaling in transgenic mice hearts overexpressing TRIM72. Exp. Cell Res. 319, 1451–1462. doi: 10.1016/j.yexcr.2013.02.016
Han, R. (2011). Muscle membrane repair and inflammatory attack in dysferlinopathy. Skelet Muscle 1:10. doi: 10.1186/2044-5040-1-10
Hatakeyama, S. (2017). TRIM family proteins: roles in autophagy, immunity, and carcinogenesis. Trends Biochem. Sci. 42, 297–311. doi: 10.1016/j.tibs.2017.01.002
He, B., Tang, R. H., Weisleder, N., Xiao, B., Yuan, Z., Cai, C., et al. (2012). Enhancing muscle membrane repair by gene delivery of MG53 ameliorates muscular dystrophy and heart failure in delta-Sarcoglycan-deficient hamsters. Mol. Ther. 20, 727–735. doi: 10.1038/mt.2012.5
Hoffmann, C., and Weigert, C. (2017). Skeletal muscle as an endocrine organ: the role of myokines in exercise adaptations. Cold Spring Harb. Perspect. Med. 7:a029793. doi: 10.1101/cshperspect.a029793
Hong, J., Park, J. S., Lee, H., Jeong, J., Hyeon Yun, H., Yun Kim, H., et al. (2016). Myosin heavy chain is stabilized by BCL-2 interacting cell death suppressor (BIS) in skeletal muscle. Exp. Mol. Med. 48:e225. doi: 10.1038/emm.2016.2
Hord, J. M., Botchlett, R., and Lawler, J. M. (2016). Age-related alterations in the sarcolemmal environment are attenuated by lifelong caloric restriction and voluntary exercise. Exp. Gerontol. 83, 148–157. doi: 10.1016/j.exger.2016.08.006
Hu, X., and Xiao, R. P. (2018). MG53 and disordered metabolism in striated muscle. Biochim. Biophys. Acta Mol. Basis Dis. 1864(5 Pt B), 1984–1990. doi: 10.1016/j.bbadis.2017.10.013
Hwang, M., Ko, J. K., Weisleder, N., Takeshima, H., and Ma, J. (2011). Redox-dependent oligomerization through a leucine zipper motif is essential for MG53-mediated cell membrane repair. Am. J. Physiol. Cell Physiol. 301, C106–C114. doi: 10.1152/ajpcell.00382.2010
Iizuka, K., Machida, T., and Hirafuji, M. (2014). Skeletal muscle is an endocrine organ. J. Pharmacol. Sci. 125, 125–131. doi: 10.1254/jphs.14r02cp
Jia, Y., Chen, K., Lin, P., Lieber, G., Nishi, M., Yan, R., et al. (2014). Treatment of acute lung injury by targeting MG53-mediated cell membrane repair. Nat. Commun. 5:4387. doi: 10.1038/ncomms5387
Jung, S. Y., and Ko, Y. G. (2010). TRIM72, a novel negative feedback regulator of myogenesis, is transcriptionally activated by the synergism of MyoD (or myogenin) and MEF2. Biochem. Biophys. Res. Commun. 396, 238–245. doi: 10.1016/j.bbrc.2010.04.072
Karstoft, K., and Pedersen, B. K. (2016). Skeletal muscle as a gene regulatory endocrine organ. Curr. Opin. Clin. Nutr. Metab. Care 19, 270–275. doi: 10.1097/mco.0000000000000283
Laustsen, P. G., Michael, M. D., Crute, B. E., Cohen, S. E., Ueki, K., Kulkarni, R. N., et al. (2002). Lipoatrophic diabetes in Irs1(-/-)/Irs3(-/-) double knockout mice. Genes Dev. 16, 3213–3222. doi: 10.1101/gad.1034802
Lee, C. S., Yi, J. S., Jung, S. Y., Kim, B. W., Lee, N. R., Choo, H. J., et al. (2010). TRIM72 negatively regulates myogenesis via targeting insulin receptor substrate-1. Cell Death Differ 17, 1254–1265. doi: 10.1038/cdd.2010.1
Lee, E. H. (2010). Ca2+ channels and skeletal muscle diseases. Prog. Biophys. Mol. Biol. 103, 35–43. doi: 10.1016/j.pbiomolbio.2010.05.003
Lee, K. J., Park, C. S., Woo, J. S., Kim, D. H., Ma, J., and Lee, E. H. (2012). Mitsugumin 53 attenuates the activity of sarcoplasmic reticulum Ca(2+)-ATPase 1a (SERCA1a) in skeletal muscle. Biochem. Biophys. Res. Commun. 428, 383–388. doi: 10.1016/j.bbrc.2012.10.063
Lek, A., Evesson, F. J., Lemckert, F. A., Redpath, G. M., Lueders, A. K., Turnbull, L., et al. (2013). Calpains, cleaved mini-dysferlinC72, and L-type channels underpin calcium-dependent muscle membrane repair. J. Neurosci. 33, 5085–5094. doi: 10.1523/jneurosci.3560-12.2013
Li, H., Duann, P., Lin, P. H., Zhao, L., Fan, Z., Tan, T., et al. (2015). Modulation of wound healing and scar formation by MG53 protein-mediated cell membrane repair. J. Biol. Chem. 290, 24592–24603. doi: 10.1074/jbc.M115.680074
Lijie, G., Yueyue, Z., Nan, Z., Ling, W., Xuan, W., and Weijie, Y. (2019). Mitsugumin 53 promotes mitochondrial autophagy through regulating Ambra1 expression in C2C12 myoblast cells. Cell Biol. Int. 43, 290–298. doi: 10.1002/cbin.11097
Lin, P., Zhu, H., Cai, C., Wang, X., Cao, C., Xiao, R., et al. (2012). Nonmuscle myosin IIA facilitates vesicle trafficking for MG53-mediated cell membrane repair. Faseb J. 26, 1875–1883. doi: 10.1096/fj.11-188599
Liu, C., Hu, Y. H., Han, Y., Wang, Y. B., Zhang, Y., Zhang, X. Q., et al. (2020). MG53 protects against contrast-induced acute kidney injury by reducing cell membrane damage and apoptosis. Acta Pharmacol. Sin. doi: 10.1038/s41401-020-0420-8 [Epub ahead of print].
Liu, F., Song, R., Feng, Y., Guo, J., Chen, Y., Zhang, Y., et al. (2015). Upregulation of MG53 induces diabetic cardiomyopathy through transcriptional activation of peroxisome proliferation-activated receptor alpha. Circulation 131, 795–804. doi: 10.1161/circulationaha.114.012285
Liu, J., Zhu, H., Zheng, Y., Xu, Z., Li, L., Tan, T., et al. (2015). Cardioprotection of recombinant human MG53 protein in a porcine model of ischemia and reperfusion injury. J. Mol. Cell Cardiol. 80, 10–19. doi: 10.1016/j.yjmcc.2014.12.010
Ma, H., Liu, J., Bian, Z., Cui, Y., Zhou, X., Zhang, B., et al. (2015). Effect of metabolic syndrome on mitsugumin 53 expression and function. PLoS One 10:e0124128. doi: 10.1371/journal.pone.0124128
Ma, L. L., Kong, F. J., Guo, J. J., Zhu, J. B., Shi, H. T., Li, Y., et al. (2017). Hypercholesterolemia abrogates remote ischemic preconditioning-induced cardioprotection: role of reperfusion injury salvage kinase signals. Shock 47, 363–369. doi: 10.1097/shk.0000000000000737
Ma, L. L., Zhang, F. J., Qian, L. B., Kong, F. J., Sun, J. F., Zhou, C., et al. (2013). Hypercholesterolemia blocked sevoflurane-induced cardioprotection against ischemia-reperfusion injury by alteration of the MG53/RISK/GSK3beta signaling. Int. J. Cardiol. 168, 3671–3678. doi: 10.1016/j.ijcard.2013.06.037
Matsuda, C., Miyake, K., Kameyama, K., Keduka, E., Takeshima, H., Imamura, T., et al. (2012). The C2A domain in dysferlin is important for association with MG53 (TRIM72). PLoS Curr. 4:e5035add8caff4. doi: 10.1371/5035add8caff4
McCroskery, S., Thomas, M., Maxwell, L., Sharma, M., and Kambadur, R. (2003). Myostatin negatively regulates satellite cell activation and self-renewal. J. Cell Biol. 162, 1135–1147. doi: 10.1083/jcb.200207056
McNeil, P. (2009). Membrane repair redux: redox of MG53. Nat. Cell Biol. 11, 7–9. doi: 10.1038/ncb0109-7
Mukund, K., and Subramaniam, S. (2020). Skeletal muscle: a review of molecular structure and function, in health and disease. Wiley Interdiscip Rev. Syst. Biol. Med. 12:e1462. doi: 10.1002/wsbm.1462
Nguyen, N., Yi, J. S., Park, H., Lee, J. S., and Ko, Y. G. (2014). Mitsugumin 53 (MG53) ligase ubiquitinates focal adhesion kinase during skeletal myogenesis. J. Biol. Chem. 289, 3209–3216. doi: 10.1074/jbc.M113.525154
Okino, R., Usui, A., Yoneyama, Y., Takahashi, S. I., and Hakuno, F. (2020). Myoblasts with higher IRS-1 levels are eliminated from the normal cell layer during differentiation. Front. Endocrinol. (Lausanne) 11:96. doi: 10.3389/fendo.2020.00096
Ozato, K., Shin, D. M., Chang, T. H., and Morse, H. C. III (2008). TRIM family proteins and their emerging roles in innate immunity. Nat. Rev. Immunol. 8, 849–860. doi: 10.1038/nri2413
Relaix, F., Montarras, D., Zaffran, S., Gayraud-Morel, B., Rocancourt, D., Tajbakhsh, S., et al. (2006). Pax3 and Pax7 have distinct and overlapping functions in adult muscle progenitor cells. J. Cell Biol. 172, 91–102. doi: 10.1083/jcb.200508044
Rocheteau, P., Vinet, M., and Chretien, F. (2015). Dormancy and quiescence of skeletal muscle stem cells. Results Probl. Cell Differ. 56, 215–235. doi: 10.1007/978-3-662-44608-9_10
Sermersheim, M., Lin, P., Kenney, A., McMichael, T., Cai, C., Gumpper, K., et al. (2020). MG53 suppresses interferon-β and inflammation via regulation of ryanodine receptor-mediated intracellular calcium signaling. Nat. Commun. 11:3624.
Shea, K. L., Xiang, W., LaPorta, V. S., Licht, J. D., Keller, C., Basson, M. A., et al. (2010). Sprouty1 regulates reversible quiescence of a self-renewing adult muscle stem cell pool during regeneration. Cell Stem Cell 6, 117–129. doi: 10.1016/j.stem.2009.12.015
Soleimani, V. D., Nguyen, D., Ramachandran, P., Palidwor, G. A., Porter, C. J., Yin, H., et al. (2018). Cis-regulatory determinants of MyoD function. Nucleic Acids Res. 46, 7221–7235. doi: 10.1093/nar/gky388
Song, R., Peng, W., Zhang, Y., Lv, F., Wu, H. K., Guo, J., et al. (2013). Central role of E3 ubiquitin ligase MG53 in insulin resistance and metabolic disorders. Nature 494, 375–379. doi: 10.1038/nature11834
Takeshima, H., Iino, M., Takekura, H., Nishi, M., Kuno, J., Minowa, O., et al. (1994). Excitation-contraction uncoupling and muscular degeneration in mice lacking functional skeletal muscle ryanodine-receptor gene. Nature 369, 556–559. doi: 10.1038/369556a0
Tamemoto, H., Kadowaki, T., Tobe, K., Yagi, T., Sakura, H., Hayakawa, T., et al. (1994). Insulin resistance and growth retardation in mice lacking insulin receptor substrate-1. Nature 372, 182–186. doi: 10.1038/372182a0
Tan, T., Ko, Y. G., and Ma, J. (2016). Dual function of MG53 in membrane repair and insulin signaling. BMB Rep. 49, 414–423. doi: 10.5483/bmbrep.2016.49.8.079
Terauchi, Y., Iwamoto, K., Tamemoto, H., Komeda, K., Ishii, C., Kanazawa, Y., et al. (1997). Development of non-insulin-dependent diabetes mellitus in the double knockout mice with disruption of insulin receptor substrate-1 and beta cell glucokinase genes. Genetic reconstitution of diabetes as a polygenic disease. J. Clin. Invest. 99, 861–866. doi: 10.1172/JCI119250
von Maltzahn, J., Jones, A. E., Parks, R. J., and Rudnicki, M. A. (2013). Pax7 is critical for the normal function of satellite cells in adult skeletal muscle. Proc. Natl. Acad. Sci. U.S.A. 110, 16474–16479. doi: 10.1073/pnas.1307680110
Wang, Q., Bian, Z., Jiang, Q., Wang, X., Zhou, X., Park, K. H., et al. (2020). MG53 does not manifest the development of diabetes in db/db mice. Diabetes 69, 1052–1064. doi: 10.2337/db19-0807
Weisleder, N., Takeshima, H., and Ma, J. (2008). Immuno-proteomic approach to excitation–contraction coupling in skeletal and cardiac muscle: molecular insights revealed by the mitsugumins. Cell Calcium 43, 1–8. doi: 10.1016/j.ceca.2007.10.006
Weisleder, N., Takeshima, H., and Ma, J. (2009). Mitsugumin 53 (MG53) facilitates vesicle trafficking in striated muscle to contribute to cell membrane repair. Commun. Integr. Biol. 2, 225–226. doi: 10.4161/cib.2.3.8077
Weisleder, N., Takizawa, N., Lin, P., Wang, X., Cao, C., Zhang, Y., et al. (2012). Recombinant MG53 protein modulates therapeutic cell membrane repair in treatment of muscular dystrophy. Sci. Transl. Med. 4:139ra185. doi: 10.1126/scitranslmed.3003921
Wu, H. K., Zhang, Y., Cao, C. M., Hu, X., Fang, M., Yao, Y., et al. (2019). Glucose-sensitive myokine/cardiokine MG53 regulates systemic insulin response and metabolic homeostasis. Circulation 139, 901–914. doi: 10.1161/circulationaha.118.037216
Xu, Y., Ma, L. L., Zhou, C., Zhang, F. J., Kong, F. J., Wang, W. N., et al. (2013). Hypercholesterolemic myocardium is vulnerable to ischemia-reperfusion injury and refractory to sevoflurane-induced protection. PLoS One 8:e76652. doi: 10.1371/journal.pone.0076652
Yao, Y., Zhang, B., Zhu, H., Li, H., Han, Y., Chen, K., et al. (2016). MG53 permeates through blood-brain barrier to protect ischemic brain injury. Oncotarget 7, 22474–22485. doi: 10.18632/oncotarget.7965
Yi, J. S., Park, J. S., Ham, Y. M., Nguyen, N., Lee, N. R., Hong, J., et al. (2013). MG53-induced IRS-1 ubiquitination negatively regulates skeletal myogenesis and insulin signalling. Nat. Commun. 4:2354. doi: 10.1038/ncomms3354
Yuan, H., Niu, Y., Liu, X., Yang, F., Niu, W., and Fu, L. (2013). Proteomic analysis of skeletal muscle in insulin-resistant mice: response to 6-week aerobic exercise. PLoS One 8:e53887. doi: 10.1371/journal.pone.0053887
Zabielski, P., Lanza, I. R., Gopala, S., Heppelmann, C. J., Bergen, H. R. III, Dasari, S., et al. (2016). Altered skeletal muscle mitochondrial proteome as the basis of disruption of mitochondrial function in diabetic mice. Diabetes 65, 561–573. doi: 10.2337/db15-0823
Zhang, Y., Wu, H. K., Lv, F., and Xiao, R. P. (2017). MG53: biological function and potential as a therapeutic target. Mol. Pharmacol. 92, 211–218. doi: 10.1124/mol.117.108241
Zhu, H., Hou, J., Roe, J. L., Park, K. H., Tan, T., Zheng, Y., et al. (2015). Amelioration of ischemia-reperfusion-induced muscle injury by the recombinant human MG53 protein. Muscle Nerve 52, 852–858. doi: 10.1002/mus.24619
Keywords: metabolic syndrome, skeletal muscle regeneration, myogenesis, insulin resistance, mitochondria, calcium homeostasis, plasma membrane repair, TRIM72
Citation: Benissan-Messan DZ, Zhu H, Zhong W, Tan T, Ma J and Lee PHU (2020) Multi-Cellular Functions of MG53 in Muscle Calcium Signaling and Regeneration. Front. Physiol. 11:583393. doi: 10.3389/fphys.2020.583393
Received: 14 July 2020; Accepted: 09 October 2020;
Published: 06 November 2020.
Edited by:
Matias Mosqueira, Heidelberg University Hospital, GermanyReviewed by:
Margaret Westfall, University of Michigan, United StatesCopyright © 2020 Benissan-Messan, Zhu, Zhong, Tan, Ma and Lee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianjie Ma, SmlhbmppZS5NYUBvc3VtYy5lZHU=; Peter H. U. Lee, UGV0ZXJfTGVlQGJyb3duLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.