
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Physiol. , 03 July 2019
Sec. Red Blood Cell Physiology
Volume 10 - 2019 | https://doi.org/10.3389/fphys.2019.00815
This article is part of the Research Topic Pathophysiology of Rare Hemolytic Anemias View all 15 articles
 Satheesh Chonat1,2
Satheesh Chonat1,2 Mary Risinger3
Mary Risinger3 Haripriya Sakthivel4
Haripriya Sakthivel4 Omar Niss4,5
Omar Niss4,5 Jennifer A. Rothman6
Jennifer A. Rothman6 Loan Hsieh7
Loan Hsieh7 Stella T. Chou8,9Janet L. Kwiatkowski8,9Eugene Khandros8,9
Stella T. Chou8,9Janet L. Kwiatkowski8,9Eugene Khandros8,9 Matthew F. Gorman10Donald T. Wells11Tamara Maghathe4Neha Dagaonkar12
Matthew F. Gorman10Donald T. Wells11Tamara Maghathe4Neha Dagaonkar12 Katie G. Seu4
Katie G. Seu4 Kejian Zhang13
Kejian Zhang13 Wenying Zhang5,14
Wenying Zhang5,14 Theodosia A. Kalfa4,5*
Theodosia A. Kalfa4,5*Hereditary spherocytosis (HS) is the most common red blood cell (RBC) membrane disorder causing hereditary hemolytic anemia. Patients with HS have defects in the genes coding for ankyrin (ANK1), band 3 (SLC4A1), protein 4.2 (EPB42), and α (SPTA1) or β-spectrin (SPTB). Severe recessive HS is most commonly due to biallelic SPTA1 mutations. α-spectrin is produced in excess in normal erythroid cells, therefore SPTA1-associated HS ensues with mutations causing significant decrease of normal protein expression from both alleles. In this study, we systematically compared genetic, rheological, and protein expression data to the varying clinical presentation in eleven patients with SPTA1-associated HS. The phenotype of HS in this group of patients ranged from moderately severe to severe transfusion-dependent anemia and up to hydrops fetalis which is typically fatal if transfusions are not initiated before term delivery. The pathogenicity of the mutations could be corroborated by reduced SPTA1 mRNA expression in the patients’ reticulocytes. The disease severity correlated to the level of α-spectrin protein in their RBC cytoskeleton but was also affected by other factors. Patients carrying the low expression αLEPRA allele in trans to a null SPTA1 mutation were not all transfusion dependent and their anemia improved or resolved with partial or total splenectomy, respectively. In contrast, patients with near-complete or complete α-spectrin deficiency have a history of having been salvaged from fatal hydrops fetalis, either because they were born prematurely and started transfusions early or because they had intrauterine transfusions. They have suboptimal reticulocytosis or reticulocytopenia and remain transfusion dependent even after splenectomy; these patients require either lifetime transfusions and iron chelation or stem cell transplant. Comprehensive genetic and phenotypic evaluation is critical to provide accurate diagnosis in patients with SPTA1-associated HS and guide toward appropriate management.
Hereditary spherocytosis (HS) is the most common red blood cell (RBC) cytoskeleton disorder causing hereditary hemolytic anemia (HHA), characterized by sphere-shaped erythrocytes (spherocytes) with increased osmotic fragility. HS can affect all ethnic groups but is more common in people of northern European ancestry where the prevalence is 1 in 1000–2500 (Gallagher, 2005). Spherocytes are formed because of loss of membrane due to quantitative defects in proteins that link the cytoskeleton to the lipid bilayer (“vertical” linkages) (Eber and Lux, 2004). The scaffolding network of the RBC cytoskeleton is assembled by α- and β-spectrin heterodimers self-associating in a head-to-head fashion to form tetramers, bound to the lipid membrane via the anchoring complex of ankyrin, protein 4.2, and band 3 (Salomao et al., 2008). In autosomal dominant HS, which accounts for approximately 75% of cases, mutations of ankyrin (ANK1), band 3 (SLC4A1), and β-spectrin (SPTB) genes predominate. Recessive HS is most often due to compound heterozygosity for defects in the genes encoding ankyrin, α-spectrin (SPTA1), or protein 4.2 (EPB42) (Eber and Lux, 2004; Gallagher, 2005; Mohandas, 2018).
Two normal SPTA1 alleles allow for overproduction of α-spectrin chains (Hanspal and Palek, 1987). Therefore, HS due to α-spectrin deficiency manifests when both of the SPTA1 alleles are affected by mutations causing significant quantitative defect. Two SPTA1 low expression alleles were identified early-on to be associated with RBC membrane disorders and their study helped to determine the quantitative requirements of the RBC cytoskeleton for α-spectrin (Wilmotte et al., 1993; Wichterle et al., 1996). αLELY (Low Expression LYon) has a minor allele frequency (MAF) of 25.5% (gnomad.broadinstitute.org) and consists of the mutation c.6531-12C>T in intron 45, causing partial skipping of exon 46 in half of the transcripts and consequently a 50% decrease in the amount of α-spectrin (Wilmotte et al., 1993; Marechal et al., 1995). αLELY in trans to an SPTA1 allele with a hereditary elliptocytosis (HE)-associated mutation modifies the phenotype from HE to hereditary pyropoikilocytosis (Niss et al., 2016). In contrast, αLELY in trans to a null SPTA1 allele causes no disease, indicating that production of ∼25% of normal α-spectrin is enough for normal RBC cytoskeleton assembly (Delaunay et al., 2004). αLEPRA (Low Expression PRAgue) is a deep intronic SPTA1 mutation (c.4339-99C > T). Positioned at -99 of intron 30, it activates an alternative acceptor splice site at position -70 of the same intron. The alternative splicing results in frameshift and premature termination of translation, leading to decreased α-spectrin production. This allele (MAF of 0.5% per gnomad.broadinstitute.org) produces only about 16% of full-length spectrin as compared to the normal SPTA1 allele, based on studies with metabolic labeling of erythroblasts in vitro (Wichterle et al., 1996). αLEPRA in trans to a null SPTA1 allele (leading to a total α-spectrin production of about 8%) has been shown to cause severe autosomal recessive HS, with anemia and jaundice that resolve with splenectomy (Wichterle et al., 1996; Delaunay et al., 2004). Complete α-spectrin deficiency has been shown to cause lethal anemia in utero (Whitfield et al., 1991).
We present here eleven patients with HS due to α-spectrin deficiency and discuss their phenotype/genotype correlation (Table 1).

Table 1. Genetic mutations and associated phenotype in HS due to SPTA1 mutations.
Patients with the clinical diagnosis of HHA and their parents were enrolled in an Institutional Review Board-approved research protocol based at Cincinnati Children’s Hospital Medical Center. DNA was isolated from peripheral blood (in the case of patient 9 from liver tissue preserved in paraffin after autopsy), and analyzed on an NGS HHA panel; the regions of interest for enrichment and DNA sequencing included the coding exons plus 20 bases of intronic boundaries for 32 genes known to be associated with RBC membrane and enzyme disorders and with congenital dyserythropoietic anemias: ABCG5, ABCG8, AK1, ALDOA, ANK1, C15orf41, CDAN1, EPB41, EPB42, G6PD, GATA1, GCLC, GPI, GPX1, GSR, GSS, HK1, KIF23, KLF1, NT5C3A, PFKM, PGK1, PIEZO1, PKLR, RHAG, SEC23B, SLC2A1(GLUT1), SLC4A1, SPTA1, SPTB, TPI1, and XK. Regulatory regions and deep intronic areas of these genes with published disease-causing mutations were included in the HHA panel design. Sanger sequencing was used to confirm all mutations found in patients and in parental samples (except for parents of patients #7 and #11) to establish the phase.
Whole blood samples were collected in K2-EDTA-containing vials from study subjects at least 3 months after last transfusion to avoid misinterpretation from the presence of donor RBCs. Samples from healthy volunteers were collected at same time and shipped as travel controls. Specimens were stored or shipped at 4°C and were analyzed within 24 h of sample collection using LoRRca® MaxSis (Mechatronics, United States LLC, Warwick, RI, United States). RBC deformation was recorded while the cells were exposed to a constant shear stress of 30 Pa and an increasing osmotic gradient (0–600 mOsm/kg) in order to generate the ektacytometry curve (Da Costa et al., 2016; Zaninoni et al., 2018).
Red blood cell membrane “ghosts” were prepared from patients #1, #6, and #7, at least 3 months after last transfusion, by hypotonic lysis (Bennett, 1983) and cytoskeletal proteins were evaluated by immunodetection using the size-based capillary electrophoresis instrument Wes (ProteinSimple, San Jose, CA, United States). Capillary immuno-electrophoresis was also performed on lysates prepared from isolated CD71+ cells from patient #10 (with a similarly prepared healthy volunteer sample as control).
Reticulocytes were magnetically isolated from whole blood collected in K2-EDTA-containing vials from patients #4 and #10, in order to validate the pathogenicity of their genotype findings, using anti-CD71 microbeads and positive selection through an AutoMACS separator (Miltenyi Biotec). A reticulocyte sample from patient 1 was prepared similarly, to be used as positive control. RNA was isolated using the QiaAmp RNA Blood Mini kit (QIAGEN) and reverse transcription was performed using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). SPTA1 mRNA expression level was determined by qPCR using a StepOnePlus Real-Time PCR System (Applied Biosystems) and FAM-labeled Taqman probes (Thermo Fisher Scientific) for SPTA1 spanning exons 48–49 (Assay ID Hs01005878_m1) and ACTB (Assay ID Hs01060665_g1) as a reference gene.
We observed three different phenotypes in patients with recessive HS due to biallelic SPTA1 mutations, verified to be in trans by parental targeted sequencing (Table 1). The heterozygous parents did not have any evidence of hemolytic anemia as expected, since even αLELY in trans to a null SPTA1 allele, producing a total of ∼25% of normal α-spectrin, causes no disease (Delaunay et al., 2004).
The first four patients listed in Table 1 (within Group I) had the well-known phenotype of SPTA1-associated HS, i.e., severe, transfusion-dependent HHA with brisk reticulocytosis (Agre et al., 1982; Wichterle et al., 1996). These patients presented with hyperbilirubinemia and non-immune hemolytic anemia soon after birth, requiring their first transfusion as neonates, before any meaningful RBC phenotypical testing could be obtained. They continued to require frequent transfusions, precluding any further erythrocyte phenotype work-up until splenectomy. Patients 1–3 had a SPTA1 nonsense mutation (expected to lead to nonsense-mediated decay of the transcript) in trans to the low expression αLEPRA mutation (c.4339-99C > T). Patient 1 required monthly transfusions for hemoglobin (Hgb) in the range of 6.4–7.5 g/dL with absolute reticulocyte count (ARC) of 130–360 × 109/L for the first four years of life, with consequent iron overload, requiring chelation treatment with deferasirox. After molecular diagnosis of SPTA1-associated HS utilizing NGS of the HHA panel, he underwent partial splenectomy at 4 years of age. He was able to remain transfusion-free for 18 months with Hgb 8.7–11 g/dL, but then his anemia gradually worsened with increasing transfusion requirement. He had a follow-up total splenectomy at 7 years of age; at that time the remaining splenic tissue had been impressively regrown to 435 g, based on the pathology report. The patient has had no further transfusion requirement for the past 4 years, with Hgb now in 12.7–15.8 g/dL range. Blood smear from a non-transfused blood sample after splenectomy shows many spherocytes and moderate poikilocytosis, as expected with α-spectrin deficiency (Eber and Lux, 2004), and ektacytometry reveals the typical HS curve (Da Costa et al., 2016; Figure 1A). Patient 2 had a similar course for the first 2.5 years. After molecular diagnosis of SPTA1-associated HS, he underwent partial splenectomy and he remains transfusion-free since then with Hgb in the range of 9–10.8 g/dL and minimal regrowth of the remaining splenic tissue based on ultrasound (108–123 ml). Patient 3 has not yet had splenectomy due to parental preference and continues to require frequent transfusions.
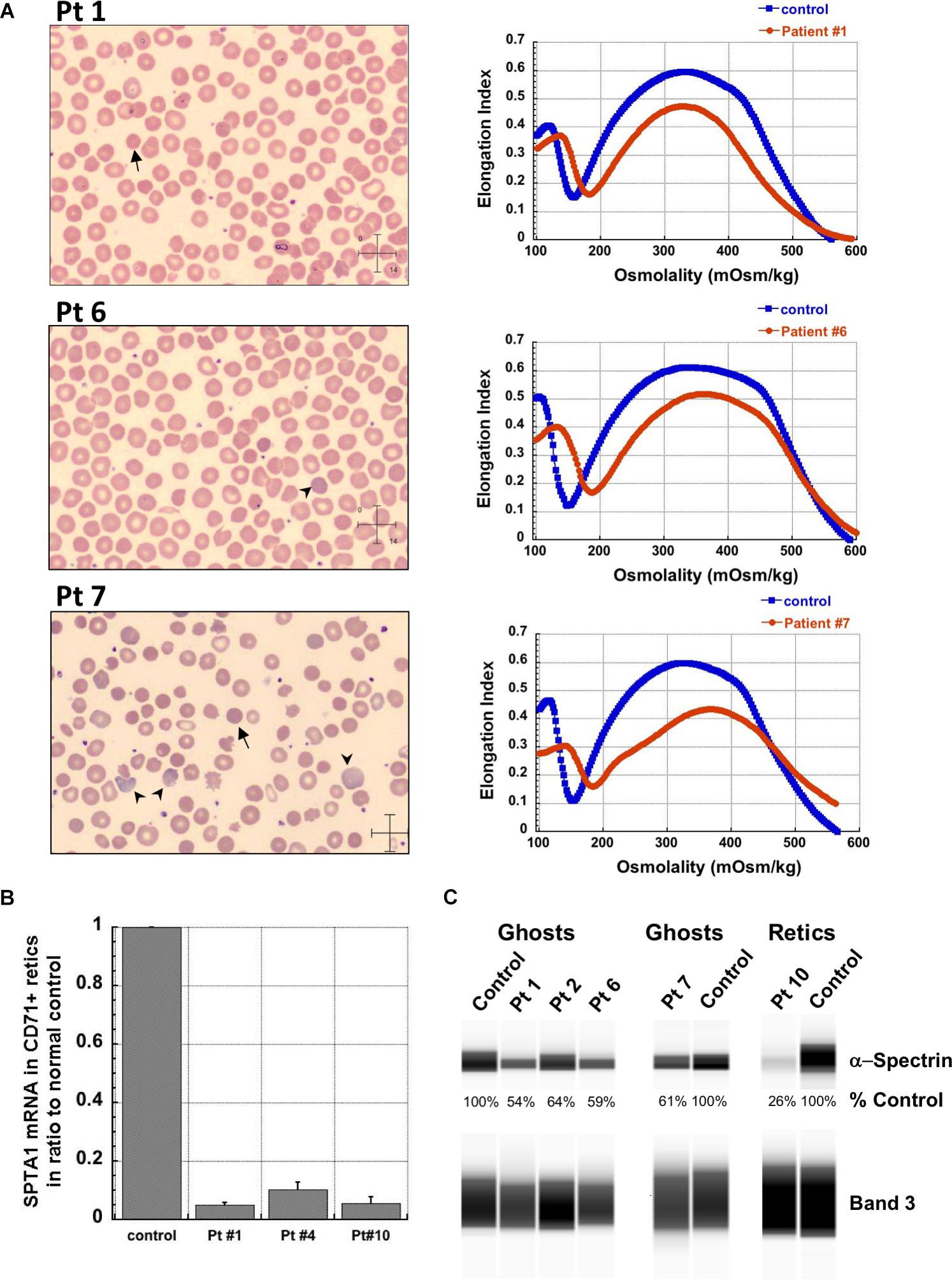
Figure 1. Studies in peripheral blood of patients with SPTA1-associated HS. (A) Peripheral blood smears on the left from patients 1, 6 and 7 showing multiple spherocytes (arrows) lacking central pallor due to decreased surface area to volume ratio and aniso-poikilocytosis. Patient 7 also has polychromasia with increased reticulocytes (arrowheads) indicating significant hemolysis prior to splenectomy. Ektacytometry, on the right, demonstrates the typical HS curve for the patients (red) vs. control (blue). HS is characterized by increased Omin indicating decreased RBC surface to volume ratio and decreased EImax (maximum Elongation Index) which depends mostly on the cytoskeleton mechanics. Frequently, the declining portion of the curve (represented by Ohyp, the osmolality value where the cells are at half of the maximum elongation) is also decreased (as in patient 1) indicating increased intracellular viscosity, however, it may also be normal as in patients 6 and 7 (Clark et al., 1983; Zaninoni et al., 2018). (B) qPCR in RNA isolated from patients’ reticulocytes demonstrated severely decreased α-spectrin expression. Patient 1 who has αLEPRA in trans to a null SPTA1 mutation was found to express α-spectrin at about 5% in comparison to normal control at levels similar to the original calculations for αLEPRA (about 16% of full-length spectrin as compared to the normal SPTA1 allele based on studies with metabolic labeling of erythroblasts in vitro (Wichterle et al., 1996), and therefore a total of 8% α-spectrin when αLEPRA is in trans to a null allele). qPCR appears to be a useful diagnostic assay when an unknown low-expression allele is suspected in trans to a null SPTA1 mutation or deletion in a disease suspected to be severe HS, such as the case of patient 4. (C) Quantitation of α-spectrin/band 3 ratio in RBC ghosts (Patients 1, 2, 6, and 7) and reticulocytes (Patient 10) expressed as percent of α-spectrin/band 3 ratio in corresponding normal control samples by immunodetection using size-based capillary electrophoresis. Agre et al. (1985) in one of the first descriptions of HS due to α-spectrin deficiency noted that the clinical severity of anemia was proportional to a degree of spectrin deficiency, ranging from 53% of normal spectrin content in severely anemic patients to 31% of normal in nearly lethal cases.
Patient 4 had a similar clinical presentation but NGS for the HHA-panel was negative. Deletion/duplication assay by Comparative Genomic Hybridization was performed and identified a heterozygous deletion at 1q23.1 involving the SPTA1 gene. No mutation was identified in trans. Since the patient remained transfusion dependent precluding phenotypical evaluation of his RBCs, α-spectrin mRNA expression in his reticulocytes was evaluated by qPCR and was found decreased at levels comparable to patient 1, confirming SPTA1-associated recessive HS (Figure 1B). Hgb normalized at ∼13 g/dL with no need for further transfusions post total splenectomy at 3 years of age.
Regrowth of the splenic remnant after partial splenectomy is a possibility, occasionally requiring a second surgery. Nevertheless, in both patients 1 and 2 who had splenectomy before 5 years of age in order to limit the transfusional iron overload, partial splenectomy was a reasonable choice with the goal to preserve splenic immune function (Englum et al., 2016). The different response of these two patients to partial splenectomy was most likely due to difference in splenic regrowth, rather than a difference in phenotypic severity. α-spectrin in the erythrocyte ghosts of patient 1 was determined by immunoelectrophoresis to be 54% vs. 64% for patient 2 in comparison to normal control (Figure 1C).
Surprisingly, we identified four patients (patients 5–8 in Table 1, comprising Group II) who, despite having equivalent genotype with the first four patients of a null SPTA1 mutation in trans to αLEPRA, appeared to have a milder phenotype, ranging from improvement in transfusion requirement after frequent transfusions in the first 2–3 years of life (patients 5 and 6) to well compensated hemolysis (patient 8). The milder severity of the disease did not seem to correlate with RBC osmotic fragility or deformability based on ektacytometry (Figure 1A) or the α-spectrin quantitation in RBC ghosts which was only slightly lower compared to the α-spectrin level in patient 2 (Figure 1C). Additional gene polymorphisms and mutations found in the HHA panel (Supplementary Table S1) did not explain the milder phenotype noted in the patients of Group II versus Group I. Heterogeneity in the level of normal α-spectrin expression from the α-LEPRA allele (c.4339-99C > T), variability in HIF-pathway, and erythropoietin response, and sometimes a different tolerance to anemia by patients and/or parents may be contributing to differences in the phenotype between Group I and Group II patients.
The third phenotype (Group III in Table 1) associated with HS due to near-complete or complete α-spectrin deficiency is less well known since it has been typically embryonal lethal causing fatal hydrops fetalis in the third trimester of pregnancy or perinatally, such as the case of patient 9. His parents requested genetic counseling after having a fetus and a newborn child die with the clinical picture of hydrops fetalis. Both parents were found to carry a frameshift SPTA1 mutation. DNA isolated from the patient’s liver tissue, preserved in paraffin after autopsy, was sequenced and revealed that the patient was compound heterozygous for these SPTA1 mutations, predicting absence of normal α-spectrin production. With the progress of fetal medicine allowing prenatal diagnosis of severe anemia and in utero transfusions, more of these patients are now surviving to term and the disease is increasingly recognized.
Patient 10 was born prematurely at 33 weeks of gestation with severe non-immune hemolytic anemia requiring transfusion soon after birth. He remained transfusion dependent and total splenectomy did not significantly improve his hemolytic anemia. NGS for the HHA-panel revealed homozygosity for an intronic SPTA1 variant, likely causing alternative splicing and severely decreased expression of α-spectrin, as verified by qPCR (Figure 1B) and immunoelectrophoresis performed in lysates prepared from isolated reticulocytes. The patient is now doing well after bone marrow transplant. Patient 11, an infant of Amish-Mennonite origin required five in-utero transfusions for severe anemia and had significant neonatal hyperbilirubinemia at birth requiring exchange transfusion. HHA gene panel revealed homozygosity for a SPTA1 frameshift mutation, explaining her severe reticulocytopenia since her reticulocytes with complete α-spectrin deficiency are extremely fragile, failing to survive the shear stress in circulation.
The genetic and phenotypic information gathered from our patient cohort demonstrates that patients with recessive SPTA1-associated HS due to the low expression SPTA1 variant αLEPRA in trans to a null SPTA1 mutation respond to either partial or total splenectomy by significant improvement or resolution of anemia, respectively. In contrast, patients with near complete or complete α-spectrin deficiency because of biallelic null or rare intronic SPTA1 mutations that result in severely decreased expression of the protein, are unlikely to have a measurable response to splenectomy and require either lifetime transfusions and iron chelation or stem cell transplant. This group of patients have typically reticulocytopenia and history of having been salvaged from fatal hydrops fetalis, either because they were born prematurely and started transfusions early or because they had intrauterine transfusions.
Next generation sequencing panels are robust and rapid diagnostic tools for HHA, especially when frequent transfusions preclude phenotypic evaluation of the patients’ RBCs. Specialized assays such as qPCR of reticulocyte mRNA can be used for verification of the diagnosis. Comprehensive genetic and phenotypic evaluation is critical to provide insights into the variable phenotypes of patients with HHA and guide toward appropriate management.
The manuscript is a retrospective case series, with no patient identifiable information, performed under two IRB-approved protocols in Cincinnati Children’s Hospital Medical Center (CCHMC) for diagnostic, non-interventional studies of Congenital Hemolytic and Dyserythropoietic Anemias: Study #2011-1510 and Study #2015-6150. Patients and families were consented either in person in CCHMC hematology clinic or over the phone in the presence of a witness.
SC, MR, KZ, WZ, and TK contributed to the conception and design of the study. SC, MR, HS, TM, ND, and KS performed the experiments. SC, ON, JR, LH, STC, JK, EK, MG, DW, and TK provided the clinical information. TK organized the database. SC and TK wrote the first draft of the manuscript. MR, HS, and KS wrote sections of the manuscript. All authors revised, read, and approved the submitted version of the manuscript.
This work was supported by the National Center for Advancing Translational Sciences of the National Institutes of Health, under the award number 1UL1TR001425-01. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We are grateful to the CCHMC hematology clinical research support team, patients, and families for their enthusiastic participation in this study.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2019.00815/full#supplementary-material
Agre, P., Casella, J. F., Zinkham, W. H., McMillan, C., and Bennett, V. (1985). Partial deficiency of erythrocyte spectrin in hereditary spherocytosis. Nature 314, 380–383. doi: 10.1038/314380a0
Agre, P., Orringer, E. P., and Bennett, V. (1982). Deficient red-cell spectrin in severe, recessively inherited spherocytosis. N. Engl. J. Med. 306, 1155–1161. doi: 10.1056/NEJM198205133061906
Bennett, V. (1983). Proteins involved in membrane–cytoskeleton association in human erythrocytes: spectrin, ankyrin, and band 3. Meth. Enzymol. 96, 313–324. doi: 10.1016/s0076-6879(83)96029-9
Bogardus, H., Schulz, V. P., Maksimova, Y., Miller, B. A., Li, P., Forget, B. G., et al. (2014). Severe nondominant hereditary spherocytosis due to uniparental isodisomy at the SPTA1 locus. Haematologica 99, e168–e170. doi: 10.3324/haematol.2014.110312
Clark, M. R., Mohandas, N., and Shohet, S. B. (1983). Osmotic gradient ektacytometry: comprehensive characterization of red cell volume and surface maintenance. Blood 61, 899–910.
Da Costa, L., Suner, L., Galimand, J., Bonnel, A., Pascreau, T., Couque, N., et al. (2016). Diagnostic tool for red blood cell membrane disorders: assessment of a new generation ektacytometer. Blood Cells Mol. Dis. 56, 9–22. doi: 10.1016/j.bcmd.2015.09.001
Delaunay, J., Nouyrigat, V., Proust, A., Schischmanoff, P. O., Cynober, T., Yvart, J., et al. (2004). Different impacts of alleles alphaLEPRA and alphaLELY as assessed versus a novel, virtually null allele of the SPTA1 gene in trans. Br. J. Haematol. 127, 118–122. doi: 10.1111/j.1365-2141.2004.05160.x
Eber, S., and Lux, S. E. (2004). Hereditary spherocytosis–defects in proteins that connect the membrane skeleton to the lipid bilayer. Semin Hematol. 41, 118–141. doi: 10.1053/j.seminhematol.2004.01.002
Englum, B. R., Rothman, J., Leonard, S., Reiter, A., Thornburg, C., Brindle, M., et al. (2016). Hematologic outcomes after total splenectomy and partial splenectomy for congenital hemolytic anemia. J. Pediatr. Surg. 51, 122–127. doi: 10.1016/j.jpedsurg.2015.10.028
Gallagher, P. G. (2005). Red cell membrane disorders. Hematology Am. Soc. Hematol. Educ. Program 2005, 13–18. doi: 10.1182/asheducation-2005.1.13
Hanspal, M., and Palek, J. (1987). Synthesis and assembly of membrane skeletal proteins in mammalian red cell precursors. J. Cell Biol. 105, 1417–1424. doi: 10.1083/jcb.105.3.1417
Marechal, J., Wilmotte, R., Kanzaki, A., Dhermy, D., Garbarz, M., Galand, C., et al. (1995). Ethnic distribution of allele alpha LELY, a low-expression allele of red-cell spectrin alpha-gene. Br. J. Haematol. 90, 553–556. doi: 10.1111/j.1365-2141.1995.tb05583.x
Mohandas, N. (2018). Inherited hemolytic anemia: a possessive beginner’s guide. Hematology Am. Soc. Hematol. Educ. Program 2018, 377–381. doi: 10.1182/asheducation-2018.1.377
Niss, O., Chonat, S., Dagaonkar, N., Almansoori, M. O., Kerr, K., Rogers, Z. R., et al. (2016). Genotype-phenotype correlations in hereditary elliptocytosis and hereditary pyropoikilocytosis. Blood Cells Mol. Dis. 61, 4–9. doi: 10.1016/j.bcmd.2016.07.003
Salomao, M., Zhang, X., Yang, Y., Lee, S., Hartwig, J. H., Chasis, J. A., et al. (2008). Protein 4.1R-dependent multiprotein complex: new insights into the structural organization of the red blood cell membrane. Proc. Natl. Acad. Sci. U.S.A. 105, 8026–8031. doi: 10.1073/pnas.0803225105
Whitfield, C. F., Follweiler, J. B., Lopresti-Morrow, L., and Miller, B. A. (1991). Deficiency of alpha-spectrin synthesis in burst-forming units-erythroid in lethal hereditary spherocytosis. Blood 78, 3043–3051.
Wichterle, H., Hanspal, M., Palek, J., and Jarolim, P. (1996). Combination of two mutant alpha spectrin alleles underlies a severe spherocytic hemolytic anemia. J. Clin. Invest. 98, 2300–2307. doi: 10.1172/JCI119041
Wilmotte, R., Marechal, J., Morle, L., Baklouti, F., Philippe, N., Kastally, R., et al. (1993). Low expression allele alpha LELY of red cell spectrin is associated with mutations in exon 40 (alpha V/41 polymorphism) and intron 45 and with partial skipping of exon 46. J. Clin. Invest. 91, 2091–2096. doi: 10.1172/JCI116432
Zaninoni, A., Fermo, E., Vercellati, C., Consonni, D., Marcello, A. P., Zanella, A., et al. (2018). Use of laser assisted optical rotational cell analyzer (lorrca maxsis) in the diagnosis of rbc membrane disorders, enzyme defects, and congenital dyserythropoietic anemias: a monocentric study on 202 patients. Front. Physiol. 9:451. doi: 10.3389/fphys.2018.00451
Keywords: SPTA1, α-spectrin, αLEPRA, hereditary spherocytosis, next generation sequencing, hemolytic anemia, hydrops fetalis
Citation: Chonat S, Risinger M, Sakthivel H, Niss O, Rothman JA, Hsieh L, Chou ST, Kwiatkowski JL, Khandros E, Gorman MF, Wells DT, Maghathe T, Dagaonkar N, Seu KG, Zhang K, Zhang W and Kalfa TA (2019) The Spectrum of SPTA1-Associated Hereditary Spherocytosis. Front. Physiol. 10:815. doi: 10.3389/fphys.2019.00815
Received: 01 February 2019; Accepted: 11 June 2019;
Published: 03 July 2019.
Edited by:
Paola Bianchi, IRCCS Ca ’Granda Foundation Maggiore Policlinico Hospital, ItalyReviewed by:
Archana Mishra Agarwal, University of Utah Hospital, United StatesCopyright © 2019 Chonat, Risinger, Sakthivel, Niss, Rothman, Hsieh, Chou, Kwiatkowski, Khandros, Gorman, Wells, Maghathe, Dagaonkar, Seu, Zhang, Zhang and Kalfa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Theodosia A. Kalfa, dGhlb2Rvc2lhLmthbGZhQGNjaG1jLm9yZw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.