 Tim Ott1Lilian Kaufmann2Martin Granzow2Katrin Hinderhofer2Claus R. Bartram2Susanne Theiß2Angelika Seitz3
Tim Ott1Lilian Kaufmann2Martin Granzow2Katrin Hinderhofer2Claus R. Bartram2Susanne Theiß2Angelika Seitz3 Nagarajan Paramasivam4,5
Nagarajan Paramasivam4,5 Angela Schulz6Ute Moog2
Angela Schulz6Ute Moog2 Martin Blum1†
Martin Blum1† Christina M. Evers2*†
Christina M. Evers2*†- 1Institute of Zoology, University of Hohenheim, Stuttgart, Germany
- 2Institute of Human Genetics, Heidelberg University, Heidelberg, Germany
- 3Department of Neuroradiology, University Hospital Heidelberg, Heidelberg, Germany
- 4Medical Faculty Heidelberg, Heidelberg University, Heidelberg, Germany
- 5Division of Theoretical Bioinformatics, German Cancer Research Center (DKFZ), Heidelberg, Germany
- 6Genomics & Proteomics Core Facility, German Cancer Research Center (DKFZ), Heidelberg, Germany
Joubert syndrome (JS) is a congenital autosomal-recessive or—in rare cases–X-linked inherited disease. The diagnostic hallmark of the so-called molar tooth sign describes the morphological manifestation of the mid- and hind-brain in axial brain scans. Affected individuals show delayed development, intellectual disability, ataxia, hyperpnea, sleep apnea, abnormal eye, and tongue movements as well as hypotonia. At the cellular level, JS is associated with the compromised biogenesis of sensory cilia, which identifies JS as a member of the large group of ciliopathies. Here we report on the identification of novel compound heterozygous variants (p.Y503C and p.Q485*) in the centrosomal gene PIBF1 in a patient with JS via trio whole exome sequencing. We have studied the underlying disease mechanism in the frog Xenopus, which offers fast assessment of cilia functions in a number of embryological contexts. Morpholino oligomer (MO) mediated knockdown of the orthologous Xenopus pibf1 gene resulted in defective mucociliary clearance in the larval epidermis, due to reduced cilia numbers and motility on multiciliated cells. To functionally assess patient alleles, mutations were analyzed in the larval skin: the p.Q485* nonsense mutation resulted in a disturbed localization of PIBF1 to the ciliary base. This mutant failed to rescue the ciliation phenotype following knockdown of endogenous pibf1. In contrast, the missense variant p.Y503C resulted in attenuated rescue capacity compared to the wild type allele. Based on these results, we conclude that in the case of this patient, JS is the result of a pathogenic combination of an amorphic and a hypomorphic PIBF1 allele. Our study underscores the versatility of the Xenopus model to study ciliopathies such as JS in a rapid and cost-effective manner, which should render this animal model attractive for future studies of human ciliopathies.
Introduction
Joubert syndrome (JS, OMIM # 213300) comprises a group of autosomal recessive or X-linked inherited disorders with a distinct cerebellar and brainstem malformation recognizable on brain imaging, the “molar tooth sign.” The typical brain malformation of JS patients gives their midbrain an appearance reminiscent of a molar or wisdom tooth on axial MRI (Figures 1E–H). The “molar tooth” appearance results from three anatomical abnormalities of brainstem and cerebellum: (a) an abnormally deep “interpeduncular fossa,” (b) prominent, thickened, and elongated “superior cerebellar peduncles,” and (c) absence or hypoplasia of the midline portion of the cerebellum, the “cerebellar vermis” (see Figures 1E–H for the “molar tooth sign” and Figures 1I–L showing the corresponding MRI of a healthy control individual) (Maria et al., 1997, 1999). Typical clinical symptoms of JS are hypotonia, global developmental delay, intellectual disability, abnormal breathing pattern, abnormal eye movements, and cerebellar ataxia. Additional features include retinal dystrophy, cystic kidney disease, liver fibrosis, polydactyly, cleft palate, and facial dysmorphism in some patients (for review see (Parisi and Glass, 1993)). The estimated birth prevalence of JS is 1:80,000–1:100,000 (Parisi and Glass, 1993), but this may represent an underestimate due to many undiagnosed cases. A higher prevalence is found in the French–Canadian population, with several founder variants noted (Badhwar et al., 2000; Srour et al., 2012a,b, 2015). Founder variants in different genes have also been identified in the Canadian Hutterite, the Ashkenazi Jewish, and the Dutch population (Edvardson et al., 2010; Valente et al., 2010; Huang et al., 2011; Shaheen et al., 2014; Kroes et al., 2016). To date, pathogenic variants in more than 30 genes are known to cause JS (for review see Parisi and Glass, 1993). The encoded proteins of all these genes localize either to the primary cilium, basal body and/or centrosome and play a role in the formation, morphology, and/or function of these organelles, rendering JS a member of the rapidly expanding family of ciliopathies (Parisi and Glass, 1993; Romani et al., 2013). Common features of many ciliopathies include brain malformation, renal disease, retinal dystrophy, and polydactyly. Pathogenic variants in genes that cause Joubert syndrome have also been identified in ciliopathies with clinical findings that overlap with JS, e.g., Meckel-Gruber syndrome (MKS), Jeune asphyxiating thoracic dystrophy (JATD), Bardet-Biedl syndrome (BBS), oral-facial-digital syndrome (OFD), and juvenile nephronophthisis. The severe end of the clinical spectrum is represented by the lethal disorder MKS (Barker et al., 2014). Most of the genes causative of MKS are also associated with JS, namely CEP290, TMEM67, RPGRIP1L, CC2D2A, CEP41, MKS1, B9D1, B9D2, TMEM138, TMEM231, TCTN2, TCTN3, TMEM237, CPLANE1, CSPP1, CEP120, TMEM107, and TMEM216 (Parisi and Glass, 1993; Valente et al., 2010; Thomas et al., 2012; Romani et al., 2014; Bachmann-Gagescu et al., 2015; Knopp et al., 2015; Shaheen et al., 2015; Roosing et al., 2016; Slaats et al., 2016). In addition, several families with occurrence of JS and MKS in siblings have been reported (Brancati et al., 2009; Valente et al., 2010). Features of the skeletal ciliopathy JATD have been reported in several children with JS caused by mutations in CSPP1 and KIAA0586 (Tuz et al., 2014; Malicdan et al., 2015). Pathogenic variants in the three BBS genes CEP290, MKS1, and NPHP1 have been shown to cause both BBS and JS (Leitch et al., 2008; Zaghloul and Katsanis, 2009; Knopp et al., 2015). Patients with oral-facial-digital syndrome (OFD) show features that overlap considerably with JS, as do several genes causative for OFD (Franco and Thauvin-Robinet, 2016). Patients with juvenile nephrophthisis can also show clinical overlap with JS: about 10% of individuals have extrarenal findings, including the molar tooth sign in some cases (Saunier et al., 2005). Conversely, nephronophthisis, can also be a renal manifestation in JS (Parisi and Glass, 1993). These examples illustrate the complex clinical and genetic background of JS and related ciliopathies. Preliminary genotype-phenotype correlation for some genes indicate that biallelic null alleles lead to MKS while at least one hypomorphic (e.g., missense) variant is associated with JS (Delous et al., 2007; Mougou-Zerelli et al., 2009; Tallila et al., 2009; Iannicelli et al., 2010; Romani et al., 2014). However, the molecular and cellular mechanisms that lead to a specific phenotype in patients with ciliopathies are not fully understood. Altered sonic hedgehog (SHH) signaling via defective cilia has been proposed to be the causative pathomechanism for the characteristic molar tooth sign in JS, but does not fully explain the mid-hindbrain phenotype (Spassky et al., 2008; Doherty, 2009).
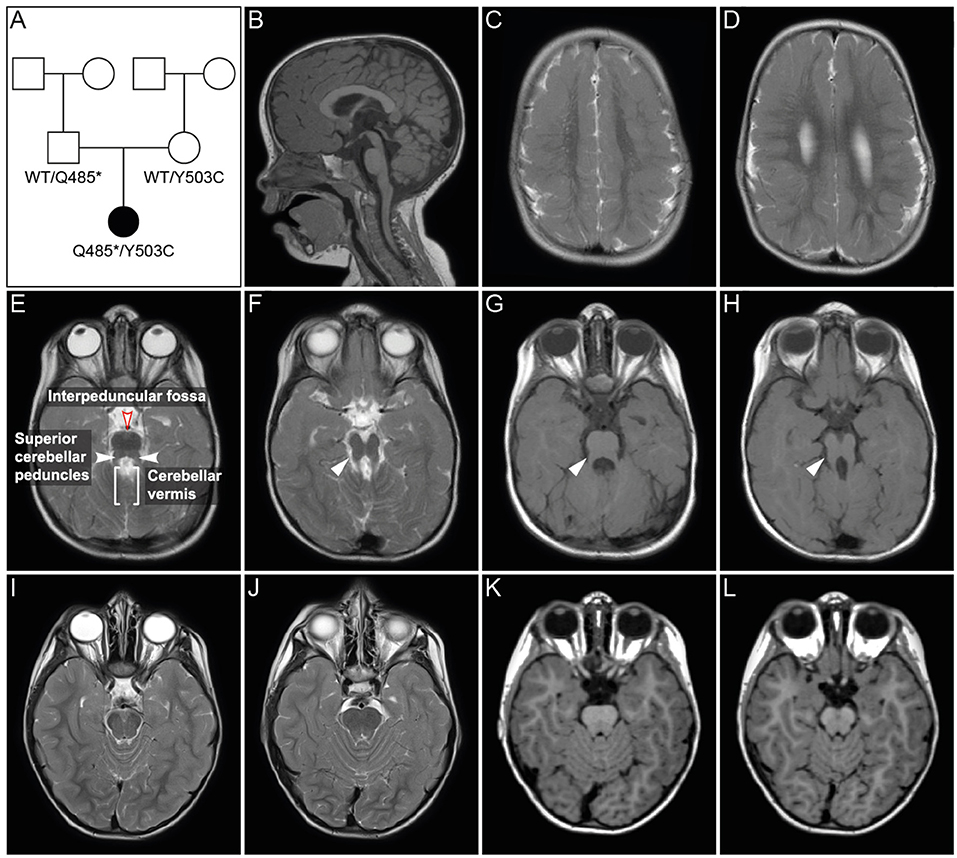
Figure 1. Pedigree and MRI scans. (A) Pedigree of patient family. Black symbol, affected individual; White symbols, unaffected individuals. (B–H) MR imaging of the patient at age 2 years and 6 months. Sagittal (B) and axial (C–H) images showed polymicrogyria in the parietal and temporal region (C,D) and hypoplasia of vermis cerebellum (B,E–H). Axial MR images of cerebellum and brainstem (E–H) showed a mild “molar tooth sign” (marked with white arrows in F–H) due to a deep interpeduncular fossa, prominent and elongated superior cerebellar peduncles and a hypoplastic cerebellar vermis. (I–L) Corresponding MR images of a healthy control individual.
Recently, mutations in PIBF1 have been identified as a cause of JS, using a combination of a siRNA-based functional genomics screen and exome sequencing data (Wheway et al., 2015). A second publication reported a girl with a biallelic 36-bp insertion in PIBF1 and clinical signs of JS (Hebbar et al., 2018). The patients presented with ataxia and developmental delay, ranging from mild to moderate. Imaging ranged from the classic molar tooth sign to moderate vermis hypoplasia with mildly thick superior cerebellar peduncles and characteristic superior cerebellar dysplasia (Wheway et al., 2015). In addition, thinning of corpus callosum, facial dysmorphism, hypotonia and enlarged cystic kidneys were observed in one patient (Hebbar et al., 2018). Polymicrogyria has not been described in association with PIBF1 variants so far.
PIBF1, also known as PIBF, CEP90, JBTS33, and C13orf24, consists of 22 coding exons and is widely expressed in different human tissues, including the brain, kidney, and liver, with the highest expression in testis and thyroid (Fagerberg et al., 2014). PIBF1 encodes the progesterone immunomodulatory binding factor 1 that is induced by the steroid hormone progesterone and overexpressed in highly proliferating cells (Lachmann et al., 2004; Cohen et al., 2016). The parent compound measures 90 kDa and is associated with the centrosome (Lachmann et al., 2004). A splice variant that is found in cytoplasm measures 34–36 kDa (Polgar et al., 2003; Lachmann et al., 2004). The protein regulates the immune system to maintain a normal pregnancy, may play a role in preterm labor and promotes the proliferation, migration, and invasion of astrozytoma/glioblastoma cells (Gonzalez-Arenas et al., 2014; Hudic et al., 2015, 2016; Gutierrez-Rodriguez et al., 2017). PIBF1 encodes a centrosomal protein that may play an important role in ciliogenesis (Wheway et al., 2015). However, the precise molecular and cellular mechanisms that cause the complex JS phenotype in individuals with PIBF1 mutations have not yet been elucidated.
Here, we report on novel PIBF1 variants in a girl with JS. The variants were identified by whole-exome sequencing (WES) and functionally assessed in the Xenopus model. Our analyses demonstrate that both PIBF1 alleles reflected loss of function variants. In general terms, the Xenopus model proves to be an excellent model to study the functional impact of rare genetic variants identified by diagnostic exome sequencing in patients with human ciliopathies.
Materials and Methods
Participants
The patient and her parents were recruited and clinically phenotyped by the Outpatient Clinic of the Institute of Human Genetics, University Hospital Heidelberg, as part of the “Genome-wide genetic analysis of rare hereditary disorders” study. Written informed consent for participation in the study and publication of study results was obtained from both parents. The study was approved by the Ethics Committee of the Faculty of Medicine at the University of Heidelberg and adhered to the tenets of the Declaration of Helsinki. A summary of the study results and its clinical implications have been published elsewhere (Evers et al., 2017). Written informed consent for the publication of this case report and parental results was obtained from the patient's parents.
Case Report
The girl was the first child of non-consanguineous healthy parents from Germany. Her pedigree is shown in Figure 1A. She was born after 40 weeks of gestation with a birth weight of 2,620 g (1st centile), length of 48 cm (3rd centile), and head circumference (OFC) of 34 cm (20th centile). Soon after birth, spastic tetraparesis, truncal hypotonia, and feeding difficulties were noted. At age 6 months, she developed abnormal eye movements. An electroencephalogram (EEG) was normal. The girl showed a severe failure to thrive and developmental delays. Routine pediatric investigations, including basic laboratory testing and metabolic screening, resulted in normal values with the exception of mildly elevated liver enzymes (GOT: 134U/l, GTP: 164 U/l, GGT: 493 U/l), which persisted during childhood. Regular abdominal ultrasound examinations were normal with no signs of hepatic fibrosis. Ophthalmological examination including fundoscopy at age 3 years showed no abnormalities. At her first visit to the Genetic Outpatient Department at age 4 years 2 month, she presented with global developmental delay, no speech, spastic tetraplegia and a submucosal cleft palate. Her height was 85 cm (<1st centile, −4.99 SDS), her weight 11.47 kg (<1st centile, −3.08 SDS) and her OFC 49.5 cm (8th centile, −1.43 SDS). At follow up examination at age 6 years 9 months, she had a height of 98.0 cm (<1st centile, −5.49 SDS), a weight of 13.6 kg (<1st centile, −4.30 SDS) and an OFC of 50 cm (2nd centile, −2.09 SDS). cMRI at age 6 months revealed bilateral polymicrogyria in the parietal and temporal areas. Follow up MRIs at age 23 months and 2 years and 6 months showed polymicrogyria, hypoplasia of vermis cerebelli, and a mild molar tooth sign (Figures 1E–H). Chromosomal analysis and molecular karyotyping (array analysis) gave normal results. Gene panel diagnostics for Joubert syndrome by next generation sequencing of 129 known and potentially ciliopathy genes showed no pathogenic mutation. The gene panel did not include PIBF1, which was not known to cause JS at the time of analysis. A single gene test by Sanger sequencing of GPR5, a gene associated with polymicrogyria, gave normal results.
Exome Sequencing
Genomic DNA was isolated from leukocytes of the patient and both parents by standard procedures (Miller et al., 1988). Whole exome sequencing (WES) and analysis of the sequence data of the patient and her parents was performed at the German Cancer Research Center (DKFZ) in Heidelberg, Germany, as described previously (Paramasivam et al., 2018). Variants with a minor allele frequency (MAF) >1% in the 1000 genome phase III and Exome Aggregation Consortium (ExAC) database (Lek et al., 2016) were considered common alleles and discarded, as were variants detected in 328 WES and 177 whole genome sequencing (WGS) local control samples with a frequency above 2%. Gene-based annotations from Gencode V19 were added using ANNOVAR (Wang et al., 2010). All single nucleotide variants (SNVs) and indels affecting protein sequences and variants within ±2 bases around the intron-exon junction were considered as functional. Variants were further assessed by the seven different variant effect prediction tools SIFT, PolyPhen2, LRT, MutationTaster, MutationAssessor, FATHMM, and PROVEAN from dbNSFP (Ng and Henikoff, 2003; Chun and Fay, 2009; Adzhubei et al., 2010; Schwarz et al., 2010; Reva et al., 2011; Choi et al., 2012; Liu et al., 2013; Shihab et al., 2013) and CADD scores (Kircher et al., 2014). Variants were classified according to standards and guidelines of the American College of Medical Genetics and Genomics (ACMG) (Richards et al., 2015). To confirm WES data by Sanger sequencing, exons 11 and 12 and adjacent intron boundaries of PIBF1 (RefSeq NM_006346.2, ensemble transcript ENST00000326291.6) were sequenced using Big Dye Terminator V1.1 cycle sequencing kit and ABI 3130xl genetic analyzer. Primer sequences and PCR conditions are available upon request.
RT PCR, qPCR, and Sequencing
Total RNA from patients, parents, and control blood was extracted using the MasterPure RNA Purification Kit (Epicentre Biotechnologies). cDNA was synthesized using random hexamer primers and reverse transcriptase RT Maxima (Fermentas). qPCR was carried out using SybrGreen mix (Thermo Scientific). Expression levels using primer pairs for the three regions of PIBF1 (exon 2–4, exon 10–12, exon 15–17) were normalized to ADP-ribosylation factor 1 (ARF1). PCR products of patient, parents and control were sequenced by Sanger sequencing (GATC).
Western Blot Analysis of Overexpressed Protein
To analyze the expression of overexpressed PIBF1 variants, Hek293T cells were transfected in 6-well plates with 1 μg of the corresponding plasmids using Turbofect transfection reagent (Thermo Scientific. Cells were lysed 24 h post transfection and proteins were separated by 10% SDS-PAGE. For Western blot analysis, a rabbit anti-GFP antibody (1:1,000, Adgene) and a mouse anti-PIBF1 antibody (1:500, Biozol) were used.
Protein Structure Analysis
For analyzing putative protein domains, the following algorithms were used: NCBI conserved domain search (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi), InterProScan (https://www.ebi.ac.uk/interpro/search/sequence-search), WoLF PSORT (https://wolfpsort.hgc.jp), and epestfind (http://emboss.bioinformatics.nl/cgi-bin/emboss/epestfind).
Xenopus Injection Experiments
Adult Xenopus laevis frogs were obtained from Nasco (U.S.A.; https://www.enasco.com/c/Education-Supplies/Xenopus-Frogs). Xenopus laevis embryos were injected at the 4-cell stage into the ventral marginal zone to target the epidermal cell lineage (Moody, 2000). Translation blocking morpholino oligomere (TBMO; 5′-CCGGGACATCTTTACACTTTACATA-3′) was injected at 4 pmol per embryo. mRNAs of EGFP or PIBF1 fusion constructs were injected at a dose of 0.4 pmol per embryo. Lineage tracer Fluorescein Dextran (FD, 10,000 MW, anionic, lysine fixable) was used at 50 ng per injection. Embryos were cultured until stage 30 and subsequently processed for analyses.
RNA in situ Hybridization and Immunofluorescence Staining
Xenopus embryos were fixed using 4% paraformaldehyde solution (for in situ hybridization and acetylated Tuba4a staining) or Dent's (for Pibf1 and Tjp1 staining). RNA in situ hybridization was performed as described previously (Belo et al., 1997) using a full length digoxigenin labeled pibf1 probe. The following reagents were used for immunofluorescence staining: anti-acetylated Tuba4a (T6793, Sigma; 1:800), anti-Pibf1 (SAB1401526, Sigma; 1:200), and anti-Tjp1 (21773-1-AP, Proteintech Europe; 1:400).
High-Speed Video-Microscopy
Capturing of ciliary beating required flat mounting of the specimens in a chamber constructed on a slide with tape and a cover slip. Only the most ventral cells allowed differential interference contrast microscopy of the ciliary tufts, which were recorded at 600 fps using a Hamamatsu X high speed video camera. Kymographs were generated using ImageJ (https://imagej.net/Generate_and_exploit_Kymographs).
Results
The phenotypic features of the patient (cf. section Case Report), including developmental delay, hypotonia, polymicrogyria, vermian hypoplasia, and mild molar tooth sign (Figure 1), led to the clinical diagnosis of JS. The observed liver involvement with elevated GOT/GRP is also a typical finding of this ciliopathy. A cleft palate reported here is a rare finding in JS and demonstrates its clinical overlap with oral-facial-digital syndrome. The microcephaly of the patient is not part of the classical JS spectrum, but has been reported in a patient with a PIBF1 missense mutation (Kodani et al., 2015).
Exome Sequencing
Exome sequencing variants were filtered as described above and only heterozygous de novo variants and variants being consistent with an autosomal recessive disease model were considered. Applying these filter criteria, 10 variants remained (Table 1). These were further assessed by in silico predicted effects on protein function, as described above (Table 1). Subsequently, a literature search was performed to gain further information about gene function and to determine if the gene had been previously associated with intellectual disability, neurological or developmental disorders in humans. This narrowed the candidate list to PIBF1 variants c.1453C>T; p.(Q485*) and c.1508A>G; p.(Y503C). The variant c.1453C>T; p.(Q485*) was classified as pathogenic [class 5, according to ACMG criteria; (Richards et al., 2015)]. The variant c.1508A>G; p.(Y503C) was classified as variant of unknown significance [class 3, according to (Richards et al., 2015)].

Table 1. De novo and compound heterozygous variants and prediction of their functional effects.
Expression Analysis of PIBF1 Variants in the Patient
cDNA fragments of three different regions of PIBF1 (exons 2–4, 10–12, and 15–17) were amplified from the patient and both parents, showing a higher expression in the patient compared to mother, father, adult, and infant control (Figure S1A). Interestingly, the cDNA of exons 15–17, which are localized 3′ of the predicted premature stop codon of the variant p.(Q485*), showed a higher expression in the patient as well. However, whether both variants are transcribed in the patient was still unclear. Sequencing of the PCR product of the patient showed that both PIBF1 variants could be detected in the patient (Figure 2A). The parents' cDNAs carried either the missense or the nonsense variant in heterozygous state (data not shown). This indicated that mRNA harboring the predicted pathogenic variant was not degraded by nonsense-mediated mRNA decay (NMD) and that both variants resulted in stable mRNAs. The enhanced transcription of both PIBF1 variants in the patient could be a compensatory response to decreased PIBF1 protein levels due to protein instability of the mutants. The PIBF1 nonsense variant p.(Q485*) was expected to result in the synthesis of a truncated protein lacking the C-terminal 273 amino acids. Expression analysis of wildtype (WT) and mutated EGFP-PIBF1 constructs using an anti GFP antibody showed expression of WT and both mutated proteins, the one with the missense and the one with the truncating variant at expected size (Figure S1B). This result indicated that both, missense and the nonsense variant, resulted in the synthesis of stable proteins with the nonsense variant being smaller than the WT and the missense, as expected. Analyses using an antibody against the full length PIBF1 protein could detect the WT and the missense variant, but not the truncated nonsense variant. However, the sensitivity of the antibody was far lower than that of the GFP antibody and it has not been tested on truncated PIBF1 proteins so far. Whether its epitope is in the lacking C-terminus has to be determined.
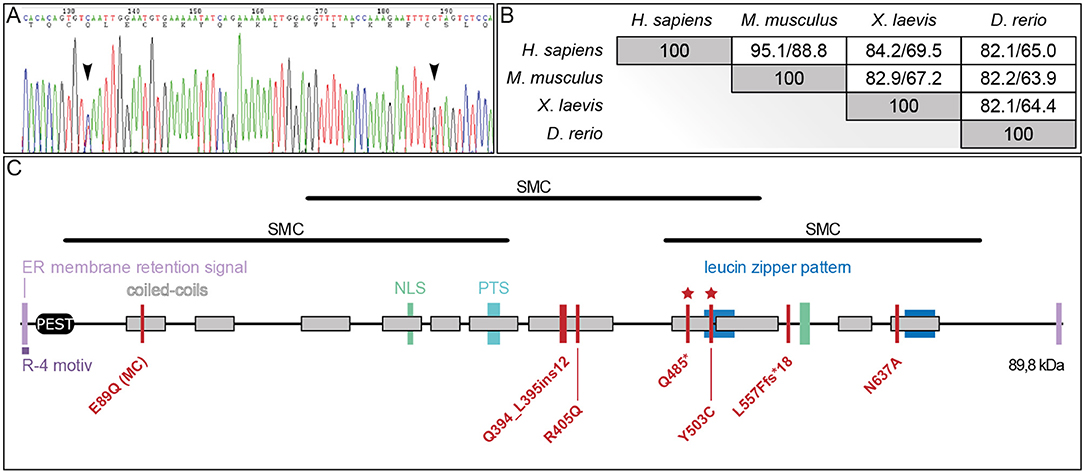
Figure 2. PIBF1 protein conservation and domain structure. (A) Sanger sequencing of PIBF1 from RT-PCR product of the patient mRNA extracted from blood. Black arrow heads indicate position of mutations. (B) Conservation of amino acid sequences between human, mouse, Xenopus, and zebrafish PIBF1. (C) Putative domain structure. JS and microcephaly (MC) mutations are indicated as red bars. For details see text. *Novel mutations identified in this study.
Protein Structure and Evolutionary Conservation of Pibf1
A comparison of PIBF1 protein sequences from various vertebrate species revealed a high degree of conservation (Figure 2B). The Xenopus sequence showed 69.5% identity (84.2% similarity) to human PIBF1, very close to the mouse (Figure 2B). Functional studies at the protein level have not been reported for PIBF1 so far. Applying the NCBI conserved domain search algorithm highlighted three possible SMC-related domains (structural maintenance of chromosomes; Figure 2C). Additional searches identified a putative PEST domain, two ER membrane retention signals including one R-4 motiv, two nuclear localization sequences (NLS), a peroxisomal targeting signal (PTS), and two leucine zipper sequences as well as 11 coiled-coil domains along the 757 amino acids (Figure 2C). The three known JS mutations as well as the two novel alleles reported here localize to the C-terminal half of the protein and within coil-coiled domain, while a 6th mutation, which caused microcephaly, was found at the N-terminus, again in a coiled-coil domain (Figure 2C).
The above in silico analyses thus showed that the two novel mutations are located in important regions of a highly conserved JS candidate gene. In order to prove that these alleles indeed were causative for JS in the patient, they needed to be functionally tested in a relevant vertebrate model organism, particularly because the variant p.(Y503C) did not fulfill the ACMG-criteria to be classified as pathogenic or likely pathogenic (Richards et al., 2015). We chose to apply the Xenopus model, because of its suitability for studying ciliopathies: speed, high-throughput, and low cost of analyses (Johnston et al., 2017; Blum and Ott, 2018).
Expression of pibf1 in Ciliated Tissues During Xenopus Embryonic Development
As a prerequisite to functionally analyzing the putative JS alleles, we analyzed whether the endogenous pibf1 mRNA was expressed in tissues related to cilia. Transcription of pibf1 in embryos of defined developmental stages was analyzed using whole-mount in situ hybridization. Maternally deposited pibf1 mRNA was present in the cytoplasm of the animal hemisphere in cleavage stage embryos (Figure 3A). At the onset of gastrulation, signals were found in future mesodermal tissues (Figure 3B; involuting marginal zone). From neurulation onwards, expression was found in the axial (notochord) and paraxial (somites) mesoderm (Figures 3C,D). Additionally, a dotted epidermal pattern was obvious which resembled the distribution of multiciliated cells (MCCs) in the larval skin and was maintained until the end of neurulation (Figures 3C,D). In the 2-day larva (stage 25, Figure 3E), mRNA transcripts were seen in the ciliated otic vesicle (Figure 3E′) as well as in the forming ciliated nephrostomes of the embryonic kidney. The latter staining intensified and was prominently visible in sections of stage 30 tadpoles (Figures 3F,F′). A persistent staining in the head region became more pronounced from stage 30 onwards (Figures 3F–H). Enrichment of pibf1 transcripts in the head region as well as in the spinal cord was in agreement with the expected function of Pibf1 during neural development (Figures 3E–G). At these stages, the pibf1 signal became less discrete, appeared more diffuse and was present at low levels in most tissues (Figures 3E–G). Histological sectioning revealed enrichment in the retina and inner nuclear cell layer (Figure 3H′). In summary, pibf1 was expressed in many tissues harboring cilia, namely otic vesicle, nephrostomes, brain, retina, and possibly MCCs.
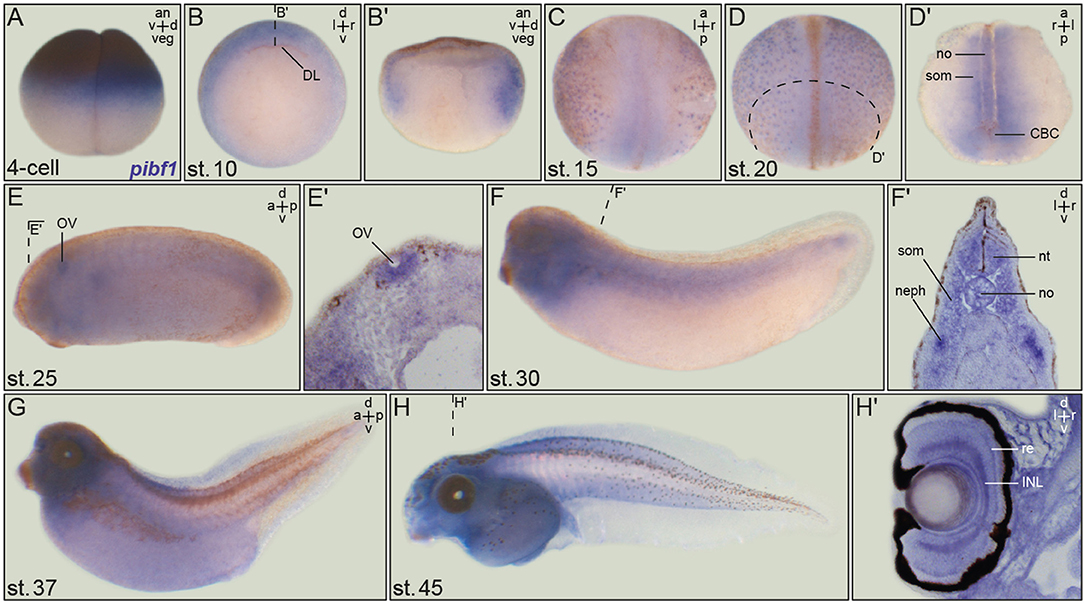
Figure 3. Embryonic pibf1 expression correlates with ciliated tissues. Embryos of defined stages were analyzed for pibf1 mRNA by whole-mount in situ hybridization using a digoxigenin-labeled antisense probe. (A) Maternal transcripts in the animal hemisphere of the 4-cell embryo. (B) Expression in the involuting marginal zone tissue of the early gastrula embryo. (C) Staining of pibf1 in the axial and paraxial mesoderm at early neurula stages. (D) Pibf1 signals in skin MCCs and axial/paraxial mesoderm. (E) Expression in the otic vesicle at stage 25. (F–H) Expression pattern in tadpoles, including the head region, somites, and nephrostomes. Histological section of the eye (H′) revealed signals in the retina as well as the inner nuclear cell layer. Planes of histological sections in (B′,D′,E′,F′,H′) are indicated in the respective panels. an, animal; a, anterior; d, dorsal; DL, dorsal lip; INL, inner nuclear layer; l, left; neph, nephrostomes; no, notochord; nt, neural tube; ov, otic vesicle; p, posterior; r, right; re, retina; som, somite; v, ventral; veg, vegetal.
Pibf1 Protein Localization in Larval Skin MCCs
JS is associated with dysfunctional primary, i.e., immotile and sensory cilia. The unexpected mRNA expression in the larval skin hinted at localization in MCCs, which harbor hundreds of motile cilia. In order to investigate the possible expression of Pibf1 in MCCs, immunofluorescence staining using a monoclonal mouse PIBF1 antibody raised against the human protein was applied. This antibody detected hundreds of spots on individual cells dotted on the larval skin (Figures 4A,A′). Co-staining with the basal body marker (Cetn1; Park et al., 2008) unequivocally demonstrated that Pibf1 indeed localized to the base of individual cilia on MCCs, specifically to basal bodies, as Pibf1 and Cetn1 partially overlapped (Figures 4A–C). Therefore, Pibf1 seemed to be a bona fide component of all basal bodies in ciliated cells in Xenopus.
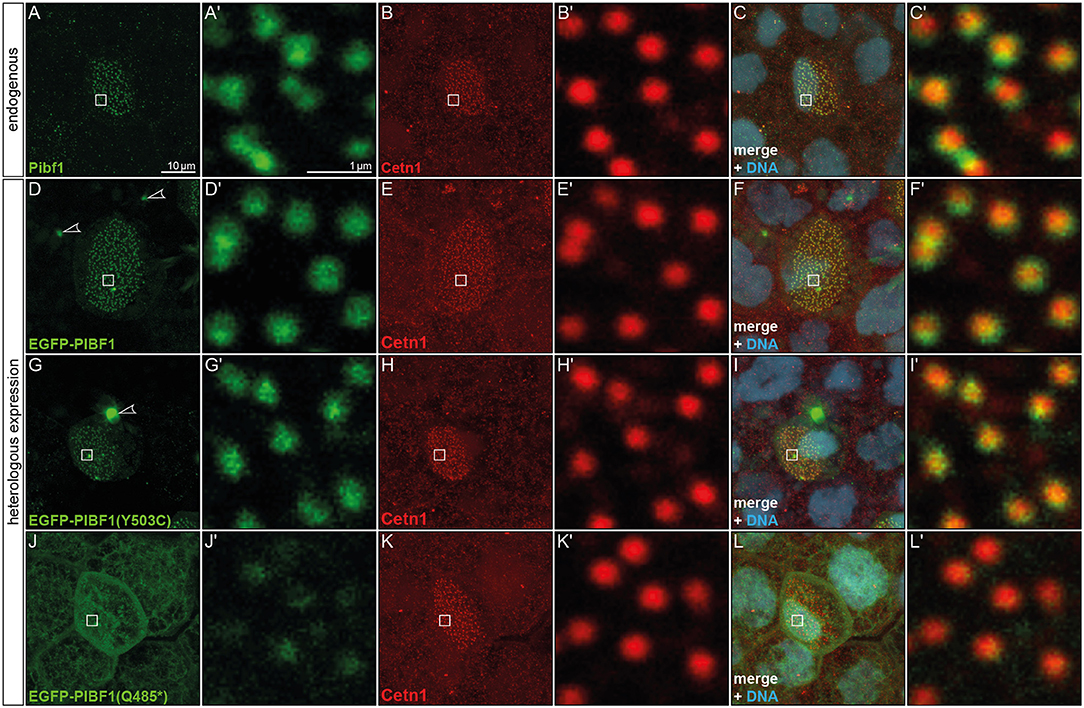
Figure 4. Pibf1 localization to MCC basal bodies is attenuated in nonsense JS variant. (A–C) Localization of the endogenous Pibf1 protein, using an anti-PIBF1 antibody (A) and co-staining with Centrin 1 (B) and Hoechst 33342 to highlight the nucleus (C). (D–F) An EGFP-PIBF1 fusion protein recapitulates the staining of the endogenous protein. (G–I) Unaltered expression of the missense allele Y503C. (J–L) Attenuated basal body localization of the truncated Q485* variant of PIBF1. Arrowheads mark protein aggregates observed upon overexpression of EGFP fusion proteins.
In order to ascertain whether or not the missense or nonsense mutations have impact on the localization of PIBF1 to basal bodies, we cloned fusion constructs in which the N-terminus of the human WT or mutant ORFs of PIBF1 were linked to EGFP. Injection of the WT PIBF1 construct into the Xenopus epidermis recapitulated the endogenous distribution at the basal bodies of MCCs (Figures 4D–F), demonstrating that ectopic expression of a fusion protein did not interfere with correct localization of the protein. In many cases, aggregates of fusion protein were additionally found in targeted cells (Figures 4D,G). The missense construct fully phenocopied this localization (Figures 4G–I). In contrast, the signal of the nonsense variant was much attenuated at basal bodies and was additionally found in a non-localized manner throughout the cell (Figures 4J–L). Taken together, these analyses showed that (1) Pibf1 was unexpectedly expressed in motile cilia of the larval skin; (2) Pibf1 localized to basal bodies, in agreement with its centrosomal expression in other contexts (Kim and Rhee, 2011; Kim et al., 2012); (3) the nonsense allele was much reduced in its localization to ciliary basal bodies, indicative of a ciliary function; (4) the missense allele appeared unaffected in its ciliary localization, raising questions as to the underlying mechanism of JS in the patient.
Functional Analysis of Wildtype and Mutant PIBF1 Alleles in the Xenopus Larval Skin
Although JS is not related to motile cilia, the expression and localization of Pibf1 in basal bodies of MCCs afforded the opportunity of testing whether this protein played a role in motile cilia as well and whether the mutant alleles were affected in this function. Skin MCCs of Xenopus larvae function in much the same way as human airway epithelia, namely in mucociliary clearance as a first line of defense against pathogens (Dubaissi and Papalopulu, 2011; Walentek et al., 2014; Blum and Ott, 2018). MCC cilia beat in a coordinated manner to move mucus, produced by goblet cells, from anterior to posterior (head to tail), and to thereby remove environmental particles and pathogens caught by the mucus layer (Brooks and Wallingford, 2014). In order to assess a possible role of pibf1 in this process, an antisense morpholino oligomer (MO) targeting the translational start site of the mRNA (translation blocking MO, TBMO) was designed. In retinal epithelial cells (RPE-1) and inner medullary collecting duct cells (IMCD3), loss of Pibf1 resulted in fewer or absent cilia (Kim et al., 2012; Wheway et al., 2015). We therefore analyzed the presence of MCC cilia in morphant larvae that were injected with TBMO into the skin lineage at the 4-cell stage. Successful gene knockdown was proven by immunofluorescence staining for Pibf1, which demonstrated the efficient depletion of the protein from MCCs of morphant specimens (Figure 5). Immunofluorescence staining of cilia in morphant specimens was performed by staining the ciliary axoneme with an antibody against acetylated tubulin (Tuba4a). This analysis clearly demonstrated markedly reduced numbers of cilia on morphant MCCs (Figures 6A–E). Functional consequences of reduced cilia numbers were assessed by high-speed video microscopy of larval skin at stage 30. Movie S1 shows that coordinated ciliary beating was lost in morphants as compared to WT specimens. The loss of cilia was also apparent in kympgraphs from high-speed movies (Figure 6L,M). Ciliary tufts on WT and morphant MCCs were grouped into three classes, representing normal, mild, or strong reduction of cilia numbers. No unaffected ciliary tufts were retained in morphant MCCs, as displayed in Figure 6M.
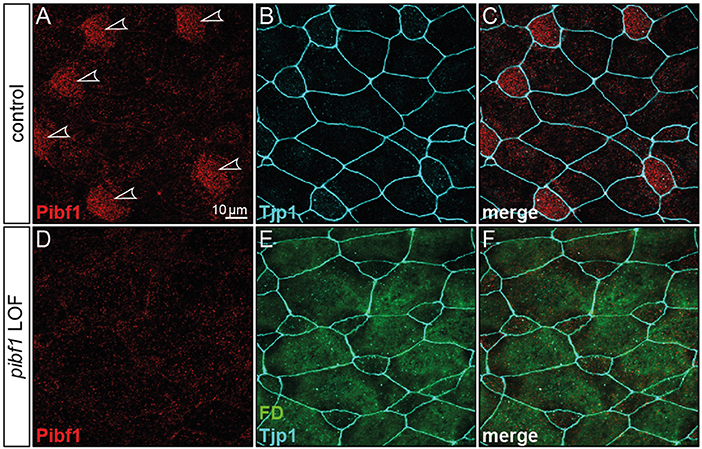
Figure 5. Loss of Pibf1 protein in pibf1 morphant Xenopus skin MCCs. Basal body staining of Pibf1 (A–C) was lost in TBMO injected specimen (D–F). Tjp1 immunofluorescence was used to mark cell boundaries (B,C,E,F). Fluorescent dextrane (green) was co-injected as lineage tracer to control targeting of injections (E,F). Arrowheads highlight MCC cilia in WT embryo.
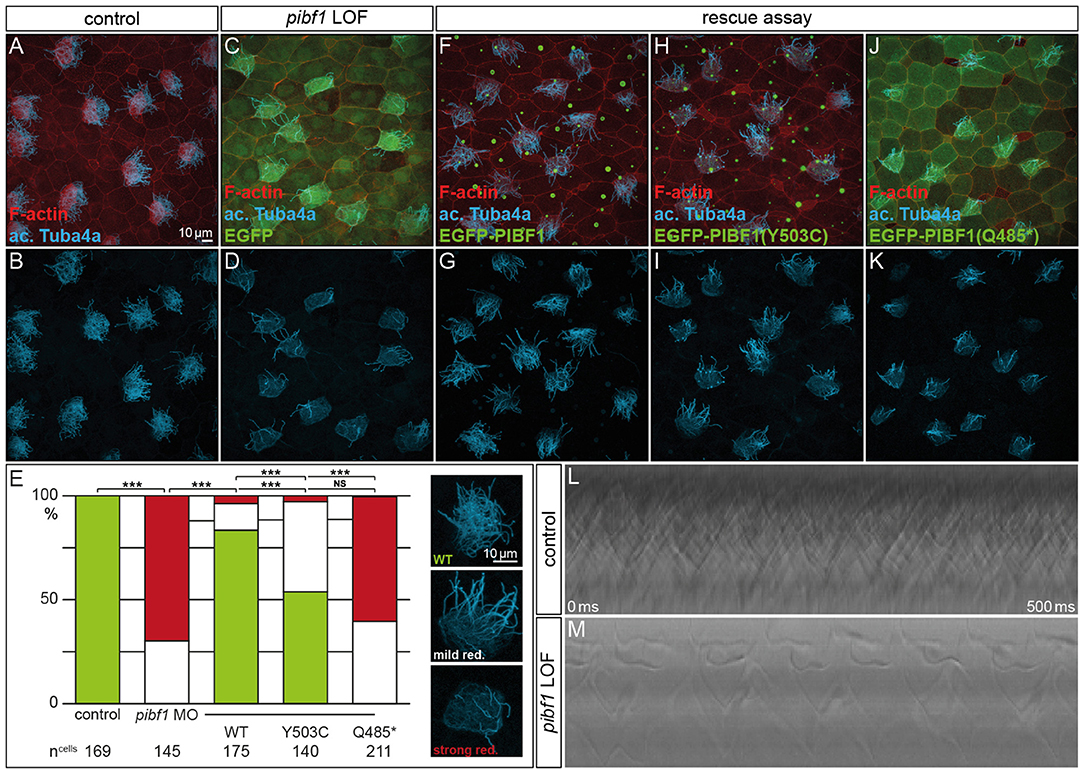
Figure 6. Rescue of morphant MCC ciliation is lost with mutant PIBF1 alleles. (A–E) MCC ciliation in WT embryos was lost in pibf1 TBMO-injected specimens. (E–G) Rescue of ciliation upon co-injection of TBMO and EGFP-PIBF1. (E,H,I) Attenuated rescue in embryos co-injected with missense PIBF1 allele. (E,J,K) Defective rescue upon co-injection of the nonsense PIBF1 allele. (E) Quantification of results. (L,M) Kymographs representing ciliary beating in WT and TBMO-injected stage 30 larval skin MCCs. ***Very highly significant, p < 0.01.
Next, we asked whether heterologous expression of a WT human PIBF1 construct was able to rescue cilia numbers in morphants. To that end, TBMO and WT PIBF1 were co-injected into 4-cell embryos and targeted to the larval skin. As shown in Figures 6E–G, the WT human gene rescued cilia numbers in a highly significant manner. In a last set of experiments, we analyzed the rescue capacity of equimolar amounts of the two novel Pibf1 alleles identified in our JS patient. While the nonsense mutant was unable to rescue the gene knockdown (Figures 6E,H,I), a residual and attenuated rescue capability was observed when the missense mutant allele was co-injected (Figures 6E,J,K). In summary, our functional analysis of mutant Pibf1 alleles demonstrated a role of Pibf1 in motile cilia of larval skin MCCs in Xenopus, and identified both nonsense and missense allele as non-functional, in agreement with the manifested JS in the patient girl.
Discussion
Mutant gene alleles identified in patient DNAs represent a valuable resource for studying protein function and are a prerequisite for the elucidation of pathomechanisms at the molecular level. A given mutation may not, however, reveal its pathogenicity at first glance. In the case of the compound heterozygous JS patient analyzed here, one of the mutations, the truncation variant p.Q485*, was highlighted as pathogenic according to ACMG criteria (Richards et al., 2015). Our analysis in ciliated Xenopus cells confirmed its predicted pathogenicity. The disturbed localization of this variant at basal bodies in combination with the inability to rescue the ciliation phenotype of Pibf1 deficient frog MCCs identifies it as an amorphic allele. Clinically even more important, our studies of the missense-variant p.Y503C indicated a pathogenic effect of this variant, too. A careful cell by cell analysis of mutant MCCs revealed that this mutant showed an about 50% rescue capacity, significantly below the >80% achieved with the WT allele (Figure 6). It should be noted, that this analysis was performed in a tissue that is not relevant in JS patients and on motile cilia, while JS is caused by defects of primary, immotile cilia. Therefore, in the context of a bona fide JS target tissues, the combination of these mutations may give rise to even more pronounced defects at high frequency. Recapitulating the patient gene setup in any animal model would be experimentally more challenging, require more time and be more expensive. The attenuated rescue ability, however, unequivocally demonstrates that the missense variant is hypomorphic in nature, leading to a re-classification of the allele from “unkown significant” to “likely pathogenic,” according to ACMG criteria (Richards et al., 2015). The finding of a hypomorphic allele due to a missense mutation in combination with a null allele due to a truncating mutation is a typical finding in patients with JS and has been reported for a number of other causative genes (such as RPGRIP1L, TMEM67, CCD2D2A, and TCTN3), whereas biallelic null alleles in these genes are associated with a more severe phenotype (Delous et al., 2007; Mougou-Zerelli et al., 2009; Tallila et al., 2009; Iannicelli et al., 2010; Romani et al., 2014).
The precise role of PIBF1 in the context of ciliary biogenesis is not well-understood. All JS-associated mutations in PIBF1 identified so far cluster in the C-terminal region of the protein [Figure 2; (Wheway et al., 2015; Hebbar et al., 2018)]. In contrast, a homozygous missense mutation that is linked to microcephaly is present within the N-terminus [Figure 2; (Kodani et al., 2015)]. The large structural maintenance of chromosomes (SMC) domains constitute almost 90% of the protein, suggesting that PIBF serves as a scaffolding factor which may dimerize with ciliary SMC proteins such as SMC1A or SMC3 (Khanna et al., 2005). A search of the protein interaction database IntAct (https://www.ebi.ac.uk/intact/) revealed a number of SMC proteins that were shown to interact with PIBF1, for example CEP63 and PCM1 (Kim et al., 2012; Gupta et al., 2015; Yachie et al., 2016). As PCM1 is not relevant for cilia formation in multiciliated mouse tracheal epithelial cells, it is not a promising candidate to explain the loss of cilia in PIBF1 depleted MCCs (Vladar and Stearns, 2007). CEP63, in contrast, is required during the centriolar duplication cycle, acts in parallel with its paralog deup1 in basal body formation in MCCs and harbors an SMC related domain (Zhao et al., 2013). The reduced number of cilia on morphant MCCs may result from such a mechanism. Xenopus MCC offer themselves for in-depth analyses of potential interaction partners, which are beyond the scope of the present study.
The advent of high-throughput sequencing technology, in particular whole exome sequencing (WES), has led to a revolution of genetic diagnostics of rare diseases, e.g., developmental disorders. Before the era of WES many patients had undergone a long and frustrating “diagnostic odyssey” to obtain an accurate diagnosis. This has been widely overcome with the introduction of WES, which has emerged as an effective diagnostic tool leading to diagnostic rates of around 40% in patients with previously undiagnosed neurodevelopmental or pediatric neurologic disorders [for review see (Wright et al., 2018)]. A diagnosis is essential for an optimal clinical management of the particular patient, e.g., initiation of a specific therapy or surveillance program, and appropriate access to education, social care and patient support groups (Boycott et al., 2017). A molecular diagnosis is also important for the patients' parents and other family members, in particular for informed decision-making with regard to family planning, and possibly prenatal diagnosis (PD) or preimplantation genetic diagnosis (PGD). In the case reported here, a molecular diagnosis could not have been established without the Xenopus analysis of the PIBF1 missense variant and the resulting re-classification as likely pathogenic. Important consequence for the parents, who previously had decided against further children for fear of another disabled child, was that they now opt for a further pregnancy with PD.
Furthermore, identifying the molecular genetic cause of a disease is essential for a better understanding of its pathogenesis and the development of novel treatment strategies. However, interpretation of high-throughput sequencing data can be difficult. WES also uncovers many rare variants of which the functional impact is not known. Thus, a molecular diagnosis may be missed. Furthermore, recent studies in the field of cancer genetics and prenatal diagnosis indicate that unambiguous genetic results such as the finding of unclassified variants can lead to false treatment decisions and dissatisfaction with genomic testing (Kurian et al., 2017; Desai et al., 2018). Therefore, animal models are needed to verify or discard candidate disease alleles. Because of its genetic closeness, the mouse has been the model of choice to assess human genetic diseases. However, analyses in mice are costly and slow; in addition, the mouse is not suited for high-throughput analysis and cannot possibly keep up with the pace at which candidate variants keep being identified by WES. Therefore, additional and complementing animal models need to be promoted. Among the non-mammalian models, the zebrafish is widely used, while Xenopus is less well-known among clinical scientists. The frog, however, offers unique advantages particularly when investigating ciliopathies (Blum and Ott, 2018). The developing embryo presents its ciliated skin for a total of 3 days and allows easy and straightforward observation (including video microscopy) and functional analyses. Manipulations can be performed in a unilateral fashion, such that the non-manipulated side serves as an internal control. Given the inter-individual variability of phenotypes, this represents a unique advantage of the Xenopus model. The present analysis demonstrates that a syndrome like JS, which is caused by defects of primary, immotile cilia, can be successfully dissected in the context of motile cilia, as the expression analysis revealed a pan-ciliary presence of pibf1 mRNA. This is likely true for most ciliopathies of primary cilia. Other organs that are easily addressed in the Xenopus model include the kidney (Getwan and Lienkamp, 2017) and heart (Duncan and Khokha, 2016). It seems, therefore, warranted to promote Xenopus among clinical scientist as a complementing model to mouse and zebrafish, in order to allow for the most efficient assessment of disease alleles.
Author's Note
The authors dedicate this paper to the memory of the late Herbert Steinbeißer, who has been an inspiration to all of us and who has brought this group of people together.
Author Contributions
TO conceived Xenopus experiments together with MB, performed and evaluated all Xenopus experiments, and wrote the manuscript together with MB and CE. MB conceived and evaluated experiments and wrote the paper. LK and ST performed mRNA/cDNA analysis of the patient and parents and westernblots. LK wrote sections of the manuscript. WES study coordination was done by CB and UM. NP and ASc performed WES analysis. Bioinformatic WES data analysis was done by NP, ASc, and MG, further evaluation of WES data was done by CE and UM. KH performed confirmation of WES variants by Sanger-Sequencing. ASe performed MRI images and their interpretation. Genotype–Phenotype correlation was done by CE, MG, KH, LK, and UM. All authors contributed to manuscript revision, read and approved the submitted version.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2019.00134/full#supplementary-material
Movie S1. Uncoordinated beating of MCC cilia upon pibf1 gene knockdown. Coordinated ciliary beating of MCC (left) was lost in pibf1 morphant specimen (right). Note the reduced number of cilia on morphant cell. Time lapse movie was recorded at 600 fps and plays at 30 fps (0.05 × real time).
Figure S1. Expression anlayses of PIBF1 variants. (A) Relative expression levels of PIBF1 exons analyzed by qPCR from patient, parents' and control blood. Shown are the mean ± SD of three independent RT-PCRs and subsequent qPCRs. ARF1 expression was used for normalization. (B) Western blot analyses of overexpressed EGFP-PIBF1 constructs in HEK293 cells, using anti-GFP and anti-PIBF1 antibodies, respectively.
References
Adzhubei, I. A., Schmidt, S., Peshkin, L., Ramensky, V. E., Gerasimova, A., Bork, P., et al. (2010). A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249. doi: 10.1038/nmeth0410-248
Bachmann-Gagescu, R., Dempsey, J. C., Phelps, I. G., O'Roak, B. J., Knutzen, D. M., Rue, T. C., et al. (2015). Joubert syndrome: a model for untangling recessive disorders with extreme genetic heterogeneity. J. Med. Genet. 52, 514–522. doi: 10.1136/jmedgenet-2015-103087
Badhwar, A., Andermann, F., Valerio, R. M., and Andermann, E. (2000). Founder effect in Joubert Syndrome. Ann. Neurol. 48, 435–436.
Barker, A. R., Thomas, R., and Dawe, H. R. (2014). Meckel-Gruber syndrome and the role of primary cilia in kidney, skeleton, and central nervous system development. Organogenesis 10, 96–107. doi: 10.4161/org.27375
Belo, J. A., Bouwmeester, T., Leyns, L., Kertesz, N., Gallo, M., Follettie, M., et al. (1997). Cerberus-like is a secreted factor with neutralizing activity expressed in the anterior primitive endoderm of the mouse gastrula. Mech. Dev. 68, 45–57. doi: 10.1016/S0925-4773(97)00125-1
Blum, M., and Ott, T. (2018). Xenopus: an undervalued model organism to study and model human genetic disease. Cells Tissues Organs. doi: 10.1159/000490898. [Epub ahead of print].
Boycott, K. M., Rath, A., Chong, J. X., Hartley, T., Alkuraya, F. S., Baynam, G., et al. (2017). International cooperation to enable the diagnosis of all rare genetic diseases. Am. J. Hum. Genet. 100, 695–705. doi: 10.1016/j.ajhg.2017.04.003
Brancati, F., Iannicelli, M., Travaglini, L., Mazzotta, A., Bertini, E., Boltshauser, E., et al. (2009). MKS3/TMEM67 mutations are a major cause of COACH Syndrome, a Joubert Syndrome related disorder with liver involvement. Hum. Mutat. 30, E432–E442. doi: 10.1002/humu.20924
Brooks, E. R., and Wallingford, J. B. (2014). Multiciliated cells. Curr. Biol. 24, R973–R982. doi: 10.1016/j.cub.2014.08.047
Choi, Y., Sims, G. E., Murphy, S., Miller, J. R., and Chan, A. P. (2012). Predicting the functional effect of amino acid substitutions and indels. PLoS ONE 7:e46688. doi: 10.1371/journal.pone.0046688
Chun, S., and Fay, J. C. (2009). Identification of deleterious mutations within three human genomes. Genome Res. 19, 1553–1561. doi: 10.1101/gr.092619.109
Cohen, R. A., Check, J. H., and Dougherty, M. P. (2016). Evidence that exposure to progesterone alone is a sufficient stimulus to cause a precipitous rise in the immunomodulatory protein the progesterone induced blocking factor (PIBF). J. Assist. Reprod. Genet. 33, 221–229. doi: 10.1007/s10815-015-0619-7
Delous, M., Baala, L., Salomon, R., Laclef, C., Vierkotten, J., Tory, K., et al. (2007). The ciliary gene RPGRIP1L is mutated in cerebello-oculo-renal syndrome (Joubert syndrome type B) and Meckel syndrome. Nat. Genet. 39, 875–881. doi: 10.1038/ng2039
Desai, P., Haber, H., Bulafka, J., Russell, A., Clifton, R., Zachary, J., et al. (2018). Impacts of variants of uncertain significance on parental perceptions of children after prenatal chromosome microarray testing. Prenat. Diagn. 38, 740–747. doi: 10.1002/pd.5323
Doherty, D. (2009). Joubert syndrome: insights into brain development, cilium biology, and complex disease. Semin. Pediatr. Neurol. 16, 143–154. doi: 10.1016/j.spen.2009.06.002
Dubaissi, E., and Papalopulu, N. (2011). Embryonic frog epidermis: a model for the study of cell-cell interactions in the development of mucociliary disease. Dis. Model. Mech. 4, 179–192. doi: 10.1242/dmm.006494
Duncan, A. R., and Khokha, M. K. (2016). Xenopus as a model organism for birth defects-Congenital heart disease and heterotaxy. Semin. Cell Dev. Biol. 51, 73–79. doi: 10.1016/j.semcdb.2016.02.022
Edvardson, S., Shaag, A., Zenvirt, S., Erlich, Y., Hannon, G. J., Shanske, A. L., et al. (2010). Joubert syndrome 2 (JBTS2) in Ashkenazi Jews is associated with a TMEM216 mutation. Am. J. Hum. Genet. 86, 93–97. doi: 10.1016/j.ajhg.2009.12.007
Evers, C., Staufner, C., Granzow, M., Paramasivam, N., Hinderhofer, K., Kaufmann, L., et al. (2017). Impact of clinical exomes in neurodevelopmental and neurometabolic disorders. Mol. Genet. Metab. 121, 297–307. doi: 10.1016/j.ymgme.2017.06.014
Fagerberg, L., Hallstrom, B. M., Oksvold, P., Kampf, C., Djureinovic, D., Odeberg, J., et al. (2014). Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteomics 13, 397–406. doi: 10.1074/mcp.M113.035600
Franco, B., and Thauvin-Robinet, C. (2016). Update on oral-facial-digital syndromes (OFDS). Cilia 5:12. doi: 10.1186/s13630-016-0034-4
Getwan, M., and Lienkamp, S. S. (2017). Toolbox in a tadpole: Xenopus for kidney research. Cell Tissue Res. 369, 143–157. doi: 10.1007/s00441-017-2611-2
Gonzalez-Arenas, A., Valadez-Cosmes, P., Jimenez-Arellano, C., Lopez-Sanchez, M., and Camacho-Arroyo, I. (2014). Progesterone-induced blocking factor is hormonally regulated in human astrocytoma cells, and increases their growth through the IL-4R/JAK1/STAT6 pathway. J. Steroid Biochem. Mol. Biol. 144(Pt B), 463–46470. doi: 10.1016/j.jsbmb.2014.09.007
Gupta, G. D., Coyaud, E., Goncalves, J., Mojarad, B. A., Liu, Y., Wu, Q., et al. (2015). A Dynamic protein interaction landscape of the human centrosome-cilium interface. Cell 163, 1484–1499. doi: 10.1016/j.cell.2015.10.065
Gutierrez-Rodriguez, A., Hansberg-Pastor, V., and Camacho-Arroyo, I. (2017). Proliferative and invasive effects of progesterone-induced blocking factor in human glioblastoma cells. Biomed. Res. Int. 2017:1295087. doi: 10.1155/2017/1295087
Hebbar, M., Kanthi, A., Shukla, A., Bielas, S., and Girisha, K. M. (2018). A biallelic 36-bp insertion in PIBF1 is associated with Joubert syndrome. J. Hum. Genet. 63, 935–939. doi: 10.1038/s10038-018-0462-7
Huang, L., Szymanska, K., Jensen, V. L., Janecke, A. R., Innes, A. M., Davis, E. E., et al. (2011). TMEM237 is mutated in individuals with a Joubert syndrome related disorder and expands the role of the TMEM family at the ciliary transition zone. Am. J. Hum. Genet. 89, 713–730. doi: 10.1016/j.ajhg.2011.11.005
Hudic, I., Stray-Pedersen, B., Szekeres-Bartho, J., Fatusic, Z., Dizdarevic-Hudic, L., Tomic, V., et al. (2015). Maternal serum progesterone-induced blocking factor (PIBF) in the prediction of preterm birth. J. Reprod. Immunol. 109, 36–40. doi: 10.1016/j.jri.2015.02.006
Hudic, I., Szekeres-Bartho, J., Stray-Pedersen, B., Fatusic, Z., Polgar, B., and Ecim-Zlojutro, V. (2016). Lower urinary and serum progesterone-induced blocking factor in women with preterm birth. J. Reprod. Immunol. 117, 66–69. doi: 10.1016/j.jri.2016.07.003
Iannicelli, M., Brancati, F., Mougou-Zerelli, S., Mazzotta, A., Thomas, S., Elkhartoufi, N., et al. (2010). Novel TMEM67 mutations and genotype-phenotype correlates in meckelin-related ciliopathies. Hum. Mutat. 31, E1319–E1331. doi: 10.1002/humu.21239
Johnston, J. J., Lee, C., Wentzensen, I. M., Parisi, M. A., Crenshaw, M. M., Sapp, J. C., et al. (2017). Compound heterozygous alterations in intraflagellar transport protein CLUAP1 in a child with a novel Joubert and oral-facial-digital overlap syndrome. Cold Spring Harb. Mol. Case Stud. 3:a001321. doi: 10.1101/mcs.a001321
Khanna, H., Hurd, T. W., Lillo, C., Shu, X., Parapuram, S. K., He, S., et al. (2005). RPGR-ORF15, which is mutated in retinitis pigmentosa, associates with SMC1, SMC3, and microtubule transport proteins. J. Biol. Chem. 280, 33580–33587. doi: 10.1074/jbc.M505827200
Kim, K., Lee, K., and Rhee, K. (2012). CEP90 is required for the assembly and centrosomal accumulation of centriolar satellites, which is essential for primary cilia formation. PLoS ONE 7:e48196. doi: 10.1371/journal.pone.0048196
Kim, K., and Rhee, K. (2011). The pericentriolar satellite protein CEP90 is crucial for integrity of the mitotic spindle pole. J. Cell Sci. 124(Pt 3), 338–347. doi: 10.1242/jcs.078329
Kircher, M., Witten, D. M., Jain, P., O'Roak, B. J., Cooper, G. M., and Shendure, J. (2014). A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 46, 310–315. doi: 10.1038/ng.2892
Knopp, C., Rudnik-Schoneborn, S., Eggermann, T., Bergmann, C., Begemann, M., Schoner, K., et al. (2015). Syndromic ciliopathies: from single gene to multi gene analysis by SNP arrays and next generation sequencing. Mol. Cell. Probes 29, 299–307. doi: 10.1016/j.mcp.2015.05.008
Kodani, A., Yu, T. W., Johnson, J. R., Jayaraman, D., Johnson, T. L., Al-Gazali, L., et al. (2015). Centriolar satellites assemble centrosomal microcephaly proteins to recruit CDK2 and promote centriole duplication. eLife 4:7519. doi: 10.7554/eLife.07519
Kroes, H. Y., Monroe, G. R., van der Zwaag, B., Duran, K. J., de Kovel, C. G., van Roosmalen, M. J., et al. (2016). Joubert syndrome: genotyping a Northern European patient cohort. Eur. J. Hum. Genet. 24, 214–220. doi: 10.1038/ejhg.2015.84
Kurian, A. W., Li, Y., Hamilton, A. S., Ward, K. C., Hawley, S. T., Morrow, M., et al. (2017). Gaps in incorporating germline genetic testing into treatment decision-making for early-stage breast cancer. J. Clin. Oncol. 35, 2232–2239. doi: 10.1200/JCO.2016.71.6480
Lachmann, M., Gelbmann, D., Kalman, E., Polgar, B., Buschle, M., Von Gabain, A., et al. (2004). PIBF (progesterone induced blocking factor) is overexpressed in highly proliferating cells and associated with the centrosome. Int. J. Cancer 112, 51–60. doi: 10.1002/ijc.20326
Leitch, C. C., Zaghloul, N. A., Davis, E. E., Stoetzel, C., Diaz-Font, A., Rix, S., et al. (2008). Hypomorphic mutations in syndromic encephalocele genes are associated with Bardet-Biedl syndrome. Nat. Genet. 40, 443–448. doi: 10.1038/ng.97
Lek, M., Karczewski, K. J., Minikel, E. V., Samocha, K. E., Banks, E., Fennell, T., et al. (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291. doi: 10.1038/nature19057
Liu, X., Jian, X., and Boerwinkle, E. (2013). dbNSFP v2.0: a database of human non-synonymous SNVs and their functional predictions and annotations. Hum. Mutat. 34, E2393–E2402. doi: 10.1002/humu.22376
Malicdan, M. C., Vilboux, T., Stephen, J., Maglic, D., Mian, L., Konzman, D., et al. (2015). Mutations in human homologue of chicken talpid3 gene (KIAA0586) cause a hybrid ciliopathy with overlapping features of Jeune and Joubert syndromes. J. Med. Genet. 52, 830–839. doi: 10.1136/jmedgenet-2015-103316
Maria, B. L., Hoang, K. B., Tusa, R. J., Mancuso, A. A., Hamed, L. M., Quisling, R. G., et al. (1997). “Joubert syndrome” revisited: key ocular motor signs with magnetic resonance imaging correlation. J. Child Neurol. 12, 423–430. doi: 10.1177/088307389701200703
Maria, B. L., Quisling, R. G., Rosainz, L. C., Yachnis, A. T., Gitten, J., Dede, D., et al. (1999). Molar tooth sign in Joubert syndrome: clinical, radiologic, and pathologic significance. J. Child Neurol. 14, 368–376. doi: 10.1177/088307389901400605
Miller, S. A., Dykes, D. D., and Polesky, H. F. (1988). A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 16:1215. doi: 10.1093/nar/16.3.1215
Moody, S. A. (2000). Cell lineage analysis in Xenopus embryos. Methods Mol. Biol. 135, 331–347. doi: 10.1385/1-59259-685-1:331
Mougou-Zerelli, S., Thomas, S., Szenker, E., Audollent, S., Elkhartoufi, N., Babarit, C., et al. (2009). CC2D2A mutations in Meckel and Joubert syndromes indicate a genotype-phenotype correlation. Hum. Mutat. 30, 1574–1582. doi: 10.1002/humu.21116
Ng, P. C., and Henikoff, S. (2003). SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 31, 3812–3814. doi: 10.1093/nar/gkg509
Paramasivam, N., Granzow, M., Evers, C., Hinderhofer, K., Wiemann, S., Bartram, C. R., et al. (2018). Identification and prioritization of causal variants of human genetic disorders from exome or whole genome sequencing data. OBM Genet. doi: 10.1101/209882. [Epub ahead of print].
Parisi, M., and Glass, I. (1993). “Joubert syndrome,” in GeneReviewsⓇ, eds M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. H. Bean, K. Stephens, and A. Amemiya (Seattle, WA).
Park, T. J., Mitchell, B. J., Abitua, P. B., Kintner, C., and Wallingford, J. B. (2008). Dishevelled controls apical docking and planar polarization of basal bodies in ciliated epithelial cells. Nat. Genet. 40, 871–879. doi: 10.1038/ng.104
Polgar, B., Kispal, G., Lachmann, M., Paar, C., Nagy, E., Csere, P., et al. (2003). Molecular cloning and immunologic characterization of a novel cDNA coding for progesterone-induced blocking factor. J. Immunol. 171, 5956–5963. doi: 10.4049/jimmunol.171.11.5956
Reva, B., Antipin, Y., and Sander, C. (2011). Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 39:e118. doi: 10.1093/nar/gkr407
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Romani, M., Micalizzi, A., Kraoua, I., Dotti, M. T., Cavallin, M., Sztriha, L., et al. (2014). Mutations in B9D1 and MKS1 cause mild Joubert syndrome: expanding the genetic overlap with the lethal ciliopathy Meckel syndrome. Orphanet J. Rare Dis. 9:72. doi: 10.1186/1750-1172-9-72
Romani, M., Micalizzi, A., and Valente, E. M. (2013). Joubert syndrome: congenital cerebellar ataxia with the molar tooth. Lancet Neurol. 12, 894–905. doi: 10.1016/S1474-4422(13)70136-4
Roosing, S., Romani, M., Isrie, M., Rosti, R. O., Micalizzi, A., Musaev, D., et al. (2016). Mutations in CEP120 cause Joubert syndrome as well as complex ciliopathy phenotypes. J. Med. Genet. 53, 608–615. doi: 10.1136/jmedgenet-2016-103832
Saunier, S., Salomon, R., and Antignac, C. (2005). Nephronophthisis. Curr. Opin. Genet. Dev. 15, 324–331. doi: 10.1016/j.gde.2005.04.012
Schwarz, J. M., Rodelsperger, C., Schuelke, M., and Seelow, D. (2010). MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 7, 575–576. doi: 10.1038/nmeth0810-575
Shaheen, R., Almoisheer, A., Faqeih, E., Babay, Z., Monies, D., Tassan, N., et al. (2015). Identification of a novel MKS locus defined by TMEM107 mutation. Hum. Mol. Genet. 24, 5211–5218. doi: 10.1093/hmg/ddv242
Shaheen, R., Shamseldin, H. E., Loucks, C. M., Seidahmed, M. Z., Ansari, S., Ibrahim Khalil, M., et al. (2014). Mutations in CSPP1, encoding a core centrosomal protein, cause a range of ciliopathy phenotypes in humans. Am. J. Hum. Genet. 94, 73–79. doi: 10.1016/j.ajhg.2013.11.010
Shihab, H. A., Gough, J., Cooper, D. N., Stenson, P. D., Barker, G. L., Edwards, K. J., et al. (2013). Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum. Mutat. 34, 57–65. doi: 10.1002/humu.22225
Slaats, G. G., Isabella, C. R., Kroes, H. Y., Dempsey, J. C., Gremmels, H., Monroe, G. R., et al. (2016). MKS1 regulates ciliary INPP5E levels in Joubert syndrome. J. Med. Genet. 53, 62–72. doi: 10.1136/jmedgenet-2015-103250
Spassky, N., Han, Y. G., Aguilar, A., Strehl, L., Besse, L., Laclef, C., et al. (2008). Primary cilia are required for cerebellar development and Shh-dependent expansion of progenitor pool. Dev. Biol. 317, 246–259. doi: 10.1016/j.ydbio.2008.02.026
Srour, M., Hamdan, F. F., McKnight, D., Davis, E., Mandel, H., Schwartzentruber, J., et al. (2015). Joubert syndrome in French Canadians and identification of mutations in CEP104. Am. J. Hum. Genet. 97, 744–753. doi: 10.1016/j.ajhg.2015.09.009
Srour, M., Hamdan, F. F., Schwartzentruber, J. A., Patry, L., Ospina, L. H., Shevell, M. I., et al. (2012a). Mutations in TMEM231 cause Joubert syndrome in French Canadians. J. Med. Genet. 49, 636–641. doi: 10.1136/jmedgenet-2012-101132
Srour, M., Schwartzentruber, J., Hamdan, F. F., Ospina, L. H., Patry, L., Labuda, D., et al. (2012b). Mutations in C5ORF42 cause Joubert syndrome in the French Canadian population. Am. J. Hum. Genet. 90, 693–700. doi: 10.1016/j.ajhg.2012.02.011
Tallila, J., Salonen, R., Kohlschmidt, N., Peltonen, L., and Kestila, M. (2009). Mutation spectrum of Meckel syndrome genes: one group of syndromes or several distinct groups? Hum. Mutat. 30, E813–E830. doi: 10.1002/humu.21057
Thomas, S., Legendre, M., Saunier, S., Bessieres, B., Alby, C., Bonniere, M., et al. (2012). TCTN3 mutations cause Mohr-Majewski syndrome. Am. J. Hum. Genet. 91, 372–378. doi: 10.1016/j.ajhg.2012.06.017
Tuz, K., Bachmann-Gagescu, R., O'Day, D. R., Hua, K., Isabella, C. R., Phelps, I. G., et al. (2014). Mutations in CSPP1 cause primary cilia abnormalities and Joubert syndrome with or without Jeune asphyxiating thoracic dystrophy. Am. J. Hum. Genet. 94, 62–72. doi: 10.1016/j.ajhg.2013.11.019
Valente, E. M., Logan, C. V., Mougou-Zerelli, S., Lee, J. H., Silhavy, J. L., Brancati, F., et al. (2010). Mutations in TMEM216 perturb ciliogenesis and cause Joubert, Meckel and related syndromes. Nat. Genet. 42, 619–625. doi: 10.1038/ng.594
Vladar, E. K., and Stearns, T. (2007). Molecular characterization of centriole assembly in ciliated epithelial cells. J. Cell Biol. 178, 31–42. doi: 10.1083/jcb.200703064
Walentek, P., Bogusch, S., Thumberger, T., Vick, P., Dubaissi, E., Beyer, T., et al. (2014). A novel serotonin-secreting cell type regulates ciliary motility in the mucociliary epidermis of Xenopus tadpoles. Development 141, 1526–1533. doi: 10.1242/dev.102343
Wang, K., Li, M., and Hakonarson, H. (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38:e164. doi: 10.1093/nar/gkq603
Wheway, G., Schmidts, M., Mans, D. A., Szymanska, K., Nguyen, T. T., Racher, H., et al. (2015). An siRNA-based functional genomics screen for the identification of regulators of ciliogenesis and ciliopathy genes. Nat. Cell Biol. 17, 1074–1087. doi: 10.1038/ncb3201
Wright, C. F., McRae, J. F., Clayton, S., Gallone, G., Aitken, S., FitzGerald, T. W., et al. (2018). Making new genetic diagnoses with old data: iterative reanalysis and reporting from genome-wide data in 1,133 families with developmental disorders. Genet. Med. 20, 1216–1223. doi: 10.1038/gim.2017.246
Yachie, N., Petsalaki, E., Mellor, J. C., Weile, J., Jacob, Y., Verby, M., et al. (2016). Pooled-matrix protein interaction screens using Barcode Fusion Genetics. Mol. Syst. Biol. 12:863. doi: 10.15252/msb.20156660
Zaghloul, N. A., and Katsanis, N. (2009). Mechanistic insights into Bardet-Biedl syndrome, a model ciliopathy. J. Clin. Invest. 119, 428–437. doi: 10.1172/JCI37041
Keywords: PIBF1, Joubert syndrome, Xenopus, molar tooth sign, cilia, ciliopathy
Citation: Ott T, Kaufmann L, Granzow M, Hinderhofer K, Bartram CR, Theiß S, Seitz A, Paramasivam N, Schulz A, Moog U, Blum M and Evers CM (2019) The Frog Xenopus as a Model to Study Joubert Syndrome: The Case of a Human Patient With Compound Heterozygous Variants in PIBF1. Front. Physiol. 10:134. doi: 10.3389/fphys.2019.00134
Received: 14 September 2018; Accepted: 04 February 2019;
Published: 25 February 2019.
Edited by:
Emily Sempou, Yale University, United StatesReviewed by:
Mustafa Khokha, Yale University, United StatesSally Ann Moody, George Washington University, United States
Copyright © 2019 Ott, Kaufmann, Granzow, Hinderhofer, Bartram, Theiß, Seitz, Paramasivam, Schulz, Moog, Blum and Evers. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christina M. Evers, Y2hyaXN0aW5hLmV2ZXJzQG1lZC51bmktaGVpZGVsYmVyZy5kZQ==
†These authors have contributed equally to this work