 Jie Ji1
Jie Ji1 Yi Tang2*
Yi Tang2*- 1Department of Comprehensive (VIP) Inpatient Ward, Sichuan Clinical Research Center for Cancer, Sichuan Cancer Hospital & Institute, Sichuan Cancer Center, Affiliated Cancer Hospital of University of Electronic Science and Technology of China, Chengdu, China
- 2Department of Pharmacy, Sichuan Clinical Research Center for Cancer, Sichuan Cancer Hospital & Institute, Sichuan Cancer Center, Affiliated Cancer Hospital of University of Electronic Science and Technology of China, Chengdu, China
Drug addiction is a chronic and potentially deadly disease that is considered a global health problem and describes the alteration of brain function by psychostimulant drugs through changes in the reward system. However, there is still no ideal strategy for the management of drug addiction. Previous studies have suggested that microglia are involved in events associated with neuroplasticity and memory, which are also related to drug addiction. Many studies have shown that psychoactive substances may act directly on immune cells, altering their function and inducing the production of various inflammatory mediators. In recent years, a ketogenic diet (KD) was shown to have therapeutic benefits as a dietary therapy for a variety of neurological disorders. With respect to drug addiction, studies have shown that a KD can alleviate glucose metabolism disorders caused by alcohol use disorders by increasing ketone metabolism, thereby reducing withdrawal symptoms. This finding indicates the potential of a KD as a treatment for drug addiction, since a KD may promote the transition of microglia to a predominantly anti-inflammatory state through several mechanisms. Here, we discuss recent research showing that a KD plays a variety of roles in controlling microglia-mediated inflammation, opening new treatment avenues to treat drug addiction. This succinct analysis offers evidence of the enormous potential of a KD to treat drug addiction through the inhibition of microglial activation.
Introduction
Drug addiction is a chronic, recurrent condition that has recently gained recognition as a public health issue in numerous nations (Schottenfeld and O'Malley, 2016). The hallmarks of this illness are obsessively seeking and using a substance; an inability to control one’s intake of the substance; and a depressive mood, with little consideration for social, physical, or individual repercussions (Koob and Volkow, 2016).
Current studies have shown that signalling pathways involving dopamine receptors may be responsible for the adaptive cellular responses in the central nervous system (CNS) caused by prolonged exposure to psychoactive drugs. For example, D1 activation increases cAMP levels, which in turn activates extracellular signal-regulated kinase (ERK) and protein kinase A (PKA) (Andrianarivelo et al., 2019). The activation of these molecules leads to the activation of transcription factors such as delta FosB (ΔFosB), factors regulated by cAMP response element-binding protein (CREB), myocyte enhancer factor 2 (MEF2), and nuclear factor kappa B (NF-κB), which are related to changes in gene and protein expression (Nestler, 2012), receptor expression, neuronal excitability and cell morphology (Teague and Nestler, 2022). Psychostimulants can alter neuroplasticity-related signalling cascades, which are essential for the desire for psychostimulants and associated drug seeking and relapse (Krasnova et al., 2013).
Recently, it was demonstrated that microglia may regulate some of these molecular pathways. For example, microglia can change the shape and function of dopaminergic neurons by altering the expression of receptors and the levels of tyrosine hydroxylase (TH) and dopamine transporter (DAT); it has also been demonstrated the fluorescence intensity of DAT and TH in the ventral tegmental area (VTA) is decreased in male mice exposed to social stress compared with unhandled control male mice (Catale et al., 2022; Smith et al., 2020). Microglia also participate in glutamate-induced synaptic alterations by altering AMPA receptor expression, the AMPAR/NMDAR ratio, and glutamate release (Basilico et al., 2022; Ji et al., 2013). Finally, neurotransmission mediated by glutamate and dopamine also affects microglial activation (Yan et al., 2015), including in the context of psychostimulant abuse (Canedo et al., 2021). In this review, we found that the addiction-related behavioural alterations caused by binge methamphetamine exposure are mediated by astrocyte‒microglia crosstalk, in which the release of glutamate from astrocytes in a TNF/IP3 receptor (IP3R)/SNARE-dependent manner leads to microglial activation, neuroinflammation, and ultimately changes in addictive behaviour in mice (Canedo et al., 2021).
Encouragingly, multiple studies have revealed a strong correlation between diet-induced ketosis and the primarily anti-inflammatory polarization of microglia in animals (Fedorovich et al., 2018; Ghosh et al., 2018). Moreover, several researchers have reported that nutritional ketosis results in the inhibition of molecules such as mitogen-activated protein kinase (MAPK), p38, NF-κB and the nucleotide-binding, leucine-rich repeat, pyrin-domain-containing 3 (NLRP3) inflammasome (Guo et al., 2018; Trotta et al., 2019) in addition to an increase in the synthesis of substances such as peroxisome proliferator-activated receptor (PPAR) and interleukin (IL)-10 (Newman et al., 2017). These changes may favour a switch to predominantly anti-inflammatory/neurorestorative microglial polarization (Xu et al., 2018; Yang et al., 2018). Therefore, these findings suggest that diet-induced ketosis may have pleiotropic effects on important mediators of microglial function. If this is also true in human patients, new avenues for treating neurological disorders could be explored.
A ketogenic diet (KD) is a diet with a reduced proportion of carbohydrates and an increased proportion of fat (Veech, 2004). A KD is characterized by adequate energy and protein intake and the restriction of carbohydrates, typically to less than 30–50 g/day (Trimboli et al., 2020). In a high-fat diet, calories from fat typically constitute 30%–75% of the total daily caloric intake. However, some nutritional protocols may aim for an even higher percentage of calories from fat, reaching up to 90%. High-fat diets are usually characterized by excessive intake of saturated fatty acids and calories, which can lead to an increase in health problems, particularly heart disease, weight gain, and obesity (Han et al., 2023).
A KD induces a state of ketosis that is similar to that caused by fasting. Normally, carbohydrates are converted to glucose, which the brain uses as its primary energy source. However, when no carbohydrates are available, the body uses other energy sources, such as acetyl coenzyme A (ac-CoA), which produces excess ketone molecules, including acetoacetate (ACA), β-hydroxybutyrate (BHB), and acetone, through a process called ketogenesis (Fukao et al., 2004). Unlike pathological ketoacidosis, ketosis is a normal process, as ketone bodies can be utilized effectively without reaching dangerous levels (Paoli et al., 2013). In the past, a KD was widely and successfully used to treat epileptic disorders (Neal et al., 2008). According to recent studies, a KD may also be beneficial for treating alcohol or drug addiction (Li et al., 2022; Wiers et al., 2021). For example, research has demonstrated that a KD with a ratio of fats to carbohydrates and proteins of approximately 6:1 (93% fat, 2% carbohydrates and 5% protein; 5 weeks) interferes with the sensitization of ambulatory responses in cocaine-treated animals and reduces the stereotyped responses elicited by cocaine in rats (Martinez et al., 2019);. In clinical studies, a KD (80% fat, 15% protein, and 5% carbohydrates; 3 weeks) reduced alcohol craving and withdrawal symptoms in patients with alcohol use disorders. In addition, BHB, a KD metabolite and the most abundant ketone body, can β-hydroxybutyrylate CaMKII-α, resulting in significant inhibition of T286 autophosphorylation and decreased CaMKII activity, which plays a critical role in mediating the effect of KD consumption in reducing cocaine reinstatement (Li et al., 2022).
The biochemistry of ketogenesis
Under physiological conditions, fatty acid oxidation produces acetyl-CoA, which enters the tricarboxylic acid (TCA) cycle and reacts chemically with oxaloacetate to form citrate. However, under the metabolic conditions induced by a KD, oxaloacetate is released from mitochondria and used in gluconeogenesis (Elamin et al., 2017). Under these conditions, the amount of oxaloacetate in the mitochondrial environment is far less than the amount of acetyl-CoA produced, and oxaloacetate undergoes a series of condensation processes that are characteristic of ketogenesis (Kim et al., 2010). First, acetoacetyl-CoA is generated by the combination of two acetyl-CoA molecules. HMG-CoA synthase 2 facilitates a functionally irreversible and rate-limiting interaction between acetoacetyl-CoA and another acetyl-CoA molecule to generate HMG-CoA (Hyatt et al., 2016). After HMG-CoA is produced, it dissociates to form ACA, which is then further reduced to BHB via a process facilitated by BHB dehydrogenase in which nicotinamide adenine dinucleotide (NAD)/NADH acts as a hydrogen donor (Bentourkia et al., 2009). Notably, BHB is the major ketone body (KB) because the level of BHB in the circulation and tissues is significantly greater than the level of ACA (Pifferi et al., 2008).
The liver releases BHB and ACA into the bloodstream, where they are subsequently taken up by the heart, brain, skeletal muscle, and other tissues with high metabolic needs (Kim et al., 2010). Once BHB reaches these tissues, BHB dehydrogenase converts BHB to ACA, which functions as a major regulator of the mitochondrial NAD+/NADH ratio (Pifferi et al., 2011). The enzyme succinyl-CoA:3-oxoacid CoA transferase then catalyses the hydrolysis of ACA to produce acetoacetyl-CoA and succinate, and thiolase cleaves acetoacetyl-CoA to yield acetyl-CoA. Acetyl-CoA and succinate serve as substrates for the TCA cycle and complex II of the electron transfer chain (ETC.), respectively (Roy et al., 2012) (Figure 1). This mechanism may explain the increased succinate dehydrogenase activity in rodents fed a KD for extended periods (Hashim and VanItallie, 2014). The effects of a KD can be replicated with KB supplements, and while not widely accepted, there is evidence that the normal ability of the liver to produce KBs may be impeded by KB supplementation (Hugo et al., 2012).
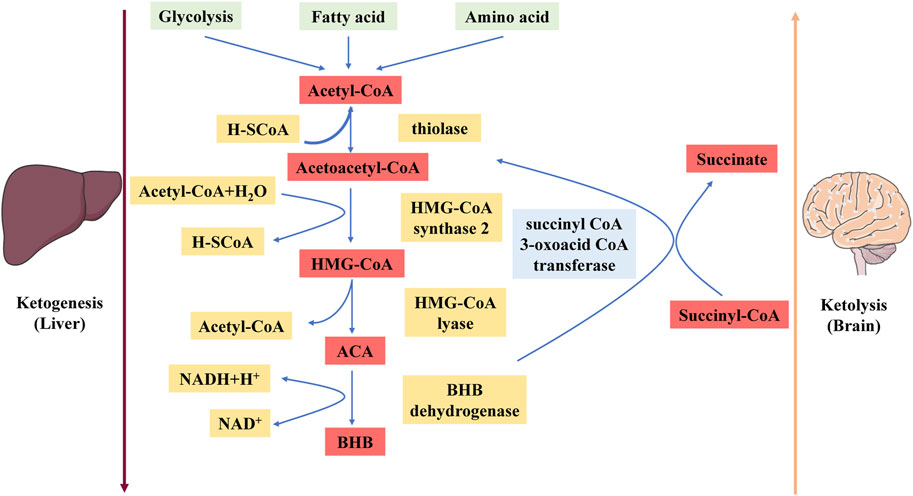
Figure 1. Schematic diagram depicting the ketogenesis and ketolysis reactions. ACA, acetoacetate; BHB, β-hydroxybutyrate. Under physiological conditions, acetyl-CoA produced by fatty acid oxidation enters the tricarboxylic acid (TCA) cycle and subsequently engages in a chemical reaction with oxaloacetate to produce citrate. The level of synthesized acetyl-CoA greatly exceeds the amount of oxaloacetate in the mitochondrial environment, and acetyl-CoA engages in a series of condensation reactions, which are hallmarks of ketogenesis. First, two acetyl-CoA molecules combine to produce acetoacetyl-CoA. This molecule reacts with another acetyl-CoA molecule to form HMG-CoA via a functionally irreversible and rate-limiting reaction facilitated by HMG-CoA synthase 2. Once formed, this compound dissociates to form the KB acetoacetate (ACA), which is further reduced to BHB via a reaction facilitated by BHB dehydrogenase in which NAD+/NADH acts as the hydrogen donor. BHB is released into the circulation from the liver and ultimately enters the brain.
Role of microglia in neural function and CNS homeostasis
Microglia surveil and quickly react to foreign substances under physiological conditions, actively observing and controlling alterations in neuronal activity (Schafer et al., 2012). Microglia take on this role because they briefly come into contact with synapses and extrasynaptic regions through highly coordinated movement (Salter and Beggs, 2014), and they express homologous receptors for neurotransmitters such as glutamate and gamma-aminobutyric acid (GABA) as well as receptors for a variety of neuronal mediators, such as CD200 and fractalkine (CX3CL1), on their surfaces (Koizumi et al., 2013). Additionally, microglia express receptors for purinergic neurotransmitters such as adenosine triphosphate (ATP), serotonin, and acetylcholine (Schafer et al., 2012). An overwhelming body of research suggests that ATP, which is generated by neurons, is a key regulator and the most effective inducer of microglial activity (Orr et al., 2009).
Numerous research groups have reported that microglia may control GABAergic and glutamatergic neurotransmission (Pascual et al., 2012). ATP, which binds to cognate receptors on astrocytes and increases glutamatergic activity by upregulating metabotropic receptor 5, is the most likely regulator of glutamatergic neurotransmission (Pascual et al., 2012). The ability of microglia to regulate GABAergic transmission is particularly significant under pathological conditions. This regulatory process involves the release of ATP from injured neurons, which elicits the release of brain-derived neurotrophic factor (BDNF) from activated microglia. The released BDNF has the potential to reverse the direction of GABAergic neurotransmission (Ferrini and De Koninck, 2013).
Responses of microglia to cocaine
Although changes in the glutamatergic and dopaminergic systems are thought to be the primary neurobiological mechanisms governing motivated behaviour, it is widely recognized that psychoactive substances can also modify the glutamatergic, serotonergic, and GABAergic neurotransmitter systems as well as the levels of various molecules, such as cytokines and neurotrophic factors (Doggui et al., 2021). Furthermore, microglia can respond to neurochemical alterations caused by psychoactive substances because they express ion channels and neurotransmitter receptors, which are likewise expressed by neurons (Figure 2). Notably, microglial activation has been demonstrated to alter the reward system (Taylor et al., 2015), mostly due to the high susceptibility of dopaminergic neurons to neuroinflammatory signals (Douma and de Kloet, 2020).
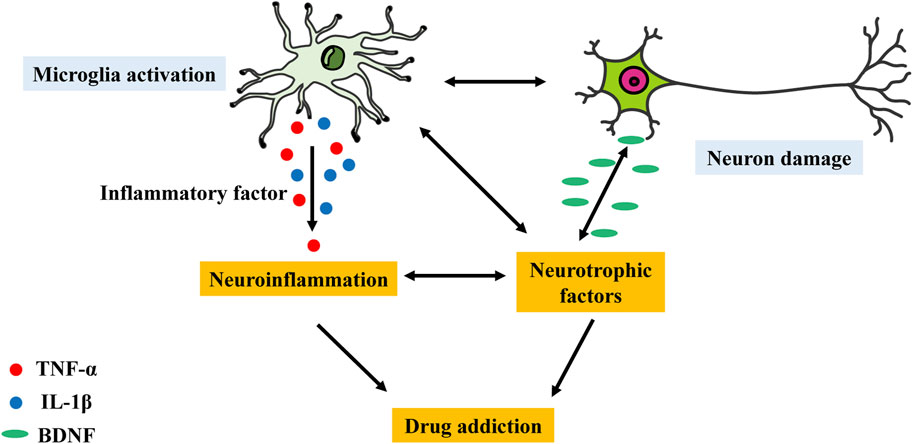
Figure 2. Possible effects through which the microglial activation–neuronal damage interaction may lead to neuroinflammation and further mediate drug addiction. Disruption of the interaction between microglial activation and neurons via inflammatory factors or neurotrophic factors may lead to neuroinflammation and drug addiction.
Microglia and cocaine
The crucial role of microglia in the development and maturation of synapses is widely acknowledged. Drugs of abuse impair synaptic and neuronal function over time. Thus, increasing amounts of data point to a possible link between drug addiction and glial function abnormalities (Kierdorf and Prinz, 2017; Schmidt and Engel, 2021). In particular, the abuse of opioids, alcohol, and psychostimulants is thought to be influenced by the glial and neuroimmune systems. It is hypothesized that substances of abuse activate microglia by stimulating innate immune receptors, causing the release of cytokines and chemokines, which have an impact on neuronal function (Schmidt and Engel, 2021). Moreover, innate immune receptor stimulation may cause synaptic remodelling directly (Schmidt and Engel, 2021). A theory known as the “xenobiotic hypothesis” has emerged recently. This hypothesis suggests that because substances of abuse are exogenous, they are viewed as foreign “invaders” that activate the immune system’s defence mechanism. Microglia, the main resident immune cells in the brain, act as the first line of defence. However, repeated and continuous administration of substances of abuse results in hyperactivation of microglia and a neuroinflammatory state, which can further exacerbate drug addiction by altering neuronal function (Deng et al., 2020). Microglia play an important role in drug addiction; a recent study showing that microglia are required for synaptic alterations during cocaine withdrawal lends further credence to this speculation (Cadet and Bisagno, 2014). More research on microglial overactivation might improve our knowledge of the mechanism of drug addiction and possibly lead to the development of innovative treatments.
Preclinical research has shown that cocaine upregulates the expression of the traditional microglial marker ionized calcium-binding adaptor molecule-1 (Iba-1) in the hippocampus, frontal cortex, and nucleus accumbens (NAc) (Costa et al., 2013; Jarvis et al., 2019), as well as in sections of the mouse and rat brain (Chivero et al., 2020). Furthermore, cocaine upregulates the expression of CD11 in the striatum, cortical areas, and VTA (Chivero et al., 2021). Interestingly, research has shown that acute cocaine injection elevates CD11 expression only in animals with a history of cocaine self-administration (Brown et al., 2018). Moreover, cocaine increases the expression of CD68, a transmembrane glycoprotein expressed by microglia, indicating increased phagocytic activity (da Silva et al., 2021). Cocaine affects the number and shape of microglia in addition to their protein expression. For example, research has shown that after cocaine is used for 7 days in a row, more cells express allograft inflammatory factor 1 (AIF1) (Thangaraj et al., 2020). Additionally, previous studies have shown that cocaine exposure decreases the number of microglial branches and increases the size of microglial cell bodies (Burkovetskaya et al., 2020; da Silva et al., 2021). In other words, cocaine causes morphologic changes that could be linked to heightened microglial activation.
Finally, the behavioural and molecular effects of psychostimulants can be altered by microglial activity (Linker et al., 2020). The inhibition of microglia by minocycline, a common tetracycline antibiotic, reverses the effects of cocaine on behaviour, conditioned place preference (CPP), and dopamine release (Northcutt et al., 2015). Moreover, minocycline also modifies cocaine reward after exposure to morphine and nicotine (Taylor et al., 2016). Furthermore, the glial modulator and phosphodiesterase 4 (PDE4) inhibitor ibudilast ameliorates cocaine addiction in both humans and animals (Mu et al., 2021), and the colony stimulating factor-1 (CSF-1) inhibitor PLX3397 also reduces cocaine-induced behavioural changes by depleting microglia (Linker et al., 2020).
There have been few clinical studies on the effects of cocaine on microglia in humans, and those that exist have generated controversial results. The only study that was performed to assess cocaine-induced microglial activation in a clinical population exploited the binding of the PET radioligand [11C]PBR28 to TSPO (Narendran et al., 2014). However, the authors failed to demonstrate an alteration in [11C]PBR28 binding to TSPO in abstinent (for a minimum of 14 days) patients who met the DSM-IV criteria for cocaine dependence compared with healthy controls (Narendran et al., 2014).
Positive effects of induced ketosis on anti-inflammatory microglial polarization
Increased NAD+ production and GPR109A receptor activation
Numerous studies have confirmed that, after prolonged ketosis, NAD+ levels are increased in the CNS of animals (Xin et al., 2018). Other researchers have reported the inhibition of glycolysis in the brains of study participants (Courchesne-Loyer et al., 2017). These findings may have therapeutic implications since the elevation of NAD+ levels and simultaneous reduction in NADH levels, along with the inhibition of glycolysis, may cause the anti-inflammatory effects of a ketotic state on microglia in the brain. An increase in the NAD/NADH ratio has an important effect, as it alters the activity of the transcriptional inhibitor C-terminal-binding protein (CtBP), which binds to the acetyltransferase p300 in microglia and other myeloid lineage cells to regulate the transcription of NF-κB and the expression of other proinflammatory genes. However, this hypothesis needs to be empirically confirmed (Shen et al., 2017). One of the defining characteristics of proinflammatory microglial polarization is increased glycolysis, which increases NADH levels and prevents CtBP from dimerizing, eliminating its ability to function as a transcriptional repressor. The opposite is true for decreased glycolysis and increased oxidative phosphorylation, which increase NAD+ levels (Ghosh et al., 2018; Shen et al., 2017). Notably, an increase in NAD+ levels in the brain due to diet-induced ketosis may have a beneficial anti-inflammatory effect through the binding of CtBP to the promoter regions of genes that promote inflammation in microglia. Moreover, research suggests that a more direct way to produce the same effect is through the binding of KBs, particularly BHB (Pinto et al., 2018). Furthermore, because KBs promote the primarily anti-inflammatory polarization of microglia, they may represent potential therapeutic options.
According to previous research, BHB inhibits the generation of COX-2 and iNOS by activating microglia both in vivo and in vitro, partly through the activation of G protein-coupled receptor 109A (GPR109A) (Fu et al., 2015). This receptor decreases the degradation of IkBa and prevents NF-κB from translocating to the nucleus, which allows the NF-κB to induce the transcription of inflammatory molecules (Fu et al., 2015). Studies have also shown that rats that have ingested or been injected with BHB have lower NLRP3 activity in microglia and other cells of the CNS. This decrease in activity could contribute independently to a reduction in neuroinflammation by lowering IL-1 and IL-18 levels (Yamanashi et al., 2017). Additionally, this effect may be partially mediated by binding to GPR109A and the consequent suppression of NLRP3 assembly due to endoplasmic reticulum (ER) stress (Youm et al., 2015). Although clearly important, this mechanism is not the only one through which BHB administration may decrease ER stress and, subsequently, the in vivo activation of the inflammasome. For example, BHB can reduce ER stress by blocking mitochondrial fragmentation mediated by dynamin-related protein 1 (DRP-1) and stimulating AMP-activated protein kinase (AMPK) (Bae et al., 2016). From the perspective of anxiety and depression treatment, the potential of diet-induced ketosis to inhibit NLRP3 activity in the brain is interesting because there is increasing evidence that stress-mediated activation of this inflammasome precipitates or exacerbates anxiety and depression symptoms, whereas its inhibition results in their amelioration or, in certain cases, termination (Iwata et al., 2016).
Suppression of histone deacetylase
Nuclear factor erythroid 2-related factor 2 (Nrf-2) in the brain is also upregulated by ketosis (Milder et al., 2010). Although the underlying mechanisms are unknown, this change appears to be directly caused by BHB (Izuta et al., 2018) and probably occurs via histone deacetylase inhibition (Cai et al., 2015). In animal models of various neurological diseases or traumatic brain injury, Nrf-2 upregulation has been shown to be positively correlated with the predominantly anti-inflammatory polarization of microglia, with concomitant decreases in iNOS and IL-6 production as well as the induction of anti-inflammatory M2 polarization in vivo. Studies have explored the potential of Nrf-2 upregulation as a therapeutic approach for reducing neuroinflammation (Li et al., 2018). Although the mechanisms underlying these beneficial effects of Nrf-2 upregulation in ameliorating microglial activity and neuroinflammation involve the inhibition of inflammatory factor secretion and NLRP3 activity, they are complementary to the mechanisms underlying the effects of KD consumption and BHB administration. However, they are sufficiently different to suggest a level of synergy that would not be possible by modifying a single biochemical pathway (Ahmed et al., 2017). For example, the anti-inflammatory effect of Nrf-2 activity results from conserved cross-talk between Nrf-2 and NLRP3 in vivo, which is facilitated by a sophisticated mechanism that works antagonistically with the Rho family kinase RAK to limit the development of inflammation and oxidative stress (Cuadrado et al., 2014).
Conclusion and outlook
In this article, we summarize the intriguing potential of a KD to treat drug addiction through the modification of microglial activation. Interestingly, the possible therapeutic benefits of KD consumption and other strategies for fostering ketosis for CNS diseases have not received much attention in the field of neuropsychiatry. The neurobiological mechanisms of various mental disorders, especially drug addiction, may be significantly influenced by primarily proinflammatory microglial polarization, as indicated by accumulating evidence from preclinical, postmortem, and in vivo human studies. A KD was shown to be beneficial in various neurological disorders, which suggests that its potential to treat drug addiction cannot be disregarded. Decreasing microglial activation via consumption of a KD might be beneficial for numerous illnesses caused by drug addiction.
KD consumption may be promoted recovery from drug addiction through various effects. However, some researchers have shown concerns regarding the consumption of a KD in individuals neurological diseases, such as decreased appetite, increased risk of malnutrition, and several adverse effects (Włodarek, 2019). The common adverse effects of KD consumption include metabolic abnormalities, gastrointestinal symptoms, kidney stones, and slow growth in children (Kossoff et al., 2009). However, most of these adverse effects were observed in children. Therefore, a KD should be applied with caution in people with drug addiction, as this particular population often suffers from multisystem disorders such as increased risk of malnutrition (Jeynes and Gibson, 2017) and gastrointestinal symptoms (Su et al., 2020). Finally, further preclinical studies and randomized controlled clinical trials are needed to optimize KD strategies, such as the timing of intervention and nutrient composition, and assess the suitability, effectiveness, and safety of a KD in the treatment of drug addiction.
Author contributions
JJ: Writing - original draft. YT: Writing - review and editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
We are very grateful to the Sichuan Cancer Hospital and Institute, Sichuan Cancer Center, Affiliate Cancer Hospital of University of Electronic Science and Technology of China (UESTC) for their support.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Ahmed, S. M., Luo, L., Namani, A., Wang, X. J., and Tang, X. (2017). Nrf2 signaling pathway: pivotal roles in inflammation. Biochim. Biophys. Acta Mol. Basis Dis. 1863, 585–597. doi:10.1016/j.bbadis.2016.11.005
Andrianarivelo, A., Saint-Jour, E., Walle, R., Trifilieff, P., and Vanhoutte, P. (2019). Modulation and functions of dopamine receptor heteromers in drugs of abuse-induced adaptations. Neuropharmacology 152, 42–50. doi:10.1016/j.neuropharm.2018.12.003
Bae, H. R., Kim, D. H., Park, M. H., Lee, B., Kim, M. J., Lee, E. K., et al. (2016). β-Hydroxybutyrate suppresses inflammasome formation by ameliorating endoplasmic reticulum stress via AMPK activation. Oncotarget 7, 66444–66454. doi:10.18632/oncotarget.12119
Basilico, B., Ferrucci, L., Ratano, P., Golia, M. T., Grimaldi, A., Rosito, M., et al. (2022). Microglia control glutamatergic synapses in the adult mouse hippocampus. Glia 70, 173–195. doi:10.1002/glia.24101
Bentourkia, M., Tremblay, S., Pifferi, F., Rousseau, J., Lecomte, R., and Cunnane, S. (2009). PET study of 11C-acetoacetate kinetics in rat brain during dietary treatments affecting ketosis. Am. J. Physiol. Endocrinol. Metab. 296, E796–E801. doi:10.1152/ajpendo.90644.2008
Brown, K. T., Levis, S. C., O'Neill, C. E., Northcutt, A. L., Fabisiak, T. J., Watkins, L. R., et al. (2018). Innate immune signaling in the ventral tegmental area contributes to drug-primed reinstatement of cocaine seeking. Brain Behav. Immun. 67, 130–138. doi:10.1016/j.bbi.2017.08.012
Burkovetskaya, M. E., Small, R., Guo, L., Buch, S., and Guo, M. L. (2020). Cocaine self-administration differentially activates microglia in the mouse brain. Neurosci. Lett. 728, 134951. doi:10.1016/j.neulet.2020.134951
Cadet, J. L., and Bisagno, V. (2014). Glial-neuronal ensembles: partners in drug addiction-associated synaptic plasticity. Front. Pharmacol. 5, 204. doi:10.3389/fphar.2014.00204
Cai, D., Yin, S., Yang, J., Jiang, Q., and Cao, W. (2015). Histone deacetylase inhibition activates Nrf2 and protects against osteoarthritis. Arthritis Res. Ther. 17, 269. doi:10.1186/s13075-015-0774-3
Canedo, T., Portugal, C. C., Socodato, R., Almeida, T. O., Terceiro, A. F., Bravo, J., et al. (2021). Astrocyte-derived TNF and glutamate critically modulate microglia activation by methamphetamine. Neuropsychopharmacology 46, 2358–2370. doi:10.1038/s41386-021-01139-7
Catale, C., Lo Iacono, L., Martini, A., Heil, C., Guatteo, E., Mercuri, N. B., et al. (2022). Early life social stress causes sex- and region-dependent dopaminergic changes that are prevented by minocycline. Mol. Neurobiol. 59, 3913–3932. doi:10.1007/s12035-022-02830-6
Chivero, E. T., Liao, K., Niu, F., Tripathi, A., Tian, C., Buch, S., et al. (2020). Engineered extracellular vesicles loaded with miR-124 attenuate cocaine-mediated activation of microglia. Front. Cell Dev. Biol. 8, 573. doi:10.3389/fcell.2020.00573
Chivero, E. T., Thangaraj, A., Tripathi, A., Periyasamy, P., Guo, M. L., and Buch, S. (2021). NLRP3 inflammasome blockade reduces cocaine-induced microglial activation and neuroinflammation. Mol. Neurobiol. 58, 2215–2230. doi:10.1007/s12035-020-02184-x
Costa, B. M., Yao, H., Yang, L., and Buch, S. (2013). Role of endoplasmic reticulum (ER) stress in cocaine-induced microglial cell death. J. Neuroimmune Pharmacol. 8, 705–714. doi:10.1007/s11481-013-9438-8
Courchesne-Loyer, A., Croteau, E., Castellano, C. A., St-Pierre, V., Hennebelle, M., and Cunnane, S. C. (2017). Inverse relationship between brain glucose and ketone metabolism in adults during short-term moderate dietary ketosis: a dual tracer quantitative positron emission tomography study. J. Cereb. Blood Flow. Metab. 37, 2485–2493. doi:10.1177/0271678X16669366
Cuadrado, A., Martín-Moldes, Z., Ye, J., and Lastres-Becker, I. (2014). Transcription factors NRF2 and NF-κB are coordinated effectors of the Rho family, GTP-binding protein RAC1 during inflammation. J. Biol. Chem. 289, 15244–15258. doi:10.1074/jbc.M113.540633
da Silva, M. C. M., Gomes, G. F., de Barros Fernandes, H., da Silva, A. M., Teixeira, A. L., Moreira, F. A., et al. (2021). Inhibition of CSF1R, a receptor involved in microglia viability, alters behavioral and molecular changes induced by cocaine. Sci. Rep. 11, 15989. doi:10.1038/s41598-021-95059-7
Deng, S. L., Chen, J. G., and Wang, F. (2020). Microglia: a central player in depression. Curr. Med. Sci. 40, 391–400. doi:10.1007/s11596-020-2193-1
Doggui, R., Elsawy, W., Conti, A. A., and Baldacchino, A. (2021). Association between chronic psychoactive substances use and systemic inflammation: a systematic review and meta-analysis. Neurosci. Biobehav Rev. 125, 208–220. doi:10.1016/j.neubiorev.2021.02.031
Douma, E. H., and de Kloet, E. R. (2020). Stress-induced plasticity and functioning of ventral tegmental dopamine neurons. Neurosci. Biobehav Rev. 108, 48–77. doi:10.1016/j.neubiorev.2019.10.015
Elamin, M., Ruskin, D. N., Masino, S. A., and Sacchetti, P. (2017). Ketone-based metabolic therapy: is increased NAD(+) a primary mechanism? Front. Mol. Neurosci. 10, 377. doi:10.3389/fnmol.2017.00377
Fedorovich, S. V., Voronina, P. P., and Waseem, T. V. (2018). Ketogenic diet versus ketoacidosis: what determines the influence of ketone bodies on neurons? Neural Regen. Res. 13, 2060–2063. doi:10.4103/1673-5374.241442
Ferrini, F., and De Koninck, Y. (2013). Microglia control neuronal network excitability via BDNF signalling. Neural Plast. 2013, 429815. doi:10.1155/2013/429815
Fu, S. P., Wang, J. F., Xue, W. J., Liu, H. M., Liu, B. R., Zeng, Y. L., et al. (2015). Anti-inflammatory effects of BHBA in both in vivo and in vitro Parkinson's disease models are mediated by GPR109A-dependent mechanisms. J. Neuroinflammation 12, 9. doi:10.1186/s12974-014-0230-3
Fukao, T., Lopaschuk, G. D., and Mitchell, G. A. (2004). Pathways and control of ketone body metabolism: on the fringe of lipid biochemistry. Prostagl. Leukot. Essent. Fat. Acids 70, 243–251. doi:10.1016/j.plefa.2003.11.001
Ghosh, S., Castillo, E., Frias, E. S., and Swanson, R. A. (2018). Bioenergetic regulation of microglia. Glia 66, 1200–1212. doi:10.1002/glia.23271
Guo, M., Wang, X., Zhao, Y., Yang, Q., Ding, H., Dong, Q., et al. (2018). Ketogenic diet improves brain ischemic tolerance and inhibits NLRP3 inflammasome activation by preventing drp1-mediated mitochondrial fission and endoplasmic reticulum stress. Front. Mol. Neurosci. 11, 86. doi:10.3389/fnmol.2018.00086
Han, Y., Wu, H., Sun, S., Zhao, R., Deng, Y., Zeng, S., et al. (2023). Effect of high fat diet on disease development of polycystic ovary syndrome and lifestyle intervention strategies. Nutrients 15, 2230. doi:10.3390/nu15092230
Hashim, S. A., and VanItallie, T. B. (2014). Ketone body therapy: from the ketogenic diet to the oral administration of ketone ester. J. Lipid Res. 55, 1818–1826. doi:10.1194/jlr.R046599
Hugo, S. E., Cruz-Garcia, L., Karanth, S., Anderson, R. M., Stainier, D. Y., and Schlegel, A. (2012). A monocarboxylate transporter required for hepatocyte secretion of ketone bodies during fasting. Genes Dev. 26, 282–293. doi:10.1101/gad.180968.111
Hyatt, H. W., Kephart, W. C., Holland, A. M., Mumford, P., Mobley, C. B., Lowery, R. P., et al. (2016). A ketogenic diet in rodents elicits improved mitochondrial adaptations in response to resistance exercise training compared to an isocaloric western diet. Front. Physiol. 7, 533. doi:10.3389/fphys.2016.00533
Iwata, M., Ota, K. T., Li, X. Y., Sakaue, F., Li, N., Dutheil, S., et al. (2016). Psychological stress activates the inflammasome via release of adenosine triphosphate and stimulation of the purinergic type 2X7 receptor. Biol. Psychiatry 80, 12–22. doi:10.1016/j.biopsych.2015.11.026
Izuta, Y., Imada, T., Hisamura, R., Oonishi, E., Nakamura, S., Inagaki, E., et al. (2018). Ketone body 3-hydroxybutyrate mimics calorie restriction via the Nrf2 activator, fumarate, in the retina. Aging Cell 17, e12699. doi:10.1111/acel.12699
Jarvis, R., Tamashiro-Orrego, A., Promes, V., Tu, L., Shi, J., and Yang, Y. (2019). Cocaine self-administration and extinction inversely alter neuron to glia exosomal dynamics in the nucleus accumbens. Front. Cell Neurosci. 13, 581. doi:10.3389/fncel.2019.00581
Jeynes, K. D., and Gibson, E. L. (2017). The importance of nutrition in aiding recovery from substance use disorders: a review. Drug Alcohol Depend. 179, 229–239. doi:10.1016/j.drugalcdep.2017.07.006
Ji, K., Akgul, G., Wollmuth, L. P., and Tsirka, S. E. (2013). Microglia actively regulate the number of functional synapses. PLoS One 8, e56293. doi:10.1371/journal.pone.0056293
Kierdorf, K., and Prinz, M. (2017). Microglia in steady state. J. Clin. Invest. 127, 3201–3209. doi:10.1172/JCI90602
Kim, D. Y., Vallejo, J., and Rho, J. M. (2010). Ketones prevent synaptic dysfunction induced by mitochondrial respiratory complex inhibitors. J. Neurochem. 114, 130–141. doi:10.1111/j.1471-4159.2010.06728.x
Koizumi, S., Ohsawa, K., Inoue, K., and Kohsaka, S. (2013). Purinergic receptors in microglia: functional modal shifts of microglia mediated by P2 and P1 receptors. Glia 61, 47–54. doi:10.1002/glia.22358
Koob, G. F., and Volkow, N. D. (2016). Neurobiology of addiction: a neurocircuitry analysis. Lancet Psychiatry 3, 760–773. doi:10.1016/S2215-0366(16)00104-8
Kossoff, E. H., Zupec-Kania, B. A., Amark, P. E., Ballaban-Gil, K. R., Christina Bergqvist, A. G., Blackford, R., et al. (2009). Optimal clinical management of children receiving the ketogenic diet: recommendations of the International Ketogenic Diet Study Group. Epilepsia 50, 304–317. doi:10.1111/j.1528-1167.2008.01765.x
Krasnova, I. N., Chiflikyan, M., Justinova, Z., McCoy, M. T., Ladenheim, B., Jayanthi, S., et al. (2013). CREB phosphorylation regulates striatal transcriptional responses in the self-administration model of methamphetamine addiction in the rat. Neurobiol. Dis. 58, 132–143. doi:10.1016/j.nbd.2013.05.009
Li, C., Chen, T., Zhou, H., Zhang, C., Feng, Y., Tang, F., et al. (2018). Schisantherin A attenuates neuroinflammation in activated microglia: role of Nrf2 activation through ERK phosphorylation. Cell Physiol. Biochem. 47, 1769–1784. doi:10.1159/000491059
Li, H., Wan, X., Wu, Z., Zhou, Y., Chen, R., Xu, W., et al. (2022). β-hydroxybutyrate reduces reinstatement of cocaine conditioned place preference through hippocampal CaMKII-α β-hydroxybutyrylation. Cell Rep. 41, 111724. doi:10.1016/j.celrep.2022.111724
Linker, K. E., Gad, M., Tawadrous, P., Cano, M., Green, K. N., Wood, M. A., et al. (2020). Microglial activation increases cocaine self-administration following adolescent nicotine exposure. Nat. Commun. 11, 306. doi:10.1038/s41467-019-14173-3
Martinez, L. A., Lees, M. E., Ruskin, D. N., and Masino, S. A. (2019). A ketogenic diet diminishes behavioral responses to cocaine in young adult male and female rats. Neuropharmacology 149, 27–34. doi:10.1016/j.neuropharm.2019.02.001
Milder, J. B., Liang, L. P., and Patel, M. (2010). Acute oxidative stress and systemic Nrf2 activation by the ketogenic diet. Neurobiol. Dis. 40, 238–244. doi:10.1016/j.nbd.2010.05.030
Mu, L., Liu, X., Yu, H., Hu, M., Friedman, V., Kelly, T. J., et al. (2021). Ibudilast attenuates cocaine self-administration and prime- and cue-induced reinstatement of cocaine seeking in rats. Neuropharmacology 201, 108830. doi:10.1016/j.neuropharm.2021.108830
Narendran, R., Lopresti, B. J., Mason, N. S., Deuitch, L., Paris, J., Himes, M. L., et al. (2014). Cocaine abuse in humans is not associated with increased microglial activation: an 18-kDa translocator protein positron emission tomography imaging study with [11C]PBR28. J. Neurosci. 34, 9945–9950. doi:10.1523/JNEUROSCI.0928-14.2014
Neal, E. G., Chaffe, H., Schwartz, R. H., Lawson, M. S., Edwards, N., Fitzsimmons, G., et al. (2008). The ketogenic diet for the treatment of childhood epilepsy: a randomised controlled trial. Lancet Neurol. 7, 500–506. doi:10.1016/S1474-4422(08)70092-9
Nestler, E. J. (2012). Transcriptional mechanisms of drug addiction. Clin. Psychopharmacol. Neurosci. 10, 136–143. doi:10.9758/cpn.2012.10.3.136
Newman, J. C., Covarrubias, A. J., Zhao, M., Yu, X., Gut, P., Ng, C. P., et al. (2017). Ketogenic diet reduces midlife mortality and improves memory in aging mice. Cell Metab. 26, 547–557. doi:10.1016/j.cmet.2017.08.004
Northcutt, A. L., Hutchinson, M. R., Wang, X., Baratta, M. V., Hiranita, T., Cochran, T. A., et al. (2015). DAT isn't all that: cocaine reward and reinforcement require Toll-like receptor 4 signaling. Mol. Psychiatry 20, 1525–1537. doi:10.1038/mp.2014.177
Orr, A. G., Orr, A. L., Li, X. J., Gross, R. E., and Traynelis, S. F. (2009). Adenosine A(2A) receptor mediates microglial process retraction. Nat. Neurosci. 12, 872–878. doi:10.1038/nn.2341
Paoli, A., Rubini, A., Volek, J. S., and Grimaldi, K. A. (2013). Beyond weight loss: a review of the therapeutic uses of very-low-carbohydrate (ketogenic) diets. Eur. J. Clin. Nutr. 67, 789–796. doi:10.1038/ejcn.2013.116
Pascual, O., Ben Achour, S., Rostaing, P., Triller, A., and Bessis, A. (2012). Microglia activation triggers astrocyte-mediated modulation of excitatory neurotransmission. Proc. Natl. Acad. Sci. U. S. A. 109, E197–E205. doi:10.1073/pnas.1111098109
Pifferi, F., Tremblay, S., Croteau, E., Fortier, M., Tremblay-Mercier, J., Lecomte, R., et al. (2011). Mild experimental ketosis increases brain uptake of 11C-acetoacetate and 18F-fluorodeoxyglucose: a dual-tracer PET imaging study in rats. Nutr. Neurosci. 14, 51–58. doi:10.1179/1476830510Y.0000000001
Pifferi, F., Tremblay, S., Plourde, M., Tremblay-Mercier, J., Bentourkia, M., and Cunnane, S. C. (2008). Ketones and brain function: possible link to polyunsaturated fatty acids and availability of a new brain PET tracer, 11C-acetoacetate. Epilepsia 49 (Suppl. 8), 76–79. doi:10.1111/j.1528-1167.2008.01842.x
Pinto, A., Bonucci, A., Maggi, E., Corsi, M., and Businaro, R. (2018). Anti-oxidant and anti-inflammatory activity of ketogenic diet: new perspectives for neuroprotection in alzheimer's disease. Antioxidants (Basel) 7, 63. doi:10.3390/antiox7050063
Roy, M., Nugent, S., Tremblay-Mercier, J., Tremblay, S., Courchesne-Loyer, A., Beaudoin, J. F., et al. (2012). The ketogenic diet increases brain glucose and ketone uptake in aged rats: a dual tracer PET and volumetric MRI study. Brain Res. 1488, 14–23. doi:10.1016/j.brainres.2012.10.008
Salter, M. W., and Beggs, S. (2014). Sublime microglia: expanding roles for the guardians of the CNS. Cell 158, 15–24. doi:10.1016/j.cell.2014.06.008
Schafer, D. P., Lehrman, E. K., Kautzman, A. G., Koyama, R., Mardinly, A. R., Yamasaki, R., et al. (2012). Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74, 691–705. doi:10.1016/j.neuron.2012.03.026
Schmidt, K., and Engel, P. (2021). Mechanisms underlying gut microbiota-host interactions in insects. J. Exp. Biol. 224, jeb207696. doi:10.1242/jeb.207696
Schottenfeld, R. S., and O'Malley, S. S. (2016). Meeting the growing need for heroin addiction treatment. JAMA Psychiatry 73, 437–438. doi:10.1001/jamapsychiatry.2016.0139
Shen, Y., Kapfhamer, D., Minnella, A. M., Kim, J. E., Won, S. J., Chen, Y., et al. (2017). Bioenergetic state regulates innate inflammatory responses through the transcriptional co-repressor CtBP. Nat. Commun. 8, 624. doi:10.1038/s41467-017-00707-0
Smith, B. L., Laaker, C. J., Lloyd, K. R., Hiltz, A. R., and Reyes, T. M. (2020). Adolescent microglia play a role in executive function in male mice exposed to perinatal high fat diet. Brain Behav. Immun. 84, 80–89. doi:10.1016/j.bbi.2019.11.010
Su, H., Chen, T., Zhong, N., Jiang, H., Du, J., Xiao, K., et al. (2020). Decreased GABA concentrations in left prefrontal cortex of methamphetamine dependent patients: a proton magnetic resonance spectroscopy study. J. Clin. Neurosci. 71, 15–20. doi:10.1016/j.jocn.2019.11.021
Taylor, A. M., Castonguay, A., Ghogha, A., Vayssiere, P., Pradhan, A. A., Xue, L., et al. (2016). Neuroimmune regulation of GABAergic neurons within the ventral tegmental area during withdrawal from chronic morphine. Neuropsychopharmacology 41, 949–959. doi:10.1038/npp.2015.221
Taylor, A. M., Castonguay, A., Taylor, A. J., Murphy, N. P., Ghogha, A., Cook, C., et al. (2015). Microglia disrupt mesolimbic reward circuitry in chronic pain. J. Neurosci. 35, 8442–8450. doi:10.1523/JNEUROSCI.4036-14.2015
Teague, C. D., and Nestler, E. J. (2022). Key transcription factors mediating cocaine-induced plasticity in the nucleus accumbens. Mol. Psychiatry 27, 687–709. doi:10.1038/s41380-021-01163-5
Thangaraj, A., Periyasamy, P., Guo, M. L., Chivero, E. T., Callen, S., and Buch, S. (2020). Mitigation of cocaine-mediated mitochondrial damage, defective mitophagy and microglial activation by superoxide dismutase mimetics. Autophagy 16, 289–312. doi:10.1080/15548627.2019.1607686
Trimboli, P., Castellana, M., Bellido, D., and Casanueva, F. F. (2020). Confusion in the nomenclature of ketogenic diets blurs evidence. Rev. Endocr. Metab. Disord. 21, 1–3. doi:10.1007/s11154-020-09546-9
Trotta, M. C., Maisto, R., Guida, F., Boccella, S., Luongo, L., Balta, C., et al. (2019). The activation of retinal HCA2 receptors by systemic beta-hydroxybutyrate inhibits diabetic retinal damage through reduction of endoplasmic reticulum stress and the NLRP3 inflammasome. PLoS One 14, e0211005. doi:10.1371/journal.pone.0211005
Veech, R. L. (2004). The therapeutic implications of ketone bodies: the effects of ketone bodies in pathological conditions: ketosis, ketogenic diet, redox states, insulin resistance, and mitochondrial metabolism. Prostagl. Leukot. Essent. Fat. Acids 70, 309–319. doi:10.1016/j.plefa.2003.09.007
Wiers, C. E., Vendruscolo, L. F., van der Veen, J. W., Manza, P., Shokri-Kojori, E., Kroll, D. S., et al. (2021). Ketogenic diet reduces alcohol withdrawal symptoms in humans and alcohol intake in rodents. Sci. Adv. 7, eabf6780. doi:10.1126/sciadv.abf6780
Włodarek, D. (2019). Role of ketogenic diets in neurodegenerative diseases (Alzheimer's Disease and Parkinson's Disease). Nutrients 11, 169. doi:10.3390/nu11010169
Xin, L., Ipek, Ö., Beaumont, M., Shevlyakova, M., Christinat, N., Masoodi, M., et al. (2018). Nutritional Ketosis Increases NAD(+)/NADH Ratio in Healthy Human Brain: An in vivo Study by (31)P-MRS. Front. Nutr. 5, 62. doi:10.3389/fnut.2018.00062
Xu, X., Yin, D., Ren, H., Gao, W., Li, F., Sun, D., et al. (2018). Selective NLRP3 inflammasome inhibitor reduces neuroinflammation and improves long-term neurological outcomes in a murine model of traumatic brain injury. Neurobiol. Dis. 117, 15–27. doi:10.1016/j.nbd.2018.05.016
Yamanashi, T., Iwata, M., Kamiya, N., Tsunetomi, K., Kajitani, N., Wada, N., et al. (2017). Beta-hydroxybutyrate, an endogenic NLRP3 inflammasome inhibitor, attenuates stress-induced behavioral and inflammatory responses. Sci. Rep. 7, 7677. doi:10.1038/s41598-017-08055-1
Yan, Y., Jiang, W., Liu, L., Wang, X., Ding, C., Tian, Z., et al. (2015). Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell 160, 62–73. doi:10.1016/j.cell.2014.11.047
Yang, S. J., Shao, G. F., Chen, J. L., and Gong, J. (2018). The NLRP3 Inflammasome: An Important Driver of Neuroinflammation in Hemorrhagic Stroke. Cell Mol. Neurobiol. 38, 595–603. doi:10.1007/s10571-017-0526-9
Keywords: ketogenic diet, microglia, inflammation, drug addiction, βhydroxybutyrate
Citation: Ji J and Tang Y (2025) A ketogenic diet regulates microglial activation to treat drug addiction. Front. Pharmacol. 16:1462699. doi: 10.3389/fphar.2025.1462699
Received: 10 July 2024; Accepted: 07 January 2025;
Published: 23 January 2025.
Edited by:
Sijie Tan, University of South China, ChinaReviewed by:
Ramón Sotomayor-Zárate, Universidad de Valparaiso, ChileFu Gao, Yale University, United States
Copyright © 2025 Ji and Tang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yi Tang, MTM2NjgyNDMyMTlAMTYzLmNvbQ==