 Heng Jiang1†
Heng Jiang1† Qiang Ye
Qiang Ye Yanfei Du
Yanfei Du- 1Department of Cardiology, The Affiliated Hospital of Southwest Medical University, Luzhou, Sichuan, China
- 2Key Laboratory of Medical Electrophysiology, Ministry of Education and Medical Electrophysiological Key Laboratory of Sichuan Province, Institute of Cardiovascular Research, Southwest Medical University, Luzhou, China
Cardiac fibrosis is a serious health problem because it is a common pathological change in almost all forms of cardiovascular diseases. Cardiac fibrosis is characterized by the transdifferentiation of cardiac fibroblasts (CFs) into cardiac myofibroblasts and the excessive deposition of extracellular matrix (ECM) components produced by activated myofibroblasts, which leads to fibrotic scar formation and subsequent cardiac dysfunction. However, there are currently few effective therapeutic strategies protecting against fibrogenesis. This lack is largely because the molecular mechanisms of cardiac fibrosis remain unclear despite extensive research. The Janus kinase/signal transducer and activator of transcription (JAK/STAT) signaling cascade is an extensively present intracellular signal transduction pathway and can regulate a wide range of biological processes, including cell proliferation, migration, differentiation, apoptosis, and immune response. Various upstream mediators such as cytokines, growth factors and hormones can initiate signal transmission via this pathway and play corresponding regulatory roles. STAT3 is a crucial player of the JAK/STAT pathway and its activation is related to inflammation, malignant tumors and autoimmune illnesses. Recently, the JAK/STAT3 signaling has been in the spotlight for its role in the occurrence and development of cardiac fibrosis and its activation can promote the proliferation and activation of CFs and the production of ECM proteins, thus leading to cardiac fibrosis. In this manuscript, we discuss the structure, transactivation and regulation of the JAK/STAT3 signaling pathway and review recent progress on the role of this pathway in cardiac fibrosis. Moreover, we summarize the current challenges and opportunities of targeting the JAK/STAT3 signaling for the treatment of fibrosis. In summary, the information presented in this article is critical for comprehending the role of the JAK/STAT3 pathway in cardiac fibrosis, and will also contribute to future research aimed at the development of effective anti-fibrotic therapeutic strategies targeting the JAK/STAT3 signaling.
1 Introduction
Cardiovascular disease is still the major cause of global death despite great progress in treatment methods. Myocardial fibrosis is a common pathology of most cardiovascular diseases at the end stage (Rockey et al., 2015). It can destroy the cardiac structure, impair cardiac excitation-contraction coupling, and impede cardiac function of both contraction and relaxation, thereby promoting the development of cardiovascular disease into heart failure (Gyöngyösi et al., 2017; Nguyen et al., 2017). The order of severity of cardiac fibrosis is related to higher long-term mortality of cardiovascular disease, particularly heart failure (Azevedo et al., 2010; Aoki et al., 2011). Due to the complex and incompletely elucidated mechanisms of fibrosis, there is currently no specific antifibrotic treatment available for cardiac fibrosis.
The Janus kinase/signal transducer and activator of transcription (JAK/STAT) signaling pathway, as a central communication node within cells, plays an essential role in a variety of pathophysiological activities like cell division, differentiation, immune regulation and tumorigenesis (Zhang J. Q. et al., 2022). It has been reported that many upstream mediators can activate this pathway to exert their biological functions, comprising growth factors, hormones, and cytokines (Darnell et al., 1994; Liu J. et al., 2023). The JAK/STAT pathway consists of three parts: ligand-receptor complexes, JAKs, along with transcription factors STATs. Among the STAT protein family, STAT3 is the most well-studied member and its activation can play beneficial or detrimental roles in various diseases. On the one hand, STAT3 shows highly activated in most cancers and cardiac injuries (Xian et al., 2021; Zhuang et al., 2022) and is demonstrated to be a pathogenic regulator (Yu and Jove, 2004). On the other hand, STAT3 is also recognized as a protective molecule, and its activation may confer cardioprotection against several cardiovascular diseases including ischemia and ischemia-reperfusion injury (Negoro et al., 2000; Fuglesteg et al., 2008; Harhous et al., 2019) and cardiac hypertrophy (Enomoto et al., 2015). Recently, accumulating evidence has confirmed a novel profibrotic role of the JAK/STAT3 signaling activation in multiple tissues and organs, including the heart (Bao et al., 2020), liver (Ogata et al., 2006), kidney (Zheng et al., 2019), lung (Celada et al., 2018), and skin (Dees et al., 2020). In this regard, the JAK/STAT3 pathway may emerge as a potential therapeutic target for treating fibrotic diseases (Barry et al., 2007). However, there is a lack of a comprehensive summary on the role of the JAK/STAT3 signaling in mediating cardiac fibrosis. In this review, we discuss the structure, transactivation and regulation of the JAK/STAT3 signaling pathway and review current progress on the role of this pathway in cardiac fibrosis and challenges and opportunities of targeting the JAK/STAT3 signaling for the treatment of fibrosis.
2 The cellular and molecular mechanisms of cardiac fibrosis
Cardiac fibrosis usually occurs when myocardial tissue is suffering from a pathological stimulus such as ischemia, hypoxia, overload, inflammation or other pathogenic factors. It serves a dual role: it protects myocardial tissue integrity as a normal reparative response during injury, yet persistent and excessive scar formation greatly impairs the heart’s systolic and diastolic functions (Leask, 2015). Cardiac fibrosis not only increases ventricular stiffness but also induces the secretion of growth factors and cytokines to promote cardiomyocyte hypertrophy, ultimately leading to a decline in myocardial compliance, heart failure, and even sudden death (Mohammed et al., 2015; Francis Stuart et al., 2016).
Cardiac fibrosis is a common pathological feature manifested by multiple cardiovascular diseases, such as heart failure, hypertension, arrhythmia, cardiomyopathy, and myocardial infarction, and also plays a significant role in their onset and progression (Tao et al., 2014; Chen et al., 2015; Chung et al., 2021; Qi et al., 2022). Cardiac fibrosis manifests as the over-proliferation and differentiation of CFs and massive accumulation of extracellular matrix (ECM) components in the myocardium, like fibronectin, type I collagen, and type III collagen (Schafer et al., 2017). Myofibroblasts differentiated from CFs can synthesize contractile proteins like α-smooth muscle actin (α-SMA), leading to the distortion of tissue and cell structure (Hinz, 2007; Hinz, 2010). On the other hand, myofibroblasts can express excessive amounts of ECM proteins, thus leading to the substitution of permanent fibrotic scars for normal tissues, increased cardiac stiffness, and varying degrees of cardiac diastolic and systolic dysfunction (Weber, 1989; Cleutjens et al., 1995; Dobaczewski et al., 2006; Liu et al., 2017; Wang et al., 2022b).
The source of myofibroblasts in fibrotic hearts remains a disputed matter. Although some studies indicate that a significant proportion of myofibroblasts may originate from endothelial cells, epithelial cells or hematopoietic fibroblast progenitors (Möllmann et al., 2006; Zeisberg et al., 2007; Aisagbonhi et al., 2011), prevailing evidence confirms that the primary source of myofibroblasts in fibrotic heart tissue could be the activation of resident CFs (Ali et al., 2014; Moore-Morris et al., 2014; Kanisicak et al., 2016; Shinde and Frangogiannis, 2017; Moore-Morris et al., 2018). Furthermore, it has been suggested that pericytes could potentially serve as a reservoir of myofibroblasts, but the precise mechanism by which they operate remains uncertain, and there may be an overlap between pericytes and resident fibroblast subsets (Humphreys et al., 2010).
Although the molecular mechanisms involved in cardiac fibrosis are complex and variable, the transformation of CFs to myofibroblasts plays a central role in the process of cardiac fibrosis. Acute cardiac injury initiates a robust inflammatory response. This process involves the infiltration of immune cells into the cardiac tissue, which subsequently release inflammatory cytokines such as transforming growth factor (TGF)-β1, tumor necrosis factor-α (TNF-α) and interleukins (ILs) (Bujak and Frangogiannis, 2007; Christia et al., 2013). These cytokines activate CFs and instigate ECM remodeling through diverse signaling cascades. Concurrently, neurohormones within the renin-angiotensin-aldosterone system (RAAS) and the sympathetic nervous system, particularly Angiotensin II (Ang II), aldosterone, and catecholamines, are upregulated (Zou et al., 2004; Ferreira et al., 2016; Azushima et al., 2020). Their activation compels myofibroblasts to ramp up collagen production, culminating in the deposition of fibrotic tissue in the heart, which is a hallmark of cardiac remodeling. Additionally, mechanical stress, often a consequence of increased cardiac afterload in conditions like hypertension or valvular disease, prompts cardiomyocytes and fibroblasts to adapt by modifying their ECM, which alters their size, shape, and function (Li et al., 2018). Moreover, oxidative stress in the cardiac environment, primarily characterized by the overproduction of reactive oxygen species (ROS), inflicts direct cellular damage and fosters inflammation and apoptosis. These effects collectively trigger signaling pathways that exacerbate myocardial fibrosis (Grosche et al., 2018). Lastly, metabolic imbalances, including the production of advanced glycation end-products (AGEs) and lipotoxicity in cardiomyocytes, along with vascular implications like endothelial dysfunction, significantly contribute to the progression of cardiac fibrosis (Huby et al., 2015; Chen et al., 2016; Marciniec et al., 2017).
Among the aforementioned mediators, TGF-β1 is regarded as a central and potent profibrotic factor and evokes cardiac fibrosis mainly through activation of downstream classic small mother against decapentaplegic (Smad) signaling pathway. This process involves the binding of extracellular TGF-β1 ligand to TGF-β type II receptor (TGF-βRII), which phosphorylates TGF-β type I receptor (TGF-βRI). Activated TGF-βRI then phosphorylates and activates R-Smads (mainly Smad2 and Smad3), which further form a complex with Smad4. The complex moves to the nucleus and interacts with other co-activators to induce the transcription of fibrosis-related genes such as fibronectin, α-SMA and collagens (Shi and Massagué, 2003; Działo et al., 2018; Hu et al., 2018). Additionally, TGF-β1 also leads to cardiac fibrosis through activating several noncanonical (also called Smad-independent) signaling pathways, like phosphatidylinositol 3-kinase/protein kinase B (PI3K/Akt), mitogen-activated protein kinase [MAPK, mainly comprising p38, c-Jun NH2-terminal kinase (JNK) and extracellular signal-regulated kinase (ERK)] or Rho-like GTPases signaling pathways. In addition to the most common TGF-β signaling, the pathogenesis of cardiac fibrosis also involves a variety of other intracellular molecular pathways, including the JAK/STAT3 signaling (Zhang et al., 2019b), Wnt/β-Catenin signaling (Mizutani et al., 2016), integrin/focal adhesion kinase (FAK) signaling (Zhao et al., 2016; Molkentin et al., 2017), Hippo signaling (Singh et al., 2016), and myocardial related transcription factor (MRTF)/serum response factor (SRF) signaling (Tomasek et al., 2005; Lighthouse and Small, 2016). Therefore, targeting these fibrotic mediators or cascades could provide promising therapeutic approaches for treating fibrotic diseases.
3 Structure, function, transcriptional activity and regulation of the JAK/STAT3 signaling pathway
3.1 Molecular structure of STAT3
In mammals, there are seven proteins belonging to the STAT family, which consists of cytoplasmic transcription factors named STAT1-STAT4, STAT5a, STAT5b, and STAT6 (Hu et al., 2020b). Among these, STAT3 is the most extensively studied and plays pivotal roles in controlling various cellular biological processes. STAT3 was originally discovered in 1994 through a series of studies on cytokine-induced acute responses of target genes. Unlike other family members, global deletion of STAT3 can cause embryonic death. The STAT3 protein consists of 770 amino acid residues and, similar to other members of the STAT family, it can be divided into six distinct functional domains (Figure 1): an NH2-terminal domain (NTD), a coiled-coil domain (CCD), a DNA binding domain (DBD), a linker domain (LD), an Src homology 2 (SH2) domain, and a COOH-terminal transactivation domain (TAD). Each domain has a specific function (Hu et al., 2021) (Table 1).
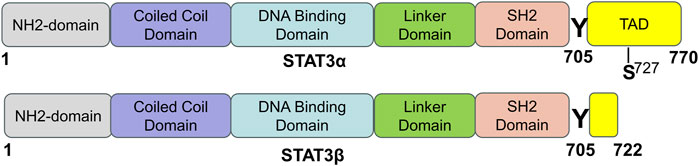
FIGURE 1. The domain structure and phosphorylation sites of STAT3 protein. STAT3 has two splicing isoforms, STAT3α and STAT3β, and they are comprised of 770 and 722 amino acids, respectively. STAT3 contains six different functional domains, including the NH2-terminal domain, coiled-coil domain, DNA binding domain, linker domain, SH2 domain, and COOH-terminal transactivation domain (TAD). “Y” means a tyrosine phosphorylation site, and “S” means a serine phosphorylation site [adapted from ref. (Hu et al., 2021).

TABLE 1. Function of STAT3 domains.
STAT3 is expressed widely in different cell types within the heart, such as cardiomyocytes, fibroblasts, immune cells, and endothelial cells. Two isoforms of the STAT3 protein, STAT3α (92 kDa) and STAT3β (83 kDa), are produced through alternative splicing of the identical gene. STAT3β is missing the COOH-terminal 55 amino acids, which are correspondingly replaced by seven distinct amino acid residues (Schaefer et al., 1995; Caldenhoven et al., 1996). Research has shown that while STAT3β is not vital for survival, mice deficient in STAT3α do not survive past birth (Maritano et al., 2004). STAT3α possesses two phosphorylation sites, namely, Tyr705 and Ser727, whereas STAT3β only possesses one phosphorylation site, specifically Tyr705. When either Tyr705 or Ser727 is phosphorylated, STAT3 is activated and exerts its function. STAT3 can be activated by more than 50 extracellular ligands, which are commonly some cytokines, hormones, growth factors, and chemokines, such as ILs, interferons, colony-stimulating factors, epidermal growth factor (EGF), and platelet-derived growth factor (PDGF) (Darnell, 1997; Hu et al., 2021). STAT3’s biological functions are complicated and diverse and its main physiological roles under normal conditions are summarized in the following section.
STAT3 is an important intracellular signaling molecule that has multiple functions under normal physiological conditions. These functions include: (1) Regulating the proliferation and differentiation of various cell types by binding to specific DNA sequences and affecting gene expression. For example, STAT3 promotes the proliferation of corneal limbal keratinocytes via a ΔNp63-dependent mechanism, and inhibiting this pathway can increase cell differentiation (Hsueh et al., 2011). STAT3 also mediates megakaryocyte differentiation induced by RAD001 (Su et al., 2013). (2) Regulating the activation, proliferation, and secretion of cytokines by immune cells, which can modulate immune responses and inflammation. For instance, STAT3 inhibition can induce apoptosis and/or activate effective immune responses in colon cancer cells, overcoming cancer-induced immune tolerance (Jahangiri et al., 2020). Likewise, systemic injection of penetrating c-Myc and gp130 peptides can inhibit pancreatic tumor growth and induce anti-tumor immunity (Aftabizadeh et al., 2021). (3) Mediating the expression of inflammation-related genes in response to various cytokines and growth factors. One of the most prominent examples is IL-6, which we will discuss in detail later. (4) Maintaining the self-renewal and differentiation of stem cells by regulating the transcription of target genes. Phosphorylated STAT3 is functionally associated with the expression of self-renewal genes in embryonic stem cells (Bourillot et al., 2009). Moreover, constitutively activated STAT3 can sustain the self-renewal process in the absence of leukemia inhibitory factor (LIF) (Matsuda et al., 1999). (5) Participating in tissue repair and regeneration processes by modulating cell survival and growth. For instance, Transmembrane and ubiquitin like domain containing 1 (Tmub1) inhibits the phosphorylation and activation of STAT3, impairing liver regeneration in mice after partial hepatectomy (Fu et al., 2019). Conversely, Krüppel-like factor 4 (KLF4) deletion in vivo induces axonal regeneration in adult retinal ganglion cells (RGCs) through the JAK/STAT3 signaling pathway. This regeneration can be further enhanced by removing the endogenous JAK/STAT3 pathway inhibitor SOCS3 (Qin et al., 2013). (6) Regulating the energy metabolism of cells by influencing the expression of mitochondrial oxidative phosphorylation-related genes. For example, icaritin inhibits the survival and glycolysis of glioblastoma (GBM) cells through the IL-6/STAT3 pathway (Li et al., 2019a). Additionally, STAT3 promotes mitochondrial respiration and reduces the production of ROS in neural precursor cells (Su et al., 2020). (7) Playing an essential role in early embryonic development, as embryos with STAT3 gene defects will die in the early stages of development. In humans, LIF and STAT3 are expressed in decidual tissue during early pregnancy. LIF can induce STAT3 phosphorylation in non-decidualized and decidualized human endometrial stromal cells in vitro, suggesting that LIF/STAT3 signaling is involved in human embryo implantation and decidualization (Shuya et al., 2011). Furthermore, conditional ablation of STAT3 in the uterus can result in embryo implantation failure (Lee et al., 2013).
3.2 Molecular structure of JAK
In mammals, the JAK family consists of four main members (JAK1-JAK3 and Tyk2), which are non-receptor tyrosine protein kinases (Schindler and Darnell, 1995). JAK1, JAK2, and Tyk2 have broad expression, whereas JAK3 is mainly present in cells of the hematopoietic lineage (Speirs et al., 2018). Upon interaction of cytokines or growth factors with their corresponding receptors, JAK tyrosine kinases are activated, thereby facilitating intracellular signal transduction.
The JAK protein is made up of seven similar regions (JH1-JH7) and includes four functional domains: a domain for tyrosine kinase, a domain for pseudokinase, an SH2 domain, and an NH2-terminal FERM domain (Four-point-one protein, Ezrin, Radixin, Moesin) (Figure 2) (Banerjee et al., 2017). The carboxy-terminal portion of each JAK includes the catalytic kinase domain (JH1) and the pseudokinase domain (JH2). JH1, containing nearly 250 amino acid residues, is the active phosphotransferase domain needed for phosphorylation of cytokine receptors and downstream STAT proteins. JH2 is similar to JH1 in structure, but it is generally considered to have no catalytic activity and can regulate the kinase activity of JH1 (Zhao et al., 2018; Xin et al., 2020). According to reports, the JAK2 protein’s JH2 exhibits a minimal level of kinase activity as stated by Ungureanu et al. (2011). The N-terminal region of each JAK contains the SH2 (JH3 with half of JH4) and FERM (JH5-JH7 and one-half of JH4) domains, which collectively facilitate the interaction between JAK proteins and the box1/2 regions of cytokine receptors located near the cell membrane (Saharinen et al., 2000; Wallweber et al., 2014; Hubbard, 2017; Morris et al., 2018; Xin et al., 2020; Raivola et al., 2021).
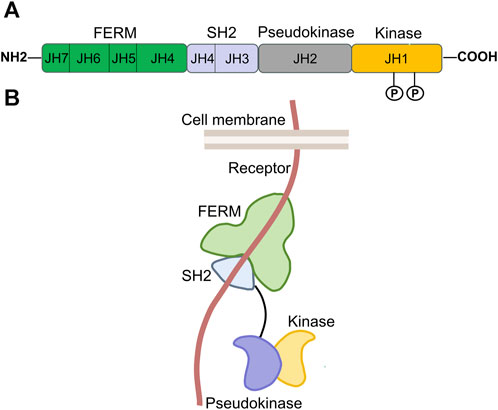
FIGURE 2. Structure of JAK. (A). Domains and conserved phosphorylation sites of the JAK protein. The JAK protein family contains four members, JAK1-3, and TYK2. Each is composed of seven homologous regions, labeled JH1-JH7. These regions make up four distinct functional domains, of which, JH1 corresponds to the kinase domain; JH2 is the pseudokinase domain; JH3 and a portion of JH4 together form the SH2 domain; and the combination of JH5, JH6, JH7, and the rest of JH4 constitutes the FERM domain. “P” represents conserved tyrosine phosphorylation sites of the JAK protein. (B). Three-dimensional spatial structure of JAK in cells [adapted from ref. (Hu et al., 2021).
3.3 Canonical JAK/STAT3 signaling pathway
The JAK/STAT signaling pathway is activated by more than 50 cytokines and growth factors, including hormones, interferons (IFN), ILs, and colony stimulating factors (Darnell, 1997). These molecules regulate various cellular events, such as hematopoiesis, immune adaptability, tissue repair, inflammation, cell apoptosis, and adipogenesis (Owen et al., 2019). The JAK/STAT3 pathway is activated when these extracellular ligands bind to their dedicated transmembrane receptors (Figure 3). The cytosolic domains of these receptors are constitutively interacting with receptor-related JAK tyrosine kinases. These JAK kinases are nonactivated before the ligand stimulation, while the coupling of the ligand with its receptor results in auto-phosphorylation of JAK kinases (Feng et al., 1997). Upon activation, the JAK molecules phosphorylate the cytoplasmic segment of the receptors at particular tyrosine residues, subsequently serving as binding sites for cytoplasmic STAT3 protein and attracting the recruitment of the STAT3 protein. After docking, STAT3 is phosphorylated by JAK kinase and subsequently associates with itself or other phosphorylated STAT monomers to create homodimers or heterodimers upon separation from the receptor. Ultimately, these dynamic molecular pairs migrate from the cytoplasm to the nucleus, where they attach to target gene promoters and stimulate the expression of target genes (O'Shea et al., 2015; Durham et al., 2019), often causing proliferation, differentiation, and apoptosis.
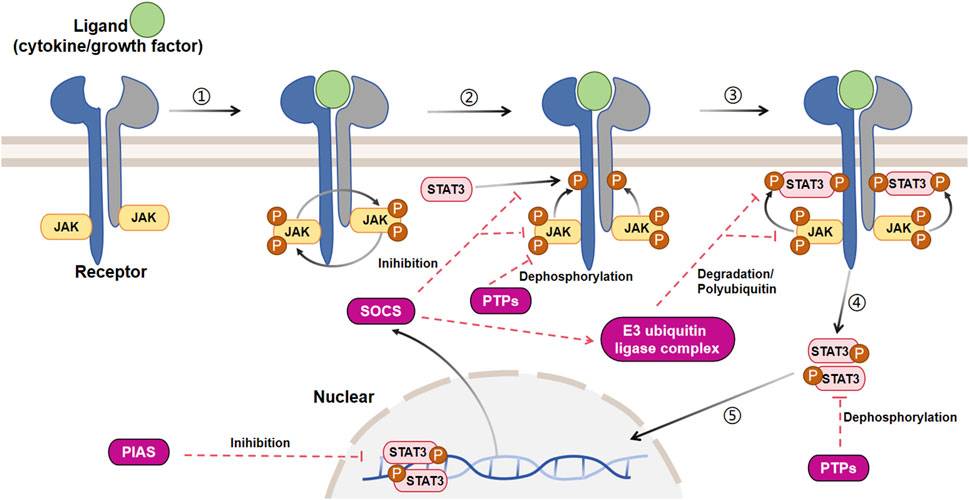
FIGURE 3. Signal transduction and negative regulation of the canonical JAK/STAT3 pathway. The JAK/STAT3 cascade is initiated by the interaction between a ligand and its corresponding receptor. This interaction leads to the auto-phosphorylation of the JAK kinase bound to the receptor. Once activated, JAK phosphorylates a tyrosine residue on the receptor, creating a docking site for cytoplasmic STAT3 and recruiting STAT3. At this docking site, JAK phosphorylates STAT3. The phosphorylated STAT3 then dissociates from the receptor and forms dimers. These STAT3 dimers move to the nucleus, where they bind to promoters and regulate transcription. The JAK/STAT3 cascade is controlled by three primary types of negative regulators: PTPs (protein tyrosine phosphatases), PIAS (protein inhibitor of activated STAT), and CIS/SOCS (suppressor of cytokine signaling). PTPs block the JAK/STAT3 signaling mainly by interacting directly with the STAT3 dimers and JAK to dephosphorylate them. PIAS prevents the JAK/STAT3 signaling principally by inhibiting the binding of STAT3 to DNA. As a common objective caused by the activation of JAK/STAT3, CIS/SOCS mainly hinders the JAK/STAT3 cascade through the following methods: (1) obstructing the recruitment of STAT3 to the phosphorylated receptor; (2) directly interacting with JAK to suppress its kinase function; (3) prompting the creation of an E3 ubiquitin ligase complex that breaks down JAK or prevents STAT3 from binding to the SOCS protein [adapted from refs. (Gurzov et al., 2016; Hu et al., 2021).
3.4 Noncanonical JAK/STAT3 signaling pathway
The function of STAT3 is influenced by different post-translational modifications, including phosphorylation, methylation, acetylation, and ubiquitination, occurring at various amino acid sites. In addition to classical signal transduction, JAK/STAT3 may also play a role in nonclassical signal transduction. Research has indicated that STAT3, which is not phosphorylated on Tyr705, has the ability to move from cytoplasm to the nucleus and can activate various STAT3 target genes in the absence of Ser727 phosphorylation (Bharadwaj et al., 2020). Additionally, the process can be facilitated by Lys685 acetylation and NF-kB signaling activation, as suggested by previous studies (Yang et al., 2007; Dasgupta et al., 2014). Besides being activated in the cytosol, all STAT proteins (excluding STAT4) have the ability to localize to the mitochondrion, leading to an enhancement in oxidative phosphorylation and membrane polarization. For example, STAT3 monomers phosphorylated on Ser727 can translocate into the mitochondrion without dimerization to increase membrane polarization and ATP synthesis, and inhibit ROS production and mitochondrial permeability transition pore (MPTP) opening, thus exerting a protective role (Boengler et al., 2010; Garama et al., 2016; Avalle and Poli, 2018). Besides, STAT3 has also been reported to translocate to the endoplasmic reticulum and contribute to reduce oxidative stress-induced apoptosis (Avalle et al., 2019). In the nucleus, certain STAT molecules that are not phosphorylated interact with heterochromatin protein 1 (HP1) located on heterochromatin. Phosphorylation of STAT by JAK or other kinases can cause the detachment of HP1 from heterochromatin, leading to its destabilization. Subsequently, phospho-STAT can interact with particular regions on autosomes and regulate the expression of target genes (Shi et al., 2006; Shi et al., 2008b; Li, 2008). This noncanonical JAK/STAT signaling is critical for sustaining heterochromatin stability. Moreover, increasing evidence has shown that activation of JAK/STAT signaling can cause chromatin remodeling in mammals (Christova et al., 2007; Shi et al., 2008a). Besides being triggered by JAK, STAT3 can also be activated by alternative non-receptor tyrosine kinases or JAK-independent receptors. As an example, the c-Src enzyme is capable of phosphorylating STAT3, which then can promote the expression of oncogenes (Yu et al., 1995). EGF receptor and PDGF receptor can directly activate STAT3 (Ruff-Jamison et al., 1994; Liu et al., 2023a).
3.5 Cross-talk between the STAT3 signaling and other pathways
Besides the prevalent JAK/STAT3 signaling pathway, STAT3 also engages in alternative signaling pathways or establishes communication with these pathways, thereby producing biological impacts. STAT3 is involved in the classic TGF-β/Smad signaling pathway (Pedroza et al., 2018; Chen et al., 2019b; Sun et al., 2022) and Smad-independent TGF-β signaling pathways, such as the ERK-mediated MAPK (Park et al., 2020; Shen et al., 2021), JNK (Park et al., 2020), and PI3K/Akt signaling pathways (Zhu et al., 2018; Lee et al., 2019). In addition to TGF-β-related signaling pathways, STAT3 also participates in many other signaling cascades, such as Fyn (a member of the Src kinase family) (Seo et al., 2016; Zhu et al., 2018; Zhu et al., 2023), peroxisome proliferator-activated receptor (PPAR) (Lo et al., 2017b; Németh et al., 2019), and Notch signaling (Chen et al., 2019c).
3.6 Negative regulation of canonical JAK/STAT3 signaling
The inhibition of canonical JAK/STAT3 signaling involves three primary categories of negative regulators (Figure 3): protein inhibitor of activated STAT (PIAS), protein tyrosine phosphatases (PTPs), and suppressor of cytokine signaling (SOCS/CIS). These regulators, as described by Liongue et al., play a crucial role in preventing the excessive phosphorylation of STAT3 (Liongue et al., 2016; Villarino et al., 2017; Yang et al., 2017).
The process of JAK/STAT signal transduction contains a series of intracellular tyrosine phosphorylation, so PTPs have a key role in regulating this pathway. PTPs can directly dephosphorylate and inactivate the STAT dimers, and block the JAK/STAT cascade. For instance, a receptor tyrosine phosphatase PTPRTR can bind to and dephosphorylate the tyrosine residue at site 705 in STAT3 (Zhang et al., 2007). SHP-2, a significant member of the PTP family and also a target gene for activated STAT3, can decrease the phosphorylation level of STAT3 (Schmitz et al., 2000). In addition, PTPs can dephosphorylate JAK and prevent the JAK/STAT signaling.
The PIAS family comprises four transcription regulatory factors, namely, PIAS1-PIAS4. PIAS was originally identified to be a suppressor of STAT, and PIAS3 can combine with STAT3. PIAS only binds to phosphorylated STAT dimers rather than STAT monomers (Hu et al., 2021). PIAS mainly suppresses the transcriptional activity of STAT by means of three mechanisms. (1) Preventing the DNA-binding activity of STAT and blocking STAT-DNA interactions (Sonnenblick et al., 2004). (2) Recruiting transcriptional co-inhibitory factor such as histone deacetylase (Tussié-Luna et al., 2002). (3) Promoting STAT SUMOylation (Yuan et al., 2015).
SOCS family proteins are considered as major triggers of the JAK/STAT signaling attenuation, and there are eight members in this family: SOCS1-7 and cytokine-inducible SH2 protein (CIS) (Minamoto et al., 1997; Piessevaux et al., 2008; Kazi et al., 2014). Cytokine-stimulated JAK/STAT signaling activation induces the SOCS proteins, which act as negative feedback suppressors to regulate this pathway (Naka et al., 1997; Kershaw et al., 2013b). For example, SOCS3 gene is quickly induced by phosphorylated STAT3 dimers in the nucleus, and in turn SOCS3 protein interacts with activated JAK and its receptor to suppress JAK activity, thus preventing further JAK/STAT3 signaling activation (Babon et al., 2012; Kershaw et al., 2013a). SOCS primarily inhibits the JAK/STAT cascade in the following ways. (1) It competes with STAT for binding to the phosphorylated receptor and prevents STAT recruitment. (2) It forms an E3 ubiquitin ligase complex via the COOH-terminal SOCS box and degrades JAK or STAT that binds to SOCS (Kamran et al., 2013). (3) The SOCS protein has the ability to directly and specifically interact with either JAK or its receptor in order to inhibit the activity of JAK kinase. An example is the presence of a distinct brief pattern known as the kinase inhibitory region (KIR) in SOCS1 and SOCS3. This pattern enables these two proteins to hinder the catalytic activity of JAK by directly binding to JAK or its receptor (Sasaki et al., 1999; Yasukawa et al., 1999; Alexander, 2002).
3.7 The JAK/STAT3 pathway induces fibrosis
Studies have indicated that the JAK/STAT3 pathway plays a key role in the process of fibrosis. It can be activated by various pro-fibrotic mediators, such as TGF-β1, PDGF, vascular endothelial growth factor (VEGF), IL-6, Ang II, serotonin (5-HT), and endothelin (ET-1), and then leads to fibrogenesis (Rane and Reddy, 2000; Zhang et al., 2015; Roskoski, 2016) (Figure 4A). The JAK/STAT3 pathway is also demonstrated to be a central integrator of multiple pro-fibrotic pathways and its activation can promote the activation of fibroblasts and the expression of fibrosis-related genes, such as α-SMA, collagens, and fibronectin (Zhang et al., 2015; Chakraborty et al., 2017; Dees et al., 2020). In addition, once activated, STAT3 can induce the expression of hypoxia-inducible factor-1α (HIF-1α), a transcription factor that responds to hypoxic conditions and stimulates the production of ECM (Yang et al., 2021) (Figure 4A). Activated STAT3 can also trigger epithelial to mesenchymal transition (EMT), a cellular process that allows epithelial cells to transform into mesenchymal cells with more power in migration and invasion, and facilitates the progression of fibrosis (Montero et al., 2021; Yang et al., 2021) (Figure 4B).
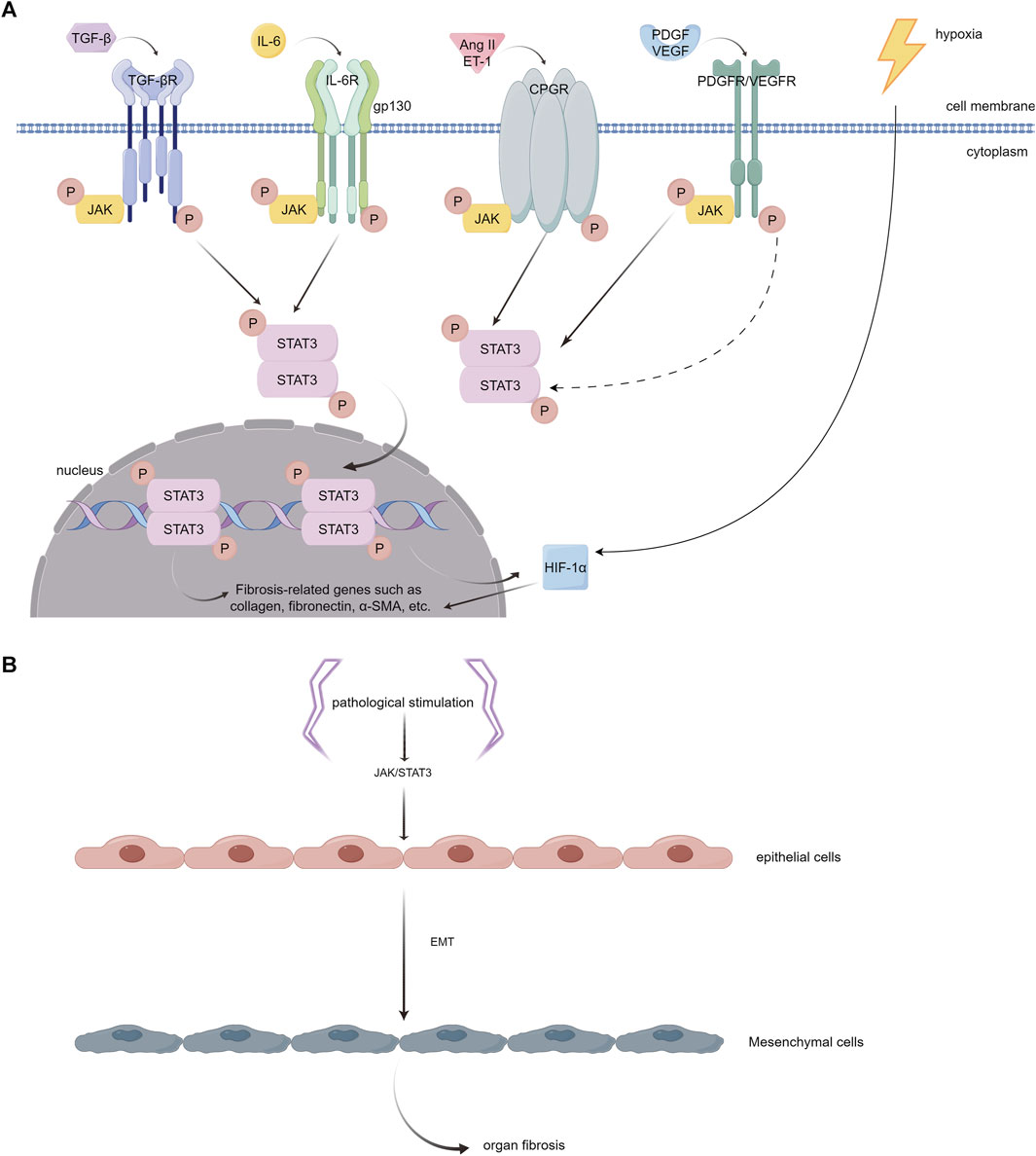
FIGURE 4. (A). Different JAK/STAT3 activators that play important roles in the pathophysiology of myocardial fibrosis. (1) TGF-β interacts with its receptor (TGF-βR) on the cell surface, initiating receptor kinase activity. This activity leads to JAK phosphorylation and subsequent activation of STAT3. However, the precise mechanism underlying this process remains to be fully elucidated. (2) IL-6 binds to its specific receptor, IL-6R, forming a complex. This complex then associates with the membrane protein gp130. Activation of JAKs, which are associated with gp130, is critical for phosphorylating specific tyrosine residues on gp130. These residues act as anchoring points for STAT3. (3) Ang II and ET-1 engage with the GPCR family, triggering the phosphorylation of tyrosine in JAK kinase and consequently activating STAT3. (4) PDGF and VEGF each bind to their respective tyrosine kinase receptors. This binding results in the phosphorylation of tyrosine residues on the receptors, which can indirectly or transactivate JAK, leading to the activation of the STAT3 pathway. Once phosphorylated, STAT3 dimerizes and moves into the nucleus. In the nucleus, these STAT3 dimers attach to specific DNA sequences, enhancing the transcription of genes that are pivotal in driving inflammation and fibrosis, including collagen, fibronectin, α-SMA, etc. In addition, the activation of STAT3 has the capability to stimulate the expression of HIF-1α and enhance the production of ECM in hypoxic environments. (B). Epithelial to mesenchymal transition (EMT). The activation of JAK/STAT3 signaling by pathological stimuli has the potential to induce a phenotypic transition of epithelial cells into mesenchymal cells. These mesenchymal cells exhibit enhanced migration and invasion capabilities. (By Figdraw).
3.8 The effects of the JAK/STAT3 pathway on different types of cardiac injury
The JAK/STAT3 pathway plays a pivotal role in various aspects of cardiac physiology and pathology, exhibiting multifaceted roles in the heart (Figure 5). It mediates protective effects in different stages of ischemia, including ischemia pre-, post-, and remote conditioning (Hattori et al., 2001; You et al., 2011; Gao et al., 2017). Agents such as N-acetylcysteine (NAC) and allopurinol (Wang et al., 2013), and insulin (Fuglesteg et al., 2008) are known to protect against myocardial ischemia-reperfusion injury through activation of the JAK/STAT3 pathway. Their protective mechanism likely involves the reduction of ROS production, decrease in cardiomyocyte apoptosis, promotion of angiogenesis, and delay in MPTP opening. In the context of myocardial infarction, molecular factors like miR-124, IL-10, and growth arrest and DNA damage-inducible α (GADD45A) exert beneficial effects through the STAT3 pathway. Specifically, miR-124 offers anti-apoptotic benefits, IL-10 provides anti-inflammatory effects, and GADD45A enhances VEGF-mediated angiogenesis, collectively improving prognosis (He et al., 2018; Wang et al., 2022a; Tesoro et al., 2022). Conversely, conditional deletion of STAT3 in cardiomyocytes exacerbates cardiac remodeling during the subacute phase of myocardial infarction or under chronic β-adrenergic stimulation (Enomoto et al., 2015; Zhang et al., 2016). Furthermore, cardiomyocyte-specific transgenic expression of SOCS1 inhibits JAK/STAT3 activation in enterovirus-induced myocarditis, but this is associated with increased mortality in mice, highlighting a complex interplay (Yasukawa et al., 2003).
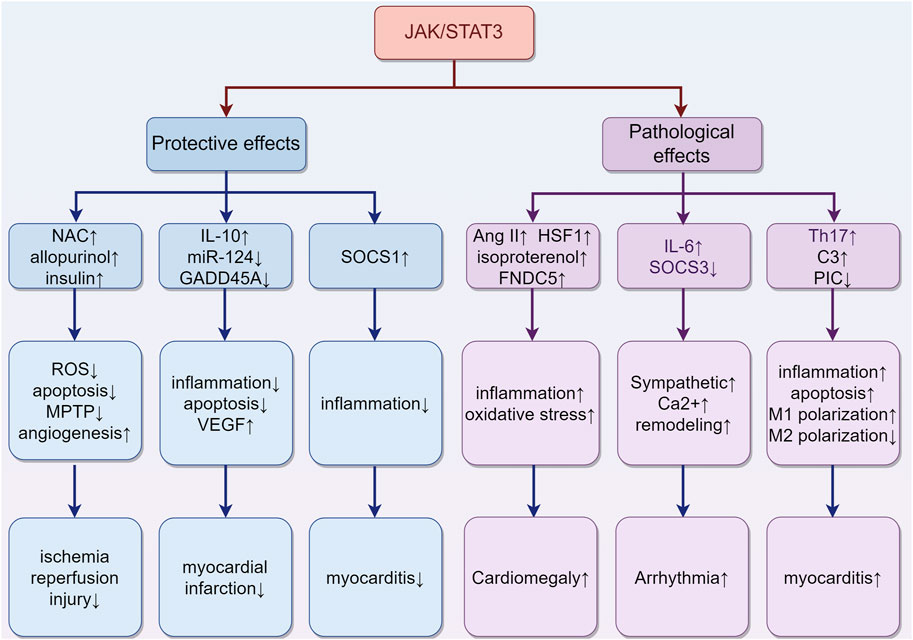
FIGURE 5. The role of activation of the JAK/STAT3 pathway in different types of cardiac damage. (1) In ischemia-reperfusion injury, agents such as NAC, allopurinol, and insulin may confer protective effects. They achieve this by reducing ROS production and cardiomyocyte apoptosis, promoting angiogenesis, and delaying the opening of the MPTP. (2) In the case of myocardial infarction, certain molecular factors like miR-124, IL-10, and GADD45A exert beneficial effects through the STAT3 pathway. These include anti-apoptotic (miR-124), anti-inflammatory (IL-10), and VEGF-mediated angiogenic effects (GADD45A), collectively contributing to improved prognosis. (3) The situation of myocarditis is more complex. The upregulation of SOCS1 can inhibit inflammation. Meanwhile, the upregulation of complement C3 and Th17 cells, along with the downregulation of Piceatannol, may exacerbate inflammation. These findings highlight the multifaceted impact on the progression of myocarditis. (4) Cardiac hypertrophy is influenced by Ang II, HSF1, isoproterenol, and FNDC5, which collaboratively induce hypertrophy through increased oxidative stress and inflammation. (5) Arrhythmias are closely associated with JAK/STAT3 activity, which contributes to myocardial sarcoplasmic reticulum Ca2+ overload, increased cardiac sympathetic nerve activity, and ventricular remodeling. “↑” represents activation, upregulation or exacerbation, and “↓” represents inhibition, downregulation or relief. (By Figdraw).
Despite its protective roles, the JAK/STAT3 pathway also has detrimental effects. For instance, in myocarditis, IL-6-triggered increases in liver complement C3 and Th17 cells may exacerbate inflammation (Camporeale et al., 2013; Wang et al., 2020). Additionally, inhibiting the JAK/STAT3 signaling with piceatannol could improve sepsis-induced cardiac dysfunction by relieving cell apoptosis and inflammation in septic mice and H9C2 cardiomyocytes, suggesting a critical role of the JAK/STAT3 pathway in sepsis-related myocardial injury (Xie et al., 2021). This pathway also skews macrophage polarization towards M1 and away from M2, contributing to coxsackievirus B3 (CVB3)-induced myocardial inflammation and injury (Wang et al., 2023). Chronic activation of JAK/STAT3 can induce cardiac hypertrophy, as evidenced by Ang II-induced activation of TLR4 and STAT3, promoting hypertrophy via the IL-6/JAK2/STAT3 pathway (Han et al., 2018). Other activators like Heat-shock transcription factor 1 (HSF1), isoproterenol, and Fibronectin type III domain containing 5 (FNDC5) also trigger this pathway, resulting in increased cardiac inflammation, oxidative stress, and pathological hypertrophy (Zhao et al., 2017; Yuan et al., 2018; Geng et al., 2019). Moreover, JAK/STAT3 is implicated in cardiac arrhythmias. Inhibiting JAK2/STAT3 phosphorylation reduces malignant ventricular arrhythmias post-myocardial infarction by attenuating ventricular remodeling (Gao et al., 2020). Cardiac-specific SOCS3 gene knockout mice exhibit myocardial sarcoplasmic reticulum Ca2+ overload and subsequent ventricular arrhythmias because of the activation of cardiac gp130 signaling (Yajima et al., 2011). Additionally, IL-6 overexpression, via the STAT3 pathway, promotes cardiac sympathetic nerve activity, increasing the incidence of ventricular arrhythmias (Peng et al., 2023).
4 Multiple mediators regulate cardiac fibrosis through the STAT3 signaling pathway
4.1 ILs
ILs are a type of cytokine proteins that various cells, mainly immune ones, produce. Cytokines modulate cellular functions such as growth, maturation, movement, adhesion, activation and differentiation (Zhang and An, 2007; Brocker et al., 2010). ILs are a large family of cytokines with more than 60 members, which can be grouped into four categories: IL-1 related, type 1 helical (IL-4 related, γ chain and IL-6/IL-12 related), type 2 helical (IL-10 related and IL-28 related), and IL-17 related (Brocker et al., 2010). ILs regulate homeostasis by influencing the cardiovascular, neuroendocrine and metabolic systems in the human body (Corwin, 2000).
Recent research has demonstrated that ILs contribute to myocardial fibrosis via the STAT3 pathway. Some ILs play proinflammatory and fibrotic roles, and IL-6 is the most representative (Figure 6). In the absence of NF-E2-related factor 2 (Nrf2), IL-6 levels further increase in response to Ang II, thereby activating the IL-6/STAT3 pathway, which causes cardiomegaly and inflammation (Chen et al., 2019a). In addition, Ang II can induce Toll-like receptor phosphorylation of STAT3, increase IL-6 production, and continuously activate the JAK/STAT pathway, thereby providing positive feedback and promoting myocardial hypertrophy, fibrosis, and ventricular remodeling (Chen et al., 2017a; Han et al., 2018; Zhang et al., 2019b). IL-6 enhances STAT3 phosphorylation in cultured CFs, whereas inhibiting STAT3 reduces IL6-induced collagen synthesis and reverses pressure overload-induced cardiac hypertrophy (Mir et al., 2012). In a transverse aortic constriction (TAC)-induced mouse heart failure model, inhibiting IL6/gp130/STAT3 with raloxifene alleviated TAC-induced myocarditis, cardiac remodeling and dysfunction (Huo et al., 2021). In mice with CVB3-induced dilated cardiomyopathy (DCM), IL-6 knockout reduced the phosphorylation level of STAT3 in myocardial tissue, thereby improving myocardial remodeling induced by DCM (Li et al., 2019b).
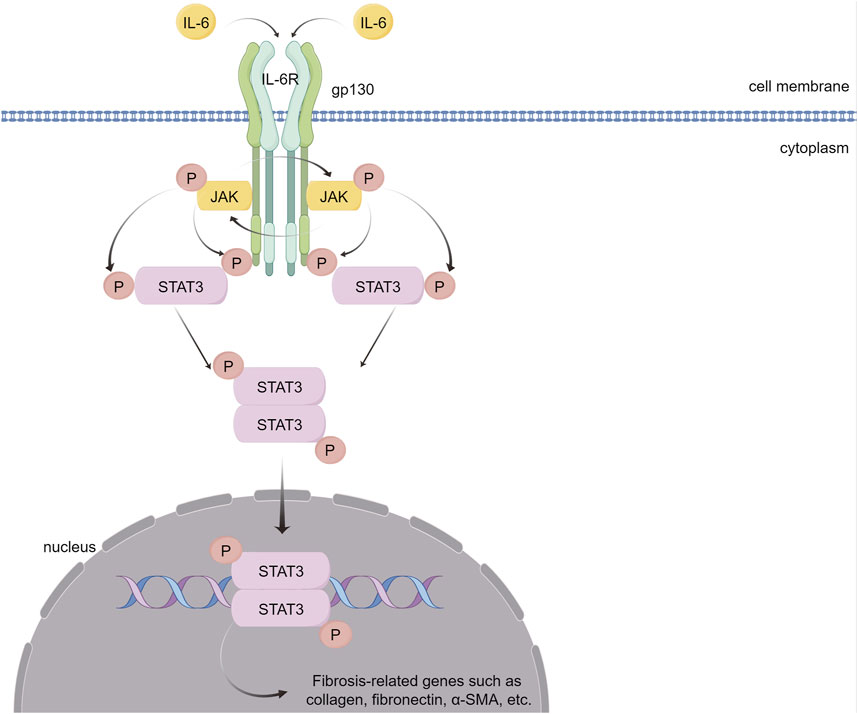
FIGURE 6. IL-6 causes myocardial fibrosis through the JAK/STAT3 signaling pathway. IL-6 binds to its receptor, IL-6R, forming a complex that activates the gp130 receptor. This activation triggers the JAK family of tyrosine kinases. Once activated, these JAKs phosphorylate STAT3, a crucial step in the signaling pathway. Phosphorylated STAT3 dimerizes and translocates into the nucleus. There, STAT3 dimers bind to specific DNA sequences, promoting the transcription of genes that are pivotal in mediating inflammation and fibrosis. (By Figdraw).
4.2 TGF-β
The TGF-β and STAT3 signaling pathways have a feedback loop that regulates the acute/chronic stress response in the heart. TGF-β signaling affects STAT3 as an important target in its downstream pathway (Pedroza et al., 2018; Chen et al., 2019b; Sun et al., 2022). Several studies have demonstrated the interaction between TGF-β and STAT3 in cardiac fibrosis. For instance, it has been reported that TGF-β-induced CD44/STAT3 signaling plays a crucial part in atrial fibrosis and fibrillation formation. CD44 is a membrane receptor that modulates fibrosis. Blocking CD44 signaling can reduce TGF-β-induced STAT3 activation and collagen expression in atrial fibroblasts, implicating a potential approach for treating atrial fibrosis and fibrillation (Chang et al., 2017). Moreover, Ephrinb2-mediated myocardial fibrosis involves the activation of the TGF-β/Smad3 and STAT3 pathways. Further study revealed that Ephrinb2 could enhance the interaction of TGF-β/Smad3 and STAT3 signaling to promote cardiac fibrosis (Su et al., 2017). Furthermore, tyrosine mutation at site 705 to glutamic acid constitutively activated STAT3, which could further enhance the interaction between Smad3 and STAT3 (Su et al., 2017). One previous study showed that a high-fat diet could activate the left ventricular renin–angiotensin system (RAS) and JAK1/2-STAT1/3 pathways in rats by increasing ROS and IL-6 production, ultimately causing cardiac fibrosis. This creates a positive feedback loop that activates the TGF-β1/Smad3 fibrotic pathway and enhances left ventricular collagen synthesis (Eid et al., 2019). In cultured CFs, TGF-β1 can activate STAT3 phosphorylation, increasing fibrosis-related protein expression, and relaxin can block STAT3 phosphorylation and reverse TGF-β1-induced fibrosis (Yuan et al., 2017). These results suggest that STAT3 either acts as a separate signal molecule downstream of TGF-β or interacts with the TGF-β/Smad pathway to regulate cardiac fibrosis (Figure 7).
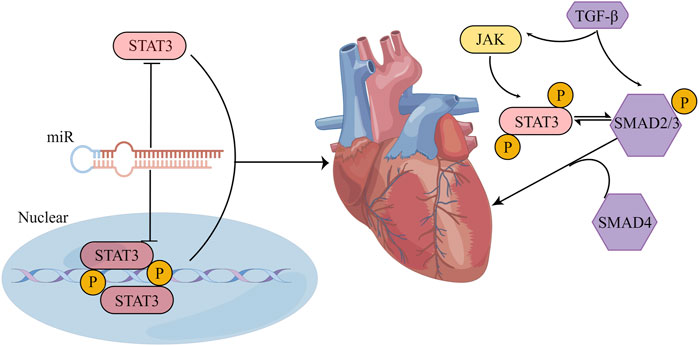
FIGURE 7. STAT3 influences cardiac fibrosis through multiple pathways. (1) The crosstalk between STAT3 and miR manifests in several ways: STAT3 can form either a direct feedback or an indirect feedback loop by binding with miR; it can also mediate the transcription of downstream miR; meanwhile, miR can influence the translation of STAT3 mRNA. (2) Positioned downstream of the TGF-β/SMAD signaling cascade, STAT3 might collaboratively regulate myocardial fibrosis with TGF-β. Their synergistic action could potentially be associated with the phosphorylation of STAT3. (By Figdraw).
4.3 MicroRNAs (miRs)
MiRs are a class of endogenous noncoding single-stranded RNAs that are about 19–25 nucleotides long. First, within the nucleus, RNA polymerase II transcribes the gene encoding the miR into the primary transcript (pri-miR). Then, the pri-miR is transported to the cytoplasm under the cooperative action of the Ran-GTP enzyme and transporter Exportin5, and the double-stranded RNA-specific nuclease Dicer enzyme cleaves the pri-miR, which is transported to the cytoplasm to form double-stranded miR of 21–25 nucleotides. The helicase unwinds the double-stranded miR, leading to degradation of one strand and the formation of a mature miR with a hydroxyl group at the 3′-end and a phosphate group at the 5′-end. Finally, the RNA-induced gene silencing complex binds the mature miR, thereby regulating target gene silencing post-transcriptionally (Lu and Rothenberg, 2018). In recent years, the relationship between miRs and pathological fibrosis has been examined, but the specific mechanisms by which miRs regulate fibrosis are still worth exploring. During the development of liver fibrosis induced by viral hepatitis, the levels of miR-16, miR-146a, miR-221, and miR-222 were markedly increased in the serum of patients with chronic hepatitis C (Abdel-Al et al., 2018). In the livers of mice treated with CCl4, miR-30c and miR-193 were specifically downregulated (Roy et al., 2015). Interestingly, other studies indicated that miR-29 could promote apoptosis in cardiomyocytes by downregulating antiapoptotic genes such as Bcl-2, CDC42 and Tcl-1, while miR-29 could prevent fibrosis by inhibiting the release of collagen from the ECM (Pekarsky et al., 2006; Mott et al., 2007; van Rooij et al., 2008). These results indicate that different miRs may have opposite effects on fibrosis regulation, and the same miR may have significant differences in fibrosis regulation.
STAT3 and miRs have crosstalk that is crucial for maintaining cardiac function under normal and pathological conditions. This STAT3-miR crosstalk can mediate cardiac disease in several ways. First, STAT3 can directly bind to miRs to mediate a feedback regulatory relationship or mediate an indirect feedback regulatory relationship with miRs through a long noncoding RNA (lncRNA)/protein. As an example, in oxygen-glucose deprivation-induced cardiomyocyte injury, lncRNA MIAT, which is associated with myocardial infarction, captures miR-181a-5p and boosts the expression of JAK2. This, in turn, amplifies myocardial inflammation and apoptosis through the JAK2/STAT3 signaling pathway (Tan et al., 2021). In addition, miR-21 activates the STAT3 signaling by targeting tumor suppressor cell adhesion molecule 1 (CADM1) and enhances cardiac fibrosis (Cao et al., 2017). Second, STAT3 can directly mediate the transcription of downstream miRs, and phosphorylated STAT3 can cooperate with other transcription factors to promote or inhibit the transcription of miRs. In diabetic hearts exposed to ischaemia/reperfusion, STAT3 has the ability to attach to the miR-17–92 promoter and stimulate the targeted inhibition of pro-apoptotic prolyl hydroxylase 3 (PHD3) by miR-17/20a, resulting in a decrease in apoptosis (Samidurai et al., 2020). Moreover, phosphorylated STAT3 can interact with NF-κB and inhibit miR-188-3p expression (Kuo et al., 2017; Sp et al., 2018; Masoumi-Dehghi et al., 2020). Third, miRs specifically recognize the 3′UTR of STAT3 mRNA and form incomplete complementary pairing, resulting in the inhibition of STAT3 mRNA translation, thereby blocking STAT3 expression. Following myocardial infarction, the expression of STAT3 mRNA is reduced by miR-17-5p and miR-124, which leads to the deterioration of autophagy, inflammation, myocardial remodeling, and apoptosis. These miRs bind to the 3′UTR of STAT3 mRNA (He et al., 2018; Chen et al., 2022). In summary, multiple miRs can interact with STAT3 through different mechanisms to enhance or inhibit cardiac fibrosis (Figure 7).
4.4 Other mediators impact cardiac fibrosis through the STAT3 signaling pathway
In addition to the above mediators that can affect cardiac fibrosis through the STAT3 signaling pathway, there are other mediators that can affect myocardial fibrosis caused by ischemia/reperfusion, atrial fibrillation, diabetic heart disease, DCM, and hypertensive heart damage through the STAT3 signaling pathway (Table 2).
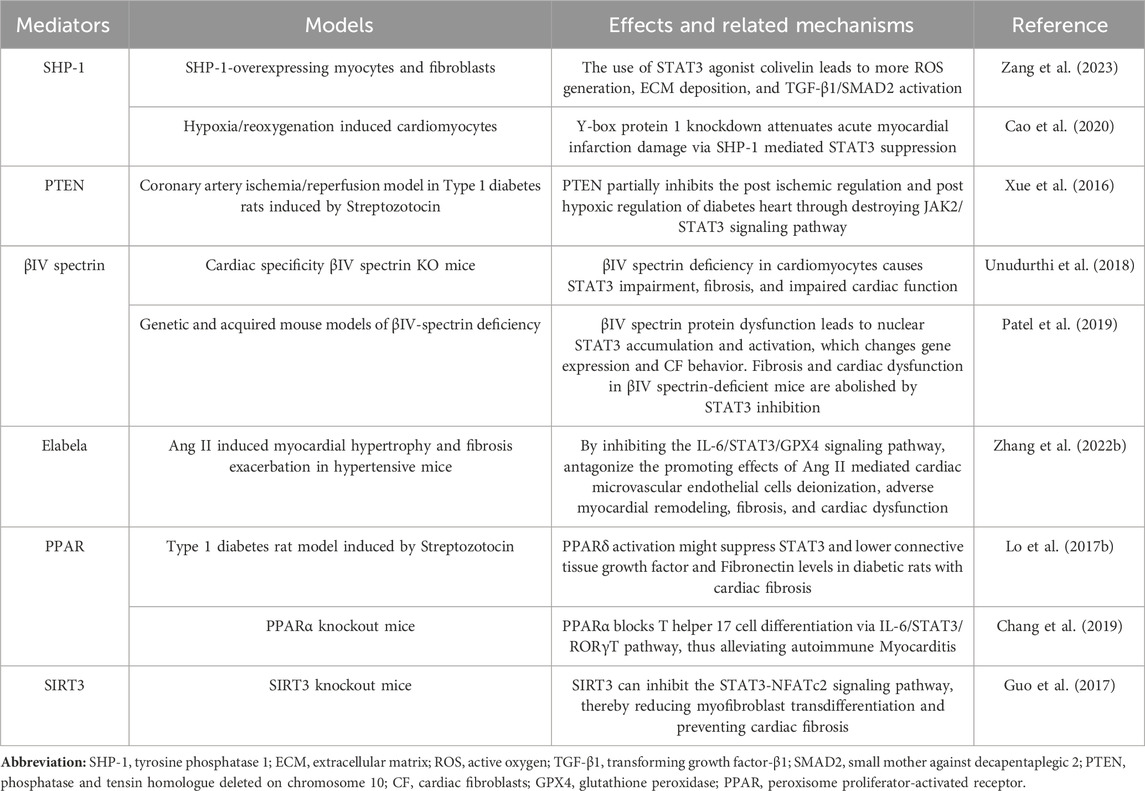
TABLE 2. Mediators regulate fibrosis through the STAT3 signaling pathway.
5 The regulatory role of STAT3 and autophagy in cardiac fibrosis
Autophagy is widely present in eukaryotic organisms and is a process that degrades harmful substances in cells and promotes their recycling through the lysosome pathway. In general, moderate autophagy can maintain the stability of the internal environment, while excessive autophagy can induce cell damage (Kuma et al., 2017). The process is mainly divided into four stages: induction, initiation, elongation, and mature degradation, which are regulated by complex molecular mechanisms (Estrada-Navarrete et al., 2016; Liu et al., 2016; Lin et al., 2019; Kaushal et al., 2020). Autophagy recovers and removes damaged proteins and organelles, playing an important role in maintaining the normal function of myocardial cells (Mialet-Perez and Vindis, 2017). Interestingly, the role of autophagy in fibrosis may vary with fibrosis progression. Zhang et al. found that inhibiting autophagy could improve myocardial fibrosis in mice subjected to TAC surgery (Zhang et al., 2021). At 20 weeks after TAC in mice with endothelial leptin receptor gene knockout, myocardial fibrosis in these mice was improved by autophagy activation (Gogiraju et al., 2019). These research results demonstrate that the activation or inhibition of autophagy may occur during the process of cardiac fibrosis, and the role of autophagy in fibrosis has a dual nature.
Autophagy could potentially be linked to numerous signaling pathways, one of which is the STAT3 signaling pathway that governs the fate of cells, determining whether they survive or perish. Yuan et al.'s research indicates that relaxin attenuates TGF-β1-induced autophagy in primary CFs by suppressing the phosphorylation of STAT3, thereby reducing cardiac fibrosis (Yuan et al., 2017). In septic cardiomyopathy, the reduced expression of miR-125b leads to excessive activation of STAT3/high mobility group box protein 1 (HMGB1), resulting in elevated ROS generation and impaired autophagic flow, ultimately leading to myocardial dysfunction (Yu et al., 2021). Additionally, the overexpression of Src-associated in mitosis 68 (Sam68) promotes the osteogenic differentiation of human valvular interstitial cells (hVICs) through the STAT3 signaling-mediated autophagy inhibition, thus inducing aortic valve calcification, while knockdown of Sam68 reduces the phosphorylation of TNF-α-activated STAT3 and the expression of downstream genes, thereby affecting autophagic flow in hVICs (Liu et al., 2023b). The activation of STAT3 is crucial for reducing cardiac autophagy and inhibiting cardiac ischemia/reperfusion injury, as demonstrated by the inhibition of soluble receptor for advanced glycation end-products on cardiac ischemia/reperfusion injury (Dang et al., 2019).
6 Challenges and opportunities for targeting the STAT3 signaling pathway for the treatment of fibrosis
Targeting STAT3 for heart disease treatment presents significant challenges. STAT3 is widely recognized for its role in promoting myocardial fibrosis. However, myocardial fibrosis may not always be detrimental in certain heart diseases. Excessive fibrosis, for instance, can lead to adverse remodeling in myocardial infarction patients, potentially resulting in heart failure. Yet, in the early stages of myocardial infarction, fibrosis is crucial in maintaining the structural integrity of the infarcted ventricle (Prabhu and Frangogiannis, 2016). Moreover, STAT3 actively participates in the activation and proliferation of CFs, fostering fibrotic remodeling. In cardiomyocytes, STAT3 exhibits a dual nature. It can offer protective or adverse effects, such as enhancing survival and mitigating oxidative stress or mediating cardiac hypertrophy (Wang et al., 2021; Li et al., 2022). Despite cardiomyocytes not being directly involved in ECM production, they can influence the fibrotic response through paracrine signals (Qu et al., 2017). Additionally, the STAT3 signaling pathway interacts with other pathways, playing varying roles. JAK1, for example, binds to TGF-βR1, while JAKs also associate with gp130 and get activated by TGF-β (Itoh et al., 2018). Previous studies have shown that STAT3 works in tandem with Smad3 to induce connective tissue growth factor, contributing to fibrosis (Liu et al., 2013; Tang et al., 2017). Conversely, overactivated STAT3 signaling in lung fibroblasts diminishes SMAD signaling by reducing Smad3 phosphorylation, potentially due to Smad7 induction, although this theory requires experimental validation (O'Donoghue et al., 2012). Thus, identifying the optimal timing for STAT3 inhibition is crucial for maximizing therapeutic benefits and minimizing side effects. Targeting STAT3 in CFs could effectively reduce fibrosis, but its protective potential in cardiomyocytes warrants consideration. Overall, STAT3’s role in cardiac biology is multifaceted. A thorough understanding of its function across various cell types and disease stages is essential for developing effective treatments.
Despite the complexities in targeting STAT3 signaling for fibrosis treatment, recent advancements have yielded promising results (Table 3). Presently, methods to directly inhibit STAT3, aimed at targeting fibrosis, are categorized based on various target domains. These include the SH2, DBD, NTD, and TAD. In this section, we highlight key STAT3 inhibitors that specifically target these domains of the STAT3 protein.
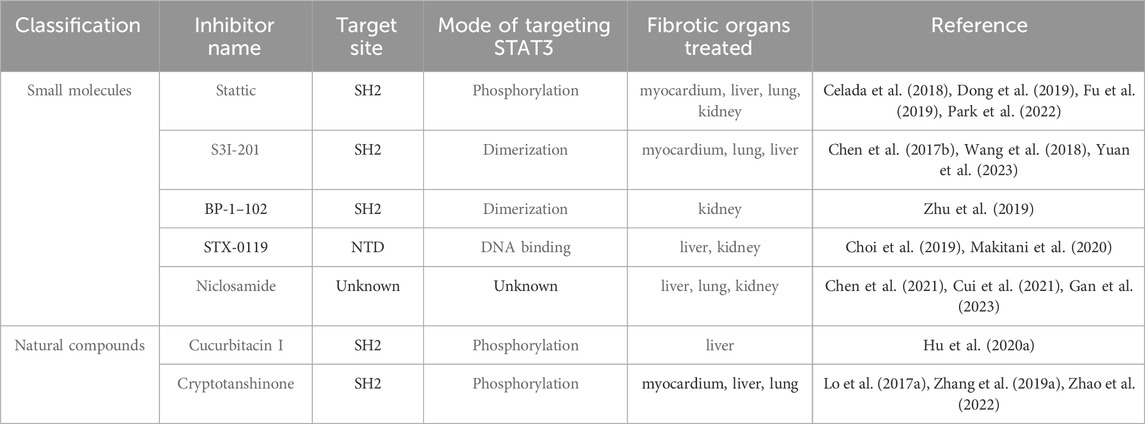
TABLE 3. STAT3 inhibitors for treating organ fibrosis.
6.1 Inhibitors targeting the SH2 domain
STAT3 homodimerization is facilitated by protein-protein interactions between the SH2 domains of the individual monomers, particularly via phosphorylation at Tyr705. This pivotal molecular interaction has been harnessed to develop inhibitors targeting STAT3 directly (Furtek et al., 2016). Inhibiting the SH2 domain not only disrupts STAT3 activation and dimerization but also impedes its subsequent nuclear translocation and the expression of genes regulated by STAT3.
Several small molecule STAT3 inhibitors, notably Stattic, S3I-201, and S3I-201 analogs, play a significant role in mitigating myocardial fibrosis. These inhibitors function by binding to the SH2 domain of STAT3, thereby curtailing its activity. Elevated levels of fibroblast growth factor 23 (FGF23) are reported to induce atrial fibrosis in atrial fibrillation patients through enhancing ROS production and subsequent STAT3 and Smad3 phosphorylation. Stattic has been shown to counteract these effects (Dong et al., 2019). Moreover, administering S3I-201 to mice with myocardial infarction has demonstrated reduced left atrial fibrosis in vivo (Chen et al., 2017b).
Another category of inhibitors targeting STAT3’s SH2 domain comprises derivatives of natural compounds. Cryptotanshinone, a primary active component extracted from Salvia miltiorrhiza, suppresses the STAT3 pathway to reduce cardiac fibrosis and improve cardiac function in diabetic rats (Lo et al., 2017a). In vitro studies reveal that cryptotanshinone significantly curbs Ang II-induced cardiomyocyte hypertrophy and TGF-β-induced myofibroblast activation by impeding STAT3 phosphorylation and nuclear translocation (Li et al., 2023). Additionally, natural compounds like curcumin and resveratrol have been identified to possess properties beneficial in combating atherosclerosis (Zordoky et al., 2015; Ganjali et al., 2017).
These inhibitors are crucial for their anti-inflammatory and anti-atherosclerotic properties, suggesting their potential as therapeutic agents for ameliorating fibrosis. However, these inhibitors are not without drawbacks. A primary issue is that most inhibitors targeting the SH2 domain lack specificity to STAT3, making it challenging to exclude the involvement of other STAT proteins in fibrosis (Szelag et al., 2016). Additionally, STAT3 monomers or unphosphorylated STAT3 proteins can interact with other proteins to transcribe downstream target genes, which limits the efficacy of targeting the SH2 domain. Further complicating matters, activating mutations in the SH domain have been identified in somatic cells. The impact of these somatic mutations on the binding efficiency of SH2 domain inhibitors to STAT3, and consequently on their effectiveness, remains to be fully understood (Qiu and Fan, 2016). Therefore, the precise targeting of STAT3’s SH2 domain warrants further research focus.
6.2 Inhibitors targeting the DBD domain
The DBD of STAT3 specifically recognizes and binds to distinct DNA elements in target genes. This selective interaction facilitates the precise induction of target gene expression, characterized by high specificity.
Research has uncovered that platinum compounds, including IS3-295, CPA-1, CPA-7, and platinum tetrachloride (IV), effectively block the DNA-binding activity of STAT3. These compounds can inhibit cell growth and induce apoptosis, while not affecting normal cells and avoiding prolonged STAT3 activation (Beebe et al., 2018). Additionally, Galiellalactone, a natural product, impedes STAT3’s DNA-binding activity by interacting with its DBD domain. To enhance its oral bioavailability, N-acetyl L-cysteine methyl ester has been added to the thiol group, resulting in the creation of the prodrug GPA512. However, GPA512’s lack of specificity, as it also disrupts other signaling pathways like NF-κB and TGF-β, could pose challenges in its future development (Don-Doncow et al., 2014; Escobar et al., 2016). InS3-54, discovered through an advanced computer screening method, selectively binds to STAT3’s DBD domain in vitro, inhibiting its DNA-binding activity. Its analog, InS3-54A18, exhibits improved solubility, specificity, and pharmacological properties, while showing minimal side effects in animal models (Huang et al., 2016).
While virtual screening techniques, including molecular modeling, have demonstrated that certain inhibitors can directly bind to the DBD domain of STAT3, the scarcity of adequate assay systems has limited the identification of small molecule inhibitors in this category. This constraint has significantly impeded the drug development process. Additionally, inhibitors targeting the STAT3 DBD encounter similar challenges to those faced by SH2 domain-targeting inhibitors in terms of therapeutic application.
6.3 Inhibitors targeting NTD and TAD domains
Inhibitors targeting the NTDs and TAD of STAT3 can modulate the binding of STAT3 dimers and regulate DNA transcription, potentially contributing to anti-fibrotic effects. In the study of the selective STAT3 NTD inhibitor ST3-H2A2, Timofeeva et al. observed that this compound robustly activated apoptosis genes, leading to the induction of apoptosis in cancer cells (Timofeeva et al., 2013). Moreover, researchers have successfully identified the allosterically active small molecule K116, which binds to the TAD of STAT3 and effectively inhibits its activity (Huang et al., 2018).
In summary, while numerous STAT3 inhibitors have demonstrated anti-fibrotic properties, identifying inhibitors that are highly efficient, low in toxicity, and have minimal side effects remains a challenge. Additionally, there is a scarcity of extensive animal studies on the pharmacology and toxicology of these inhibitors. Furthermore, only a limited number of these inhibitors have progressed to clinical evaluation. However, the integration of STAT3 inhibitors with other targeted therapeutic agents, particularly in combination with immunotherapy agents, offers promising potential. It is hoped that future research will lead to significant advancements, enabling the broader clinical application of STAT3 inhibitors.
7 Conclusion
Cardiac fibrosis results from the excessive accumulation of ECM in the myocardium and is central to many cardiac pathologies. Since JAK/STAT3 activation can increase fibrotic effector cells and ECM deposition through various pathways, it may be a potential target of antifibrotic therapy. As mentioned previously, we emphasized the promoting effects of various mediators on cardiac fibrosis through activation of the JAK/STAT3 signaling pathway. However, there may be many other mediators that have not yet been identified, and modern proteomics technology and protein identification will speed up the discovery. Regarding fibrosis, the antifibrotic effect of STAT3 inhibitors is receiving attention, but there has been little research on their ability to inhibit myocardial fibrosis. While further research is required to elucidate its role in various types of myocardial fibrosis, the JAK/STAT3 signaling holds promise as a therapeutic target for cardiac fibrosis due to its connection between cardiac inflammation and fibrosis.
Author contributions
HJ: Formal Analysis, Writing–original draft, Writing–review and editing. JY: Data curation, Formal Analysis, Writing–original draft. TL: Formal Analysis, Funding acquisition, Writing–review and editing. XW: Data curation, Writing–review and editing. ZF: Data curation, Visualization, Writing–review and editing. QY: Conceptualization, Software, Writing–review and editing. YD: Conceptualization, Funding acquisition, Writing–original draft, Writing–review and editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by Grants from the National Natural Science Foundation of China (Grant No. 82200287 to YD), Sichuan Science and Technology Program (Grant Nos. 2023NSFSC1653 to YD and 2022NSFSC0674 to TL), Luzhou Science and Technology Project (Grant No. 2022-JYJ-106 to YD), and Southwest Medical University Science and Technology Project (Grant No. 2021ZKMS034 to TL).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abdel-Al, A., El-Ahwany, E., Zoheiry, M., Hassan, M., Ouf, A., Abu-Taleb, H., et al. (2018). miRNA-221 and miRNA-222 are promising biomarkers for progression of liver fibrosis in HCV Egyptian patients. Virus Res. 253, 135–139. doi:10.1016/j.virusres.2018.06.007
Aftabizadeh, M., Li, Y. J., Zhao, Q., Zhang, C., Ambaye, N., Song, J., et al. (2021). Potent antitumor effects of cell-penetrating peptides targeting STAT3 axis. JCI Insight 6 (2), e136176. doi:10.1172/jci.insight.136176
Aisagbonhi, O., Rai, M., Ryzhov, S., Atria, N., Feoktistov, I., and Hatzopoulos, A. K. (2011). Experimental myocardial infarction triggers canonical Wnt signaling and endothelial-to-mesenchymal transition. Dis. Model Mech. 4 (4), 469–483. doi:10.1242/dmm.006510
Alexander, W. S. (2002). Suppressors of cytokine signalling (SOCS) in the immune system. Nat. Rev. Immunol. 2 (6), 410–416. doi:10.1038/nri818
Ali, S. R., Ranjbarvaziri, S., Talkhabi, M., Zhao, P., Subat, A., Hojjat, A., et al. (2014). Developmental heterogeneity of cardiac fibroblasts does not predict pathological proliferation and activation. Circ. Res. 115 (7), 625–635. doi:10.1161/circresaha.115.303794
Aoki, T., Fukumoto, Y., Sugimura, K., Oikawa, M., Satoh, K., Nakano, M., et al. (2011). Prognostic impact of myocardial interstitial fibrosis in non-ischemic heart failure. -Comparison between preserved and reduced ejection fraction heart failure. Circ. J. 75 (11), 2605–2613. doi:10.1253/circj.cj-11-0568
Avalle, L., Camporeale, A., Morciano, G., Caroccia, N., Ghetti, E., Orecchia, V., et al. (2019). STAT3 localizes to the ER, acting as a gatekeeper for ER-mitochondrion Ca(2+) fluxes and apoptotic responses. Cell Death Differ. 26 (5), 932–942. doi:10.1038/s41418-018-0171-y
Avalle, L., and Poli, V. (2018). Nucleus, Mitochondrion, or Reticulum? STAT3 à La Carte. Int. J. Mol. Sci. 19 (9), 2820. doi:10.3390/ijms19092820
Azevedo, C. F., Nigri, M., Higuchi, M. L., Pomerantzeff, P. M., Spina, G. S., Sampaio, R. O., et al. (2010). Prognostic significance of myocardial fibrosis quantification by histopathology and magnetic resonance imaging in patients with severe aortic valve disease. J. Am. Coll. Cardiol. 56 (4), 278–287. doi:10.1016/j.jacc.2009.12.074
Azushima, K., Morisawa, N., Tamura, K., and Nishiyama, A. (2020). Recent research advances in renin-angiotensin-aldosterone system receptors. Curr. Hypertens. Rep. 22 (3), 22. doi:10.1007/s11906-020-1028-6
Babon, J. J., Kershaw, N. J., Murphy, J. M., Varghese, L. N., Laktyushin, A., Young, S. N., et al. (2012). Suppression of cytokine signaling by SOCS3: characterization of the mode of inhibition and the basis of its specificity. Immunity 36 (2), 239–250. doi:10.1016/j.immuni.2011.12.015
Banerjee, S., Biehl, A., Gadina, M., Hasni, S., and Schwartz, D. M. (2017). JAK-STAT signaling as a target for inflammatory and autoimmune diseases: current and future prospects. Drugs 77 (5), 521–546. doi:10.1007/s40265-017-0701-9
Bao, Q., Zhang, B., Suo, Y., Liu, C., Yang, Q., Zhang, K., et al. (2020). Intermittent hypoxia mediated by TSP1 dependent on STAT3 induces cardiac fibroblast activation and cardiac fibrosis. Elife 9, e49923. doi:10.7554/eLife.49923
Barry, S. P., Townsend, P. A., Latchman, D. S., and Stephanou, A. (2007). Role of the JAK-STAT pathway in myocardial injury. Trends Mol. Med. 13 (2), 82–89. doi:10.1016/j.molmed.2006.12.002
Beebe, J. D., Liu, J. Y., and Zhang, J. T. (2018). Two decades of research in discovery of anticancer drugs targeting STAT3, how close are we? Pharmacol. Ther. 191, 74–91. doi:10.1016/j.pharmthera.2018.06.006
Bharadwaj, U., Kasembeli, M. M., Robinson, P., and Tweardy, D. J. (2020). Targeting Janus kinases and signal transducer and activator of transcription 3 to treat inflammation, fibrosis, and cancer: rationale, progress, and caution. Pharmacol. Rev. 72 (2), 486–526. doi:10.1124/pr.119.018440
Boengler, K., Hilfiker-Kleiner, D., Heusch, G., and Schulz, R. (2010). Inhibition of permeability transition pore opening by mitochondrial STAT3 and its role in myocardial ischemia/reperfusion. Basic Res. Cardiol. 105 (6), 771–785. doi:10.1007/s00395-010-0124-1
Bourillot, P. Y., Aksoy, I., Schreiber, V., Wianny, F., Schulz, H., Hummel, O., et al. (2009). Novel STAT3 target genes exert distinct roles in the inhibition of mesoderm and endoderm differentiation in cooperation with Nanog. Stem Cells 27 (8), 1760–1771. doi:10.1002/stem.110
Brocker, C., Thompson, D., Matsumoto, A., Nebert, D. W., and Vasiliou, V. (2010). Evolutionary divergence and functions of the human interleukin (IL) gene family. Hum. Genomics 5 (1), 30–55. doi:10.1186/1479-7364-5-1-30
Bujak, M., and Frangogiannis, N. G. (2007). The role of TGF-beta signaling in myocardial infarction and cardiac remodeling. Cardiovasc Res. 74 (2), 184–195. doi:10.1016/j.cardiores.2006.10.002
Caldenhoven, E., van Dijk, T. B., Solari, R., Armstrong, J., Raaijmakers, J. A., Lammers, J. W., et al. (1996). STAT3beta, a splice variant of transcription factor STAT3, is a dominant negative regulator of transcription. J. Biol. Chem. 271 (22), 13221–13227. doi:10.1074/jbc.271.22.13221
Camporeale, A., Marino, F., Papageorgiou, A., Carai, P., Fornero, S., Fletcher, S., et al. (2013). STAT3 activity is necessary and sufficient for the development of immune-mediated myocarditis in mice and promotes progression to dilated cardiomyopathy. EMBO Mol. Med. 5 (4), 572–590. doi:10.1002/emmm.201201876
Cao, W., Shi, P., and Ge, J. J. (2017). miR-21 enhances cardiac fibrotic remodeling and fibroblast proliferation via CADM1/STAT3 pathway. BMC Cardiovasc Disord. 17 (1), 88. doi:10.1186/s12872-017-0520-7
Cao, X., Zhu, N., Zhang, Y., Chen, Y., Zhang, J., Li, J., et al. (2020). Y-box protein 1 promotes hypoxia/reoxygenation- or ischemia/reperfusion-induced cardiomyocyte apoptosis via SHP-1-dependent STAT3 inactivation. J. Cell Physiol. 235 (11), 8187–8198. doi:10.1002/jcp.29474
Celada, L. J., Kropski, J. A., Herazo-Maya, J. D., Luo, W., Creecy, A., Abad, A. T., et al. (2018). PD-1 up-regulation on CD4+ T cells promotes pulmonary fibrosis through STAT3-mediated IL-17A and TGF-β1 production. Sci. Transl. Med. 10 (460), eaar8356. doi:10.1126/scitranslmed.aar8356
Chakraborty, D., Šumová, B., Mallano, T., Chen, C. W., Distler, A., Bergmann, C., et al. (2017). Activation of STAT3 integrates common profibrotic pathways to promote fibroblast activation and tissue fibrosis. Nat. Commun. 8 (1), 1130. doi:10.1038/s41467-017-01236-6
Chang, H., Zhao, F., Xie, X., Liao, Y., Song, Y., Liu, C., et al. (2019). PPARα suppresses Th17 cell differentiation through IL-6/STAT3/RORγt pathway in experimental autoimmune myocarditis. Exp. Cell Res. 375 (1), 22–30. doi:10.1016/j.yexcr.2018.12.005
Chang, S. H., Yeh, Y. H., Lee, J. L., Hsu, Y. J., Kuo, C. T., and Chen, W. J. (2017). Transforming growth factor-β-mediated CD44/STAT3 signaling contributes to the development of atrial fibrosis and fibrillation. Basic Res. Cardiol. 112 (5), 58. doi:10.1007/s00395-017-0647-9
Chen, B., Yang, Y., Wu, J., Song, J., and Lu, J. (2022). microRNA-17-5p downregulation inhibits autophagy and myocardial remodelling after myocardial infarction by targeting STAT3. Autoimmunity 55 (1), 43–51. doi:10.1080/08916934.2021.1992754
Chen, D., Li, Z., Bao, P., Chen, M., Zhang, M., Yan, F., et al. (2019a). Nrf2 deficiency aggravates Angiotensin II-induced cardiac injury by increasing hypertrophy and enhancing IL-6/STAT3-dependent inflammation. Biochim. Biophys. Acta Mol. Basis Dis. 1865 (6), 1253–1264. doi:10.1016/j.bbadis.2019.01.020
Chen, F., Chen, D., Zhang, Y., Jin, L., Zhang, H., Wan, M., et al. (2017a). Interleukin-6 deficiency attenuates angiotensin II-induced cardiac pathogenesis with increased myocyte hypertrophy. Biochem. Biophys. Res. Commun. 494 (3-4), 534–541. doi:10.1016/j.bbrc.2017.10.119
Chen, M., Li, H., Wang, G., Shen, X., Zhao, S., and Su, W. (2016). Atorvastatin prevents advanced glycation end products (AGEs)-induced cardiac fibrosis via activating peroxisome proliferator-activated receptor gamma (PPAR-γ). Metabolism 65 (4), 441–453. doi:10.1016/j.metabol.2015.11.007
Chen, Q., Gao, C., Wang, M., Fei, X., and Zhao, N. (2021). TRIM18-Regulated STAT3 signaling pathway via PTP1B promotes renal epithelial-mesenchymal transition, inflammation, and fibrosis in diabetic kidney disease. Front. Physiol. 12, 709506. doi:10.3389/fphys.2021.709506
Chen, W., Yuan, H., Cao, W., Wang, T., Chen, W., Yu, H., et al. (2019b). Blocking interleukin-6 trans-signaling protects against renal fibrosis by suppressing STAT3 activation. Theranostics 9 (14), 3980–3991. doi:10.7150/thno.32352
Chen, X., Su, J., Feng, J., Cheng, L., Li, Q., Qiu, C., et al. (2019c). TRIM72 contributes to cardiac fibrosis via regulating STAT3/Notch-1 signaling. J. Cell Physiol. 234 (10), 17749–17756. doi:10.1002/jcp.28400
Chen, X., Tang, Y., Gao, M., Qin, S., Zhou, J., and Li, X. (2015). Prenatal exposure to lipopolysaccharide results in myocardial fibrosis in rat offspring. Int. J. Mol. Sci. 16 (5), 10986–10996. doi:10.3390/ijms160510986
Chen, Y., Surinkaew, S., Naud, P., Qi, X. Y., Gillis, M. A., Shi, Y. F., et al. (2017b). JAK-STAT signalling and the atrial fibrillation promoting fibrotic substrate. Cardiovasc Res. 113 (3), 310–320. doi:10.1093/cvr/cvx004
Choi, S., Jung, H. J., Kim, M. W., Kang, J. H., Shin, D., Jang, Y. S., et al. (2019). A novel STAT3 inhibitor, STX-0119, attenuates liver fibrosis by inactivating hepatic stellate cells in mice. Biochem. Biophys. Res. Commun. 513 (1), 49–55. doi:10.1016/j.bbrc.2019.03.156
Christia, P., Bujak, M., Gonzalez-Quesada, C., Chen, W., Dobaczewski, M., Reddy, A., et al. (2013). Systematic characterization of myocardial inflammation, repair, and remodeling in a mouse model of reperfused myocardial infarction. J. Histochem Cytochem 61 (8), 555–570. doi:10.1369/0022155413493912
Christova, R., Jones, T., Wu, P. J., Bolzer, A., Costa-Pereira, A. P., Watling, D., et al. (2007). P-STAT1 mediates higher-order chromatin remodelling of the human MHC in response to IFNgamma. J. Cell Sci. 120 (18), 3262–3270. doi:10.1242/jcs.012328
Chung, H., Kim, Y., Park, C. H., Kim, J. Y., Min, P. K., Yoon, Y. W., et al. (2021). Effect of sarcomere and mitochondria-related mutations on myocardial fibrosis in patients with hypertrophic cardiomyopathy. J. Cardiovasc Magn. Reson 23 (1), 18. doi:10.1186/s12968-021-00718-3
Cleutjens, J. P., Verluyten, M. J., Smiths, J. F., and Daemen, M. J. (1995). Collagen remodeling after myocardial infarction in the rat heart. Am. J. Pathol. 147 (2), 325–338.
Corwin, E. J. (2000). Understanding cytokines. Part I: physiology and mechanism of action. Biol. Res. Nurs. 2 (1), 30–40. doi:10.1177/109980040000200104
Cui, Z. Y., Wang, G., Zhang, J., Song, J., Jiang, Y. C., Dou, J. Y., et al. (2021). Parthenolide, bioactive compound of Chrysanthemum parthenium L., ameliorates fibrogenesis and inflammation in hepatic fibrosis via regulating the crosstalk of TLR4 and STAT3 signaling pathway. Phytother. Res. 35 (10), 5680–5693. doi:10.1002/ptr.7214
Dang, M., Zeng, X., Chen, B., Wang, H., Li, H., Liu, Y., et al. (2019). Soluble receptor for advance glycation end-products inhibits ischemia/reperfusion-induced myocardial autophagy via the STAT3 pathway. Free Radic. Biol. Med. 130, 107–119. doi:10.1016/j.freeradbiomed.2018.10.437
Darnell, J. E. (1997). STATs and gene regulation. Science 277 (5332), 1630–1635. doi:10.1126/science.277.5332.1630
Darnell, J. E., Kerr, I. M., and Stark, G. R. (1994). Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 264 (5164), 1415–1421. doi:10.1126/science.8197455
Dasgupta, M., Unal, H., Willard, B., Yang, J., Karnik, S. S., and Stark, G. R. (2014). Critical role for lysine 685 in gene expression mediated by transcription factor unphosphorylated STAT3. J. Biol. Chem. 289 (44), 30763–30771. doi:10.1074/jbc.M114.603894
Dees, C., Pötter, S., Zhang, Y., Bergmann, C., Zhou, X., Luber, M., et al. (2020). TGF-β-induced epigenetic deregulation of SOCS3 facilitates STAT3 signaling to promote fibrosis. J. Clin. Invest. 130 (5), 2347–2363. doi:10.1172/jci122462
Dobaczewski, M., Bujak, M., Zymek, P., Ren, G., Entman, M. L., and Frangogiannis, N. G. (2006). Extracellular matrix remodeling in canine and mouse myocardial infarcts. Cell Tissue Res. 324 (3), 475–488. doi:10.1007/s00441-005-0144-6
Don-Doncow, N., Escobar, Z., Johansson, M., Kjellström, S., Garcia, V., Munoz, E., et al. (2014). Galiellalactone is a direct inhibitor of the transcription factor STAT3 in prostate cancer cells. J. Biol. Chem. 289 (23), 15969–15978. doi:10.1074/jbc.M114.564252
Dong, Q., Li, S., Wang, W., Han, L., Xia, Z., Wu, Y., et al. (2019). FGF23 regulates atrial fibrosis in atrial fibrillation by mediating the STAT3 and SMAD3 pathways. J. Cell Physiol. 234 (11), 19502–19510. doi:10.1002/jcp.28548
Durham, G. A., Williams, J. J. L., Nasim, M. T., and Palmer, T. M. (2019). Targeting SOCS proteins to control JAK-STAT signalling in disease. Trends Pharmacol. Sci. 40 (5), 298–308. doi:10.1016/j.tips.2019.03.001
Działo, E., Tkacz, K., and Błyszczuk, P. (2018). Crosstalk between the TGF-β and WNT signalling pathways during cardiac fibrogenesis. Acta Biochim. Pol. 65 (3), 341–349. doi:10.18388/abp.2018_2635
Eid, R. A., Alkhateeb, M. A., El-Kott, A. F., Eleawa, S. M., Zaki, M. S. A., Alaboodi, S. A., et al. (2019). A high-fat diet rich in corn oil induces cardiac fibrosis in rats by activating JAK2/STAT3 and subsequent activation of ANG II/TGF-1β/Smad3 pathway: the role of ROS and IL-6 trans-signaling. J. Food Biochem. 43 (8), e12952. doi:10.1111/jfbc.12952
Enomoto, D., Obana, M., Miyawaki, A., Maeda, M., Nakayama, H., and Fujio, Y. (2015). Cardiac-specific ablation of the STAT3 gene in the subacute phase of myocardial infarction exacerbated cardiac remodeling. Am. J. Physiol. Heart Circ. Physiol. 309 (3), H471–H480. doi:10.1152/ajpheart.00730.2014
Escobar, Z., Bjartell, A., Canesin, G., Evans-Axelsson, S., Sterner, O., Hellsten, R., et al. (2016). Preclinical characterization of 3β-(N-acetyl l-cysteine methyl ester)-2aβ,3-dihydrogaliellalactone (GPA512), a prodrug of a direct STAT3 inhibitor for the treatment of prostate cancer. J. Med. Chem. 59 (10), 4551–4562. doi:10.1021/acs.jmedchem.5b01814
Estrada-Navarrete, G., Cruz-Mireles, N., Lascano, R., Alvarado-Affantranger, X., Hernández-Barrera, A., Barraza, A., et al. (2016). An autophagy-related kinase is essential for the symbiotic relationship between Phaseolus vulgaris and both rhizobia and arbuscular mycorrhizal fungi. Plant Cell 28 (9), 2326–2341. doi:10.1105/tpc.15.01012
Feng, J., Witthuhn, B. A., Matsuda, T., Kohlhuber, F., Kerr, I. M., and Ihle, J. N. (1997). Activation of Jak2 catalytic activity requires phosphorylation of Y1007 in the kinase activation loop. Mol. Cell Biol. 17 (5), 2497–2501. doi:10.1128/mcb.17.5.2497
Ferreira, V. M., Marcelino, M., Piechnik, S. K., Marini, C., Karamitsos, T. D., Ntusi, N. A. B., et al. (2016). Pheochromocytoma is characterized by catecholamine-mediated myocarditis, focal and diffuse myocardial fibrosis, and myocardial dysfunction. J. Am. Coll. Cardiol. 67 (20), 2364–2374. doi:10.1016/j.jacc.2016.03.543
Francis Stuart, S. D., De Jesus, N. M., Lindsey, M. L., and Ripplinger, C. M. (2016). The crossroads of inflammation, fibrosis, and arrhythmia following myocardial infarction. J. Mol. Cell Cardiol. 91, 114–122. doi:10.1016/j.yjmcc.2015.12.024
Fu, H., Dong, R., Zhang, Y., Xu, J., Liu, M., and Chen, P. (2019). Tmub1 negatively regulates liver regeneration via inhibiting STAT3 phosphorylation. Cell Signal 55, 65–72. doi:10.1016/j.cellsig.2018.12.013
Fuglesteg, B. N., Suleman, N., Tiron, C., Kanhema, T., Lacerda, L., Andreasen, T. V., et al. (2008). Signal transducer and activator of transcription 3 is involved in the cardioprotective signalling pathway activated by insulin therapy at reperfusion. Basic Res. Cardiol. 103 (5), 444–453. doi:10.1007/s00395-008-0728-x
Furtek, S. L., Backos, D. S., Matheson, C. J., and Reigan, P. (2016). Strategies and approaches of targeting STAT3 for cancer treatment. ACS Chem. Biol. 11 (2), 308–318. doi:10.1021/acschembio.5b00945
Gan, C., Wang, Y., Xiang, Z., Liu, H., Tan, Z., Xie, Y., et al. (2023). Niclosamide-loaded nanoparticles (Ncl-NPs) reverse pulmonary fibrosis in vivo and in vitro. J. Adv. Res. 51, 109–120. doi:10.1016/j.jare.2022.10.018
Ganjali, S., Blesso, C. N., Banach, M., Pirro, M., Majeed, M., and Sahebkar, A. (2017). Effects of curcumin on HDL functionality. Pharmacol. Res. 119, 208–218. doi:10.1016/j.phrs.2017.02.008
Gao, H., Huang, X., Tong, Y., and Jiang, X. (2020). Urolithin B improves cardiac function and reduces susceptibility to ventricular arrhythmias in rats after myocardial infarction. Eur. J. Pharmacol. 871, 172936. doi:10.1016/j.ejphar.2020.172936
Gao, S., Zhan, L., Yang, Z., Shi, R., Li, H., Xia, Z., et al. (2017). Remote limb ischaemic postconditioning protects against myocardial ischaemia/reperfusion injury in mice: activation of JAK/STAT3-Mediated nrf2-antioxidant signalling. Cell Physiol. Biochem. 43 (3), 1140–1151. doi:10.1159/000481755
Garama, D. J., White, C. L., Balic, J. J., and Gough, D. J. (2016). Mitochondrial STAT3: powering up a potent factor. Cytokine 87, 20–25. doi:10.1016/j.cyto.2016.05.019
Geng, Z., Fan, W. Y., Zhou, B., Ye, C., Tong, Y., Zhou, Y. B., et al. (2019). FNDC5 attenuates obesity-induced cardiac hypertrophy by inactivating JAK2/STAT3-associated inflammation and oxidative stress. J. Transl. Med. 17 (1), 107. doi:10.1186/s12967-019-1857-8
Gogiraju, R., Hubert, A., Fahrer, J., Straub, B. K., Brandt, M., Wenzel, P., et al. (2019). Endothelial leptin receptor deletion promotes cardiac autophagy and angiogenesis following pressure overload by suppressing akt/mTOR signaling. Circ. Heart Fail 12 (1), e005622. doi:10.1161/circheartfailure.118.005622
Grosche, J., Meißner, J., and Eble, J. A. (2018). More than a syllable in fib-ROS-is: the role of ROS on the fibrotic extracellular matrix and on cellular contacts. Mol. Asp. Med. 63, 30–46. doi:10.1016/j.mam.2018.03.005
Guo, X., Yan, F., Li, J., Zhang, C., and Bu, P. (2017). SIRT3 attenuates AngII-induced cardiac fibrosis by inhibiting myofibroblasts transdifferentiation via STAT3-NFATc2 pathway. Am. J. Transl. Res. 9 (7), 3258–3269.
Gurzov, E. N., Stanley, W. J., Pappas, E. G., Thomas, H. E., and Gough, D. J. (2016). The JAK/STAT pathway in obesity and diabetes. Febs J. 283 (16), 3002–3015. doi:10.1111/febs.13709
Gyöngyösi, M., Winkler, J., Ramos, I., Do, Q. T., Firat, H., McDonald, K., et al. (2017). Myocardial fibrosis: biomedical research from bench to bedside. Eur. J. Heart Fail 19 (2), 177–191. doi:10.1002/ejhf.696
Haghikia, A., Ricke-Hoch, M., Stapel, B., Gorst, I., and Hilfiker-Kleiner, D. (2014). STAT3, a key regulator of cell-to-cell communication in the heart. Cardiovasc Res. 102 (2), 281–289. doi:10.1093/cvr/cvu034
Han, J., Ye, S., Zou, C., Chen, T., Wang, J., Li, J., et al. (2018). Angiotensin II causes biphasic STAT3 activation through TLR4 to initiate cardiac remodeling. Hypertension 72 (6), 1301–1311. doi:10.1161/hypertensionaha.118.11860
Harhous, Z., Booz, G. W., Ovize, M., Bidaux, G., and Kurdi, M. (2019). An update on the multifaceted roles of STAT3 in the heart. Front. Cardiovasc Med. 6, 150. doi:10.3389/fcvm.2019.00150
Hattori, R., Maulik, N., Otani, H., Zhu, L., Cordis, G., Engelman, R. M., et al. (2001). Role of STAT3 in ischemic preconditioning. J. Mol. Cell Cardiol. 33 (11), 1929–1936. doi:10.1006/jmcc.2001.1456
He, F., Liu, H., Guo, J., Yang, D., Yu, Y., Yu, J., et al. (2018). Inhibition of MicroRNA-124 reduces cardiomyocyte apoptosis following myocardial infarction via targeting STAT3. Cell Physiol. Biochem. 51 (1), 186–200. doi:10.1159/000495173
Hinz, B. (2007). Formation and function of the myofibroblast during tissue repair. J. Invest. Dermatol 127 (3), 526–537. doi:10.1038/sj.jid.5700613
Hinz, B. (2010). The myofibroblast: paradigm for a mechanically active cell. J. Biomech. 43 (1), 146–155. doi:10.1016/j.jbiomech.2009.09.020
Hsueh, Y. J., Chen, H. C., Chu, W. K., Cheng, C. C., Kuo, P. C., Lin, L. Y., et al. (2011). STAT3 regulates the proliferation and differentiation of rabbit limbal epithelial cells via a ΔNp63-dependent mechanism. Invest. Ophthalmol. Vis. Sci. 52 (7), 4685–4693. doi:10.1167/iovs.10-6103
Hu, H. H., Chen, D. Q., Wang, Y. N., Feng, Y. L., Cao, G., Vaziri, N. D., et al. (2018). New insights into TGF-β/Smad signaling in tissue fibrosis. Chem. Biol. Interact. 292, 76–83. doi:10.1016/j.cbi.2018.07.008
Hu, X., Li, J., Fu, M., Zhao, X., and Wang, W. (2021). The JAK/STAT signaling pathway: from bench to clinic. Signal Transduct. Target Ther. 6 (1), 402. doi:10.1038/s41392-021-00791-1
Hu, Z. G., Han, Y., Liu, Y., Zhao, Z., Ma, F., Cui, A., et al. (2020a). CREBZF as a key regulator of STAT3 pathway in the control of liver regeneration in mice. Hepatology 71 (4), 1421–1436. doi:10.1002/hep.30919
Hu, Z. G., Zhang, S., Chen, Y. B., Cao, W., Zhou, Z. Y., Zhang, J. N., et al. (2020b). DTNA promotes HBV-induced hepatocellular carcinoma progression by activating STAT3 and regulating TGFβ1 and P53 signaling. Life Sci. 258, 118029. doi:10.1016/j.lfs.2020.118029
Huang, M., Song, K., Liu, X., Lu, S., Shen, Q., Wang, R., et al. (2018). AlloFinder: a strategy for allosteric modulator discovery and allosterome analyses. Nucleic Acids Res. 46 (1), W451–w458. doi:10.1093/nar/gky374
Huang, W., Dong, Z., Chen, Y., Wang, F., Wang, C. J., Peng, H., et al. (2016). Small-molecule inhibitors targeting the DNA-binding domain of STAT3 suppress tumor growth, metastasis and STAT3 target gene expression in vivo. Oncogene 35 (6), 783–792. doi:10.1038/onc.2015.215
Hubbard, S. R. (2017). Mechanistic insights into regulation of JAK2 tyrosine kinase. Front. Endocrinol. (Lausanne) 8, 361. doi:10.3389/fendo.2017.00361
Huby, A. C., Antonova, G., Groenendyk, J., Gomez-Sanchez, C. E., Bollag, W. B., Filosa, J. A., et al. (2015). Adipocyte-Derived hormone leptin is a direct regulator of aldosterone secretion, which promotes endothelial dysfunction and cardiac fibrosis. Circulation 132 (22), 2134–2145. doi:10.1161/circulationaha.115.018226
Humphreys, B. D., Lin, S. L., Kobayashi, A., Hudson, T. E., Nowlin, B. T., Bonventre, J. V., et al. (2010). Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am. J. Pathol. 176 (1), 85–97. doi:10.2353/ajpath.2010.090517
Huo, S., Shi, W., Ma, H., Yan, D., Luo, P., Guo, J., et al. (2021). Alleviation of inflammation and oxidative stress in pressure overload-induced cardiac remodeling and heart failure via IL-6/STAT3 inhibition by raloxifene. Oxid. Med. Cell Longev. 2021, 6699054. doi:10.1155/2021/6699054
Itoh, Y., Saitoh, M., and Miyazawa, K. (2018). Smad3-STAT3 crosstalk in pathophysiological contexts. Acta Biochim. Biophys. Sin. (Shanghai) 50 (1), 82–90. doi:10.1093/abbs/gmx118
Jahangiri, A., Dadmanesh, M., and Ghorban, K. (2020). STAT3 inhibition reduced PD-L1 expression and enhanced antitumor immune responses. J. Cell Physiol. 235 (12), 9457–9463. doi:10.1002/jcp.29750
Kamran, M. Z., Patil, P., and Gude, R. P. (2013). Role of STAT3 in cancer metastasis and translational advances. Biomed. Res. Int. 2013, 421821. doi:10.1155/2013/421821
Kanisicak, O., Khalil, H., Ivey, M. J., Karch, J., Maliken, B. D., Correll, R. N., et al. (2016). Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat. Commun. 7, 12260. doi:10.1038/ncomms12260
Kaushal, G. P., Chandrashekar, K., Juncos, L. A., and Shah, S. V. (2020). Autophagy function and regulation in kidney disease. Biomolecules 10 (1), 100. doi:10.3390/biom10010100
Kazi, J. U., Kabir, N. N., Flores-Morales, A., and Rönnstrand, L. (2014). SOCS proteins in regulation of receptor tyrosine kinase signaling. Cell Mol. Life Sci. 71 (17), 3297–3310. doi:10.1007/s00018-014-1619-y
Kershaw, N. J., Murphy, J. M., Liau, N. P., Varghese, L. N., Laktyushin, A., Whitlock, E. L., et al. (2013a). SOCS3 binds specific receptor-JAK complexes to control cytokine signaling by direct kinase inhibition. Nat. Struct. Mol. Biol. 20 (4), 469–476. doi:10.1038/nsmb.2519
Kershaw, N. J., Murphy, J. M., Lucet, I. S., Nicola, N. A., and Babon, J. J. (2013b). Regulation of Janus kinases by SOCS proteins. Biochem. Soc. Trans. 41 (4), 1042–1047. doi:10.1042/bst20130077
Kishore, R., and Verma, S. K. (2012). Roles of STATs signaling in cardiovascular diseases. Jakstat 1 (2), 118–124. doi:10.4161/jkst.20115
Kuma, A., Komatsu, M., and Mizushima, N. (2017). Autophagy-monitoring and autophagy-deficient mice. Autophagy 13 (10), 1619–1628. doi:10.1080/15548627.2017.1343770
Kuo, W. Y., Hwu, L., Wu, C. Y., Lee, J. S., Chang, C. W., and Liu, R. S. (2017). STAT3/NF-κB-Regulated lentiviral TK/GCV suicide gene therapy for cisplatin-resistant triple-negative breast cancer. Theranostics 7 (3), 647–663. doi:10.7150/thno.16827
Leask, A. (2015). Getting to the heart of the matter: new insights into cardiac fibrosis. Circ. Res. 116 (7), 1269–1276. doi:10.1161/circresaha.116.305381
Lee, J. H., Kim, T. H., Oh, S. J., Yoo, J. Y., Akira, S., Ku, B. J., et al. (2013). Signal transducer and activator of transcription-3 (Stat3) plays a critical role in implantation via progesterone receptor in uterus. Faseb J. 27 (7), 2553–2563. doi:10.1096/fj.12-225664
Lee, T. M., Harn, H. J., Chiou, T. W., Chuang, M. H., Chen, C. H., Chuang, C. H., et al. (2019). Preconditioned adipose-derived stem cells ameliorate cardiac fibrosis by regulating macrophage polarization in infarcted rat hearts through the PI3K/STAT3 pathway. Lab. Invest. 99 (5), 634–647. doi:10.1038/s41374-018-0181-x
Li, H., Liang, Q., and Wang, L. (2019a). Icaritin inhibits glioblastoma cell viability and glycolysis by blocking the IL-6/Stat3 pathway. J. Cell Biochem. 120 (5), 7257–7264. doi:10.1002/jcb.28000
Li, Q., Ye, W. X., Huang, Z. J., Zhang, Q., and He, Y. F. (2019b). Effect of IL-6-mediated STAT3 signaling pathway on myocardial apoptosis in mice with dilated cardiomyopathy. Eur. Rev. Med. Pharmacol. Sci. 23 (7), 3042–3050. doi:10.26355/eurrev_201904_17586
Li, W., Qu, X., Kang, X., Zhang, H., Zhang, X., Hu, H., et al. (2022). Silibinin eliminates mitochondrial ROS and restores autophagy through IL6ST/JAK2/STAT3 signaling pathway to protect cardiomyocytes from doxorubicin-induced injury. Eur. J. Pharmacol. 929, 175153. doi:10.1016/j.ejphar.2022.175153
Li, W. J., Yan, H., Zhou, Z. Y., Zhang, N., Ding, W., Liao, H. H., et al. (2023). Cryptotanshinone attenuated pathological cardiac remodeling in vivo and in vitro experiments. Oxid. Med. Cell Longev. 2023, 4015199. doi:10.1155/2023/4015199
Li, W. X. (2008). Canonical and non-canonical JAK-STAT signaling. Trends Cell Biol. 18 (11), 545–551. doi:10.1016/j.tcb.2008.08.008
Li, Y., Zhang, X., Li, L., Wang, X., Chen, Z., Wang, X., et al. (2018). Mechanical stresses induce paracrine β-2 microglobulin from cardiomyocytes to activate cardiac fibroblasts through epidermal growth factor receptor. Clin. Sci. (Lond) 132 (16), 1855–1874. doi:10.1042/cs20180486
Lighthouse, J. K., and Small, E. M. (2016). Transcriptional control of cardiac fibroblast plasticity. J. Mol. Cell Cardiol. 91, 52–60. doi:10.1016/j.yjmcc.2015.12.016
Lin, T. A., Wu, V. C., and Wang, C. Y. (2019). Autophagy in chronic kidney diseases. Cells 8 (1), 61. doi:10.3390/cells8010061
Liongue, C., Taznin, T., and Ward, A. C. (2016). Signaling via the CytoR/JAK/STAT/SOCS pathway: emergence during evolution. Mol. Immunol. 71, 166–175. doi:10.1016/j.molimm.2016.02.002
Liu, J., Wang, F., and Luo, F. (2023a). The role of JAK/STAT pathway in fibrotic diseases: molecular and cellular mechanisms. Biomolecules 13 (1), 119. doi:10.3390/biom13010119
Liu, N., Shi, Y., and Zhuang, S. (2016). Autophagy in chronic kidney diseases. Kidney Dis. (Basel) 2 (1), 37–45. doi:10.1159/000444841
Liu, T., Song, D., Dong, J., Zhu, P., Liu, J., Liu, W., et al. (2017). Current understanding of the pathophysiology of myocardial fibrosis and its quantitative assessment in heart failure. Front. Physiol. 8, 238. doi:10.3389/fphys.2017.00238
Liu, X., Zheng, Q., Wang, K., Luo, J., Wang, Z., Li, H., et al. (2023b). Sam68 promotes osteogenic differentiation of aortic valvular interstitial cells by TNF-α/STAT3/autophagy axis. J. Cell Commun. Signal 17, 863–879. doi:10.1007/s12079-023-00733-2
Liu, Y., Liu, H., Meyer, C., Li, J., Nadalin, S., Königsrainer, A., et al. (2013). Transforming growth factor-β (TGF-β)-mediated connective tissue growth factor (CTGF) expression in hepatic stellate cells requires Stat3 signaling activation. J. Biol. Chem. 288 (42), 30708–30719. doi:10.1074/jbc.M113.478685
Lo, S. H., Hsu, C. T., Niu, H. S., Niu, C. S., Cheng, J. T., and Chen, Z. C. (2017a). Cryptotanshinone inhibits STAT3 signaling to alleviate cardiac fibrosis in type 1-like diabetic rats. Phytother. Res. 31 (4), 638–646. doi:10.1002/ptr.5777
Lo, S. H., Hsu, C. T., Niu, H. S., Niu, C. S., Cheng, J. T., and Chen, Z. C. (2017b). Ginsenoside Rh2 improves cardiac fibrosis via pparδ-STAT3 signaling in type 1-like diabetic rats. Int. J. Mol. Sci. 18 (7), 1364. doi:10.3390/ijms18071364
Lu, T. X., and Rothenberg, M. E. (2018). MicroRNA. J. Allergy Clin. Immunol. 141 (4), 1202–1207. doi:10.1016/j.jaci.2017.08.034
Makitani, K., Ogo, N., and Asai, A. (2020). STX-0119, a novel STAT3 dimerization inhibitor, prevents fibrotic gene expression in a mouse model of kidney fibrosis by regulating Cxcr4 and Ccr1 expression. Physiol. Rep. 8 (20), e14627. doi:10.14814/phy2.14627
Marciniec, K., Pawełczak, B., Latocha, M., Skrzypek, L., Maciążek-Jurczyk, M., and Boryczka, S. (2017). Synthesis, anti-breast cancer activity, and molecular docking study of a new group of acetylenic quinolinesulfonamide derivatives. Molecules 22 (2), 300. doi:10.3390/molecules22020300
Maritano, D., Sugrue, M. L., Tininini, S., Dewilde, S., Strobl, B., Fu, X., et al. (2004). The STAT3 isoforms alpha and beta have unique and specific functions. Nat. Immunol. 5 (4), 401–409. doi:10.1038/ni1052
Masoumi-Dehghi, S., Babashah, S., and Sadeghizadeh, M. (2020). microRNA-141-3p-containing small extracellular vesicles derived from epithelial ovarian cancer cells promote endothelial cell angiogenesis through activating the JAK/STAT3 and NF-κB signaling pathways. J. Cell Commun. Signal 14 (2), 233–244. doi:10.1007/s12079-020-00548-5
Matsuda, T., Nakamura, T., Nakao, K., Arai, T., Katsuki, M., Heike, T., et al. (1999). STAT3 activation is sufficient to maintain an undifferentiated state of mouse embryonic stem cells. Embo J. 18 (15), 4261–4269. doi:10.1093/emboj/18.15.4261
Mialet-Perez, J., and Vindis, C. (2017). Autophagy in health and disease: focus on the cardiovascular system. Essays Biochem. 61 (6), 721–732. doi:10.1042/ebc20170022
Minamoto, S., Ikegame, K., Ueno, K., Narazaki, M., Naka, T., Yamamoto, H., et al. (1997). Cloning and functional analysis of new members of STAT induced STAT inhibitor (SSI) family: SSI-2 and SSI-3. Biochem. Biophys. Res. Commun. 237 (1), 79–83. doi:10.1006/bbrc.1997.7080
Mir, S. A., Chatterjee, A., Mitra, A., Pathak, K., Mahata, S. K., and Sarkar, S. (2012). Inhibition of signal transducer and activator of transcription 3 (STAT3) attenuates interleukin-6 (IL-6)-induced collagen synthesis and resultant hypertrophy in rat heart. J. Biol. Chem. 287 (4), 2666–2677. doi:10.1074/jbc.M111.246173
Mizutani, M., Wu, J. C., and Nusse, R. (2016). Fibrosis of the neonatal mouse heart after cryoinjury is accompanied by Wnt signaling activation and epicardial-to-mesenchymal transition. J. Am. Heart Assoc. 5 (3), e002457. doi:10.1161/jaha.115.002457
Mohammed, S. F., Hussain, S., Mirzoyev, S. A., Edwards, W. D., Maleszewski, J. J., and Redfield, M. M. (2015). Coronary microvascular rarefaction and myocardial fibrosis in heart failure with preserved ejection fraction. Circulation 131 (6), 550–559. doi:10.1161/circulationaha.114.009625
Molkentin, J. D., Bugg, D., Ghearing, N., Dorn, L. E., Kim, P., Sargent, M. A., et al. (2017). Fibroblast-specific genetic manipulation of p38 mitogen-activated protein kinase in vivo reveals its central regulatory role in fibrosis. Circulation 136 (6), 549–561. doi:10.1161/circulationaha.116.026238
Möllmann, H., Nef, H. M., Kostin, S., von Kalle, C., Pilz, I., Weber, M., et al. (2006). Bone marrow-derived cells contribute to infarct remodelling. Cardiovasc Res. 71 (4), 661–671. doi:10.1016/j.cardiores.2006.06.013
Montero, P., Milara, J., Roger, I., and Cortijo, J. (2021). Role of JAK/STAT in interstitial lung diseases; molecular and cellular mechanisms. Int. J. Mol. Sci. 22 (12), 6211. doi:10.3390/ijms22126211
Moore-Morris, T., Cattaneo, P., Guimarães-Camboa, N., Bogomolovas, J., Cedenilla, M., Banerjee, I., et al. (2018). Infarct fibroblasts do not derive from bone marrow lineages. Circ. Res. 122 (4), 583–590. doi:10.1161/circresaha.117.311490
Moore-Morris, T., Guimarães-Camboa, N., Banerjee, I., Zambon, A. C., Kisseleva, T., Velayoudon, A., et al. (2014). Resident fibroblast lineages mediate pressure overload-induced cardiac fibrosis. J. Clin. Invest. 124 (7), 2921–2934. doi:10.1172/jci74783
Morris, R., Kershaw, N. J., and Babon, J. J. (2018). The molecular details of cytokine signaling via the JAK/STAT pathway. Protein Sci. 27 (12), 1984–2009. doi:10.1002/pro.3519
Mott, J. L., Kobayashi, S., Bronk, S. F., and Gores, G. J. (2007). mir-29 regulates Mcl-1 protein expression and apoptosis. Oncogene 26 (42), 6133–6140. doi:10.1038/sj.onc.1210436
Naka, T., Narazaki, M., Hirata, M., Matsumoto, T., Minamoto, S., Aono, A., et al. (1997). Structure and function of a new STAT-induced STAT inhibitor. Nature 387 (6636), 924–929. doi:10.1038/43219
Negoro, S., Kunisada, K., Tone, E., Funamoto, M., Oh, H., Kishimoto, T., et al. (2000). Activation of JAK/STAT pathway transduces cytoprotective signal in rat acute myocardial infarction. Cardiovasc Res. 47 (4), 797–805. doi:10.1016/s0008-6363(00)00138-3
Németh, Á., Mózes, M. M., Calvier, L., Hansmann, G., and Kökény, G. (2019). The PPARγ agonist pioglitazone prevents TGF-β induced renal fibrosis by repressing EGR-1 and STAT3. BMC Nephrol. 20 (1), 245. doi:10.1186/s12882-019-1431-x
Nguyen, M. N., Kiriazis, H., Gao, X. M., and Du, X. J. (2017). Cardiac fibrosis and arrhythmogenesis. Compr. Physiol. 7 (3), 1009–1049. doi:10.1002/cphy.c160046
O'Donoghue, R. J., Knight, D. A., Richards, C. D., Prêle, C. M., Lau, H. L., Jarnicki, A. G., et al. (2012). Genetic partitioning of interleukin-6 signalling in mice dissociates Stat3 from Smad3-mediated lung fibrosis. EMBO Mol. Med. 4 (9), 939–951. doi:10.1002/emmm.201100604
Ogata, H., Chinen, T., Yoshida, T., Kinjyo, I., Takaesu, G., Shiraishi, H., et al. (2006). Loss of SOCS3 in the liver promotes fibrosis by enhancing STAT3-mediated TGF-beta1 production. Oncogene 25 (17), 2520–2530. doi:10.1038/sj.onc.1209281
O'Shea, J. J., Schwartz, D. M., Villarino, A. V., Gadina, M., McInnes, I. B., and Laurence, A. (2015). The JAK-STAT pathway: impact on human disease and therapeutic intervention. Annu. Rev. Med. 66, 311–328. doi:10.1146/annurev-med-051113-024537
Owen, K. L., Brockwell, N. K., and Parker, B. S. (2019). JAK-STAT signaling: a double-edged sword of immune regulation and cancer progression. Cancers (Basel) 11 (12), 2002. doi:10.3390/cancers11122002
Park, J. Y., Yoo, K. D., Bae, E., Kim, K. H., Lee, J. W., Shin, S. J., et al. (2022). Blockade of STAT3 signaling alleviates the progression of acute kidney injury to chronic kidney disease through antiapoptosis. Am. J. Physiol. Ren. Physiol. 322 (5), F553–f572. doi:10.1152/ajprenal.00595.2020
Park, Y. J., Lee, K. H., Jeon, M. S., Lee, Y. H., Ko, Y. J., Pang, C., et al. (2020). Hepatoprotective potency of chrysophanol 8-O-glucoside from rheum palmatum L. Against hepatic fibrosis via regulation of the STAT3 signaling pathway. Int. J. Mol. Sci. 21 (23), 9044. doi:10.3390/ijms21239044
Patel, N. J., Nassal, D. M., Greer-Short, A. D., Unudurthi, S. D., Scandling, B. W., Gratz, D., et al. (2019). βIV-Spectrin/STAT3 complex regulates fibroblast phenotype, fibrosis, and cardiac function. JCI Insight 4 (20), e131046. doi:10.1172/jci.insight.131046
Pedroza, M., To, S., Assassi, S., Wu, M., Tweardy, D., and Agarwal, S. K. (2018). Role of STAT3 in skin fibrosis and transforming growth factor beta signalling. Rheumatol. Oxf. 57 (10), 1838–1850. doi:10.1093/rheumatology/kex347
Pekarsky, Y., Santanam, U., Cimmino, A., Palamarchuk, A., Efanov, A., Maximov, V., et al. (2006). Tcl1 expression in chronic lymphocytic leukemia is regulated by miR-29 and miR-181. Cancer Res. 66 (24), 11590–11593. doi:10.1158/0008-5472.Can-06-3613
Peng, C., Lu, Y., Li, R., Zhang, L., Liu, Z., Xu, X., et al. (2023). Neuroimmune modulation mediated by il-6: a potential target for the treatment of ischemia-induced ventricular arrhythmias. Heart Rhythm. 1547-5271, 03071. doi:10.1016/j.hrthm.2023.12.020
Piessevaux, J., Lavens, D., Peelman, F., and Tavernier, J. (2008). The many faces of the SOCS box. Cytokine Growth Factor Rev. 19 (5-6), 371–381. doi:10.1016/j.cytogfr.2008.08.006
Prabhu, S. D., and Frangogiannis, N. G. (2016). The biological basis for cardiac repair after myocardial infarction: from inflammation to fibrosis. Circ. Res. 119 (1), 91–112. doi:10.1161/circresaha.116.303577
Qi, W., Li, X., Ren, Y., Liu, X., Fu, H., Wang, X., et al. (2022). Downregulation of lncRNA Miat contributes to the protective effect of electroacupuncture against myocardial fibrosis. Chin. Med. 17 (1), 57. doi:10.1186/s13020-022-00615-6
Qin, S., Zou, Y., and Zhang, C. L. (2013). Cross-talk between KLF4 and STAT3 regulates axon regeneration. Nat. Commun. 4, 2633. doi:10.1038/ncomms3633
Qiu, Z. Y., and Fan, Y. (2016). Large granular lymphocytic leukemia and JAK/STAT signaling pathway--review. Zhongguo Shi Yan Xue Ye Xue Za Zhi 24 (1), 254–260. doi:10.7534/j.issn.1009-2137.2016.01.049
Qu, H., Wang, Y., Wang, Y., Yang, T., Feng, Z., Qu, Y., et al. (2017). Luhong formula inhibits myocardial fibrosis in a paracrine manner by activating the gp130/JAK2/STAT3 pathway in cardiomyocytes. J. Ethnopharmacol. 202, 28–37. doi:10.1016/j.jep.2017.01.033
Raivola, J., Haikarainen, T., Abraham, B. G., and Silvennoinen, O. (2021). Janus kinases in leukemia. Cancers (Basel) 13 (4), 800. doi:10.3390/cancers13040800
Rane, S. G., and Reddy, E. P. (2000). Janus kinases: components of multiple signaling pathways. Oncogene 19 (49), 5662–5679. doi:10.1038/sj.onc.1203925
Rockey, D. C., Bell, P. D., and Hill, J. A. (2015). Fibrosis--a common pathway to organ injury and failure. N. Engl. J. Med. 372 (12), 1138–1149. doi:10.1056/NEJMra1300575
Roskoski, R. (2016). Janus kinase (JAK) inhibitors in the treatment of inflammatory and neoplastic diseases. Pharmacol. Res. 111, 784–803. doi:10.1016/j.phrs.2016.07.038
Roy, S., Benz, F., Vargas Cardenas, D., Vucur, M., Gautheron, J., Schneider, A., et al. (2015). miR-30c and miR-193 are a part of the TGF-β-dependent regulatory network controlling extracellular matrix genes in liver fibrosis. J. Dig. Dis. 16 (9), 513–524. doi:10.1111/1751-2980.12266
Ruff-Jamison, S., Zhong, Z., Wen, Z., Chen, K., Darnell, J. E., and Cohen, S. (1994). Epidermal growth factor and lipopolysaccharide activate Stat3 transcription factor in mouse liver. J. Biol. Chem. 269 (35), 21933–21935. doi:10.1016/s0021-9258(17)31735-0
Saharinen, P., Takaluoma, K., and Silvennoinen, O. (2000). Regulation of the Jak2 tyrosine kinase by its pseudokinase domain. Mol. Cell Biol. 20 (10), 3387–3395. doi:10.1128/mcb.20.10.3387-3395.2000
Samidurai, A., Roh, S. K., Prakash, M., Durrant, D., Salloum, F. N., Kukreja, R. C., et al. (2020). STAT3-miR-17/20 signalling axis plays a critical role in attenuating myocardial infarction following rapamycin treatment in diabetic mice. Cardiovasc Res. 116 (13), 2103–2115. doi:10.1093/cvr/cvz315
Sasaki, A., Yasukawa, H., Suzuki, A., Kamizono, S., Syoda, T., Kinjyo, I., et al. (1999). Cytokine-inducible SH2 protein-3 (CIS3/SOCS3) inhibits Janus tyrosine kinase by binding through the N-terminal kinase inhibitory region as well as SH2 domain. Genes Cells 4 (6), 339–351. doi:10.1046/j.1365-2443.1999.00263.x
Schaefer, T. S., Sanders, L. K., and Nathans, D. (1995). Cooperative transcriptional activity of Jun and Stat3 beta, a short form of Stat3. Proc. Natl. Acad. Sci. U. S. A. 92 (20), 9097–9101. doi:10.1073/pnas.92.20.9097
Schafer, S., Viswanathan, S., Widjaja, A. A., Lim, W. W., Moreno-Moral, A., DeLaughter, D. M., et al. (2017). IL-11 is a crucial determinant of cardiovascular fibrosis. Nature 552 (7683), 110–115. doi:10.1038/nature24676
Schindler, C., and Darnell, J. E. (1995). Transcriptional responses to polypeptide ligands: the JAK-STAT pathway. Annu. Rev. Biochem. 64, 621–651. doi:10.1146/annurev.bi.64.070195.003201
Schmitz, J., Dahmen, H., Grimm, C., Gendo, C., Müller-Newen, G., Heinrich, P. C., et al. (2000). The cytoplasmic tyrosine motifs in full-length glycoprotein 130 have different roles in IL-6 signal transduction. J. Immunol. 164 (2), 848–854. doi:10.4049/jimmunol.164.2.848
Seo, H. Y., Jeon, J. H., Jung, Y. A., Jung, G. S., Lee, E. J., Choi, Y. K., et al. (2016). Fyn deficiency attenuates renal fibrosis by inhibition of phospho-STAT3. Kidney Int. 90 (6), 1285–1297. doi:10.1016/j.kint.2016.06.038
Shen, K., Li, R., Zhang, X., Qu, G., Li, R., Wang, Y., et al. (2021). Acetyl oxygen benzoate engeletin ester promotes KLF4 degradation leading to the attenuation of pulmonary fibrosis via inhibiting TGFβ1-smad/p38MAPK-lnc865/lnc556-miR-29b-2-5p-STAT3 signal pathway. Aging (Albany NY) 13 (10), 13807–13821. doi:10.18632/aging.202975
Shi, M., Lin, T. H., Appell, K. C., and Berg, L. J. (2008a). Janus-kinase-3-dependent signals induce chromatin remodeling at the Ifng locus during T helper 1 cell differentiation. Immunity 28 (6), 763–773. doi:10.1016/j.immuni.2008.04.016
Shi, S., Calhoun, H. C., Xia, F., Li, J., Le, L., and Li, W. X. (2006). JAK signaling globally counteracts heterochromatic gene silencing. Nat. Genet. 38 (9), 1071–1076. doi:10.1038/ng1860
Shi, S., Larson, K., Guo, D., Lim, S. J., Dutta, P., Yan, S. J., et al. (2008b). Drosophila STAT is required for directly maintaining HP1 localization and heterochromatin stability. Nat. Cell Biol. 10 (4), 489–496. doi:10.1038/ncb1713
Shi, Y., and Massagué, J. (2003). Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 113 (6), 685–700. doi:10.1016/s0092-8674(03)00432-x
Shinde, A. V., and Frangogiannis, N. G. (2017). Mechanisms of fibroblast activation in the remodeling myocardium. Curr. Pathobiol. Rep. 5 (2), 145–152. doi:10.1007/s40139-017-0132-z
Shuya, L. L., Menkhorst, E. M., Yap, J., Li, P., Lane, N., and Dimitriadis, E. (2011). Leukemia inhibitory factor enhances endometrial stromal cell decidualization in humans and mice. PLoS One 6 (9), e25288. doi:10.1371/journal.pone.0025288
Singh, A., Ramesh, S., Cibi, D. M., Yun, L. S., Li, J., Li, L., et al. (2016). Hippo signaling mediators yap and taz are required in the epicardium for coronary vasculature development. Cell Rep. 15 (7), 1384–1393. doi:10.1016/j.celrep.2016.04.027
Sonnenblick, A., Levy, C., and Razin, E. (2004). Interplay between MITF, PIAS3, and STAT3 in mast cells and melanocytes. Mol. Cell Biol. 24 (24), 10584–10592. doi:10.1128/mcb.24.24.10584-10592.2004
Sp, N., Kang, D. Y., Kim, D. H., Park, J. H., Lee, H. G., Kim, H. J., et al. (2018). Nobiletin inhibits CD36-dependent tumor angiogenesis, migration, invasion, and sphere formation through the Cd36/stat3/nf-κb signaling Axis. Nutrients 10 (6), 772. doi:10.3390/nu10060772
Speirs, C., Williams, J. J. L., Riches, K., Salt, I. P., and Palmer, T. M. (2018). Linking energy sensing to suppression of JAK-STAT signalling: a potential route for repurposing AMPK activators? Pharmacol. Res. 128, 88–100. doi:10.1016/j.phrs.2017.10.001
Su, Y. C., Li, S. C., Peng, H. Y., Ho, Y. H., Chen, L. J., and Liao, H. F. (2013). RAD001-mediated STAT3 upregulation and megakaryocytic differentiation. Thromb. Haemost. 109 (3), 540–549. doi:10.1160/th12-10-0734
Su, S. A., Yang, D., Wu, Y., Xie, Y., Zhu, W., Cai, Z., et al. (2017). EphrinB2 regulates cardiac fibrosis through modulating the interaction of Stat3 and TGF-β/smad3 signaling. Circ. Res. 121 (6), 617–627. doi:10.1161/circresaha.117.311045
Su, Y., Zhang, W., Patro, C. P. K., Zhao, J., Mu, T., Ma, Z., et al. (2020). STAT3 regulates mouse neural progenitor proliferation and differentiation by promoting mitochondrial metabolism. Front. Cell Dev. Biol. 8, 362. doi:10.3389/fcell.2020.00362
Sun, W., Kim, D. H., Byon, C. H., Choi, H. I., Park, J. S., Bae, E. H., et al. (2022). β-Elemene attenuates renal fibrosis in the unilateral ureteral obstruction model by inhibition of STAT3 and Smad3 signaling via suppressing MyD88 expression. Int. J. Mol. Sci. 23 (10), 5553. doi:10.3390/ijms23105553
Szelag, M., Piaszyk-Borychowska, A., Plens-Galaska, M., Wesoly, J., and Bluyssen, H. A. (2016). Targeted inhibition of STATs and IRFs as a potential treatment strategy in cardiovascular disease. Oncotarget 7 (30), 48788–48812. doi:10.18632/oncotarget.9195
Tan, J. K., Ma, X. F., Wang, G. N., Jiang, C. R., Gong, H. Q., and Liu, H. (2021). LncRNA MIAT knockdown alleviates oxygen-glucose deprivation-induced cardiomyocyte injury by regulating JAK2/STAT3 pathway via miR-181a-5p. J. Cardiol. 78 (6), 586–597. doi:10.1016/j.jjcc.2021.08.018
Tang, L. Y., Heller, M., Meng, Z., Yu, L. R., Tang, Y., Zhou, M., et al. (2017). Transforming growth factor-β (TGF-β) directly activates the JAK1-STAT3 Axis to induce hepatic fibrosis in coordination with the SMAD pathway. J. Biol. Chem. 292 (10), 4302–4312. doi:10.1074/jbc.M116.773085
Tao, H., Yang, J. J., Shi, K. H., Deng, Z. Y., and Li, J. (2014). DNA methylation in cardiac fibrosis: new advances and perspectives. Toxicology 323, 125–129. doi:10.1016/j.tox.2014.07.002
Tesoro, L., Hernández, I., Ramírez-Carracedo, R., Díez-Mata, J., Alcharani, N., Jiménez-Guirado, B., et al. (2022). NIL10: a new IL10-receptor binding nanoparticle that induces cardiac protection in mice and pigs subjected to acute myocardial infarction through STAT3/NF-κB activation. Pharmaceutics 14 (10), 2044. doi:10.3390/pharmaceutics14102044
Timofeeva, O. A., Tarasova, N. I., Zhang, X., Chasovskikh, S., Cheema, A. K., Wang, H., et al. (2013). STAT3 suppresses transcription of proapoptotic genes in cancer cells with the involvement of its N-terminal domain. Proc. Natl. Acad. Sci. U. S. A. 110 (4), 1267–1272. doi:10.1073/pnas.1211805110
Tomasek, J. J., McRae, J., Owens, G. K., and Haaksma, C. J. (2005). Regulation of alpha-smooth muscle actin expression in granulation tissue myofibroblasts is dependent on the intronic CArG element and the transforming growth factor-beta1 control element. Am. J. Pathol. 166 (5), 1343–1351. doi:10.1016/s0002-9440(10)62353-x
Tussié-Luna, M. I., Bayarsaihan, D., Seto, E., Ruddle, F. H., and Roy, A. L. (2002). Physical and functional interactions of histone deacetylase 3 with TFII-I family proteins and PIASxbeta. Proc. Natl. Acad. Sci. U. S. A. 99 (20), 12807–12812. doi:10.1073/pnas.192464499
Ungureanu, D., Wu, J., Pekkala, T., Niranjan, Y., Young, C., Jensen, O. N., et al. (2011). The pseudokinase domain of JAK2 is a dual-specificity protein kinase that negatively regulates cytokine signaling. Nat. Struct. Mol. Biol. 18 (9), 971–976. doi:10.1038/nsmb.2099
Unudurthi, S. D., Nassal, D., Greer-Short, A., Patel, N., Howard, T., Xu, X., et al. (2018). βIV-Spectrin regulates STAT3 targeting to tune cardiac response to pressure overload. J. Clin. Invest. 128 (12), 5561–5572. doi:10.1172/jci99245
van Rooij, E., Sutherland, L. B., Thatcher, J. E., DiMaio, J. M., Naseem, R. H., Marshall, W. S., et al. (2008). Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc. Natl. Acad. Sci. U. S. A. 105 (35), 13027–13032. doi:10.1073/pnas.0805038105
Villarino, A. V., Kanno, Y., and O'Shea, J. J. (2017). Mechanisms and consequences of Jak-STAT signaling in the immune system. Nat. Immunol. 18 (4), 374–384. doi:10.1038/ni.3691
Wallweber, H. J., Tam, C., Franke, Y., Starovasnik, M. A., and Lupardus, P. J. (2014). Structural basis of recognition of interferon-α receptor by tyrosine kinase 2. Nat. Struct. Mol. Biol. 21 (5), 443–448. doi:10.1038/nsmb.2807
Wang, J., Liu, T., Chen, X., Jin, Q., Chen, Y., Zhang, L., et al. (2020). Bazedoxifene regulates Th17 immune response to ameliorate experimental autoimmune myocarditis via inhibition of STAT3 activation. Front. Pharmacol. 11, 613160. doi:10.3389/fphar.2020.613160
Wang, M., Li, J., Ding, Y., Cai, S., Li, Z., and Liu, P. (2021). PEX5 prevents cardiomyocyte hypertrophy via suppressing the redox-sensitive signaling pathways MAPKs and STAT3. Eur. J. Pharmacol. 906, 174283. doi:10.1016/j.ejphar.2021.174283
Wang, T., Mao, X., Li, H., Qiao, S., Xu, A., Wang, J., et al. (2013). N-Acetylcysteine and allopurinol up-regulated the Jak/STAT3 and PI3K/Akt pathways via adiponectin and attenuated myocardial postischemic injury in diabetes. Free Radic. Biol. Med. 63, 291–303. doi:10.1016/j.freeradbiomed.2013.05.043
Wang, Y., Gao, H., Cao, X., Li, Z., Kuang, Y., Ji, Y., et al. (2022a). Role of GADD45A in myocardial ischemia/reperfusion through mediation of the JNK/p38 MAPK and STAT3/VEGF pathways. Int. J. Mol. Med. 50 (6), 144. doi:10.3892/ijmm.2022.5200
Wang, Y., Li, M., Chen, J., Yu, Y., Yu, Y., Shi, H., et al. (2023). Macrophage CAPN4 regulates CVB3-induced cardiac inflammation and injury by promoting NLRP3 inflammasome activation and phenotypic transformation to the inflammatory subtype. Free Radic. Biol. Med. 208, 430–444. doi:10.1016/j.freeradbiomed.2023.08.032
Wang, Y., Wang, M., Samuel, C. S., and Widdop, R. E. (2022b). Preclinical rodent models of cardiac fibrosis. Br. J. Pharmacol. 179 (5), 882–899. doi:10.1111/bph.15450
Wang, Z., Li, J., Xiao, W., Long, J., and Zhang, H. (2018). The STAT3 inhibitor S3I-201 suppresses fibrogenesis and angiogenesis in liver fibrosis. Lab. Invest. 98 (12), 1600–1613. doi:10.1038/s41374-018-0127-3
Weber, K. T. (1989). Cardiac interstitium in health and disease: the fibrillar collagen network. J. Am. Coll. Cardiol. 13 (7), 1637–1652. doi:10.1016/0735-1097(89)90360-4
Xian, S., Chen, A., Wu, Y., Wen, H., Lu, C., Huang, F., et al. (2021). Interference with the expression of S1PR1 or STAT3 attenuates valvular damage due to rheumatic heart disease. Int. J. Mol. Med. 48 (3), 179. doi:10.3892/ijmm.2021.5012
Xie, L., Wu, Y., Zhou, C., Tan, Z., Xu, H., Chen, G., et al. (2021). Piceatannol protects against sepsis-induced myocardial dysfunction via direct inhibition of JAK2. Int. Immunopharmacol. 96, 107639. doi:10.1016/j.intimp.2021.107639
Xin, P., Xu, X., Deng, C., Liu, S., Wang, Y., Zhou, X., et al. (2020). The role of JAK/STAT signaling pathway and its inhibitors in diseases. Int. Immunopharmacol. 80, 106210. doi:10.1016/j.intimp.2020.106210
Xue, R., Lei, S., Xia, Z. Y., Wu, Y., Meng, Q., Zhan, L., et al. (2016). Selective inhibition of PTEN preserves ischaemic post-conditioning cardioprotection in STZ-induced Type 1 diabetic rats: role of the PI3K/Akt and JAK2/STAT3 pathways. Clin. Sci. (Lond) 130 (5), 377–392. doi:10.1042/cs20150496
Yajima, T., Murofushi, Y., Zhou, H., Park, S., Housman, J., Zhong, Z. H., et al. (2011). Absence of SOCS3 in the cardiomyocyte increases mortality in a gp130-dependent manner accompanied by contractile dysfunction and ventricular arrhythmias. Circulation 124 (24), 2690–2701. doi:10.1161/circulationaha.111.028498
Yang, J., Liao, X., Agarwal, M. K., Barnes, L., Auron, P. E., and Stark, G. R. (2007). Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NFkappaB. Genes Dev. 21 (11), 1396–1408. doi:10.1101/gad.1553707
Yang, X., Bao, M., Fang, Y., Yu, X., Ji, J., and Ding, X. (2021). STAT3/HIF-1α signaling activation mediates peritoneal fibrosis induced by high glucose. J. Transl. Med. 19 (1), 283. doi:10.1186/s12967-021-02946-8
Yang, Y., Hu, W., Di, S., Ma, Z., Fan, C., Wang, D., et al. (2017). Tackling myocardial ischemic injury: the signal transducer and activator of transcription 3 (STAT3) at a good site. Expert Opin. Ther. Targets 21 (2), 215–228. doi:10.1080/14728222.2017.1275566
Yasukawa, H., Misawa, H., Sakamoto, H., Masuhara, M., Sasaki, A., Wakioka, T., et al. (1999). The JAK-binding protein JAB inhibits Janus tyrosine kinase activity through binding in the activation loop. Embo J. 18 (5), 1309–1320. doi:10.1093/emboj/18.5.1309
Yasukawa, H., Yajima, T., Duplain, H., Iwatate, M., Kido, M., Hoshijima, M., et al. (2003). The suppressor of cytokine signaling-1 (SOCS1) is a novel therapeutic target for enterovirus-induced cardiac injury. J. Clin. Invest. 111 (4), 469–478. doi:10.1172/jci16491
You, L., Li, L., Xu, Q., Ren, J., and Zhang, F. (2011). Postconditioning reduces infarct size and cardiac myocyte apoptosis via the opioid receptor and JAK-STAT signaling pathway. Mol. Biol. Rep. 38 (1), 437–443. doi:10.1007/s11033-010-0126-y
Yu, C. L., Meyer, D. J., Campbell, G. S., Larner, A. C., Carter-Su, C., Schwartz, J., et al. (1995). Enhanced DNA-binding activity of a Stat3-related protein in cells transformed by the Src oncoprotein. Science 269 (5220), 81–83. doi:10.1126/science.7541555
Yu, H., and Jove, R. (2004). The STATs of cancer--new molecular targets come of age. Nat. Rev. Cancer 4 (2), 97–105. doi:10.1038/nrc1275
Yu, Y., Ou-Yang, W. X., Zhang, H., Jiang, T., Tang, L., Tan, Y. F., et al. (2021). MiR-125b enhances autophagic flux to improve septic cardiomyopathy via targeting STAT3/HMGB1. Exp. Cell Res. 409 (2), 112842. doi:10.1016/j.yexcr.2021.112842
Yuan, H. X., Chen, Y. T., Li, Y. Q., Wang, Y. S., Ou, Z. J., Li, Y., et al. (2023). Endothelial extracellular vesicles induce acute lung injury via follistatin-like protein 1. Sci. China Life Sci. 22, 1–15. doi:10.1007/s11427-022-2328-x
Yuan, J., Zhang, F., and Niu, R. (2015). Multiple regulation pathways and pivotal biological functions of STAT3 in cancer. Sci. Rep. 5, 17663. doi:10.1038/srep17663
Yuan, L., Qiu, L., Ye, Y., Wu, J., Wang, S., Wang, X., et al. (2018). Heat-shock transcription factor 1 is critically involved in the ischaemia-induced cardiac hypertrophy via JAK2/STAT3 pathway. J. Cell Mol. Med. 22 (9), 4292–4303. doi:10.1111/jcmm.13713
Yuan, Y., Zhang, Y., Han, X., Li, Y., Zhao, X., Sheng, L., et al. (2017). Relaxin alleviates TGFβ1-induced cardiac fibrosis via inhibition of Stat3-dependent autophagy. Biochem. Biophys. Res. Commun. 493 (4), 1601–1607. doi:10.1016/j.bbrc.2017.09.110
Zang, X., Zhao, Z., Chen, K., Song, W., Ma, J., Fu, H., et al. (2023). SHP-1 alleviates atrial fibrosis in atrial fibrillation by modulating STAT3 activation. Exp. Biol. Med. (Maywood) 248, 979–990. doi:10.1177/15353702231165717
Zeisberg, E. M., Tarnavski, O., Zeisberg, M., Dorfman, A. L., McMullen, J. R., Gustafsson, E., et al. (2007). Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 13 (8), 952–961. doi:10.1038/nm1613
Zhang, J. M., and An, J. (2007). Cytokines, inflammation, and pain. Int. Anesthesiol. Clin. 45 (2), 27–37. doi:10.1097/AIA.0b013e318034194e
Zhang, J. Q., Li, R., Dong, X. Y., He, N., Yin, R. J., Yang, M. K., et al. (2022a). Design, synthesis and structure-activity relationship studies of meridianin derivatives as novel JAK/STAT3 signaling inhibitors. Int. J. Mol. Sci. 23 (4), 2199. doi:10.3390/ijms23042199
Zhang, L., He, J., Wang, J., Liu, J., Chen, Z., Deng, B., et al. (2019). Knockout RAGE alleviates cardiac fibrosis through repressing endothelial-to-mesenchymal transition (EndMT) mediated by autophagy. Cell Death Dis. 12 (5), 470. doi:10.1038/s41419-021-03750-4
Zhang, W., Qu, X., Chen, B., Snyder, M., Wang, M., Li, B., et al. (2016). Critical roles of STAT3 in β-adrenergic functions in the heart. Circulation 133 (1), 48–61. doi:10.1161/circulationaha.115.017472
Zhang, X., Guo, A., Yu, J., Possemato, A., Chen, Y., Zheng, W., et al. (2007). Identification of STAT3 as a substrate of receptor protein tyrosine phosphatase T. Proc. Natl. Acad. Sci. U. S. A. 104 (10), 4060–4064. doi:10.1073/pnas.0611665104
Zhang, Y., Dees, C., Beyer, C., Lin, N. Y., Distler, A., Zerr, P., et al. (2015). Inhibition of casein kinase II reduces TGFβ induced fibroblast activation and ameliorates experimental fibrosis. Ann. Rheum. Dis. 74 (5), 936–943. doi:10.1136/annrheumdis-2013-204256
Zhang, Y., Lu, W., Zhang, X., Lu, J., Xu, S., Chen, S., et al. (2019a). Cryptotanshinone protects against pulmonary fibrosis through inhibiting Smad and STAT3 signaling pathways. Pharmacol. Res. 147, 104307. doi:10.1016/j.phrs.2019.104307
Zhang, Y., Zhang, L., Fan, X., Yang, W., Yu, B., Kou, J., et al. (2019b). Captopril attenuates TAC-induced heart failure via inhibiting Wnt3a/β-catenin and Jak2/Stat3 pathways. Biomed. Pharmacother. 113, 108780. doi:10.1016/j.biopha.2019.108780
Zhang, Z., Tang, J., Song, J., Xie, M., Liu, Y., Dong, Z., et al. (2022b). Elabela alleviates ferroptosis, myocardial remodeling, fibrosis and heart dysfunction in hypertensive mice by modulating the IL-6/STAT3/GPX4 signaling. Free Radic. Biol. Med. 181, 130–142. doi:10.1016/j.freeradbiomed.2022.01.020
Zhao, L., Wu, D., Sang, M., Xu, Y., Liu, Z., and Wu, Q. (2017). Stachydrine ameliorates isoproterenol-induced cardiac hypertrophy and fibrosis by suppressing inflammation and oxidative stress through inhibiting NF-κB and JAK/STAT signaling pathways in rats. Int. Immunopharmacol. 48, 102–109. doi:10.1016/j.intimp.2017.05.002
Zhao, Q., Bai, J., Chen, Y., Liu, X., Zhao, S., Ling, G., et al. (2022). An optimized herbal combination for the treatment of liver fibrosis: hub genes, bioactive ingredients, and molecular mechanisms. J. Ethnopharmacol. 297, 115567. doi:10.1016/j.jep.2022.115567
Zhao, X. B., Qin, Y., Niu, Y. L., and Yang, J. (2018). Matrine inhibits hypoxia/reoxygenation-induced apoptosis of cardiac microvascular endothelial cells in rats via the JAK2/STAT3 signaling pathway. Biomed. Pharmacother. 106, 117–124. doi:10.1016/j.biopha.2018.06.003
Zhao, X. K., Cheng, Y., Liang Cheng, M., Yu, L., Mu, M., Li, H., et al. (2016). Focal adhesion kinase regulates fibroblast migration via integrin beta-1 and plays a central role in fibrosis. Sci. Rep. 6, 19276. doi:10.1038/srep19276
Zheng, C., Huang, L., Luo, W., Yu, W., Hu, X., Guan, X., et al. (2019). Inhibition of STAT3 in tubular epithelial cells prevents kidney fibrosis and nephropathy in STZ-induced diabetic mice. Cell Death Dis. 10 (11), 848. doi:10.1038/s41419-019-2085-0
Zhu, B., Han, R., Ni, Y., Guo, H., Liu, X., Li, J., et al. (2023). Podocarpusflavone alleviated renal fibrosis in obstructive nephropathy by inhibiting Fyn/Stat3 signaling pathway. J. Nat. Med. 77 (3), 464–475. doi:10.1007/s11418-023-01685-y
Zhu, F., Bai, X., Hong, Q., Cui, S., Wang, X., Xiao, F., et al. (2019). STAT3 inhibition partly abolishes IL-33-induced bone marrow-derived monocyte phenotypic transition into fibroblast precursor and alleviates experimental renal interstitial fibrosis. J. Immunol. 203 (10), 2644–2654. doi:10.4049/jimmunol.1801273
Zhu, X., Shi, D., Cao, K., Ru, D., Ren, J., Rao, Z., et al. (2018). Sphingosine kinase 2 cooperating with Fyn promotes kidney fibroblast activation and fibrosis via STAT3 and AKT. Biochim. Biophys. Acta Mol. Basis Dis. 1864 (11), 3824–3836. doi:10.1016/j.bbadis.2018.09.007
Zhuang, L., Jia, K., Chen, C., Li, Z., Zhao, J., Hu, J., et al. (2022). DYRK1B-STAT3 drives cardiac hypertrophy and heart failure by impairing mitochondrial bioenergetics. Circulation 145 (11), 829–846. doi:10.1161/circulationaha.121.055727
Zordoky, B. N., Robertson, I. M., and Dyck, J. R. (2015). Preclinical and clinical evidence for the role of resveratrol in the treatment of cardiovascular diseases. Biochim. Biophys. Acta 1852 (6), 1155–1177. doi:10.1016/j.bbadis.2014.10.016
Keywords: cardiovascular diseases, JAK/STAT3 signaling, cardiac fibrosis, cardiac fibroblast proliferation and activation, signal transduction and regulation, upstream mediators, anti-fibrotic therapies
Citation: Jiang H, Yang J, Li T, Wang X, Fan Z, Ye Q and Du Y (2024) JAK/STAT3 signaling in cardiac fibrosis: a promising therapeutic target. Front. Pharmacol. 15:1336102. doi: 10.3389/fphar.2024.1336102
Received: 10 November 2023; Accepted: 18 January 2024;
Published: 01 March 2024.
Edited by:
Yan Sanders, Eastern Virginia Medical School, United StatesReviewed by:
Dongze Qin, Albert Einstein College of Medicine, United StatesScott M. MacDonnell, Regeneron Pharmaceuticals, Inc., United States
Copyright © 2024 Jiang, Yang, Li, Wang, Fan, Ye and Du. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qiang Ye, YXJ0MDA2MDIzQHllYWgubmV0; Yanfei Du, ZHlmc3dtdTAzMDRAc3dtdS5lZHUuY24=
†These authors have contributed equally to this work