 Xiaoying Yin1,2,3,4†
Xiaoying Yin1,2,3,4† Xinxin Yin1,2,3,4†Xin Pan1,2,3,4Jingyu Zhang1,2,3,4
Xinxin Yin1,2,3,4†Xin Pan1,2,3,4Jingyu Zhang1,2,3,4 Xinhui Fan1,2,3,4
Xinhui Fan1,2,3,4 Jiaxin Li1,2,3,4
Jiaxin Li1,2,3,4 Xiaoxuan Zhai1,2,3,4Lijun Jiang1,2,3,4
Xiaoxuan Zhai1,2,3,4Lijun Jiang1,2,3,4 Panpan Hao4
Panpan Hao4 Jiali Wang1,2,3,4*Yuguo Chen1,2,3,4*
Jiali Wang1,2,3,4*Yuguo Chen1,2,3,4*- 1Department of Emergency and Chest Pain Center, Qilu Hospital of Shandong University, Jinan, China
- 2Clinical Research Center for Emergency and Critical Care Medicine of Shandong Province, Institute of Emergency and Critical Care Medicine of Shandong University, Qilu Hospital of Shandong University, Jinan, China
- 3Key Laboratory of Emergency and Critical Care Medicine of Shandong Province, Qilu Hospital of Shandong University, Jinan, China
- 4Key Laboratory of Cardiovascular Remodeling and Function Research, Chinese Ministry of Education, Chinese Ministry of Health and Chinese Academy of Medical Sciences, The State and Shandong Province Joint Key Laboratory of Translational Cardiovascular Medicine, Qilu Hospital of Shandong University, Jinan, China
Cardiac fibrosis plays an indispensable role in cardiac tissue homeostasis and repair after myocardial infarction (MI). The cardiac fibroblast-to-myofibroblast differentiation and extracellular matrix collagen deposition are the hallmarks of cardiac fibrosis, which are modulated by multiple signaling pathways and various types of cells in time-dependent manners. Our understanding of the development of cardiac fibrosis after MI has evolved in basic and clinical researches, and the regulation of fibrotic remodeling may facilitate novel diagnostic and therapeutic strategies, and finally improve outcomes. Here, we aim to elaborate pathophysiology, examination and intervention of cardiac fibrosis after MI.
1 Introduction
Myocardial infarction (MI) is a leading cause of global morbidity and mortality and the primary contributor to heart failure (HF) (Groenewegen et al., 2020). The limited regenerative capacity leads to massive loss of cardiomyocytes (CMs) and excessive deposition of extracellular matrix (ECM) after MI (Prabhu and Frangogiannis, 2016; Dattagupta and Immaneni, 2018), which is called cardiac remodeling. Cardiac fibrosis is a pathological process of cardiac remodeling. Although timely and effective reperfusion can reverse this adverse effect, the incidence of cardiac fibrosis is increasing. During a life span of post-MI patients, fibrotic tissue accumulates in the process of left ventricular remodeling, and expands over time to remote non-infarcted region, which significantly alters cardiac structure and deteriorates cardiac function (Gil et al., 2022). Many patients survive with long-term adverse prognosis created strain on already overstretched healthcare systems and hampered medical management.
Abundant interstitial and perivascular fibroblasts in the adult heart play an essential role in maladaptive repair and fibrosis. Resident cardiac fibroblasts (CFs) are considered the primary cells that maintain ECM homeostasis by overseeing its quantity and quality, even if they are activated by pathological signals. To prevent the catastrophic outcomes of MI, CFs and myofibroblasts (MFs) deposit ECM to replace necrotic CMs and maintain the structural integrity of the heart coming along with viable CMs hypertrophy, whereas excessive ECM accumulation forms a fibrotic scar that provokes cardiac dysfunction and lethal arrhythmias (Burke et al., 2021a). Moreover, cardiac systolic dysfunction can be induced via scar with low tensile strength after disordered healing, while diastolic dysfunction increases after excessively fibrogenic activation and collagen deposition (Venugopal et al., 2022). Unfortunately, interventions for post-MI fibrotic remodeling have been limited.
With the limitation of therapeutic effects of drugs and surgeries, cardiac fibrosis is normally in MI patients. Despite various studies now addressing myocardial fibrosis, the understanding of its pathogenesis, clinical implications, and managements remains limited.
2 Pathophysiology of post-MI cardiac fibrosis
Fibrosis is a crucial determinant of cardiac function, stiffness, and conduction, with cardiac elasticity and compliance decreasing as fibrosis increases, contributing to systolic and diastolic dysfunction and even lethal arrhythmia and impairment of oxygen utilization (Fan et al., 2012). Thereby, it is very important to note that the complex pathophysiology and multiple mechanisms have been implicated in the fibrotic process following AMI. Also, the related molecular signaling network is complex and sophisticated, which involves various inflammatory mediators, inflammatory cells, and activating stromal fibrogenic effector cells, such as fibroblasts (Rockey et al., 2015). Additionally, the cardiac stromal cells exert profibrotic action via secreting cardiokines, which can predict adverse fibrotic remodeling after MI. (Masurkar et al., 2023).
The post-MI remodeling has three phases: the inflammatory phase (the first 3 days), proliferative phase (3–14days), and maturation phase (2weeks–2months) accompanying the dilation of non-infarcted zone, hypertrophy of CMs, and phenotypic transformation of CFs (Venugopal et al., 2022). During cardiac remodeling, inflammation, oxidative stress, disordered ECM, and CFs collectively cause cardiac fibrosis. According to the features and location of ECM protein deposition, there are usually two species of post-MI fibrosis: reparative and reactive fibrosis, with the former directly replacing necrotic cardiac tissue after MI and the latter being the pathological consequence of over-activated CFs and including perivascular and interstitial fibrosis (de Boer et al., 2019). Since adverse events and mortality are regarded as being related to the severity of cardiac fibrosis (Benjamin et al., 2018), uncovering these mechanisms is critical to help source novel therapeutic targets, diagnostic or prognostic performance.
2.1 Fibroblasts and myofibroblasts
Noteworthily, there are relatively static CFs and no MFs in a healthy heart (Hall et al., 2021). Recent single-cell multi-omics studies have elevated our knowledge of CFs in cardiac fibrosis (Forte et al., 2021a). For example, new CFs have been found, presenting as early as 1 day after MI (Shi et al., 2021), which can promote inflammation and recruit leukocytes. Then, CFs transfer to a proliferative, reparative, and proangiogenic phenotype, with maximum proliferation within 2–4 days. Moreover, the DNA damage response-associated CFs are up-regulated from day 3 up to day 7, as well as some senescence-associated CFs at day 7, indicating that cardiac fibrosis can be limited via activating DNA damage response and senescence (Shibamoto et al., 2019). Finally, CFs downregulate angiogenesis and convert to MFs at week 1 (Mouton et al., 2019) under new baseline conditions that the ultimate profibrotic culprits MFs produce excessive ECM, consisting principally of collagen I and III accompanying proteoglycans and elastin fibers (Kruszewska et al., 2022). Interestingly, although collagen V is minimally expressed, its deficiency increases scar size and cardiac dysfunction in an MI mouse model (Yokota et al., 2020). Recent research also finds that CFs interact with platelet leading to the alteration of collagen composition and content, and platelets mediate the reduction of inflammation after 24 h and scar formation after 21 days post AMI (Reusswig et al., 2022).
In a fibroblast-ablated mice model, there is a pronounced downregulation of CFs and the network formed by the collagen VI, microfibrillar collagen and the basement membrane, without accompanying overtly alteration of fibrillar collagen and the ECM proteome. Surprisingly, cardiac function is better preserved after MI, which suggests that controlled fibroblast reduction may have cardioprotective and therapeutic value in heart disease (Kuwabara et al., 2022). Moreover, as the mainly producers of ECM, CFs also produce cytokines, together with macrophages (Blythe et al., 2019), while, inactivating transcription factor sex-determining region Y box nine in CFs reduces cardiac fibrosis and late inflammation (Scharf et al., 2019). Thus, it is very important to distinguish the various phenotypes and functions of CFs under different conditions through detecting the markers of CFs. For instance, a study has identified proangiogenic and fibroblast-specific protein 1 (FSP1)-positive CFs that were distinct from profibrotic MFs (Saraswati et al., 2019). Moreover, CFs with growth factor receptor platelet-derived growth factor receptor α (PDGFRα) deficiency are responsible for the 50% reduction in CFs quantity (Ivey et al., 2019), while CFs with smad3 deficiency produce a decreased level of collagen (Huang et al., 2020). Further, there are several other markers of CFs, such as vimentin, transcription factor Tcf21, and MEFSK4 (mouse embryonic fibroblasts) (Venugopal et al., 2022). However, except for Tcf21 and PDGFRα, these markers are insufficiently sensitive and due to a lack of specific features (Acharya et al., 2012; Alex et al., 2022).
MFs have the specific markers as well, such as periostin and α-smooth muscle actin (α-SMA) (Kanisicak et al., 2016; Venugopal et al., 2022). In addition, after 1 week of MI, MFs increase in number and express periostin, collagen triple helix repeat containing 1, and dimethylarginine dimethylaminohydrolase 1, while the last is also expressed in activated CFs (Zhuang et al., 2020). Further, MFs are heterogeneous, i.e., they have different phenotypes; for instance, some are proliferating cells, and others express different levels of transforming growth factor β1 (TGF-β1), thrombospondin 4, and periostin (Farbehi et al., 2019).
There is, therefore, a clear and pressing need to identify additional novel mediators of cardiac fibrosis presentation and progression in response to pathological stimuli to facilitate the development of alternative therapeutic strategies targeting cardiac fibrosis.
During the process of cardiac fibrosis, CFs and CF-to-MF transformation are pathologically activated by many damage stimuli, such as TGF-β, platelet-derived growth factor (PDGF), epidermal growth factor (EGF), fibroblast growth factor (FGF), tumor necrosis factor α (TNF-α), angiotensin II (Ang II), interleukin-1 (IL-1), IL-4, and aldosterone (Fan et al., 2012). Recent research finds G-protein-coupled receptor kinase (GRK)-5 regulates fibroblast activation in vitro and in vivo, which suggests the inhibitor of GRK5 may be a novel target (Eguchi et al., 2021).
2.2 Extracellular matrix
In the working hearts, the relative glide movement of CMs and blood flow generates shear forces, constituting mechanical force with stretch and strain constrains, which is an important regulator of cells activation in fibrosis. ECM provides the heart with a structural scaffold and interacts with cells via adhesion molecules, such as integrins and cadherins, and distributes mechanical force through the cardiac tissue to individual cell (Souders et al., 2009; Zheng et al., 2016). In brief, collagen binding is responsible for load transfer and unnormal stretching limitation (Weber et al., 1994), which suggests the amount, distribution, and organization of ECM components modulate cardiac morphology and function (Spinale, 2007; Chute et al., 2019). For example, a pure scar in a rat MI model has lower wall thickness reduction over time before and after the decellularization, indicating excess collagen deposition during scar maturation and overall stiffening (Brazile et al., 2021). Moreover, the CMs fusion significantly decreases along with massive CMs death producing space and unnormal mechanical forces, which alters activation patterns of the cells and promotes CF-to-MF transformation (Kachanova et al., 2022).
The production and degradation of ECM are commonly regulated by MFs, together with macrophages and other cell types (Yap et al., 2023), such as CFs produce matrix metalloproteinases (MMPs) degrading collagens and tissue inhibitors of MMPs (TIMPs) inducing collagen synthesis (Nikolov and Popovski, 2022). There are four major ECM-associated peptides: the N-terminal propeptide of collagen type III (PIIINP) (indicating collagen III synthesis), the C-terminal telopeptide of collagen type I (ICTP) (indicating collagen I degradation), N-terminal propeptide of collagen type I (PINP), and C-terminal propeptide of collagen type I (PICP) (both PINP and PICP indicate collagen I synthesis) (Nikolov and Popovski, 2022). A recent study finds that the single mutation of thioredoxin-interacting protein cysteine 247 reduces collagen I α1 chain in MI mice (Nakayama et al., 2021). During scar formation, optimal collagen crosslinks require disintegrin and metalloproteinases (Chute et al., 2022), whereas PXS-5153A, the lysyl oxidase-like 2/3 enzymatic inhibitor, reduces collagen crosslinking and fibrosis (Schilter et al., 2019). Additionally, anti-integrin α(v) therapy also diminishes cardiac fibrosis via suppressing integrin-ECM interactions and cell adhesin (Bouvet et al., 2020). Also, there are several other fibrosis-associated non-collagen components, such as osteopontin, periostin, and galectin-3 (Li et al., 2022b).
The imbalance of ECM production and degradation induces adverse cardiac remodeling and dysfunction, with the insufficient repair disrupting cardiac tissue integrity, and excessive scar diminishing therapeutic efficacy (Yap et al., 2023).
2.3 Neuroendocrine system
2.3.1 Renin-angiotensin-aldosterone system
The activation of renin-angiotensin-aldosterone system (RAAS) plays a crucial role in development and progression of MI (Shigemura et al., 2019), having independently association with a higher risk of adverse cardiovascular events and mortality (Ivanes et al., 2012). After MI, the cardiac overload causes chamber dilation and elevates cardiac wall stress, followed by activating RAAS and inflammation response, which promotes the formation of MFs and excessive fibrosis (Fan and Guan, 2016; Zhou et al., 2019).
The essential process of RAAS is activated as described below. As the initial and rate-limiting step of the classical RAAS, renin is an aspartyl protease mainly produced by the juxtaglomerular cells of the renal afferent arteriole (Shigemura et al., 2019). Its plasma activity is associated with greater burden of coronary artery disease (Unkart et al., 2020). The synthesis and release of renin are stimulated by three major mechanisms: the decrease of sodium chloride concentration in the macula dense and perfusion pressure as sensed by renal baroreceptors; and the activation of β-adrenergic receptors in juxtaglomerular cells by catecholamines (Gomez and Sequeira-Lopez, 2018).
The liver produces angiotensinogen in the circulation, which is then activated by renin in juxtaglomerular cells to generate inactive angiotensin I (Ang I) (AlQudah et al., 2020). Subsequently, angiotensin-converting enzyme (ACE) converts Ang I into biologically active octapeptide Ang II (AlQudah et al., 2020), which is degraded to angiotensin III and several other angiotensins (Boorsma et al., 2021). For example, Ang II can be converted to angiotensin (1–7) via ACE2 with subsequently activating Mas receptor to decrease myocardial fibrosis (Boorsma et al., 2021), alternatively converted to Ang III with binding type 1 receptor by aminopeptidase A (Boitard et al., 2019). Moreover, both Ang II and Ang (1–7) are cleaved enzymatically to Ang (3–7) and Ang (5–7) via fibroblast growth factor-23 (FGF-23) stimulating dipeptidyl peptidase 3, with countering the therapeutic benefits from angiotensin-converting enzyme inhibitors (ACEI) and angiotensin receptor blockers (ARB) binding Mas receptor, which can be suppressed via the specific dipeptidyl peptidase three antibody procizumab (Boorsma et al., 2021). Interestingly, Ang II stimulates osteopontin synthesis and increases the concentrations of FGF-23 to negatively regulate ACE2 concentrations (Boorsma et al., 2021). As the main RAAS effector peptide, Ang II usually has 2 G protein-coupled receptors (GPCRs): type 1 Ang II receptor (AT1R) and the type 2 receptor (AT2R) (Pugliese et al., 2020), with the activation of the former increasing proinflammatory response and aldosterone levels, and the later having anti-fibrotic and anti-inflammatory effects (Ziaja et al., 2021). At the meantime, Ang II causes CMs hypertrophy, CFs hyperplasia (Leancă et al., 2022), and the secretion of molecular mediators (e.g., norepinephrine and endothelin) to promote cardiac remodeling (Williams, 2001; Gajarsa and Kloner, 2011). For example, endothelin 1 (ET-1), a potent vasoconstrictor peptide, promotes inflammatory and CMs hypertrophy to cause adverse remodeling (Zhang et al., 2019a; Haryono et al., 2022). Interestingly, alamandine, a substance with only one amino acid residue difference from Ang II, alleviates cardiac dysfunction and fibrosis via inhibiting oxidative stress in vivo and vitro models (Zhao et al., 2022a). In addition, the upregulation of left ventricle voltage-dependent anion channel one in MI patients is regulated via the RAAS activation, and inhibition of the anion channel reduces atrial fibrosis (Klapper-Goldstein et al., 2020).
The regulation of RAAS is relative with multiple signaling pathways (e.g., extracellular signal-regulated kinase (ERK) and janus kinase (JAK) pathways) (Chen et al., 2020b); for instance, RAAS promotes collagen secretion via TGF-β1/smad2/3 pathway (Li et al., 2022b). Moreover, microRNA (miRNA) can also regulate RAAS, such as miR-181/Adamts1/neutrophil gelatinase-associated lipocalin pathway (Garg et al., 2020). Further, Ang II promotes CFs viability, activation, and migration through the circCELF1/miR-636/dickkopf WNT signaling pathway inhibitor 2 pathway in AMI mice (Li et al., 2022g).
2.3.2 Sympathetic nervous system
After AMI, sympathetic nervous system (SNS) is activated immediately as a compensatory mechanism to increase cardiac output and maintain blood pressure. The norepinephrine is primarily synthesized and secreted from adrenal medulla and is modulated through β-adrenergic receptors (β-ARs) on the heart (Lymperopoulos et al., 2007).
The heart expresses various ARs belonging to GPCRs that the most predominant subtype belongs to β1-AR, 15% to β2-AR, and the remainder to β3-AR and α1-AR. Briefly, β-ARs regulate cardiac function via impacting myocardial contractility (Tanner et al., 2021), such as β1-AR and β2-AR with chronotropic and ionotropic effects, contrarily β3-AR with negative inotropic properties (Yoshikawa et al., 1996; Capote et al., 2015; Lymperopoulos, 2018). Noteworthily, the β2-AR, not the β1-AR, is the predominant subtype in the non-cardiocytes (e.g., fibroblasts, endothelial cells, immune cells) (Lymperopoulos et al., 2021). For example, CMs death, hypertrophy, and cardiac fibrosis are decreased in β2-AR knockout bone marrow transplantation mice following isoproterenol treatment, which suggests β2-AR expresses in the heart’s immune cells (Atsuki et al., 2019; Tanner et al., 2021). As a GPCR, β2-AR couples to both Gs and Gi proteins, and the β2-AR-stimulated cardioprotective Gi signaling depends on the heterodimerization of β2-ARs and 5-hydroxytryptamine receptors 2B (Song et al., 2021c).
Recent years, β3-AR has become a therapeutic target for cardiac fibrosis. Mechanistically, the reduced reactive oxygen species (ROS) levels following β3-AR activation attenuate fibrosis through reduced release of paracrine profibrotic agents in β3-AR expressing myocytes. Moreover, β3-AR/protein kinase G (PKG) signaling emerged as a promising therapeutic target in heart failure with preserved ejection fraction (HFpEF). Diastolic dysfunction in patients with HFpEF mainly results from the combination of increased cardiomyocyte stiffness with left ventricle (LV) hypertrophic remodeling and interstitial fibrosis. Cardiomyocyte stiffness results from both increased myofilaments Ca2+ sensitivity and higher titin stiffness, related to reduced PKG activity in the myocardium of HFpEF patients and subsequent lower phosphorylation of these targets. Therefore, β3-AR stimulation, by improving nitric oxide synthase (NOS)/PKG signaling, should restore the phosphorylation of sarcomeric proteins but also improve the regulation of Ca2+ handling for cardiac myocytes relaxation (Michel et al., 2020). For example, treatment with β3-AR agonists can potentially address this because they stimulate cardiac myocyte Na+/K+-ATPase. This occurs via downstream activation of cyclic guanosine monophosphate-dependent signaling pathways which the β3-AR has in common with NO donors, GC-1 activators15 and the angiotensin receptor-neprilysin inhibitor (ARNI) (Bundgaard et al., 2022).
After MI, upregulated SNS activity induces hematopoietic stem and progenitor cells proliferation and release in the bone marrow. In the early stage, sympathetic overdrive activates apoptotic pathway and promotes neutrophil influx in the necrotic area and infarct expansion (Amin et al., 2011). Subsequently, the stimulation of β1-AR promotes CMs hypertrophy and renin release (Gajarsa and Kloner, 2011). In MI mice, exchange protein activated by cyclic-adenosine, which could be upregulated by β-AR activation, prevents left atrial fibrosis (Surinkaew et al., 2019), while β-AR mainly modulates collagen production by CFs and causes proliferation of human CFs and fibrotic remodeling (Turner et al., 2003; Humeres and Frangogiannis, 2019). Further, β2-AR upregulates CFs proliferation via the secretion of IL-6, depending on Gαs/ERK1/2 (Tanner et al., 2020). To sum up, the regulation of sympathetic neurohormone expression is a key therapeutic option in attenuating cardiac remodeling.
2.3.3 Natriuretic peptides
Natriuretic peptides (NPs), primarily as endocrine hormones, are secreted by atrial and ventricular CMs under upregulated wall stress and stretching in MI and regulate diuresis, natriuresis, and vasodilation, as well as inhibit SNS, RAAS, and ET-1 (Kuhn, 2016). NPs have three isoforms: A-type NP (ANP, which inhibits collagen synthesis and is a main fibrotic driver), B-type NP (BNP; a prognosis predictor after MI), and C-type NP (CNP) (Kangawa et al., 1984; Kuhn, 2016). NPs have anti-apoptotic, anti-inflammatory, and anti-fibrotic effects to prevent myocardial ischemic reperfusion injury (MIRI) and adverse remodeling, with their concentrations reflecting profibrotic environments and identifying patients at risk for remodeling, while ANP and BNP reduce vasoconstriction, CFs proliferation, CF-to-MF transformation, collagen synthesis, and MMP release by activating the cyclic guanosine monophosphate (cGMP) pathway (Goetze et al., 2020). The inhibition of phosphodiesterase-9 activity dose-dependently increases cGMP and the cGMP/NP ratio of plasma and urinary, which suggests that its inhibition may constitute a novel therapeutic approach in clinical HF. (Scott et al., 2019).
2.4 Immunity
The immune system plays an important role in maintaining homeostasis after MI, in which dying CMs release many damage-associated molecular patterns (DAMPs) and activate the cascade of inflammatory mediators (e.g., inflammatory cytokines and chemokines) (Simões and Riley, 2022). Noteworthily, the severity of inflammatory responses dictates the degree of MI (Ong et al., 2018). Thus, it is important to strike an appropriate balance between inflammatory and anti-inflammatory response, and timely regulate inflammatory signals during cardiac remodeling (Yap et al., 2023). After MI, immune response is rapidly initiated via mobilizing distinct immune cell populations (Yap et al., 2023). The differentially altering immune cells infiltration affects cardiac fibrosis, for instance, allografts suppression of tumorigenicity 2 (ST-2) deficiency reduces infiltration of F4/80 (+) macrophages, CD3 (+) T cells and CD20 (+) B cells and thus alleviates vascular occlusion and fibrosis of allografts (Zhang et al., 2021e). In this section, we focus on different immune cell populations, cytokines, chemokines and growth factors to improve mechanistic understanding of immune responses. Then, later on in this article, we will focus in particular on preventing fibrosis with immune treatment. In what follows, we first describe the key features of different immune cells.
2.4.1 Neutrophils and macrophages
As the most abundant peripheral blood granulocytes, neutrophils constitute 50%–70% of the total amount of blood granulocytes in most mammals and 20%–30% in mice (Mestas and Hughes, 2004) in a physiologically circadian pattern (Aziz et al., 2021), exiting blood with night-time peaking (fresh neutrophils) and day-time peaking (aged neutrophils) (Adrover et al., 2019). Recent studies have shown that neutrophils modulate cardiac remodeling via the initiation and termination of an inflammatory response and largely infiltrate the infarct zone 1 day after MI and the peri-infarct zone 4 weeks after MI, with disturbed circadian rhythm, in mice (Zhong et al., 2022). However, neutrophil-mediated MIRI is limited by selectin-targeting glycosaminoglycan-peptide conjugate (Dehghani et al., 2022). Normally, massive leucocytes remain in the bone marrow, and only a low number of leucocytes stay in the circulation, whereas 2-arachidonoylglycerol induces massive cardiac infiltration by neutrophils and monocytes and increases cardiac dysfunction and fibrosis in a MIRI mouse model (Schloss et al., 2019). In MI patients, immature CD10neg neutrophils and CD14+HLA-DRneg/low monocytes increase in number (Fraccarollo et al., 2021), and there are still around 10-fold more leukocytes in the scar than in remote zone at 6 weeks after MI in mice (Scharf et al., 2019).
Recent evidence has associated monocytes/macrophages with the etiopathology of cardiac fibrosis, and the interventions targeting these cells have been challenging due to the heterogeneity and the antagonizing roles of different subtypes (Chen et al., 2022b). Remarkably, as the largest population of immune cells, macrophages can be classified into two general subtypes based on cell surface with or without C-C chemokine receptor 2 (CCR2): monocyte-derived CCR2+ macrophages, and yolk-sac-derived CCR2– macrophages. The former exerts pro-inflammatory effects and recruits monocytes, whereas the latter is considered to prevent excessive inflammation (Dick et al., 2019; Yap et al., 2023). At the meantime, macrophages also can be classified as classically activated (M1) or alternatively activated (M2) based on stimulatory environment. M1 macrophages degrade ECM and clear cell debris, and M2 macrophages promote angiogenesis and collagen deposition (Yang et al., 2021b). Further, M2b macrophages promote lymphangiogenesis to reduce myocardial fibrosis and cardiac dysfunction (Wang et al., 2022b). At the early stage following MI, macrophages and monocytes are almost exclusively derived from haematopoietic stem cells or extramedullary splenic reservoirs. They usually infiltrate the infarct area with pro-inflammatory phenotypes (Halade et al., 2018; Bajpai et al., 2019; Yap et al., 2023). Neonatally activated macrophages modulate angiogenesis, inflammation, and CMs proliferation, which is contrary to adult macrophages with different metabolites of oxygenation and nutrients (Lantz et al., 2021).
Herein, we describe the process of immune response after MI based on recent literature. At the beginning, these immune cells accumulate within hours, known as the inflammatory phase, and the peak recruitment of these immune cells occurs approximately 3 days after MI (Yap et al., 2023). Neutrophils are firstly recruited after MI and then phagocytized by proinflammatory CCR2+ Ly6Chigh monocyte-derived macrophages with the replacement of some CCR2(-) resident macrophages, which increase anti-inflammatory and profibrotic cytokines (e.g., IL-10 and TGF-β) and decrease proinflammatory cytokines (e.g., IL-1β and TNFα) (Simões and Riley, 2022). Concurrently, macrophages phagocytize dead cells, and anti-inflammatory T cells (e.g., regulatory T cells/Tregs) are recruited (Dobaczewski et al., 2010; Hofmann and Frantz, 2015). After 1–2 days of MI, the anti-inflammatory phase (also known as proliferative phase) ensues, in which the macrophages undergo rapid proliferation with anti-inflammatory or reparative properties (Dick et al., 2019). At this stage, cytokines also promote CF-to-MF transformation. Briefly, the resolution of inflammation and reparative tissue remodeling are initiated. Further, the recovery phase follows after 3–7 days of MI, MFs and CCR2+ macrophages mediate fibrosis and scar formation (Yap et al., 2023). Moreover, the macrophages subsequently transform into reparative CCR2+ Ly6Clow macrophages via the transcriptional program dependent on a nuclear receptor subfamily four group A member 1, which promotes fibrotic scar formation via collagen deposition and MFs transformation (Simões and Riley, 2022). Additionally, histamine deficiency promotes monocyte/macrophage-to-MF transformation in MI-induced cardiac fibrosis (Zhu et al., 2022). In contrast, specifically marked Gata6(+) pericardial macrophages accumulate on the cardiac surface after MI and prevent fibrosis (Jin et al., 2022).
2.4.2 Other immune cells
Due to the regulative functions of immunity, it presents an intriguing direction for therapeutic intervention about cardiac fibrosis. Therefore, it is very important to comprehensively understand phenotypes and behaviours of different immune cells after MI.
Currently, there are also the up-to-date knowledge about other immune cells, such as B cells, T cells, and eosinophils. For example, B cells infiltrate into damaged myocardium within 1–7 days (Adamo et al., 2020), and B cells deficiency downregulates cytokines (e.g., TNF-α, IL-1β, IL-6, and TGF-1β) and collagen synthesis to alleviate fibrosis after MI (Mo et al., 2021). Additionally, MMP-2 increases the cytotoxicity of CD8+ T cells in acute MI patients (Li et al., 2021g). Cross-priming dendritic cells activate cytotoxic CD8+ T cells to exacerbate inflammatory damage and fibrosis (Forte et al., 2021b). Moreover, OSU-ERb-012, an estrogen receptor-β agonist, inhibits CD4+ T cells and improves cardiac remodeling in the MI-induced HF mouse model (Rosenzweig et al., 2022). Tregs promote Ly6Chigh monocyte conversion into M2 macrophages by secreting cytokines (e.g., IL-10, IL-13, and TGF-β) to initiate the anti-inflammatory or regenerative phase (Kino et al., 2020).
Furthermore, Tregs can directly regulate CFs (Kino et al., 2020). Eosinophils are increased in the blood and heart (mostly in the infarct area) in MI patients and mice, and eosinophil depletion promotes cardiac dysfunction and fibrosis (Liu et al., 2020a).
2.4.3 Cytokines
Massive researches have demonstrated the role of cytokines in MI that cytokines not only form a complex network to regulate inflammatory response, but also can form cytokine storm to worse myocardium injure following cardiac decompensation (He et al., 2022). Upon cardiac injury, the inflammatory signaling molecules immediately increased.
DAMPs bind toll-like receptors (TLRs), activate inflammasomes (e.g., nod-like receptor protein 3 (NLRP3)), and promote cytokines/chemokines synthesis to induce activation and recruitment of immune cells and engage immune defenses (Schroder and Tschopp, 2010). Most commonly, IL-1 family includes pro-inflammatory and anti-inflammatory members. For example, IL-1α reduces the remodeling in border zone CFs by upregulating steroidogenic acute regulatory protein (Razin et al., 2021). Moreover, IL-33 mediates the shift in the inflammatory phase toward its resolution through IL-1R4 (Fearon and Fearon, 2008). MFs with physiological stretching release IL-33 to bind the ST-2 receptor on the CMs membrane to promote cell survival and integrity (Vianello et al., 2019), whereas IL-33 worsens cardiac remodeling by recruiting eosinophils (Ghali et al., 2020). In a myeloid IL-4 receptor-α deficiency model, insufficient fibrotic remodeling is induced via downregulated TIMPs and collagen I deposition (Song et al., 2021a; Song et al., 2021b). IL-21 induces apoptosis of Ly6Clow macrophages and prevents cardiac repair (Kubota et al., 2021). Furthermore, IL-38 influences dendritic cells to reduce inflammation and fibrosis (Wei et al., 2020).
2.4.4 Chemokines
Extensive evidence also implicates chemokines in the pathogenesis of cardiac fibrosis. Chemokines are key regulators controlling the migration and positioning of immune cells, and various cells proliferation to promote structural remodeling and functional recovery of the heart with inflammation quickly subsiding (Frangogiannis, 2014; Ma, 2021). However, persistent cytokines induce late cardiac contractility and adverse outcome (Wang et al., 2018). In the affected myocardium and heart-draining lymph nodes, MI induces complementary B-cell responses, while B cells infiltrate the infarct zone via the CXC-motif chemokine ligand 13 (CXCL13, the ligand of CXCR5-CXC-chemokine receptor type 5 (CXCR5)) axis and induce TGF-β1 expression (Heinrichs et al., 2021). CXCL8 induces neutrophil infiltration, whereas CC chemokines, such as chemokine CC-motif ligand 2 (CCL2), mediate the recruitment of mononuclear cells (Chen and Frangogiannis, 2021). In addition, CCL2, also known as monocyte chemoattractant protein-1 (MCP-1), has a higher serum level in ST-segment elevation myocardial infarction (STEMI) patients, but lower plasma levels in MI patients without collateral circulation (Kobusiak-Prokopowicz et al., 2007; Sahinarslan et al., 2010). Furthermore, CXCL10 and CXCL12 have leukocyte-independent mediatory effects, directly modulating CFs (Chen and Frangogiannis, 2021). Additionally, single-cell sequencing has found different immune cell abundance (resting and activated mast cells, activated CD4 memory T cells) and high expression of chemokines in MI patients (CCL3, CXCL3, CXCL8, and CXCL16 in CD1C-CD141-dendritic cells and CCL4 and CCL5 in natural killer cells) (Zhou et al., 2022).
2.4.5 Growth factors
To explore novel treatments targeting cardiac fibrosis, it is very important to identify and elucidate precise mechanisms of growth factors, such as PDGF, FGF, and TGF have been best studied.
The PDGF family is composed of cell division stimulators and has five subunits (PDGF-AA, PDGF-BB, PDGF-AB, PDGF-CC and PDGF-DD), as well as two receptors, PDGFRα and PDGFRβ. All PDGF members have been shown to play a role in cardiac fibrosis (Bertaud et al., 2023). For example, overexpression of PDGF-A modulates scar content, reduces scar size, and increases capillary and arteriolar density in the infarct border zone (Rashid et al., 2021), whereas PDGF-AB enhances angiogenesis and increases scar anisotropy (high fiber alignment) without affecting overall scar size or stiffness (Thavapalachandran et al., 2020).
The FGF family has 22 members and pleiotropic effects (Bertaud et al., 2023). For example, FGF21 inhibits inflammation and fibrosis by downregulating early growth response protein 1 (Li et al., 2021c; Li et al., 2021d). Additionally, FGF12 overexpression decreases collagen I and III and fibronectin in Ang II-induced CFs (Liu et al., 2022). FGF10 increases cardiomyocyte renewal and limits fibrosis to promote cardiac regeneration and repair, which suggests FGF10 may be a clinically relevant target for heart repair (Hubert et al., 2022). Conversely, FGF23 increases myocardial fibrosis and dysfunction via activating β-catenin and promoting the pro-fibrotic crosstalk between CMs and CFs in a paracrine manner (Hao et al., 2016; Leifheit-Nestler et al., 2018).
There are also other growth factors involved in cardiac fibrosis, such as neuregulin-1, a paracrine growth factor secreted by cardiac endothelial cells, modulates hypertrophic and fibrotic processes during early cardiac remodeling via the neuregulin-1/erythroblastic leukemia viral oncogene homolog (ERBB) four axis (Dugaucquier et al., 2020).
In this section, we mainly focus on the multifaceted contributions of diverse immune cells populations and mediators after MI. As immunity affecting the prognosis of MI patients has become an area of substantial therapeutic interest, we discuss novel interventions regarding immunity in a later section.
2.5 Molecular mechanisms
Many mechanisms (e.g., oxidative stress, inflammation, and mechanical stress) are known to affect cardiac fibrosis. Additionally, a large amount of antifibrotic therapy researches targeting underlying molecular mechanisms have been implemented with technological advancement (Figure 1) (The figure is drawn by figdraw). Due to the various signaling pathways involved in the process, an understanding of the precise mechanisms of cardiac fibrosis is still limited. From this viewpoint, it is essential to summarize and comprehend current evidence of cell signaling pathways associated with cardiac fibrosis. Hence, in the following sections, we summarize the recent data about the roles and crucial functions of several key signaling pathways in post-MI fibrosis: TGF-β, phosphatidylinositol 3-kinase (PI3K)/protein kinase B (Akt), nuclear factor erythroid 2-related factor 2 (Nrf2), mitogen-activated protein kinase (MAPK) and other molecular mechanisms. The molecularly targeted drugs may further decline the death rate of MI. Herein, it is indispensable and promising to target these aberrant signaling pathways and improve the pathological manifestations in post-MI fibrosis.
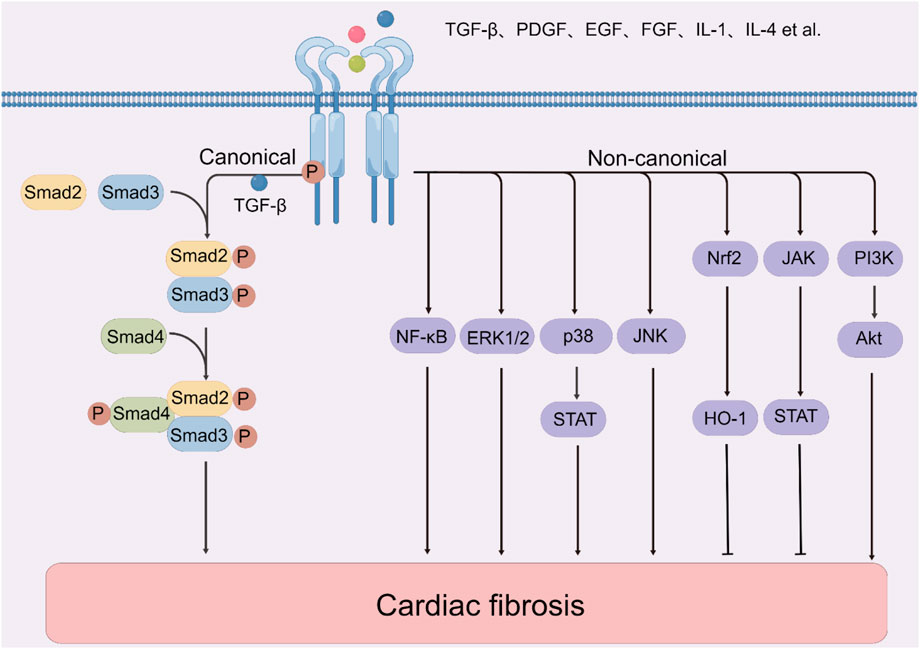
FIGURE 1. The molecular mechanisms of cardiac fibrosis. Note: Akt, protein kinase B; EGF, epidermal growth factor; ERK, extracellular signal-regulated kinase; FGF, fibroblast growth factor; HO-1, heme oxygenase-1; IL-1, interleukin-1; IL-4, interleukin-4; JAK, janus kinase; JNK, c-Jun N-terminal kinase; NF-κB, nuclear factor kappa-B; PDGF, platelet-derived growth factor; PI3K, phosphatidylinositol 3-kinase; STAT, signal transducer and activator of transcription 3; TGF-β, transforming growth factor β.
2.5.1 TGF-β signaling pathway
TGF-β is the most typical cytokine regulating fibrosis and inducing collagen synthesis, which is secreted by macrophages into the ECM in an inactive form (pro-TGF-β) and then activated by proteases (e.g., plasmin, MMP-2, MMP-9, and ROS) (Takawale et al., 2015; Hata and Chen, 2016). Firstly, it binds TGF-β receptor(TGFβR)II, and then TGFβRI is phosphorylated and forms a receptor heterocomplex to react with smads protein. Bruton’s tyrosine kinase is up-regulated in MI, with directly binding and phosphorylating TGFβRI at tyrosine 182, and then activating the downstream to promote CF-to-MF transformation and the excessive ECM gene expression. And its second-generation inhibitor Acalabrutinib attenuates cardiac fibrosis (Wang et al., 2022a). Smads proteins have three types: receptor-regulated smad (smad1, smad2, smad3, smad5, and smad8), common smad (smad4), and inhibitory smad (smad6 and smad7). The TGF-β complex binds to R-smads (smad2 and smad3) and Co-smads (smad4), and then transfers into nucleus to regulate the transcription of target genes (Zhang et al., 2022c). The smad-dependent canonical pathway, coordinating with non-canonical pathways, transduces TGF-β signals (Zhang et al., 2022f).
TGF-β, as a critical molecule of MFs, promotes collagen synthesis, CF-to-MF transformation, and other fibrotic factors production, as well as activates multiple signaling pathways (Li et al., 2022b), while CFs and inflammatory cells express cystine knot protein gremlin-1 colocalizing with TGF-β and reduce collagen deposition (Müller et al., 2021). Further, TGF-β1 can activate CFs to increase collagen deposition, and sustained TGF-β1 expression subsequently leads to cardiac dysfunction (Xue et al., 2019). Moreover, TGF-β1 regulates ECM remodeling by promoting MMP/TIMP imbalance (Hodges et al., 2019). Additionally, a study has found that TGF-β3 expression gradually attained its peak in 1 month after MI with an opposite expression trend of TGF-β1 and TGF-β2 in MI patients, while TGF-β3 downregulated proliferation, migration, and collagen synthesis and upregulated lysyl oxidase and osteopontin in Ang II-induced human CFs and MI patients by promoting smad7 expression (Xue et al., 2019). The recombinant osteopontin activates cell-cycle re-entry in CMs, stimulates multiple cardiac cells, and improves scar formation, LV remodeling, and regional and global function after MI (Rotem et al., 2022).
Even if TGF-β2 and TGF-β3 are involved in cardiac fibrosis, the fibrotic effects triggered by the TGF-β family have primarily been attributed to TGF-β1 (Dewald et al., 2004). For example, phosphoglycerate mutase one deficiency suppresses inflammation, apoptosis, and fibrosis in post-MI by targeting TGF-β1 (Wu et al., 2021c). High serum tissue non-specific alkaline phosphatase (TNAP) level in MI patients can serve as a fibrotic biomarker and is positively correlated with mortality risk (Cheng et al., 2021), while TNAP inhibition provokes an antifibrotic effect through adenosine monophosphate-activated protein kinase (AMPK)/TGF-β1/smads and p53 (Gao et al., 2020b). Two inhibitory smads (smad6 and smad7) can prevent R-smad phosphorylation (Bertaud et al., 2023). Further, smad7 also restrains MFs activation by suppressing profibrotic ERBB2 in a TGF-β-independent manner (Humeres et al., 2022) but does not restrain the anti-inflammatory function of TGF-β in macrophages (Li et al., 2022d). Another member of the TGF-β superfamily, lefty1 alleviates post-MI CFs proliferation, differentiation, and secretion by suppressing the p-smad2 and p-ERK1/2 axis (Li et al., 2021a). Moreover, SH2 domain-containing protein tyrosine phosphatase-2 inhibits fibrosis via the ERK/smad pathway (Lu et al., 2021). Chordin-like one inhibits extracellular bone morphogenetic protein 4 (BMP4) to inhibit smad1/5/8 activation and autophagy in CMs and suppresses TGF-β1-induced fibrosis and CF-to-MF transformation (Ruozi et al., 2022).
Noteworthily, many non-coding miRNAs have been reported to be involved in cardiac fibrosis via regulating TGF-β signaling pathway (Zhao et al., 2022b). In this section, we summarize recent studies focusing on this pathway. Briefly, post-MI repair requires tight regulation of the TGF-β signaling pathway in case of excessive fibrosis and adverse remodeling leading to heart failure.
2.5.2 PI3K/Akt signaling pathway
The PI3K/Akt/protein kinase B signaling pathway is one of the important intracellular signal transduction pathways. PI3K converts phosphatidylinositol 4,5-bisphosphate (PIP2) into phosphatidylinositol 3,4,5-trisphosphate (PIP3). Then PIP3 binds to the pleckstrin homology domain of Akt to alter its conformation and activate the downstream molecules, such as vascular endothelial growth factor (VEGF), endothelial nitric oxide synthase, while inhibiting mammalian target of rapamycin (mTOR) complex 1, glycogen synthase kinase 3β, forkhead box subfamily O, respectively (Zhang et al., 2022c). In recent years, basic research finds that targeting PI3K/Akt pathway is a beneficial signaling mechanism for anti-fibrotic treatments following AMI regulating cell proliferation, differentiation, migration and apoptosis. For example, Apelin-13 inhibits the PI3K/Akt axis to attenuate fibrosis in HF rats and AngII-induced CFs (Zhong et al., 2020). Further, visceral adipose tissue-derived serine protease inhibitor vaspin alleviates fibrotic remodeling and oxidative stress and decreases ANP, BNP, and collagen I and III by inhibiting the PI3K/Akt axis (Ji et al., 2022b). Additionally, inhibition of calcium and integrin binding protein one reduces cardiac fibrosis and levels of α-SMA, vimentin, and collagen I and III by inhibiting the PI3K/Akt pathway (Hu et al., 2022a). Since an essential requirement for the post-MI repair is recovering the capillary network in the injured area due to new vessel sprouting from existing ones (Gao et al., 2020a), transcription factor Yin-Yang one represses CMs apoptosis and fibrosis and promotes angiogenesis by enhancing Akt phosphorylation and increasing VEGF (Huang et al., 2021). The lysyl oxidase-like protein two increases MFs transformation, collagen fiber production and mechanical strength via the PI3K/Akt/mTOR pathway (Yang et al., 2016). By contrast, ivabradine prevents fibrosis and cardiac hypertrophy via suppressing PI3K/Akt/mTOR/p70S6K signaling (Yu et al., 2019; Dai et al., 2021). Klotho significantly reduces cardiac fibrosis and suppresses myocardial inflammation and apoptosis in MI-induced HF model via inducing autophagy through the inhibition of PI3k/Akt/mTOR signaling pathway (Wang et al., 2022d).
2.5.3 Nrf2 signaling pathway
As a transcription factor and the product of the nuclear factor erythroid-derived 2-like 2 gene, Nrf2 consists of seven functional domains and participates in regulating oxidative stress and antioxidant genes (Ray et al., 2012; Hayes and Dinkova-Kostova, 2014). It transfers signaling molecules to the nucleus and initiates antioxidant gene transcription (Kensler et al., 2007). And its downstream target, heme oxygenase-1 (HO-1), is a rate-limiting enzyme that catalyzes heme to biliverdin Ixα, carbon monoxide, and iron (Wu et al., 2021a). Nrf2 signaling pathway plays a crucial role in post-MI remodeling. For example, in the MI rat model and Ang II-treated CFs, ghrelin ameliorates cardiac fibrosis by activating Nrf2 to inhibit the nicotinamide adenine dinucleotide phosphate (NADPH)/ROS pathway (Wang et al., 2021c; Wang et al., 2021d). In addition, Pinocembrin ameliorates cardiac remodeling by ROS clearance and Nrf2/HO-1 pathway activation, which further suppresses collagen fibers deposition and apoptosis and promotes angiogenesis (Chen et al., 2021d). Moreover, corosolic acid regulates the AMPK-α/Nrf2/HO-1 axis to inhibit cardiac fibrosis, oxidative stress, inflammation, and apoptosis (Wang et al., 2020i). Furthermore, Nrf2 reduces innate immune response in MI mice (Bromage et al., 2022). Thus, the protective effect of Nrf2/HO-1 following AMI should not be ignored. And it constitutes an appealing target for anti-fibrotic treatments. Plantarum activates Nrf2 antioxidant defense pathway and ameliorates cardiac dysfunction and collagen expression (Aboulgheit et al., 2021).
2.5.4 MAPK signaling pathway
As a class of highly conserved serine/threonine protein kinases, MAPKs have four primary branches: ERK, c-jun N-terminal kinase (c-JNK), p38/MAPK and ERK5 (Gallo and Johnson, 2002; Cargnello and Roux, 2011). During variously physiological and pathological processes, these kinases can be sequentially activated and regulate proliferation, growth, and differentiation of cardiac cells, such as CMs, CFs, endothelial cells and macrophages (Muslin, 2008). In this section, we mainly introduce some recent studies of the MAPK pathway from the aspects of molecular regulation.
Calcium-activated chloride channels protein anoctamin-1 promotes CFs proliferation and secretion via the MAPK pathway (Tian et al., 2020). The upregulation of OUT domain-containing 7B suppresses phosphorylated focal adhesion kinase and ERK/p38 activities and reduces the levels of α-SMA and collagen I (Zhang et al., 2022b), while nicotinamide riboside kinase-2 regulates the p38 pathway to alleviate post-MI scar size and fibrosis (Ahmad et al., 2020). Further, zinc finger protein ZBTB20 protects the heart by inhibiting the JNK pathway (Li et al., 2020a; Li et al., 2020b), while melatonin improves myocardial fibrosis in the infarct border zone and apoptosis via the JNK/p53 pathway after MI in a diabetic mouse model (Lu et al., 2020). In oxygen-glucose deprivation/reoxygenation (OGD/R)-induced H9c2 cells and myocardial fibrosis model of mice, protocatechualdehyde, a major component from Salvia miltiorrhiza, against ischemic injury by suppressing endoplasmic reticulum stress-associated protein kinase R-like endoplasmic reticulum kinase/transcription factor 6α/inositol-requiring enzyme1α pathways (Wan et al., 2021).
In short, comprehensive study and understanding the mechanism of the MAPK pathway, taking this signaling pathway as the anti-fibrotic target are the keys to address challenges of post-MI fibrosis.
2.5.5 Other molecular mechanisms
In addition to the above signaling pathways, other pathways have also been shown to be related to cardiac fibrosis. For example, ELABELA peptide increases angiogenesis and reduces cardiac interstitial fibrosis through activating ERK/hypoxia-inducible factor-1alpha/VEGF pathway in MIRI rat model (Rakhshan et al., 2022). A transcriptional complex (A-kinase anchoring protein 2, protein kinase A, and steroid receptor coactivator 3) modulates proangiogenic and antiapoptotic processes via protein kinase A-mediated phosphorylation and estrogen receptor α activation (Maric et al., 2021).
As a family of signal-dependent transcription factors, nuclear factor kappa B (NF-κB) is located in the cytoplasm in an inactive form, but it migrates to the nucleus following stimulation, and regulates its targets via binding to NF-κB response elements on the DNA (Li and Verma, 2002). As a typical pro-inflammatory signaling pathway, NF-κB regulates gene transcription and promotes inflammatory responses (He et al., 2022), for example, exendin-4 regulates the NF-κB axis to prevent inflammation and cardiac remodeling (Eid et al., 2020), and Nur77 improves cardiac fibrosis by inhibiting the NF-κB-dependent pathway (Chen et al., 2021a). Further, hippo pathways are vital mechanisms of cardiac repair. For example, hippo pathway kinases Lats1/2 inhibit yes associated protein (YAP)-induced injury response, while conditional deletion of Lats1/2 in adult resting CFs initiates CF-to-MF transformation (Xiao et al., 2019). Moreover, platelet-activating factor receptor and YAP1 are significantly increased in MI mice, accompanying with its positive feedback loop in cardiac fibrosis (Li et al., 2022f).
In addition to the common signaling pathways noted above, other recent studies of molecular mechanisms are summarized. For example, researchers have found that fibrosis is associated with calmodulin/p38/signal transducer and activator of transcription (STAT) 3, wnt/β-catenin, TLR4/calmodulin-dependent protein kinase II and B lymphoma Mo-MLV insertion region 1 homolog/p15/retinoblastoma pathways et al. (Li et al., 2020g; Han et al., 2020; Yang et al., 2021c; Fu et al., 2021; Bugg et al., 2022; Zhang et al., 2022e; Shi et al., 2022).
Crosstalk also exists in different signaling pathways; for instance, endogenous TGF-β1 repressor SKI activates the hippo pathway via LIM domain-containing protein one to inhibit CFs activation (Landry et al., 2021). There are some studies about ion channel; for instance, the mechanosensitive ion channel transient receptor potential vanilloid four deletion regulates CF-to-MF transformation to improve harmful remodeling after MI (Adapala et al., 2020; Adapala et al., 2021). Piezo1 activates CFs to induce MFs recruitment and excessive ECM deposit (Braidotti et al., 2022), while coronary vascular endothelial sodium channel activation promotes cardiac fibrosis and dysfunction (Hill et al., 2022). Further, embryonic CFs of mice with mitochondrial Ca2+ uniporter deletion are more sensitive to Ca2+ overload than normal CFs (Huo et al., 2020). Briefly, it is necessary to comprehensively understand the molecular mechanisms of cardiac fibrosis after MI before performing specific interventions.
To sum up, it is critical to develop and optimize therapeutic strategies according to fundamental mechanisms and pathophysiology of cardiac fibrosis. To date, research in basic science has disclosed a range of pathophysiological mechanisms of post-MI cardiac fibrosis, and many attractive inhibitors and antagonists have been developed based on the molecular mechanisms. However, a lot of them have not currently been launched in human clinical trials to advance toward clinical application, and investigations remain challenging and need to be studied further.
3 Evaluations of cardiac fibrosis
3.1 Cardiac magnetic resonance late gadolinium enhancement
Cardiac magnetic resonance (CMR) uses different sequences and modalities to assess the heart, such as extracellular volume, derived from T1-weighted imaging, examination of total interstitial space, T2 mapping and weighted imaging examination of edema, and late gadolinium enhancement (LGE) examination of scar and fibrosis (Kali et al., 2014; Gupta et al., 2021). Because extracellular space is enlarged by dead CMs and post-infarct fibrosis, CMR-LGE imaging with excessively retained gadolinium-based contrast agents represents a non-invasive standard for assessing myocardial viability and fibrosis (Holtackers et al., 2022); for instance, a subendocardial scar can be detected via dark-blood LGE-CMR (Holtackers et al., 2021). Feature tracking of CMR accurately quantifies cardiac strain, and a retrospective study has found that almost 75% of acute scars and 80% of subacute scars could be detected by CMR with a segmental circumferential strain of native cine sequences (Polacin et al., 2022). In the mouse MI models with monocyte populations deletion, elastin deposition, as an inflammatory response and a potential fibrotic biomarker, could be detected via CMR with an elastin/tropoelastin-specific contrast agent (Elkenhans et al., 2021). However, the use of CMR is limited by availability, time, cost, and severe renal insufficiency as an adverse effect of contrast administration, which can be solved by segmental peak circumferential strain calculation (Polacin et al., 2022). Additionally, CMR screens post-MI patients at risk of ventricular tachycardia by identifying and quantifying a heterogeneous scar zone and substrate features (Merino-Caviedes et al., 2021). A case-control study has retrospectively reviewed LGE-CMR data of chronic post-MI and found that border zone channel mass was the strongest independent scar-derived variable and precision risk stratification relevant to sustained monomorphic ventricular tachycardia, while border zone channel mass was associated with the qualitative structure, heterogeneity, spatial distribution, and slow conducting channels within the scar (Jáuregui et al., 2022).
3.2 Echocardiography
Early detection of myocardial fibrosis can identify patients at risk of adverse events, which is independently associated with a measure of scar between echocardiography and defibrillator intervention (Gaibazzi et al., 2020). A large single-center clinical cohort study has found that diastolic dysfunction effectively identified mortality risk with relevance to higher incidence and extent of scar via echocardiography and LGE (Wang et al., 2020a; Wang et al., 2020b; Wang et al., 2020c; Wang et al., 2020d). Speckle tracking echocardiography (STE) can evaluate MI via end-systolic radial strain peak to reflect segmental scar with very high sensitivity and specificity, and when combined with blood pressure, non-invasive myocardial work parameters (e.g., myocardial work index, constructive work, and myocardial work efficiency) can be obtained and are significantly lower in the segment with the largest LGE than without LGE after contrasting with gadolinium (Mahdiui et al., 2021). These parameters are emerging potential markers of segmental myocardial viability, prognostic markers, and therapeutic targets in STEMI patients with primary percutaneous coronary intervention (PCI) (Mahdiui et al., 2021). Moreover, LV mechanical dispersion measured by CMR and STE is correlated with scar burden as a prognostic parameter (Holtackers et al., 2021).
3.3 Computed tomography
Cardiac computed tomography (CT) estimates the extracellular volume and macroscopic scar via CT delayed enhancement (CT-DE) with relatively low iodine contrast compared to CMR-LGE (Gupta et al., 2021). X-ray microCT implements quantitative 3D analysis and visualization of cardiac fibrosis in MI mice (Janbandhu et al., 2022).
3.4 Molecular imaging
CFs activation is promising for targeted therapy, which can be detected and tracked by fibroblast activation protein (FAP) imaging with novel radiotracer [68Ga]MHLL1 (Langer et al., 2021). FAP imaging has also detected activated CFs in the non-edematous and non-infarcted area in a prospective study of reperfused STEMI patients (Xie et al., 2022). Moreover, FAP-α deletion attenuates cardiac dilation in MI mice (Hoffmann et al., 2021). Additionally, a retrospective study has found a strong correlation between CFs activation volume with cardiac function and peak creatine kinase via 68Ga-FAP-α inhibitor positron-emission tomography (PET) (Kessler et al., 2021), while PET and single-photon emission computed tomography (SPECT) indirectly assessed cardiac fibrosis via myocardial perfusion imaging (Gupta et al., 2021). PET immunoimaging DOTATATE tracers can find high expression of somatostatin receptor two in M1 inflammatory macrophages (Toner et al., 2022). Multiparametric imaging characterizes the immune response transforming to tissue repair after MI (Hess et al., 2022).
3.5 Biomarkers
Rapidly advancing technologies favor post-MI fibrotic biomarkers identification, such as MMP, collagen peptides, galectin-3, and ST-2 (Bostan et al., 2020), which can be combined with MI biomarkers, such as creatine kinase-myocardial band (CK-MB), troponin, and N-terminal-pro type brain natriuretic peptides (NT-proBNP). A prospective study of 92 patients over 70 years old with MI finds that patients over 70 years old with MI and fragility have significantly higher levels of myocardial stress and fibrosis (Aidumova et al., 2022). Galectin-3, a β-galactoside-binding lectin mainly synthesized by macrophages, maintains cardiac structure and function in the early MI stage, and promotes tissue fibrosis and scar formation in the late stage (Leancă et al., 2022). The TNF-α, soluble tumour necrosis factor-α receptor-1 and 2 and oxidative stress could be considered as potential non-invasive diagnostic and therapeutic biomarkers for coronary chronic total occlusion in the oldest patients with coronary heart disease (Li et al., 2020e). Additionally, low miR-26a plasma level is highly correlated with certain markers (e.g., CK-MB and troponin I) in STEMI patients (Chiang et al., 2020), and immunoreactivity of Nε-(carboxymethyl)lysine is positively correlated with NT-proBNP and cardiac fibrosis (Nogami et al., 2020). Furthermore, abnormal myocardial collagen I and III release certain peptides in the circulation as fibrotic markers (Nikolov and Popovski, 2022), such as PICP and PIIINP, directly correlating with indexes of cardiac diastolic function (Osokina et al., 2020). Furthermore, serum PIIINP of ≥381.4 ng/ml on the 12th day increases the risk of cardiac fibrosis 1 year after the disease onset in STEMI patients with preserved ejection fraction (EF) of I–III degree (Osokina et al., 2021). Moreover, human epididymis factor-4 is an independent predictor of low EF as a diagnostic marker and therapeutic target in cardiac fibrosis (Kilci et al., 2021). ST-segment resolution (STR) is a marker for severe myocardial fibrosis and is associated with scar thickness and size, while STEMI patients with STR of <40.15% easily develop transmural scars (Dong et al., 2021b). The combination of the ICTP/PIIINP ratio and ST2 might aid in risk stratification and serve as prognosis biomarkers in HF patients (Dupuy et al., 2019).
In addition to the above, more and more new technologies have emerged, such as Bayesian cardiac strain imaging assessing murine cardiac fibrosis (Al Mukaddim et al., 2022), single-cell mRNA sequencing inspecting dynamic interstitial cell response in MI mice (Forte et al., 2020), stereological method quantifying CMs (Mühlfeld and Schipke, 2022), and high-throughput screening differential genes expression of monocytes-CFs communication (Wu et al., 2022b; Wu et al., 2022d).
Therefore, developing new tools that allow both an early detection of cardiac fibrosis and the determination of its origin and characteristics will potentially lead to the rapid and efficient treatment of patients.
4 Interventions for cardiac fibrosis
The therapies against cardiac fibrosis are still a focus of clinical attention, and how to reverse fibrosis is always a hot topic. Unfortunately, the effective measures are still lacking and lead to the devastating clinical outcomes, despite the various encouraging results from experimental studies (Morfino et al., 2022). In the studies of clinical drugs, RAAS antagonists have been shown to attenuate cardiac fibrosis and dysfunction, with clinical applications limited by their hypotensive effects and inability to stop the fibrotic progression (AlQudah et al., 2020). Conversely, TGF-β inhibitors (e.g., pirfenidone) improve fibrosis, without affecting blood pressure, but with unexpected side effects (e.g., liver toxicity). At the meantime, some known drugs are going through different phases of clinical trials (Table 1), such as RAAS inhibitors, sodium-glucose cotransporter-2 inhibitors (SGLT2is), BNP, and GRK2 inhibitors, in forms of monotherapy alone or combined with other drugs. Furthermore, fibrosis involves multiple molecules and processes (e.g., inflammatory cells recruitment, molecular mediators release, collagen synthesis, cells differention), which suggests small molecules targeting fibrosis would be the promising interventions. However, these novel therapies are still limited in preclinical studies without the validation of clinical efficacies against fibrosis. For the development of anti-fibrotic drugs, it is great important to apply novel molecular targets or drug repurposing via screening drugs tested and approved for other indications. In this section, we focus on the known drugs, novel compounds and other treatments with anti-fibrotic effects, which are shown in Figure 2 (The figure is drawn by figdraw).
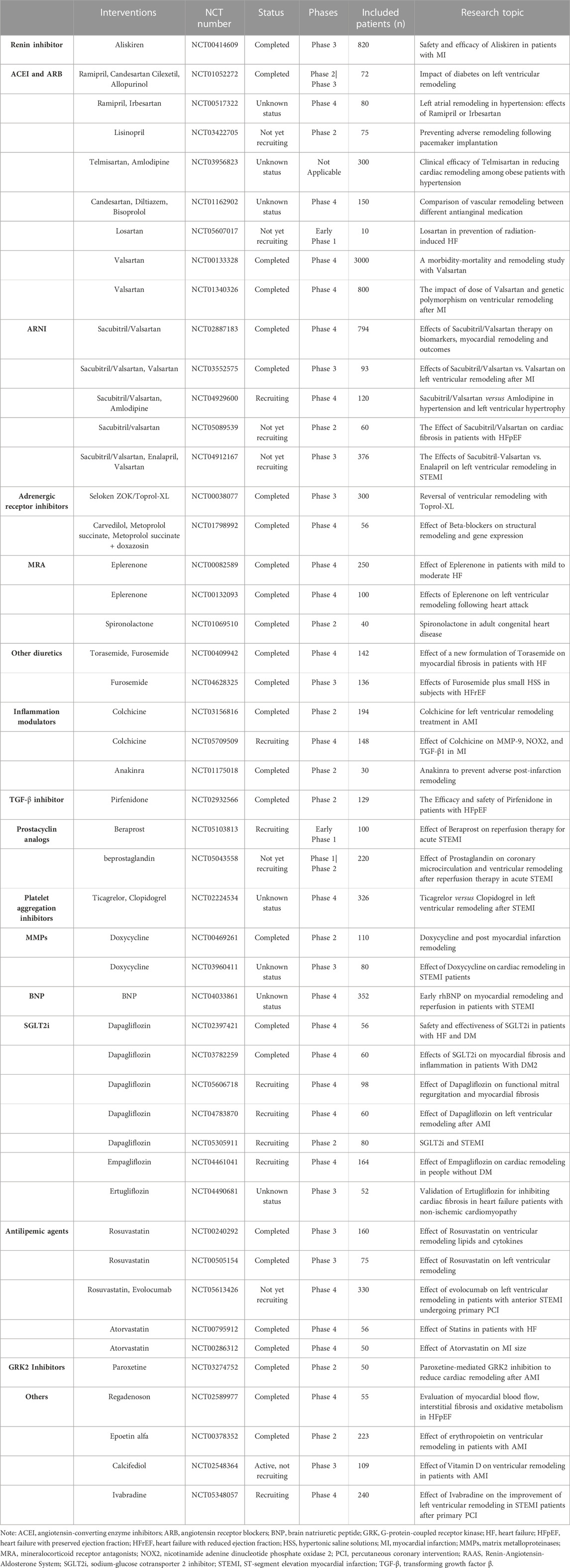
TABLE 1. Recent progress in clinical trials for treating fibrosis and its complications.
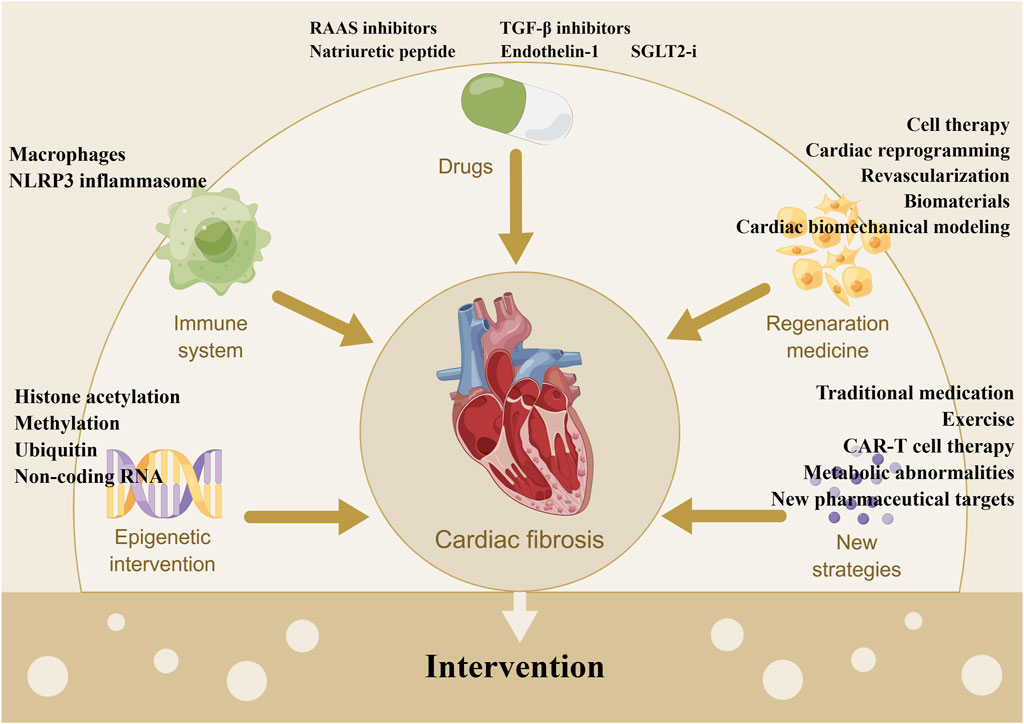
FIGURE 2. Summary of the interventions of cardiac fibrosis. Note: CAR, chimeric antigen receptor; NLRP3: nod-like receptor protein three; RAAS: renin-angiotensin-aldosterone system; SGLT2-i: sodium-glucose cotransporter-2 inhibitor; TGF-β: transforming growth factor β.
4.1 Pharmaceutical interventions
4.1.1 RAAS inhibitors
Several clinical trials of RAAS inhibitors are currently progressing in different phases, which are presented in Table 1. Other non-clinical trials have been reported in the recent years. For example, sliskiren is not only the first Food and Drug Administration-approved and orally active renin inhibitor to treat hypertension, but also regulates collagen metabolism and cardiac fibrosis in vivo and in vitro (Kleinert, 1996; Zhi et al., 2013). Additionally, compound 21 is a non-peptide AT2R agonist with antifibrotic effect (Wang et al., 2017). Targeting RAAS represents a promising therapeutic approach to combat fibrosis, however, conventional RAAS inhibitors cannot completely hamper the fibrotic progression. Together with ACEI, ARNI sacubitril/valsartan suppresses cardiac dysfunction and fibrosis via the downregulation of TGF-β1, BNP, α-SMA, vimentin (Liu et al., 2021b). Furthermore, valsartan and sacubitril/valsartan prevent adverse remodeling in MI rats by reducing oxidative stress, inflammation, and fibrosis (Raj et al., 2021). In MI with hypertensive rats model, the mineralocorticoid receptor antagonist (MRA) spironolactone reduces CFs, MFs, and macrophages infiltration in the heart and kidney (Leader et al., 2021); however, it also binds other steroid receptors (e.g., progesterone and androgen receptors) causing side effects (e.g., gynecomastia and galactorrhea) (Weldon and Brown, 2019). Moreover, eplerenone, a well-tolerated selective MRA, decreases PIIINP with good efficacy when baseline PIIINP is ≥3.6 mmol/L after MI with HF or diabetes (Stienen et al., 2020). As a centrally acting aminopeptidase A inhibitor prodrug, QGC606 inhibits the overactivation of the brain renin-angiotensin system and fibrotic remodeling without lowering blood pressure (Boitard et al., 2022).
4.1.2 TGF-β inhibitors
Antifibrotic drug pirfenidone, a TGF-β1 inhibitor, has been identified from molecular information and transcriptomic data in a swine MI model (Aimo et al., 2022a), whereas evidence in humans has been limited to a phase 2 study evaluating extracellular volume changes with CMR (Aimo et al., 2022b). The clinical use of pirfenidone is limited by the high doses and various side effects. In an article, the total releasing duration of pirfenidone is prolonged by using acellular peritoneal matrix-loaded pirfenidone nanodroplets, which alleviates cardiac fibrosis (Fu et al., 2022b). Except pirfenidone, there are several interventions targeting TGF-β signaling pathway against cardiac fibrosis, such as dihydrolycorine, choline, indole alkaloids, and indole derivatives (Qin et al., 2022). Moreover, 2,5-dimethylcelecoxib inhibits the TGF-β axis and suppresses CF-to-MF transformation in a cryoinjury-induced MI model (Ikushima et al., 2022). The caffeic acid p-nitro phenethyl ester-pNO2 suppresses fibrosis, inflammation, and apoptosis via the TGF-β1/Gal-3 pathway (Wan et al., 2022b). Additionally, thymosin β4 decreases MFs growth and TGF-β1-induced activation to reduce fibrosis (Wang et al., 2022c). Salinomycin inhibits CFs activation and ECM secretion via the inhibition of TGF-β1-dependent p38/MAPK and Rho-kinase pathway in CFs Ang II-infused mice (Burke et al., 2021b). Nintedanib, another antifibrotic agent, was approved to improve pulmonary fibrosis. But the evidence for its role in the treatment of cardiac fibrosis is still lacking (Yvette, 2021).
4.1.3 Drugs targeting the natriuretic peptide family
Strong evidence shows that NPs treatment has beneficial effects on post-MI cardiac remodeling. For example, a previous study has demonstrated that intravenous administration of ANP inhibited RAAS, SNS activity, MIRI, and cardiac remodeling in MI patients (Kasama et al., 2008). Another study has found that continuous CNP infusion (0.1 mg/kg/min) by osmotic mini-pump for 2 weeks after permanent coronary artery occlusion prevents cardiac remodeling (Wang et al., 2007). Further, when BNP (15 mg/kg/day) is intravenously injected over 8 weeks in rats with permanent coronary occlusion, BNP treatment prevents cardiac hypertrophy and EF decline and decreases plasma Ang II level and collagen content in the myocardium (He et al., 2009). Furthermore, after the genetic knockout of Npr1 gene encoding NPR-A in mice, blood pressure rises, and cardiac hypertrophy develops. Blood pressure becomes elevated by 41 mmHg in Npr1−/− mice, together with a 60% increased heart weight/body weight ratio and CM hypertrophy. These findings indicate that endogenous NPs can prevent the development of cardiac hypertrophy (Vellaichamy et al., 2014; Pandey, 2018). A fourth phase clinical trial is ongoing to evaluate early rhBNP intervention in myocardial remodeling and reperfusion in patients with STEMI (Table 1). However, the current evidence mainly exists at the level of animal experiments, and future clinical applications still need to be further explored.
4.1.4 Endothelin-1
ET-1 plays a major role in regulating myocardial fibrosis in several pathological conditions, and its receptor blocker might be beneficial in attenuating biventricular remodeling (Ramos et al., 2018). A few decades ago, studies found that ET-1 A and ET-1 A/B receptor antagonists substantially improved survival, cardiac function, and adverse cardiac remodeling (Sakai et al., 1996; Fraccarollo et al., 2002). Current evidence suggests that ET-1 is not only a molecular marker of cardiac fibrosis but also a novel therapeutic target (Hoffman et al., 2019). However, there is still no clinical evidence that drugs such as bosentan and enversentan have therapeutic effects on cardiac fibrosis.
4.1.5 Sodium-glucose cotransporter-2 inhibitors
In experimental studies and clinical trials, it has been demonstrated that SGLT2is are cardioprotective independently from controlling glucose, for instance, canagliflozin attenuates fibrosis via reducing JAK/STAT signaling, activating adenosine monophosphate-activated protein kinase, and antioxidant signaling (Sabe et al., 2023). In clinical practice, empagliflozin can significantly reduce mortality and hospitalization of HF patients (Kang et al., 2020). It reduces collagen deposit and fibrosis without improving cardiac function via the inhibition of the TGF-β1/smad3 pathway during the early post-MI period (Daud et al., 2021). However, another study reports that the short-term and low-dose empagliflozin increases cardiac systolic function by downregulating MMP-9 and sodium hydrogen exchanger one and upregulating sarco/endoplasmic reticulum Ca2+-ATPase without changing arterial stiffness, blood pressure, fibrotic markers levels, and necroptosis (Goerg et al., 2021). Another selective SGLT1 inhibitor KGA-2727 improves fibrotic remodeling in MI mice (Sawa et al., 2020). Moreover, the DELIVER trial finds dapagliflozin reduces the combined risk of worsening heart failure or cardiovascular death among patients with mildly HFpEF (Solomon et al., 2022). In HFpEF pigs, it also decreases hypertension and reverses concentric remodeling of the heart, with the inhibition of inflammatory response and NO-cGMP-PKG pathway activation (Zhang et al., 2019b). Regrettably, SGLT2i cannot reduce extracellular volume expansion expanded by myocardial interstitial fibrosis (Bojer et al., 2022). Except SGLT2i, there are also other hypoglycemic agents associated with cardiac fibrosis. For example, metformin reduces collagen IIIA1, α-SMA, and CD68 levels after 2 weeks of reperfusion and improves fibrotic remodeling (Loi et al., 2021). Furthermore, metformin and cyclosporin A exert cardiac protection by regulating the balance between AMPK and apoptosis in the mitochondria of bile duct-ligated rats (Moheimani et al., 2021).
4.2 Targeting the immune system
The immune system is activated by MI, which can be a therapeutic target. Thus, immune-related interventions have been a study hotspot with the advance of precision medicine; for instance, the injection of human vascular cell adhesion molecule 1-expressing CFs restores cardiac function by promoting lymphangiogenesis (Iwamiya et al., 2020). Additionally, CD34 cells, isolated from mobilized human mononuclear peripheral blood cells, reduce cardiac scar and fibrosis in MI mice (Tripathi et al., 2020). A ligand-binding blocking anti-CD28 monoclonal antibody improves post-MI healing in mice (Gladow et al., 2020). Furthermore, the immune checkpoint programmed cell death protein one inhibits immune response to prevent damage, and its depletion increases T-cell infiltration in reperfused MI (Michel et al., 2022). In addition, high B cell counts are correlated with enhanced EF in MI patients with PCI, and empagliflozin can treat the MI-induced B cell developmental arrest (Xu et al., 2022b). Moreover, glucocorticoids released by the neuroendocrine system induce Na+/H+-exchanger 1-mediated autophagic death of bone marrow B cells and reduce B cell progenitor proliferation and differentiation (Xu et al., 2022b). Another study reports that adoptive transfer of atorvastatin-induced tolerogenic dendritic cells alleviates CMs apoptosis, fibrosis, inflammatory cells infiltration, and oxidative stress by suppressing TLR-4/NF-κB pathway in MI (Wang et al., 2023).
4.2.1 Targeting the macrophages
Recently, massive studies focus on the detrimental macrophages as the antifibrotic therapeutic targets. For example, cardiac rupture may be induced by macrophage-induced inflammation and downregulated activation of reparative MFs. In the macrophage protease-activated receptor two knockout mice, there are down-regulation of proinflammatory cytokines, recruitment of macrophages, fibrosis in a remote area, and macrophage-derived interferon-β expression, which stimulate the JAK/STAT3 pathway in CFs (Zuo et al., 2020). In addition, granulocyte colony-stimulating factor (G-CSF) improves cardiac remodeling by upregulating JAK2/STAT3 axis (Wang et al., 2020h). Further, N-Propargyl caffeate amide promotes pro-resolving macrophage polarization and prevents cardiac fibrosis by activating peroxisome proliferator-activated receptors-γ (PPAR-γ) pathway (Cheng et al., 2020). Interestingly, cortical bone stem cells can induce a novel macrophage phenotype to modify cardiac inflammation after MI (Hobby et al., 2021). Moreover, hypoxia-induced mitogenic factor deletion promotes M2 macrophages and inhibits M1 macrophages polarization to improve cardiac repair (Li et al., 2021f). M2b macrophages reduce the amount of collagen I and α-SMA, proliferation and migration of CFs, and differentiation of CFs into MFs, whereas M2a macrophages are profibrotic macrophages with opposite effects (Yue et al., 2020). The activation of M2-like macrophage-derived neuregulin-1/ERBB/PI3K/Akt signaling attenuates apoptosis and senescence of CFs in mice (Shiraishi et al., 2022). In infiltrated macrophages, a selective STING inhibitor H-151, alleviates cardiac fibrosis in the MI mouse model via the inhibition of cardiac dsDNA-triggered type I interferon response (Hu et al., 2022b). Furthermore, the knockdown of interferon-induced protein with tetratricopeptide repeats three in MI reduces the amount of CD68+ macrophages, TNF-α, IL-1β and IL-6 levels, infarct size, fibrosis, and collagen content (Sun et al., 2021a; Sun et al., 2021b). 5-methoxytryptophan reduces fibrosis by downregulating macrophages and T-cells infiltration (Hsu et al., 2021). Additionally, 2-benzylidene-3-cyclohexylamino-2,3-dihydro-1H-inden-1-one, the dual-specificity phosphatase six inhibitor, improves cardiac dysfunction and fibrosis in MI rats by inhibiting macrophages formation and inflammation after MI (Zhang et al., 2023). Taken together, the future studies might focus on modulating different populations and phenotypes of macrophages to improve patient prognosis and cardiac remodeling.
4.2.2 Targeting the NLRP3 inflammasome
Several studies about NLRP3 inflammasome in the process of post-MI fibrosis have been reported. NLRP3 inflammasome is a proteolytic complex of the NLRP3 protein, procaspase-1, and apoptosis-associated speck-like protein (Zhang et al., 2022c), which regulates inflammatory response, pyroptosis and mitochondria and MF differentiation in cardiac fibrosis. Thus, it may represent a new therapeutical target, and its inhibitor oridonin decreases IL-1β and IL-18 levels and ameliorates myocardial fibrosis in MI mice (Gao et al., 2021). Moreover, calcium-sensing receptor activates the NLRP3 inflammasome in neutrophils and promotes apoptosis and fibrosis after MI, which is inhibited by Calhex231 (Liu et al., 2020b). Further, the N-butylidenephthalide-pretreated aging MI rats improves human adipose-derived stem cell engraftment and attenuates NLRP3 inflammasome-mediated cardiac fibrosis (Lee et al., 2020). Additionally, glycogen synthase kinase-3 inhibition suppresses the activation of NLRP3 inflammasome in CFs but not in CMs (Wang et al., 2020g). Therapeutic hypothermia attenuates MIRI via regulating sirtuin 3/NLRP3 signalling pathway (Zhang et al., 2022a). Thereby, the NLRP3 inflammasome is a key anti-fibrotic mediator, and its inhibition has beneficial effects on cardiac remodeling. Furthermore, its non-specific inhibitor colchicine is going on some clinical trials (Table 1).
4.3 Interventions for epigenetic regulation
4.3.1 Histone acetylation and methylation
The challenge of epigenetic editing regarding specific cellular targets can provide promising therapeutic options in the coming years. Large preclinical studies have demonstrated the cardioprotective effects of histone deacetylase inhibitors via various mechanisms, such as the suppression of cardiac fibrosis, enhancement of angiogenesis and mitochondrial biogenesis, and prevention of electrical remodeling (Chun, 2020). Moreover, polyunsaturated fatty acids, eicosapentaenoic acid, and docosahexaenoic acid prevent cardiac remodeling by inhibiting p300-histone acetyl-transferase activity in MI rats (Sunagawa et al., 2022a), with the same efficacy as the inhibition of jumonji domain-containing protein three histone demethylase (Long et al., 2020). The inhibition of the disruptor of telomeric silencing 1-like expression reduces methylation modification of histone H3 on spleen tyrosine kinase promoter, which can inhibit the TGF-β1/smad3 axis and prevent myocardial fibrosis and CFs proliferation (Li et al., 2022a). Moreover, silence of methyltransferase-like 3 decreases m6A modification on fibrotic genes and reduces CFs proliferation and TGF-β1-induced collagen production (Li et al., 2021e).
4.3.2 Intervention for ubiquitin
Conserved small molecular protein ubiquitin regulates protein turnover via the ubiquitin-proteasome system. Post-ischemic ubiquitin treatment attenuates cardiac dysfunction, myocardial fibrosis, apoptosis, hypertrophy, and serum cytokine/chemokine levels (Dalal et al., 2023), which suggests ubiquitin has a protective role in cardiac remodeling. Conversely, an endogenous E3 ubiquitin ligase ring-finger protein four knockdown induces extensive interstitial fibrosis after MI (Qiu et al., 2020). Additionally, ubiquitin C-terminal hydrolase L1 regulates cardiac fibrosis through glucose-regulated protein (Lei et al., 2020).
4.3.3 Non-coding RNAs
The protein-coding genes are rare, meanwhile the majority of the transcribed genome are non-coding RNAs that mainly including microRNA, circular RNA (circRNA), long non-coding RNA (lncRNA). Numerous studies suggest that non-coding RNAs participate in pathophysiological process of post-MI fibrosis as epigenetic regulators, with the specialization of tissues and cells, so it is necessary to study the role of non-coding RNAs in fibrotic regulation and molecular mechanisms (Supplemental Table S1). In addition, non-coding RNAs can be carried into target cells by extracellular vesicles (EVs) (e.g., exosomes) with capacity to escape from immunogen clearance, and emerging studies show that they regulate fibrosis as diagnostic markers and therapeutic targets in MI (Table 2). With technical advances, extrusion filters produce massive EVs from live cells with native effects (Wang et al., 2021e) and the modulation of exosome imprinting repairs damaged tissue without immune rejection (Hill et al., 2022). Based on the above information, future research might be focusing on their biodistribution and precise delivery to target cells against cardiac fibrosis.
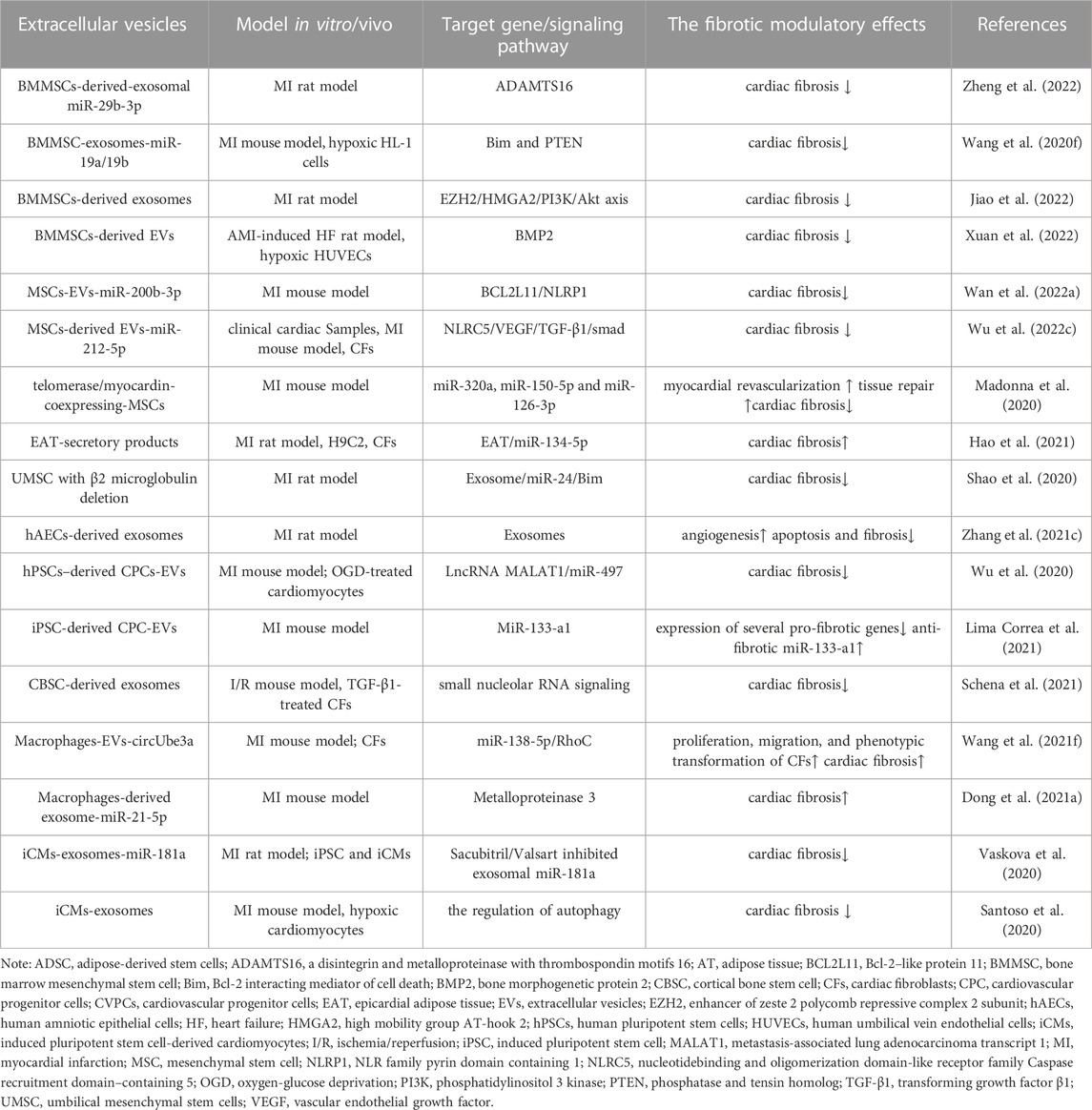
TABLE 2. The fibrotic modulatory effects of extracellular vesicles in myocardial infarction.
4.4 Regenerative medicine
Regenerative medicine is a challenging and broad interesting topic with enormous potential to regenerate novel cells by injecting cells, growth factors, and biomaterials and reprogramming, considering fibrotic scar structure and mechanics, to facilitate novel cell differentiation, maintenance, and function in the extracellular microenvironment (French and Holmes, 2019).
4.4.1 Cell therapy
It’s a novel direction for anti-fibrotic treatment based on cell therapy in recent years. Given revascularization after MI as a cornerstone of current treatment, cell therapy seems like an ideal therapy. Because of the paracrine effects promoting repair of mesenchymal stem cells (MSCs), preconditioned MSCs in situ may be promising for post-MI repair (Sim et al., 2021). The paracrine therapeutic anti-inflammatory and antifibrotic effects of human amniotic MSCs are increased by S100A8/A9 and calcium-binding proteins after MI (Chen et al., 2021c). Moreover, subcutaneous implantation of TheraCyte devices encapsulating human W8B2+ cardiac stem cells improves cardiac remodeling and function after MI (Kompa et al., 2021), and stem cells-derived CMs patches restore normal electrical propagation without the risk of arrhythmia (Fassina et al., 2022). Induced cardiosphere (iCS) can be produced by self-replicative RNA approach, differentiating into CMs, while intravenous and intramyocardial injection of C-X-C chemokine receptor four positive subpopulation of iCS-derived cells has similar therapeutic effects in the mice MI model (Xu et al., 2021a). Additionally, intracoronary injection of allogeneic cardiosphere-derived cells immediately prior to reperfusion in an AMI pig model impedes adverse remodeling (Sousonis et al., 2021). The human amniotic membrane MSCs-derived conditioned medium modulates autophagy via mTOR/ULK1 pathway to against MIRI. (Mokhtari et al., 2021). The skeletal muscle-derived Sca-1+/PW1+/Pax7− interstitial cells attenuate cardiac remodeling after transplanted into the infarcted myocardium (Ruchaya et al., 2022).
4.4.2 Cardiac reprogramming
Pluripotent stem cells generate functional CMs for post-MI regeneration, and cardiac reprogramming in vivo generates chamber-matched new CMs (Zhang et al., 2021d). Additionally, CFs can be reprogrammed into CMs and cardiovascular progenitor cells via lentiviral packaging techniques (Isomi et al., 2021) and CRISPR (Jiang et al., 2022), respectively. Moreover, microRNA-delivery platforms efficiently reprogram CFs into induced CMs (Yang et al., 2021a) or non-CMs into CMs-like cells (Kaur et al., 2021). Further, photo biomodulation therapy modulates gene transcription and miRNA expression to reverse the profibrotic signaling pathway (Feliciano et al., 2022), and fibroblast cilia regulates cardiac regeneration (Djenoune et al., 2022). A study has used miR-208b-3p mimic, ascorbic acid, and BMP4 to reprogram mouse tail-tip CFs into different cells (CMs, endothelial cells, and smooth muscle cells) and form cardiovascular tissue-like structure (Cho et al., 2021). Additionally, anti-BMP 1.3 monoclonal antibody inhibits the TGF-β pathway to reduce MFs activation and scar formation and exerts cardiac protection through BMP 5 (Vukicevic et al., 2022).
4.4.3 Revascularization
As the main goal of treatment in MI, revascularization restores myocardial perfusion and reduces the infarction size, and improves cardiac function (Solhpour and Yusuf, 2014). PCI is the standard therapy for patients presenting in the first 12h from symptom onset, however, early thrombolysis should be considered with the absence of PCI (Leancă et al., 2022). Despite the above therapies, cardiac remodeling is still in one-third of MI patients (Solhpour and Yusuf, 2014). Macrophages and CFs also cause vascular disintegration and capillary rarefication (Wu et al., 2021b). Fortunately, the prevascularized cell sheets use reprogrammed cardiac cells to improve adverse remodeling (Song et al., 2020). High mobility group box one protein recruits bone marrow PDGFRα+-mesenchymal cells to induce angiogenesis and antifibrotic effect (Goto et al., 2020). However, these are basic studies without clinical applications. In a transmural scar distal to total coronary occlusion, up to 2 weeks after MI, large and medium coronary arteries maintain structural integrity, but are destroyed by subsequently progressive neointimal hyperplasia, intravascular fibrosis, and inward remodeling (Dedkov, 2021). Hence, the time frame for revascularization and optimizing cell-based regenerative therapies is the first 2 weeks in MI rats, but it is longer in humans (Dedkov, 2021).
4.4.4 Biomaterials
Biomaterials (e.g., hydrogels, nanocarriers, and cardiac patches) support self-renewal in the injured heart, with reliable biosafety and moderate promise, on-demand biodegradation, and multiple biofunctions to deliver the therapeutic drugs; for instance, a type of hydrogel injected in the peri-infarcted zone promotes fibrotic healing in the infarct zone and inhibits reactive fibrosis and hypertrophy in the remote zone (Wen et al., 2020). Another injectable thermosensitive hydrogel of chitosan/dextran/β-glycerophosphate delivers umbilical cord MSCs to repair the damaged heart (Ke et al., 2020), while injectable disulfide-cross-linked chitosan hydrogel loaded with basic FGF synergistically exerts antifibrotic, antiapoptotic, and proangiogenic effects (Fu et al., 2022a). Moreover, a conductive and MMP-degradable hydrogel stabilizes hypoxia-inducible factor 1-α to reduce the infarcted area and inflammation factors, promote vascularization and the expression of junctional protein connexin 43, and recover cardiac function (Wei et al., 2022). The miR-124-3p-loaded nanoparticles activate PTEN/P13K/Akt pathway to decrease oxidative stress and myocardial injury (Cheng et al., 2022). However, there are still many matters that have not been figured out, such as the analysis of dyskinesia of the infarct zone, the mechanical properties of myocardium, and the key mechanisms underlying the cardiac benefits of a hydrogel implantation. Thus, hydrogels are still difficult to translate from preclinical studies to humans.
4.4.5 Cardiac biomechanical modeling
Cardiac biomechanical modeling is a promising new tool for MI prognosis and therapy, such as a computational model predicting multiple time-dependent paracrine and intracellular drivers of CFs phenotype and post-MI fibrosis (Zeigler et al., 2020). Additionally, a library of scar tissue mechanical properties allows for the mechanics of cardiac modeling to assess the healing stage, rate, and collagen density, which can be potentially used as valuable biomarkers and therapies (Dempsey et al., 2020).
4.5 New strategies
4.5.1 Traditional medication
Traditional medication has developed for thousands of years as an ancient wisdom, with various herbal drugs and their isolated compounds. Recently, some studies have been performed to evaluate the anti-fibrotic efficacy of traditional medication and compounds as innovative antifibrotic therapies. For example, both fasudil and aconite ameliorate myocardial fibrosis (Xiang et al., 2022; Xing et al., 2022). Danqi soft capsule inhibits CFs proliferation and migration, collagen secretion, and CF-to-MF transformation in post-MI HF rats (Ma et al., 2022). Many studies have demonstrated the signaling pathways of traditional medication. Calycosin and taohong siwu decoction suppress TGF-β1-induced CFs proliferation and collagen deposition (Tan et al., 2021; Chen et al., 2022a). Moreover, zerumbone, ganxin V, nutmeg-5, ginsenoside Rg2, jatrorrhizine, cardiotonic pill, and huoxin pill all regulate the TGF-β1/smad pathway to attenuate fibrosis (Li et al., 2020d; Wang et al., 2021b; Yan et al., 2022a; Yan et al., 2022b; Li et al., 2022c; Hao and Jiao, 2022; Liang et al., 2022). Moreover, citri reticulatae pericarpium also reduces CMs apoptosis, CFs proliferation, and CF-to-MF transformation by upregulating PPAR-γ expression (Chen et al., 2022c), while auraptene improves cardiac hypertrophy and dysfunction via activating PPARα (Sunagawa et al., 2022b). Tanshinone IIA suppresses inflammation and fibrosis via the regulation of NADPH oxidase 4 (Chen et al., 2021b). Additionally, panaxatriol saponin inhibits CFs activation and proliferation and fibrosis by regulating oxidative stress and Nrf2 pathway (Yao et al., 2022). Astragaloside IV improves fibrosis by suppressing ROS/Caspase-1/GSDMD pathway (Zhang et al., 2022d), and liquiritin play the same role via the inhibition of CCL5 expression and NF-κB pathway (Han et al., 2022). Storax effectively protects cardiomyocytes against myocardial fibrosis and cardiac dysfunction by inhibiting the AT1R/ankyrin repeat domain 1/p53 signaling pathway (Xu et al., 2022c). In addition, nutraceuticals (e.g., curcumin, berberine, hibiscus roselle, flaxseed, and garlic alliin) are useful as antifibrotic substances acting via multiple signaling pathways (Zivarpour et al., 2022).
4.5.2 Exercise
In the modern era, unhealthy lifestyle (e.g., alcohol drinking, binge eating, smoking, physical inactivity) has been established as a risk factor for MI. Except for drug therapy, exercise-based cardiac rehabilitation should be taken into consideration to prevent the progression of adverse cardiac remodeling. For example, exercise significantly improves post-MI survival in the diet-induced obesity model (Peres Valgas Da Silva et al., 2022). Furthermore, moderate resistance exercise activates CMs proliferation through follistatin-like 1 (Hu et al., 2020). Moreover, exercise training increases FGF 21 and regulates the TGF-β1-smad2/3-MMP2/9 axis (Ma et al., 2021) and expression of lncRNAs H19, GAS5, and MIAT to decrease fibrosis in MI mice (Farsangi et al., 2021). Additionally, exercise inhibits tryptase release by mast cells and cardiac fibrosis (Bayat et al., 2021). The preconditioning with high-intensity interval training decreases heart injuries by increasing G-CSF and G-CSFR in the MI mouse model (Ghanimati et al., 2020). Noteworthily, the effect of combining the exercise with dietary intervention has been well validated. For example, aerobic-resistance training combined with vitamin D3 supplement suppresses the expression of TGF-β1, smad2/3, and collagen I and III to alleviate myocardial fibrosis and dysfunction (Mehdipoor et al., 2021).
4.5.3 CAR-T cell therapy
Anti-fibrotic T-cell therapy with chimeric antigen receptor (CAR) is engineered receptors with function to redirect lymphocytes to recognize and eliminate cells expressing a specific target antigen. This interaction occurs in a specific CAR domain called “antigen binding domain” and allows endogenous activation of T cells, with subsequent elimination of target cells (Morfino et al., 2022). Aghajanian and his colleagues firstly investigated that T-cell immunotherapy could specifically target pathologic cardiac fibrosis. The endogenous cardiac fibroblasts target FAP has been shown to benefit cardiac fibrosis. Adoptive transfer of T cells expressing a CAR against FAP, results in a significant reduction in cardiac fibrosis and restoration of function after injury in mice (Aghajanian et al., 2019). Moreover, a new approach is represented by the use of CAR-T cells engineered in vivo using lipid nanoparticles containing mRNA coding for a receptor directed against the FAP protein, expressed by MFs (Rurik et al., 2022). This strategy has proved to be safe and effective in reducing myocardial fibrosis and improving cardiac function in mice. However, there are still many limitations to this approach. For example, it may lead to antigen escape and the syndrome of release of pro-inflammatory cytokines (Asnani, 2018; Majzner and Mackall, 2018; Ghosh et al., 2020). Thus, potential toxicity associated with CAR-T cell therapy has stimulated the search for alternative approaches, such as the use of CAR-natural killer cells, and safer cell programming methods. To sum up, the anti-fibrotic management following MI is still a serious challenge, and current therapies involving protecting the remaining CMs and preventing fibrosis.
4.5.4 Intervention for metabolic abnormalities
Metabolism is an essential process for the maintenance of life, and metabolic homeostasis needs the coordination of anabolism and catabolism, which have been extensively studied. Recent studies involve metabolism of glucose, lipid, nucleotide, amino acid and so on. For example, glycolysis is not only related to TGF-β and Krüppel-like factor 5 signaling (Methatham et al., 2022), but also is inhibited by kallistatin/serpina3c to reduce post-MI fibrosis by activating Nr4a1 (Ji et al., 2022a). Protein kinase R regulates inflammation, insulin resistance, and glucose balance to improve cardiac fibrosis in isoproterenol-induced MI rats (Mangali et al., 2021). The high-density lipoproteins and stimulator of steroid receptor coactivators MCB-613 induce reverse remodeling via the reduction of cardiac dysfunction, hypertrophy, and fibrosis (De Geest and Mishra, 2021). Lysophosphatidic acid-lysophosphatidic acid receptor two signaling promotes angiogenesis and maintains vascular homeostasis to reduce scar formation and cardiac dysfunction (Pei et al., 2022). Furthermore, ketone ester reprograms gene expression of ketone body utilization and normalizes ATP production in post-infarct remodeling (Yurista et al., 2021). Sphingosine kinase one inhibitor PF543 reduces α-SMA, collagen, IL-1β, IL-6, and TNF-α levels to decrease myocardial injury, fibrosis, and inflammation (Wu et al., 2022a). Notably, deoxycholic acid-G protein-coupled bile acid receptor pathway activation also decreases inflammation and fibrosis (Wang et al., 2021a). Recombinant slit2, a secretive ECM protein, regulates the level of blood lipid decreasing total cholesterol, triglycerides, and low-density lipoprotein cholesterol, and increasing high-density lipoprotein cholesterol level in rats, which relieves the myocardial fibrosis, inflammation and oxidative stress in coronary heart disease (Liu et al., 2021a).
Following, we summarize the recent studies about amino acids and nucleotides. The inhibition of cardiac thyrotropin-releasing hormone reduces post-infarct hypertrophy and fibrosis (Schuman et al., 2021). Additionally, triiodothyronine pretreatment improves post-MI dysfunction and inhibits fibrosis by activating the insulin-like growth factor-1/PI3K/Akt signaling pathway (Zeng et al., 2021). In an MI sheep model, the upregulation of 5-hydroxytryptamine induces valve fibrosis, which could be improved by cyproheptadine (Marsit et al., 2022). Oral propionate, the important components of short-chain fatty acids, modulates macrophages polarization and pro-inflammatory cytokine via reducing JNK/p38/NF-κB phosphorylation to improve post-MI chronic cardiac remodeling (Zhou et al., 2023). The upregulated expression of ectonucleotide pyrophosphatase/phosphodiesterase 1 (ENPP1) after a cardiac injury can regulate cardiac repair, whereas uridine or ENPP1 inhibitor myricetin enhances cardiac repair by targeting the ENPP1/adenosine monophosphate (AMP) pathway (Li et al., 2022e).
Lastly, there are some other metabolic researches, for instance, chronic daily alcohol uptake enhances MI-induced cardiac dysfunction, fibrosis, and mitochondrial dysfunction (Liang et al., 2020). Urolithin A, a type of gut bacterial metabolite, inhibits myocardial fibrosis by activating the Nrf2 pathway (Chen et al., 2022d). Additionally, dimethyl fumarate promotes anti-inflammatory and preparative regulation by modulating oxidative metabolism in macrophages and CFs (Mouton et al., 2021). Furthermore, atypical chemokine receptor four deletion inhibits IL-6 expression and CFs proliferation to alleviate cardiac remodeling (Zhang et al., 2021a; Zhang et al., 2021b). Copper is reduced in myocardial ischemia-induced cardiac fibrosis, while it inhibits the CF-to-MF transformation as a pro-fibrinolytic switch and improves cardiac function (Xiao et al., 2023).
Post-MI cardiac fibrosis is related to unbalance of energy substrate metabolism (e.g., glucose, lipid, and amino acid). Furthermore, it is an energy-consuming process, which suggests that interventions of cardiac fibrosis combined with metabolic abnormalities are very important, with the positive efficacies against fibrosis.
4.5.5 New pharmaceutical targets
An increasing number of new pharmaceutical targets have been developed, such as a soluble epoxide hydrolase vaccine, which improves cardiac function, and boron, as well as a new ligand of the apelin peptide jejunum receptor apela, which reduce myocardial fibrosis and apoptosis (Bouchareb et al., 2020; Pan et al., 2020; Kitsuka et al., 2022). There are also interventions targeting inflammation response, for instance, the inhibition of coagulation protein tissue factor cytoplasmic domain improves cardiac remodeling by regulating inflammation and angiogenesis (Chong et al., 2021). Resveratrol supplementation also decreases proinflammatory cytokine levels, cardiac dysfunction, and atrial interstitial fibrosis in MI-induced rats (Jiang et al., 2021).
Moreover, There are many other drugs that can improve fibrosis, such as piperine, thymoquinone, an oleanolic acid Qi-Tai-Suan, spinal cord stimulation and exogenous hydrogen sulfide (Li et al., 2020f; Viswanadha et al., 2020; Farag et al., 2021; Qian et al., 2022; He et al., 2023). Endostatin, alarin, L-carnitine, pentraxin 3 depletion, and mdivi-1 also can attenuate oxidative stress to inhibit myocardial fibrosis (Li et al., 2021b; Xu et al., 2021b; Emran et al., 2021; Xu et al., 2022a; Ding et al., 2022).
In experimental studies are useful to regulate cardiac fibrosis, even without clinical applications. For example, ephrinA1-Fc attenuates chronically non-reperfused post-MI remodeling (Whitehurst et al., 2020). Secreted frizzled protein three protects the heart via ischemic preconditioning in a pig model (Vatner et al., 2021). Upregulation of periostin regulates post-infarct fibrosis via cyclic AMP response element-binding protein 1 (Xue et al., 2020; Xue et al., 2022). Anti-proprotein convertase subtilisin/kexin type 9 intervention reduces infarct size and cardiac dysfunction (Guo et al., 2021). Epoxylipids improve cardiac fibrosis and dysfunction (Imig et al., 2022). Furthermore, menthol and HC-030031 reduces cardiac fibrosis via the regulation of cation channel (Li et al., 2020c; Wang et al., 2020e). As a pleiotropic hormone, serelaxin mitigates adverse remodeling and modulates bioactive sphingolipid signaling (Devarakonda et al., 2022). Additionally, nesfatin-1 suppresses necroptosis via regulating receptor-interacting protein kinase (RIPK) 1/RIPK3/mixed lineage kinase domain-like protein axis and RhoA/ Rho-associated coiled-coil-containing protein kinase/RIP3pathway (Sharifi et al., 2021). Pulsed-field ablation exerts ablating ventricular scar, and eliminates viable myocardium separated from the catheter by collagen and fat (Younis et al., 2022). Calpain inhibition decreases collagen formation (Potz et al., 2022). The purified human tropoelastin significantly repairs the infarcted heart in a MI rodent model (Hume et al., 2023).
Together with, researches on cardiac fibrosis have evolved with the advancement of various genetics and proteomics approaches in recent years. However, a lot of novel targets with anti-fibrotic effects are limited in basic researches, without clinical application. Advances have been made in the therapeutic field, and timely coronary reperfusion in associations with novel therapies, such as ARNI and SGLT2i, along with MRAs and beta blockers, counteract adverse ventricular remodeling and promote reverse ventricular remodeling, decreasing progression to HF and mortality. Future therapeutic perspectives, such as microRNAs, bone marrow derived-cells, and molecules targeting inflammation are currently under research, with promising results.
5 Conclusion
In summary, regional fibrotic control in infarcted areas and suppression of collagen accumulation in non-infarcted areas are vital to improve adverse remodeling and clinical outcome after MI. Even being treated according to the guideline-recommended protocols, it is still adverse fibrotic remodeling in MI patients. In the future, the study should focus on the exploring the deeper pathophysiological mechanisms underlying the onset and progression of post-MI fibrosis, then further determining therapeutic targets, and optimizing intervention strategies. In the meantime, antifibrotic precision interventions still need a clinical translation. Although clinical trials associated with anti-fibrotic drugs already have been performed, the patients included in trials are rather small. Hence, it is urgent to explore integrated and personalized therapeutic strategies to inhibit progressively fibrotic remodeling after MI. Moreover, early identification, diagnosis, and management of cardiac fibrosis are significantly important in improving the survival and prognosis of MI patients.
Author contributions
XAY and XNY wrote the first draft of the review. XP, JZ, XF, JL, XZ, and LJ did literature search and date interpretation. JW, PH and YC designed the review and revised the report carefully.
Funding
This study was supported by the State Key Program of the National Natural Science Foundation of China (82030059), National Natural Science Foundation of China (81772036, 82072144,81873950, 82172178, 81873953), National Key R&D Program of China (2020YFC1512700, 2020YFC1512705, 2020YFC1512703), National S&T Fundamental Resources Investigation Project (2018FY100600, 2018FY100602), Key R&D Program of Shandong Province (2021ZLGX02, 2021SFGC0503), Taishan Pandeng Scholar Program of Shandong Province (tspd20181220), Taishan Young Scholar Program of Shandong Province (tsqn20161065, tsqn201812129), Youth Top-Talent Project of National Ten Thousand Talents Plan, Qilu Young Scholar Program and the Fundamental Research Funds of Shandong University (2018JC011).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2023.1070973/full#supplementary-material
References
Aboulgheit, A., Karbasiafshar, C., Zhang, Z., Sabra, M., Shi, G., Tucker, A., et al. (2021). Lactobacillus plantarum probiotic induces Nrf2-mediated antioxidant signaling and eNOS expression resulting in improvement of myocardial diastolic function. Am. J. Physiol. Heart Circ. Physiol. 321, H839–h849. doi:10.1152/ajpheart.00278.2021
Acharya, A., Baek, S. T., Huang, G., Eskiocak, B., Goetsch, S., Sung, C. Y., et al. (2012). The bHLH transcription factor Tcf21 is required for lineage-specific EMT of cardiac fibroblast progenitors. Development 139, 2139–2149. doi:10.1242/dev.079970
Adamo, L., Rocha-Resende, C., and Mann, D. L. (2020). The emerging role of B lymphocytes in cardiovascular disease. Annu. Rev. Immunol. 38, 99–121. doi:10.1146/annurev-immunol-042617-053104
Adapala, R. K., Kanugula, A. K., Paruchuri, S., Chilian, W. M., and Thodeti, C. K. (2020). TRPV4 deletion protects heart from myocardial infarction-induced adverse remodeling via modulation of cardiac fibroblast differentiation. Basic Res. Cardiol. 115, 14. doi:10.1007/s00395-020-0775-5
Adapala, R. K., Katari, V., Teegala, L. R., Thodeti, S., Paruchuri, S., and Thodeti, C. K. (2021). TRPV4 mechanotransduction in fibrosis. Cells 10.
Adrover, J. M., Del Fresno, C., Crainiciuc, G., Cuartero, M. I., Casanova-Acebes, M., Weiss, L. A., et al. (2019). A neutrophil timer coordinates immune defense and vascular protection. Immunity 50, 966–967. doi:10.1016/j.immuni.2019.11.001
Aghajanian, H., Kimura, T., Rurik, J. G., Hancock, A. S., Leibowitz, M. S., Li, L., et al. (2019). Targeting cardiac fibrosis with engineered T cells. Nature 573, 430–433. doi:10.1038/s41586-019-1546-z
Ahmad, F., Tomar, D., Aryal, A. C. S., Elmoselhi, A. B., Thomas, M., Elrod, J. W., et al. (2020). Nicotinamide riboside kinase-2 alleviates ischemia-induced heart failure through P38 signaling. Biochim. Biophys. Acta Mol. Basis Dis. 1866, 165609. doi:10.1016/j.bbadis.2019.165609
Aidumova, O. Y., Shchukin, Y. V., Limareva, L. V., and Piskunov, M. V. (2022). Geriatric status of patients over 70 years of age with myocardial infarction and the relationship with the severity of myocardial stress, fibrosis and angiogenesis. Adv. Gerontol. 35, 538–543.
Aimo, A., Iborra-Egea, O., Martini, N., Galvez-Monton, C., Burchielli, S., Panichella, G., et al. (2022a). Cardiac protection by pirfenidone after myocardial infarction: A bioinformatic analysis. Sci. Rep. 12, 4691. doi:10.1038/s41598-022-08523-3
Aimo, A., Spitaleri, G., Panichella, G., LupóN, J., Emdin, M., and Bayes-Genis, A. (2022b). Pirfenidone as a novel cardiac protective treatment. Heart Fail Rev. 27, 525–532. doi:10.1007/s10741-021-10175-w
AL Mukaddim, R., Weichmann, A. M., Taylor, R., Hacker, T. A., Pier, T., Hardin, J., et al. (2022). Murine cardiac fibrosis localization using adaptive Bayesian cardiac strain imaging in vivo. Sci. Rep. 12, 8522. doi:10.1038/s41598-022-12579-6
Alex, L., Tuleta, I., Harikrishnan, V., and Frangogiannis, N. G. (2022). Validation of specific and reliable genetic tools to identify, label, and target cardiac pericytes in mice. J. Am. Heart Assoc. 11, e023171. doi:10.1161/JAHA.121.023171
Alqudah, M., Hale, T. M., and Czubryt, M. P. (2020). Targeting the renin-angiotensin-aldosterone system in fibrosis. Matrix Biol. 91-92, 92–108. doi:10.1016/j.matbio.2020.04.005
Amin, P., Singh, M., and Singh, K. (2011). β-Adrenergic receptor-stimulated cardiac myocyte apoptosis: Role of β1 integrins. J. Signal Transduct. 2011, 179057. doi:10.1155/2011/179057
Asnani, A. (2018). Cardiotoxicity of immunotherapy: Incidence, diagnosis, and management. Curr. Oncol. Rep. 20, 44. doi:10.1007/s11912-018-0690-1
Atsuki, I., Shota, T., Kota, T., Shota, F., Masanori, O., Makiko, M., et al. (2019). Myofibroblast β2 adrenergic signaling amplifies cardiac hypertrophy in mice. Biochem. Biophys. Res. Commun. 510 (1), 149–155. doi:10.1016/j.bbrc.2019.01.070
Aziz, I. S., Mcmahon, A. M., Friedman, D., Rabinovich-Nikitin, I., Kirshenbaum, L. A., and Martino, T. A. (2021). Circadian influence on inflammatory response during cardiovascular disease. Curr. Opin. Pharmacol. 57, 60–70. doi:10.1016/j.coph.2020.11.007
Bajpai, G., Bredemeyer, A., Li, W., Zaitsev, K., Koenig, A. L., Lokshina, I., et al. (2019). Tissue resident CCR2-and CCR2+ cardiac macrophages differentially orchestrate monocyte recruitment and fate specification following myocardial injury. Circ. Res. 124, 263–278. doi:10.1161/CIRCRESAHA.118.314028
Bayat, M., Chien, S., and Chehelcheraghi, F. (2021). Aerobic exercise-assisted cardiac regeneration by inhibiting tryptase release in mast cells after myocardial infarction. Biomed. Res. Int. 2021, 5521564. doi:10.1155/2021/5521564
Benjamin, E. J., Virani, S. S., Callaway, C. W., Chamberlain, A. M., Chang, A. R., Cheng, S., et al. (2018). Heart disease and stroke statistics-2018 update: A report from the American heart association. Circulation 137, e67–e492. doi:10.1161/CIR.0000000000000558
Bertaud, A., Joshkon, A., Heim, X., Bachelier, R., Bardin, N., Leroyer, A. S., et al. (2023). Signaling pathways and potential therapeutic strategies in cardiac fibrosis. Int. J. Mol. Sci. 24, 1756. doi:10.3390/ijms24021756
Blythe, N. M., Muraki, K., Ludlow, M. J., Stylianidis, V., Gilbert, H. T. J., Evans, E. L., et al. (2019). Mechanically activated Piezo1 channels of cardiac fibroblasts stimulate p38 mitogen-activated protein kinase activity and interleukin-6 secretion. J. Biol. Chem. 294, 17395–17408. doi:10.1074/jbc.RA119.009167
Boitard, S. E., Keck, M., Deloux, R., Girault-Sotias, P. E., Marc, Y., DE Mota, N., et al. (2022). QGC606: A best-in-class orally active centrally acting aminopeptidase A inhibitor prodrug for treating heart failure following myocardial infarction. Can. J. Cardiol. 38, 815–827. doi:10.1016/j.cjca.2022.01.019
Boitard, S. E., Marc, Y., Keck, M., Mougenot, N., Agbulut, O., Balavoine, F., et al. (2019). Brain renin-angiotensin system blockade with orally active aminopeptidase A inhibitor prevents cardiac dysfunction after myocardial infarction in mice. J. Mol. Cell Cardiol. 127, 215–222. doi:10.1016/j.yjmcc.2018.12.008
Bojer, A. S., SøRENSEN, M. H., GæDE, P., and Madsen, P. L. (2022). Myocardial extracellular volume expansion in type 2 diabetes is associated with ischemic heart disease, autonomic neuropathy, and active smoking. Diabetes Care 45, 3032–3039. doi:10.2337/dc22-0942
Boorsma, E. M., Ter Maaten, J. M., Damman, K., VAN Veldhuisen, D. J., Dickstein, K., Anker, S. D., et al. (2021). Dipeptidyl peptidase 3, a marker of the antagonist pathway of the renin-angiotensin-aldosterone system in patients with heart failure. Eur. J. Heart Fail 23, 947–953. doi:10.1002/ejhf.2158
Bostan, M. M., Stătescu, C., Anghel, L., Șerban, I. L., Cojocaru, E., and Sascău, R. (2020). Post-myocardial infarction ventricular remodeling biomarkers-the key link between pathophysiology and clinic. Biomolecules 10, 1587. doi:10.3390/biom10111587
Bouchareb, R., Katz, M., Saadallah, N., Sassi, Y., Ali, S., and Lebeche, D. (2020). Boron improves cardiac contractility and fibrotic remodeling following myocardial infarction injury. Sci. Rep. 10, 17138. doi:10.1038/s41598-020-73864-w
Bouvet, M., Claude, O., Roux, M., Skelly, D., Masurkar, N., Mougenot, N., et al. (2020). Anti-integrin α(v) therapy improves cardiac fibrosis after myocardial infarction by blunting cardiac PW1(+) stromal cells. Sci. Rep. 10, 11404. doi:10.1038/s41598-020-68223-8
Braidotti, N., Chen, S. N., Long, C. S., Cojoc, D., and Sbaizero, O. (2022). Piezo1 channel as a potential target for hindering cardiac fibrotic remodeling. Int. J. Mol. Sci. 23, 8065. doi:10.3390/ijms23158065
Brazile, B. L., Butler, J. R., Patnaik, S. S., Claude, A., Prabhu, R., Williams, L. N., et al. (2021). Biomechanical properties of acellular scar ECM during the acute to chronic stages of myocardial infarction. J. Mech. Behav. Biomed. Mater 116, 104342. doi:10.1016/j.jmbbm.2021.104342
Bromage, D. I., Trevelin, S. C., Huntington, J., Yang, V. X., Muthukumar, A., Mackie, S. J., et al. (2022). Nrf2 attenuates the innate immune response after experimental myocardial infarction. Biochem. Biophys. Res. Commun. 606, 10–16. doi:10.1016/j.bbrc.2022.03.043
Bugg, D., Bailey, L. R. J., Bretherton, R. C., Beach, K. E., Reichardt, I. M., Robeson, K. Z., et al. (2022). MBNL1 drives dynamic transitions between fibroblasts and myofibroblasts in cardiac wound healing. Cell Stem Cell 29, 419–433. doi:10.1016/j.stem.2022.01.012
Bundgaard, H., Axelsson Raja, A., Iversen, K., Valeur, N., TøNDER, N., Schou, M., et al. (2022). Hemodynamic effects of cyclic guanosine monophosphate-dependent signaling through β3 adrenoceptor stimulation in patients with advanced heart failure: A randomized invasive clinical trial. Circ. Heart Fail 15, e009120. doi:10.1161/CIRCHEARTFAILURE.121.009120
Burke, R. M., Burgos Villar, K. N., and Small, E. M. (2021a). Fibroblast contributions to ischemic cardiac remodeling. Cell Signal 77, 109824. doi:10.1016/j.cellsig.2020.109824
Burke, R. M., Dirkx, R. A., Quijada, P., Lighthouse, J. K., Mohan, A., O'Brien, M., et al. (2021b). Prevention of fibrosis and pathological cardiac remodeling by salinomycin. Circ. Res. 128, 1663–1678. doi:10.1161/CIRCRESAHA.120.317791
Capote, L. A., Mendez Perez, R., and Lymperopoulos, A. (2015). GPCR signaling and cardiac function. Eur. J. Pharmacol. 763, 143–148. doi:10.1016/j.ejphar.2015.05.019
Cargnello, M., and Roux, P. P. (2011). Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 75, 50–83. doi:10.1128/MMBR.00031-10
Chen, B., and Frangogiannis, N. G. (2021). Chemokines in myocardial infarction. J. Cardiovasc Transl. Res. 14, 35–52. doi:10.1007/s12265-020-10006-7
Chen, G., Huang, S., Song, F., Zhou, Y., and He, X. (2020a). Lnc-Ang362 is a pro-fibrotic long non-coding RNA promoting cardiac fibrosis after myocardial infarction by suppressing Smad7. Arch. Biochem. Biophys. 685, 108354. doi:10.1016/j.abb.2020.108354
Chen, G., Xu, H., Xu, T., Ding, W., Zhang, G., Hua, Y., et al. (2022a). Calycosin reduces myocardial fibrosis and improves cardiac function in post-myocardial infarction mice by suppressing TGFBR1 signaling pathways. Phytomedicine 104, 154277. doi:10.1016/j.phymed.2022.154277
Chen, H., Chew, G., Devapragash, N., Loh, J. Z., Huang, K. Y., Guo, J., et al. (2022b). The E3 ubiquitin ligase WWP2 regulates pro-fibrogenic monocyte infiltration and activity in heart fibrosis. Nat. Commun. 13, 7375. doi:10.1038/s41467-022-34971-6
Chen, H., Li, M., Liu, L., Zhu, D., and Tian, G. (2020b). Telmisartan improves myocardial remodeling by inhibiting leptin autocrine activity and activating PPARγ. Exp. Biol. Med. (Maywood) 245, 654–666. doi:10.1177/1535370220908215
Chen, J., Jia, J., Ma, L., Li, B., Qin, Q., Qian, J., et al. (2021a). Nur77 deficiency exacerbates cardiac fibrosis after myocardial infarction by promoting endothelial-to-mesenchymal transition. J. Cell Physiol. 236, 495–506. doi:10.1002/jcp.29877
Chen, M., Zhu, H., Zhu, Q., Wu, X., Zhou, Y., Gao, R., et al. (2022c). Citri Reticulatae Pericarpium alleviates postmyocardial infarction heart failure by upregulating PPARγ expression. Clin. Exp. Pharmacol. Physiol. 49, 661–673. doi:10.1111/1440-1681.13642
Chen, P., Pei, J., Wang, X., Tai, S., Tang, L., and Hu, X. (2022d). Gut bacterial metabolite Urolithin A inhibits myocardial fibrosis through activation of Nrf2 pathway in vitro and in vivo. Mol. Med. 28, 19. doi:10.1186/s10020-022-00444-1
Chen, R., Chen, W., Huang, X., and Rui, Q. (2021b). Tanshinone IIA attenuates heart failure via inhibiting oxidative stress in myocardial infarction rats. Mol. Med. Rep. 23, 404. doi:10.3892/mmr.2021.12043
Chen, T. J., Yeh, Y. T., Peng, F. S., Li, A. H., and Wu, S. C. (2021c). S100A8/A9 enhances immunomodulatory and tissue-repairing properties of human amniotic mesenchymal stem cells in myocardial ischemia-reperfusion injury. Int. J. Mol. Sci. 22, 11175. doi:10.3390/ijms222011175
Chen, X., Ding, Z., Li, T., Jiang, W., Zhang, J., and Deng, X. (2020c). MicroR-26b targets high mobility group, AT-hook 2 to ameliorate myocardial infarction-induced fibrosis by suppression of cardiac fibroblasts activation. Curr. Neurovasc Res. 17, 204–213. doi:10.2174/1567202617666200506101258
Chen, X., Wan, W., Guo, Y., Ye, T., Fo, Y., Sun, Y., et al. (2021d). Pinocembrin ameliorates post-infarct heart failure through activation of Nrf2/HO-1 signaling pathway. Mol. Med. 27, 100. doi:10.1186/s10020-021-00363-7
Cheng, X., Wang, L., Wen, X., Gao, L., Li, G., Chang, G., et al. (2021). TNAP is a novel regulator of cardiac fibrosis after myocardial infarction by mediating TGF-β/Smads and ERK1/2 signaling pathways. EBioMedicine 67, 103370. doi:10.1016/j.ebiom.2021.103370
Cheng, Y., He, Q., Li, N., and Luo, M. (2022). Activation of PTEN/P13K/AKT signaling pathway by miRNA-124-3p-loaded nanoparticles to regulate oxidative stress attenuates cardiomyocyte regulation and myocardial injury. Oxid. Med. Cell Longev. 2022, 8428596. doi:10.1155/2022/8428596
Cheng, Y., Luo, D., Zhao, Y., and Rong, J. (2020). N-Propargyl caffeate amide (PACA) prevents cardiac fibrosis in experimental myocardial infarction by promoting pro-resolving macrophage polarization. Aging (Albany NY) 12, 5384–5398. doi:10.18632/aging.102959
Chiang, M. H., Liang, C. J., Lin, L. C., Yang, Y. F., Huang, C. C., Chen, Y. H., et al. (2020). miR-26a attenuates cardiac apoptosis and fibrosis by targeting ataxia-telangiectasia mutated in myocardial infarction. J. Cell Physiol. 235, 6085–6102. doi:10.1002/jcp.29537
Cho, J., Kim, S., Lee, H., Rah, W., Cho, H. C., Kim, N. K., et al. (2021). Regeneration of infarcted mouse hearts by cardiovascular tissue formed via the direct reprogramming of mouse fibroblasts. Nat. Biomed. Eng. 5, 880–896. doi:10.1038/s41551-021-00783-0
Chong, S. Y., Zharkova, O., Yatim, S., Wang, X., Lim, X. C., Huang, C., et al. (2021). Tissue factor cytoplasmic domain exacerbates post-infarct left ventricular remodeling via orchestrating cardiac inflammation and angiogenesis. Theranostics 11, 9243–9261. doi:10.7150/thno.63354
Chun, P. (2020). Therapeutic effects of histone deacetylase inhibitors on heart disease. Arch. Pharm. Res. 43, 1276–1296. doi:10.1007/s12272-020-01297-0
Chute, M., Aujla, P., Jana, S., and Kassiri, Z. (2019). The non-fibrillar side of fibrosis: Contribution of the basement membrane, proteoglycans, and glycoproteins to myocardial fibrosis. J. Cardiovasc Dev. Dis. 6, 35. doi:10.3390/jcdd6040035
Chute, M., Aujla, P. K., Li, Y., Jana, S., Zhabyeyev, P., Rasmuson, J., et al. (2022). ADAM15 is required for optimal collagen cross-linking and scar formation following myocardial infarction. Matrix Biol. 105, 127–143. doi:10.1016/j.matbio.2021.12.002
Cui, S., Liu, Z., Tao, B., Fan, S., Pu, Y., Meng, X., et al. (2021). miR-145 attenuates cardiac fibrosis through the AKT/GSK-3β/β-catenin signaling pathway by directly targeting SOX9 in fibroblasts. J. Cell Biochem. 122, 209–221. doi:10.1002/jcb.29843
Dai, Y., Chen, Y., Wei, G., Zha, L., and Li, X. (2021). Ivabradine protects rats against myocardial infarction through reinforcing autophagy via inhibiting PI3K/AKT/mTOR/p70S6K pathway. Bioengineered 12, 1826–1837. doi:10.1080/21655979.2021.1925008
Dalal, S., Shook, P. L., Singh, M., and Singh, K. (2023). Post-ischemic cardioprotective potential of exogenous ubiquitin in myocardial remodeling late after ischemia/reperfusion injury. Life Sci. 312, 121216. doi:10.1016/j.lfs.2022.121216
Dattagupta, A., and Immaneni, S. (2018). ST2: Current status. Indian Heart J. 70 (1), S96–s101. doi:10.1016/j.ihj.2018.03.001
Daud, E., Ertracht, O., Bandel, N., Moady, G., Shehadeh, M., Reuveni, T., et al. (2021). The impact of empagliflozin on cardiac physiology and fibrosis early after myocardial infarction in non-diabetic rats. Cardiovasc Diabetol. 20, 132. doi:10.1186/s12933-021-01322-6
DE Boer, R. A., DE Keulenaer, G., Bauersachs, J., Brutsaert, D., Cleland, J. G., Diez, J., et al. (2019). Towards better definition, quantification and treatment of fibrosis in heart failure. A scientific roadmap by the Committee of Translational Research of the Heart Failure Association (HFA) of the European Society of Cardiology. Eur. J. Heart Fail 21, 272–285. doi:10.1002/ejhf.1406
DE Geest, B., and Mishra, M. (2021). Role of high-density lipoproteins in cardioprotection and in reverse remodeling: Therapeutic implications. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1866, 159022. doi:10.1016/j.bbalip.2021.159022
Dedkov, E. I. (2021). Large- and medium-sized arteries remaining in transmural scar distal to permanent coronary ligation undergo neointimal hyperplasia and inward remodeling. J. Histochem Cytochem 69, 321–338. doi:10.1369/00221554211004297
Dehghani, T., Thai, P. N., Sodhi, H., Ren, L., Sirish, P., Nader, C. E., et al. (2022). Selectin-targeting glycosaminoglycan-peptide conjugate limits neutrophil-mediated cardiac reperfusion injury. Cardiovasc Res. 118, 267–281. doi:10.1093/cvr/cvaa312
Dempsey, S., Jafari, P., So, A., and Samani, A. (2020). A composite material based neural network for tissue mechanical properties estimation toward stage assessment of infarction. Annu. Int. Conf. IEEE Eng. Med. Biol. Soc. 2020, 2800–2803. doi:10.1109/EMBC44109.2020.9176151
Devarakonda, T., Valle Raleigh, J., Mauro, A. G., Lambert, J. M., Cowart, L. A., and Salloum, F. N. (2022). Chronic treatment with serelaxin mitigates adverse remodeling in a murine model of ischemic heart failure and modulates bioactive sphingolipid signaling. Sci. Rep. 12, 8897. doi:10.1038/s41598-022-12930-x
Dewald, O., Ren, G., Duerr, G. D., Zoerlein, M., Klemm, C., Gersch, C., et al. (2004). Of mice and dogs: Species-specific differences in the inflammatory response following myocardial infarction. Am. J. Pathol. 164, 665–677. doi:10.1016/S0002-9440(10)63154-9
Dick, S. A., Macklin, J. A., Nejat, S., Momen, A., Clemente-Casares, X., Althagafi, M. G., et al. (2019). Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat. Immunol. 20, 29–39. doi:10.1038/s41590-018-0272-2
Ding, J., Zhang, Z., Li, S., Wang, W., DU, T., Fang, Q., et al. (2022). Mdivi-1 alleviates cardiac fibrosis post myocardial infarction at infarcted border zone, possibly via inhibition of Drp1-Activated mitochondrial fission and oxidative stress. Arch. Biochem. Biophys. 718, 109147. doi:10.1016/j.abb.2022.109147
Djenoune, L., Berg, K., Brueckner, M., and Yuan, S. (2022). A change of heart: New roles for cilia in cardiac development and disease. Nat. Rev. Cardiol. 19, 211–227. doi:10.1038/s41569-021-00635-z
Dobaczewski, M., Xia, Y., Bujak, M., Gonzalez-Quesada, C., and Frangogiannis, N. G. (2010). CCR5 signaling suppresses inflammation and reduces adverse remodeling of the infarcted heart, mediating recruitment of regulatory T cells. Am. J. Pathol. 176, 2177–2187. doi:10.2353/ajpath.2010.090759
Dong, J., Zhu, W., and Wan, D. (2021a). Downregulation of microRNA-21-5p from macrophages-derived exosomes represses ventricular remodeling after myocardial infarction via inhibiting tissue inhibitors of metalloproteinase 3. Int. Immunopharmacol. 96, 107611. doi:10.1016/j.intimp.2021.107611
Dong, Q., Wen, X., Chang, G., Xia, R., Wang, S., Yang, Y., et al. (2021b). ST-segment resolution as a marker for severe myocardial fibrosis in ST-segment elevation myocardial infarction. BMC Cardiovasc Disord. 21, 455. doi:10.1186/s12872-021-02269-y
Dufeys, C., Daskalopoulos, E. P., Castanares-Zapatero, D., Conway, S. J., Ginion, A., Bouzin, C., et al. (2021). AMPKα1 deletion in myofibroblasts exacerbates post-myocardial infarction fibrosis by a connexin 43 mechanism. Basic Res. Cardiol. 116, 10. doi:10.1007/s00395-021-00846-y
Dugaucquier, L., Feyen, E., Mateiu, L., Bruyns, T. A. M., DE Keulenaer, G. W., and Segers, V. F. M. (2020). The role of endothelial autocrine NRG1/ERBB4 signaling in cardiac remodeling. Am. J. Physiol. Heart Circ. Physiol. 319, H443–h455. doi:10.1152/ajpheart.00176.2020
Dupuy, A. M., Kuster, N., Curinier, C., Huet, F., Plawecki, M., Solecki, K., et al. (2019). Exploring collagen remodeling and regulation as prognosis biomarkers in stable heart failure. Clin. Chim. Acta 490, 167–171. doi:10.1016/j.cca.2018.08.042
Eguchi, A., Coleman, R., Gresham, K., Gao, E., Ibetti, J., Chuprun, J. K., et al. (2021). GRK5 is a regulator of fibroblast activation and cardiac fibrosis. Proc. Natl. Acad. Sci. U. S. A. 118, e2012854118. doi:10.1073/pnas.2012854118
Eid, R. A., Alharbi, S. A., EL-Kott, A. F., Eleawa, S. M., Zaki, M. S. A., EL-Sayed, F., et al. (2020). Exendin-4 ameliorates cardiac remodeling in experimentally induced myocardial infarction in rats by inhibiting PARP1/NF-κB Axis in A SIRT1-dependent mechanism. Cardiovasc Toxicol. 20, 401–418. doi:10.1007/s12012-020-09567-5
Elkenhans, B., Protti, A., Shah, A., Onthank, D., and Botnar, R. (2021). Visualization of elastin using cardiac magnetic resonance imaging after myocardial infarction as inflammatory response. Sci. Rep. 11, 11004. doi:10.1038/s41598-021-90092-y
Emran, T., Chowdhury, N. I., Sarker, M., Bepari, A. K., Hossain, M., Rahman, G. M. S., et al. (2021). L-carnitine protects cardiac damage by reducing oxidative stress and inflammatory response via inhibition of tumor necrosis factor-alpha and interleukin-1beta against isoproterenol-induced myocardial infarction. Biomed. Pharmacother. 143, 112139. doi:10.1016/j.biopha.2021.112139
Fan, D., Takawale, A., Lee, J., and Kassiri, Z. (2012). Cardiac fibroblasts, fibrosis and extracellular matrix remodeling in heart disease. Fibrogenes. Tissue Repair 5, 15. doi:10.1186/1755-1536-5-15
Fan, Z., and Guan, J. (2016). Antifibrotic therapies to control cardiac fibrosis. Biomater. Res. 20, 13. doi:10.1186/s40824-016-0060-8
Farag, M. M., Khalifa, A. A., Elhadidy, W. F., and Rashad, R. M. (2021). Thymoquinone dose-dependently attenuates myocardial injury induced by isoproterenol in rats via integrated modulations of oxidative stress, inflammation, apoptosis, autophagy, and fibrosis. Naunyn Schmiedeb. Arch. Pharmacol. 394, 1787–1801. doi:10.1007/s00210-021-02087-1
Farbehi, N., Patrick, R., Dorison, A., Xaymardan, M., Janbandhu, V., Wystub-Lis, K., et al. (2019). Single-cell expression profiling reveals dynamic flux of cardiac stromal, vascular and immune cells in health and injury. Elife 8, e43882. doi:10.7554/eLife.43882
Farsangi, S. J., Rostamzadeh, F., Sheikholeslami, M., Jafari, E., and Karimzadeh, M. (2021). Modulation of the expression of long non-coding RNAs H19, GAS5, and MIAT by endurance exercise in the hearts of rats with myocardial infarction. Cardiovasc Toxicol. 21, 162–168. doi:10.1007/s12012-020-09607-0
Fassina, D., Costa, C. M., Longobardi, S., Karabelas, E., Plank, G., Harding, S. E., et al. (2022). Modelling the interaction between stem cells derived cardiomyocytes patches and host myocardium to aid non-arrhythmic engineered heart tissue design. PLoS Comput. Biol. 18, e1010030. doi:10.1371/journal.pcbi.1010030
Fearon, W. F., and Fearon, D. T. (2008). Inflammation and cardiovascular disease: Role of the interleukin-1 receptor antagonist. Circulation 117, 2577–2579. doi:10.1161/CIRCULATIONAHA.108.772491
Feliciano, R. D. S., Manchini, M. T., Atum, A. L. B., Da Silva, G. A., AntôNIO, E. L., Serra, A. J., et al. (2022). Photobiomodulation therapy's effects on cardiac fibrosis activation after experimental myocardial infarction. Lasers Surg. Med. 54, 883–894. doi:10.1002/lsm.23544
Feng, Y., Bao, Y., Ding, J., Li, H., Liu, W., Wang, X., et al. (2022). MicroRNA-130a attenuates cardiac fibrosis after myocardial infarction through TGF-β/Smad signaling by directly targeting TGF-β receptor 1. Bioengineered 13, 5779–5791. doi:10.1080/21655979.2022.2033380
Forte, E., Mclellan, M. A., Skelly, D. A., and Rosenthal, N. A. (2021a). Ex uno, plures-from one tissue to many cells: A review of single-cell transcriptomics in cardiovascular biology. Int. J. Mol. Sci. 22, 2071. doi:10.3390/ijms22042071
Forte, E., Perkins, B., Sintou, A., Kalkat, H. S., Papanikolaou, A., Jenkins, C., et al. (2021b). Cross-priming dendritic cells exacerbate immunopathology after ischemic tissue damage in the heart. Circulation 143, 821–836. doi:10.1161/CIRCULATIONAHA.120.044581
Forte, E., Skelly, D. A., Chen, M., Daigle, S., Morelli, K. A., Hon, O., et al. (2020). Dynamic interstitial cell response during myocardial infarction predicts resilience to rupture in genetically diverse mice. Cell Rep. 30, 3149–3163.e6. doi:10.1016/j.celrep.2020.02.008
Fraccarollo, D., Bauersachs, J., Kellner, M., Galuppo, P., and Ertl, G. (2002). Cardioprotection by long-term ET(A) receptor blockade and ACE inhibition in rats with congestive heart failure: Mono-versus combination therapy. Cardiovasc Res. 54, 85–94. doi:10.1016/s0008-6363(01)00553-3
Fraccarollo, D., Neuser, J., MöLLER, J., Riehle, C., Galuppo, P., and Bauersachs, J. (2021). Expansion of CD10(neg) neutrophils and CD14(+)HLA-DR(neg/low) monocytes driving proinflammatory responses in patients with acute myocardial infarction. Elife 10, e66808. doi:10.7554/eLife.66808
Frangogiannis, N. G. (2014). The inflammatory response in myocardial injury, repair, and remodelling. Nat. Rev. Cardiol. 11, 255–265. doi:10.1038/nrcardio.2014.28
French, B. A., and Holmes, J. W. (2019). Implications of scar structure and mechanics for post-infarction cardiac repair and regeneration. Exp. Cell Res. 376, 98–103. doi:10.1016/j.yexcr.2019.01.001
Fu, B., Wang, X., Chen, Z., Jiang, N., Guo, Z., Zhang, Y., et al. (2022a). Improved myocardial performance in infarcted rat heart by injection of disulfide-cross-linked chitosan hydrogels loaded with basic fibroblast growth factor. J. Mater Chem. B 10, 656–665. doi:10.1039/d1tb01961a
Fu, H., Shuai, W., Kong, B., Jiang, X., and Huang, H. (2021). MD1 depletion predisposes to ventricular arrhythmias in the setting of myocardial infarction. Heart Lung Circ. 30, 869–881. doi:10.1016/j.hlc.2020.09.938
Fu, Y., Shi, J., Qian, H., Qin, C., Liu, L., Shen, J., et al. (2022b). Alleviation of cardiac fibrosis using acellular peritoneal matrix-loaded pirfenidone nanodroplets after myocardial infarction in rats. Eur. J. Pharmacol. 933, 175238. doi:10.1016/j.ejphar.2022.175238
Gaibazzi, N., Suma, S., Lorenzoni, V., Sartorio, D., Pressman, G., Siniscalchi, C., et al. (2020). Myocardial scar by pulse-cancellation echocardiography is independently associated with appropriate defibrillator intervention for primary prevention after myocardial infarction. J. Am. Soc. Echocardiogr. 33, 1123–1131. doi:10.1016/j.echo.2020.04.020
Gajarsa, J. J., and Kloner, R. A. (2011). Left ventricular remodeling in the post-infarction heart: A review of cellular, molecular mechanisms, and therapeutic modalities. Heart Fail Rev. 16, 13–21. doi:10.1007/s10741-010-9181-7
Gallo, K. A., and Johnson, G. L. (2002). Mixed-lineage kinase control of JNK and p38 MAPK pathways. Nat. Rev. Mol. Cell Biol. 3, 663–672. doi:10.1038/nrm906
Gao, L., Mei, S., Zhang, S., Qin, Q., Li, H., Liao, Y., et al. (2020a). Cardio-renal exosomes in myocardial infarction serum regulate proangiogenic paracrine signaling in adipose mesenchymal stem cells. Theranostics 10, 1060–1073. doi:10.7150/thno.37678
Gao, L., Wang, L. Y., Liu, Z. Q., Jiang, D., Wu, S. Y., Guo, Y. Q., et al. (2020b). TNAP inhibition attenuates cardiac fibrosis induced by myocardial infarction through deactivating TGF-β1/Smads and activating P53 signaling pathways. Cell Death Dis. 11, 44. doi:10.1038/s41419-020-2243-4
Gao, R. F., Li, X., Xiang, H. Y., Yang, H., Lv, C. Y., Sun, X. L., et al. (2021). The covalent NLRP3-inflammasome inhibitor Oridonin relieves myocardial infarction induced myocardial fibrosis and cardiac remodeling in mice. Int. Immunopharmacol. 90, 107133. doi:10.1016/j.intimp.2020.107133
Garg, A., Foinquinos, A., Jung, M., Janssen-Peters, H., Biss, S., Bauersachs, J., et al. (2020). MiRNA-181a is a novel regulator of aldosterone-mineralocorticoid receptor-mediated cardiac remodelling. Eur. J. Heart Fail 22, 1366–1377. doi:10.1002/ejhf.1813
Ghali, R., Habeichi, N. J., Kaplan, A., Tannous, C., Abidi, E., Bekdash, A., et al. (2020). IL-33 induces type-2-cytokine phenotype but exacerbates cardiac remodeling post-myocardial infarction with eosinophil recruitment, worsened systolic dysfunction, and ventricular wall rupture. Clin. Sci. (Lond) 134, 1191–1218. doi:10.1042/CS20200402
Ghanimati, R., Rajabi, H., Ramezani, F., Ramez, M., Bapiran, M., and Nasirinezhad, F. (2020). The effect of preconditioning with high-intensity training on tissue levels of G-CSF, its receptor and C-kit after an acute myocardial infarction in male rats. BMC Cardiovasc Disord. 20, 75. doi:10.1186/s12872-020-01380-w
Ghosh, A. K., Chen, D. H., Guha, A., Mackenzie, S., Walker, J. M., and Roddie, C. (2020). CAR T cell therapy-related cardiovascular outcomes and management: Systemic disease or direct cardiotoxicity? JACC CardioOncol 2, 97–109. doi:10.1016/j.jaccao.2020.02.011
Gil, H., Goldshtein, M., Etzion, S., Elyagon, S., Hadad, U., Etzion, Y., et al. (2022). Defining the timeline of periostin upregulation in cardiac fibrosis following acute myocardial infarction in mice. Sci. Rep. 12, 21863.
Gladow, N., Hollmann, C., Ramos, G., Frantz, S., Kerkau, T., Beyersdorf, N., et al. (2020). Treatment of mice with a ligand binding blocking anti-CD28 monoclonal antibody improves healing after myocardial infarction. PLoS One 15, e0227734. doi:10.1371/journal.pone.0227734
Goerg, J., Sommerfeld, M., Greiner, B., Lauer, D., Seckin, Y., Kulikov, A., et al. (2021). Low-dose empagliflozin improves systolic heart function after myocardial infarction in rats: Regulation of MMP9, NHE1, and SERCA2a. Int. J. Mol. Sci. 22, 5437. doi:10.3390/ijms22115437
Goetze, J. P., Bruneau, B. G., Ramos, H. R., Ogawa, T., DE Bold, M. K., and DE Bold, A. J. (2020). Cardiac natriuretic peptides. Nat. Rev. Cardiol. 17, 698–717. doi:10.1038/s41569-020-0381-0
Gomez, R. A., and Sequeira-Lopez, M. L. S. (2018). Renin cells in homeostasis, regeneration and immune defence mechanisms. Nat. Rev. Nephrol. 14, 231–245. doi:10.1038/nrneph.2017.186
Goto, T., Miyagawa, S., Tamai, K., Matsuura, R., Kido, T., Kuratani, T., et al. (2020). High-mobility group box 1 fragment suppresses adverse post-infarction remodeling by recruiting PDGFRα-positive bone marrow cells. PLoS One 15, e0230392. doi:10.1371/journal.pone.0230392
Groenewegen, A., Rutten, F. H., Mosterd, A., and Hoes, A. W. (2020). Epidemiology of heart failure. Eur. J. Heart Fail 22, 1342–1356. doi:10.1002/ejhf.1858
Guo, Y., Yan, B., Tai, S., Zhou, S., and Zheng, X. L. (2021). PCSK9: Associated with cardiac diseases and their risk factors? Arch. Biochem. Biophys. 704, 108717. doi:10.1016/j.abb.2020.108717
Gupta, S., Ge, Y., Singh, A., GräNI, C., and Kwong, R. Y. (2021). Multimodality imaging assessment of myocardial fibrosis. JACC Cardiovasc Imaging 14, 2457–2469. doi:10.1016/j.jcmg.2021.01.027
Halade, G. V., Norris, P. C., Kain, V., Serhan, C. N., and Ingle, K. A. (2018). Splenic leukocytes define the resolution of inflammation in heart failure. Sci. Signal 11, eaao1818. doi:10.1126/scisignal.aao1818
Hall, C., Gehmlich, K., Denning, C., and Pavlovic, D. (2021). Complex relationship between cardiac fibroblasts and cardiomyocytes in health and disease. J. Am. Heart Assoc. 10, e019338. doi:10.1161/JAHA.120.019338
Han, X., Yang, Y., Zhang, M., Li, L., Xue, Y., Jia, Q., et al. (2022). Liquiritin protects against cardiac fibrosis after myocardial infarction by inhibiting CCL5 expression and the NF-κB signaling pathway. Drug Des. Devel Ther. 16, 4111–4125. doi:10.2147/DDDT.S386805
Han, X., Zhao, Z. A., Yan, S., Lei, W., Wu, H., Lu, X. A., et al. (2020). CXADR-like membrane protein protects against heart injury by preventing excessive pyroptosis after myocardial infarction. J. Cell Mol. Med. 24, 13775–13788. doi:10.1111/jcmm.15955
Hao, H., Li, X., Li, Q., Lin, H., Chen, Z., Xie, J., et al. (2016). FGF23 promotes myocardial fibrosis in mice through activation of β-catenin. Oncotarget 7, 64649–64664. doi:10.18632/oncotarget.11623
Hao, M., and Jiao, K. (2022). Jatrorrhizine reduces myocardial infarction-induced apoptosis and fibrosis through inhibiting p53 and TGF-β1/Smad2/3 pathways in mice. Acta Cir. Bras. 37, e370705. doi:10.1590/acb370705
Hao, S., Sui, X., Wang, J., Zhang, J., Pei, Y., Guo, L., et al. (2021). Secretory products from epicardial adipose tissue induce adverse myocardial remodeling after myocardial infarction by promoting reactive oxygen species accumulation. Cell Death Dis. 12, 848. doi:10.1038/s41419-021-04111-x
Haryono, A., Ramadhiani, R., Ryanto, G. R. T., and Emoto, N. (2022). Endothelin and the cardiovascular system: The long journey and where we are going. Biol. (Basel) 11.
Hata, A., and Chen, Y. G. (2016). TGF-Β signaling from receptors to smads. Cold Spring Harb. Perspect. Biol. 8, a022061. doi:10.1101/cshperspect.a022061
Hayes, J. D., and Dinkova-Kostova, A. T. (2014). The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 39, 199–218. doi:10.1016/j.tibs.2014.02.002
He, J., Chen, Y., Huang, Y., Yao, F., Wu, Z., Chen, S., et al. (2009). Effect of long-term B-type natriuretic peptide treatment on left ventricular remodeling and function after myocardial infarction in rats. Eur. J. Pharmacol. 602, 132–137. doi:10.1016/j.ejphar.2008.10.064
He, W., Chen, P., Chen, Q., Cai, Z., and Zhang, P. (2022). Cytokine storm: Behind the scenes of the collateral circulation after acute myocardial infarction. Inflamm. Res. 71, 1143–1158. doi:10.1007/s00011-022-01611-0
He, Y., Sun, Z., Jiang, J., Yin, X., Han, J., Zhang, Y., et al. (2023). Spinal cord stimulation attenuates neural remodeling, inflammation, and fibrosis after myocardial infarction. Neuromodulation 26, 57–67. doi:10.1016/j.neurom.2021.09.005
Heinrichs, M., Ashour, D., Siegel, J., BüCHNER, L., Wedekind, G., Heinze, K. G., et al. (2021). The healing myocardium mobilizes a distinct B-cell subset through a CXCL13-CXCR5-dependent mechanism. Cardiovasc Res. 117, 2664–2676. doi:10.1093/cvr/cvab181
Hess, A., Borchert, T., Ross, T. L., Bengel, F. M., and Thackeray, J. T. (2022). Characterizing the transition from immune response to tissue repair after myocardial infarction by multiparametric imaging. Basic Res. Cardiol. 117, 14. doi:10.1007/s00395-022-00922-x
Hill, M. A., Jaisser, F., and Sowers, J. R. (2022). Role of the vascular endothelial sodium channel activation in the Genesis of pathologically increased cardiovascular stiffness. Cardiovasc Res. 118, 130–140. doi:10.1093/cvr/cvaa326
Hobby, A. R. H., Berretta, R. M., Eaton, D. M., Kubo, H., Feldsott, E., Yang, Y., et al. (2021). Cortical bone stem cells modify cardiac inflammation after myocardial infarction by inducing a novel macrophage phenotype. Am. J. Physiol. Heart Circ. Physiol. 321, H684–h701. doi:10.1152/ajpheart.00304.2021
Hodges, M. M., Zgheib, C., Xu, J., Hu, J., Dewberry, L. C., Hilton, S. A., et al. (2019). Differential expression of transforming growth factor-β1 is associated with fetal regeneration after myocardial infarction. Ann. Thorac. Surg. 108, 59–66. doi:10.1016/j.athoracsur.2018.12.042
Hoffman, K. A., Reynolds, C., Bottazzi, M. E., Hotez, P., and Jones, K. (2019). Improved biomarker and imaging analysis for characterizing progressive cardiac fibrosis in a mouse model of chronic chagasic cardiomyopathy. J. Am. Heart Assoc. 8, e013365. doi:10.1161/JAHA.119.013365
Hoffmann, D. B., Fraccarollo, D., Galuppo, P., Frantz, S., Bauersachs, J., and Tillmanns, J. (2021). Genetic ablation of fibroblast activation protein alpha attenuates left ventricular dilation after myocardial infarction. PLoS One 16, e0248196. doi:10.1371/journal.pone.0248196
Hofmann, U., and Frantz, S. (2015). Role of lymphocytes in myocardial injury, healing, and remodeling after myocardial infarction. Circ. Res. 116, 354–367. doi:10.1161/CIRCRESAHA.116.304072
Holtackers, R. J., Emrich, T., Botnar, R. M., Kooi, M. E., Wildberger, J. E., and Kreitner, K. F. (2022). Late gadolinium enhancement cardiac magnetic resonance imaging: From basic concepts to emerging methods. Rofo 194, 491–504. doi:10.1055/a-1718-4355
Holtackers, R. J., VAN DE Heyning, C. M., Chiribiri, A., Wildberger, J. E., Botnar, R. M., and Kooi, M. E. (2021). Dark-blood late gadolinium enhancement cardiovascular magnetic resonance for improved detection of subendocardial scar: A review of current techniques. J. Cardiovasc Magn. Reson 23, 96. doi:10.1186/s12968-021-00777-6
Hsu, W. T., Tseng, Y. H., Jui, H. Y., Kuo, C. C., Wu, K. K., and Lee, C. M. (2021). 5-Methoxytryptophan attenuates postinfarct cardiac injury by controlling oxidative stress and immune activation. J. Mol. Cell Cardiol. 158, 101–114. doi:10.1016/j.yjmcc.2021.05.014
Hu, G., Ding, X., Gao, F., and Li, J. (2022a). Calcium and integrin binding protein 1 (CIB1) induces myocardial fibrosis in myocardial infarction via regulating the PI3K/Akt pathway. Exp. Anim. 71, 1–13. doi:10.1538/expanim.21-0063
Hu, S., Gao, Y., Gao, R., Wang, Y., Qu, Y., Yang, J., et al. (2022b). The selective STING inhibitor H-151 preserves myocardial function and ameliorates cardiac fibrosis in murine myocardial infarction. Int. Immunopharmacol. 107, 108658. doi:10.1016/j.intimp.2022.108658
Hu, S., Liu, H., Hu, Z., Li, L., and Yang, Y. (2020). Follistatin-like 1: A dual regulator that promotes cardiomyocyte proliferation and fibrosis. J. Cell Physiol. 235, 5893–5902. doi:10.1002/jcp.29588
Huang, S., Chen, B., Humeres, C., Alex, L., Hanna, A., and Frangogiannis, N. G. (2020). The role of Smad2 and Smad3 in regulating homeostatic functions of fibroblasts in vitro and in adult mice. Biochim. Biophys. Acta Mol. Cell Res. 1867, 118703. doi:10.1016/j.bbamcr.2020.118703
Huang, Y., Li, L., Chen, H., Liao, Q., Yang, X., Yang, D., et al. (2021). The protective role of yin-yang 1 in cardiac injury and remodeling after myocardial infarction. J. Am. Heart Assoc. 10, e021895. doi:10.1161/JAHA.121.021895
Hubert, F., Payan, S. M., Pelce, E., Bouchard, L., Sturny, R., Lenfant, N., et al. (2022). FGF10 promotes cardiac repair through a dual cellular mechanism increasing cardiomyocyte renewal and inhibiting fibrosis. Cardiovasc Res. 118, 2625–2637. doi:10.1093/cvr/cvab340
Hume, R. D., Kanagalingam, S., Deshmukh, T., Chen, S., Mithieux, S. M., Rashid, F. N., et al. (2023). Tropoelastin improves post-infarct cardiac function. Circ. Res. 132, 72–86. doi:10.1161/CIRCRESAHA.122.321123
Humeres, C., and Frangogiannis, N. G. (2019). Fibroblasts in the infarcted, remodeling, and failing heart. JACC Basic Transl. Sci. 4, 449–467. doi:10.1016/j.jacbts.2019.02.006
Humeres, C., Shinde, A. V., Hanna, A., Alex, L., HernáNDEZ, S. C., Li, R., et al. (2022). Smad7 effects on TGF-β and ErbB2 restrain myofibroblast activation and protect from postinfarction heart failure. J. Clin. Invest 132, e146926. doi:10.1172/JCI146926
Huo, J., Lu, S., Kwong, J. Q., Bround, M. J., Grimes, K. M., Sargent, M. A., et al. (2020). MCUb induction protects the heart from postischemic remodeling. Circ. Res. 127, 379–390. doi:10.1161/CIRCRESAHA.119.316369
Ikushima, E., Ishikane, S., Kishigami, T., Matsunaga, H., Igawa, K., Tomooka, K., et al. (2022). 2,5-Dimethylcelecoxib attenuates cardiac fibrosis caused by cryoinjury-induced myocardial infarction by suppressing the fibroblast-to-myofibroblast transformation via inhibition of the TGF-β signaling pathway. Biochem. Pharmacol. 197, 114950. doi:10.1016/j.bcp.2022.114950
Imig, J. D., Cervenka, L., and Neckar, J. (2022). Epoxylipids and soluble epoxide hydrolase in heart diseases. Biochem. Pharmacol. 195, 114866. doi:10.1016/j.bcp.2021.114866
Isomi, M., Sadahiro, T., Fujita, R., Abe, Y., Yamada, Y., Akiyama, T., et al. (2021). Direct reprogramming with Sendai virus vectors repaired infarct hearts at the chronic stage. Biochem. Biophys. Res. Commun. 560, 87–92. doi:10.1016/j.bbrc.2021.04.121
Ivanes, F., Susen, S., Mouquet, F., Pigny, P., Cuilleret, F., SautièRE, K., et al. (2012). Aldosterone, mortality, and acute ischaemic events in coronary artery disease patients outside the setting of acute myocardial infarction or heart failure. Eur. Heart J. 33, 191–202. doi:10.1093/eurheartj/ehr176
Ivey, M. J., Kuwabara, J. T., Riggsbee, K. L., and Tallquist, M. D. (2019). Platelet-derived growth factor receptor-α is essential for cardiac fibroblast survival. Am. J. Physiol. Heart Circ. Physiol. 317, H330–h344. doi:10.1152/ajpheart.00054.2019
Iwamiya, T., Segard, B. D., Matsuoka, Y., and Imamura, T. (2020). Human cardiac fibroblasts expressing VCAM1 improve heart function in postinfarct heart failure rat models by stimulating lymphangiogenesis. PLoS One 15, e0237810. doi:10.1371/journal.pone.0237810
Janbandhu, V., Martin, E., Chapman, G., Dunwoodie, S. L., and Harvey, R. P. (2022). Quantitative 3D analysis and visualization of cardiac fibrosis by microcomputed tomography. Star. Protoc. 3, 101055. doi:10.1016/j.xpro.2021.101055
JáUREGUI, B., Soto-Iglesias, D., Penela, D., Acosta, J., FernáNDEZ-Armenta, J., Linhart, M., et al. (2022). Cardiovascular magnetic resonance determinants of ventricular arrhythmic events after myocardial infarction. Europace 24, 938–947. doi:10.1093/europace/euab275
Ji, J. J., Qian, L. L., Zhu, Y., Jiang, Y., Guo, J. Q., Wu, Y., et al. (2022a). Kallistatin/Serpina3c inhibits cardiac fibrosis after myocardial infarction by regulating glycolysis via Nr4a1 activation. Biochim. Biophys. Acta Mol. Basis Dis. 1868, 166441. doi:10.1016/j.bbadis.2022.166441
Ji, M., Li, Y., Liu, Y., and Ma, G. (2022b). Vaspin ameliorates cardiac remodeling by suppressing phosphoinositide 3-kinase/protein kinase B pathway to improve oxidative stress in heart failure rats. J. Cardiovasc Pharmacol. 80, 442–452. doi:10.1097/FJC.0000000000001291
Jiang, J., Gu, X., Wang, H., and Ding, S. (2021). Resveratrol improves cardiac function and left ventricular fibrosis after myocardial infarction in rats by inhibiting NLRP3 inflammasome activity and the TGF-β1/SMAD2 signaling pathway. PeerJ 9, e11501. doi:10.7717/peerj.11501
Jiang, L., Liang, J., Huang, W., Ma, J., Park, K. H., Wu, Z., et al. (2022). CRISPR activation of endogenous genes reprograms fibroblasts into cardiovascular progenitor cells for myocardial infarction therapy. Mol. Ther. 30, 54–74. doi:10.1016/j.ymthe.2021.10.015
Jiao, W., Hao, J., Xie, Y., Meng, M., and Gao, W. (2022). EZH2 mitigates the cardioprotective effects of mesenchymal stem cell-secreted exosomes against infarction via HMGA2-mediated PI3K/AKT signaling. BMC Cardiovasc Disord. 22, 95. doi:10.1186/s12872-022-02533-9
Jin, H., Liu, K., Huang, X., Huo, H., Mou, J., Qiao, Z., et al. (2022). Genetic lineage tracing of pericardial cavity macrophages in the injured heart. Circ. Res. 130, 1682–1697. doi:10.1161/CIRCRESAHA.122.320567
Kachanova, O., Lobov, A., and Malashicheva, A. (2022). The role of the notch signaling pathway in recovery of cardiac function after myocardial infarction. Int. J. Mol. Sci. 23, 12509. doi:10.3390/ijms232012509
Kali, A., Cokic, I., Tang, R. L., Yang, H. J., Sharif, B., MarbáN, E., et al. (2014). Determination of location, size, and transmurality of chronic myocardial infarction without exogenous contrast media by using cardiac magnetic resonance imaging at 3 T. Circ. Cardiovasc Imaging 7, 471–481. doi:10.1161/CIRCIMAGING.113.001541
Kang, S., Verma, S., Hassanabad, A. F., Teng, G., Belke, D. D., Dundas, J. A., et al. (2020). Direct effects of empagliflozin on extracellular matrix remodelling in human cardiac myofibroblasts: Novel translational clues to explain EMPA-REG OUTCOME results. Can. J. Cardiol. 36, 543–553. doi:10.1016/j.cjca.2019.08.033
Kangawa, K., Fukuda, A., Kubota, I., Hayashi, Y., Minamitake, Y., and Matsuo, H. (1984). Human atrial natriuretic polypeptides (hANP): Purification, structure synthesis and biological activity. J. Hypertens. Suppl. 2 (2), S321–S323.
Kanisicak, O., Khalil, H., Ivey, M. J., Karch, J., Maliken, B. D., Correll, R. N., et al. (2016). Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat. Commun. 7, 12260. doi:10.1038/ncomms12260
Kasama, S., Furuya, M., Toyama, T., Ichikawa, S., and Kurabayashi, M. (2008). Effect of atrial natriuretic peptide on left ventricular remodelling in patients with acute myocardial infarction. Eur. Heart J. 29, 1485–1494. doi:10.1093/eurheartj/ehn206
Kaur, K., Hadas, Y., Kurian, A. A., Żak, M. M., Yoo, J., Mahmood, A., et al. (2021). Direct reprogramming induces vascular regeneration post muscle ischemic injury. Mol. Ther. 29, 3042–3058. doi:10.1016/j.ymthe.2021.07.014
Ke, X., Li, M., Wang, X., Liang, J., Wang, X., Wu, S., et al. (2020). An injectable chitosan/dextran/β -glycerophosphate hydrogel as cell delivery carrier for therapy of myocardial infarction. Carbohydr. Polym. 229, 115516. doi:10.1016/j.carbpol.2019.115516
Kensler, T. W., Wakabayashi, N., and Biswal, S. (2007). Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu. Rev. Pharmacol. Toxicol. 47, 89–116. doi:10.1146/annurev.pharmtox.46.120604.141046
Kessler, L., Kupusovic, J., Ferdinandus, J., Hirmas, N., Umutlu, L., Zarrad, F., et al. (2021). Visualization of fibroblast activation after myocardial infarction using 68Ga-fapi PET. Clin. Nucl. Med. 46, 807–813. doi:10.1097/RLU.0000000000003745
Kilci, H., AltıNBILEK, E., Çetinkal, G., SıĞıRCı, S., KoçAŞ, B. B., YıLDıZ, S. S., et al. (2021). Relation of a novel fibrosis marker and post-myocardial infarction left ventricular ejection fraction in revascularized patients. Biomark. Med. 15, 1651–1658. doi:10.2217/bmm-2021-0003
Kino, T., Khan, M., and Mohsin, S. (2020). The regulatory role of T cell responses in cardiac remodeling following myocardial infarction. Int. J. Mol. Sci. 21, 5013. doi:10.3390/ijms21145013
Kitsuka, T., Shiraki, A., Oyama, J. I., Nakagami, H., Tanaka, A., and Node, K. (2022). A novel soluble epoxide hydrolase vaccine protects murine cardiac muscle against myocardial infarction. Sci. Rep. 12, 6923. doi:10.1038/s41598-022-10641-x
Klapper-Goldstein, H., Verma, A., Elyagon, S., Gillis, R., Murninkas, M., Pittala, S., et al. (2020). VDAC1 in the diseased myocardium and the effect of VDAC1-interacting compound on atrial fibrosis induced by hyperaldosteronism. Sci. Rep. 10, 22101. doi:10.1038/s41598-020-79056-w
Kleinert, H. D. (1996). Hemodynamic effects of renin inhibitors. Am. J. Nephrol. 16, 252–260. doi:10.1159/000169005
Kobusiak-Prokopowicz, M., Orzeszko, J., Mazur, G., Mysiak, A., Orda, A., Poreba, R., et al. (2007). Chemokines and left ventricular function in patients with acute myocardial infarction. Eur. J. Intern Med. 18, 288–294. doi:10.1016/j.ejim.2007.02.001
Kompa, A. R., Greening, D. W., Kong, A. M., Mcmillan, P. J., Fang, H., Saxena, R., et al. (2021). Sustained subcutaneous delivery of secretome of human cardiac stem cells promotes cardiac repair following myocardial infarction. Cardiovasc Res. 117, 918–929. doi:10.1093/cvr/cvaa088
Kruszewska, J., Cudnoch-Jedrzejewska, A., and Czarzasta, K. (2022). Remodeling and fibrosis of the cardiac muscle in the course of obesity-pathogenesis and involvement of the extracellular matrix. Int. J. Mol. Sci. 23, 4195. doi:10.3390/ijms23084195
Kubota, A., Suto, A., Suga, K., Iwata, A., Tanaka, S., Suzuki, K., et al. (2021). Inhibition of Interleukin-21 prolongs the survival through the promotion of wound healing after myocardial infarction. J. Mol. Cell Cardiol. 159, 48–61. doi:10.1016/j.yjmcc.2021.06.006
Kuhn, M. (2016). Molecular physiology of membrane guanylyl cyclase receptors. Physiol. Rev. 96, 751–804. doi:10.1152/physrev.00022.2015
Kuwabara, J. T., Hara, A., Bhutada, S., Gojanovich, G. S., Chen, J., Hokutan, K., et al. (2022). Consequences of PDGFRα(+) fibroblast reduction in adult murine hearts. Elife 11, e69854. doi:10.7554/eLife.69854
Landry, N. M., Rattan, S. G., Filomeno, K. L., Meier, T. W., Meier, S. C., Foran, S. J., et al. (2021). SKI activates the Hippo pathway via LIMD1 to inhibit cardiac fibroblast activation. Basic Res. Cardiol. 116, 25. doi:10.1007/s00395-021-00865-9
Lang, M., Ou, D., Liu, Z., Li, Y., Zhang, X., and Zhang, F. (2021). LncRNA MHRT promotes cardiac fibrosis via miR-3185 pathway following myocardial infarction. Int. Heart J. 62, 891–899. doi:10.1536/ihj.20-298
Langer, L. B. N., Hess, A., Korkmaz, Z., Tillmanns, J., Reffert, L. M., Bankstahl, J. P., et al. (2021). Molecular imaging of fibroblast activation protein after myocardial infarction using the novel radiotracer [(68)Ga]MHLL1. Theranostics 11, 7755–7766. doi:10.7150/thno.51419
Lantz, C., Becker, A., and Thorp, E. B. (2021). Can polarization of macrophage metabolism enhance cardiac regeneration? J. Mol. Cell Cardiol. 160, 87–96. doi:10.1016/j.yjmcc.2021.07.003
Leader, C. J., Wilkins, G. T., and Walker, R. J. (2021). The effect of spironolactone on cardiac and renal fibrosis following myocardial infarction in established hypertension in the transgenic Cyp1a1Ren2 rat. PLoS One 16, e0260554. doi:10.1371/journal.pone.0260554
Leancă, S. A., Crișu, D., Petriș, A. O., AfrăsâNIE, I., Genes, A., Costache, A. D., et al. (2022). Left ventricular remodeling after myocardial infarction: From physiopathology to treatment. Life 12.
Lee, T. L., Lai, T. C., Lin, S. R., Lin, S. W., Chen, Y. C., Pu, C. M., et al. (2021). Conditioned medium from adipose-derived stem cells attenuates ischemia/reperfusion-induced cardiac injury through the microRNA-221/222/PUMA/ETS-1 pathway. Theranostics 11, 3131–3149. doi:10.7150/thno.52677
Lee, T. M., Harn, H. J., Chiou, T. W., Chuang, M. H., Chen, C. H., Chuang, C. H., et al. (2020). Host pre-conditioning improves human adipose-derived stem cell transplantation in ageing rats after myocardial infarction: Role of NLRP3 inflammasome. J. Cell Mol. Med. 24, 12272–12284. doi:10.1111/jcmm.15403
Lei, Q., Yi, T., Li, H., Yan, Z., Lv, Z., Li, G., et al. (2020). Ubiquitin C-terminal hydrolase L1 (UCHL1) regulates post-myocardial infarction cardiac fibrosis through glucose-regulated protein of 78 kDa (GRP78). Sci. Rep. 10, 10604. doi:10.1038/s41598-020-67746-4
Leifheit-Nestler, M., Kirchhoff, F., Nespor, J., Richter, B., Soetje, B., Klintschar, M., et al. (2018). Fibroblast growth factor 23 is induced by an activated renin-angiotensin-aldosterone system in cardiac myocytes and promotes the pro-fibrotic crosstalk between cardiac myocytes and fibroblasts. Nephrol. Dial. Transpl. 33, 1722–1734. doi:10.1093/ndt/gfy006
Li, C. Y., Zhang, J. R., Li, X. X., Zhao, L., XI, H., Hu, W. N., et al. (2021a). Lefty1 ameliorates post-infarction fibrosis by suppressing p-smad2 and p-ERK1/2 signaling pathways. J. Cardiovasc Transl. Res. 14, 636–646. doi:10.1007/s12265-020-10089-2
Li, F., Li, L., Zhang, J., Yang, X., and Liu, Y. (2022a). Histone methyltransferase DOT1L mediates the TGF-β1/Smad3 signaling pathway through epigenetic modification of SYK in myocardial infarction. Hum. Cell 35, 98–110. doi:10.1007/s13577-021-00625-w
Li, F., Long, T. Y., Bi, S. S., Sheikh, S. A., and Zhang, C. L. (2020a). circPAN3 exerts a profibrotic role via sponging miR-221 through FoxO3/ATG7-activated autophagy in a rat model of myocardial infarction. Life Sci. 257, 118015. doi:10.1016/j.lfs.2020.118015
Li, F., Yang, Y., Xue, C., Tan, M., Xu, L., Gao, J., et al. (2020b). Zinc Finger Protein ZBTB20 protects against cardiac remodelling post-myocardial infarction via ROS-TNFα/ASK1/JNK pathway regulation. J. Cell Mol. Med. 24, 13383–13396. doi:10.1111/jcmm.15961
Li, G., Yang, J., Zhang, D., Wang, X., Han, J., and Guo, X. (2022b). Research progress of myocardial fibrosis and atrial fibrillation. Front. Cardiovasc Med. 9, 889706. doi:10.3389/fcvm.2022.889706
Li, J., Ding, H., Li, Y., Zhou, H., Wang, W., Mei, Y., et al. (2021b). Alarin alleviated cardiac fibrosis via attenuating oxidative stress in heart failure rats. Amino Acids 53, 1079–1089. doi:10.1007/s00726-021-03005-8
Li, J., Ge, F., Wuken, S., Jiao, S., Chen, P., Huang, M., et al. (2022c). Zerumbone, a humulane sesquiterpene from Syringa pinnatifolia, attenuates cardiac fibrosis by inhibiting of the TGF-β1/Smad signaling pathway after myocardial infarction in mice. Phytomedicine 100, 154078. doi:10.1016/j.phymed.2022.154078
Li, J., Gong, L., Zhang, R., Li, S., Yu, H., Liu, Y., et al. (2021c). Fibroblast growth factor 21 inhibited inflammation and fibrosis after myocardial infarction via EGR1. Eur. J. Pharmacol. 910, 174470. doi:10.1016/j.ejphar.2021.174470
Li, J., Li, R., Tuleta, I., Hernandez, S. C., Humeres, C., Hanna, A., et al. (2022d). The role of endogenous Smad7 in regulating macrophage phenotype following myocardial infarction. Faseb J. 36, e22400. doi:10.1096/fj.202101956RR
Li, J., Salvador, A. M., Li, G., Valkov, N., Ziegler, O., Yeri, A., et al. (2021d). Mir-30d regulates cardiac remodeling by intracellular and paracrine signaling. Circ. Res. 128, e1–e23. doi:10.1161/CIRCRESAHA.120.317244
Li, Q., and Verma, I. M. (2002). NF-kappaB regulation in the immune system. Nat. Rev. Immunol. 2, 725–734. doi:10.1038/nri910
Li, R., Liu, R., Yan, F., Zhuang, X., Shi, H., and Gao, X. (2020c). Inhibition of TRPA1 promotes cardiac repair in mice after myocardial infarction. J. Cardiovasc Pharmacol. 75, 240–249. doi:10.1097/FJC.0000000000000783
Li, S., Yokota, T., Wang, P., Ten Hoeve, J., Ma, F., LE, T. M., et al. (2022e). Cardiomyocytes disrupt pyrimidine biosynthesis in nonmyocytes to regulate heart repair. J. Clin. Invest 132, e149711. doi:10.1172/JCI149711
Li, T. Y., Su, W., Li, L. L., Zhao, X. G., Yang, N., Gai, J. X., et al. (2022f). Critical role of PAFR/YAP1 positive feedback loop in cardiac fibrosis. Acta Pharmacol. Sin. 43, 2862–2872. doi:10.1038/s41401-022-00903-9
Li, T., Zhuang, Y., Yang, W., Xie, Y., Shang, W., Su, S., et al. (2021e). Silencing of METTL3 attenuates cardiac fibrosis induced by myocardial infarction via inhibiting the activation of cardiac fibroblasts. Faseb J. 35, e21162. doi:10.1096/fj.201903169R
Li, X., Xiang, N., and Wang, Z. (2020d). Ginsenoside Rg2 attenuates myocardial fibrosis and improves cardiac function after myocardial infarction via AKT signaling pathway. Biosci. Biotechnol. Biochem. 84, 2199–2206. doi:10.1080/09168451.2020.1793292
Li, X. X., Mu, B., Li, X., and Bie, Z. D. (2022g). circCELF1 inhibits myocardial fibrosis by regulating the expression of DKK2 through FTO/m(6)A and miR-636. J. Cardiovasc Transl. Res. 15, 998–1009. doi:10.1007/s12265-022-10209-0
Li, X., Zhang, F., Zhou, H., Hu, Y., Guo, D., Fang, X., et al. (2020e). Interplay of TNF-α, soluble TNF receptors and oxidative stress in coronary chronic total occlusion of the oldest patients with coronary heart disease. Cytokine 125, 154836. doi:10.1016/j.cyto.2019.154836
Li, Y., Dong, M., Wang, Q., Kumar, S., Zhang, R., Cheng, W., et al. (2021f). HIMF deletion ameliorates acute myocardial ischemic injury by promoting macrophage transformation to reparative subtype. Basic Res. Cardiol. 116, 30. doi:10.1007/s00395-021-00867-7
Li, Y., Liu, M., Yi, J., Song, X., Zheng, X., Liu, D., et al. (2020f). Exogenous hydrogen sulfide inhibits apoptosis by regulating endoplasmic reticulum stress-autophagy axis and improves myocardial reconstruction after acute myocardial infarction. Acta Biochim. Biophys. Sin. (Shanghai) 52, 1325–1336. doi:10.1093/abbs/gmaa133
Li, Y., Qi, Q., Yang, W. C., Zhang, T. L., Lu, C. C., Yao, Y. J., et al. (2020g). Sphingosylphosphorylcholine alleviates hypoxia-caused apoptosis in cardiac myofibroblasts via CaM/p38/STAT3 pathway. Apoptosis 25, 853–863. doi:10.1007/s10495-020-01639-9
Li, Y., Qin, L., Bai, Q., Zhang, J., Chen, R., and Song, K. (2021g). CD100 modulates cytotoxicity of CD8(+) T cells in patients with acute myocardial infarction. BMC Immunol. 22, 13. doi:10.1186/s12865-021-00406-y
Liang, B., Zhang, X. X., Li, R., Zhu, Y. C., Tian, X. J., and Gu, N. (2022). Guanxin V alleviates acute myocardial infarction by restraining oxidative stress damage, apoptosis, and fibrosis through the TGF-β1 signalling pathway. Phytomedicine 100, 154077. doi:10.1016/j.phymed.2022.154077
Liang, Y., Xu, X., Li, Q., Deng, Y., Xie, M., Zheng, Y., et al. (2020). Chronic alcohol intake exacerbates cardiac dysfunction after myocardial infarction. Alcohol Alcohol 55, 524–530. doi:10.1093/alcalc/agaa055
Lima Correa, B., EL Harane, N., Desgres, M., Perotto, M., Alayrac, P., Guillas, C., et al. (2021). Extracellular vesicles fail to trigger the generation of new cardiomyocytes in chronically infarcted hearts. Theranostics 11, 10114–10124. doi:10.7150/thno.62304
Liu, A., Zhang, Y., Xun, S., Zhou, G., Lin, L., and Mei, Y. (2022). Fibroblast growth factor 12 attenuated cardiac remodeling via suppressing oxidative stress. Peptides 153, 170786. doi:10.1016/j.peptides.2022.170786
Liu, J. W., Liu, H. T., and Chen, L. (2021a). The therapeutic role of Slit2 in anti-fibrosis, anti-inflammation and anti-oxidative stress in rats with coronary heart disease. Cardiovasc Toxicol. 21, 973–983. doi:10.1007/s12012-021-09688-5
Liu, J., Yang, C., Liu, T., Deng, Z., Fang, W., Zhang, X., et al. (2020a). Eosinophils improve cardiac function after myocardial infarction. Nat. Commun. 11, 6396. doi:10.1038/s41467-020-19297-5
Liu, W., Sun, J., Guo, Y., Liu, N., Ding, X., Zhang, X., et al. (2020b). Calhex231 ameliorates myocardial fibrosis post myocardial infarction in rats through the autophagy-NLRP3 inflammasome pathway in macrophages. J. Cell Mol. Med. 24, 13440–13453. doi:10.1111/jcmm.15969
Liu, Y., Fan, Y., Li, J., Chen, M., Chen, A., Yang, D., et al. (2021b). Combination of LCZ696 and ACEI further improves heart failure and myocardial fibrosis after acute myocardial infarction in mice. Biomed. Pharmacother. 133, 110824. doi:10.1016/j.biopha.2020.110824
Loi, H., Kramar, S., Laborde, C., Marsal, D., Pizzinat, N., Cussac, D., et al. (2021). Metformin attenuates postinfarction myocardial fibrosis and inflammation in mice. Int. J. Mol. Sci. 22, 9393. doi:10.3390/ijms22179393
Long, F., Wang, Q., Yang, D., Zhu, M., Wang, J., Zhu, Y., et al. (2020). Targeting JMJD3 histone demethylase mediates cardiac fibrosis and cardiac function following myocardial infarction. Biochem. Biophys. Res. Commun. 528, 671–677. doi:10.1016/j.bbrc.2020.05.115
Lu, L., Ma, J., Sun, M., Wang, X., Gao, E., Lu, L., et al. (2020). Melatonin ameliorates MI-induced cardiac remodeling and apoptosis through a JNK/p53-Dependent mechanism in diabetes mellitus. Oxid. Med. Cell Longev. 2020, 1535201. doi:10.1155/2020/1535201
Lu, Y. G., Tan, H., Ma, Q., Li, X. X., Cui, J., Zhang, X., et al. (2021). SH2 domain-containing protein tyrosine phosphatase-2 (SHP-2) prevents cardiac remodeling after myocardial infarction through ERK/SMAD signaling pathway. Hum. Cell 34, 325–334. doi:10.1007/s13577-020-00430-x
Luo, S., Zhang, M., Wu, H., Ding, X., Li, D., Dong, X., et al. (2021). Sail: A new conserved anti-fibrotic lncRNA in the heart. Basic Res. Cardiol. 116, 15. doi:10.1007/s00395-021-00854-y
Lymperopoulos, A. (2018). Arrestins in the cardiovascular system: An update. Prog. Mol. Biol. Transl. Sci. 159, 27–57. doi:10.1016/bs.pmbts.2018.07.003
Lymperopoulos, A., Cora, N., Maning, J., Brill, A. R., and Sizova, A. (2021). Signaling and function of cardiac autonomic nervous system receptors: Insights from the GPCR signalling universe. Febs J. 288, 2645–2659. doi:10.1111/febs.15771
Lymperopoulos, A., Rengo, G., and Koch, W. J. (2007). Adrenal adrenoceptors in heart failure: Fine-tuning cardiac stimulation. Trends Mol. Med. 13, 503–511. doi:10.1016/j.molmed.2007.10.005
Ma, J., Ren, M., Li, J., Zheng, C., Chen, Q., and Ma, S. (2022). Danqi soft caspule prevents atrial fibrillation by ameliorating left atrial remodeling through inhibiting cardiac fibroblasts differentiation and function. Phytomedicine 101, 154134. doi:10.1016/j.phymed.2022.154134
Ma, Y., Kuang, Y., Bo, W., Liang, Q., Zhu, W., Cai, M., et al. (2021). Exercise training alleviates cardiac fibrosis through increasing fibroblast growth factor 21 and regulating TGF-β1-smad2/3-MMP2/9 signaling in mice with myocardial infarction. Int. J. Mol. Sci. 22, 12341. doi:10.3390/ijms222212341
Ma, Y. (2021). Role of neutrophils in cardiac injury and repair following myocardial infarction. Cells 10, 1676. doi:10.3390/cells10071676
Madonna, R., Pieragostino, D., Rossi, C., Guarnieri, S., Nagy, C. T., Giricz, Z., et al. (2020). Transplantation of telomerase/myocardin-co-expressing mesenchymal cells in the mouse promotes myocardial revascularization and tissue repair. Vasc. Pharmacol. 135, 106807. doi:10.1016/j.vph.2020.106807
Mahdiui, M. E., VAN DER Bijl, P., Abou, R., DE Paula Lustosa, R., VAN DER Geest, R., Ajmone Marsan, N., et al. (2021). Myocardial work, an echocardiographic measure of post myocardial infarct scar on contrast-enhanced cardiac magnetic resonance. Am. J. Cardiol. 151, 1–9. doi:10.1016/j.amjcard.2021.04.009
Majzner, R. G., and Mackall, C. L. (2018). Tumor antigen escape from CAR T-cell therapy. Cancer Discov. 8, 1219–1226. doi:10.1158/2159-8290.CD-18-0442
Mangali, S., Bhat, A., Dasari, D., Sriram, D., and Dhar, A. (2021). Inhibition of double stranded RNA dependent protein kinase (PKR) abrogates isoproterenol induced myocardial ischemia in vitro in cultured cardiomyocytes and in vivo in wistar rats. Eur. J. Pharmacol. 906, 174223. doi:10.1016/j.ejphar.2021.174223
Maric, D., Paterek, A., Delaunay, M., LóPEZ, I. P., Arambasic, M., and Diviani, D. (2021). A-kinase anchoring protein 2 promotes protection against myocardial infarction. Cells 10, 2861. doi:10.3390/cells10112861
Marsit, O., Clavel, M. A., Paquin, A., DeschêNES, V., Hadjadj, S., SéNéCHAL-Dumais, I., et al. (2022). Effects of cyproheptadine on mitral valve remodeling and regurgitation after myocardial infarction. J. Am. Coll. Cardiol. 80, 500–510. doi:10.1016/j.jacc.2022.05.025
Masurkar, N., Bouvet, M., Logeart, D., Jouve, C., Dramé, F., Claude, O., et al. (2023). Novel cardiokine GDF3 predicts adverse fibrotic remodeling after myocardial infarction. Circulation 147, 498–511. doi:10.1161/CIRCULATIONAHA.121.056272
Mehdipoor, M., Damirchi, A., Razavi Tousi, S. M. T., and Babaei, P. (2021). Concurrent vitamin D supplementation and exercise training improve cardiac fibrosis via TGF-β/Smad signaling in myocardial infarction model of rats. J. Physiol. Biochem. 77, 75–84. doi:10.1007/s13105-020-00778-6
Merino-Caviedes, S., Gutierrez, L. K., Alfonso-AlmazáN, J. M., Sanz-EstéBANEZ, S., Cordero-Grande, L., Quintanilla, J. G., et al. (2021). Time-efficient three-dimensional transmural scar assessment provides relevant substrate characterization for ventricular tachycardia features and long-term recurrences in ischemic cardiomyopathy. Sci. Rep. 11, 18722. doi:10.1038/s41598-021-97399-w
Mestas, J., and Hughes, C. C. (2004). Of mice and not men: Differences between mouse and human immunology. J. Immunol. 172, 2731–2738. doi:10.4049/jimmunol.172.5.2731
Methatham, T., Nagai, R., and Aizawa, K. (2022). A new hypothetical concept in metabolic understanding of cardiac fibrosis: Glycolysis combined with TGF-β and KLF5 signaling. Int. J. Mol. Sci. 23, 4302. doi:10.3390/ijms23084302
Michel, L., Korste, S., Spomer, A., Hendgen-Cotta, U. B., Rassaf, T., and Totzeck, M. (2022). PD1 deficiency modifies cardiac immunity during baseline conditions and in reperfused acute myocardial infarction. Int. J. Mol. Sci. 23, 7533. doi:10.3390/ijms23147533
Michel, L. Y. M., Farah, C., and Balligand, J. L. (2020). The Beta3 adrenergic receptor in healthy and pathological cardiovascular tissues. Cells 9, 2584. doi:10.3390/cells9122584
Mo, F., Luo, Y., Yan, Y., Li, J., Lai, S., and Wu, W. (2021). Are activated B cells involved in the process of myocardial fibrosis after acute myocardial infarction? An in vivo experiment. BMC Cardiovasc Disord. 21, 5. doi:10.1186/s12872-020-01775-9
Moheimani, H. R., Amiriani, T., Alizadeh, A. M., Jand, Y., Shakiba, D., Ensan, P. S., et al. (2021). Preconditioning and anti-apoptotic effects of Metformin and Cyclosporine-A in an isolated bile duct-ligated rat heart. Eur. J. Pharmacol. 893, 173807. doi:10.1016/j.ejphar.2020.173807
Mokhtari, B., Badalzadeh, R., and Aboutaleb, N. (2021). Modulation of autophagy as the target of mesenchymal stem cells-derived conditioned medium in rat model of myocardial ischemia/reperfusion injury. Mol. Biol. Rep. 48, 3337–3348. doi:10.1007/s11033-021-06359-0
Morfino, P., Aimo, A., Castiglione, V., GáLVEZ-MontóN, C., Emdin, M., and Bayes-Genis, A. (2022). Treatment of cardiac fibrosis: From neuro-hormonal inhibitors to CAR-T cell therapy. Heart Fail Rev. 28, 555–569. doi:10.1007/s10741-022-10279-x
Mouton, A. J., Flynn, E. R., Moak, S. P., Aitken, N. M., Omoto, A. C. M., Li, X., et al. (2021). Dimethyl fumarate preserves left ventricular infarct integrity following myocardial infarction via modulation of cardiac macrophage and fibroblast oxidative metabolism. J. Mol. Cell Cardiol. 158, 38–48. doi:10.1016/j.yjmcc.2021.05.008
Mouton, A. J., Ma, Y., Rivera Gonzalez, O. J., Daseke, M. J., 2Nd, Flynn, E. R., Freeman, T. C., et al. (2019). Fibroblast polarization over the myocardial infarction time continuum shifts roles from inflammation to angiogenesis. Basic Res. Cardiol. 114, 6. doi:10.1007/s00395-019-0715-4
MüHLFELD, C., and Schipke, J. (2022)., 11. Cells.Methodological progress of stereology in cardiac research and its application to normal and pathological heart development
MüLLER, B., Schneider, M., MüLLER, K. A. L., Lunov, O., Borst, O., Simmet, T., et al. (2021). Protective role of Gremlin-1 in myocardial function. Eur. J. Clin. Invest 51, e13539. doi:10.1111/eci.13539
Muslin, A. J. (2008). MAPK signalling in cardiovascular health and disease: Molecular mechanisms and therapeutic targets. Clin. Sci. (Lond) 115, 203–218. doi:10.1042/CS20070430
Nakayama, Y., Mukai, N., Wang, B. F., Yang, K., Patwari, P., Kitsis, R. N., et al. (2021). Txnip C247S mutation protects the heart against acute myocardial infarction. J. Mol. Cell Cardiol. 155, 36–49. doi:10.1016/j.yjmcc.2021.02.013
Nikolov, A., and Popovski, N. (2022). Extracellular matrix in heart disease: Focus on circulating collagen type I and III derived peptides as biomarkers of myocardial fibrosis and their potential in the prognosis of heart failure: A concise review. Metabolites 12, 297. doi:10.3390/metabo12040297
Nogami, M., Hoshi, T., Toukairin, Y., Arai, T., and Nishio, T. (2020). Immunohistochemistry of advanced glycation end product N(ε)-(carboxymethyl)lysine in coronary arteries in relation to cardiac fibrosis and serum N-terminal-pro basic natriuretic peptide in forensic autopsy cases. BMC Res. Notes 13, 239. doi:10.1186/s13104-020-05082-6
Ong, S. B., HernáNDEZ-ReséNDIZ, S., Crespo-Avilan, G. E., Mukhametshina, R. T., Kwek, X. Y., Cabrera-Fuentes, H. A., et al. (2018). Inflammation following acute myocardial infarction: Multiple players, dynamic roles, and novel therapeutic opportunities. Pharmacol. Ther. 186, 73–87. doi:10.1016/j.pharmthera.2018.01.001
Osokina, A., Karetnikova, V., Polikutina, O., Ivanova, A., Gruzdeva, O., Dyleva, Y., et al. (2021). Prognostic potential of cardiac structural and functional parameters and N-terminal propeptide of type III procollagen in predicting cardiac fibrosis one year after myocardial infarction with preserved left ventricular ejection fraction. Aging (Albany NY) 13, 194–203. doi:10.18632/aging.202495
Osokina, A. V., Karetnikova, V. N., Polikutina, O. M., Ivanova, A. V., Artemova, T. P., Ryzhenkova, S. N., et al. (2020). Dynamics of parameters of transmitral blood flow and markers of myocardial fibrosis in patients with myocardial infarction. Kardiologiia 60, 84–91. doi:10.18087/cardio.2020.6.n994
Pan, Y., Li, Q., Yan, H., Huang, J., and Wang, Z. (2020). Apela improves cardiac and renal function in mice with acute myocardial infarction. J. Cell Mol. Med. 24, 10382–10390. doi:10.1111/jcmm.15651
Pandey, K. N. (2018). Molecular and genetic aspects of guanylyl cyclase natriuretic peptide receptor-A in regulation of blood pressure and renal function. Physiol. Genomics 50, 913–928. doi:10.1152/physiolgenomics.00083.2018
Pei, J., Cai, L., Wang, F., Xu, C., Pei, S., Guo, H., et al. (2022). LPA(2) contributes to vascular endothelium homeostasis and cardiac remodeling after myocardial infarction. Circ. Res. 131, 388–403. doi:10.1161/CIRCRESAHA.122.321036
Peres Valgas Da Silva, C., Shettigar, V. K., Baer, L. A., Abay, E., Pinckard, K. M., Vinales, J., et al. (2022). Exercise training after myocardial infarction increases survival but does not prevent adverse left ventricle remodeling and dysfunction in high-fat diet fed mice. Life Sci. 311, 121181. doi:10.1016/j.lfs.2022.121181
Polacin, M., Karolyi, M., Eberhard, M., Matziris, I., Alkadhi, H., Kozerke, S., et al. (2022). Segmental strain for scar detection in acute myocardial infarcts and in follow-up exams using non-contrast CMR cine sequences. BMC Cardiovasc Disord. 22, 226. doi:10.1186/s12872-022-02664-z
Potz, B. A., Sabe, A. A., Sabe, S. A., Lawandy, I. J., Abid, M. R., Clements, R. T., et al. (2022). Calpain inhibition decreases myocardial fibrosis in chronically ischemic hypercholesterolemic swine. J. Thorac. Cardiovasc Surg. 163, e11–e27. doi:10.1016/j.jtcvs.2019.11.150
Prabhu, S. D., and Frangogiannis, N. G. (2016). The biological basis for cardiac repair after myocardial infarction: From inflammation to fibrosis. Circ. Res. 119, 91–112. doi:10.1161/CIRCRESAHA.116.303577
Pugliese, N. R., Masi, S., and Taddei, S. (2020). The renin-angiotensin-aldosterone system: A crossroad from arterial hypertension to heart failure. Heart Fail Rev. 25, 31–42. doi:10.1007/s10741-019-09855-5
Qian, M., Feng, Z. Q., Zheng, R. N., Hu, K. W., Sun, J. Z., Sun, H. B., et al. (2022). Qi-Tai-Suan, an oleanolic acid derivative, ameliorates ischemic heart failure via suppression of cardiac apoptosis, inflammation and fibrosis. Chin. J. Nat. Med. 20, 432–442. doi:10.1016/S1875-5364(22)60156-0
Qin, R., Zhao, Q., Han, B., Zhu, H. P., Peng, C., Zhan, G., et al. (2022). Indole-based small molecules as potential therapeutic agents for the treatment of fibrosis. Front. Pharmacol. 13, 845892. doi:10.3389/fphar.2022.845892
Qiu, F., Han, Y., Shao, X., Paulo, P., Li, W., Zhu, M., et al. (2020). Knockdown of endogenous RNF4 exacerbates ischaemia-induced cardiomyocyte apoptosis in mice. J. Cell Mol. Med. 24, 9545–9559. doi:10.1111/jcmm.15363
Raj, P., Sayfee, K., Parikh, M., Yu, L., Wigle, J., Netticadan, T., et al. (2021). Comparative and combinatorial effects of resveratrol and sacubitril/valsartan alongside valsartan on cardiac remodeling and dysfunction in MI-induced rats. Molecules 26, 5006. doi:10.3390/molecules26165006
Rakhshan, K., Sharifi, M., Ramezani, F., Azizi, Y., and Aboutaleb, N. (2022). ERK/HIF-1α/VEGF pathway: A molecular target of ELABELA (ELA) peptide for attenuating cardiac ischemia-reperfusion injury in rats by promoting angiogenesis. Mol. Biol. Rep. 49, 10509–10519. doi:10.1007/s11033-022-07818-y
Ramos, S. R., Pieles, G., Sun, M., Slorach, C., Hui, W., and Friedberg, M. K. (2018). Early versus late cardiac remodeling during right ventricular pressure load and impact of preventive versus rescue therapy with endothelin-1 receptor blockers. J. Appl. Physiol. 124, 1349–1362. doi:10.1152/japplphysiol.00975.2017
Rashid, F. N., Clayton, Z. E., Ogawa, M., Perdomo, J., Hume, R. D., Kizana, E., et al. (2021). Platelet derived growth factor-A (Pdgf-a) gene transfer modulates scar composition and improves left ventricular function after myocardial infarction. Int. J. Cardiol. 341, 24–30. doi:10.1016/j.ijcard.2021.07.021
Ray, P. D., Huang, B. W., and Tsuji, Y. (2012). Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal 24, 981–990. doi:10.1016/j.cellsig.2012.01.008
Razin, T., Melamed-Book, N., Argaman, J., Galin, I., Lowy, Y., Anuka, E., et al. (2021). Interleukin-1α dependent survival of cardiac fibroblasts is associated with StAR/STARD1 expression and improved cardiac remodeling and function after myocardial infarction. J. Mol. Cell Cardiol. 155, 125–137. doi:10.1016/j.yjmcc.2020.10.013
Reusswig, F., Polzin, A., Klier, M., Dille, M. A., Ayhan, A., Benkhoff, M., et al. (2022)., 11. Cells.Only acute but not chronic thrombocytopenia protects mice against left ventricular dysfunction after acute myocardial infarction
Rockey, D. C., Bell, P. D., and Hill, J. A. (2015). Fibrosis-A common pathway to organ injury and failure. N. Engl. J. Med. 373, 96. doi:10.1056/NEJMc1504848
Rosenzweig, R., Kumar, V., Gupta, S., Bermeo-Blanco, O., Stratton, M. S., Gumina, R. J., et al. (2022). Estrogen receptor-β agonists modulate T-lymphocyte activation and ameliorate left ventricular remodeling during chronic heart failure. Circ. Heart Fail 15, e008997. doi:10.1161/CIRCHEARTFAILURE.121.008997
Rotem, I., Konfino, T., Caller, T., Schary, Y., Shaihov-Teper, O., Palevski, D., et al. (2022). Osteopontin promotes infarct repair. Basic Res. Cardiol. 117, 51. doi:10.1007/s00395-022-00957-0
Ruchaya, P. J., Lewis-Mcdougall, F. C., Sornkarn, N., Amin, S., Grimsdell, B., Shaalan, A., et al. (2022). Transplantation of skeletal muscle-derived sca-1(+)/PW1(+)/pax7(-) interstitial cells (PICs) improves cardiac function and attenuates remodeling in mice subjected to myocardial infarction. Cells 11, 4050. doi:10.3390/cells11244050
Ruozi, G., Bortolotti, F., Mura, A., Tomczyk, M., Falcione, A., Martinelli, V., et al. (2022). Cardioprotective factors against myocardial infarction selected in vivo from an AAV secretome library. Sci. Transl. Med. 14, eabo0699. doi:10.1126/scitranslmed.abo0699
Rurik, J. G., TombáCZ, I., Yadegari, A., MéNDEZ FernáNDEZ, P. O., Shewale, S. V., Li, L., et al. (2022). CAR T cells produced in vivo to treat cardiac injury. Science 375, 91–96. doi:10.1126/science.abm0594
Sim, W. S., Park, B. W., Ban, K., and Park, H. J. (2021). In Situ Preconditioning of Human Mesenchymal Stem Cells Elicits Comprehensive Cardiac Repair Following Myocardial Infarction. Int. J. Mol. Sci. 22.
Sabe, S. A., Xu, C. M., Sabra, M., Harris, D. D., Malhotra, A., Aboulgheit, A., et al. (2023). Canagliflozin improves myocardial perfusion, fibrosis, and function in a swine model of chronic myocardial ischemia. J. Am. Heart Assoc. 12, e028623. doi:10.1161/JAHA.122.028623
Sahinarslan, A., Kocaman, S. A., Topal, S., Ercin, U., Bukan, N., Yalcin, R., et al. (2010). Relation between serum monocyte chemoattractant protein-1 and coronary collateral development. Coron. Artery Dis. 21, 455–459. doi:10.1097/MCA.0b013e32833fd29b
Sakai, S., Miyauchi, T., Kobayashi, M., Yamaguchi, I., Goto, K., and Sugishita, Y. (1996). Inhibition of myocardial endothelin pathway improves long-term survival in heart failure. Nature 384, 353–355. doi:10.1038/384353a0
Santoso, M. R., Ikeda, G., Tada, Y., Jung, J. H., Vaskova, E., Sierra, R. G., et al. (2020). Exosomes from induced pluripotent stem cell-derived cardiomyocytes promote autophagy for myocardial repair. J. Am. Heart Assoc. 9, e014345. doi:10.1161/JAHA.119.014345
Saraswati, S., Marrow, S. M. W., Watch, L. A., and Young, P. P. (2019). Identification of a pro-angiogenic functional role for FSP1-positive fibroblast subtype in wound healing. Nat. Commun. 10, 3027. doi:10.1038/s41467-019-10965-9
Sawa, Y., Saito, M., Ishida, N., Ibi, M., Matsushita, N., Morino, Y., et al. (2020). Pretreatment with KGA-2727, a selective SGLT1 inhibitor, is protective against myocardial infarction-induced ventricular remodeling and heart failure in mice. J. Pharmacol. Sci. 142, 16–25. doi:10.1016/j.jphs.2019.11.001
Scharf, G. M., Kilian, K., Cordero, J., Wang, Y., Grund, A., Hofmann, M., et al. (2019). Inactivation of Sox9 in fibroblasts reduces cardiac fibrosis and inflammation. JCI Insight 5, e126721. doi:10.1172/jci.insight.126721
Schena, G. J., Murray, E. K., Hildebrand, A. N., Headrick, A. L., Yang, Y., Koch, K. A., et al. (2021). Cortical bone stem cell-derived exosomes' therapeutic effect on myocardial ischemia-reperfusion and cardiac remodeling. Am. J. Physiol. Heart Circ. Physiol. 321, H1014–h1029. doi:10.1152/ajpheart.00197.2021
Schilter, H., Findlay, A. D., Perryman, L., Yow, T. T., Moses, J., Zahoor, A., et al. (2019). The lysyl oxidase like 2/3 enzymatic inhibitor, PXS-5153A, reduces crosslinks and ameliorates fibrosis. J. Cell Mol. Med. 23, 1759–1770. doi:10.1111/jcmm.14074
Schloss, M. J., Horckmans, M., Guillamat-Prats, R., Hering, D., Lauer, E., Lenglet, S., et al. (2019). 2-Arachidonoylglycerol mobilizes myeloid cells and worsens heart function after acute myocardial infarction. Cardiovasc Res. 115, 602–613. doi:10.1093/cvr/cvy242
Schroder, K., and Tschopp, J. (2010). The inflammasomes. Cell 140, 821–832. doi:10.1016/j.cell.2010.01.040
Schuman, M. L., Peres Diaz, L. S., Aisicovich, M., Ingallina, F., Toblli, J. E., Landa, M. S., et al. (2021). Cardiac thyrotropin-releasing hormone inhibition improves ventricular function and reduces hypertrophy and fibrosis after myocardial infarction in rats. J. Card. Fail 27, 796–807. doi:10.1016/j.cardfail.2021.04.003
Scott, N. J. A., Rademaker, M. T., Charles, C. J., Espiner, E. A., and Richards, A. M. (2019). Hemodynamic, hormonal, and renal actions of phosphodiesterase-9 inhibition in experimental heart failure. J. Am. Coll. Cardiol. 74, 889–901. doi:10.1016/j.jacc.2019.05.067
Shao, L., Zhang, Y., Pan, X., Liu, B., Liang, C., Zhang, Y., et al. (2020). Knockout of beta-2 microglobulin enhances cardiac repair by modulating exosome imprinting and inhibiting stem cell-induced immune rejection. Cell Mol. Life Sci. 77, 937–952. doi:10.1007/s00018-019-03220-3
Sharifi, M., Nazarinia, D., Ramezani, F., Azizi, Y., Naderi, N., and Aboutaleb, N. (2021). Necroptosis and RhoA/ROCK pathways: Molecular targets of nesfatin-1 in cardioprotection against myocardial ischemia/reperfusion injury in a rat model. Mol. Biol. Rep. 48, 2507–2518. doi:10.1007/s11033-021-06289-x
Shi, J., Sun, J., Liu, L., Shan, T., Meng, H., Yang, T., et al. (2022). P16ink4a overexpression ameliorates cardiac remodeling of mouse following myocardial infarction via CDK4/pRb pathway. Biochem. Biophys. Res. Commun. 595, 62–68. doi:10.1016/j.bbrc.2022.01.077
Shi, S. Y., Luo, X., Yamawaki, T. M., Li, C. M., Ason, B., and Furtado, M. B. (2021). Recent advances in single-cell profiling and multispecific therapeutics: Paving the way for a new era of precision medicine targeting cardiac fibroblasts. Curr. Cardiol. Rep. 23, 82. doi:10.1007/s11886-021-01517-z
Shibamoto, M., Higo, T., Naito, A. T., Nakagawa, A., Sumida, T., Okada, K., et al. (2019). Activation of DNA damage response and cellular senescence in cardiac fibroblasts limit cardiac fibrosis after myocardial infarction. Int. Heart J. 60, 944–957. doi:10.1536/ihj.18-701
Shigemura, N., Takai, S., Hirose, F., Yoshida, R., Sanematsu, K., and Ninomiya, Y. (2019). Expression of renin-angiotensin system components in the taste organ of mice. Nutrients 11, 2251. doi:10.3390/nu11092251
Shiraishi, M., Yamaguchi, A., and Suzuki, K. (2022). Nrg1/ErbB signaling-mediated regulation of fibrosis after myocardial infarction. Faseb J. 36, e22150. doi:10.1096/fj.202101428RR
SimõES, F. C., and Riley, P. R. (2022). Immune cells in cardiac repair and regeneration. Development 149.
Solhpour, A., and Yusuf, S. W. (2014). Fibrinolytic therapy in patients with ST-elevation myocardial infarction. Expert Rev. Cardiovasc Ther. 12, 201–215. doi:10.1586/14779072.2014.867805
Solomon, S. D., Mcmurray, J. J. V., Claggett, B., DE Boer, R. A., Demets, D., Hernandez, A. F., et al. (2022). Dapagliflozin in heart failure with mildly reduced or preserved ejection fraction. N. Engl. J. Med. 387, 1089–1098. doi:10.1056/NEJMoa2206286
Song, J., Frieler, R. A., Whitesall, S. E., Chung, Y., Vigil, T. M., Muir, L. A., et al. (2021a). Myeloid interleukin-4 receptor α is essential in postmyocardial infarction healing by regulating inflammation and fibrotic remodeling. Am. J. Physiol. Heart Circ. Physiol. 320, H323–h337. doi:10.1152/ajpheart.00251.2020
Song, L., Duan, X., Zeng, X., Duan, X., and Li, L. (2021b). Regulatory mechanism of LINC00152 on aggravating heart failure through triggering fibrosis in an infarcted myocardium. Dis. Markers 2021, 2607358. doi:10.1155/2021/2607358
Song, S. Y., Kim, H., Yoo, J., Kwon, S. P., Park, B. W., Kim, J. J., et al. (2020). Prevascularized, multiple-layered cell sheets of direct cardiac reprogrammed cells for cardiac repair. Biomater. Sci. 8, 4508–4520. doi:10.1039/d0bm00701c
Song, Y., Xu, C., Liu, J., Li, Y., Wang, H., Shan, D., et al. (2021c). Heterodimerization with 5-HT(2B)R is indispensable for β(2)ar-mediated cardioprotection. Circ. Res. 128, 262–277. doi:10.1161/CIRCRESAHA.120.317011
Souders, C. A., Bowers, S. L., and Baudino, T. A. (2009). Cardiac fibroblast: The renaissance cell. Circ. Res. 105, 1164–1176. doi:10.1161/CIRCRESAHA.109.209809
Sousonis, V., Sfakianaki, T., Ntalianis, A., Nanas, I., Kontogiannis, C., Aravantinos, D., et al. (2021). Intracoronary administration of allogeneic cardiosphere-derived cells immediately prior to reperfusion in pigs with acute myocardial infarction reduces infarct size and attenuates adverse cardiac remodeling. J. Cardiovasc Pharmacol. Ther. 26, 88–99. doi:10.1177/1074248420941672
Spinale, F. G. (2007). Myocardial matrix remodeling and the matrix metalloproteinases: Influence on cardiac form and function. Physiol. Rev. 87, 1285–1342. doi:10.1152/physrev.00012.2007
Stienen, S., Rossignol, P., Barros, A., Girerd, N., Pitt, B., Zannad, F., et al. (2020). Determinants of anti-fibrotic response to mineralocorticoid receptor antagonist therapy: Insights from the eplerenone post-acute myocardial infarction heart failure efficacy and survival study (EPHESUS) and early eplerenone treatment in patients with acute ST-elevation myocardial infarction without heart failure (REMINDER) trials. Clin. Res. Cardiol. 109, 194–204. doi:10.1007/s00392-019-01500-3
Sun, B., Zhao, C., and Mao, Y. (2021a). MiR-218-5p mediates myocardial fibrosis after myocardial infarction by targeting CX43. Curr. Pharm. Des. 27, 4504–4512. doi:10.2174/1381612827666210929111622
Sun, J., Zhang, Q., Liu, X., and Shang, X. (2021b). Downregulation of interferon-induced protein with tetratricopeptide repeats 3 relieves the inflammatory response and myocardial fibrosis of mice with myocardial infarction and improves their cardiac function. Exp. Anim. 70, 522–531. doi:10.1538/expanim.21-0060
Sunagawa, Y., Katayama, A., Funamoto, M., Shimizu, K., Shimizu, S., Sari, N., et al. (2022a). The polyunsaturated fatty acids, EPA and DHA, ameliorate myocardial infarction-induced heart failure by inhibiting p300-HAT activity in rats. J. Nutr. Biochem. 106, 109031. doi:10.1016/j.jnutbio.2022.109031
Sunagawa, Y., Kawaguchi, S., Miyazaki, Y., Katanasaka, Y., Funamoto, M., Shimizu, K., et al. (2022b). Auraptene, a citrus peel-derived natural product, prevents myocardial infarction-induced heart failure by activating PPARα in rats. Phytomedicine 107, 154457. doi:10.1016/j.phymed.2022.154457
Surinkaew, S., Aflaki, M., Takawale, A., Chen, Y., Qi, X. Y., Gillis, M. A., et al. (2019). Exchange protein activated by cyclic-adenosine monophosphate (Epac) regulates atrial fibroblast function and controls cardiac remodelling. Cardiovasc Res. 115, 94–106. doi:10.1093/cvr/cvy173
Takawale, A., Sakamuri, S. S., and Kassiri, Z. (2015). Extracellular matrix communication and turnover in cardiac physiology and pathology. Compr. Physiol. 5, 687–719. doi:10.1002/cphy.c140045
Tan, Z., Jiang, X., Zhou, W., Deng, B., Cai, M., Deng, S., et al. (2021). Taohong siwu decoction attenuates myocardial fibrosis by inhibiting fibrosis proliferation and collagen deposition via TGFBR1 signaling pathway. J. Ethnopharmacol. 270, 113838. doi:10.1016/j.jep.2021.113838
Tanner, M. A., Maitz, C. A., and Grisanti, L. A. (2021). Immune cell β(2)-adrenergic receptors contribute to the development of heart failure. Am. J. Physiol. Heart Circ. Physiol. 321, H633–h649. doi:10.1152/ajpheart.00243.2021
Tanner, M. A., Thomas, T. P., Maitz, C. A., and Grisanti, L. A. (2020). β2-Adrenergic receptors increase cardiac fibroblast proliferation through the gαs/ERK1/2-dependent secretion of interleukin-6. Int. J. Mol. Sci. 21, 8507. doi:10.3390/ijms21228507
Thavapalachandran, S., Grieve, S. M., Hume, R. D., LE, T. Y. L., Raguram, K., Hudson, J. E., et al. (2020). Platelet-derived growth factor-AB improves scar mechanics and vascularity after myocardial infarction. Sci. Transl. Med. 12, eaay2140. doi:10.1126/scitranslmed.aay2140
Tian, X., Sun, C., Wang, X., Ma, K., Chang, Y., Guo, Z., et al. (2020). ANO1 regulates cardiac fibrosis via ATI-mediated MAPK pathway. Cell Calcium 92, 102306. doi:10.1016/j.ceca.2020.102306
Tian, Z., Zhang, Y., and Lyu, X. (2021). Promoting roles of KLF5 in myocardial infarction in mice involving microRNA-27a suppression and the following GFPT2/TGF-β/Smad2/3 axis activation. Cell Cycle 20, 874–893. doi:10.1080/15384101.2021.1907512
Toner, Y. C., Ghotbi, A. A., Naidu, S., Sakurai, K., VAN Leent, M. M. T., Jordan, S., et al. (2022). Systematically evaluating DOTATATE and FDG as PET immuno-imaging tracers of cardiovascular inflammation. Sci. Rep. 12, 6185. doi:10.1038/s41598-022-09590-2
Tripathi, H., Peng, H., Donahue, R., Chelvarajan, L., Gottipati, A., Levitan, B., et al. (2020). Isolation methods for human CD34 subsets using fluorescent and magnetic activated cell sorting: An in vivo comparative study. Stem Cell Rev. Rep. 16, 413–423. doi:10.1007/s12015-019-09939-7
Turner, N. A., Porter, K. E., Smith, W. H., White, H. L., Ball, S. G., and Balmforth, A. J. (2003). Chronic beta2-adrenergic receptor stimulation increases proliferation of human cardiac fibroblasts via an autocrine mechanism. Cardiovasc Res. 57, 784–792. doi:10.1016/s0008-6363(02)00729-0
Unkart, J. T., Allison, M. A., Abdelmalek, J. A., Jenny, N. S., Mcclelland, R. L., Budoff, M., et al. (2020). Relation of plasma renin activity to subclinical peripheral and coronary artery disease (from the multiethnic study of atherosclerosis). Am. J. Cardiol. 125, 1794–1800. doi:10.1016/j.amjcard.2020.03.022
Vaskova, E., Ikeda, G., Tada, Y., Wahlquist, C., Mercola, M., and Yang, P. C. (2020). Sacubitril/valsartan improves cardiac function and decreases myocardial fibrosis via downregulation of exosomal miR-181a in a rodent chronic myocardial infarction model. J. Am. Heart Assoc. 9, e015640. doi:10.1161/JAHA.119.015640
Vatner, D. E., Zhang, J., Zhao, X., Yan, L., Kudej, R., and Vatner, S. F. (2021). Secreted frizzled protein 3 is a novel cardioprotective mechanism unique to the clinically relevant fourth window of ischemic preconditioning. Am. J. Physiol. Heart Circ. Physiol. 320, H798–h804. doi:10.1152/ajpheart.00849.2020
Vellaichamy, E., Das, S., Subramanian, U., Maeda, N., and Pandey, K. N. (2014). Genetically altered mutant mouse models of guanylyl cyclase/natriuretic peptide receptor-A exhibit the cardiac expression of proinflammatory mediators in a gene-dose-dependent manner. Endocrinology 155, 1045–1056. doi:10.1210/en.2013-1416
Venugopal, H., Hanna, A., Humeres, C., and Frangogiannis, N. G. (2022)., 11. Cells, 1386. doi:10.3390/cells11091386Properties and functions of fibroblasts and myofibroblasts in myocardial infarctionCells
Vianello, E., Dozio, E., Tacchini, L., Frati, L., and Corsi Romanelli, M. M. (2019). ST2/IL-33 signaling in cardiac fibrosis. Int. J. Biochem. Cell Biol. 116, 105619. doi:10.1016/j.biocel.2019.105619
Viswanadha, V. P., Dhivya, V., Beeraka, N. M., Huang, C. Y., Gavryushova, L. V., Minyaeva, N. N., et al. (2020). The protective effect of piperine against isoproterenol-induced inflammation in experimental models of myocardial toxicity. Eur. J. Pharmacol. 885, 173524. doi:10.1016/j.ejphar.2020.173524
Vukicevic, S., Colliva, A., Kufner, V., Martinelli, V., Moimas, S., Vodret, S., et al. (2022). Bone morphogenetic protein 1.3 inhibition decreases scar formation and supports cardiomyocyte survival after myocardial infarction. Nat. Commun. 13, 81. doi:10.1038/s41467-021-27622-9
Wan, J., Lin, S., Yu, Z., Song, Z., Lin, X., Xu, R., et al. (2022a). Protective effects of MicroRNA-200b-3p encapsulated by mesenchymal stem cells-secreted extracellular vesicles in myocardial infarction via regulating BCL2L11. J. Am. Heart Assoc. 11, e024330. doi:10.1161/JAHA.121.024330
Wan, Q., Zhang, L., Zhou, Q., Han, Y., Li, Z., and Li, B. (2022b). Protection of CAPE-pNO(2) against chronic myocardial ischemia by the TGF-β1/galectin-3 pathway in vivo and in vitro. Inflammation 45, 1039–1058. doi:10.1007/s10753-021-01600-1
Wan, Y. J., Wang, Y. H., Guo, Q., Jiang, Y., Tu, P. F., and Zeng, K. W. (2021). Protocatechualdehyde protects oxygen-glucose deprivation/reoxygenation-induced myocardial injury via inhibiting PERK/ATF6α/IRE1α pathway. Eur. J. Pharmacol. 891, 173723. doi:10.1016/j.ejphar.2020.173723
Wang, B., Tan, Y., Zhou, W., Yang, J., Jiang, Y., Liu, X., et al. (2022a). Loss of BTK ameliorates the pathological cardiac fibrosis and dysfunction. Matrix Biol. 112, 171–189. doi:10.1016/j.matbio.2022.08.010
Wang, C., Yue, Y., Huang, S., Wang, K., Yang, X., Chen, J., et al. (2022b). M2b macrophages stimulate lymphangiogenesis to reduce myocardial fibrosis after myocardial ischaemia/reperfusion injury. Pharm. Biol. 60, 384–393. doi:10.1080/13880209.2022.2033798
Wang, D. M., Jin, J. J., Tian, L. M., and Zhang, Z. (2020a). MiR-195 promotes myocardial fibrosis in MI rats via targeting TGF-β1/Smad. J. Biol. Regul. Homeost. Agents 34, 1325–1332. doi:10.23812/20-201-A
Wang, F., He, Y., Yao, N., Ruan, L., and Tian, Z. (2022c). Thymosin β4 protects against cardiac damage and subsequent cardiac fibrosis in mice with myocardial infarction. Cardiovasc Ther. 2022, 1308651. doi:10.1155/2022/1308651
Wang, G., Wang, R., Ruan, Z., Liu, L., Li, Y., and Zhu, L. (2020b). MicroRNA-132 attenuated cardiac fibrosis in myocardial infarction-induced heart failure rats. Biosci. Rep. 40. doi:10.1042/BSR20201696
Wang, J., Zhang, J., Lin, X., Wang, Y., Wu, X., Yang, F., et al. (2021a). DCA-TGR5 signaling activation alleviates inflammatory response and improves cardiac function in myocardial infarction. J. Mol. Cell Cardiol. 151, 3–14. doi:10.1016/j.yjmcc.2020.10.014
Wang, J., Zhang, S., Li, X., and Gong, M. (2020c). LncRNA SNHG7 promotes cardiac remodeling by upregulating ROCK1 via sponging miR-34-5p. Aging (Albany NY) 12, 10441–10456. doi:10.18632/aging.103269
Wang, K., Li, Z., Li, Y., Liu, X., Sun, Y., Hong, J., et al. (2022d). Cardioprotection of Klotho against myocardial infarction-induced heart failure through inducing autophagy. Mech. Ageing Dev. 207, 111714. doi:10.1016/j.mad.2022.111714
Wang, L., Singh, H., Mulyala, R. R., Weber, J., Barasch, E., and Cao, J. J. (2020d). The association between left ventricular diastolic dysfunction and myocardial scar and their collective impact on all-cause mortality. J. Am. Soc. Echocardiogr. 33, 161–170. doi:10.1016/j.echo.2019.09.022
Wang, Q., Chen, Z., Guo, J., Peng, X., Zheng, Z., Chen, H., et al. (2023). Atorvastatin-induced tolerogenic dendritic cells improve cardiac remodeling by suppressing TLR-4/NF-κB activation after myocardial infarction. Inflamm. Res. 72, 13–25. doi:10.1007/s00011-022-01654-3
Wang, Q., Fu, W., Yu, X., Xu, H., Sui, D., and Wang, Y. (2021b). Ginsenoside Rg2 alleviates myocardial fibrosis by regulating TGF-β1/Smad signalling pathway. Pharm. Biol. 59, 106–113. doi:10.1080/13880209.2020.1867197
Wang, Q., Liu, A. D., Li, T. S., Tang, Q., Wang, X. C., and Chen, X. B. (2021c). Ghrelin ameliorates cardiac fibrosis after myocardial infarction by regulating the Nrf2/NADPH/ROS pathway. Peptides 144, 170613. doi:10.1016/j.peptides.2021.170613
Wang, Q., Yang, Y., Chen, K., Li, D., Tang, B., Peng, K., et al. (2020e). Dietary menthol attenuates inflammation and cardiac remodeling after myocardial infarction via the transient receptor potential melastatin 8. Am. J. Hypertens. 33, 223–233. doi:10.1093/ajh/hpz162
Wang, R., Peng, L., Lv, D., Shang, F., Yan, J., Li, G., et al. (2021d). Leonurine attenuates myocardial fibrosis through upregulation of miR-29a-3p in mice post-myocardial infarction. J. Cardiovasc Pharmacol. 77, 189–199. doi:10.1097/FJC.0000000000000957
Wang, S., Li, L., Liu, T., Jiang, W., and Hu, X. (2020f). miR-19a/19b-loaded exosomes in combination with mesenchymal stem cell transplantation in a preclinical model of myocardial infarction. Regen. Med. 15, 1749–1759. doi:10.2217/rme-2019-0136
Wang, S., Su, X., Xu, L., Chang, C., Yao, Y., Komal, S., et al. (2020g). Glycogen synthase kinase-3β inhibition alleviates activation of the NLRP3 inflammasome in myocardial infarction. J. Mol. Cell Cardiol. 149, 82–94. doi:10.1016/j.yjmcc.2020.09.009
Wang, W., Ye, S., Zhang, L., Jiang, Q., Chen, J., Chen, X., et al. (2020h). Granulocyte colony-stimulating factor attenuates myocardial remodeling and ventricular arrhythmia susceptibility via the JAK2-STAT3 pathway in a rabbit model of coronary microembolization. BMC Cardiovasc Disord. 20, 85. doi:10.1186/s12872-020-01385-5
Wang, X., Guo, Z., Ding, Z., and Mehta, J. L. (2018). Inflammation, autophagy, and apoptosis after myocardial infarction. J. Am. Heart Assoc. 7, e008024. doi:10.1161/JAHA.117.008024
Wang, X., Hu, S., Li, J., Zhu, D., Wang, Z., Cores, J., et al. (2021e). Extruded mesenchymal stem cell nanovesicles are equally potent to natural extracellular vesicles in cardiac repair. ACS Appl. Mater Interfaces 13, 55767–55779. doi:10.1021/acsami.1c08044
Wang, Y., DE Waard, M. C., Sterner-Kock, A., Stepan, H., Schultheiss, H. P., Duncker, D. J., et al. (2007). Cardiomyocyte-restricted over-expression of C-type natriuretic peptide prevents cardiac hypertrophy induced by myocardial infarction in mice. Eur. J. Heart Fail 9, 548–557. doi:10.1016/j.ejheart.2007.02.006
Wang, Y., Del Borgo, M., Lee, H. W., Baraldi, D., Hirmiz, B., Gaspari, T. A., et al. (2017). Anti-fibrotic potential of AT2 receptor agonists. Front. Pharmacol. 8, 564. doi:10.3389/fphar.2017.00564
Wang, Y., Li, C., Zhao, R., Qiu, Z., Shen, C., Wang, Z., et al. (2021f). CircUbe3a from M2 macrophage-derived small extracellular vesicles mediates myocardial fibrosis after acute myocardial infarction. Theranostics 11, 6315–6333. doi:10.7150/thno.52843
Wang, Z. P., Che, Y., Zhou, H., Meng, Y. Y., Wu, H. M., Jin, Y. G., et al. (2020i). Corosolic acid attenuates cardiac fibrosis following myocardial infarction in mice. Int. J. Mol. Med. 45, 1425–1435. doi:10.3892/ijmm.2020.4531
Weber, K. T., Sun, Y., Tyagi, S. C., and Cleutjens, J. P. (1994). Collagen network of the myocardium: Function, structural remodeling and regulatory mechanisms. J. Mol. Cell Cardiol. 26, 279–292. doi:10.1006/jmcc.1994.1036
Wei, X., Chen, S., Xie, T., Chen, H., Jin, X., Yang, J., et al. (2022). An MMP-degradable and conductive hydrogel to stabilize HIF-1α for recovering cardiac functions. Theranostics 12, 127–142. doi:10.7150/thno.63481
Wei, Y., Lan, Y., Zhong, Y., Yu, K., Xu, W., Zhu, R., et al. (2020). Interleukin-38 alleviates cardiac remodelling after myocardial infarction. J. Cell Mol. Med. 24, 371–384. doi:10.1111/jcmm.14741
Weldon, S. M., and Brown, N. F. (2019). Inhibitors of aldosterone synthase. Vitam. Horm. 109, 211–239. doi:10.1016/bs.vh.2018.10.002
Wen, Y., Li, X. Y., Li, Z. Y., Wang, M. L., Chen, P. P., Liu, Y., et al. (2020). Intra-myocardial delivery of a novel thermosensitive hydrogel inhibits post-infarct heart failure after degradation in rat. J. Cardiovasc Transl. Res. 13, 677–685. doi:10.1007/s12265-019-09941-x
Whitehurst, K. S., Chan, V. A., Estes, H. K., Valsaraj, S., Kent, S., Sharma, U. M., et al. (2020). EphrinA1-Fc attenuates ventricular remodeling and dysfunction in chronically nonreperfused WT but not EphA2-R-M mice. Int. J. Mol. Sci. 21, 5811. doi:10.3390/ijms21165811
Williams, B. (2001). Angiotensin II and the pathophysiology of cardiovascular remodeling. Am. J. Cardiol. 87, 10c–17c. doi:10.1016/s0002-9149(01)01507-7
Wu, J., Li, S., Li, C., Cui, L., Ma, J., and Hui, Y. (2021a). The non-canonical effects of heme oxygenase-1, a classical fighter against oxidative stress. Redox Biol. 47, 102170. doi:10.1016/j.redox.2021.102170
Wu, Q., Wang, J., Tan, W. L. W., Jiang, Y., Wang, S., Li, Q., et al. (2020). Extracellular vesicles from human embryonic stem cell-derived cardiovascular progenitor cells promote cardiac infarct healing through reducing cardiomyocyte death and promoting angiogenesis. Cell Death Dis. 11, 354. doi:10.1038/s41419-020-2508-y
Wu, X., Reboll, M. R., Korf-Klingebiel, M., and Wollert, K. C. (2021b). Angiogenesis after acute myocardial infarction. Cardiovasc Res. 117, 1257–1273. doi:10.1093/cvr/cvaa287
Wu, X., Xu, J., Li, X., Dai, J., and Wang, L. (2022a). Inhibition of SphK1/S1P signaling pathway alleviates fibrosis and inflammation of rat myocardium after myocardial infarction. Comput. Math. Methods Med. 2022, 5985375. doi:10.1155/2022/5985375
Wu, X., Zhao, X., Xiong, Y., Zheng, M., Zhong, C., and Zhou, Y. (2022b). Deciphering cell-type-specific gene expression signatures of cardiac diseases through reconstruction of bulk transcriptomes. Front. Cell Dev. Biol. 10, 792774. doi:10.3389/fcell.2022.792774
Wu, Y., Chen, S., Wen, P., Wu, M., Wu, Y., Mai, M., et al. (2021c). PGAM1 deficiency ameliorates myocardial infarction remodeling by targeting TGF-β via the suppression of inflammation, apoptosis and fibrosis. Biochem. Biophys. Res. Commun. 534, 933–940. doi:10.1016/j.bbrc.2020.10.070
Wu, Y., Peng, W., Fang, M., Wu, M., and Wu, M. (2022c). MSCs-derived extracellular vesicles carrying miR-212-5p alleviate myocardial infarction-induced cardiac fibrosis via NLRC5/VEGF/TGF-β1/SMAD Axis. J. Cardiovasc Transl. Res. 15, 302–316. doi:10.1007/s12265-021-10156-2
Wu, Y., Wang, T., Qiao, L., and Lin, H. (2022d). Upregulated microRNA-210-3p improves sevoflurane-induced protective effect on ventricular remodeling in rats with myocardial infarction by inhibiting ADCY9. Funct. Integr. Genomics 22, 279–289. doi:10.1007/s10142-021-00816-6
Xiang, C., Zhu, Y., Xu, M., and Zhang, D. (2022). Fasudil ameliorates osteoporosis following myocardial infarction by regulating cardiac calcitonin secretion. J. Cardiovasc Transl. Res. 15, 1352–1365. doi:10.1007/s12265-022-10271-8
Xiao, Y., Feng, Q., Huang, L., Meng, X., Han, P., Zhang, W., et al. (2023). Copper promotes cardiac functional recovery via suppressing the transformation of fibroblasts to myofibroblasts in ischemia-infarcted monkey hearts. J. Nutr. Biochem. 111, 109180. doi:10.1016/j.jnutbio.2022.109180
Xiao, Y., Hill, M. C., Li, L., Deshmukh, V., Martin, T. J., Wang, J., et al. (2019). Hippo pathway deletion in adult resting cardiac fibroblasts initiates a cell state transition with spontaneous and self-sustaining fibrosis. Genes Dev. 33, 1491–1505. doi:10.1101/gad.329763.119
Xie, B., Wang, J., XI, X. Y., Guo, X., Chen, B. X., Li, L., et al. (2022). Fibroblast activation protein imaging in reperfused ST-elevation myocardial infarction: Comparison with cardiac magnetic resonance imaging. Eur. J. Nucl. Med. Mol. Imaging 49, 2786–2797. doi:10.1007/s00259-021-05674-9
Xing, Z., Yang, C., He, J., Feng, Y., Li, X., Peng, C., et al. (2022). Cardioprotective effects of aconite in isoproterenol-induced myocardial infarction in rats. Oxid. Med. Cell Longev. 2022, 1090893. doi:10.1155/2022/1090893
Xiong, X., Liu, J., He, Q., Dai, R., Zhang, H., Cao, Z., et al. (2021). Long non-coding RNA NORAD aggravates acute myocardial infarction by promoting fibrosis and apoptosis via miR-577/COBLL1 axis. Environ. Toxicol. 36, 2256–2265. doi:10.1002/tox.23339
Xu, J., Wu, H., Mai, Z., Yi, J., Wang, X., Li, L., et al. (2021a). Therapeutic effects of CXCR4(+) subpopulation of transgene-free induced cardiosphere-derived cells on experimental myocardial infarction. Cell Prolif. 54, e13041. doi:10.1111/cpr.13041
Xu, X., Jiang, T., Li, Y., and Kong, L. (2021b). Endostatin attenuates heart failure via inhibiting reactive oxygen species in myocardial infarction rats. Biosci. Rep. 41. doi:10.1042/BSR20200787
Xu, Y., Hu, Y., Geng, Y., Zhao, N., Jia, C., Song, H., et al. (2022a). Pentraxin 3 depletion (PTX3 KD) inhibited myocardial fibrosis in heart failure after myocardial infarction. Aging (Albany NY) 14, 4036–4049. doi:10.18632/aging.204070
Xu, Y., Jiang, K., Chen, F., Qian, J., Wang, D., Wu, Y., et al. (2022b). Bone marrow-derived naïve B lymphocytes improve heart function after myocardial infarction: A novel cardioprotective mechanism for empagliflozin. Basic Res. Cardiol. 117, 47. doi:10.1007/s00395-022-00956-1
Xu, Z., Lu, D., Yuan, J., Wang, L., Wang, J., Lei, Z., et al. (2022c). Storax attenuates cardiac fibrosis following acute myocardial infarction in rats via suppression of at1r-ankrd1-P53 signaling pathway. Int. J. Mol. Sci. 23, 13161. doi:10.3390/ijms232113161
Xuan, L., Fu, D., Zhen, D., Wei, C., Bai, D., Yu, L., et al. (2022). Extracellular vesicles derived from human bone marrow mesenchymal stem cells protect rats against acute myocardial infarction-induced heart failure. Cell Tissue Res. 389, 23–40. doi:10.1007/s00441-022-03612-1
Xue, K., Chen, S., Chai, J., Yan, W., Zhu, X., Dai, H., et al. (2022). Upregulation of periostin through CREB participates in myocardial infarction-induced myocardial fibrosis. J. Cardiovasc Pharmacol. 79, 687–697. doi:10.1097/FJC.0000000000001244
Xue, K., Zhang, J., Li, C., Li, J., Wang, C., Zhang, Q., et al. (2019). The role and mechanism of transforming growth factor beta 3 in human myocardial infarction-induced myocardial fibrosis. J. Cell Mol. Med. 23, 4229–4243. doi:10.1111/jcmm.14313
Xue, Y., Fan, X., Yang, R., Jiao, Y., and Li, Y. (2020). miR-29b-3p inhibits post-infarct cardiac fibrosis by targeting FOS. Biosci. Rep. 40. doi:10.1042/BSR20201227
Yan, L. L., Wei, X. H., Shi, Q. P., Pan, C. S., Li, K. Y., Zhang, B., et al. (2022a). Cardiotonic Pills® protects from myocardial fibrosis caused by in stent restenosis in miniature pigs. Phytomedicine 106, 154405. doi:10.1016/j.phymed.2022.154405
Yan, M., Liu, Q., Jiang, Y., Wang, B., Ji, Y., Liu, H., et al. (2020). Long noncoding RNA LNC_000898 alleviates cardiomyocyte apoptosis and promotes cardiac repair after myocardial infarction through modulating the miR-375/PDK1 Axis. J. Cardiovasc Pharmacol. 76, 77–85. doi:10.1097/FJC.0000000000000845
Yan, T., Zhu, X., Zhang, X., Jia, X., Liu, J., Wang, X., et al. (2022b). The application of proteomics and metabolomics to reveal the molecular mechanism of Nutmeg-5 in ameliorating cardiac fibrosis following myocardial infarction. Phytomedicine 105, 154382. doi:10.1016/j.phymed.2022.154382
Yang, J., Savvatis, K., Kang, J. S., Fan, P., Zhong, H., Schwartz, K., et al. (2016). Targeting LOXL2 for cardiac interstitial fibrosis and heart failure treatment. Nat. Commun. 7, 13710. doi:10.1038/ncomms13710
Yang, L., Xue, S., DU, M., and Lian, F. (2021a). Highly efficient MicroRNA delivery using functionalized carbon dots for enhanced conversion of fibroblasts to cardiomyocytes. Int. J. Nanomedicine 16, 3741–3754. doi:10.2147/IJN.S304873
Yang, W., Lin, J., Zhou, J., Zheng, Y., Jiang, S., He, S., et al. (2021b). Innate lymphoid cells and myocardial infarction. Front. Immunol. 12, 758272. doi:10.3389/fimmu.2021.758272
Yang, W., Zhang, A., Han, Y., Su, X., Chen, Y., Zhao, W., et al. (2021c). Cyclin-dependent kinase inhibitor 2b controls fibrosis and functional changes in ischemia-induced heart failure via the BMI1-p15-Rb signalling pathway. Can. J. Cardiol. 37, 655–664. doi:10.1016/j.cjca.2020.05.016
Yao, H., He, Q., Huang, C., Wei, S., Gong, Y., Li, X., et al. (2022). Panaxatriol saponin ameliorates myocardial infarction-induced cardiac fibrosis by targeting Keap1/Nrf2 to regulate oxidative stress and inhibit cardiac-fibroblast activation and proliferation. Free Radic. Biol. Med. 190, 264–275. doi:10.1016/j.freeradbiomed.2022.08.016
Yap, J., Irei, J., Lozano-Gerona, J., Vanapruks, S., Bishop, T., and Boisvert, W. (2023). Macrophages in cardiac remodelling after myocardial infarction. Nat. Rev. Cardiol. 2023. doi:10.1038/s41569-022-00823-5
Yokota, T., Mccourt, J., Ma, F., Ren, S., Li, S., Kim, T. H., et al. (2020). Type V collagen in scar tissue regulates the size of scar after heart injury. Cell 182, 545–562.e23. doi:10.1016/j.cell.2020.06.030
Yoshikawa, T., Port, J. D., Asano, K., Chidiak, P., Bouvier, M., Dutcher, D., et al. (1996). Cardiac adrenergic receptor effects of carvedilol. Eur. Heart J. 17 (2), 8–16. doi:10.1093/eurheartj/17.suppl_b.8
Younis, A., Zilberman, I., Krywanczyk, A., Higuchi, K., Yavin, H. D., Sroubek, J., et al. (2022). Effect of pulsed-field and radiofrequency ablation on heterogeneous ventricular scar in a swine model of healed myocardial infarction. Circ. Arrhythm. Electrophysiol. 15, e011209. doi:10.1161/CIRCEP.122.011209
Yu, Y., Hu, Z., Li, B., Wang, Z., and Chen, S. (2019). Ivabradine improved left ventricular function and pressure overload-induced cardiomyocyte apoptosis in a transverse aortic constriction mouse model. Mol. Cell Biochem. 450, 25–34. doi:10.1007/s11010-018-3369-x
Yue, Y., Huang, S., Wang, L., Wu, Z., Liang, M., Li, H., et al. (2020). M2b macrophages regulate cardiac fibroblast activation and alleviate cardiac fibrosis after reperfusion injury. Circ. J. 84, 626–635. doi:10.1253/circj.CJ-19-0959
Yurista, S. R., Matsuura, T. R., Silljé, H. H. W., Nijholt, K. T., Mcdaid, K. S., Shewale, S. V., et al. (2021). Ketone ester treatment improves cardiac function and reduces pathologic remodeling in preclinical models of heart failure. Circ. Heart Fail 14, e007684. doi:10.1161/CIRCHEARTFAILURE.120.007684
Yvette, N. L. (2021). Nintedanib: A review in fibrotic interstitial lung diseases. Drugs 81 (5), 575–586. doi:10.1007/s40265-021-01487-0
Zeigler, A. C., Nelson, A. R., Chandrabhatla, A. S., Brazhkina, O., Holmes, J. W., and Saucerman, J. J. (2020). Computational model predicts paracrine and intracellular drivers of fibroblast phenotype after myocardial infarction. Matrix Biol. 91-92, 136–151. doi:10.1016/j.matbio.2020.03.007
Zeng, B., Liao, X., Liu, L., Zhang, C., Ruan, H., and Yang, B. (2021). Thyroid hormone mediates cardioprotection against postinfarction remodeling and dysfunction through the IGF-1/PI3K/AKT signaling pathway. Life Sci. 267, 118977. doi:10.1016/j.lfs.2020.118977
Zhang, J., Lu, Y., Yu, P., Li, Z., Liu, Y., Zhang, J., et al. (2022a). Therapeutic hypothermia alleviates myocardial ischaemia-reperfusion injury by inhibiting inflammation and fibrosis via the mediation of the SIRT3/NLRP3 signalling pathway. J. Cell Mol. Med. 26, 4995–5007. doi:10.1111/jcmm.17523
Zhang, J., Wang, Y. J., Wang, X., Xu, L., Yang, X. C., and Zhao, W. S. (2019a). PKC-mediated endothelin-1 expression in endothelial cell promotes macrophage activation in atherogenesis. Am. J. Hypertens. 32, 880–889. doi:10.1093/ajh/hpz069
Zhang, J., Zha, Y., Jiao, Y., Li, Y., Wang, J., and Zhang, S. (2022b). OTUD7B (Cezanne) ameliorates fibrosis after myocardial infarction via FAK-ERK/P38 MAPK signaling pathway. Arch. Biochem. Biophys. 724, 109266. doi:10.1016/j.abb.2022.109266
Zhang, M., Zhang, M., Zhou, T., Liu, M., Xia, N., Gu, M., et al. (2021a). Inhibition of fibroblast IL-6 production by ACKR4 deletion alleviates cardiac remodeling after myocardial infarction. Biochem. Biophys. Res. Commun. 547, 139–147. doi:10.1016/j.bbrc.2021.02.013
Zhang, N., Feng, B., Ma, X., Sun, K., Xu, G., and Zhou, Y. (2019b). Dapagliflozin improves left ventricular remodeling and aorta sympathetic tone in a pig model of heart failure with preserved ejection fraction. Cardiovasc Diabetol. 18, 107. doi:10.1186/s12933-019-0914-1
Zhang, Q., Wang, L., Wang, S., Cheng, H., Xu, L., Pei, G., et al. (2022c). Signaling pathways and targeted therapy for myocardial infarction. Signal Transduct. Target Ther. 7, 78. doi:10.1038/s41392-022-00925-z
Zhang, S., Wang, N., Ma, Q., Fan, F., and Ma, X. (2021b). LncRNA TUG1 acts as a competing endogenous RNA to mediate CTGF expression by sponging miR-133b in myocardial fibrosis after myocardial infarction. Cell Biol. Int. 45, 2534–2543. doi:10.1002/cbin.11707
Zhang, X., Qu, H., Yang, T., Liu, Q., and Zhou, H. (2022d). Astragaloside IV attenuate MI-induced myocardial fibrosis and cardiac remodeling by inhibiting ROS/caspase-1/GSDMD signaling pathway. Cell Cycle 21, 2309–2322. doi:10.1080/15384101.2022.2093598
Zhang, Y., Fu, C., Zhao, S., Jiang, H., Li, W., and Liu, X. (2022e). PRELP promotes myocardial fibrosis and ventricular remodelling after acute myocardial infarction by the wnt/β-catenin signalling pathway. Cardiovasc J. Afr. 33, 228–233. doi:10.5830/CVJA-2022-001
Zhang, Y. Q., Hong, L., Jiang, Y. F., Hu, S. D., Zhang, N. N., Xu, L. B., et al. (2021c). hAECs and their exosomes improve cardiac function after acute myocardial infarction in rats. Aging (Albany NY) 13, 15032–15043. doi:10.18632/aging.203066
Zhang, Y., Zhang, H., Yang, Z., Zhang, X. H., Miao, Q., Li, M., et al. (2022f). miR-155 down-regulation protects the heart from hypoxic damage by activating fructose metabolism in cardiac fibroblasts. J. Adv. Res. 39, 103–117. doi:10.1016/j.jare.2021.10.007
Zhang, Z., Chen, Y., Zheng, L., DU, J., Wei, S., Zhu, X., et al. (2023). A DUSP6 inhibitor suppresses inflammatory cardiac remodeling and improves heart function after myocardial infarction. Dis. Model Mech. 16, dmm049662. doi:10.1242/dmm.049662
Zhang, Z., Villalpando, J., Zhang, W., and Nam, Y. J. (2021d). Chamber-specific protein expression during direct cardiac reprogramming. Cells 10, 1513. doi:10.3390/cells10061513
Zhang, Z., Zhang, N., Shi, J., Dai, C., Wu, S., Jiao, M., et al. (2021e). Allograft or recipient ST2 deficiency oppositely affected cardiac allograft vasculopathy via differentially altering immune cells infiltration. Front. Immunol. 12, 657803. doi:10.3389/fimmu.2021.657803
Zhao, K., Xu, T., Mao, Y., Wu, X., Hua, D., Sheng, Y., et al. (2022a). Alamandine alleviated heart failure and fibrosis in myocardial infarction mice. Biol. Direct 17, 25. doi:10.1186/s13062-022-00338-6
Zhao, Y., DU, D., Chen, S., Chen, Z., and Zhao, J. (2022b)., 13. Genes (Basel), 1390. doi:10.3390/genes13081390New insights into the functions of MicroRNAs in cardiac fibrosis: From mechanisms to therapeutic strategiesGenes
Zheng, G., Zhang, J., Zhao, H., Wang, H., Pang, M., Qiao, X., et al. (2016). α3 integrin of cell-cell contact mediates kidney fibrosis by integrin-linked kinase in proximal tubular E-cadherin deficient mice. Am. J. Pathol. 186, 1847–1860. doi:10.1016/j.ajpath.2016.03.015
Zheng, J., Zhang, X., Cai, W., Yang, Y., Guo, T., Li, J., et al. (2022). Bone marrow mesenchymal stem cell-derived exosomal microRNA-29b-3p promotes angiogenesis and ventricular remodeling in rats with myocardial infarction by targeting ADAMTS16. Cardiovasc Toxicol. 22, 689–700. doi:10.1007/s12012-022-09745-7
Zhi, H., Luptak, I., Alreja, G., Shi, J., Guan, J., Metes-Kosik, N., et al. (2013). Effects of direct Renin inhibition on myocardial fibrosis and cardiac fibroblast function. PLoS One 8, e81612. doi:10.1371/journal.pone.0081612
Zhong, S., Guo, H., Wang, H., Xing, D., Lu, T., Yang, J., et al. (2020). Apelin-13 alleviated cardiac fibrosis via inhibiting the PI3K/Akt pathway to attenuate oxidative stress in rats with myocardial infarction-induced heart failure. Biosci. Rep. 40. doi:10.1042/BSR20200040
Zhong, Y., Yu, X., Li, X., Zhou, H., and Wang, Y. (2022). Augmented early aged neutrophil infiltration contributes to late remodeling post myocardial infarction. Microvasc. Res. 139, 104268. doi:10.1016/j.mvr.2021.104268
Zhou, J., Wen, T., Li, Q., Chen, Z., Peng, X., Wei, C., et al. (2022). Single-cell sequencing revealed pivotal genes related to prognosis of myocardial infarction patients. Comput. Math. Methods Med. 2022, 6534126. doi:10.1155/2022/6534126
Zhou, M. M., Li, D. W., Xu, L., Kong, B., Wang, X., Tang, Y. H., et al. (2023). Propionate alleviated post-infarction cardiac dysfunction by macrophage polarization in a rat model. Int. Immunopharmacol. 115, 109618. doi:10.1016/j.intimp.2022.109618
Zhou, X. L., Fang, Y. H., Wan, L., Xu, Q. R., Huang, H., Zhu, R. R., et al. (2019). Notch signaling inhibits cardiac fibroblast to myofibroblast transformation by antagonizing TGF-β1/Smad3 signaling. J. Cell Physiol. 234, 8834–8845. doi:10.1002/jcp.27543
Zhu, B., Zhu, X., Wang, X., Wu, J., Ding, S., Zhang, W., et al. (2022). Histamine deficiency promotes myofibroblasts transformation from HDC-expressing CD11b(+) myeloid cells in injured hearts post myocardial infarction. J. Cardiovasc Transl. Res. 15, 621–634. doi:10.1007/s12265-021-10172-2
Zhuang, L., Lu, L., Zhang, R., Chen, K., and Yan, X. (2020). Comprehensive integration of single-cell transcriptional profiling reveals the heterogeneities of non-cardiomyocytes in healthy and ischemic hearts. Front. Cardiovasc Med. 7, 615161. doi:10.3389/fcvm.2020.615161
Ziaja, M., Urbanek, K. A., Kowalska, K., and Piastowska-Ciesielska, A. W. (2021). Angiotensin II and angiotensin receptors 1 and 2-multifunctional system in cells biology, what do we know? Cells 10, 381. doi:10.3390/cells10020381
Zivarpour, P., Reiner, Ž., Hallajzadeh, J., Mirsafaei, L., and Asemi, Z. (2022). The effect of nutraceuticals on multiple signaling pathways in cardiac fibrosis injury and repair. Heart Fail Rev. 27, 321–336. doi:10.1007/s10741-020-09980-6
Zuo, P., Zhu, Y., Li, Y., Zuo, Z., Sheng, Z., Yan, G., et al. (2020). Protease-activated receptor 2 deficiency in hematopoietic lineage protects against myocardial infarction through attenuated inflammatory response and fibrosis. Biochem. Biophys. Res. Commun. 526, 253–260. doi:10.1016/j.bbrc.2020.03.077
Keywords: myocardial infarction, fibrosis, cardiac remodeling, antifibrotic therapy, extracellular matrix, fibroblast, myofibroblast
Citation: Yin X, Yin X, Pan X, Zhang J, Fan X, Li J, Zhai X, Jiang L, Hao P, Wang J and Chen Y (2023) Post-myocardial infarction fibrosis: Pathophysiology, examination, and intervention. Front. Pharmacol. 14:1070973. doi: 10.3389/fphar.2023.1070973
Received: 15 October 2022; Accepted: 13 March 2023;
Published: 28 March 2023.
Edited by:
Zhe-Sheng Chen, St. John’s University, United StatesReviewed by:
Camila Hochman-Mendez, Texas Heart Institute, United StatesKshitiz Kz, UCONN Health, United States
Hitoshi Kurose, Kyushu University, Japan
Copyright © 2023 Yin, Yin, Pan, Zhang, Fan, Li, Zhai, Jiang, Hao, Wang and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiali Wang, d2FuZ2ppYWxpXzIwMDBAMTI2LmNvbQ==; Yuguo Chen, Y2hlbjkxOTA4NUBzZHUuZWR1LmNu
†These authors have contributed equally to this work and share first authorship