
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol. , 20 April 2020
Sec. Cardiovascular and Smooth Muscle Pharmacology
Volume 11 - 2020 | https://doi.org/10.3389/fphar.2020.00413
This article is part of the Research Topic Perspectives on Antiarrhythmic Drug Therapy: Disappointing Past, Current Efforts and Faint Hopes View all 15 articles
 Balázs Horváth1,2*
Balázs Horváth1,2* Tamás Hézső1
Tamás Hézső1 Dénes Kiss1
Dénes Kiss1 Kornél Kistamás1
Kornél Kistamás1 János Magyar1,3
János Magyar1,3 Péter P. Nánási1,4
Péter P. Nánási1,4 Tamás Bányász1
Tamás Bányász1Based on recent findings, an increased late sodium current (INa,late) plays an important pathophysiological role in cardiac diseases, including rhythm disorders. The article first describes what is INa,late and how it functions under physiological circumstances. Next, it shows the wide range of cellular mechanisms that can contribute to an increased INa,late in heart diseases, and also discusses how the upregulated INa,late can play a role in the generation of cardiac arrhythmias. The last part of the article is about INa,late inhibiting drugs as potential antiarrhythmic agents, based on experimental and preclinical data as well as in the light of clinical trials.
During the non-pacemaker action potential (AP) in the heart, depolarization of the cell membrane opens voltage gated sodium channels (Nav) for a short period of time (Scanley et al., 1990; Mitsuiye and Noma, 2002) giving rise to the early sodium current peak (INa,early). This INa,early causes the upstroke of the non-pacemaker AP. Through the course of the AP Nav channels may recover from inactivation and reopen, generating a sustained current component, called late sodium current (INa,late). INa,late flows throughout the plateau phase of the AP therefore it significantly contributes to AP morphology, even though its magnitude is only a fraction of INa,early (Figure 1A).
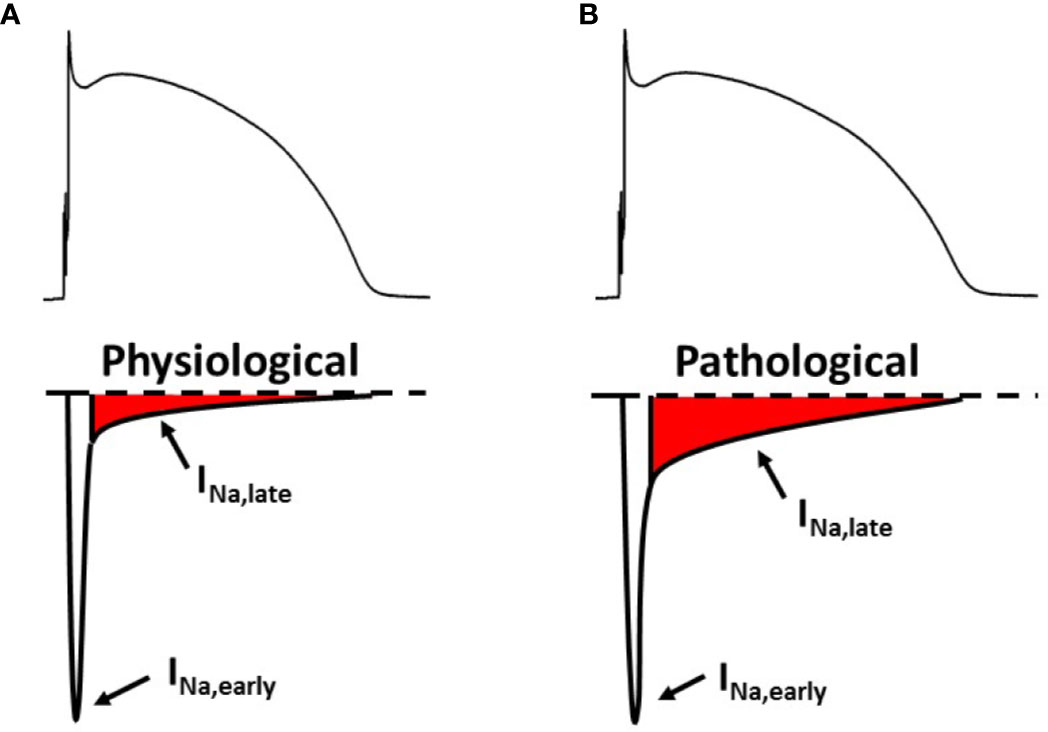
Figure 1 The early and the late component of the sodium current under physiological (A) and pathological (B) conditions. Upper panels: membrane potential; lower panels: sodium current. INa,early, early (peak) component of the sodium current; INa,late, late (sustained) component of the sodium current.
If INa,late is increased, it might play a pathophysiological role in acquired cardiac diseases (Figure 1B) such as myocardial ischemia (Maier and Sossalla, 2013) and heart failure (Coppini et al., 2013; Pourrier et al., 2014). In the cardiomyocytes, an upregulated INa,late hinders repolarization and causes a larger sodium entry, therefore increasing intracellular sodium concentration ([Na+]i). An increased [Na+]i, in turn, leads to a larger intracellular calcium content. These factors together can possibly cause contractile dysfunction (Sossalla et al., 2011), disturbed myocardial energetics (Liu and O'Rourke, 2008) and cardiac arrhythmias (Antzelevitch et al., 2014).
Mammalian cardiac cells express a wide variety of Nav isoforms, differing in unit conductance, voltage sensitivity, kinetics, and drug sensitivity. In the majority of cardiac tissues, the dominant isoform of the pore-forming subunit is Nav1.5, which is relatively insensitive to the sodium channel toxin tetrodotoxin (TTX) (Gellens et al., 1992; Catterall et al., 2005). Many of the TTX-sensitive (“non-cardiac”) Nav channels (Nav1.1, Nav1.2, Nav1.3, Nav1.4, and Nav1.6) are also shown to be present in cardiac tissue (Maier et al., 2002; Haufe et al., 2005; Valdivia et al., 2005; Biet et al., 2012; Yang et al., 2012). In nodal tissue Nav1.1 and Nav1.6 are expressed in the largest quantities. Besides the pore-forming subunit, four auxiliary subunits (ß1, ß2, ß3, and ß4) and certain scaffolding proteins also participate in building up the whole complex, which also attaches to the cytoskeleton. These molecules can interact with each other and may modify the kinetics and voltage dependence of the actual channel (Malhotra et al., 2001).
Mechanisms that are discussed in the followings may contribute to the profile of INa,late during the AP. Understanding these mechanisms better might be helpful in developing new antiarrhythmic therapeutic strategies targeting INa,late.
At the resting membrane potential, the vast majority of Nav1.5 channels are in their closed state. Upon depolarization, Nav1.5 channels open up within 1–2 ms after which they inactivate rapidly (Scanley et al., 1990; Mitsuiye and Noma, 2002). This produces INa,early and the upstroke of the non-pacemaker cardiac AP. During a sustained depolarization, Nav1.5 channels can reopen with a small probability. In ventricular myocytes, three modes of Nav1.5 channel activity have been characterized in single-channel experiments: transient mode (TM), burst mode (BM), and late scattered mode (LSM) (Maltsev, 2006).
INa,early is mainly the result of TM activity, while BM and LSM are responsible for the sustained sodium current, INa,late (Figure 1A). The magnitude of the sustained current component is only about 0.5–1 % of INa,early measured 50 ms after the onset of the depolarizing pulse (Maltsev, 2006). During a sustained depolarization BM openings rapidly decline in the first tens of milliseconds therefore leaving LSM as the gating mode being mainly responsible for INa,late toward the end of the plateau phase.
Mutations of the channel protein and certain diseases can change the contribution of different Nav1.5 channel activity patterns to the macroscopic current, therefore increasing INa,late (Bezzina et al., 1999; Valdivia et al., 2005; Wu et al., 2006; Maltsev et al., 2007; Maltsev and Undrovinas, 2008; Song et al., 2008; Maltsev et al., 2009; Xi et al., 2009; Guo et al., 2010; Trenor et al., 2012) (Figure 1B). Apparently, each gating mode has a distinct drug sensitivity or drug affinity as well (Belardinelli et al., 2004; Ravens et al., 2004; Belardinelli et al., 2006). Based on this, selective pharmacological targeting of certain gating modes might have potential antiarrhythmic and/or cardioprotective effects (Belardinelli et al., 2006; Hoyer et al., 2011; Morita et al., 2011).
The voltage dependence of the steady state activation and inactivation of most Nav channels overlaps with each other (Zaza and Rocchetti, 2013). This overlap provides a voltage range (“window”) where inactivated Nav channels are able to recover from inactivation and then might reopen. When the actual membrane potential falls within this “window” of overlap, a sustained current is evoked. Under physiological circumstances this “window current” mechanism likely plays a limited role in INa,late, because the Nav1.5 voltage “window” is around −70 mV, falling quite far from the AP plateau. Additionally, in the window voltage range, the current density is less than 5 % of the maximum current density in healthy myocytes (Maltsev et al., 1998; Wang et al., 2002; Liu et al., 2007). Hence, the “window current” mechanism is unlikely to be a major determinant of INa,late in healthy myocytes. Mutations of channel proteins or altered regulation in certain diseases may shift either the steady-state activation or inactivation curves of Nav channels to significantly change this voltage window, therefore increasing INa,late under these pathological conditions (Wang et al., 1996; Ruan et al., 2009).
During the AP of cardiac myocytes, the membrane potential changes continuously. Nav channels are incorporated into this dynamic system. It has been proposed by Clancy et al. (2003) that the voltage “history” of the cell membrane can modulate the transition between Nav channel states, termed “non-equilibrium gating”. As a result, recovery from inactivation is also modulated by the dynamics of voltage change. The theory is supported by experimental data showing that the application of repolarizing voltage ramps or AP shape voltage commands evoke a larger INa,late compared to conventional square pulses or model simulations where “non-equilibrium gating” is not incorporated into the numerical model (Clancy et al., 2003; Magyar et al., 2004; Horvath et al., 2013).
Epilepsy (Alekov et al., 2000; Akalin et al., 2003) and certain skeletal muscle diseases (Komajda et al., 1980; Pereon et al., 2003) has been associated with pathological ECG recordings. Therefore it seemed possible that non-cardiac sodium channel mutations might cause electrical alterations in the heart. Later, Nav1.1, Nav1.2, Nav1.3, Nav1.4, Nav1.6, and Nav1.8 isoforms have been identified in cardiac tissue (Maier et al., 2002; Haufe et al., 2005; Valdivia et al., 2005; Biet et al., 2012; Yang et al., 2012). Based on the findings of Biet et al., as much as 44 % of INa,late is due to non-cardiac sodium channels (Biet et al., 2012) in canine ventricular cardiomyocytes. Furthermore, Yang et al. have shown that in mice and rabbit the TTX-resistant Nav1.8 provides a substantial amount of INa,late (Yang et al., 2012). Based on these experimental data, isoform specific sodium channel modulators might provide a valid approach in pharmacological antiarrhythmic therapy (See Non-Cardiac Sodium Channel Inhibitors as Potential Antiarrhythmic Agent for further details).
Contribution of INa,late to cardiac APs was questioned because of its small density. However, the plateau phase of the cardiac AP is shaped by a delicate balance between minuscule inward and outward current fluxes. Therefore even a small change in these currents may significantly alter the duration of the AP (Horvath et al., 2006). Inhibition of INa,late substantially shortens the cardiac AP in the conductive system (Coraboeuf et al., 1979) and in ventricular cells (Kiyosue and Arita, 1989) as well, indicating that INa,late significantly contributes to determining the duration of the non-pacemaker AP in cardiac myocytes. Recent AP voltage clamp experiments show that the density of INa,late is of similar magnitude as the major potassium currents in guinea pig (Horvath et al., 2013) and rabbit (Hegyi et al., 2018) ventricular myocytes. There is a characteristic interspecies difference in the shape of INa,late as shown in the case of guinea pig, canine, and human ventricular myocytes (Horvath et al., 2020).
The sustained sodium current is also an important factor in determining electrophysiological properties of sinoatrial node cells (Maier et al., 2003; Lei et al., 2004). Tetrodotoxin, applied in lower than 1 µM concentrations, reduces the rate of spontaneous depolarization in sinoatrial node cells (Huang et al., 2015), clearly indicating that non-cardiac Nav isoforms also contribute to cardiac automaticity.
Cardiac Purkinje cells have the largest rate-dependence of their AP duration (APD) among cardiomyocytes with fast response APs. Purkinje cell APs are longer at lower stimulation rates, while shorter at higher rates than APs of ventricular cells. It has been shown that INa,late contributes to this feature by possessing much slower decay and recovery kinetics in Purkinje cells than in ventricular cells. As a result Purkinje cell INa,late is significantly larger at low heart rates, while smaller at high heart rates compared to ventricular cells. This unique feature predisposes Purkinje cells to serve as triggers in generating arrhythmias (Li et al., 2017).
INa,late plays a role in forming the atrial AP as well (Burashnikov and Antzelevitch, 2013; Luo et al., 2014). INa,late is expected to be larger in atria than in ventricles because INa, early density is greater in atrial cells under similar conditions (Li et al., 2002; Burashnikov et al., 2007), suggesting a higher sodium channel expression in atrial cells. On the other hand, an overall more positive membrane potential, and a more negative steady-state inactivation voltage of the sodium current (Li et al., 2002; Burashnikov et al., 2007) in the atrial cells reduce the availability of the sodium channels (Burashnikov and Antzelevitch, 2008). In one set of experiments by Luo et al. maximum INa,late density has been reported to be greater in rabbit left atrial myocytes than in ventricular cells (Luo et al., 2014) and in a different investigation the two cell types seemed to be similar in this matter (Persson et al., 2007). APs are shorter in the atria compared to the ventricles reducing the amount of Na+ influx through INa,late in the former (Burashnikov and Antzelevitch, 2013).
[Na+]i is set by a dynamic equilibrium of the influx of Na+ into the cell and efflux of Na+ to the interstitial space. The [Na+]i of non-paced ventricular myocytes is around 4–8 mM in guinea-pig, rabbit, and canine; and about twice as high in rat and mouse (9–14 mM) (Despa and Bers, 2013). In non-paced human myocytes [Na+]i is thought to be in the 4–10 mM range.
Na+ can enter into the cell through Na+ channels, Na+/Ca2+ exchanger (NCX) and Na+/H+ exchanger (NHE). Na+ leaves the cell mainly via the Na+/K+ pump (NKP), but the reverse mode NCX is also responsible for a moderate Na+ efflux during the first few milliseconds of the cardiac AP. Furthermore, Na+/HCO3− cotransport, Na+/Mg2+ exchange, and Na+/K+/2Cl− cotransport can play a role in the sodium homeostasis of cardiomyocytes to a small extent (Despa and Bers, 2013). It also has to be mentioned that Na+ concentrations between the cytosol and intracellular organelles are continuously balanced.
Upon pacing, [Na+]i increases with increasing stimulation frequency, caused by the larger Na+ entry through Na+ channels and NCX. In paced, single cardiac cells approximately 25 % of the Na+ entry is mediated by Nav channels (Despa and Bers, 2013). The Na+ entry through Nav channels is about equally distributed between INa,early and INa,late (Makielski and Farley, 2006; Zaza and Rocchetti, 2013; Despa and Bers, 2013; Shryock et al., 2013), however this contribution can change at different heart rates (see Heart Rate and AP Duration Influences INa,late for details). The higher Na+ influx into paced cells is matched by an increased efflux through an elevated NKP activity. This is mainly caused by the increased [Na+]i itself, but nitric oxide-, and phospholemman-dependent mechanisms can also add to this effect (Despa and Bers, 2013).
The NCX is a secondarily active transporter that carries 1 Ca2+ and 3 Na+ at the same time (Janvier and Boyett, 1996; Fujioka et al., 2000; Sipido et al., 2007; Despa and Bers, 2013; Ginsburg et al., 2013). The NCX function is determined by the relation of the actual membrane voltage and the sum of the actual electrochemical gradients of Ca2+ and Na+. The main role of NCX is to remove Ca2+ from the cells by utilizing the potential energy present in the form of Na+ gradient (“forward mode”). Besides this mode, in the first few milliseconds of the AP, NCX mediates Na+ extrusion from the cell and Ca2+ entry into the cytosol (“reverse mode”).
Being an inward current, INa,late depolarizes the membrane, causing an increased membrane potential throughout the plateau phase and a longer AP. The more time the membrane spends in a depolarized state (above +40 mV) the higher the possibility that L-type calcium channels can open or re-open. It is well documented with AP voltage clamp technique that the L-type calcium current is flowing throughout the AP plateau (Linz and Meyer, 1998; Linz and Meyer, 2000; Banyasz et al., 2003; Fulop et al., 2004; Banyasz et al., 2012). Therefore a longer AP inevitably results in a larger Ca2+ entry to the myocyte.
Heart rate determines the magnitude of INa,late. Like many electrophysiological characteristics of cardiac cells (Banyasz et al., 2009), INa,late is reverse-rate dependent, so the faster the stimulation rate the smaller the current density will be (Nagatomo et al., 2002; Wu et al., 2011). However, with increasing heart rate the density of INa,early and maximum rate of depolarization during the AP upstroke (Vmax; an AP parameter determined by INa,early) does not decrease that much (Nagatomo et al., 2002). This is because recovery of INa,late is much slower than INa,early (Carmeliet, 2006). At higher heart rates this feature of the two sodium current components also results in a decreasing contribution of INa,late to the overall Na+ influx. Under these conditions, the more frequent AP upstrokes cause a greater Na+ entry through INa,early, and there is a reduction of INa,late density because of the very slow INa,late recovery kinetics. Moreover, rate-dependent changes of the AP length also influence Na+ entry. At high heart rates APs are shorter, therefore INa,late is active for a shorter time, accounting for a further reduction of Na+ influx through the already smaller INa,late. At the same time, extrusion of Na+ by the NKP is reduced at high pacing rates (Despa and Bers, 2013) leading to a rate-dependent [Na+]i loading in isolated cells. It must also be noted that this phenomenon is largely offset or may not occur at all during β-adrenergic stimulation because it augments NKP activity through phospholemman (Cheung et al., 2010).).
As it is described in the previous section, APD influences INa,late: the shorter the AP the smaller the Na+ flux through INa,late is. Therefore under any conditions that result in a shorter AP the contribution of INa,late to the overall Na+ influx will be smaller. This fact, together with significant differences in heart rate underlies differences in INa,late between species having short APs (e.g.: rats or mice) and long APs (guinea pig, rabbit, pig, human, etc.). In rats and mice both INa,late and Na+ influx driven by INa,late should be much smaller than in species having long APs.
Ca2+ is the key player in the excitation-contraction coupling of cardiac cells and it also regulates many other cellular functions including sarcolemmal transport mechanisms. Nav channels are regulated by the individual and cooperative actions of Ca2+, calmodulin (CaM), and Ca2+-CaM dependent protein kinase II (CaMKII) as well (Bers and Grandi, 2009; Maier, 2011; Scheuer, 2011). Signaling through the Ca2+—CaM—CaMKII pathway is thought to facilitate the sodium current, especially INa,late (Maltsev et al., 2008; Maltsev et al., 2009; Bers and Grandi, 2009).
Motifs with Ca2+ binding (EF hand) as well as CaM binding (IQ motifs) capabilities are present in the Nav1.5 channel structure. Some groups have shown that Ca2+ alone can regulate sodium channels (Wingo et al., 2004), while other results support that Ca2+ is not capable of regulating Nav channels directly; the regulation is mediated via Ca2+-CaM complex (Tan et al., 2002; Kim et al., 2004). Besides the exact regulatory mechanism, the general agreement is that when Ca2+ is elevated the SSI curve shifts toward more positive voltages (Sarhan et al., 2012), although this is a largely negligible effect at physiologically relevant Ca2+ concentrations in wild type channels. However, under conditions when Ca2+ or CaM sensing regions are mutated or when the Ca2+ sensitivity of Nav channels are severely altered, diverse functional disturbances may arise leading to an increased INa,late.
Besides the direct regulation of Nav channels, the Ca2+-CaM complex activates CaMKIIδC that also modulates these channels (Zhang and Brown, 2004; Anderson, 2005; Bers and Grandi, 2009). The active CaMKII is a Ser/Thr kinase that can phosphorylate Nav1.5 channels on at least three amino acid residues (Grandi and Herren, 2014). While there is an ongoing debate about the exact role of these phosphorylation sites in channel gating, all the studies agree on that activation of CaMKII increases INa,late.
In a meticulous set of AP voltage clamp experiments, Hegyi et al. (Hegyi et al., 2018) showed how different downstream elements of the β-adrenergic pathway regulate INa,late in rabbit ventricular myocytes. Protein kinase A, CaMKII, Epac, nitrosylation, as well as reactive oxygen species (ROS) contributed to the upregulation of INa,late during different phases of the ventricular AP.
ROS and H2O2 increase INa,late (Song et al., 2004; Song et al., 2006; Sossalla et al., 2008). Some results suggest that CaMKII can be involved in INa,late facilitation observed in the presence of oxygen free radicals (Wagner et al., 2011), because ROS can also activate CaMKII (Erickson et al., 2008). See (Wagner et al., 2013) for a detailed review.
Acidosis also modulates Nav channels (Murphy et al., 2011; Jones et al., 2011; Jones et al., 2013a; Jones et al., 2013b). Acidosis caused a rightward shift in steady-state activation, but not in steady-state inactivation in isolated canine ventricular myocytes therefore reducing INa,late (Murphy et al., 2011).
Many studies have found that hypoxia increases INa,late (Ju et al., 1996; Carmeliet, 1999; Harnmarstrom and Gage, 2002; Wang et al., 2007; Shimoda and Polak, 2011; Tang et al., 2012). Following a 15 minute hypoxic period, Wang et al. reported an increased BM channel activity, a plausible explanation of the increased INa,late.
Intermediary lipid metabolites shown to increase INa,late. Nav channels treated with lysophosphatidylcholine exhibited a sustained BM channel activity (Burnashev et al., 1991; Undrovinas et al., 1992), while palmitoylcarnitine induced a slowly inactivating sodium current (Wu and Corr, 1994). According to more recent data, poly-unsaturated fatty acids (docosahexaenoic acid and eicosapentaenoic acid) reduce both INa,early and INa,late (Pignier et al., 2007). According to the authors, the reduction is caused by a decreased overlap between the steady-state activation and inactivation voltage range.
Nitric oxide (NO) has been shown to enhance INa,late (Ahern et al., 2000). The neural NO synthase (nNOS) belongs to the huge macromolecular complex of Nav1.5, with caveolin-3 and α1-syntrophin among some additional proteins (Cheng et al., 2013).
The possible promoter regions and their role in the regulation of human SCN5A gene transcription has already been reported. (Yang et al., 2004; van Stuijvenberg et al., 2010) Recent studies have shown that the zinc-finger transcription factor, GATA4 (Tarradas et al., 2017), and the myocyte enhancing factor-2C (MEF2C) enhances SCN5A transcription (Zhou et al., 2018). However, most likely many other transcription factors are involved in the transcriptional regulation of the SCN5A gene.
Some amino acid motifs found in the Nav1.5 protein are subject to N-glycosylation. Carbohydrates account for an about 5 % of the total mass of Nav channels in the rat heart (Cohen and Levitt, 1993). The lack of channel glycosylation caused shifts toward positive voltages in both steady state activation and inactivation curves when naturally sialic-acid deficient channels were used (Zhang et al., 1999), or when these carbohydrate residues were removed by enzymatic treatment (Ufret-Vincenty et al., 2001) Glycosylation also seem to be involved in channel trafficking (Mercier et al., 2015; Cortada and Brugada, 2019)
Upon protein kinase C activation, Na+ channels are internalized from the plasma membrane (Hallaq et al., 2012). For the process, both channel phosphorylation on S1503 and ROS are required (Liu et al., 2017).
The “Fyn” tyrosine kinase phosphorylates Nav1.5 channels on the Y1495 Tyr residue, located in the III–IV linker domain. This tyrosine residue helps with anchoring Ca2+/CaM to the inactivation gate of the channel (Sarhan and Van Petegem, 2009). When Fyn phosphorylates the channel on Y1495, it increases the window voltage range by shifting the steady-state inactivation toward more positive potentials (Ahern et al., 2005), therefore resulting in an enhanced INa,late.
There are three known arginine residues in Nav1.5 (R513, R526, and R680), that are subject to methylation (Beltran-Alvarez et al., 2011). These residues are found in the domain I and domain II linker region. There are two known mutations of these arginines (namely R526H and R680H) that cause Brugada (Kapplinger et al., 2010) and LQT3 syndromes (Wang et al., 2007), respectively.
Mechanical stimuli also affect channel gating in Nav1.5 channels. Beyder et al. investigated this phenomenon both in an expression system (Beyder et al., 2010) and in isolated mouse ventricular cells (Beyder et al., 2012). The pressure ramp applied by the authors caused a 235 % increase in LSM Nav1.5 channel openings suggesting that INa,late is enhanced by mechanical stress. Similar mechanical effects can modify certain signal transduction mechanisms like nNOS and CaMKII (Jian et al., 2014), which can, in turn, increase INa,late.
The pathophysiology of cardiac arrhythmias is based on the classical concept of “arrhythmic triad”; combination of a proarrhythmic substrate, a trigger, and the modulating effect of the autonomic nervous system (Merchant and Armoundas, 2012). The exact combination depends on etiology, cardiac-, and extracardiac comorbidities. Abnormal [Na+]i homeostasis can play a role in creating an arrhythmia-prone substrate as well as in generating a trigger for the rhythm disorder. The discussed mechanisms are summarized on Figure 2.
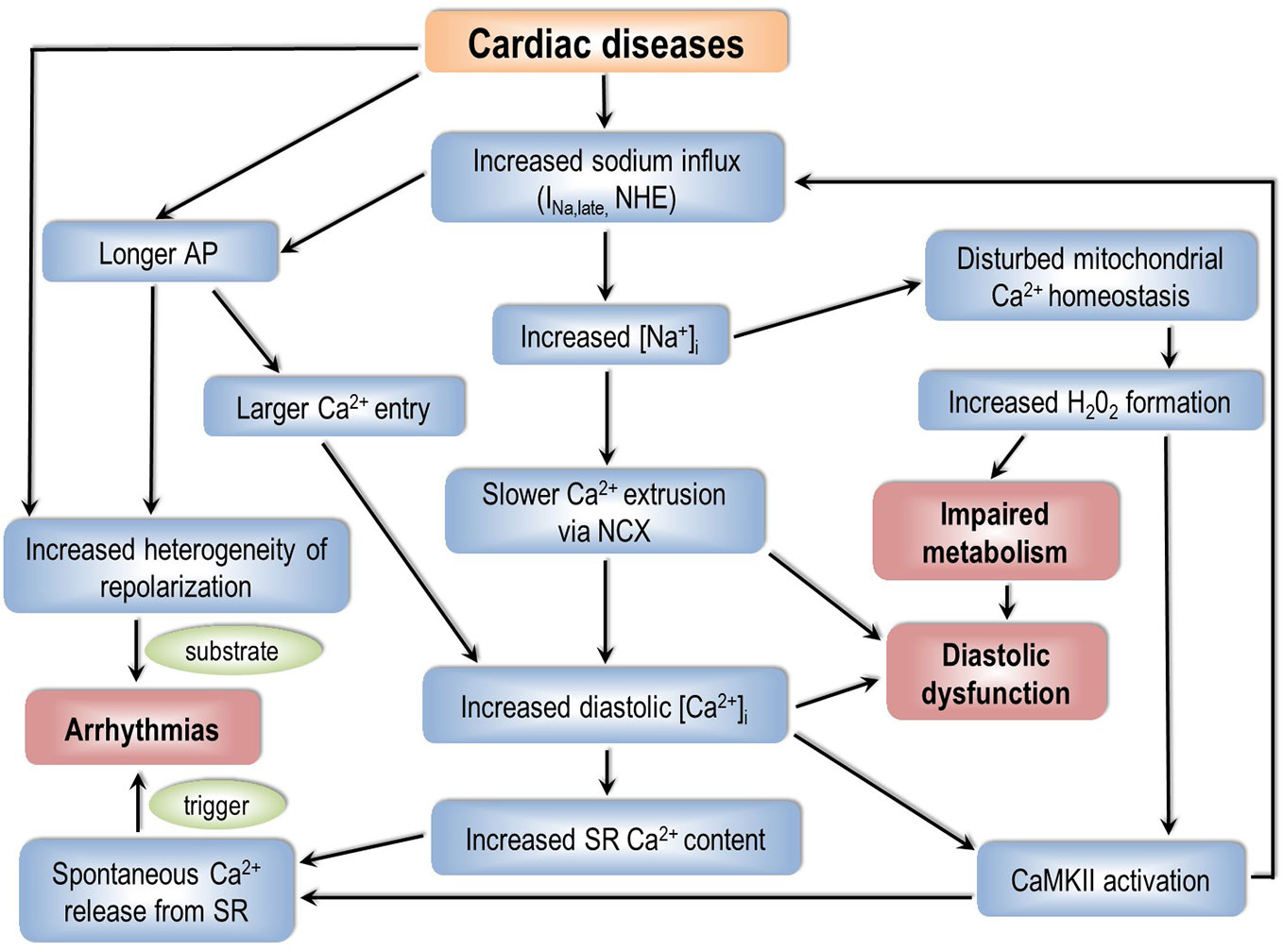
Figure 2 How can an impaired sodium homeostasis of cardiac myocytes lead to arrhythmias? AP, action potential; NHE, Na+/H+ exchanger; [Na+]i, intracellular sodium concentration; NCX, Na+/Ca2+ exchanger; [Ca2+]i, intracellular calcium concentration; SR, sarcoplasmic reticulum; CaMKII, calcium/calmodulin dependent protein kinase II.
Compared to non-failing myocytes, [Na+]i is about 2–6 mM larger in myocytes from failing hearts (Pieske et al., 2002; Despa et al., 2002; Schillinger et al., 2006; Louch et al., 2010). In a pressure- and volume-overload rabbit HF model, Despa et al. have found an increased TTX-sensitive Na+ influx (Despa et al., 2002). Interestingly, this larger influx was present not only in electrically stimulated myocytes, but in non-paced cells as well. In paced cells the most plausible candidate of this increased TTX-sensitive Na+ influx is INa,late. However, the underlying mechanism of this influx is not yet understood completely in resting myocytes.
Many cardiac diseases are associated with an increased INa,late. The list contains cardiac myocytes originating from end-stage HF (Maltsev et al., 1998; Maltsev et al., 2007) and post-myocardial infarction (Huang et al., 2001) preparations as well as animal HF models (Valdivia et al., 2005; Maltsev et al., 2007). The larger INa,late can be caused by several pathophysiologic factors including oxidative stress (ROS (Song et al., 2006; Sossalla et al., 2008) and NO (Ahern et al., 2000) mainly by S-nitrosylation of the Nav1.5 channels (Cheng et al., 2013)), hypoxia (Carmeliet, 1999; Tang et al., 2012), mechanical stress (Beyder et al., 2012), and certain ischemic metabolites, for example oxidized lipids (Burnashev et al., 1991). Looking at gating modes in single Nav1.5 channels, enhanced INa,late is likely underlain by an increased number of BM and LSM openings (Undrovinas et al., 2002; Maltsev, 2006) in HF.
The Ca2+—CaM—CaMKII signal transduction pathway is upregulated in HF (Bers, 2010), and this pathway has been shown to increase INa,late (Tan et al., 2002; Wagner et al., 2006; Ashpole et al., 2012; Ma et al., 2012). Oxidation activates CaMKII (Wagner et al., 2011) and keeps it constitutively active. The enhanced CaMKII-mediated Nav1.5 phosphorylation, therefore, certainly takes part in increasing INa,late under oxidative stress. Recent studies have found that Nav1.8 expression is significantly up-regulated, while Nav1.5 is reduced in human left ventricular hypertrophy (Ahmad et al., 2019) and HF (Dybkova et al., 2018).
When [Na+]i is elevated, it makes the NCX forward mode energetically less favorable, therefore a smaller amount of Ca2+ will leave the cell through NCX. This causes an increased [Ca2+]i load, and therefore further activates CaMKII, leading to enhanced phosphorylation of CaMKII targets such as Nav1.5. This, in turn, increases INa,late, which further elevates [Na+]i finally creating an arrhythmogenic vicious circle (Grandi and Herren, 2014). By using genetic (LQT3 mutation) as well as pharmacological (anemone toxin-II, ATX-II) approaches to increase INa,late, and therefore achieve [Na+]i loading, Yao et al. described this feedback (Yao et al., 2011). These conditions lead to the vicious circle described above, and as a result, arrhythmias can be generated because of an increase in the CaMKII-dependent phosphorylation of phospholamban and RyRs.
The mitochondrial NCX dynamically equilibrate concentrations of Ca2+ and Na+ of the mitochondrion and the cytosol. Ca2+ in the mitochondrion plays a role in determining the production of ATP and ROS by regulating the expression of enzymes involved in oxidative phosphorylation (Yang et al., 2014). If [Na+]i is elevated, it will impair Ca2+ accumulation in the mitochondrion at high pacing rates, leading to a decrease in NADH/NAD+ redox potential. This increases H2O2 generation in the cells (Liu and O'Rourke, 2008), causing oxidative stress and thereby directly and indirectly (through CaMKII (Erickson et al., 2008)) activating INa,late. Finally, the process leads to a further increase in [Na+]i (Wagner et al., 2011). This shows that, similar to an elevated [Na+]i, CaMKII activation can be caused by and can also lead to an increased ROS production.
Many inherited and acquired diseases can lead to a longer ventricular repolarization, presented as long QT (LQT) syndromes (El-Sherif et al., 2019; Locati et al., 2019). The inherited LQT3 syndrome is caused by an increased INa,late because of a mutant, much slower inactivating Nav1.5 channel. Acquired LQTs include for example heart failure (Maltsev et al., 1998; Maltsev et al., 2007; Coppini et al., 2013), myocardial ischemia and post-infarction state (Huang et al., 2001; Rivera-Fernandez et al., 2016), and type 2 diabetes mellitus (Ninkovic et al., 2016).
Under physiological conditions there is a fine balance between the inward and outward currents during the AP plateau. During the plateau phase the impedance of the membrane is large, therefore even a small change in the delicate balance can lead to a marked change in AP duration. In this setting, the depolarizing drive caused by an increased INa,late causes a longer AP (Studenik et al., 2001; Horvath et al., 2013), as well as under a longer AP, INa,late will generate a larger Na+ influx. Even in normal hearts, both APD and INa,late is greater in Purkinje fibers and in “M” cells than in the rest of the myocardium contributing to the physiological heterogeneity of repolarization. LQT syndromes increase both the spatial heterogeneity of repolarization (Maltsev et al., 2007) and the temporal variability of repolarization (El-Sherif et al., 2019) and therefore can present an arrhythmogenic substrate. This can be further exaggerated by bradycardia, where the APs are already long, and having larger heterogeneity (Szentandrassy et al., 2015). Cardiac diseases can also provide the proarrhythmic substrate in the form of temporal repolarization heterogeneity, “repolarization alternans” (Bonatti et al., 2014; Justo et al., 2016) which phenomenon is more pronounced in tachycardia.
The trigger is also highly rate-dependent. At low heart rates, where the cardiac APs are already long even under physiological conditions, an augmented INa,late can further prolong repolarization therefore increasing the probability of early afterdepolarizations (EADs), and the risk for (fatal) ventricular arrhythmias (Wang et al., 1995; Wang et al., 1996; Makita et al., 2002; Hedley et al., 2009; Cardona et al., 2010; Yamamura et al., 2010; Lowe et al., 2012). Severe bradycardia together with an enhanced INa,late and a long APD may also promote delayed afterdepolarization (DAD)-mediated triggered activities (Song et al., 2008; Coppini et al., 2013; Horvath et al., 2013). These triggered activities seem to heavily depend on an increased [Ca2+]i.
As described previously, an increased [Na+]i offsets NCX, decreasing Ca2+ removal from the cytosol (Bers, 2002; Nagy et al., 2004; Despa and Bers, 2013). This elevates diastolic [Ca2+]i and therefore increasing SR Ca2+ content; leading to spontaneous Ca2+ release events from the Ca2+-overloaded SR (Gyorke and Terentyev, 2008). This can generate DADs and therefore possibly triggering arrhythmias. At high heart rates this can further be aggravated by the two feedback loops involving CaMKII, as described in the previous sections, resulting in an enhanced CaMKII mediated phosphorylation of RyR2 therefore increasing the probability of spontaneous SR Ca2+ release events. It must be noted again that in vivo, there is no high heart rate without β-adrenergic stimulation. Adrenergic stimulation on one hand further activates CaMKII (Hegyi et al., 2018) but on the other hand, it also reduces or even diminishes [Na+]i loading of the cells by enhancing NKP activity (Cheung et al., 2010). This makes the role of INa,late in DAD-mediated arrhythmias occurring at high heart rates questionable.
In the diseased heart, however, rate-dependent properties of INa,late and [Na+]i are quite poorly investigated. At high pacing rates, INa,late decreases in LQT3 ΔKPQ mutant cells (Nagatomo et al., 2002) and an increased [Na+]i load was reported in hypertrophied feline cells (Mills et al., 2007) as well as in human cardiomyocytes from failing hearts (Pieske et al., 2002).
Pharmacologically enhanced INa,late increases repolarization heterogeneity in intact, isolated rabbit and guinea pig hearts (Restivo et al., 2004; Milberg et al., 2005), as well as in canine left ventricular wedge preparations giving rise to TdP (Shimizu and Antzelevitch, 1999a; Shimizu and Antzelevitch, 1999b). ATX-II also induces AF in a wide range of experimental conditions (Lu et al., 2012; Liang et al., 2016). Many gain-of-function SCN5A mutations (including LQT3) have been associated with atrial fibrillation (AF) (Benito et al., 2008). Also, in cases of chronic (permanent) AF, larger INa,late was found (Sossalla et al., 2010; Poulet et al., 2015). These data suggest that enhancement of INa,late might play a role in generating or maintaining AF most likely because of [Na+]i overload dependent Ca2+ overload (Nattel and Dobrev, 2012).
Natural products of peptide and non-peptide structure can inhibit sodium channels, although these compounds have negligible therapeutical relevance. Clinically relevant small-molecule sodium channel inhibitors include local anesthetics, anticonvulsants, and antiarrhythmic agents such as lidocaine, carbamazepine, phenytoin, lamotrigine, and mexiletine. These small-molecule inhibitors all bind to the so-called “local anesthetic site” of sodium channels where amino acid residues are highly conserved among different Nav subtypes (de Lera Ruiz and Kraus, 2015). Because of this, the “classic” Nav blockers are not subtype specific, they inhibit all subtypes to a certain extent. Also, these compounds somewhat inhibit both INa,early and INa,late, usually having a higher inhibitory effect on INa,late. Therefore most Nav blockers reduce excitability and impulse propagation (parameters associated with INa,early) together with the plateau sodium current (INa,late).
A few sodium channel blockers differ from the “classic” inhibitors, because they inhibit INa,late more potently than INa,early. The molecular mechanism of the preferential INa,late inhibition is still not completely understood. Even though ranolazine was used for most of the experimental and clinical studies, other selective INa,late inhibitors also exist such as lidocaine, GS-458967, GS-462808, F15845, and GS-6615 (eleclazine). The half-maximal inhibitory concentration (IC50) values of these inhibitors for the late and the early sodium current component are summarized in Table 1. For a more thorough data summary on this, see Table 2 in the review of Antzelevitch et al. (2014).
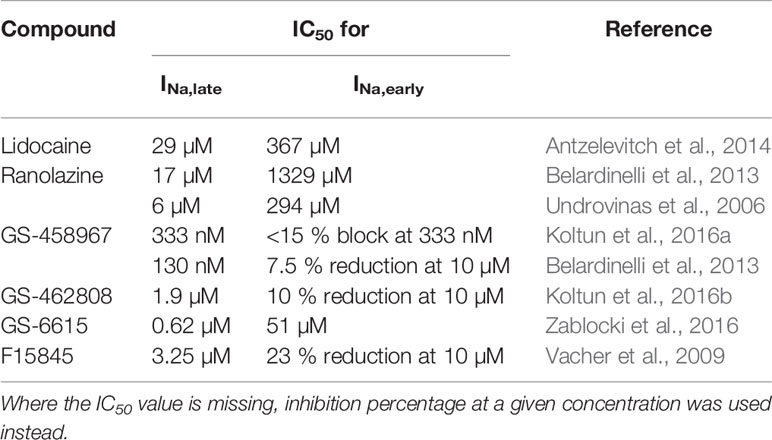
Table 1 IC50 values of selective late sodium current inhibitors for the late and the early sodium current component.
For lidocaine, IC50 values of around 25 and 300 µM were determined for INa,late and INa,early, respectively (Antzelevitch et al., 2014).
In case of ranolazine, the IC50 values are 17 µM for INa,late and 1,329 µM for INa,early in rabbit (Belardinelli et al., 2013), whereas 6 µM for INa,late (Antzelevitch et al., 2004; Undrovinas et al., 2006) and 294 µM for INa,early (Undrovinas et al., 2006) in canine ventricular myocytes.
GS-458967 was found to have an IC50 of 333 nM for INa,late inhibition while exhibiting smaller than 15% block of INa,early at the same concentration at 1 and 3 Hz pacing frequencies (Koltun et al., 2016a) measured on Nav1.5 channels expressed in HEK-293 cells with automated patch-clamp. In rabbit ventricular cardiomyocytes, the IC50 was 130 nM for INa,late, and at 10 µM, GS-458967 caused an approximately 7.5 % reduction in INa,early. (Belardinelli et al., 2013). Unfortunately for the developer, GS-458967 had a high brain penetration and a profound use dependent block on all the various sodium channel isoforms, making the compound prone for possible central nervous system side effects (Koltun et al., 2016a).
GS-462808 has an IC50 of 1.9 µM for INa,late inhibition while blocking 10 % of INa,early at 10 µM and it is also more cardiac isoform selective than GS-458967 blocking only 8 % of the Nav1.1 peak current. The problem with GS-462808 is that it caused liver lesions during the acute animal toxicity tests (Koltun et al., 2016b).
For GS-6615 the IC50 values of 0.62 and 51 µM were reported for INa,late and INa,early blockade, respectively, in manual patch-clamp experiments, with practically no effect on Nav1.1 channels (Zablocki et al., 2016).
F15845 has an IC50 of 3.25 µM for the inhibition of veratridine-induced INa,late while blocking 23 % of INa,early at 10 µM (Vacher et al., 2009). Last experimental data about F15845 were published in 2010, where it prevented ischemia-induced arrhythmias in rats (Pignier et al., 2010). Since then no new results came out regarding this agent.
Selectivity of these specific INa,late inhibitors is usually voltage-dependent, these blockers have very little effect on INa,early at more negative (quite unphysiological, for example −120 mV) holding potentials. As the holding potential gets closer to physiological resting membrane potentials, the selectivity of these compounds decrease, they start to inhibit INa,early more. Also, most inhibitors block the sodium channels in a rate-dependent (“use-dependent”) fashion; the blockers are more effective at rapid than at slow heart rates. This is because most inhibitors preferentially bind to the open and/or inactivated channels rather than the closed channel. This effect is especially strong in sodium channel blockers having fast association and dissociation kinetics (Pless et al., 2011) (Vaughan-Williams class Ib agents).
In case of 1 µM GS-458967 for example, INa,early did not change in rabbit ventricular myocytes held at −120 mV at pacing rates of 0.1, 1, or 3 Hz. When the holding potential was −80 mV, however, 1 µM GS-458967 reduced INa,early by 48 ± 7%, 50 ± 7%, and 56 ± 8% at rates of 0.1, 1, and 3 Hz, respectively (Belardinelli et al., 2013).
Ranolazine also inhibits sodium channels in a voltage-, and use-dependent fashion, moreover this blockade is also significantly larger in atria compared to ventricles (Zygmunt et al., 2011). With 50 ms long depolarizing pulses and 250 ms diastolic intervals (at 3.33 Hz), the use-dependent block by ranolazine at −120 mV was 21 % in ventricular, versus 32 % in atrial cells, whereas at −100 mV the block was 47 % versus 56 %, respectively. This data suggest that the rate dependency (use-dependency) is very pronounced in case of INa,early inhibition, but much smaller with INa,late. Therefore, based on the rate-dependent physiological (see Heart Rate and AP Duration Influences INa,late) and pharmacological characteristics of INa, early and INa,late, a quite selective inhibition of INa,late might be achieved at slow heart rates and with long APs, but at fast rates, with short AP duration, sodium channel blockers similarly inhibit both INa, early and INa,late.
At therapeutical plasma concentrations, ranolazine inhibits other ionic currents besides INa,late. This includes IKr (approximately 40 % inhibition at 6 µM), and ICa,L (around 25 % inhibition at 6 µM) (Antzelevitch et al., 2004). Consequently, inhibiting INa,late and applying ranolazine are very far from being identical concepts. When ranolazine is used to inhibit INa,late, effects on other channels must not be forgotten. Besides the previous features, ranolazine is also a weak β-adrenergic antagonist (Letienne et al., 2001) and an inhibitor of fatty acid oxidation (Chaitman, 2006), even though this latter effect only becomes prominent at supratherapeutical plasma concentrations.
Riluzole blocks TTX-sensitive sodium channels preferentially, which are associated with damaged neurons (Song et al., 1997). Riluzole also directly inhibits the kainate and NMDA receptors (Debono et al., 1993) as well as potentiates GABAA receptors (He et al., 2002). In anaesthetized pigs, myocardial damage and arrhythmias induced by coronary occlusion has been reduced by riluzole (Weiss et al., 2013). Riluzole has also been found to be anti-ischemic and antiarrhythmic in a pig model of acute myocardial infarction. (Weiss and Saint, 2010).
Targeting Nav1.8 with specific inhibitors might provide a potential novel approach in the future in antiarrhythmic drug therapy, because recent studies have found that Nav1.8 expression is significantly up-regulated in human left ventricular hypertrophy (Ahmad et al., 2019) and HF (Dybkova et al., 2018). By using Nav1.8-specific blockers [either A-803467 (30 nM) or PF-01247324 (1 μM)] the authors managed to reduce INa,late and APD in these experiments. Other Nav1.8 specific inhibitors include funapide and VX-150, however these compounds have not been tested in relation to cardiac pathophysiology so far.
Because of the pronounced use-dependent effect of specific INa,late inhibitors, interpretation of experimental studies conducted on rats and mice (having resting heart rates around 400 bpm) are very difficult. Therefore this review will focus on experimental data originating from larger mammalian species.
As it was demonstrated in the previous sections, INa,late has quite different characteristics under different heart rates. Therefore it is worthwhile to split the ventricular arrhythmia topic into two subtopics accordingly.
Many in vitro experimental studies have shown that at low pacing rates with prolonged APs and increased repolarization heterogeneity (LQT3 syndrome, heart failure, hypertrophic cardiomyopathy), inhibition of INa,late effectively reduces the burden of arrhythmic episodes [EADs, DADs, triggered APs, Torsade de Pointes (TdP) (Shimizu and Antzelevitch, 1997; Song et al., 2004; Coppini et al., 2013; Belardinelli et al., 2013; Rajamani et al., 2016)].
Ranolazine and GS-458967 has been shown to suppress dofetilide-induced TdP in a canine in vivo model (Antoons et al., 2010; Bossu et al., 2018).
There was one experimental study about the potential antiarrhythmic role of F15845, where it prevented ischemia-induced arrhythmias in rats (Pignier et al., 2010). However the use of a rat model makes it hard to extrapolate this study to humans.
Under the pathological conditions listed above, the fine balance between the inward and outward currents during the AP plateau is shifted toward the depolarizing inward currents, resulting in a longer AP. Therefore, theoretically, any intervention that reduces the depolarizing currents (e.g.: L-type calcium current, NCX current, INa,late) could be effective in bringing the repolarization closer to normal. In this setting, therefore, inhibiting INa,late will reduce the depolarization drive resulting in a significantly shorter APD and the suppression of arrhythmogenic events such as EADs, even if the magnitude of INa,late is not increased. Under similar conditions, other interventions such as L-type calcium channel blockade (Abrahamsson et al., 1996) or potassium channel activation (Carlsson et al., 1992) can also shorten APD, reduce repolarization heterogeneity, and suppress the occurrence of arrhythmogenic events even if INa,late is upregulated. In LQT syndromes INa,late-mediated increase in Ca2+i is just a fraction of the total Ca2+i, and even total Ca2+i just contributes to rather than determines the arrhythmogenic events (Carlsson et al., 1996).
INa,late blockers seem to effectively prevent or terminate tachycardia-induced ventricular tachycardia, and ventricular fibrillation in healthy animal models in the presence of a β-adrenergic agonist (Alves Bento et al., 2015; Carneiro et al., 2015; Bacic et al., 2017).
However, inhibition of INa,late does not likely play a crucial role here, based on the following theoretical considerations. To start with, in healthy ventricular tissue at high heart rates INa,late is quite small, as it was discussed in Heart Rate and AP Duration Influences INa,late. Furthermore, at rapid heart rates with β-adrenergic stimulation the major arrhythmogenic mechanism is likely to be the increased L-type calcium current, the increased SR Ca2+ content, and the leaky RyR together (Merchant and Armoundas, 2012). The third but similarly important factor is that these VT/VF episodes are likely underlain by a reentry mechanism, therefore heavily depending on the fast conduction provided by INa,early. At high pacing rates the “specific” INa,late inhibitors will also block a considerable amount of INa,early as well (see Selective INa,late Inhibitors for details), and this might just be enough to break the reentry circuit (Burashnikov and Antzelevitch, 2017).
Based on the experimental data, INa,late inhibition seems to be a valid therapeutic approach to tackle ventricular arrhythmias especially at low heart rates. These experimental studies also suggest that INa,late inhibition mainly affects the arrhythmogenic substrate by making the repolarization less heterogenous (Carneiro et al., 2015), with only low impact on suppressing the triggers (Bossu et al., 2018).
GS-458967 was shown to suppress isoproterenol-, and high Ca2+-induced DADs in healthy canine pulmonary-, and superior vena cava preparations (Sicouri et al., 2013). GS-458967 also suppressed autonomically triggered AF in an intact porcine model (Carneiro et al., 2015). In other experimental studies, “classic” sodium channel inhibitors (eg, lidocaine, flecainide) also prevented and terminated AF (Wang et al., 1992; Comtois et al., 2008). However, these agents were used at concentrations causing a suppression of INa,early. Experimental data about ranolazine shows an effective reduction of AF burden (AFB) only at concentrations that potently inhibit both INa,early (Burashnikov et al., 2007; Kumar et al., 2009; Burashnikov et al., 2014) and IKr (Burashnikov et al., 2007) Suppressing IKr reduces the diastolic interval between APs therefore promoting rate-dependent INa,early inhibition.
Based on these data, specific INa,late blockade alone is not a clear and straightforward approach in AF, except for cases when a longer atrial AP is the pathogenetic factor in the initiation of AF.
So far, ranolazine has been used in the vast majority of clinical studies involving INa,late blockers. When interpreting these trials, it has to be considered that ranolazine has other effects besides inhibiting INa,late. With the use of ranolazine, the first favorable results from phase 2 clinical trials were published in the 1990s (Cocco et al., 1992; Thadani et al., 1994). In 2006, following the outcome of the MARISA (Chaitman et al., 2004a), CARISA (Chaitman et al., 2004b), and ERICA (Stone et al., 2006) trials, the Food and Drug Administration approved ranolazine as an anti-anginal agent.
The effect of clinical outcome and safety of ranolazine therapy was investigated in more than 6,500 patients with non-ST-elevation acute coronary syndrome in the MERLIN TIMI-36 trial (Morrow et al., 2007). Although cardiovascular death or myocardial infarction has not been significantly reduced by ranolazine when compared to standard therapy; but recurrent ischemia (Morrow et al., 2007) and the incidence of arrhythmias (Scirica et al., 2007) were significantly lower with ranolazine. Treatment with ranolazine, compared to placebo, resulted in significantly lower incidences of arrhythmias. Fewer patients had episodes of ventricular tachycardia lasting more than eight beats [166 (5.3%) versus 265 (8.3%); p<0.001], supraventricular tachycardia [1413 (44.7%) versus 1752 (55.0%); p<0.001], or new-onset AF [55 (1.7%) versus 75 (2.4%); p=0.08]. Moreover, longer than 3 s pauses were less frequent with ranolazine [97 (3.1%) versus 136 (4.3%); p=0.01].
In the double-blind HARMONY (ClinicalTrials.gov ID: NCT01522651) phase 2 trial (Reiffel et al., 2015), patients with paroxysmal AF and implanted pacemakers were enrolled, so that AFB could continuously be monitored over the 12 weeks of treatment period. Patients were randomized to placebo, ranolazine alone (750 mg twice a day—BID), dronedarone alone (225 mg BID), or ranolazine (750 mg BID) combined with dronedarone (either 150 mg BID or 225 mg BID). The idea behind the combination was to reduce the dose of dronedarone, and therefore the negative inotropic effect associated with dronedarone. Placebo or the drugs alone did not significantly reduce AFB. In the combination therapies, however, ranolazine with dronedarone 225 mg BID reduced AFB by 59% vs placebo (p=0.008), and ranolazine with dronedarone 150 mg BID reduced AFB by 43% (p=0.072). Also, patients tolerated both combinations well.
Into the RAFFAELLO phase 2 trial (De Ferrari et al., 2015) patients with persistent AF (7 days to 6 months) were enrolled. Two hours after successful electrical cardioversion participants were randomized to either placebo, or ranolazine 375 mg, 500 mg, or 750 mg BID. Patients were monitored daily by transtelephonic ECG. The primary end-point was the time to first AF recurrence. No dose of the ranolazine prolonged time to AF recurrence significantly compared to placebo. Of the 238 patients who took at least one dose of the study drug, AF recurred in 56.4%, 56.9%, 41.7%, and 39.7% of patients in the placebo and ranolazine 375 mg/500 mg/750 mg groups, respectively. The reduction in overall AF recurrence in the combined 500-mg and 750-mg groups was of borderline significance compared to the placebo group (p=0.053) and significant compared to 375-mg group (p=0.035).
The RAID clinical trial (NCT01534962) (Zareba et al., 2018) investigated high-risk cardiomyopathy patients who received an implantable cardioverter-defibrillator (ICD). The subjects received either ranolazine (1,000 mg BID) or placebo. The primary endpoints were VT or VF requiring ICD shock or death. Among 1,012 ICD patients the ranolazine versus placebo hazard ratio was 0.84 (95% confidence interval: 0.67 to 1.05; p=0.117) for the primary endpoint. In the ranolazine group the risk of ICD therapies for recurrent VT or VF were significantly lower (hazard ratio: 0.70; 95% confidence interval: 0.51 to 0.96; p=0.028). Other effects of ranolazine treatment however has not been significant. These included individual components of the primary endpoint, quality of life, cardiac hospitalizations, and inappropriate ICD shocks as well.
In a smaller group of participants of the RAND-CFR trial (NCT01754259) (Evaristo et al., 2018) where symptomatic diabetic patients participated with non-flow-limiting coronary artery stenosis with diffuse atherosclerosis and/or microvascular dysfunction, effect of ranolazine on T-wave heterogeneity was evaluated. At physical rest, in the ranolazine group T-wave heterogeneity was 28 % smaller (placebo: 47±6 μV, ranolazine: 39±5 μV, p=0.002), however ranolazine did not differ from the placebo group during exercise. The trial also suggested that reduction in repolarization abnormalities seemed to be independent of alterations in myocardial blood flow.
In a meta-analysis of eight randomized clinical trials (Gong et al., 2017) Gong et al. found that ranolazine significantly reduced AF incidence in different clinical settings, such as in acute coronary syndromes, after cardiac surgery and after electrical cardioversion of AF (relative risk=0.67, 95% confidence interval: 0.52–0.87, p=0.002). Moreover, the combined use of ranolazine and amiodarone compared to amiodarone alone showed a 1.23-times higher conversion rate of AF (95% confidence interval: 1.08–1.40), together with a significantly, about 10 h shorter conversion time.
Based on the evidence above, ranolazine may have therapeutic role in the treatment of cardiac rhythm disorders, of both atrial and ventricular origin. For stronger evidence, more phase 3 clinical investigations are necessary.
Besides ranolazine, until now eleclazine was the only other selective INa,late inhibitor drug candidate that made it to phase 3 clinical trials. In the first trial (NCT02300558) eleclazine was tested for safety, tolerability, and its effect on shortening of the QT interval in LQT3 patients. The primary outcome of the study showed that after 24 weeks the mean daytime corrected QT interval was significantly, 8.5 ms shorter than at baseline, and there was only one patient with a serious adverse event (nephrolithiasis). The other trial (LIBERTY-HCM; NCT02291237) targeted HCM patients for the effect of eleclazine on exercise capacity. In this trial, eleclazine has not been proven to be superior to placebo.
The last moment in the development of eleclazine came after results of the phase 2 TEMPO study (NCT02104583) were analyzed. In the trial, subjects with ventricular tachycardia/ventricular fibrillation and ICD participated. Results of the study have shown that the rate of ICD shocks was higher in subjects who received eleclazine compared to the placebo arm. Therefore in late 2016, the further development of eleclazine was terminated for all indications.
An increased INa,late is present in many heart diseases. The upregulated INa,late lengthens the cardiac AP, increases [Na+]i, and causes Ca2+ overload of cardiomyocytes by offsetting the forward mode NCX. The elevated Ca2+, in turn, mainly through CaMKII, can further increase INa,late in a vicious circle. These pathophysiological mechanisms together may result in impaired cardiac energetics and contractile dysfunction of the heart as well as cardiac arrhythmias. The prolonged AP can serve as a substrate that is prone to rhythm disorders, whereas Ca2+ overload can be the trigger. INa,late seems to possess a pathogenetic role especially in AF and in ventricular arrhythmias occurring under bradycardic conditions.
Multitude of pathophysiology studies have drawn the consequence that selective INa,late inhibition is a favorable antiarrhythmic tool in many experimental settings. Despite all these studies, the one and only drug on the market that selectively inhibits INa,late is ranolazine, although it significantly affects other ionic currents as well. Ranolazine has been a safe and effective antianginal medication since 2006 based on large randomized studies. Some recent clinical evidence also proves that ranolazine shows favorable effects in AF and in ventricular arrhythmias. For stronger evidence, more phase 3 clinical investigations are necessary. Targeting Nav1.8 with specific inhibitors is also an interesting novel approach in the future of antiarrhythmic drug therapy.
BH: conception, design and drafting the manuscript TH, DK, KK: writing sections of the manuscript JM, PN, TB: conception and final review of the manuscript. All authors agreed on publishing the manuscript in the current form.
Funding was obtained from the National Research, Development and Innovation Fund for research projects FK-128116 and PD-120794, and the Thematic Excellence Programme ED_18-1-2019-0028. Further funding was obtained from the GINOP-2.3.2-15-2016-00040 and EFOP-3.6.2-16-2017-00006 projects, which are co-financed by the European Union and the European Regional Development Fund. Research of BH and DK was supported by the Ministry of Human Capacities (ÚNKP-19-4-DE-284 to BH, ÚNKP-19-3-II-DE-288 to DK). The work was also supported by the Hungarian Academy of Sciences (János Bolyai Research Scholarship to BH).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abrahamsson, C., Carlsson, L., Duker, G. (1996). Lidocaine and nisoldipine attenuate almokalant-induced dispersion of repolarization and early afterdepolarizations in vitro. J. Cardiovasc. Electrophysiol. 7 (11), 1074–1081. doi: 10.1111/j.1540-8167.1996.tb00483.x
Ahern, G. P., Hsu, S. F., Klyachko, V. A., Jackson, M. B. (2000). Induction of persistent sodium current by exogenous and endogenous nitric oxide. J. Biol. Chem. 275 (37), 28810–28815. doi: 10.1074/jbc.M003090200
Ahern, C. A., Zhang, J.-F., Wookalis, M. J., Horn, R. (2005). Modulation of the Cardiac Sodium Channel NaV1.5 by Fyn, a Src Family Tyrosine Kinase. Circ. Res. 96 (9), 991–998. doi: 10.1161/01.RES.0000166324.00524.dd
Ahmad, S., Tirilomis, P., Pabel, S., Dybkova, N., Hartmann, N., Molina, C. E., et al. (2019). The functional consequences of sodium channel NaV 1.8 in human left ventricular hypertrophy. ESC Heart Fail 6 (1), 154–163. doi: 10.1002/ehf2.12378
Akalin, F., Tirtir, A., Yilmaz, Y. (2003). Increased QT dispersion in epileptic children. Acta Paediatr. 92 (8), 916–920. doi: 10.1080/08035250310003550
Alekov, A. K., Rahman, M. M., Mitrovic, N., Lehmann-Horn, F., Lerche, H. (2000). A sodium channel mutation causing epilepsy in man exhibits subtle defects in fast inactivation and activation in vitro. J. Physiol. (Lond.) 529 (3), 533–539. doi: 10.1111/j.1469-7793.2000.00533.x
Alves Bento, A. S., Bacic, D., Saran Carneiro, J., Nearing, B. D., Fuller, H., Justo, F. A., et al. (2015). Selective late INa inhibition by GS-458967 exerts parallel suppression of catecholamine-induced hemodynamically significant ventricular tachycardia and T-wave alternans in an intact porcine model. Heart Rhythm 12 (12), 2508–2514. doi: 10.1016/j.hrthm.2015.07.025
Anderson, M. E. (2005). Calmodulin kinase signaling in heart: an intriguing candidate target for therapy of myocardial dysfunction and arrhythmias. Pharmacol. Ther. 106 (1), 39–55. doi: 10.1016/j.pharmthera.2004.11.002
Antoons, G., Oros, A., Beekman, J. D., Engelen, M. A., Houtman, M. J., Belardinelli, L., et al. (2010). Late na(+) current inhibition by ranolazine reduces torsades de pointes in the chronic atrioventricular block dog model. J. Am. Coll. Cardiol. 55 (8), 801–809. doi: 10.1016/j.jacc.2009.10.033
Antzelevitch, C., Belardinelli, L., Zygmunt, A. C., Burashnikov, A., Di Diego, J. M., Fish, J. M., et al. (2004). Electrophysiological effects of ranolazine, a novel antianginal agent with antiarrhythmic properties. Circulation 110 (8), 904–910. doi: 10.1161/01.CIR.0000139333.83620.5D
Antzelevitch, C., Nesterenko, V., Shryock, J. C., Rajamani, S., Song, Y., Belardinelli, L. (2014). The role of late I na in development of cardiac arrhythmias. Handb. Exp. Pharmacol. 221, 137–168. doi: 10.1007/978-3-642-41588-3_7
Ashpole, N. M., Herren, A. W., Ginsburg, K. S., Brogan, J. D., Johnson, D. E., Cummins, T. R., et al. (2012). Ca2+/calmodulin-dependent protein kinase II (CaMKII) regulates cardiac sodium channel NaV1.5 gating by multiple phosphorylation sites. J. Biol. Chem. 288, 19856–19869. doi: 10.1074/jbc.M111.322537
Bacic, D., Carneiro, J. S., Bento, A. A., Nearing, B. D., Rajamani, S., Belardinelli, L., et al. (2017). Eleclazine, an inhibitor of the cardiac late sodium current, is superior to flecainide in suppressing catecholamine-induced ventricular tachycardia and T-wave alternans in an intact porcine model. Heart Rhythm. 14 (3), 448–454. doi: 10.1016/j.hrthm.2016.10.021
Banyasz, T., Fulop, L., Magyar, J., Szentandrassy, N., Varro, A., Nanasi, P. P. (2003). Endocardial versus epicardial differences in L-type calcium current in canine ventricular myocytes studied by action potential voltage clamp. Cardiovasc. Res. 58 (1), 66–75. doi: 10.1016/s0008-6363(02)00853-2
Banyasz, T., Horvath, B., Virag, L., Barandi, L., Szentandrassy, N., Harmati, G., et al. (2009). Reverse rate dependency is an intrinsic property of canine cardiac preparations. Cardiovasc. Res. 84 (2), 237–244. doi: 10.1093/cvr/cvp213
Banyasz, T., Horvath, B., Jian, Z., Izu, L. T., Ye, C.-I. (2012). Profile of L-type Ca2+ current and Na+/Ca2+ exchange current during cardiac action potential in ventricular myocytes. Heart Rhythm 9 (1), 134–142. doi: 10.1016/j.hrthm.2011.08.029
Belardinelli, L., Antzelevitch, C., Fraser, H. (2004). Inhibition of late (sustained/persistent) sodium current: a potential drug target to reduce intracellular sodium-dependent calcium overload and its detrimental effects on cardiomyocyte function. Eur. Heart J. Suppl. 6 (I), I3–I7. doi: 10.1093/eurheartj/6.suppl_i.i3
Belardinelli, L., Shryock, J. C., Fraser, H. (2006). Inhibition of the late sodium current as a potential cardioprotective principle: effects of the late sodium current inhibitor ranolazine. Heart 92, 6–14. doi: 10.1136/hrt.2005.078790
Belardinelli, L., Liu, G., Smith-Maxwell, C., Wang, W. Q., El-Bizri, N., Hirakawa, R., et al. (2013). A novel, potent, and selective inhibitor of cardiac late sodium current suppresses experimental arrhythmias. J. Pharmacol. Exp. Ther. 344 (1), 23–32. doi: 10.1124/jpet.112.198887
Beltran-Alvarez, P., Pagans, S., Brugada, R. (2011). The cardiac sodium channel is post-translationally modified by arginine methylation. J. Proteome Res. 10 (8), 3712–3719. doi: 10.1021/pr200339n
Benito, B., Brugada, R., Perich, R. M., Lizotte, E., Cinca, J., Mont, L., et al. (2008). A mutation in the sodium channel is responsible for the association of long QT syndrome and familial atrial fibrillation. Heart Rhythm 5 (10), 1434–1440. doi: 10.1016/j.hrthm.2008.07.013
Bers, D. M., Grandi, E. (2009). Calcium/Calmodulin-dependent Kinase II Regulation of Cardiac Ion Channels. J. Cardiovasc. Pharmacol. 54 (3), 180–187. doi: 10.1097/FJC.0b013e3181a25078
Bers, D. M. (2002). Cardiac excitation-contraction coupling. Nature 415 (6868), 198–205. doi: 10.1038/415198a
Bers, D. M. (2010). CaMKII Inhibition in Heart Failure Makes Jump to Human. Circ. Res. 107 (9), 1044–1046. doi: 10.1161/circresaha.110.231902
Beyder, A., Rae, J. L., Bernard, C., Strege, P. R., Sachs, F., Farrugia, G. (2010). Mechanosensitivity of Na(v)1.5, a voltage-sensitive sodium channel. J. Physiol. (Lond.) 588 (24), 4969–4985. doi: 10.1113/jphysiol.2010.199034
Beyder, A., Strege, P. R., Reyes, S., Bernard, C. E., Terzic, A., Makielski, J., et al. (2012). Ranolazine Decreases Mechanosensitivity of the Voltage-Gated Sodium Ion Channel Na(V)1.5 A Novel Mechanism of Drug Action. Circulation 125 (22), 2698–U2695. doi: 10.1161/circulationaha.112.094714
Bezzina, C., Veldkamp, M. W., van den Berg, M. P., Postma, A. V., Rook, M. B., Viersma, J. W., et al. (1999). A single Na+ channel mutation causing both long-QT and Brugada syndromes. Circ. Res. 85 (12), 1206–1213. doi: 10.1161/01.RES.85.12.1206
Biet, M., Barajas-Martinez, H., Ton, A. T., Delabre, J. F., Morin, N., Dumaine, R. (2012). About half of the late sodium current in cardiac myocytes from dog ventricle is due to non-cardiac-type Na+ channels. J. Mol. Cell. Cardiol. 53 (5), 593–598. doi: 10.1016/j.yjmcc.2012.06.012
Bonatti, R., Silva, A. F., Batatinha, J. A., Sobrado, L. F., Machado, A. D., Varone, B. B., et al. (2014). Selective late sodium current blockade with GS-458967 markedly reduces ischemia-induced atrial and ventricular repolarization alternans and ECG heterogeneity. Heart Rhythm 11 (10), 1827–1835. doi: 10.1016/j.hrthm.2014.06.017
Bossu, A., Houtman, M. J. C., Meijborg, V. M. F., Varkevisser, R., Beekman, H. D. M., Dunnink, A., et al. (2018). Selective late sodium current inhibitor GS-458967 suppresses Torsades de Pointes by mostly affecting perpetuation but not initiation of the arrhythmia. Br. J. Pharmacol. 175 (12), 2470–2482. doi: 10.1111/bph.14217
Burashnikov, A., Antzelevitch, C. (2008). Atrial-selective sodium channel blockers: do they exist? J. Cardiovasc. Pharmacol. 52 (2), 121–128. doi: 10.1097/FJC.0b013e31817618eb
Burashnikov, A., Antzelevitch, C. (2013). Role of late sodium channel current block in the management of atrial fibrillation. Cardiovasc. Drugs Ther. 27 (1), 79–89. doi: 10.1007/s10557-012-6421-1
Burashnikov, A., Antzelevitch, C. (2017). Effectiveness of Late INa Versus Peak INa Block in the Setting of Ventricular Fibrillation. Circ. Arrhythm. Electrophysiol. 10 (3), e005111. doi: 10.1161/CIRCEP.117.005111
Burashnikov, A., Di Diego, J. M., Zygmunt, A. C., Belardinelli, L., Antzelevitch, C. (2007). Atrium-selective sodium channel block as a strategy for suppression of atrial fibrillation: differences in sodium channel inactivation between atria and ventricles and the role of ranolazine. Circulation 116 (13), 1449–1457. doi: 10.1161/CIRCULATIONAHA.107.704890
Burashnikov, A., Di Diego, J. M., Barajas-Martinez, H., Hu, D., Cordeiro, J. M., Moise, N. S., et al. (2014). Ranolazine effectively suppresses atrial fibrillation in the setting of heart failure. Circ. Heart Fail 7 (4), 627–633. doi: 10.1161/CIRCHEARTFAILURE.114.001129
Burnashev, N. A., Undrovinas, A. I., Fleidervish, I. A., Makielski, J. C., Rosenshtraukh, L. V. (1991). Modulation of cardiac sodium-channel gating by lysophosphatidylcholine. J. Mol. Cell Cardiol. 23, 23–30. doi: 10.1016/0022-2828(91)90020-m
Cardona, K., Trenor, B., Rajamani, S., Romero, L., Ferrero, J. M., Saiz, J. (2010). “Effects of late sodium current enhancement during LQT-related arrhythmias. A simulation study,” in Conference proceedings: Annual International Conference of the IEEE Engineering in Medicine and Biology Society. [Buenos Aires: IEEE (Institute of Electrical and Electronics Engineers)] 2010, 3237–3240. doi: 10.1109/iembs.2010.5627184
Carlsson, L., Abrahamsson, C., Drews, L., Duker, G. (1992). Antiarrhythmic effects of potassium channel openers in rhythm abnormalities related to delayed repolarization. Circulation 85 (4), 1491–1500. doi: 10.1161/01.cir.85.4.1491
Carlsson, L., Drews, L., Duker, G. (1996). Rhythm anomalies related to delayed repolarization in vivo: influence of sarcolemmal Ca++ entry and intracellular Ca++ overload. J. Pharmacol. Exp. Ther. 279 (1), 231–239.
Carmeliet, E. (1999). Cardiac ionic currents and acute ischemia: From channels to arrhythmias. Physiol. Rev. 79 (3), 917–1017. doi: 10.1152/physrev.1999.79.3.917
Carmeliet, E. (2006). Action potential duration, rate of stimulation, and intracellular sodium. J. Cardiovasc. Electrophysiol. 17 Suppl 1, S2–S7. doi: 10.1111/j.1540-8167.2006.00378.x
Carneiro, J. S., Bento, A. S., Bacic, D., Nearing, B. D., Rajamani, S., Belardinelli, L., et al. (2015). The Selective Cardiac Late Sodium Current Inhibitor GS-458967 Suppresses Autonomically Triggered Atrial Fibrillation in an Intact Porcine Model. J. Cardiovasc. Electrophysiol. 26 (12), 1364–1369. doi: 10.1111/jce.12824
Catterall, W. A., Goldin, A. L., Waxman, S. G. (2005). International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol. Rev. 57 (4), 397–409. doi: 10.1124/pr.57.4.4
Chaitman, B. R., Skettino, S. L., Parker, J. O., Hanley, P., Meluzin, J., Kuch, J., et al. (2004a). Anti-ischemic effects and long-term survival during ranolazine monotherapy in patients with chronic severe angina. J. Am. Coll. Cardiol. 43 (8), 1375–1382. doi: 10.1016/j.jacc.2003.11.045
Chaitman, B. R., Pepine, C. J., Parker, J. O., Skopal, J., Chumakova, G., Kuch, J., et al. (2004b). Effects of ranolazine with atenolol, amlodipine, or diltiazem on exercise tolerance and angina frequency in patients with severe chronic angina: a randomized controlled trial. JAMA 291 (3), 309–316. doi: 10.1001/jama.291.3.309
Chaitman, B. R. (2006). Ranolazine for the treatment of chronic angina and potential use in other cardiovascular conditions. Circulation 113 (20), 2462–2472. doi: 10.1161/CIRCULATIONAHA.105.597500
Cheng, J., Valdivia, C. R., Vaidyanathan, R., Balijepalli, R. C., Ackerman, M. J., Makielski, J. C. (2013). Caveolin-3 suppresses late sodium current by inhibiting nNOS-dependent S-nitrosylation of SCN5A. J. Mol. Cell Cardiol. 61, 102–110. doi: 10.1016/j.yjmcc.2013.03.013
Cheung, J. Y., Zhang, X. Q., Song, J., Gao, E., Rabinowitz, J. E., Chan, T. O., et al. (2010). Phospholemman: a novel cardiac stress protein. Clin. Transl. Sci. 3 (4), 189–196. doi: 10.1111/j.1752-8062.2010.00213.x
Clancy, C. E., Tateyama, M., Liu, H., Wehrens, X. H., Kass, R. S. (2003). Non-equilibrium gating in cardiac Na+ channels: an original mechanism of arrhythmia. Circulation 107 (17), 2233–2237. doi: 10.1161/01.CIR.0000069273.51375.BD
Cocco, G., Rousseau, M. F., Bouvy, T., Cheron, P., Williams, G., Detry, J. M., et al. (1992). Effects of a new metabolic modulator, ranolazine, on exercise tolerance in angina pectoris patients treated with beta-blocker or diltiazem. J. Cardiovasc. Pharmacol. 20 (1), 131–138.
Cohen, S. A., Levitt, L. K. (1993). Partial characterization of the rH1 sodium channel protein from rat heart using subtype-specific antibodies. Circ. Res. 73 (4), 735–742. doi: 10.1161/01.res.73.4.735
Comtois, P., Sakabe, M., Vigmond, E. J., Munoz, M., Texier, A., Shiroshita-Takeshita, A., et al. (2008). Mechanisms of atrial fibrillation termination by rapidly unbinding Na+ channel blockers: insights from mathematical models and experimental correlates. Am. J. Physiol. Heart Circ. Physiol. 295 (4), H1489–H1504. doi: 10.1152/ajpheart.01054.2007
Coppini, R., Ferrantini, C., Yao, L., Fan, P., Del Lungo, M., Stillitano, F., et al. (2013). Late sodium current inhibition reverses electromechanical dysfunction in human hypertrophic cardiomyopathy. Circulation 127 (5), 575–584. doi: 10.1161/circulationaha.112.134932
Coraboeuf, E., Deroubaix, E., Coulombe, A. (1979). Effect of tetrodotoxin on action potentials of the conducting system in the dog heart. Am. J. Physiol. 236 (4), H561–H567. doi: 10.1152/ajpheart.1979.236.4.H561
Cortada, E., Brugada, R. (2019). Verges M. N-Glycosylation of the voltage-gated sodium channel beta2 subunit is required for efficient trafficking of NaV1.5/beta2 to the plasma membrane. J. Biol. Chem. 294 (44), 16123–16140. doi: 10.1074/jbc.RA119.007903
De Ferrari, G. M., Maier, L. S., Mont, L., Schwartz, P. J., Simonis, G., Leschke, M., et al. (2015). Ranolazine in the treatment of atrial fibrillation: Results of the dose-ranging RAFFAELLO (Ranolazine in Atrial Fibrillation Following An ELectricaL CardiOversion) study. Heart Rhythm 12 (5), 872–878. doi: 10.1016/j.hrthm.2015.01.021
de Lera Ruiz, M., Kraus, R. L. (2015). Voltage-Gated Sodium Channels: Structure, Function, Pharmacology, and Clinical Indications. J. Med. Chem. 58 (18), 7093–7118. doi: 10.1021/jm501981g
Debono, M. W., Le Guern, J., Canton, T., Doble, A., Pradier, L. (1993). Inhibition by riluzole of electrophysiological responses mediated by rat kainate and NMDA receptors expressed in Xenopus oocytes. Eur. J. Pharmacol. 235 (2-3), 283–289. doi: 10.1016/0014-2999(93)90147-a
Despa, S., Bers, D. M. (2013). Na+ transport in the normal and failing heart - Remember the balance. J. Mol. Cell Cardiol. 61, 2–10. doi: 10.1016/j.yjmcc.2013.04.011
Despa, S., Islam, M. A., Weber, C. R., Pogwizd, S. M., Bers, D. M. (2002). Intracellular Na+ Concentration Is Elevated in Heart Failure But Na/K Pump Function Is Unchanged. Circulation 105 (21), 2543–2548. doi: 10.1161/01.cir.0000016701.85760.97
Dybkova, N., Ahmad, S., Pabel, S., Tirilomis, P., Hartmann, N., Fischer, T. H., et al. (2018). Differential regulation of sodium channels as a novel proarrhythmic mechanism in the human failing heart. Cardiovasc. Res. 114 (13), 1728–1737. doi: 10.1093/cvr/cvy152
El-Sherif, N., Turitto, G., Boutjdir, M. (2019). Acquired Long QT Syndrome and Electrophysiology of Torsade de Pointes. Arrhythm. Electrophysiol. Rev. 8 (2), 122–130. doi: 10.15420/aer.2019.8.3
Erickson, J. R., Joiner, M. L. A., Guan, X., Kutschke, W., Yang, J. Y., Oddis, C. V., et al. (2008). A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell 133 (3), 462–474. doi: 10.1016/j.cell.2008.02.048
Evaristo, E., Stocco, F. G., Shah, N. R., Cheezum, M. K., Hainer, J., Foster, C., et al. (2018). Ranolazine reduces repolarization heterogeneity in symptomatic patients with diabetes and non-flow-limiting coronary artery stenosis. Ann. Noninvas. Electrocardiol. 23 (1), e12480. doi: 10.1111/anec.12480
Fujioka, Y., Hiroe, K., Matsuoka, S. (2000). Regulation kinetics of Na+-Ca2+ exchange current in guinea-pig ventricular myocytes. J. Physiol. (Lond.) 529 (3), 611–624. doi: 10.1111/j.1469-7793.2000.00611.x
Fulop, L., Banyasz, T., Magyar, J., Szentandrassy, N., Varro, A., Nanasi, P. P. (2004). Reopening of L-type calcium channels in human ventricular myocytes during applied epicardial action potentials. Acta Physiol. Scand. 180 (1), 39–47. doi: 10.1046/j.0001-6772.2003.01223.x
Gellens, M. E., George, A. L., Chen, L. Q., Chahine, M., Horn, R., Barchi, R. L., et al. (1992). Primary structure and functional expression of the human cardiac tetrodotoxin-insensitive voltage-dependent sodium-channel. Proc. Natl. Acad. Sci. U. S. A. 89 (2), 554–558. doi: 10.1073/pnas.89.2.554
Ginsburg, K. S., Weber, C. R., Bers, D. M. (2013). Cardiac Na+Ca2+exchanger: dynamics of Ca2+-dependent activation and deactivation in intact myocytes. J. Physiol. (Lond.) 591 (8), 2067–2086. doi: 10.1113/jphysiol.2013.252080
Gong, M., Zhang, Z., Fragakis, N., Korantzopoulos, P., Letsas, K. P., Li, G., et al. (2017). Role of ranolazine in the prevention and treatment of atrial fibrillation: A meta-analysis of randomized clinical trials. Heart Rhythm 14 (1), 3–11. doi: 10.1016/j.hrthm.2016.10.008
Grandi, E., Herren, A. W. (2014). CaMKII-dependent regulation of cardiac Na homeostasis. Front. Pharmacol. 5, 41. doi: 10.3389/fphar.2014.00041
Guo, D., Young, L., Wu, Y., Belardinelli, L., Kowey, P. R., Yan, G.-X. (2010). Increased late sodium current in left atrial myocytes of rabbits with left ventricular hypertrophy: its role in the genesis of atrial arrhythmias. Am. J. Physiol.-Heart Circ. Physiol. 298 (5), H1375–H1381. doi: 10.1152/ajpheart.01145.2009
Gyorke, S., Terentyev, D. (2008). Modulation of ryanodine receptor by luminal calcium and accessory proteins in health and cardiac disease. Cardiovasc. Res. 77 (2), 245–255. doi: 10.1093/cvr/cvm038
Hallaq, H., Wang, D. W., Kunic, J. D., George, A. L., Jr., Wells, K. S., Murray, K. T. (2012). Activation of protein kinase C alters the intracellular distribution and mobility of cardiac Na+ channels. Am. J. Physiol. Heart Circulatory Physiol. 302 (3), H782–H789. doi: 10.1152/ajpheart.00817.2010
Harnmarstrom, A. K. M., Gage, P. W. (2002). Hypoxia and persistent sodium current. Eur. Biophys. J. Biophys. Lett. 31 (5), 323–330. doi: 10.1007/s00249-002-0218-2
Haufe, V., Cordeiro, J. M., Zimmer, T., Wu, Y. S., Schiccitano, S., Benndorf, K., et al. (2005). Contribution of neuronal sodium channels to the cardiac fast sodium current I-Na is greater in dog heart Purkinje fibers than in ventricles. Cardiovasc. Res. 65 (1), 117–127. doi: 10.1016/j.cardiores.2004.08.017
He, Y., Benz, A., Fu, T., Wang, M., Covey, D. F., Zorumski, C. F., et al. (2002). Neuroprotective agent riluzole potentiates postsynaptic GABA(A) receptor function. Neuropharmacology 42 (2), 199–209. doi: 10.1016/s0028-3908(01)00175-7
Hedley, P. L., Jorgensen, P., Schlamowitz, S., Wangari, R., Moolman-Smook, J., Brink, P. A., et al. (2009). The Genetic Basis of Long QT and Short QT Syndromes: A Mutation Update. Hum. Mutat. 30 (11), 1486–1511. doi: 10.1002/humu.21106
Hegyi, B., Banyasz, T., Izu, L. T., Belardinelli, L., Bers, D. M., Chen-Izu, Y. (2018). beta-adrenergic regulation of late Na(+) current during cardiac action potential is mediated by both PKA and CaMKII. J. Mol. Cell. Cardiol. 123, 168–179. doi: 10.1016/j.yjmcc.2018.09.006
Horvath, B., Magyar, J., Szentandrassy, N., Birinyi, P., Nanasi, P. P., Banyasz, T. (2006). Contribution of I-Ks to ventricular repolarization in canine myocytes. Pflug. Arch. Eur. J. Physiol. 452 (6), 698–706. doi: 10.1007/s00424-006-0077-2
Horvath, B., Banyasz, T., Jian, Z., Hegyi, B., Kistamas, K., Nanasi, P. P., et al. (2013). Dynamics of the late Na+ current during cardiac action potential and its contribution to afterdepolarizations. J. Mol. Cell Cardiol. 64, 59–68. doi: 10.1016/j.yjmcc.2013.08.010
Horvath, B., Hezso, T., Szentandrassy, N., Kistamas, K., Arpadffy-Lovas, T., Varga, R., et al. (2020). Late sodium current in human, canine and guinea pig ventricular myocardium. J. Mol. Cell. Cardiol. 139, 14–23. doi: 10.1016/j.yjmcc.2019.12.015
Hoyer, K., Song, Y. J., Wang, D. S., Phan, D., Balschi, J., Ingwall, J. S., et al. (2011). Reducing the Late Sodium Current Improves Cardiac Function during Sodium Pump Inhibition by Ouabain. J. Pharmacol. Exp. Ther. 337 (2), 513–523. doi: 10.1124/jpet.110.176776
Huang, B., El-Sherif, T., Gidh-Jain, M., Qin, D., El-Sherif, N. (2001). Alterations of sodium channel kinetics and gene expression in the postinfarction remodeled myocardium. J. Cardiovasc. Electrophysiol. 12 (2), 218–225. doi: 10.1046/j.1540-8167.2001.00218.x
Huang, X., Du, Y., Yang, P., Lin, S., Xi, Y., Yang, Z., et al. (2015). Age-dependent alterations of voltage-gated Na+ channel isoforms in rat sinoatrial node. Mech. Ageing Dev. 152, 80–90. doi: 10.1016/j.mad.2015.10.003
Janvier, N. C., Boyett, M. R. (1996). The role of Na-Ca exchange current in the cardiac action potential. Cardiovasc. Res. 32 (1), 69–84. doi: 10.1016/s0008-6363(96)00017-x
Jian, Z., Han, H., Zhang, T., Puglisi, J., Izu, L. T., Shaw, J. A., et al. (2014). Mechanochemotransduction during cardiomyocyte contraction is mediated by localized nitric oxide signaling. Sci. Signal. 7 (317), ra27. doi: 10.1126/scisignal.2005046
Jones, D. K., Peters, C. H., Tolhurst, S. A., Claydon, T. W., Ruben, P. C. (2011). Extracellular Proton Modulation of the Cardiac Voltage-Gated Sodium Channel, Na(v)1.5. Biophys. J. 101 (9), 2147–2156. doi: 10.1016/j.bpj.2011.08.056
Jones, D. K., Peters, C. H., Allard, C. R., Claydon, T. W., Ruben, P. C. (2013a). Proton Sensors in the Pore Domain of the Cardiac Voltage-gated Sodium Channel. J. Biol. Chem. 288 (7), 4782–4791. doi: 10.1074/jbc.M112.434266
Jones, D. K., Claydon, T. W., Ruben, P. C. (2013b). Extracellular Protons Inhibit Charge Immobilization in the Cardiac Voltage-Gated Sodium Channel. Biophys. J. 105 (1), 101–107. doi: 10.1016/j.bpj.2013.04.022
Ju, Y. K., Saint, D. A., Gage, P. W. (1996). Hypoxia increases persistent sodium current in rat ventricular myocytes. J. Physiol. (Lond.) 497 (2), 337–347. doi: 10.1113/jphysiol.1996.sp021772
Justo, F., Fuller, H., Nearing, B. D., Rajamani, S., Belardinelli, L., Verrier, R. L. (2016). Inhibition of the cardiac late sodium current with eleclazine protects against ischemia-induced vulnerability to atrial fibrillation and reduces atrial and ventricular repolarization abnormalities in the absence and presence of concurrent adrenergic stimulation. Heart Rhythm 13 (9), 1860–1867. doi: 10.1016/j.hrthm.2016.06.020
Kapplinger, J. D., Tester, D. J., Alders, M., Benito, B., Berthet, M., Brugada, J., et al. (2010). An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm 7 (1), 33–46. doi: 10.1016/j.hrthm.2009.09.069
Kim, J., Ghosh, S., Liu, H. J., Tateyama, M., Kass, R. S., Pitt, G. S. (2004). Calmodulin mediates Ca2+ sensitivity of sodium channels. J. Biol. Chem. 279 (43), 45004–45012. doi: 10.1074/jbc.M407286200
Kiyosue, T., Arita, M. (1989). Late sodium current and its contribution to action potential configuration in guinea pig ventricular myocytes. Circ. Res. 64 (2), 389–397. doi: 10.1161/01.res.64.2.389
Koltun, D. O., Parkhill, E. Q., Elzein, E., Kobayashi, T., Notte, G. T., Kalla, R., et al. (2016a). Discovery of triazolopyridine GS-458967, a late sodium current inhibitor (Late INai) of the cardiac NaV 1.5 channel with improved efficacy and potency relative to ranolazine. Bioorg. Med. Chem. Lett. 26 (13), 3202–3206. doi: 10.1016/j.bmcl.2016.03.101
Koltun, D. O., Parkhill, E. Q., Elzein, E., Kobayashi, T., Jiang, R. H., Li, X., et al. (2016b). Discovery of triazolopyridinone GS-462808, a late sodium current inhibitor (Late INai) of the cardiac Nav1.5 channel with improved efficacy and potency relative to ranolazine. Bioorg. Med. Chem. Lett. 26 (13), 3207–3211. doi: 10.1016/j.bmcl.2016.03.096
Komajda, M., Frank, R., Vedel, J., Fontaine, G., Petitot, J. C., Grosgogeat, Y. (1980). Intracardiac conduction defects in dystrophia myotonica - electro-physiological study of 12 cases. Br. Heart J. 43 (3), 315–320. doi: 10.1136/hrt.43.3.315
Kumar, K., Nearing, B. D., Carvas, M., Nascimento, B. C., Acar, M., Belardinelli, L., et al. (2009). Ranolazine exerts potent effects on atrial electrical properties and abbreviates atrial fibrillation duration in the intact porcine heart. J. Cardiovasc. Electrophysiol. 20 (7), 796–802. doi: 10.1111/j.1540-8167.2009.01437.x
Lei, M., Jones, S. A., Liu, J., Lancaster, M. K., Fung, S. S., Dobrzynski, H., et al. (2004). Requirement of neuronal- and cardiac-type sodium channels for murine sinoatrial node pacemaking. J. Physiol. 559 (Pt 3), 835–848. doi: 10.1113/jphysiol.2004.068643
Letienne, R., Vie, B., Puech, A., Vieu, S., Le Grand, B., John, G. W. (2001). Evidence that ranolazine behaves as a weak beta1- and beta2-adrenoceptor antagonist in the rat [correction of cat] cardiovascular system. Naunyn-Schmiedeberg's Arch. Pharmacol. 363 (4), 464–471. doi: 10.1007/s002100000378
Li, G. R., Lau, C. P., Shrier, A. (2002). Heterogeneity of sodium current in atrial vs epicardial ventricular myocytes of adult guinea pig hearts. J. Mol. Cell. Cardiol. 34 (9), 1185–1194. doi: 10.1006/jmcc.2002.2053
Li, W., Yu, Y., Hou, J. W., Zhou, Z. W., Guo, K., Zhang, P. P., et al. (2017). Larger rate dependence of late sodium current in cardiac Purkinje cells: A potential link to arrhythmogenesis. Heart Rhythm 14 (3), 422–431. doi: 10.1016/j.hrthm.2016.11.036
Liang, F., Fan, P., Jia, J., Yang, S., Jiang, Z., Karpinski, S., et al. (2016). Inhibitions of late INa and CaMKII act synergistically to prevent ATX-II-induced atrial fibrillation in isolated rat right atria. J. Mol. Cell. Cardiol. 94, 122–130. doi: 10.1016/j.yjmcc.2016.04.001
Linz, K. W., Meyer, R. (1998). Control of L-type calcium current during the action potential of guinea-pig ventricular myocytes. J. Physiol. (Lond.) 513 (2), 425–442. doi: 10.1111/j.1469-7793.1998.425bb.x
Linz, K. W., Meyer, R. (2000). Profile and kinetics of L-type calcium current during the cardiac ventricular action potential compared in guinea-pigs, rats and rabbits. Pflug. Arch.-Eur. J. Physiol. 439 (5), 588–599. doi: 10.1007/s004240050982
Liu, T., O'Rourke, B. (2008). Enhancing Mitochondrial Ca2+ Uptake in Myocytes From Failing Hearts Restores Energy Supply and Demand Matching. Circ. Res. 103 (3), 279–288. doi: 10.1161/circresaha.108.175919
Liu, H., Sun, H. Y., Lau, C. P., Li, G. R. (2007). Regulation of voltage-gated cardiac sodium current by epidermal growth factor receptor kinase in guinea pig ventricular myocytes. J. Mol. Cell Cardiol. 42 (4), 760–768. doi: 10.1016/j.yjmcc.2006.10.013
Liu, M., Shi, G., Yang, K. C., Gu, L., Kanthasamy, A. G., Anantharam, V., et al. (2017). Role of protein kinase C in metabolic regulation of the cardiac Na(+) channel. Heart Rhythm 14 (3), 440–447. doi: 10.1016/j.hrthm.2016.12.026
Locati, E. T., Bagliani, G., Cecchi, F., Johny, H., Lunati, M., Pappone, C. (2019). Arrhythmias due to Inherited and Acquired Abnormalities of Ventricular Repolarization. Card. Electrophysiol. Clin. 11 (2), 345–362. doi: 10.1016/j.ccep.2019.02.009
Louch, W. E., Hougen, K., Mørk, H. K., Swift, F., Aronsen, J. M., Sjaastad, I., et al. (2010). Sodium accumulation promotes diastolic dysfunction in end-stage heart failure following Serca2 knockout. J. Physiol. 588 (3), 465–478. doi: 10.1113/jphysiol.2009.183517
Lowe, J. S., Stroud, D. M., Yang, T., Hall, L., Atack, T. C., Roden, D. M. (2012). Increased late sodium current contributes to long QT-related arrhythmia susceptibility in female mice. Cardiovasc. Res. 95 (3), 300–307. doi: 10.1093/cvr/cvs160
Lu, Y. Y., Cheng, C. C., Chen, Y. C., Chen, S. A., Chen, Y. J. (2012). ATX-II-induced pulmonary vein arrhythmogenesis related to atrial fibrillation and long QT syndrome. Eur. J. Clin. Invest. 42 (8), 823–831. doi: 10.1111/j.1365-2362.2012.02655.x
Luo, A., Ma, J., Song, Y., Qian, C., Wu, Y., Zhang, P., et al. (2014). Larger late sodium current density as well as greater sensitivities to ATX II and ranolazine in rabbit left atrial than left ventricular myocytes. Am. J. Physiol. Heart Circ. Physiol. 306 (3), H455–H461. doi: 10.1152/ajpheart.00727.2013
Ma, J. H., Luo, A. T., Wu, L., Wan, W., Zhang, P. H., Ren, Z. Q., et al. (2012). Calmodulin kinase II and protein kinase C mediate the effect of increased intracellular calcium to augment late sodium current in rabbit ventricular myocytes. Am. J. Physiol. Cell Physiol. 302 (8), C1141–C1151. doi: 10.1152/ajpcell.00374.2011
Magyar, J., Kiper, C. E., Dumaine, R., Burgess, D. E., Banyasz, T., Satin, J. (2004). Divergent action potential morphologies reveal nonequilibrium properties of human cardiac Na channels. Cardiovasc. Res. 64 (3), 477–487. doi: 10.1016/j.cardiores.2004.07.014
Maier, L. S., Sossalla, S. (2013). The late Na current as a therapeutic target: Where are we? J. Mol. Cell Cardiol. 61, 44–50. doi: 10.1016/j.yjmcc.2013.03.001
Maier, S. K., Westenbroek, R. E., Schenkman, K. A., Feigl, E. O., Scheuer, T., Catterall, W. A. (2002). An unexpected role for brain-type sodium channels in coupling of cell surface depolarization to contraction in the heart. Proc. Natl. Acad. Sci. United States America 99 (6), 4073–4078. doi: 10.1073/pnas.261705699
Maier, S. K. G., Westenbroek, R. E., Yamanushi, T. T., Dobrzynski, H., Boyett, M. R., Catterall, W. A., et al. (2003). An unexpected requirement for brain-type sodium channels for control of heart rate in the mouse sinoatrial node. Proc. Natl. Acad. Sci. 100 (6), 3507–3512. doi: 10.1073/pnas.2627986100
Maier, L. S. (2011). CaMKII regulation of voltage-gated sodium channels and cell excitability. Heart Rhythm 8 (3), 474–477. doi: 10.1016/j.hrthm.2010.09.080
Makielski, J. C., Farley, A. L. (2006). Na(+) current in human ventricle: implications for sodium loading and homeostasis. J. Cardiovasc. Electrophysiol. 17 (Suppl 1), S15–S20. doi: 10.1111/j.1540-8167.2006.00380.x
Makita, N., Horie, M., Nakamura, T., Ai, T., Sasaki, K., Yokoi, H., et al. (2002). Drug-induced Long-QT syndrome associated with a subclinical SCN5A mutation. Circulation 106 (10), 1269–1274. doi: 10.1161/01.cir.0000027139.42087.b6
Malhotra, J. D., Chen, C. L., Rivolta, I., Abriel, H., Malhotra, R., Mattei, L. N., et al. (2001). Characterization of sodium channel alpha- and beta-subunits in rat and mouse cardiac myocytes. Circulation 103 (9), 1303–1310. doi: 10.1161/01.CIR.103.9.1303
Maltsev, V. A., Undrovinas, A. (2008). Late sodium current in failing heart: friend or foe? Prog. Biophys. Mol. Biol. 96 (1-3), 421–451. doi: 10.1016/j.pbiomolbio.2007.07.010
Maltsev, V. A., Sabbah, H. N., Higgins, R. S., Silverman, N., Lesch, M., Undrovinas, A. I. (1998). Novel, ultraslow inactivating sodium current in human ventricular cardiomyocytes. Circulation 98 (23), 2545–2552. doi: 10.1161/01.CIR.98.23.2545
Maltsev, V. A., Silverman, N., Sabbah, H. N., Undrovinas, A. I. (2007). Chronic heart failure slows late sodium current in human and canine ventricular myocytes: Implications for repolarization variability. Eur. J. Heart Fail. 9 (3), 219–227. doi: 10.1016/j.ejheart.2006.08.007
Maltsev, V. A., Reznikov, V., Undrovinas, N. A., Sabbah, H. N., Undrovinas, A. (2008). Modulation of late sodium current by Ca2+, calmodulin, and CaMKII in normal and failing dog cardiomyocytes: similarities and differences. Am. J. Physiol. Heart Circulatory Physiol. 294 (4), H1597–H1608. doi: 10.1152/ajpheart.00484.2007
Maltsev, V. A., Kyle, J. W., Undrovinas, A. (2009). Late Na(+) current produced by human cardiac Na(+) channel isoform Na(v)1.5 is modulated by its beta(1) subunit. J. Physiol. Sci. 59 (3), 217–225. doi: 10.1007/s12576-009-0029-7
Maltsev, V. A. (2006). Undrovinas AI. A multi-modal composition of the late Na+ current in human ventricular cardiomyocytes. Cardiovasc. Res. 69 (1), 116–127. doi: 10.1016/j.cardiores.2005.08.015
Merchant, F. M., Armoundas, A. A. (2012). Role of substrate and triggers in the genesis of cardiac alternans, from the myocyte to the whole heart: implications for therapy. Circulation 125 (3), 539–549. doi: 10.1161/CIRCULATIONAHA.111.033563
Mercier, A., Clement, R., Harnois, T., Bourmeyster, N., Bois, P., Chatelier, A. (2015). Nav1.5 channels can reach the plasma membrane through distinct N-glycosylation states. Biochim. Biophys. Acta 1850 (6), 1215–1223. doi: 10.1016/j.bbagen.2015.02.009
Milberg, P., Reinsch, N., Wasmer, K., Monnig, G., Stypmann, J., Osada, N., et al. (2005). Transmural dispersion of repolarization as a key factor of arrhythmogenicity in a novel intact heart model of LQT3. Cardiovasc. Res. 65 (2), 397–404. doi: 10.1016/j.cardiores.2004.10.016
Mills, G. D., Harris, D. M., Chen, X., Houser, S. R. (2007). Intracellular sodium determines frequency-dependent alterations in contractility in hypertrophied feline ventricular myocytes. Am. J. Physiol. Heart Circ. Physiol. 292 (2), H1129–H1138. doi: 10.1152/ajpheart.00375.2006
Mitsuiye, T., Noma, A. (2002). Inactivation of cardiac Na+ channel simply through open states as revealed by single-channel analysis in guinea pig ventricular myocytes. Jpn. J. Physiol. 52 (5), 457–469. doi: 10.2170/jjphysiol.52.457
Morita, N., Lee, J. H., Xie, Y., Sovari, A., Qu, Z., Weiss, J. N., et al. (2011). Suppression of re-entrant and multifocal ventricular fibrillation by the late sodium current blocker ranolazine. J. Am. Coll. Cardiol. 57 (3), 366–375. doi: 10.1016/j.jacc.2010.07.045
Morrow, D. A., Scirica, B. M., Karwatowska-Prokopczuk, E., Murphy, S. A., Budaj, A., Varshavsky, S., et al. (2007). Effects of ranolazine on recurrent cardiovascular events in patients with non-ST-elevation acute coronary syndromes: the MERLIN-TIMI 36 randomized trial. JAMA 297 (16), 1775–1783. doi: 10.1001/jama.297.16.1775
Murphy, L., Renodin, D., Antzelevitch, C., Di Diego, J. M., Cordeiro, J. M. (2011). Extracellular proton depression of peak and late Na current in the canine left ventricle. Am. J. Physiol. Heart Circ. Physiol. 301 (3), H936–H944. doi: 10.1152/ajpheart.00204.2011
Nagatomo, T., January, C. T., Ye, B., Abe, H., Nakashima, Y., Makielski, J. C. (2002). Rate-dependent QT shortening mechanism for the LQT3 deltaKPQ mutant. Cardiovasc. Res. 54 (3), 624–629. doi: 10.1016/s0008-6363(02)00265-1
Nagy, Z. A., Virag, L., Toth, A., Biliczki, P., Acsai, K., Banyasz, T., et al. (2004). Selective inhibition of sodium-calcium exchanger by SEA-0400 decreases early and delayed afterdepolarization in canine heart. Br. J. Pharmacol. 143 (7), 827–831. doi: 10.1038/sj.bjp.0706026
Nattel, S., Dobrev, D. (2012). The multidimensional role of calcium in atrial fibrillation pathophysiology: mechanistic insights and therapeutic opportunities. Eur. Heart J. 33 (15), 1870–1877. doi: 10.1093/eurheartj/ehs079
Ninkovic, V. M., Ninkovic, S. M., Miloradovic, V., Stanojevic, D., Babic, M., Giga, V., et al. (2016). Prevalence and risk factors for prolonged QT interval and QT dispersion in patients with type 2 diabetes. Acta Diabetol. 53 (5), 737–744. doi: 10.1007/s00592-016-0864-y
Pereon, Y., Lande, G., Demolombe, S., Tich, S. N. T., Sternberg, D., Le Marec, H., et al. (2003). Paramyotonia congenita with an SCN4A mutation affecting cardiac repolarization. Neurology 60 (2), 340–342. doi: 10.1212/01.WNL.0000042093.96309.5A
Persson, F., Andersson, B., Duker, G., Jacobson, I., Carlsson, L. (2007). Functional effects of the late sodium current inhibition by AZD7009 and lidocaine in rabbit isolated atrial and ventricular tissue and Purkinje fibre. Eur. J. Pharmacol. 558 (1-3), 133–143. doi: 10.1016/j.ejphar.2006.11.040
Pieske, B., Maier, L. S., Piacentino, V., Weisser, J., Hasenfuss, G., Houser, S. (2002). Rate Dependence of [Na+]i and Contractility in Nonfailing and Failing Human Myocardium. Circulation 106 (4), 447–453. doi: 10.1161/01.cir.0000023042.50192.f4
Pignier, C., Revenaz, C., Rauly-Lestienne, I., Cussac, D., Delhon, A., Gardette, J., et al. (2007). Direct protective effects of poly-unsaturated fatty acids, DHA and EPA, against activation of cardiac late sodium current. Basic Res. Cardiol. 102 (6), 553–564. doi: 10.1007/s00395-007-0676-x
Pignier, C., Rougier, J. S., Vie, B., Culie, C., Verscheure, Y., Vacher, B., et al. (2010). Selective inhibition of persistent sodium current by F 15845 prevents ischaemia-induced arrhythmias. Br. J. Pharmacol. 161 (1), 79–91. doi: 10.1111/j.1476-5381.2010.00884.x
Pless, S. A., Galpin, J. D., Frankel, A., Ahern, C. A. (2011). Molecular basis for class Ib anti-arrhythmic inhibition of cardiac sodium channels. Nat. Commun. 2, 351. doi: 10.1038/ncomms1351
Poulet, C., Wettwer, E., Grunnet, M., Jespersen, T., Fabritz, L., Matschke, K., et al. (2015). Late Sodium Current in Human Atrial Cardiomyocytes from Patients in Sinus Rhythm and Atrial Fibrillation. PloS One 10 (6), e0131432. doi: 10.1371/journal.pone.0131432
Pourrier, M., Williams, S., McAfee, D., Belardinelli, L., Fedida, D. (2014). CrossTalk proposal: The late sodium current is an important player in the development of diastolic heart failure (heart failure with a preserved ejection fraction). J. Physiol. 592 (Pt 3), 411–414. doi: 10.1113/jphysiol.2013.262261
Rajamani, S., Liu, G., El-Bizri, N., Guo, D., Li, C., Chen, X. L., et al. (2016). The novel late Na(+) current inhibitor, GS-6615 (eleclazine) and its anti-arrhythmic effects in rabbit isolated heart preparations. Br. J. Pharmacol. 173 (21), 3088–3098. doi: 10.1111/bph.13563
Ravens, U., Wettwer, E., Hala, O. (2004). Pharmacological modulation of ion channels and transporters. Cell Calcium 35 (6), 575–582. doi: 10.1016/j.ceca.2004.01.011
Reiffel, J. A., Camm, A. J., Belardinelli, L., Zeng, D., Karwatowska-Prokopczuk, E., Olmsted, A., et al. (2015). The HARMONY Trial: Combined Ranolazine and Dronedarone in the Management of Paroxysmal Atrial Fibrillation: Mechanistic and Therapeutic Synergism. Circ. Arrhythm. Electrophysiol. 8 (5), 1048–1056. doi: 10.1161/CIRCEP.115.002856
Restivo, M., Caref, E. B., Kozhevnikov, D. O., El-Sherif, N. (2004). Spatial dispersion of repolarization is a key factor in the arrhythmogenicity of long QT syndrome. J. Cardiovasc. Electrophysiol. 15 (3), 323–331. doi: 10.1046/j.1540-8167.2004.03493.x
Rivera-Fernandez, R., Arias-Verdu, M. D., Garcia-Paredes, T., Delgado-Rodriguez, M., Arboleda-Sanchez, J. A., Aguilar-Alonso, E., et al. (2016). Prolonged QT interval in ST-elevation myocardial infarction and mortality: new prognostic scale with QT, Killip and age. J. Cardiovasc. Med. (Hagerstown) 17 (1), 11–19. doi: 10.2459/JCM.0000000000000015
Ruan, Y., Liu, N., Priori, S. G. (2009). Sodium channel mutations and arrhythmias. Nat. Rev. Cardiol. 6 (5), 337–348. doi: 10.1038/nrcardio.2009.44
Sarhan, M. F., Van Petegem, F. (2009). Ahern CA. A Double Tyrosine Motif in the Cardiac Sodium Channel Domain III-IV Linker Couples Calcium-dependent Calmodulin Binding to Inactivation Gating. J. Biol. Chem. 284 (48), 33265–33274. doi: 10.1074/jbc.M109.052910
Sarhan, M. F., Tung, C. C., Van Petegem, F., Ahern, C. A. (2012). Crystallographic basis for calcium regulation of sodium channels. Proc. Natl. Acad. Sci. U. S. A. 109 (9), 3558–3563. doi: 10.1073/pnas.1114748109
Scanley, B. E., Hanck, D. A., Chay, T., Fozzard, H. A. (1990). Kinetic-analysis of single sodium-channels from canine cardiac purkinje-cells. J. Gen. Physiol. 95 (3), 411–437. doi: 10.1085/jgp.95.3.411
Scheuer, T. (2011). Regulation of sodium channel activity by phosphorylation. Semin. Cell Dev. Biol. 22 (2), 160–165. doi: 10.1016/j.semcdb.2010.10.002
Schillinger, W., Teucher, N., Christians, C., Kohlhaas, M., Sossalla, S., Van Nguyen, P., et al. (2006). High intracellular Na+ preserves myocardial function at low heart rates in isolated myocardium from failing hearts. Eur. J. Heart Fail. 8 (7), 673–680. doi: 10.1016/j.ejheart.2006.01.013
Scirica, B. M., Morrow, D. A., Hod, H., Murphy, S. A., Belardinelli, L., Hedgepeth, C. M., et al. (2007). Effect of ranolazine, an antianginal agent with novel electrophysiological properties, on the incidence of arrhythmias in patients with non ST-segment elevation acute coronary syndrome: results from the Metabolic Efficiency With Ranolazine for Less Ischemia in Non ST-Elevation Acute Coronary Syndrome Thrombolysis in Myocardial Infarction 36 (MERLIN-TIMI 36) randomized controlled trial. Circulation 116 (15), 1647–1652. doi: 10.1161/CIRCULATIONAHA.107.724880
Shimizu, W., Antzelevitch, C. (1997). Sodium channel block with mexiletine is effective in reducing dispersion of repolarization and preventing torsade des pointes in LQT2 and LQT3 models of the long-QT syndrome. Circulation 96 (6), 2038–2047. doi: 10.1161/01.CIR.96.6.2038
Shimizu, W., Antzelevitch, C. (1999a). Cellular basis for long QT, transmural dispersion of repolarization, and torsade de pointes in the long QT syndrome. J. Electrocardiol. 32 (Suppl), 177–184. doi: 10.1016/s0022-0736(99)90077-8
Shimizu, W., Antzelevitch, C. (1999b). Cellular and ionic basis for T-wave alternans under long-QT conditions. Circulation 99 (11), 1499–1507. doi: 10.1161/01.cir.99.11.1499
Shimoda, L. A., Polak, J. (2011). Hypoxia. 4. Hypoxia and ion channel function. Am. J. Physiol. Cell Physiol. 300 (5), C951–C967. doi: 10.1152/ajpcell.00512.2010
Shryock, J. C., Song, Y. J., Rajamani, S., Antzelevitch, C., Belardinelli, L. (2013). The arrhythmogenic consequences of increasing late I-Na in the cardiomyocyte. Cardiovasc. Res. 99 (4), 600–611. doi: 10.1093/cvr/cvt145
Sicouri, S., Belardinelli, L., Antzelevitch, C. (2013). Antiarrhythmic effects of the highly selective late sodium channel current blocker GS-458967. Heart Rhythm. 10 (7), 1036–1043. doi: 10.1016/j.hrthm.2013.03.023
Sipido, K. R., Bito, V., Antoons, G., Volders, P. G., Vos, M. A. (2007). “Na/Ca exchange and cardiac ventricular arrhythmias,” in Sodium-Calcium Exchange and the Plasma Membrane Ca2+-Atpase in Cell Function: Fifth International Conference, vol. 1099 . Eds. Herchuelz, A., Blaustein, M. P., Lytton, J., Philipson, K. D. (Annals of the New York Academy of Sciences), 339–348.
Song, J. H., Huang, C. S., Nagata, K., Yeh, J. Z., Narahashi, T. (1997). Differential action of riluzole on tetrodotoxin-sensitive and tetrodotoxin-resistant sodium channels. J. Pharmacol. Exp. Ther. 282 (2), 707–714.
Song, Y., Shryock, J. C., Wu, L., Belardinelli, L. (2004). Antagonism by ranolazine of the pro-arrhythmic effects of increasing late INa in guinea pig ventricular myocytes. J. Cardiovasc. Pharmacol. 44 (2), 192–199. doi: 10.1097/00005344-200408000-00008
Song, Y., Shryock, J. C., Wagner, S., Maier, L. S., Belardinelli, L. (2006). Blocking late sodium current reduces hydrogen peroxide-induced arrhythmogenic activity and contractile dysfunction. J. Pharmacol. Exp. Ther. 318 (1), 214–222. doi: 10.1124/jpet.106.101832
Song, Y., Shryock, J. C., Belardinelli, L. (2008). An increase of late sodium current induces delayed afterdepolarizations and sustained triggered activity in atrial myocytes. Am. J. Physiol. Heart Circ. Physiol. 294 (5), H2031–H2039. doi: 10.1152/ajpheart.01357.2007
Sossalla, S., Wagner, S., Rasenack, E. C., Ruff, H., Weber, S. L., Schondube, F. A., et al. (2008). Ranolazine improves diastolic dysfunction in isolated myocardium from failing human hearts–role of late sodium current and intracellular ion accumulation. J. Mol. Cell. Cardiol. 45 (1), 32–43. doi: 10.1016/j.yjmcc.2008.03.006
Sossalla, S., Kallmeyer, B., Wagner, S., Mazur, M., Maurer, U., Toischer, K., et al. (2010). Altered Na(+) currents in atrial fibrillation effects of ranolazine on arrhythmias and contractility in human atrial myocardium. J. Am. Coll. Cardiol. 55 (21), 2330–2342. doi: 10.1016/j.jacc.2009.12.055
Sossalla, S., Maurer, U., Schotola, H., Hartmann, N., Didie, M., Zimmermann, W. H., et al. (2011). Diastolic dysfunction and arrhythmias caused by overexpression of CaMKIIdelta(C) can be reversed by inhibition of late Na(+) current. Basic Res. Cardiol. 106 (2), 263–272. doi: 10.1007/s00395-010-0136-x
Stone, P. H., Gratsiansky, N. A., Blokhin, A., Huang, I. Z., Meng, L. (2006). Antianginal efficacy of ranolazine when added to treatment with amlodipine: the ERICA (Efficacy of Ranolazine in Chronic Angina) trial. J. Am. Coll. Cardiol. 48 (3), 566–575. doi: 10.1016/j.jacc.2006.05.044
Studenik, C. R., Zhou, Z., January, C. T. (2001). Differences in action potential and early afterdepolarization properties in LQT2 and LQT3 models of long QT syndrome. Br. J. Pharmacol. 132 (1), 85–92. doi: 10.1038/sj.bjp.0703770
Szentandrassy, N., Kistamas, K., Hegyi, B., Horvath, B., Ruzsnavszky, F., Vaczi, K., et al. (2015). Contribution of ion currents to beat-to-beat variability of action potential duration in canine ventricular myocytes. Pflug. Arch. Eur. J. Physiol. 467 (7), 1431–1443. doi: 10.1007/s00424-014-1581-4
Tan, H. L., Kupershmidt, S., Zhang, R., Stepanovic, S., Roden, D. M., Wilde, A. A. M., et al. (2002). A calcium sensor in the sodium channel modulates cardiac excitability. Nature 415 (6870), 442–447. doi: 10.1038/415442a
Tang, Q., Ma, J. H., Zhang, P. H., Wan, W., Kong, L. H., Wu, L. (2012). Persistent sodium current and Na+/H+ exchange contributes to the augmentation of the reverse Na+/Ca2+ exchange during hypoxia or acute ischemia in ventricular myocytes. Pflug. Arch. Eur. J. Physiol. 463 (4), 513–522. doi: 10.1007/s00424-011-1070-y
Tarradas, A., Pinsach-Abuin, M. L., Mackintosh, C., Llora-Batlle, O., Perez-Serra, A., Batlle, M., et al. (2017). Transcriptional regulation of the sodium channel gene (SCN5A) by GATA4 in human heart. J. Mol. Cell. Cardiol. 102, 74–82. doi: 10.1016/j.yjmcc.2016.10.013
Thadani, U., Ezekowitz, M., Fenney, L., Chiang, Y. K. (1994). Double-blind efficacy and safety study of a novel anti-ischemic agent, ranolazine, versus placebo in patients with chronic stable angina pectoris. Ranolazine Study Group. Circulation 90 (2), 726–734. doi: 10.1161/01.CIR.90.2.726
Trenor, B., Cardona, K., Gomez, J. F., Rajamani, S., Ferrero, J. M., Jr., Belardinelli, L., et al. (2012). Simulation and mechanistic investigation of the arrhythmogenic role of the late sodium current in human heart failure. PloS One 7 (3), e32659. doi: 10.1371/journal.pone.0032659
Ufret-Vincenty, C. A., Baro, D. J., Lederer, W. J., Rockman, H. A., Quiñones, L. E., Santana, L. F. (2001). Role of Sodium Channel Deglycosylation in the Genesis of Cardiac Arrhythmias in Heart Failure. J. Biol. Chem. 276 (30), 28197–28203. doi: 10.1074/jbc.M102548200
Undrovinas, A. I., Fleidervish, I. A., Makielski, J. C. (1992). Inward sodium current at resting potentials in single cardiac myocytes induced by the ischemic metabolite lysophosphatidylcholine. Circ. Res. 71 (5), 1231–1241. doi: 10.1161/01.RES.71.5.1231
Undrovinas, A. I., Maltsev, V. A., Kyle, J. W., Silverman, N., Sabbah, H. N. (2002). Gating of the late Na+ channel in normal and failing human myocardium. J. Mol. Cell Cardiol. 34 (11), 1477–1489. doi: 10.1006/jmcc.2002.2100
Undrovinas, A. I., Belardinelli, L., Undrovinas, N. A., Sabbah, H. N. (2006). Ranolazine improves abnormal repolarization and contraction in left ventricular myocytes of dogs with heart failure by inhibiting late sodium current. J. Cardiovasc. Electrophysiol. 17 (Suppl 1), S169–S177. doi: 10.1111/j.1540-8167.2006.00401.x
Vacher, B., Pignier, C., Letienne, R., Verscheure, Y. (2009). Le Grand B. F 15845 inhibits persistent sodium current in the heart and prevents angina in animal models. Br. J. Pharmacol. 156 (2), 214–225. doi: 10.1111/j.1476-5381.2008.00062.x
Valdivia, C. R., Chu, W. W., Pu, J. L., Foell, J. D., Haworth, R. A., Wolff, M. R., et al. (2005). Increased late sodium current in myocytes from a canine heart failure model and from failing human heart. J. Mol. Cell Cardiol. 38 (3), 475–483. doi: 10.1016/j.yjmcc.2004.12.012
van Stuijvenberg, L., Yildirim, C., Kok, B. G., van Veen, T. A., Varro, A., Winckels, S. K., et al. (2010). Alternative promoter usage and splicing of the human SCN5A gene contribute to transcript heterogeneity. DNA Cell Biol. 29 (10), 577–587. doi: 10.1089/dna.2009.0999
Wagner, S., Dybkova, N., Rasenack, E. C. L., Jacobshagen, C., Fabritz, L., Kirchhof, P., et al. (2006). Ca2+/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. J. Clin. Invest. 116 (12), 3127–3138. doi: 10.1172/jci26620
Wagner, S., Ruff, H. M., Weber, S. L., Bellmann, S., Sowa, T., Schulte, T., et al. (2011). Reactive oxygen species-activated Ca/calmodulin kinase IIdelta is required for late I(Na) augmentation leading to cellular Na and Ca overload. Circ. Res. 108 (5), 555–565. doi: 10.1161/CIRCRESAHA.110.221911
Wagner, S., Rokita, A. G., Anderson, M. E., Maier, L. S. (2013). Redox regulation of sodium and calcium handling. Antioxid. Redox Signal. 18 (9), 1063–1077. doi: 10.1089/ars.2012.4818
Wang, Z., Page, P., Nattel, S. (1992). Mechanism of flecainide's antiarrhythmic action in experimental atrial fibrillation. Circ. Res. 71 (2), 271–287. doi: 10.1161/01.res.71.2.271
Wang, Q., Shen, J., Splawski, I., Atkinson, D., Li, Z., Robinson, J. L., et al. (1995). SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell 80 (5), 805–811. doi: 10.1016/0092-8674(95)90359-3
Wang, D. W., Yazawa, K., George, A. L., Jr., Bennett, P. B. (1996). Characterization of human cardiac Na+ channel mutations in the congenital long QT syndrome. Proc. Natl. Acad. Sci. U. S. A. 93 (23), 13200–13205. doi: 10.1073/pnas.93.23.13200
Wang, D. W., Viswanathan, P. C., Balser, J. R., George, A. L., Benson, D. W. (2002). Clinical, genetic, and biophysical characterization of SCN5A mutations associated with atrioventricular conduction block. Circulation 105 (3), 341–346. doi: 10.1161/hc0302.102592
Wang, W. P., Ma, J. H., Zhang, P. H., Luo, A. T. (2007). Redox reaction modulates transient and persistent sodium current during hypoxia in guinea pig ventricular myocytes. Pflug. Arch. Eur. J. Physiol. 454 (3), 461–475. doi: 10.1007/s00424-007-0219-1
Wang, D. W., Desai, R. R., Crotti, L., Arnestad, M., Insolia, R., Pedrazzini, M., et al. (2007). Cardiac sodium channel dysfunction in sudden infant death syndrome. Circulation 115 (3), 368–376. doi: 10.1161/CIRCULATIONAHA.106.646513
Weiss, S. M., Saint, D. A. (2010). The persistent sodium current blocker riluzole is antiarrhythmic and anti-ischaemic in a pig model of acute myocardial infarction. PloS One 5 (11), e14103. doi: 10.1371/journal.pone.0014103
Weiss, S. M., Dahlstrom, J. E., Saint, D. A. (2013). Riluzole reduces arrhythmias and myocardial damage induced by coronary occlusion in anaesthetized pigs. Clin. Exp. Pharmacol. Physiol. 40 (12), 856–863. doi: 10.1111/1440-1681.12175
Wingo, T. L., Shah, V. N., Anderson, M. E., Lybrand, T. P., Chazin, W. J., Balser, J. R. (2004). An EF-hand in the sodium channel couples intracellular calcium to cardiac excitability. Nat. Struct. Mol. Biol. 11 (3), 219–225. doi: 10.1038/nsmb737
Wu, J. Y., Corr, P. B. (1994). Palmitoyl carnitine modifies sodium currents and induces transient inward current in ventricular myocytes. Am. J. Physiol. 266 (3), H1034–H1046. doi: 10.1152/ajpheart.1994.266.3.H1034
Wu, L., Shryock, J. C., Song, Y., Belardinelli, L. (2006). An increase in late sodium current potentiates the proarrhythmic activities of low-risk QT-prolonging drugs in female rabbit hearts. J. Pharmacol. Exp. Ther. 316 (2), 718–726. doi: 10.1124/jpet.105.094862
Wu, L., Ma, J., Li, H., Wang, C., Grandi, E., Zhang, P., et al. (2011). Late sodium current contributes to the reverse rate-dependent effect of IKr inhibition on ventricular repolarization. Circulation 123 (16), 1713–1720. doi: 10.1161/CIRCULATIONAHA.110.000661
Xi, Y. T., Wu, G. R., Yang, L., Han, K., Du, Y., Wang, T. Z., et al. (2009). Increased late sodium currents are related to transcription of neuronal isoforms in a pressure-overload model. Eur. J. Heart Fail. 11 (8), 749–757. doi: 10.1093/eurjhf/hfp092
Yamamura, K., Muneuchi, J., Uike, K., Ikeda, K., Inoue, H., Takahata, Y., et al. (2010). A novel SCN5A mutation associated with the linker between III and IV domains of Na(v)1.5 in a neonate with fatal long QT syndrome. Int. J. Cardiol. 145 (1), 61–64. doi: 10.1016/j.ijcard.2009.04.023
Yang, P., Kupershmidt, S., Roden, D. M. (2004). Cloning and initial characterization of the human cardiac sodium channel (SCN5A) promoter. Cardiovasc. Res. 61 (1), 56–65. doi: 10.1016/j.cardiores.2003.09.030
Yang, T., Atack, T. C., Stroud, D. M., Zhang, W., Hall, L., Roden, D. M. (2012). Blocking Scn10a Channels in Heart Reduces Late Sodium Current and Is Antiarrhythmic. Circ. Res. 111 (3), 322–332. doi: 10.1161/circresaha.112.265173
Yang, K. C., Bonini, M. G., Dudley, S. C., Jr. (2014). Mitochondria and arrhythmias. Free Radic. Biol. Med. 71, 351–361. doi: 10.1016/j.freeradbiomed.2014.03.033
Yao, L., Fan, P., Jiang, Z., Viatchenko-Karpinski, S., Wu, Y., Kornyeyev, D., et al. (2011). Nav1.5-dependent persistent Na+ influx activates CaMKII in rat ventricular myocytes and N1325S mice. Am. J. Physiol. Cell Physiol. 301 (3), C577–C586. doi: 10.1152/ajpcell.00125.2011
Zablocki, J. A., Elzein, E., Li, X., Koltun, D. O., Parkhill, E. Q., Kobayashi, T., et al. (2016). Discovery of Dihydrobenzoxazepinone (GS-6615) Late Sodium Current Inhibitor (Late INai), a Phase II Agent with Demonstrated Preclinical Anti-Ischemic and Antiarrhythmic Properties. J. Med. Chem. 59 (19), 9005–9017. doi: 10.1021/acs.jmedchem.6b00939
Zareba, W., Daubert, J. P., Beck, C. A., Huang, D. T., Alexis, J. D., Brown, M. W., et al. (2018). Ranolazine in High-Risk Patients With Implanted Cardioverter-Defibrillators: The RAID Trial. J. Am. Coll. Cardiol. 72 (6), 636–645. doi: 10.1016/j.jacc.2018.04.086
Zaza, A., Rocchetti, M. (2013). The Late Na+ Current - Origin and Pathophysiological Relevance. Cardiovasc. Drugs Ther. 27 (1), 61–68. doi: 10.1007/s10557-012-6430-0
Zhang, T., Brown, J. H. (2004). Role of Ca2+/calmodulin-dependent protein kinase II in cardiac hypertrophy and heart failure. Cardiovasc. Res. 63 (3), 476–486. doi: 10.1016/j.cardiores.2004.04.026
Zhang, Y., Hartmann, H. A., Satin, J. (1999). Glycosylation Influences Voltage-Dependent Gating of Cardiac and Skeletal Muscle Sodium Channels. J. Membr. Biol. 171 (3), 195–207. doi: 10.1007/s002329900571
Zhou, A., Shi, G., Kang, G. J., Xie, A., Liu, H., Jiang, N., et al. (2018). RNA Binding Protein, HuR, Regulates SCN5A Expression Through Stabilizing MEF2C transcription factor mRNA. J. Am. Heart Assoc. 7 (9), e007802. doi: 10.1161/JAHA.117.007802
Zygmunt, A. C., Nesterenko, V. V., Rajamani, S., Hu, D., Barajas-Martinez, H., Belardinelli, L., et al. (2011). Mechanisms of atrial-selective block of Na(+) channels by ranolazine: I. Experimental analysis of the use-dependent block. Am. J. Physiol. Heart Circ. Physiol. 301 (4), H1606–H1614. doi: 10.1152/ajpheart.00242.2011
Keywords: voltage gated sodium channel, late sodium current, arrhythmias, antiarrhythmic drugs, sodium channel inhibitors
Citation: Horváth B, Hézső T, Kiss D, Kistamás K, Magyar J, Nánási PP and Bányász T (2020) Late Sodium Current Inhibitors as Potential Antiarrhythmic Agents. Front. Pharmacol. 11:413. doi: 10.3389/fphar.2020.00413
Received: 31 December 2019; Accepted: 18 March 2020;
Published: 20 April 2020.
Edited by:
Annamaria De Luca, University of Bari Aldo Moro, ItalyReviewed by:
Francesco Miceli, University of Naples Federico II, ItalyCopyright © 2020 Horváth, Hézső, Kiss, Kistamás, Magyar, Nánási and Bányász. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Balázs Horváth, aG9ydmF0aC5iYWxhenNAbWVkLnVuaWRlYi5odQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.