
- Key Laboratory of Nutrition and Metabolism, Institute for Nutritional Sciences, Shanghai Institutes for Biological Sciences, Graduate School of the Chinese Academy of Sciences, Chinese Academy of Sciences, Shanghai, China
Prostaglandins (PGs), a group of key lipid mediators, are involved in numerous physiological and pathological processes including inflammation and cardiovascular homeostasis. Each PG acts on its specific and distinct cell surface G protein-coupled receptors (GPCRs) or peroxisome proliferator-activated receptors (PPARs). Prostaglandin F2α receptor (FP) is required for female reproductive function such as luteolysis and parturition. It has recently been implicated in blood pressure regulation, atherosclerosis and other inflammation-related disorders. The emerging role of FP in cardiovascular diseases is highlighted and potential therapeutic translation is discussed in the current review.
Introduction
Prostanoids, including prostaglandin (PG) E2, PGD2, prostacyclin (PGI2), thromboxane A2 (TxA2), and PGF2α, are generated through PGH synthase (PGHS) – known commonly as cyclooxygenase (COX), in response to a wide variety of stimuli acting as paracrine or autocrine manner. Non-steroidal anti-inflammatory drugs (NSAIDs) such as aspirin, ibuprofen, inhibit COX isforms to achieve antipyretic, analgesic, and anti-inflammatory actions through blocking PGs biosynthesis (Funk, 2001). Accumulating evidences demonstrate COX-derived PGs play crucial role in mediating an array of cellular processes such as cell proliferation, differentiation, and apoptosis and in regulating female reproductive function and parturition, platelet aggregation, and vascular homeostasis (Smith et al., 2000; Yu et al., 2006; Funk and FitzGerald, 2007; Yu and Funk, 2007). In addition, PGs also are involved in pathogenesis of inflammation, cancer, and cardiovascular disorders (FitzGerald and Loll, 2001; Smyth et al., 2009). The biological functions of PGs could be modulated at multiple levels such as COX, PG synthases, and downstream receptors (Narumiya and FitzGerald, 2001). Elucidating the physiological roles of COX-derived PGs in cellular and whole body homeostasis and the mechanism underlying their action will no doubt offer opportunity for developing novel therapeutics for inflammatory disease, cancer, and hypertension. Here, we summarized the recent works focusing on PGF2α/FP receptor response in cardiovascular system and reviewed the recent development of potential therapeutic target of FP receptor.
PGF2α and FP Receptor
Prostanoids are formed through COXs on arachidonic acid via a two-step enzymatic process. First the arachidonic acid is bioconverted to PGG2 through COX catalytic activity and then PGH2 through peroxidase activity (POX) of PGHS enzymes. Subsequently the PGH2 is subject to metabolize to active prostanoids through individual PG synthases (Figure 1). Diversity in expression of downstream synthases results in the generation of one or two dominant PGs by individual cells. In general, PGF2α is formed by reduction of PGH2 by PG endoperoxide synthase or reductase. It also can be also formed from other PGs (Figure 1) such as PGE2 through 9-keto reductases and PGD2 through 11-keto reductases (Watanabe et al., 1985), although relatively rare. Endogenous primary PGF2α is rapidly degraded enzymatically, half-life is less than 1 min in peripheral circulation, and its relatively stable metabolite is 15-keto-dihydro-PGF2α (Basu et al., 1992).
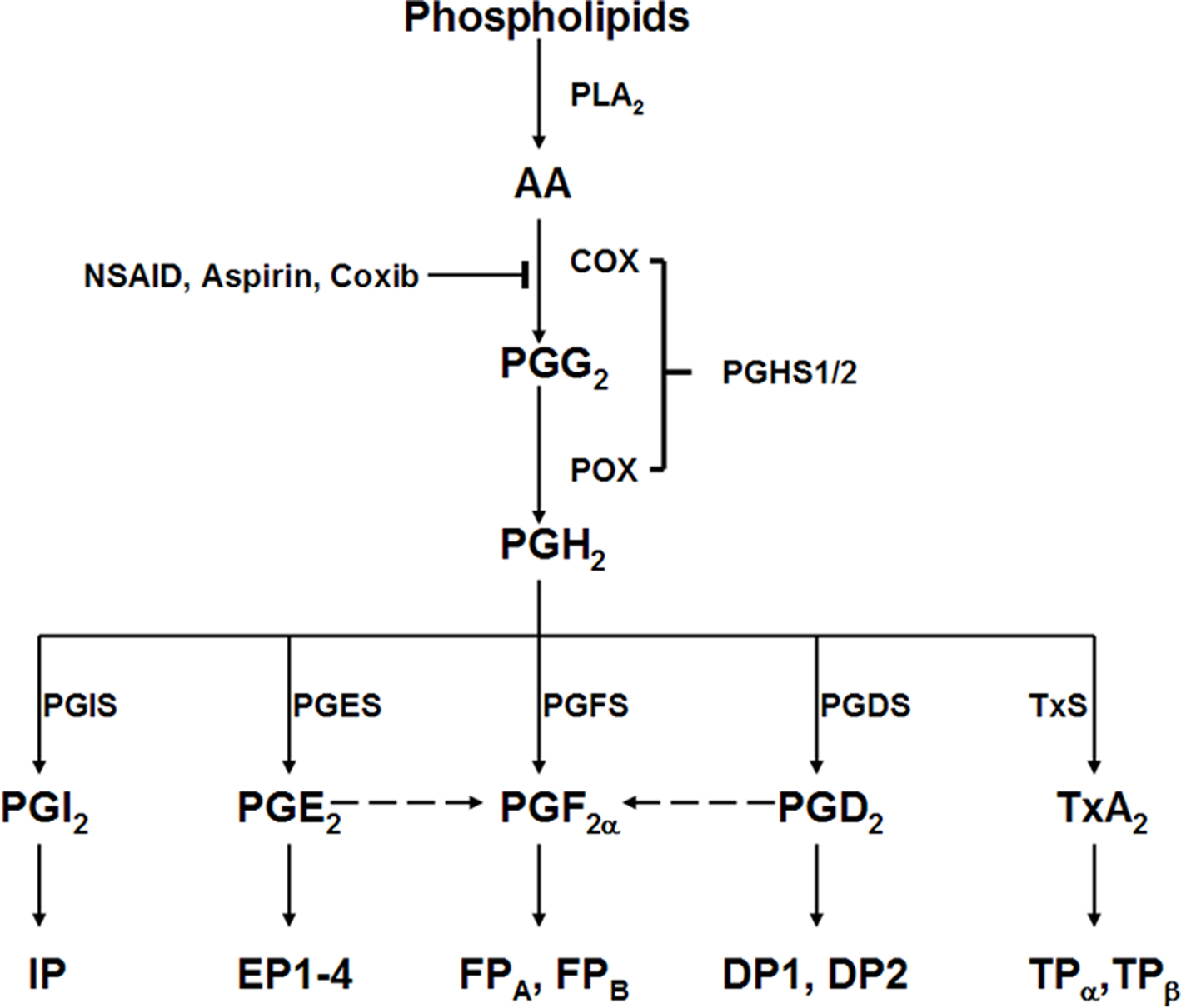
Figure 1. Prostanoid biosynthesis and response pathway. AA, arachidonic acid; PLA2, phospholipase A2; PGHS1/2, prostaglandin G/H synthase 1 or 2, which contains both cyclooxygenases (COX) and peroxidase (POX) activities; PGIS, prostaglandin I2 synthase; PGES, prostaglandin E2 synthase; PGFS, prostaglandin F synthase; PGDS, prostaglandin D2 synthase; TxS, thromboxane A2 synthase; IP, prostaglandin I2 receptor; EP, prostaglandin E2 receptor; FP, prostaglandin F2α receptor; DP, prostaglandin D2 receptor; TP, thromboxane A2 receptor.
PGF2α exits in almost all the tissues (Basu, 2007) with more abundant in the female reproductive system (Hao and Breyer, 2008); its cellular and physiological effects are mediated by a G protein-coupled receptor-the F prostanoid receptor (the FP; Narumiya et al., 1999). Two splice forms of FP (FPA and FPB) exist in human. Initially, the FP receptor was characterized as coupling to Gq protein which lead to inositol triphosphate (IP3)/diacylglycerol (DAG) generation and mobilization of intracellular calcium (Abramovitz et al., 1994; Sugimoto et al., 1994; Watanabe et al., 1994), which is linked to the proliferation of cells (Watanabe et al., 1994). Stimulation of FP also led to activation of the small G protein Rho, resulting in phosphorylation of the p125 focal adhesion kinase, cytoskeleton rearrangement and cell morphology change (Pierce et al., 1999), and phospholipase C-mediated phosphorylation of the epidermal growth factor receptor (EGFR) and mitogen-activated protein kinase (MAPK) signaling pathways in endometrial adenocarcinoma cells (Sales et al., 2004). Recently, the coupling of Gi of FP receptor has been reported, which is response for water reabsorption in renal collecting ducts in rabbit (Hebert et al., 2005).
Expression of FP receptor and its corresponding function are summarized in Table 1. FP is highly expressed in the genitourinary tract (Sugimoto et al., 1997; Saito et al., 2003). Gene manipulation studies showed that, parturition is disrupted in mice lacking either cytosolic phospholipase A2 (cPLA2; Bonventre et al., 1997), that mobilizes arachidonic acid release for COX metabolism, COX-2, the more regulated form of that enzyme (Dinchuk et al., 1995; Morham et al., 1995) or the FP receptor (Sugimoto et al., 1997). Likewise, the onset of parturition is delayed in COX-1 knock out (KO) mice (Langenbach et al., 1995) but not COX-1 knockdown (KD; Yu et al., 2005). This results in high neonatal mortality that can be rescued by PGF2α replacement (Gross et al., 1998). In the eye, the FP is expressed in the vasculature, the iris sphincter and in the anterior circular muscles, all relevant to the increased uveoscleral outflow of aqueous humor provoked by PGF2α (Mukhopadhyay et al., 2001). FP agonists are approved for local application in the treatment of glaucoma (Ishida et al., 2006). Recently, abundant FP expression has also been detected in the distal convoluted tubules (DCT) and cortical collecting ducts (CCD) of the kidney (Saito et al., 2003), implicating its role in water and electrolyte homeostasis (Hebert et al., 2005). FP is observed in lung tissue and lung fibroblasts, which facilitates bleomycin-induced pulmonary fibrosis independently of transforming growth factor β (TGFβ; Oga et al., 2009). No FP receptor seems been detected in immune system organs such as spleen and thymus (Tilley et al., 2001).
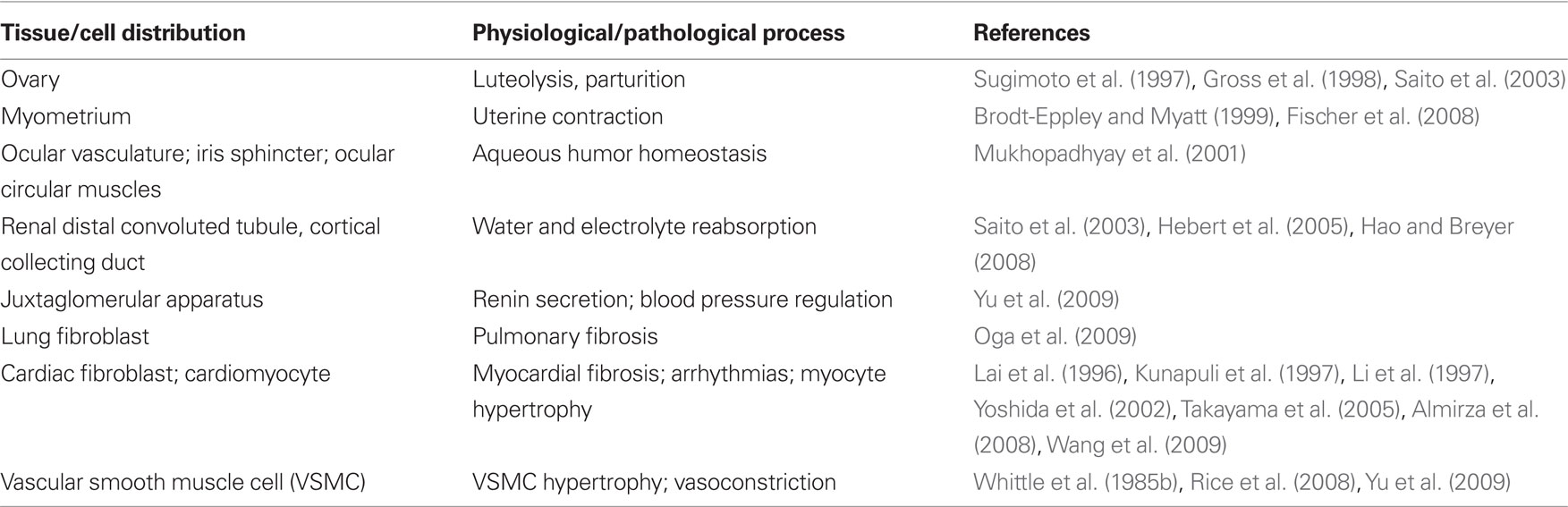
Table 1. FP expression and its physiological/pathological function.
FP in Cardiovascular Diseases
In the heart, PGF2α derives mainly from cardiac fibroblasts and its formation is increased in endocardium by ischemia (Rabinowitz et al., 1992), where it depresses contractile recovery through a mechanism associated with altered cellular energy metabolism and increased calcium accumulation (Karmazyn et al., 1993). Through FP receptor, PGF2α promotes expression of c-fos, atrial natriuretic factor (ANF), and alpha-skeletal actin in cardiomyocytes and induces cardiac myocyte hypertrophy in vitro and cardiac growth in rat (Lai et al., 1996), but does not affect myocyte proliferation in culture (Adams et al., 1996). Mechanistic studies showed PGF2α inhibits expression Ca2+-ATPase (SERCA2) via induction of Early Growth Response Protein 1 (Egr-1) in cultured neonatal cardiac myocytes (Hara et al., 2008). We have recently found that selective deletion of cardiomyocyte COX-2 releases a restraint on expression of fibroblast COX-2, thereby augmenting PGF2α formation. This, in turn, coincides with an increase in myocardial fibrosis and a predisposition to arrhythmogenesis (Wang et al., 2009). COX-2 derived PGF2α can further promote fibroblast PGF2α formation in a feed forward manner (Yoshida et al., 2002) and progressively promote fibrosis (Almirza et al., 2008). PGF2α promotes arrhythmias in cultured neonatal rat cardiac myocytes (Kunapuli et al., 1997; Li et al., 1997) and FP deletion protects against inflammatory tachycardia in mice in vivo (Takayama et al., 2005). Thus, PGF2α/FP response is involved in multiple aspects of ischemia heart disease (Figure 2), blockage of the FP may facilitate recovery from cardiac ischemia-reperfusion induced injury.
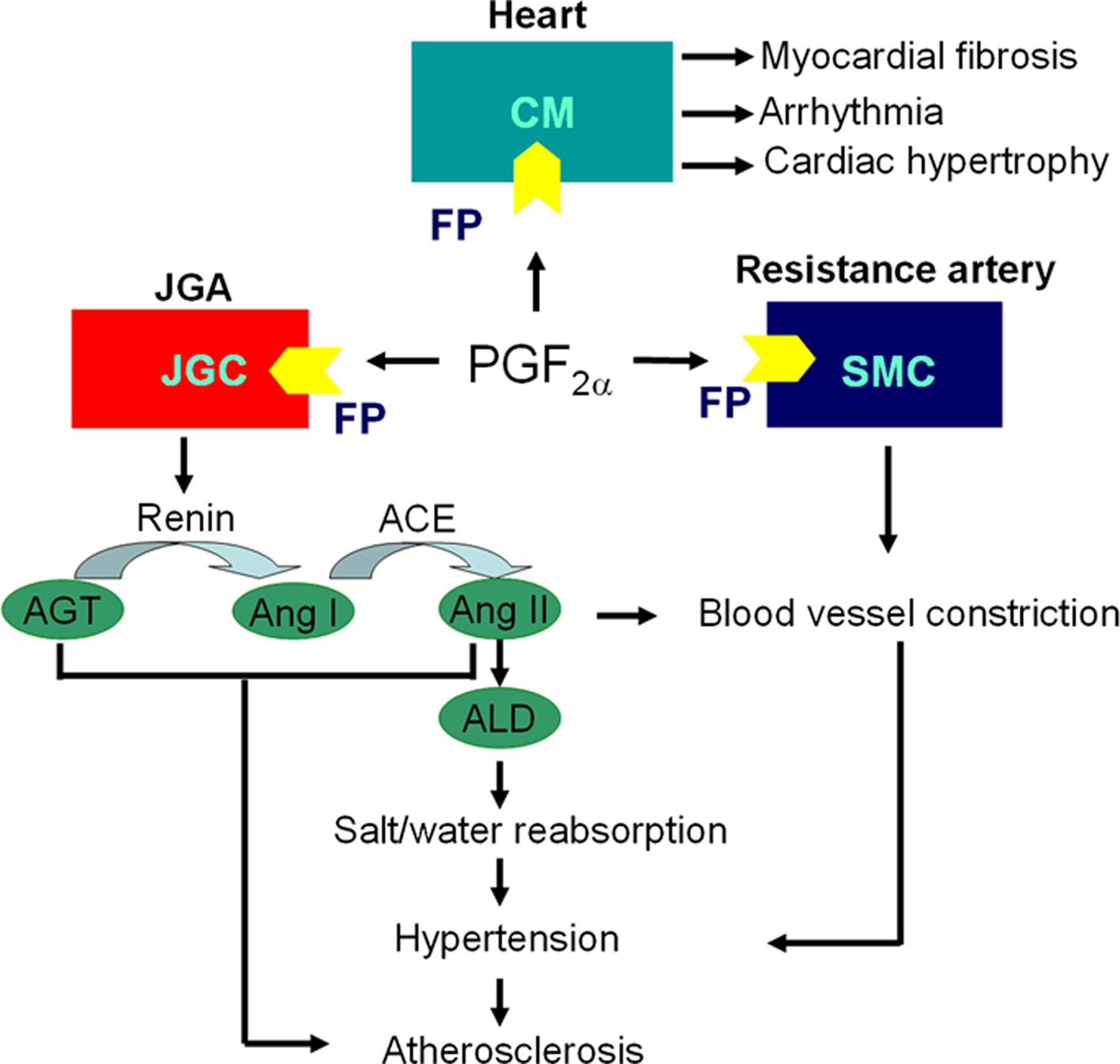
Figure 2. Scheme of PGF2α/FP pathway involved in pathogenesis of cardiovascular disease. Cardiac fibroblasts derived PGF2α induces cardiac hypertrophy, fibrosis and arrhythmia through FP receptor in adjacent cardiomyocytes (CMs); PGF2α stimulates renin release from juxtaglomerular granular cells (JGCs) by FP receptor in an autocrine fashion, and activate renin–angiotensin–aldosterone system (RAAS) to elevate blood pressure through enhancing salt/water reabsorption in kidney and constricting blood vessels directly via Angiotensin II (Ang II); PGF2α promotes resistance artery constriction through FP in smooth muscle cells (SMCs), which eventually increases blood pressure and contributes to atherosclerosis; Activated RAAS also accelerates atherosclerosis. JGA, juxtaglomerular apparatus; AGT, angiotensinogen; ACE, angiotensin-converting enzyme; ALD, aldosterone.
Vascular endothelial cells secrete surprisingly large amounts of PGF2α in response to shear stress in vitro (Di Francesco et al., 2009). The relevance of this phenomenon is poorly understood but in sufficient quantities. PGF2α may act as an incidental ligand at the TxA2 receptor-the TP (Wong et al., 2009). Furthermore, the expression of FP receptors in the medial layer of resistance vessels was observed (Yu et al., 2009), which is involved in vasoconstriction (Whittle et al., 1985a). Thus it might prove relevant to the regulation of systemic blood pressure (BP) as PGF2α direct infusion causes dose-dependent elevation of BP in anesthetized mice (Yu et al., 2009). Moreover, PGF2α increases reactive oxygen species (ROS) and induces vascular smooth muscle cells (VSMCs) hypertrophy through translocation of mammalian target of rapamycin (mTOR) from nucleus to cytoplasm and activation of phosphatidylinositol 3-kinase (PI3K) pathway (Rice et al., 2008). In mice, FP deletion reduces significantly BP in mice, both when they are placed on a regular chow diet and after manipulation of dietary fat or sodium intake. This coincides with decreased activation of renin–angiotensin–aldosterone system (RAAS; Yu et al., 2009). FP receptor expression is marked in afferent arterioles of the juxtaglomerular apparatus (JGA) and renin-containing granular cells are decreased in the FP deficient mice (FP−/−). Indeed, activation of the FP appears to regulate juxtaglomerular (JG) cell differentiation and consequent renin expression, explaining depressed activation of the RAAS in FP−/− mice. Although FP expression was not detected in the aorta or even when it was complicated by atherosclerotic lesions, FP deletion attenuates atherogenesis in hyperlipidemic mice [low-density lipoprotein (LDL) receptor knockout, Ldlr−/−]. Perhaps restraint of atherogenesis in Ldlr/FP double knockout (Ldlr−/−/FP−/−) mice merely results from disruption of renal RAAS activation with a consequent impact on systemic BP (Figure 2). Taken together, antagonism of the FP receptor may afford a strategy for the control of hypertension and its attendant vascular diseases such as atherosclerosis (Yu et al., 2009).
PGF2α in Human Inflammatory Disease
In human studies, PGF2α is one of the more abundant PGs formed at sites of inflammation (Scher and Pillinger, 2009), and is subject to inhibition by NSAIDs such as low dose aspirin (Helmersson et al., 2005b). Similar to PGE2, PGF2α is present in joint fluid collected from rheumatoid arthritis, psoriatic arthritis, osteoarthritis patients (Trang et al., 1977; Basu et al., 2001), and the levels of these PGs could also be effectively retarded by NSAIDs treatment. In addition, the synovial cells from rheumatoid arthritis patient are able to secrete PGF2α in vitro (Seppala, 1987). Along with 8-Iso-PGF2α-oxidative stress marker, PGF2α was elevated during the first hour in acute myocardial infarction (AMI) patient treated with percutaneous coronary intervention (PCI; Berg et al., 2005) and 24 h after post-surgery in elective PCI patients probably due to aspirin treatment before operation (Berg et al., 2004).
Atherosclerosis is a chronic vascular inflammation diseases characterized by the thickening of the arterial wall (Rader and Daugherty, 2008). Vascular endothelial dysfunction is believed as initial step during atherogenesis, high plasma LDL, free oxygen radicals caused by cigarette smoking, hypertension, and diabetes mellitus, and other genetic defects could cause endothelial dysfunction leading to atherosclerosis (Ross, 1999). As the major metabolite of PGF2α, 15-keto-dihydro-PGF2α is elevated in the conditions associated with those increased cardiovascular risk, such as smoking (Helmersson et al., 2005a), obese (Sinaiko et al., 2005), rheumatic disease (Basu et al., 2001), type I (Basu et al., 2005) and type II (Helmersson et al., 2004) diabetes mellitus; increased PGF2α was found in urine from population with hypercholesterolemia and smoking – the conditions associated with oxidative stress (Yin et al., 2007). Moreover, plasma PGF2α level in the elder man is positively related with common carotid artery intima-media thickness (CCA-IMT) (Wohlin et al., 2007) – a valid index of atherosclerosis. Moreover, a polymorphism in COX-1 gene (rs10306135) identified recently is associated with significantly decreased PGF2α and further lower susceptibility for cardiovascular disease (Helmersson et al., 2009). Hence, PGF2α maybe involved in initiation and progression of chronic cardiovascular diseases, such as atherosclerosis and hypertension.
Pharmacology of FP Modulation
Given the accumulating evidence pleading for the involvement of PGF2α/FP receptor response pathway in regulating ocular uveoscleral outflow and normal parturition as well as pathogenesis of hypertension and atherosclerosis, the exploration of novel compounds able to specifically stimulate or inhibit FP receptor will constitute promising therapeutic avenues.
Human FP receptors are expressed in the human ocular trabecular meshwork (Anthony et al., 1998) and topical exogenous PGF2α and FP agonists reduce intraocular pressure (IOP) in monkeys and humans without causing inflammation (Weinreb et al., 2002). Thus, FP agonists, latanoprost, bimatoprost, and travoprost, are used in the treatment of glaucoma and ocular hypertension (Ishida et al., 2006), although the precise mechanism by which they work is poorly understood. More directly relevant has been the suggestion that FP antagonism may delay luteolysis and uterine contraction during parturition (Bernal, 2001), with the potential to delay preterm birth (Olson, 2005). Until recently, AL-8810, reported 10 years ago, is the first described FP antagonist, albeit that it is a partial agonist (Griffin et al., 1999) with which there is much experience in model systems (Sharif et al., 2000; Hirst et al., 2005). Theratechnologies compound THG 113 tested as FP receptor blocker, inhibits the contractile activity of smooth muscle cells from mouse (Peri et al., 2002), sheep (Hirst et al., 2005), and human myometrium (Friel et al., 2005) in response to exogenous PGF2α in vitro probably through activating Ca2+-activated K+ channel (BKCa; Doheny et al., 2007), and delays lipopolysaccharide (LPS)-induced preterm birth in mice (Peri et al., 2002), and lowers uterine electromyographic activity and delays RU486 (a progesterone receptor blocker)-induced preterm birth in sheep (Hirst et al., 2005). More recently, AS604872, another patented FP antagonist, was shown to be effective to delay preterm parturition in rodents (Chollet et al., 2007; Cirillo et al., 2007). Thus, FP receptor could be a potential target for the pharmacological management of preterm labor. Given that renin is elevated in pregnancy-induced hypertension with decreased PGI2 biosynthesis (Fitzgerald et al., 1987), FP antagonist seems more suitable theoretically for management of pregnancy-induced hypertension with broad gestational benefits. However, further clinical investigation is required regarding therapeutic efficacy of FP antagonist in clinic.
Conclusion
In summary, PGF2α, an early focus of prostaglandin research has been quite neglected outside the field of reproductive biology in recent decades. However, emerging evidence, particularly from mice lacking its FP receptor, hint at its importance in BP regulation and atherosclerosis. PGI2 is a potent renin secretagogue, antagonism or deletion of its receptor (the IP) protects against high-renin hypertension in renoprival models of in rodents (Fujino et al., 2004), while accelerates atherogenesis (Kobayashi et al., 2004). Thus, blockade of the FP may represent a novel therapeutic strategy in syndromes of renin dependent hypertension with a more cardioprotective profile than suppressing synthesis or disrupting activation of the PGI2 receptor (IP). Given the use of FP agonists in the treatment of glaucoma, the synthesis of antagonists seems readily tractable. Such pharmacological probes will facilitate our determination of whether FP antagonists might have utility in a wide variety of cardiovascular disorders in the future.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from Chinese Academy of Sciences (Knowledge Innovation Program O914X41281 and Talent 100 Program 2010OHTP10), Clinic Research Center at Institute for Nutritional Sciences, Shanghai Institutes for Biological Sciences (CRC2010007) and the Ministry of Science and Technology of China (2011CB503906).
References
Abramovitz, M., Boie, Y., Nguyen, T., Rushmore, T. H., Bayne, M. A., Metters, K. M., Slipetz, D. M., and Grygorczyk, R. (1994). Cloning and expression of a cDNA for the human prostanoid FP receptor. J. Biol. Chem. 269, 2632–2636.
Adams, J. W., Migita, D. S., Yu, M. K., Young, R., Hellickson, M. S., Castro-Vargas, F. E., Domingo, J. D., Lee, P. H., Bui, J. S., and Henderson, S. A. (1996). Prostaglandin F2 alpha stimulates hypertrophic growth of cultured neonatal rat ventricular myocytes. J. Biol. Chem. 271, 1179–1186.
Almirza, W. H., Dernison, M. M., Peters, P. H., van Zoelen, E. J., and Theuvenet, A. P. (2008). Role of the prostanoid FP receptor in action potential generation and phenotypic transformation of NRK fibroblasts. Cell. Signal. 20, 2022–2029.
Anthony, T. L., Pierce, K. L., Stamer, W. D., and Regan, J. W. (1998). Prostaglandin F2 alpha receptors in the human trabecular meshwork. Invest. Ophthalmol. Vis. Sci. 39, 315–321.
Basu, S. (2007). Novel cyclooxygenase-catalyzed bioactive prostaglandin F2alpha from physiology to new principles in inflammation. Med. Res. Rev. 27, 435–468.
Basu, S., Larsson, A., Vessby, J., Vessby, B., and Berne, C. (2005). Type 1 diabetes is associated with increased cyclooxygenase- and cytokine-mediated inflammation. Diabetes Care 28, 1371–1375.
Basu, S., Sjoquist, B., Resul, B., and Stjernschantz, J. (1992). Presence of a 15-ketoprostaglandin delta 13-reductase in porcine cornea. Acta Chem. Scand. 46, 108–110.
Basu, S., Whiteman, M., Mattey, D. L., and Halliwell, B. (2001). Raised levels of F(2)-isoprostanes and prostaglandin F(2alpha) in different rheumatic diseases. Ann. Rheum. Dis. 60, 627–631.
Berg, K., Jynge, P., Bjerve, K., Skarra, S., Basu, S., and Wiseth, R. (2005). Oxidative stress and inflammatory response during and following coronary interventions for acute myocardial infarction. Free Radic. Res. 39, 629–636.
Berg, K., Wiseth, R., Bjerve, K., Brurok, H., Gunnes, S., Skarra, S., Jynge, P., and Basu, S. (2004). Oxidative stress and myocardial damage during elective percutaneous coronary interventions and coronary angiography. A comparison of blood-borne isoprostane and troponin release. Free Radic. Res. 38, 517–525.
Bonventre, J. V., Huang, Z., Taheri, M. R., O’Leary, E., Li, E., Moskowitz, M. A., and Sapirstein, A. (1997). Reduced fertility and postischaemic brain injury in mice deficient in cytosolic phospholipase A2. Nature 390, 622–625.
Brodt-Eppley, J., and Myatt, L. (1999). Prostaglandin receptors in lower segment myometrium during gestation and labor. Obstet. Gynecol. 93, 89–93.
Chollet, A., Tos, E. G., and Cirillo, R. (2007). Tocolytic effect of a selective FP receptor antagonist in rodent models reveals an innovative approach to the treatment of preterm labor. BMC Pregnancy Childbirth 7(Suppl. 1), S16.
Cirillo, R., Tos, E. G., Page, P., Missotten, M., Quattropani, A., Scheer, A., Schwarz, M. K., and Chollet, A. (2007). Arrest of preterm labor in rat and mouse by an oral and selective nonprostanoid antagonist of the prostaglandin F2alpha receptor (FP). Am. J. Obstet. Gynecol. 197, 54.e 1–e9.
Di Francesco, L., Totani, L., Dovizio, M., Piccoli, A., Di Francesco, A., Salvatore, T., Pandolfi, A., Evangelista, V., Dercho, R. A., Seta, F., and Patrignani, P. (2009). Induction of prostacyclin by steady laminar shear stress suppresses tumor necrosis factor-alpha biosynthesis via heme oxygenase-1 in human endothelial cells. Circ. Res. 104, 506–513.
Dinchuk, J. E., Car, B. D., Focht, R. J., Johnston, J. J., Jaffee, B. D., Covington, M. B., Contel, N. R., Eng, V. M., Collins, R. J., Czerniak, P. M., Gorry, S. A, and Trzaskos, J. M. (1995). Renal abnormalities and an altered inflammatory response in mice lacking cyclooxygenase II. Nature 378, 406–409.
Doheny, H. C., O’Reilly, M. J., Sexton, D. J., and Morrison, J. J. (2007). THG113.31, a specific PGF2alpha receptor antagonist, induces human myometrial relaxation and BKCa channel activation. Reprod. Biol. Endocrinol. 5, 10.
Fischer, D. P., Hutchinson, J. A., Farrar, D., O’Donovan, P. J., Woodward, D. F., and Marshall, K. M. (2008). Loss of prostaglandin F2alpha, but not thromboxane, responsiveness in pregnant human myometrium during labour. J. Endocrinol. 197, 171–179.
Fitzgerald, D. J., Entman, S. S., Mulloy, K., and FitzGerald, G. A. (1987). Decreased prostacyclin biosynthesis preceding the clinical manifestation of pregnancy-induced hypertension. Circulation 75, 956–963.
FitzGerald, G. A., and Loll, P. (2001). COX in a crystal ball: current status and future promise of prostaglandin research. J. Clin. Invest. 107, 1335–1337.
Friel, A. M., O’Reilly, M. W., Sexton, D. J., and Morrison, J. J. (2005). Specific PGF(2alpha) receptor (FP) antagonism and human uterine contractility in vitro. BJOG 112, 1034–1042.
Fujino, T., Nakagawa, N., Yuhki, K., Hara, A., Yamada, T., Takayama, K., Kuriyama, S., Hosoki, Y., Takahata, O., Taniguchi, T., Fukuzawa, J., Hasebe, N., Kikuchi, K., Narumiya, S., and Ushikubi, F. (2004). Decreased susceptibility to renovascular hypertension in mice lacking the prostaglandin I2 receptor IP. J. Clin. Invest. 114, 805–812.
Funk, C. D. (2001). Prostaglandins and leukotrienes: advances in eicosanoid biology. Science 294, 1871–1875.
Funk, C. D., and FitzGerald, G. A. (2007). COX-2 inhibitors and cardiovascular risk. J. Cardiovasc. Pharmacol. 50, 470–479.
Griffin, B. W., Klimko, P., Crider, J. Y., and Sharif, N. A. (1999). AL-8810: a novel prostaglandin F2 alpha analog with selective antagonist effects at the prostaglandin F2 alpha (FP) receptor. J. Pharmacol. Exp. Ther. 290, 1278–1284.
Gross, G. A., Imamura, T., Luedke, C., Vogt, S. K., Olson, L. M., Nelson, D. M., Sadovsky, Y., and Muglia, L. J. (1998). Opposing actions of prostaglandins and oxytocin determine the onset of murine labor. Proc. Natl. Acad. Sci. U.S.A. 95, 11875–11879.
Hao, C. M., and Breyer, M. D. (2008). Physiological regulation of prostaglandins in the kidney. Annu. Rev. Physiol. 70, 357–377.
Hara, S., Arai, M., Tomaru, K., Doi, H., Koitabashi, N., Iso, T., Watanabe, A., Tanaka, T., Maeno, T., Suga, T., Yokoyama, T., and Kurabayashi, M. (2008). Prostaglandin F2alpha inhibits SERCA2 gene transcription through an induction of Egr-1 in cultured neonatal rat cardiac myocytes. Int. Heart J. 49, 329–342.
Hebert, R. L., Carmosino, M., Saito, O., Yang, G., Jackson, C. A., Qi, Z., Breyer, R. M., Natarajan, C., Hata, A. N., Zhang, Y., Guan, Y., and Breyer, M. D. (2005). Characterization of a rabbit kidney prostaglandin F(2{alpha}) receptor exhibiting G(i)-restricted signaling that inhibits water absorption in the collecting duct. J. Biol. Chem. 280, 35028–35037.
Helmersson, J., Arnlov, J., Axelsson, T., and Basu, S. (2009). A polymorphism in the cyclooxygenase 1 gene is associated with decreased inflammatory prostaglandin F2alpha formation and lower risk of cardiovascular disease. Prostaglandins Leukot. Essent. Fatty Acids 80, 51–56.
Helmersson, J., Larsson, A., Vessby, B., and Basu, S. (2005a). Active smoking and a history of smoking are associated with enhanced prostaglandin F(2alpha), interleukin-6 and F2-isoprostane formation in elderly men. Atherosclerosis 181, 201–207.
Helmersson, J., Vessby, B., Larsson, A., and Basu, S. (2005b). Cyclooxygenase-mediated prostaglandin F2alpha is decreased in an elderly population treated with low-dose aspirin. Prostaglandins Leukot. Essent. Fatty Acids 72, 227–233.
Helmersson, J., Vessby, B., Larsson, A., and Basu, S. (2004). Association of type 2 diabetes with cyclooxygenase-mediated inflammation and oxidative stress in an elderly population. Circulation 109, 1729–1734.
Hirst, J. J., Parkington, H. C., Young, I. R., Palliser, H. K., Peri, K. G., and Olson, D. M. (2005). Delay of preterm birth in sheep by THG113.31, a prostaglandin F2alpha receptor antagonist. Am. J. Obstet. Gynecol. 193, 256–266.
Ishida, N., Odani-Kawabata, N., Shimazaki, A., and Hara, H. (2006). Prostanoids in the therapy of glaucoma. Cardiovasc. Drug Rev. 24, 1–10.
Karmazyn, M., Tani, M., and Neely, J. R. (1993). Effect of prostaglandins I2 (prostacyclin) and F2 alpha on function, energy metabolism, and calcium uptake in ischaemic/reperfused hearts. Cardiovasc. Res. 27, 396–402.
Kobayashi, T., Tahara, Y., Matsumoto, M., Iguchi, M., Sano, H., Murayama, T., Arai, H., Oida, H., Yurugi-Kobayashi, T., Yamashita, J. K., Katagiri, H., Majima, M., Yokode, M., Kita, T., and Narumiya, S. (2004). Roles of thromboxane A(2) and prostacyclin in the development of atherosclerosis in apoE-deficient mice. J. Clin. Invest. 114, 784–794.
Kunapuli, P., Lawson, J. A., Rokach, J., and FitzGerald, G. A. (1997). Functional characterization of the ocular prostaglandin f2alpha (PGF2alpha) receptor. Activation by the isoprostane, 12-iso-PGF2alpha. J. Biol. Chem. 272, 27147–27154.
Lai, J., Jin, H., Yang, R., Winer, J., Li, W., Yen, R., King, K. L., Zeigler, F., Ko, A., Cheng, J., Bunting, S., and Paoni, N. F. (1996). Prostaglandin F2 alpha induces cardiac myocyte hypertrophy in vitro and cardiac growth in vivo. Am. J. Physiol. 271(Pt 2), H2197–H2208.
Langenbach, R., Morham, S. G., Tiano, H. F., Loftin, C. D., Ghanayem, B. I., Chulada, P. C., Mahler, J. F., Lee, C. A., Goulding, E. H., Kluckman, K. D., Kim, H. S., and Smithies, O. (1995). Prostaglandin synthase 1 gene disruption in mice reduces arachidonic acid-induced inflammation and indomethacin-induced gastric ulceration. Cell 83, 483–492.
Li, Y., Kang, J. X., and Leaf, A. (1997). Differential effects of various eicosanoids on the production or prevention of arrhythmias in cultured neonatal rat cardiac myocytes. Prostaglandins 54, 511–530.
Morham, S. G., Langenbach, R., Loftin, C. D., Tiano, H. F., Vouloumanos, N., Jennette, J. C., Mahler, J. F., Kluckman, K. D., Ledford, A., Lee, C. A., and Smithies, O. (1995). Prostaglandin synthase 2 gene disruption causes severe renal pathology in the mouse. Cell 83, 473–482.
Mukhopadhyay, P., Bian, L., Yin, H., Bhattacherjee, P., and Paterson, C. (2001). Localization of EP(1) and FP receptors in human ocular tissues by in situ hybridization. Invest. Ophthalmol. Vis. Sci. 42, 424–428.
Narumiya, S., and FitzGerald, G. A. (2001). Genetic and pharmacological analysis of prostanoid receptor function. J. Clin. Invest. 108, 25–30.
Narumiya, S., Sugimoto, Y., and Ushikubi, F. (1999). Prostanoid receptors: structures, properties, and functions. Physiol. Rev. 79, 1193–1226.
Oga, T., Matsuoka, T., Yao, C., Nonomura, K., Kitaoka, S., Sakata, D., Kita, Y., Tanizawa, K., Taguchi, Y., Chin, K., Mishima, M., Shimizu, T., and Narumiya, S. (2009). Prostaglandin F(2alpha) receptor signaling facilitates bleomycin-induced pulmonary fibrosis independently of transforming growth factor-beta. Nat. Med. 15, 1426–1430.
Olson, D. M. (2005). The promise of prostaglandins: have they fulfilled their potential as therapeutic targets for the delay of preterm birth? J. Soc. Gynecol. Investig. 12, 466–478.
Peri, K. G., Quiniou, C., Hou, X., Abran, D., Varma, D. R., Lubell, W. D., and Chemtob, S. (2002). THG113: a novel selective FP antagonist that delays preterm labor. Semin. Perinatol. 26, 389–397.
Pierce, K. L., Fujino, H., Srinivasan, D., and Regan, J. W. (1999). Activation of FP prostanoid receptor isoforms leads to Rho-mediated changes in cell morphology and in the cell cytoskeleton. J. Biol. Chem. 274, 35944–35949.
Rabinowitz, B., Arad, M., Elazar, E., Klein, R., and Har Zahav, Y. (1992). Epicardial versus endocardial “in mirror” changes in prostaglandin synthesis after short periods of ischemia and reperfusion. Eicosanoids 5, 163–167.
Rader, D. J., and Daugherty, A. (2008). Translating molecular discoveries into new therapies for atherosclerosis. Nature 451, 904–913.
Rice, K. M., Uddemarri, S., Desai, D. H., Morrison, R. G., Harris, R., Wright, G. L., and Blough, E. R. (2008). PGF2alpha-associated vascular smooth muscle hypertrophy is ROS dependent and involves the activation of mTOR, p70S6k, and PTEN. Prostaglandins Other Lipid Mediat. 85, 49–57.
Saito, O., Guan, Y., Qi, Z., Davis, L. S., Komhoff, M., Sugimoto, Y., Narumiya, S., Breyer, R. M., and Breyer, M. D. (2003). Expression of the prostaglandin F receptor (FP) gene along the mouse genitourinary tract. Am. J. Physiol. Renal Physiol. 284, F1164–F1170.
Sales, K. J., Milne, S. A., Williams, A. R., Anderson, R. A., and Jabbour, H. N. (2004). Expression, localization, and signaling of prostaglandin F2 alpha receptor in human endometrial adenocarcinoma: regulation of proliferation by activation of the epidermal growth factor receptor and mitogen-activated protein kinase signaling pathways. J. Clin. Endocrinol. Metab. 89, 986–993.
Scher, J. U., and Pillinger, M. H. (2009). The anti-inflammatory effects of prostaglandins. J. Investig. Med. 57, 703–708.
Seppala, E. (1987). Production of prostanoids by rheumatic synovial cells in vitro: effects of anti-inflammatory drugs on arachidonic acid metabolism. Clin. Rheumatol. 6, 170–176.
Sharif, N. A., Crider, J. Y., and Davis, T. L. (2000). AL-3138 antagonizes FP prostanoid receptor-mediated inositol phosphates generation: comparison with some purported FP antagonists. J. Pharm. Pharmacol. 52, 1529–1539.
Sinaiko, A. R., Steinberger, J., Moran, A., Prineas, R. J., Vessby, B., Basu, S., Tracy, R., and Jacobs, D. R. Jr. (2005). Relation of body mass index and insulin resistance to cardiovascular risk factors, inflammatory factors, and oxidative stress during adolescence. Circulation 111, 1985–1991.
Smith, W. L., DeWitt, D. L., and Garavito, R. M. (2000). Cyclooxygenases: structural, cellular, and molecular biology. Annu. Rev. Biochem. 69, 145–182.
Smyth, E. M., Grosser, T., Wang, M., Yu, Y., and FitzGerald, G. A. (2009). Prostanoids in health and disease. J. Lipid Res. 50(Suppl.), S423–S428.
Sugimoto, Y., Hasumoto, K., Namba, T., Irie, A., Katsuyama, M., Negishi, M., Kakizuka, A., Narumiya, S., and Ichikawa, A. (1994). Cloning and expression of a cDNA for mouse prostaglandin F receptor. J. Biol. Chem. 269, 1356–1360.
Sugimoto, Y., Yamasaki, A., Segi, E., Tsuboi, K., Aze, Y., Nishimura, T., Oida, H., Yoshida, N., Tanaka, T., Katsuyama, M., Hasumoto, K., Murata, T., Hirata, M., Ushikubi, F., Negishi, M., Ichikawa, A., and Narumiya, S. (1997). Failure of parturition in mice lacking the prostaglandin F receptor. Science 277, 681–683.
Takayama, K., Yuhki, K., Ono, K., Fujino, T., Hara, A., Yamada, T., Kuriyama, S., Karibe, H., Okada, Y., Takahata, O., Taniguchi, T., Iijima, T., Iwasaki, H., Narumiya, S., and Ushikubi, F. (2005). Thromboxane A2 and prostaglandin F2alpha mediate inflammatory tachycardia. Nat. Med. 11, 562–566.
Tilley, S. L., Coffman, T. M., and Koller, B. H. (2001). Mixed messages: modulation of inflammation and immune responses by prostaglandins and thromboxanes. J. Clin. Invest. 108, 15–23.
Trang, L. E., Granstrom, E., and Lovgren, O. (1977). Levels of prostaglandins F2 alpha and E2 and thromboxane B2 in joint fluid in rheumatoid arthritis. Scand. J. Rheumatol. 6, 151–154.
Wang, D., Patel, V. V., Ricciotti, E., Zhou, R., Levin, M. D., Gao, E., Yu, Z., Ferrari, V. A., Lu, M. M., Xu, J., Zhang, H., Hui, Y., Cheng, Y., Petrenko, N., Yu, Y., and FitzGerald, G. A. (2009). Cardiomyocyte cyclooxygenase-2 influences cardiac rhythm and function. Proc. Natl. Acad. Sci. U.S.A. 106, 7548–7552.
Watanabe, K., Yoshida, R., Shimizu, T., and Hayaishi, O. (1985). Enzymatic formation of prostaglandin F2 alpha from prostaglandin H2 and D2. Purification and properties of prostaglandin F synthetase from bovine lung. J. Biol. Chem. 260, 7035–7041.
Watanabe, T., Nakao, A., Emerling, D., Hashimoto, Y., Tsukamoto, K., Horie, Y., Kinoshita, M., and Kurokawa, K. (1994). Prostaglandin F2 alpha enhances tyrosine phosphorylation and DNA synthesis through phospholipase C-coupled receptor via Ca(2+)-dependent intracellular pathway in NIH-3T3 cells. J. Biol. Chem. 269, 17619–17625.
Weinreb, R. N., Toris, C. B., Gabelt, B. T., Lindsey, J. D., and Kaufman, P. L. (2002). Effects of prostaglandins on the aqueous humor outflow pathways. Surv. Ophthalmol. 47(Suppl. 1), S53–S64.
Whittle, B. J., Hansen, D., and Salmon, J. A. (1985a). Gastric ulcer formation and cyclo-oxygenase inhibition in cat antrum follows parenteral administration of aspirin but not salicylate. Eur. J. Pharmacol. 116, 153–157.
Whittle, B. J., Oren-Wolman, N., and Guth, P. H. (1985b). Gastric vasoconstrictor actions of leukotriene C4, PGF2 alpha, and thromboxane mimetic U-46619 on rat submucosal microcirculation in vivo. Am. J. Physiol. 248(Pt 1), G580–G586.
Wohlin, M., Helmersson, J., Sundstrom, J., Arnlov, J., Vessby, B., Larsson, A., Andren, B., Lind, L., and Basu, S. (2007). Both cyclooxygenase- and cytokine-mediated inflammation are associated with carotid intima-media thickness. Cytokine 38, 130–136.
Wong, S. L., Leung, F. P., Lau, C. W., Au, C. L., Yung, L. M., Yao, X., Chen, Z. Y., Vanhoutte, P. M., Gollasch, M., and Huang, Y. (2009). Cyclooxygenase-2-derived prostaglandin F2alpha mediates endothelium-dependent contractions in the aortae of hamsters with increased impact during aging. Circ. Res. 104, 228–235.
Yin, H., Gao, L., Tai, H. H., Murphey, L. J., Porter, N. A., and Morrow, J. D. (2007). Urinary prostaglandin F2alpha is generated from the isoprostane pathway and not the cyclooxygenase in humans. J. Biol. Chem. 282, 329–336.
Yoshida, M., Sagawa, N., Itoh, H., Yura, S., Takemura, M., Wada, Y., Sato, T., Ito, A., and Fujii, S. (2002). Prostaglandin F(2alpha), cytokines and cyclic mechanical stretch augment matrix metalloproteinase-1 secretion from cultured human uterine cervical fibroblast cells. Mol. Hum. Reprod. 8, 681–687.
Yu, Y., Cheng, Y., Fan, J., Chen, X. S., Klein-Szanto, A., Fitzgerald, G. A., and Funk, C. D. (2005). Differential impact of prostaglandin H synthase 1 knockdown on platelets and parturition. J. Clin. Invest. 115, 986–995.
Yu, Y., Fan, J., Chen, X. S., Wang, D., Klein-Szanto, A. J., Campbell, R. L., FitzGerald, G. A., and Funk, C. D. (2006). Genetic model of selective COX2 inhibition reveals novel heterodimer signaling. Nat. Med. 12, 699–704.
Yu, Y., and Funk, C. D. (2007). A novel genetic model of selective COX-2 inhibition: comparison with COX-2 null mice. Prostaglandins Other Lipid Mediat. 82, 77–84.
Keywords: prostaglandin F2alpha, hypertension, atherosclerosis, FP receptor
Citation: Zhang J, Gong Y and Yu Y (2010) Prostaglandin F2α receptor: a promising therapeutic target for cardiovascular disease. Front. Pharmacol. 1:116. doi: 10.3389/fphar.2010.00116
Received: 01 May 2010;
Paper pending published: 19 May 2010;
Accepted: 17 August 2010;
Published online: 14 October 2010.
Edited by:
Colin D. Funk, Queen’s University, CanadaReviewed by:
Aida Habib, American University of Beirut, LebanonDuxin Sun, University of Michigan, USA
Copyright: © 2010 Zhang, Gong and Yu. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Ying Yu, Key Laboratory of Nutrition and Metabolism, Institute for Nutritional Sciences, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, Graduate School of the Chinese Academy of Sciences, 294 Tai Yuan Road, Shanghai 200031, China. email:eXV5aW5nQHNpYnMuYWMuY24=