 Nagham Shehade-Awwad
Nagham Shehade-Awwad
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pediatr. , 08 September 2022
Sec. Genetics of Common and Rare Diseases
Volume 10 - 2022 | https://doi.org/10.3389/fped.2022.946071
This article is part of the Research Topic Next-Generation Sequencing: An Ongoing Revolution in Pediatrics View all 13 articles
Background: Noonan syndrome (NS) is a genetic syndrome, characterized by various dysmorphic features, cardiac anomalies, short stature, and developmental delay. NS is a leading cause of cardiovascular anomalies. The syndrome results from dysregulation in the RAS-MAPK pathway and is related to the RASopathy family syndromes. Pathogenic variants in more than 20 related genes have been identified in association with NS, and several genotype-phenotype correlations were suggested. The specific severity of the same cardiovascular anomalies has not been described as linked to a specific causative gene.
Methods: For this retrospective, single-center study, data retrieved from medical charts of a multidisciplinary NS clinic included genetic diagnosis, cardiac malformations, the need for intervention, demographics, and prenatal diagnosis. We analyzed molecular genetics and the severity of cardiac malformations.
Results: The cohort comprised 74 children with NS. Consistent with previous studies, pathogenic variants in PTPN11 were the most common (62%). Cardiovascular anomalies presented in 57%; pulmonary stenosis (PS) was the most common (about 79% of anomalies). In children with pathogenic variants in PTPN11, PS tended to be more severe and required intervention in 53%, compared to 25% of children with PS and a variant in other genes.
Conclusion: This first Israeli cohort of NS showed similar rates of cardiac malformations and genetic breakdown as previously published. Variants in PTPN11 were prone to a higher risk for severe PS that requires intervention. This finding may assist in genetic counseling and cardiac treatment decisions, and stresses the importance of genetic in addition to clinical diagnosis of NS.
Noonan syndrome (NS) is a genetic disease characterized by distinctive craniofacial dysmorphic features, developmental delay, learning difficulties, short stature, and congenital cardiac anomalies (1, 2). Its prevalence is estimated as 1 in 1000 to 2500 live births (3, 4). NS is a heterogeneous condition, with variable phenotypic expression and severity (5). Clinical diagnosis is based on clinical criteria and a scoring system that includes the typical facial characteristics, typical heart defect, and short stature (6, 7). The syndrome is associated with pathogenic variants in multiple genes in the RAS-MAP kinases pathway, leading to dysregulation (8). NS shares a similar mechanism and overlapping clinical features with other syndromes such as: Noonan syndrome with multiple lentigines (previously called LEOPARD syndrome), cardiofaciocutaneous syndrome, and Costello syndrome. Collectively, these syndromes are referred to as RASopathies (9, 10). PTPN11 is the most frequent causative gene in NS, which accounts for more than 50% of the patients (11). Various pathogenic variants in PTPN11 have been identified, and attempts have been made to correlate them to syndrome severity (11, 12). More than 20 other genes related to NS have been discovered (13). Despite advances in genetic testing, many patients are diagnosed clinically without molecular testing to identify the causative variant (5).
NS is known as a leading cause of congenital heart disease (14). Pulmonary stenosis (PS) is the most common cardiac anomaly (50–60%), followed by hypertrophic cardiomyopathy (HCM) (20%) and secundum atrial septal defect (ASD) (6–32%) (15). Additional cardiac malformations, such as ventricular septal defect, peripheral PS, atrioventricular canal, aortic stenosis, mitral valve abnormalities, aortic coarctation, and coronary artery anomalies, are less common but have also been encountered (1, 16). Several studies that described phenotype-genotype correlations in NS focused on facial features relating to various genes or cardiovascular findings only within PTPN11 variants (17–20).
To date, few studies have analyzed the correlation between cardiovascular severity and the indicated genes, or compared genotype with cardiovascular phenotype. The aim of our study was to characterize the cardiovascular features in a number of causative genes within a large cohort of patients with NS, and to describe the type and severity of the cardiovascular anomalies, as well as the need for intervention by catheter or surgery during infancy and childhood.
The study was carried out at the multidisciplinary NS clinic, at Safra Children's Hospital, Israel, and was approved by the institutional Helsinki ethics committee.
A retrospective chart analysis was performed of children with NS who were followed in the clinic. Visits to specialty clinics were screened, including the cardiology and endocrine clinics; genetic test results and echocardiogram results were obtained. Inclusion criteria were children with a diagnosis of NS, either clinically or genetically. In the investigation of genotype-phenotype correlations, only those with a genetic diagnosis were included. The genetic diagnosis was based on ACMG variant classification guidelines (21).
Demographic data were described as means and standard deviations. Rates of cardiovascular malformations between genotypes were compared, and the need for intervention according to the causative genes was calculated as odds ratios and relative risks.
The diagnosis of HCM is established based on echocardiography. The hallmark finding is increased left ventricular (LV) wall thickening in the absence of a hemodynamic cause. In pediatric patients, measurements of LV wall thickness are normalized for age and body surface area using Z-score, which may be calculated by several methods (22–24). Herein, to compute the Z score for our patients with HCM, we used The Boston Hospital's Z score.
The study included 74 patients with NS; the mean age was 9.4 ± 6.03 years. In total of 40% were males; 15% had a parent who was diagnosed with NS. Fifty two patients had a molecular diagnosis and 22 had a clinical diagnosis only.
For 18 of the 52 patients with a molecular diagnosis, this was done by using various commercial RASopathy panels. 14 by exome sequencing (ES) and 4 by single-gene Sanger sequencing of PTPN11 or SOS1. The mode of genetic testing was unavailable for the remaining 16 patients.
Of the 40 patients who had undergone prenatal screening tests, 85% (n = 34) had abnormal findings. Twenty patients showed increased nuchal translucency or a cervical cyst and 6 had polyhydramnios. Eleven patients had undergone fetal echocardiography, 7 of whom had abnormal findings.
Ten causative genes were found in our cohort; their prevalence are described in Table 1. Thirty two patients (62% of those with a genetic diagnosis) had a pathogenic variants in the PTPN11 gene.

Table 1. The prevalence of gene pathogenic variants: (In total, 52 patients underwent genetic diagnosis).
In total 57% (n = 42) of all the patients had a cardiac anomaly; of them, 71% (n = 30) had a single malformation and 29% (n = 12) had multiple cardiac malformations. The most common cardiac anomaly was PS, which accounted for 79% (n = 33) of the cardiac anomalies. Cardiovascular anomalies are summarized in Table 2. The patients with PS were classified according to its severity, as defined by the peak pulmonary valve gradient (Table 3). In total 52% (n = 17) of the patients with PS required intervention, either surgical (18%, n = 3) or catheterization (82%, n = 14). In total 35% (n = 6) of these patients required an additional intervention (Figure 1). In total 21% (n = 7) of patients with PS had a dysplastic valve, 5 of them required intervention.
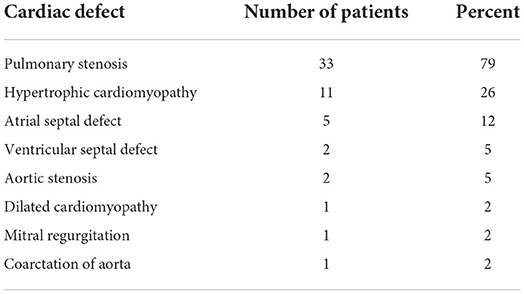
Table 2. Cardiac anomalies in 42 patients with Noonan syndrome and with cardiac involvement.
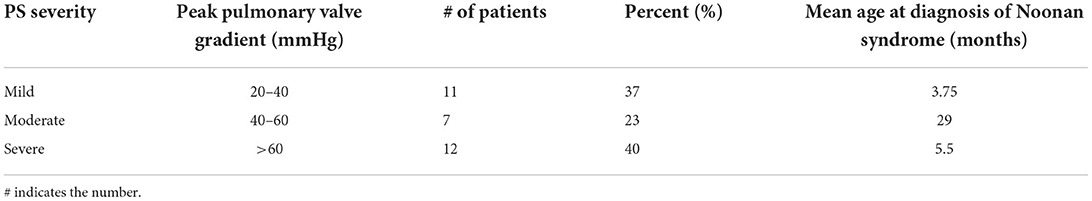
Table 3. Pulmonary stenosis severity.
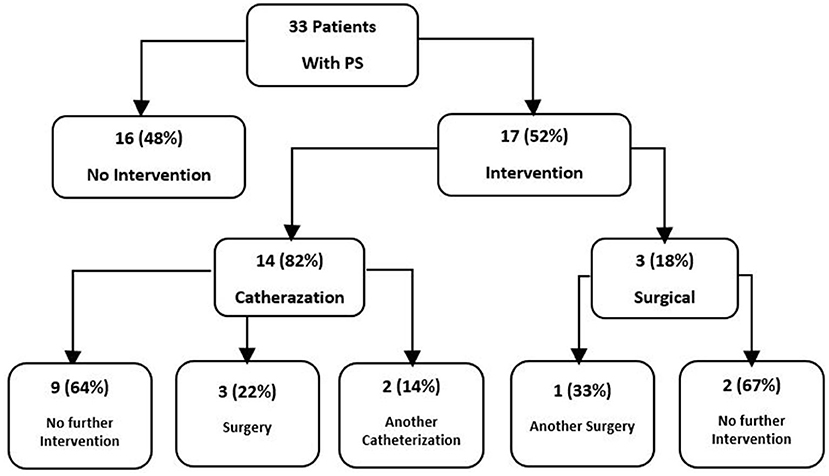
Figure 1. Intervention in 33 Noonan patients who has PS.
The second most common cardiac anomaly reported in our cohort was HCM, for which the prevalence was 26% (n = 11). In total of 64% (n = 7) were diagnosed during pregnancy or a short time after birth. The mean LV wall thickness Z score for our patients with HCM was 3.91. We calculated the mean LV mass index as 109.5 gr/m2 in males (n = 4); one of the females with a HCM diagnosis had available data for LV mass index, 69 gr/m2. These two values are considered above the normal range (>95 th percentile) (25, 26). Only 1 patient required septal myectomy. The third common cardiac anomaly in our cohort is ASD, counts for 12% (n = 5). Two of them required intervention.
The majority of patients (62%, n = 32) had pathogenic variants in the PTPN11 gene; of them, 66% (n = 21) had cardiac anomalies. In total 81% of the latter (n = 17) were diagnosed with PS. In total 53% (n = 9) of patients with PS required intervention of either catheterization (n = 8) or surgery (n = 1). Mean age of intervention was 13 months. Three patients needed additional intervention; 2 required surgical and 1 required catheterization re-intervention. One patient required a third intervention.
In addition to PS, other less frequent cardiac anomalies were seen in the PTPN11 group, as presented in Table 4. Three patients were diagnosed with both PS and HCM. This combination did not result in increased clinical severity and none of them required any intervention.
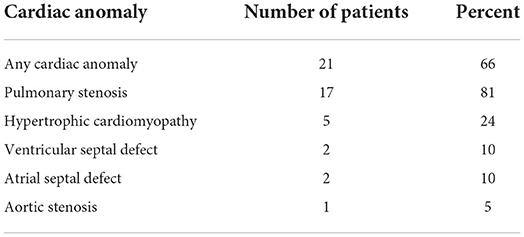
Table 4. Cardiac anomalies in 32 patients with Noonan syndrome, diagnosed with PTPN11 variants.
As most patients (62%) had a pathogenic variant in the PTPN11 gene, and the remainder were in small numbers (38%, n = 20), it was not possible to perform a subgroup analysis of every gene. Therefore, all those with pathogenic variants in genes other than PTPN11 were clustered, and this non-PTPN11 group was compared to the PTPN11 group. In total 55% (n = 11) of the non-PTPN11 group had a cardiac anomaly; of them, 73% (n = 8) were diagnosed with PS, and 25% (n = 2) required intervention of either catheterization (n = 1) or surgery (n = 1). Limited data were available regarding the age of intervention at this group with mean age of 6 months (n = 3). A subgroup analysis of the general prevalence of cardiac malformations and PS prognosis is shown in Table 5.
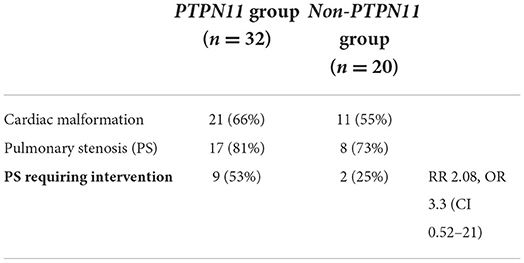
Table 5. Subgroup analysis of the general prevalence of cardiac malformations and the prognosis of pulmonary stenosis.
We compared the LV wall thickness Z-score for the PTPN11 and non-PTPN11 groups. According to The Boston Z-score system, the mean Z-score for PTPN11 patients was 0.07, and for the non-PTPN11 group, 1.14.
We examined various variants in the PTPN11 gene, in search of a correlation between the variant and the severity of cardiac anomalies in NS. Our analysis showed 7 patients with N308D pathogenic variant. In total 57% of them (n = 4) were diagnosed with cardiac defects; all had PS; 75% (n = 3) of the latter required intervention. Three patients had the M504V pathogenic variant in PTPN11. All of them were diagnosed with cardiac defects: 1 patient had PS, 1 had PS and HCM, and 1 had HCM and a ventricular septal defect.
This is the first description of an Israeli cohort of children with NS. The breakdown of causal genes was found to be similar to that previously reported. PTPN11 pathogenic variants were the leading etiology, accounting for 62% of those with a genetic diagnosis, slightly higher than previously reported (50%) (1). SOS1 variants were the second most frequent etiology, with a 12% prevalence. The rate of cardiac malformations was similar to that previously reported, including the rates of PS among those with cardiac anomalies. PS in patients with PTPN11 variants tended to be more severe and required intervention in 53% of the patients, in contrast to only 25% of those with PS and non-PTPN11 variants. This finding is different than previous descriptions of a 30% need for intervention in PS of NS, without describing the breakdown according to specific genes (1). Notably, the mean LV wall thickness was lower for patients with PTPN11 variants than for those with non-PTPN11 variants. These results are consistent with studies that showed a lower incidence of HCM with NS diagnosis and PTPN11 variants (9). The findings highlight the importance of genetic diagnosis, to enable genetic counseling in early stages, and decision making by cardiologists. Given the large spectrum of clinical features within our cohort and in NS in general, any information that may be offered to patients and their families during early genetic diagnosis may help with decision making, and appropriate clinical follow up after delivery. Parents of 11 (15%) patients in our cohort were diagnosed with NS following their children's diagnosis.
Our cohort included children who were diagnosed over several years, during which the availability and funding of genetic testing changed significantly; this had a pronounced influence on the types of genetic testing performed. When funding and financial ability of parents were lacking, and the clinical picture was very clear, several parents decided to carry out a Sanger sequencing of PTPN11 in early years. Later, when Noonan panels became more available and inexpensive, patients were recommended to undergo testing when a RASopathy was suspected. Finally, when the clinical picture was less clear, but some form of genetic syndrome was suspected, the most expensive test, namely ES, was ordered, and still funded by patients. This may explain the different ages at diagnosis observed between the three groups of children who underwent these three tests. Accordingly, the patients with the most clinically obvious signs were diagnosed earlier using Sanger sequencing, and the least obvious diagnosed later using ES. We expect that increasing availability and funding of ES will lead to earlier diagnosis of more subtle phenotypes of NS, including during prenatal screening.
Our study has several limitations. Firstly, due to the retrospective nature, the diagnosis algorithm varied, as it was carried out in a number of hospitals, and because many of our patients were referred to our specialized center following diagnosis. Therefore, some of the data were unavailable, and genetic testing differed between centers and time periods. Another limitation is that despite our relatively large sample, for a specific syndrome, especially of a small country, it was not large enough to enable statistical analysis of the clear trends observed. A multi-center or multinational study, combining multiple NS cohorts could enable subgroup analysis of the various genes, and of the various pathogenic variants within the same gene.
In summary, this is a first Israeli cohort of NS, reporting similar genetic breakdown and prevalence of cardiac anomalies as previously reported, but showing differences in severity according to gene variants. PTPN11-associated PS conferred a 2.1 relative risk for requiring intervention by surgery or catheterization, compared to PS in other gene variants. This stresses the importance of genetic testing, which enables more accurate prenatal counseling and medical decision making.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The study was carried out at the multidisciplinary NS-A clinic, at Safra Children's Hospital, Israel, and was approved by the institutional Helsinki Ethics Committee. Written informed consent was not required as per local legislation and institutional requirements.
NS-A and YY contributed equally to all stages of this manuscript, including the study conception, Helsinki approval, data collection, and the writing of the manuscript. UK contributed to cardiac data interpretation. OP-H and UK contributed to manuscript revision, reading, and approving the submitted version. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be considered as potential conflicts of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Roberts AE, Allanson JE, Tartaglia M, Gelb BD. Noonan syndrome. Lancet. (2013) 381:9863. doi: 10.1016/S0140-6736(12)61023-X
2. Sun R, Liu M, Lu L, Zheng Y, Zhang P. Congenital heart disease: causes, diagnosis, symptoms, and treatments. Cell Biochem Biophys. (2015) 72:857–60. doi: 10.1007/s12013-015-0551-6
5. Carcavilla A, Suárez-Ortega L, Rodríguez Sánchez A, Gonzalez-Casado I, Ramón-Krauel M, Labarta JI, et al. Síndrome de Noonan: actualización genética, clínica y de opciones terapéuticas. An Pediatría. (2020) 93:61.e1–61.e14. doi: 10.1016/j.anpedi.2020.04.008
6. Van Der Burgt I, Berends E, Lommen E, Van Beersum S, Hamel B, Mariman E. Clinical and molecular studies in a large Dutch family with Noonan syndrome. Am J Med Genet. (1994) 53:187–91. doi: 10.1002/ajmg.1320530213
8. Tidyman WE, Rauen KA. Noonan, Costello and cardio–facio–cutaneous syndromes: dysregulation of the Ras–MAPK pathway. Expert Rev Mol Med. (2008) 10:e37. doi: 10.1017/S1462399408000902
9. Linglart L, Gelb BD. Congenital heart defects in Noonan syndrome: Diagnosis, management, and treatment. Am J Med Genet Part C Semin Med Genet. (2020) 184:73–80. doi: 10.1002/ajmg.c.31765
10. Aoki Y, Niihori T, Inoue S, Matsubara Y. Recent advances in RASopathies. J Hum Genet. (2016) 61:33–9. doi: 10.1038/jhg.2015.114
11. Athota JP, Bhat M, Nampoothiri S, Gowrishankar K, Narayanachar SG, Puttamallesh V, et al. Molecular and clinical studies in 107 Noonan syndrome affected individuals with PTPN11 mutations. BMC Med Genet. (2020) 21:50. doi: 10.1186/s12881-020-0986-5
12. Tartaglia M, Kalidas K, Shaw A, Song X, Musat DL, van der Burgt I, et al. PTPN11 mutations in noonan syndrome: molecular spectrum, genotype-phenotype correlation, and phenotypic heterogeneity. Am J Hum Genet. (2002) 70:1555–63. doi: 10.1086/340847
13. El Bouchikhi I, Belhassan K, Moufid FZ, Iraqui Houssaini M, Bouguenouch L, Samri I, et al. Noonan syndrome-causing genes: Molecular update and an assessment of the mutation rate. Int J Pediatr Adolesc Med. (2016) 3:133–42. doi: 10.1016/j.ijpam.2016.06.003
14. Marino B, Digilio MC, Toscano A, Giannotti A, Dallapiccola B. Congenital heart diseases in children with Noonan syndrome: An expanded cardiac spectrum with high prevalence of atrioventricular canal. J Pediatr. (1999) 135:703–6. doi: 10.1016/S0022-3476(99)70088-0
15. Prendiville TW, Gauvreau K, Tworog-Dube E, Patkin L, Kucherlapati RS, Roberts AE, et al. Cardiovascular disease in Noonan syndrome. Arch Dis Child. (2014) 99:629–34. doi: 10.1136/archdischild-2013-305047
16. Pierpont ME, Digilio MC. Cardiovascular disease in Noonan syndrome. Curr Opin Pediatr. (2018) 30:601–8. doi: 10.1097/MOP.0000000000000669
17. Zenker M, Buheitel G, Rauch R, Koenig R, Bosse K, Kress W, et al. Genotype-phenotype correlations in Noonan syndrome. J Pediatr. (2004) 144:368–74. doi: 10.1016/j.jpeds.2003.11.032
18. Allanson JE, Bohring A, Dörr H-G, Dufke A, Gillessen-Kaesbach G, Horn D, et al. The face of Noonan syndrome: Does phenotype predict genotype. Am J Med Genet Part A. (2010) 152A:1960–6. doi: 10.1002/ajmg.a.33518
19. Maheshwari M, Belmont J, Fernbach S, Ho T, Molinari L, Yakub I, et al. PTPN11 Mutations in Noonan syndrome type I: detection of recurrent mutations in exons 3 and 13. Hum Mutat. (2002) 20:298–304. doi: 10.1002/humu.10129
20. Tartaglia M, Mehler EL, Goldberg R, Zampino G, Brunner HG, Kremer H, et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat Genet. (2001) 29:465–8. doi: 10.1038/ng772
21. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
22. Pettersen MD, Du W, Skeens ME, Humes RA. Regression equations for calculation of z scores of cardiac structures in a large cohort of healthy infants, children, and adolescents: an echocardiographic study. J Am Soc Echocardiogr. (2008) 21:922–34. doi: 10.1016/j.echo.2008.02.006
23. de Simone G, Daniels SR, Devereux RB, Meyer RA, Roman MJ, de Divitiis O, et al. Left ventricular mass and body size in normotensive children and adults: Assessment of allometric relations and impact of overweight. J Am Coll Cardiol. (1992) 20:1251–60. doi: 10.1016/0735-1097(92)90385-Z
24. Mawad W, Drolet C, Dahdah N, Dallaire F. A review and critique of the statistical methods used to generate reference values in pediatric echocardiography. J Am Soc Echocardiogr. (2013) 26:29–37. doi: 10.1016/j.echo.2012.09.021
25. Khoury PR, Mitsnefes M, Daniels SR, Kimball TR. Age-specific reference intervals for indexed left ventricular mass in children. J Am Soc Echocardiogr. (2009) 22:709–14. doi: 10.1016/j.echo.2009.03.003
Keywords: genetics, Noonan syndrome, cardiovascular, genotype, phenotype, PTPN11, pulmonary stenosis
Citation: Shehade-Awwad N, Yeshayahu Y, Pinhas-Hamiel O and Katz U (2022) Differences in severity of cardiovascular anomalies in children with Noonan syndrome based on the causative gene. Front. Pediatr. 10:946071. doi: 10.3389/fped.2022.946071
Received: 17 May 2022; Accepted: 18 August 2022;
Published: 08 September 2022.
Edited by:
Kathryn Weaver, Cincinnati Children's Hospital Medical Center, United StatesReviewed by:
Omar R. J. Tamimi, King Fahd Medical City, Saudi ArabiaCopyright © 2022 Shehade-Awwad, Yeshayahu, Pinhas-Hamiel and Katz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yonatan Yeshayahu, yeshayahu@assuta.co.il
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.