 Jingwen Liang
Jingwen Liang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pediatr., 09 December 2022
Sec. Genetics of Common and Rare Diseases
Volume 10 - 2022 | https://doi.org/10.3389/fped.2022.1032659
This article is part of the Research TopicGenetic and Environmental Factors in the Occurrence of Paediatric Disorders – Volume IIView all 10 articles
Background: Deafness is the most common sensory defect in humans worldwide. Approximately 50% of cases are attributed to genetic factors, and about 70% are non-syndromic hearing loss (NSHL).
Objectives: To identify clinically relevant gene variants associated with NSHL in a Chinese family using trio-based whole-exome sequencing (WES).
Materials and methods: WES was performed on the 18-month-old female proband, and her parents. Gene variants specific to the family were identified by bioinformatics analysis and evaluated for their relevance to NSHL. We verified the novel variant in this family by the next-generation sequencing.In order to elucidate the frameshift mutation of TMPRSS3 in a Chinese family, we used the Mass spectrometry to detect the gene from 1,010 healthy subjects.
Results: We identified a novel homozygous deletion (c.51delA) in exon 2 of the type II transmembrane serine protease 3 gene TMPRSS3, which resulted in a frameshift mutation just before the protein transmembrane domain (p.Q17fs). The deletion was present in the proband and her father, but not in her mother and the healthy controls. We also found mutations with potential relevance to hearing loss in DCAF17, which encodes a protein of unknown function (c. T555A: p.H185Q), and ZNF276, which encodes zinc finger protein 276 (c.1350–2A > G).
Conclusions and significance: We shown a novel frameshift mutation in TMPRSS3 associated with autosomal recessive NSHL in a Han Chinese family.
Hearing loss is the most common sensory defect in humans worldwide. Genetic factors account for at least 50% of congenital hearing loss (1). About 80% of hereditary hearing impairment is due to autosomal recessive non-syndromic prelingual sensorineural hearing loss (2). Variants of the TMRPSS3 gene have been associated with both familial and sporadic cases of autosomal recessive non-syndromic hearing loss (NSHL; DFNB8/10) (3). TMPRSS3, which encodes a transmembrane serine protease, is composed of 13 exons, with exon 2 containing the start codon (4). Many mutations in TMPRSS3 have been reported to have potential roles in NSHL, including an 8-bp deletion, an insertion of multiple beta-satellite repeat units, and a frameshift mutation (4).
In our research, we studied the frameshift mutation of TMPRSS3 that has thrown new light on the cause of the autosomal recessive NSHL in a Chinese patient.
The proband was an 18-month-old girl with NSHL. Her gestation period and birth weight (3.15 kg) were both normal, and she did not experience either asphyxia or hypoxia during delivery. Her postnatal development was normal, with head-raising at 3 months, sitting at 6 months, and walking at 14 months of age, with the exception of a small head circumference (44.5 cm) recorded at 18 months of age. General physical check-ups performed at standard times were normal. However, she was unable to speak and a hearing impairment was detected at 18 months of age. The child was diagnosed with profound sensorineural hearing loss at a local hospital. The parents have normal hearing and claim no family history of deafness or consanguinity.
To identify gene mutations that are candidates for the proband's deafness, we obtained peripheral blood samples (2 ml) from the proband, both parents, and 1,010 healthy subjects for DNA analysis. Genomic DNA was extracted using a Protease K DNA extraction kit (D3392), and the DNA was purified (OD260/280 = 1.8–2.0) and stored at −20°C until analysis.
We performed whole-exome sequencing (WES) on the proband, her parents, and detected the gene locus in a large cohort of healthy controls by mass spectrometry.Variants identified in the proband were detected using trio-based bioinformatics analysis and evaluated for their potential association with NSHL by comparison with a PubMed database search. Candidate variants were then validated by sequencing of the proband and her family (Figure 1).
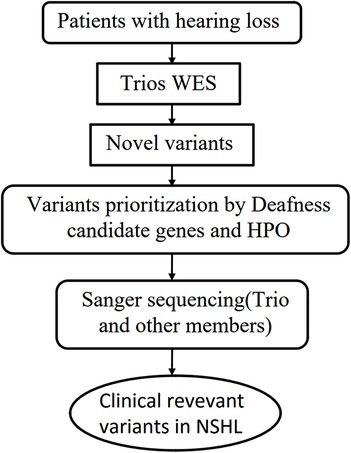
Figure 1. Flowchart to detect rare variants relevant to NSHL by WES. Abbrebiation: WES, Whole Exome Sequencing, HPO, Human Phenotype Ontology.
We conducted whole exome sequencing (WES) on the proband affected by NSHL and her corresponding parents.A novel candidate loci of TMPRSS3 was identified in this case. The variants of TMPRSS3 that were subsequently confirmed by next-generation sequencing on the Chinese Family. Family pedigree is shown in Figure 2. The proband (II-1) and her parents (I-1, I-2) carried DCAF17: c. T555 > A (p.H185Q). The proband (II-1) and her father, but not her mother and the healthy contrlos, carried TMPRSS3: c.51delA (p.Q17fs), showed profound sensorineural hearing loss. TMPRSS3: c.51delA (rs780609668) is a frameshift variant located in exon 2 and is known to be associated with autosomal recessive NSHL.
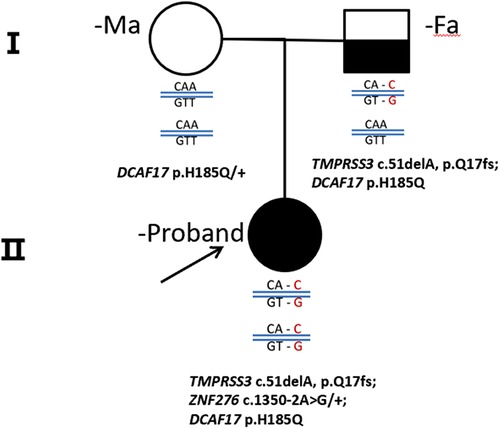
Figure 2. A pedigree drawings of the family with non-syndromic hearing imparemt. The proband's parent showed Normal hearing audiograms, while the affected gilrl, carried homozygous mutation, showed profound sensorineural hearing loss.
Mutations in a number of genes have been shown to cause severe non-syndromic deafness, including autosomal recessive NSHL (DFNB8/10), which is characterised by bilateral, severe to profound hearing loss. Such mutations have been identified in Palestinian, Tunisian, Korean, Indian, Spanish, and Greek populations (4, 8, 9, 16).
Our study found a frameshift mutation in TMPRSS3 that causes autosomal recessive NSHL in a Chinese patient. The exon 2 deletion identified, c.51del, leads to a frameshift of glutamate at position 17 (p.Q17fs) in the protein. Confirmation of the putative disease-causing variant and analysis of TMPRSS3 in all members of the Chinese family were performed by Sanger sequencing. The proband's father had a heterozygous TMPRSS3 deletion, which leads to double tracing as shown in the left to the black line on Figure 3. Figure 1 shown that the proband had the homozygous deletion. The explanation would be a novel TMPRSS3 p.Q17fs mutation that occurred in her mother. But the proband's mother did not carry this frameshift TMPRSS3 p.Q17fs mutation. Maybe the proband's mother had a deletion that is spanning over the TMPRSS3 p.Q17fs exon (or several exons, which leads to no PCR amplification), which made the proband looked like to have the homozygous deletion. Not only that, the mutation of TMPRSS3 was undetectable among 1,010 healthy subjects by Mass-spectrometric technique. These results indicate that the proband's deafness is likely to be caused by the homozygous TMPRSS3: c.51del mutation. The first mutation in a transmembrane protease associated with hearing loss was reported by Scott et al. in 2,001 (4). Since then, 24 mutations in TMPRSS3 have been identified as potentially pathogenic for inheritable deafness (Table 1). Among these, 22 are missense mutations, one is a nonsense mutation, and one is a deletion. The TMPRSS3 p.Q17fs in our study would be very rare that frameshift mutation occurs in both alleles. Our discovery therefore adds another example of NSHL caused by a deletion mutation.
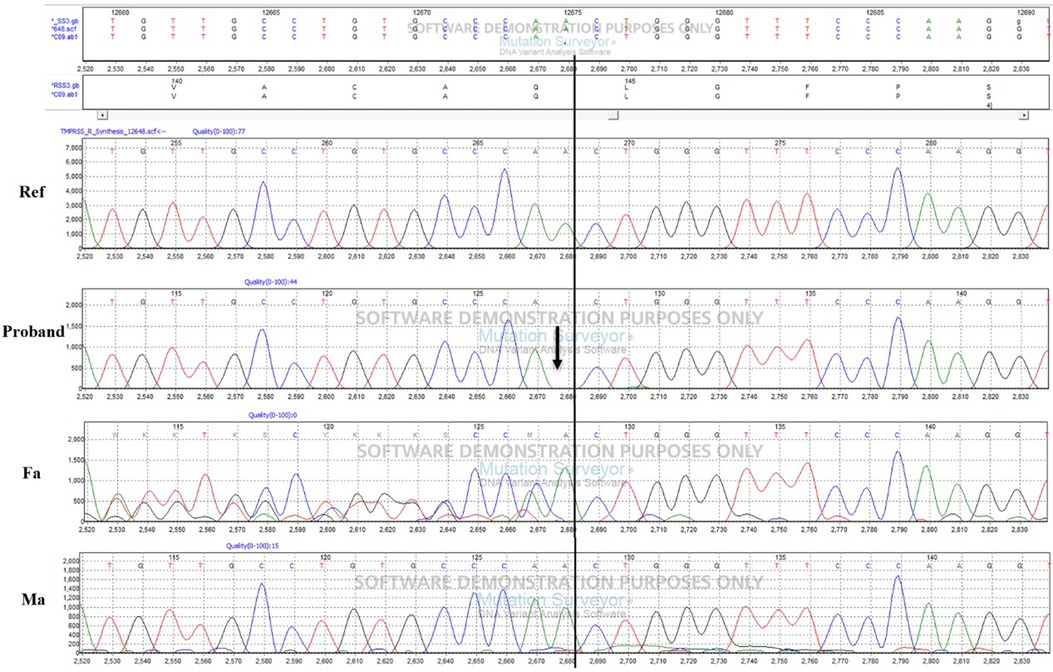
Figure 3. Validation of the frameshift mutation in TMPRSS3 by sanger sequencing.
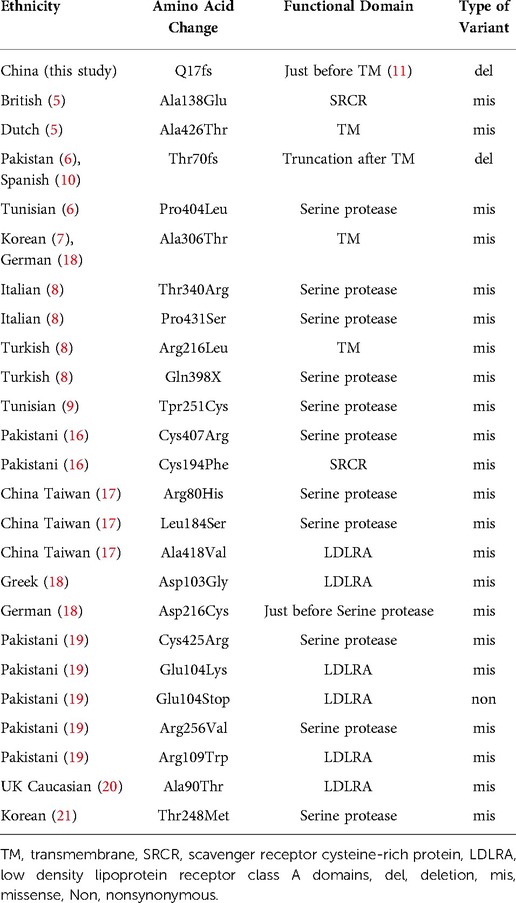
Table 1. Overview of the part of deafness-associated mutations in TMPRSS3.
We identified several other candidate deafness genes in the proband (Table 2). WES analyses showed that both parents were homozygous for DCAF17: c.T555 > A variant, and the proband was a carrier of the mutation. Mutations in DCAF17 are associated with Woodhouse–Sakati syndrome, a rare disorder characterised by alopecia, hypogonadotropic hypogonadism, sensorineural hearing loss, diabetes mellitus, and extrapyramidal movement (12). Another study suggested that DCAF17 may play as yet unexplored roles in tissue development and maintenance in adults (12). It is reported recently that DCAF17 have played an important role in gametes growth and development (13). The development of embryonic chromosomes abnormalities is directly related to the quality of gametes.We identified a homozygous missense (c. T555>A: p.H185Q) in exon 6 of DCAF17 in all members of the family (Table 2). DCAF17: c.555T > A has an allele frequency of 3% and is classified as a benign variant in the ClinVar database. Although, this variant does not associate with hearing loss. But, whether it was the homozygous missense matation in DCAF17 that caused embryonic dysplasia resulting in deafness remained to be further studied.

Table 2. Candidate genes identified in the proband with NSHL.
In contrast, the affected girl was the only carrier of the mutation detected in ZNF276 (c.1350–2A > G). The protein encoded by this gene is a 614-amino acid protein containing five C2H2-type zinc fingers and one zinc finger-associated domain. ZNF276 mutations have been identified in breast cancer (14) and may also be involved in the progression of Fanconi anaemia (15).
In conclusion, we report a identification of a frameshift mutation in the TMPRSS3 gene (c.51delA) resulting in a frameshift mutation in the protein (p.Q17fs) in the Han Chinese population. Our results thus provide a new example of autosomal recessive NSHL caused by a TMPRSS3 mutation.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
JL, ZY: These authors contributed equally to this work and should be considered co-first authorship. RX, ZY: These authors contributed equally to this work and should be considered co-corresponding authorship. ZW, JC, YL: These authors contributed equally to this work and should be considered co-senior authorship. All authors contributed to the article and approved the submitted version.
The research was funded by Special fund for National Natural Science Foundation of China (Program No. 81960290),Economic and Technological development of Longgang District, Shenzhen (Program No. LGKCYLWS2019000334 and Program No. LGKCYLWS2019000335) and High Level Project of Medicine in Longhua, ShenZhen (Program No. HLPM201907020103). None of this organizations influencede the study design, the collection, analysis, and management of data, the writing of the report, or the decision to submit to the manuscript for publication.
We thank the Chinese family for their participation and cooperation we identified a homozygous deletion. This research was partially supported by National Natural Science Foundation of China (No.81960290), District Special Fund for Economic and Technological Development in Longgang, Shenzhen (No.LGKCYLWS20190003334, No.LGKCYLWS20190003335), and High Level Project of Medicine in Longhua, ShenZhen (No.HLPM201907020103).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Morton CC, Nance WE. Newborn hearing screening-a silent revolution. N Engl J Med. (2006) 354(20):2151–64. doi: 10.1056/NEJMra050700
2. Petit C, Levilliers J, Hardelin JP. Molecular genetics of hearing loss. Annu Rev Genet. (2001) 35(35):589–646. doi: 10.1146/annurev.genet.35.102401.091224
3. Bonné-Tamir B, DeStefano AL, Briggs CE, Adair R, Franklyn B, Weiss S, et al. Linkage of congenital recessive deafness (gene DFNB10) to chromosome 21q22.3. Am J Hum Genet. (1996) 58(6):1254–9. doi: 10.1063/1.370701
4. Scott HS, Kudoh J, Wattenhofer M, Shibuya K, Berry K, Chrast AR, et al. Insertion of beta-satellite repeats identifies a transmembrane protease causing both congenital and childhood onset autosomal recessive deafness. Nat Genet. (2001) 27(1):59–63. doi: 10.1038/83768
5. Sarrias MR, Grønlund J, Padilla O, Madsen J, Holmskov U, Lozano F. The scavenger receptor cysteine-rich (SRCR) domain: an ancient and highly conserved protein module of the innate immune system. Crit Rev Immunol. (2004) 24(1):1–37. doi: 10.1615/CritRevImmunol.v24.i1.10
6. Ahmed ZM, Li XC, Powell SD, Riazuddin S, Young TL, Ramzan K, et al. Characterization of a new full length TMPRSS3 isoform and identification of mutant alleles responsible for nonsyndromic recessive deafness in Newfoundland and Pakistan. BMC Med Genet. (2004) 5(1):24. doi: 10.1186/1471-2350-5-24
7. Weegerink NJD, Margit S, Jaap O, Huygen PLM, Storm TM, Granneman S, et al. Genotype-phenotype correlation in DFNB8/10 families with TMPRSS3 mutations. J Assoc Res Otolar. (2011) 12(6):753–66. doi: 10.1007/s10162-011-0282-3
8. Wattenhofer M, Sahin-Calapoglu N, Andreasen D, Kalay E, Caylan R, Braillard B, et al. A novel TMPRSS3 missense mutation in a DFNB8/10 family prevents proteolytic activation of the protein. Hum Genet. (2005) 117(6):528–35. doi: 10.1007/s00439-005-1332-x
9. Masmoudi S, Antonarakis SE, Schwede T, Ghorbel AM, Gratri M, Pappasavas MP, et al. Novel missense mutations of TMPRSS3 in two consanguineous Tunisian families with nonsyndromic autosomal recessive deafness. Hum Mutat. (2001) 18(2):101–8. doi: 10.1002/humu.1159
10. Wattenhofer M, Di IM, Rabionet R, Dougherty L, Pampanos A, Schwede T, et al. Mutations in the TMPRSS3 gene are a rare cause of childhood nonsyndromic deafness in Caucasian patients. J Mol Med-jmm. (2002) 80(2):124–31. doi: 10.1007/s00109-001-0310-6
11. Chung J, Park SM, Chang SO, Chung T, Lee KY, Kim AR, et al. A novel mutation of TMPRSS3 related to milder auditory phenotype in Korean postlingual deafness:a possible future implication for a personalized auditory rehabilitation. J Mol Med. (2014) 92(6):651–63. doi: 10.1007/s00109-014-1128-3
12. Sheridan MB, Wohler E, Batista DAS, Applegate C, Hoover-Fong J. The use of high-density SNP array to map homozygosity in consanguineous families to efficiently identify candidate genes:application to woodhouse-sakati syndrome. Case Rep Genet. (2015) 2:169482. doi: 10.1155/2015/169482
13. Xu BL, Liu XQ, Wang L. Advances in functions of DCAF family proteins. Life Sci Res. (2020) 24(1):75–80. doi: 10.16605/j.cnki.1007-7847.2020.01.011
14. AJ Brenner,CM Aldaz. The genetics of sporadic breast cancer. Prog Clin Biol Res. (1997) 396:63–82.9108590
15. Nakamura A, Matsuura S, Tauchi H, Ohashi H, Hasegawa T, Honda K, et al. Four novel mutations of the fanconi anemia group A gene (FAA) in Japanese patients. J Hum Genet. (1999) 44(1):48–51. doi: 10.1007/s100380050106
16. Benyoseft T, Wattenhofer M, Riazuddin S, Ahmed ZM, Scott HS, Kudon J, et al. Novel mutations of TMPRSS3 in four DFNB8/B10 families segregating congenital autosomal recessive deafness. J Med Genet. (2001) 38(6):396–400. doi: 10.1136/jmg.38.6.396
17. Wong SH, Yen YC, Li SY, Yang YY. Novel mutations in the TMPRSS3 gene may contribute to Taiwanese patients with nonsyndromic hearing loss. Int J Mol Sci. (2020) 21(7):2382. doi: 10.3390/ijms21072382
18. Elbracht M, Senderek J, Eggermann T, Thürmer C, Park J, Maetin W, et al. Autosomal recessive postlingual hearing loss (DFNB8): compound heterozygosity for two novel TMPRSS3 mutations in German siblings. J Med Genet. (2007) 44:e81.17018562
19. Guipponi M, Toh MY, Tan J, Park D, Hanson K, Ballana E, et al. An integrated genetic and functional analysis of the role of type II transmembrane serine proteases (TMPRSSs) in hearing loss. Hum Mutat. (2008) 29(1):130–41.17918732
20. Lee J, Baek JI, Choi JY, Kim UK, Lee SH, Lee KY, et al. Genetic analysis of TMPRSS3 gene in the Korean population with autosomal recessive nonsyndromic hearing loss. Gene. (2013) 532(2):276–80.23958653
Keywords: TMPRSS3, homozygous, non-syndromic hearing loss, whole-exome sequencing, mutation
Citation: Liang J, Yu Z, Wang Z, Chen J, Liu Y, Yin Z and Xu R (2022) A frameshift mutation of TMPRSS3 in a Chinese family with non-syndromic hearing loss. Front. Pediatr. 10:1032659. doi: 10.3389/fped.2022.1032659
Received: 31 August 2022; Accepted: 21 November 2022;
Published: 9 December 2022.
Edited by:
Maria Elisabetta Baldassarre, University of Bari Aldo Moro, ItalyReviewed by:
Daw-Yang Hwang, National Health Research Institutes, Taiwan© 2022 Liang, Yu, Wang, Chen, Liu, Yin and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ruihuan Xu xrh69@126.com Zhangxing Wang wzx776@163.com
†These authors have contributed equally to this work and share first authorship
Specialty Section: This article was submitted to Genetics of Common and Rare Diseases, a section of the journal Frontiers in Pediatrics
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.