 Ang Li1†
Ang Li1† Weiyue Gu
Weiyue Gu Yanfang Jiang
Yanfang Jiang- 1Key Laboratory of Organ Regeneration and Transplantation of the Ministry of Education, Genetic Diagnosis Center, The First Hospital of Jilin University, Changchun, China
- 2Chigene (Beijing) Translational Medical Research Center Co., Ltd, Beijing, China
Familial non-syndromic unilateral hearing loss (NS-UHL) is rare and its genetic etiology has not been clearly elucidated. This study aimed to identify the genetic cause of NS-UHL in a three-generation Chinese family. Detailed medical history consultation and clinical examination were conducted. Further, whole-exome sequencing (WES) was performed to identify the genetic etiology of the proband, and the variant was verified by Sanger sequencing. A novel missense mutation, c.533G>C (p.Arg178Thr), in the SIX homeobox 1 gene (SIX1) was identified in four patients and co-segregated with NS-UHL in a three-generation Chinese family as a dominant trait. Using bioinformatics analyses, we show that this novel mutation is pathogenic and affects the structure of SIX1 protein. These data suggest that mutations in SIX1 gene are associated with NS-UHL. Our study added the NS-UHL phenotype associated with SIX1, and thereby improving the genetic counseling provided to individuals with SIX1 mutations.
1 Introduction
Unilateral hearing loss (UHL) is estimated to occur in 0.83 per 1,000 newborns (Prieve et al., 2000). As with bilateral hearing loss, UHL can severely affect the individuals’ lives. The etiology of approximately 35%–60% of UHL cases remains unknown (Kinney, 1953; Everberg, 1960a; Brookhouser et al., 1991). The most commonly etiologies of UHL include sequelae of bacterial meningitis, complication of viral infection, prenatal or perinatal problems, head trauma, and even genetic alterations (van Wieringen et al., 2019). With the rapid development of next-generation sequencing, genetic alterations have been identified in bilateral hearing loss, and the gene mutation spectrum has been constantly improving (https://hereditaryhearingloss.org/). However, the genetic alterations accounting for UHL have not been clearly elucidated. There is no doubt that identifying the causal genes will benefit patients in diagnosis, genetic counseling, and drug development.
UHL is often dismissed as sporadic or environmental, and the genetic basis has not been explored in depth (Dodson et al., 2012). It can be inherited as part of Pendred syndrome (PS [MIM: 274600]) or Waardenburg syndrome (WS [MIM: PS193500]). Among UHL, familial non-syndromic unilateral hearing loss (NS-UHL) (MIM:125000) is rare; only a few families are described in the literature (Everberg, 1957; 1960b; Dikkers et al., 2005). The gene KITLG has been linked to NS-UHL (Zazo Seco et al., 2015). Additionally, Dodson et al. included 34 patients with NS-UHL in a national hereditary deafness repository proved that mutations in the TECTA and COCH genes that might be causally related to the NS-UHL (Dodson et al., 2012). However, the molecular basis for other genetic causes of NS-UHL remains an important gap in current knowledge.
The absence of mutational hot genes or spots in NS-UHL hindered mutational analysis with gene panels (Dodson et al., 2012; Zazo Seco et al., 2015). Whole-exome sequencing (WES), combined with validated bioinformatics tools have facilitated the detection of variants, particularly the single nucleotide variants (SNVs) and the small insertions and deletions (InDels). Moreover, WES is being widely used in clinical practice due to its high diagnostic yields, low cost, and excellent advantages in novel genes analysis and subsequent investigation.
In this study, we performed WES to identify the genetic cause of autosomal-dominant NS-UHL in a three-generation Chinese Han pedigree. A novel missense mutation, c.533G>C (p.Arg178Thr), in the SIX1 gene was found to co-segregate with NS-UHL in this family. Our study added the NS-UHL phenotype associated with SIX1, and thereby improving the genetic counseling provided to individuals with SIX1 mutations.
2 Materials and methods
2.1 Family members
The proband (III-2) was a 7-year-old girl diagnosed with severe sensorineural hearing loss in the right ear at birth. The healthy side has normal hearing spectrum. Blood routine, urine routine, liver function and renal function showed normal parameters. The patient had no history of drug intoxication, trauma, or infection. Additionally, there was a family history of unilateral sensorineural hearing loss in her pedigree. Patients I-2, II-2, and II-4 also showed unilateral sensorineural hearing loss (Figure 1). This study was approved by the Ethics Committee of the First Hospital of Jilin University. All participants provided written informed consent.
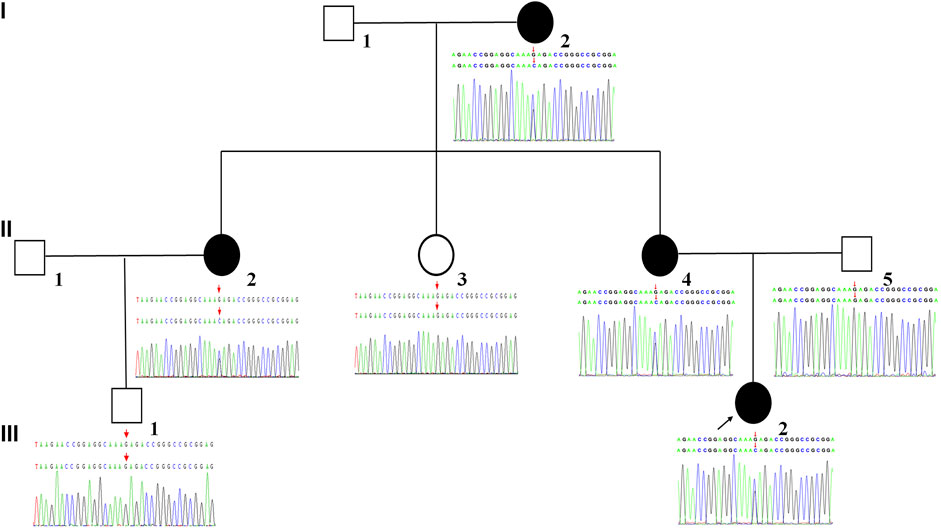
FIGURE 1. Pedigree of the family affected by NS-UHL. White symbols indicate normal individuals. The filled black symbol denotes the individual diagnosed with NS-UHL. The arrow indicates the proband of the family.
2.2 Whole exome sequencing
Genomic DNA was isolated from whole blood of all available family members using the QIAamp DNA Blood MiNi Kit (Qiagen, Germany) according to the manufacturer’s instructions. We performed genetic testing using WES technology on the proband (III-2). Whole-exome capture was xGen Exome Research Panel v2.0 (IDT, Iowa, United States). High-throughput sequencing was performed on the DNBSEQT7 (BGI, China) platform. Average sequencing depth of WES was 162.75 × with an average of 97.7% of reads covered at a depth of at least 20×. For SIX1 gene, the average depth of sequencing was 167.56×. Raw data sequenced by WES were processed by using fastp (https://github.com/OpenGene/fastp) to remove adapters and filtering low-quality reads. High-quality reads were aligned to the GRCh37/hg19 reference genome using Burrows-Wheeler Aligner (BWA, https://github.com/lh3/bwa). Base quality score recalibration and calling variant were performed using the Genome Analysis Toolkit (GATK, http://www.broadinstitute.org/gatk/). The pipeline of variants analysis is as follows. According to the sequence depth and variant quality, the high-quality and reliable variants were obtained. Variants were then annotated with minor allele frequencies (MAFs) databases (genomAD, ESP, 1,000 genomes, EXAC, dbSNP databases). The Effect of the variant on gene product was predicted by using bioinformatics softwares such as Provean (http://provean.jcvi.org/genome_submit_2.php?species=human), REVEL (https://sites.google.com/site/revelgenomics/), SIFT (http://sift.jcvi.org/), GERP (http://mendel.stanford.edu/sidowlab/downloads/gerp/index.html) and phastCons (https://varianttools.sourceforge.net/Annotation/PhastCons). According to the American College of Medical Genetics criteria (ACMG), the identified variants were classified as pathogenic, likely pathogenic, benign, likely benign, or of uncertain significance (Richards et al., 2015; Harrison et al., 2019). Online Mendelian Inheritance in Man (OMIM), Human Gene Mutation Database (HGMD), and ClinVar databases were then used as conferences of pathogenicity of every variant. Finally, the most possible pathogenic genes were identified from the screened deleterious variants combining disease correlation and clinical phenotype. The variants filtering criteria was shown in Supplementary Table S1.
2.3 Sanger sequencing
We designed specific primers (forward 5′-CGCCCACCGCCAAGTTCCGACTCC-3′ and reverse 5′-CCCGACACTCACATCCCAGAGAAACCCAC-3′) based on the variant loci detected by next-generation sequencing (NGS). DNA isolated from all available family was used as a template for PCR amplification on HEMA 9600 PCR sequencer using the specific primers for SIX1 gene. Sanger sequencing was further performed using the ABI 3730XL sequencer (Applied Biosystems). Co-segregation was analyzed in all available members.
2.4 Protein conserved prediction and 3D structure prediction
We performed multiple sequence alignments using MUSCLE (https://www.ebi.ac.uk/Tools/msa/muscle/) and prepared model diagrams of protein domain using Illustrator for Biological Sequences V1.0 (IBS). The protein sequence encoded by the transcript (NM_005982.4) and the X-ray crystal structure (PDB ID 4EGC) were used for SWISS-MODEL with 100% sequence similarity, and PyMOL (PyMol Molecular Graphics System, Schrödinger, LLC) was used to visualize the model by referring to Version 2.1.0.
3 Results
3.1 SIX1 gene mutations and sanger sequencing validation
A novel heterozygous missense variant in SIX1 gene was detected in the proband (III-2) by using WES. The missense variants SIX1 c.533G>C resulted in a threonine substitution. The frequency of the variant was absent from control database (gnomAD database, ESP database, 1,000 genomes database, EXAC database, dbSNP database). The variant were then validated in all available members by Sanger sequencing and revealed that the variant was maternally inherited. The family members (I-2, II-2, and II-4) with NS-UHL shared the same variant with the proband. Whereas, other family members (II-3, II-5, and III-1) without NS-UHL did not have any variants detected by Sanger sequencing. We verified that NS-UHL cosegregated with the SIX1 c.533G>C mutation (Figure 1).
3.2 Bioinformatics analyses of the identified mutation
All scores generated by different prediction softwares suggested deleterious effect: Provean (score -4.92), REVEL (score 0.947), SIFT (score 0.0) GERP (score 5.96) and phastCons (score 1.0). The amino acids in SIX1 protein, including arginine at position 178 (p.Arg178), are conserved across vertebrates from human to Gallus (Figure 2) and are located in the highly conserved homologous domain (HD) (Figure 2). Using PyMOL software to analyze the 3D structure of wild-type and mutant SIX1 proteins, the results (Figure 3) showed that compared with the wild-type, the c.533G>C variant substituted the neutral amino acid threonine for basic amino acid arginine at position 178 of the protein (p.Arg178Thr). In addition, compared with wild-type, the hydrogen bond between the 178th amino acid and the 123th and 127th amino acid of protein disappeared after mutation, which might affect the protein structure and function. According to the ACMG criteria, this mutation is pathogenic (Richards et al., 2015).
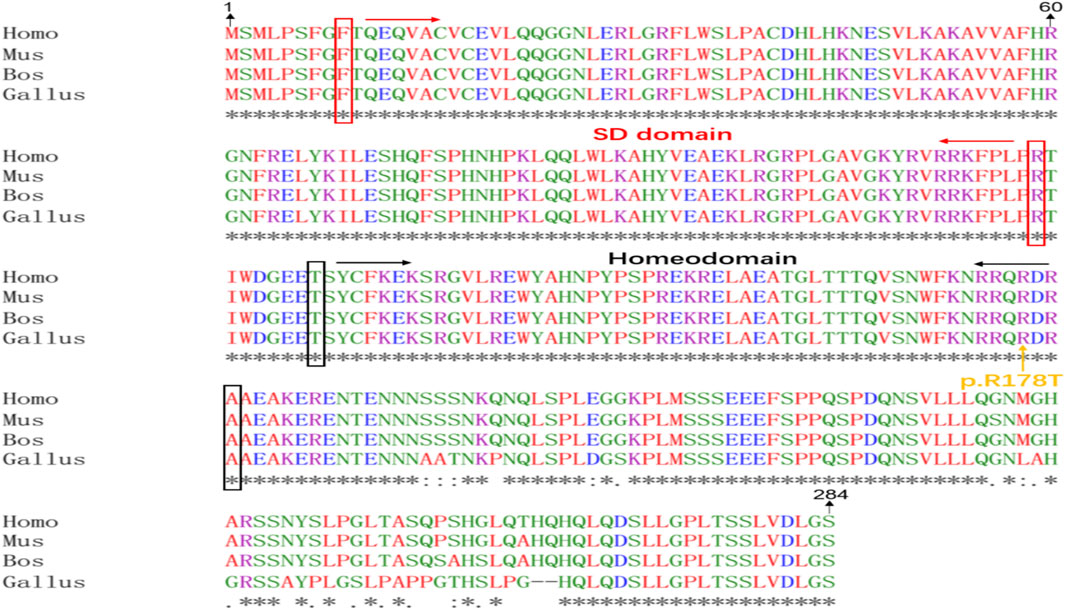
FIGURE 2. Alignment of SIX1 protein sequences from different species. Homo:Homo sapiens (NM_005982.4), Mus: Mus musculus (NM_009189.3), Bos:Bos Taurus (XM_002691019.6), Gallus:Gallus gallus (NM_001044685.2). Red: amino acid containing non-polar and hydrophobic R group, Green: amino acid containing polar and neutral R group, Blue: amino acid containing acidic R group, Purple: amino acid containing basic R group. “*”: a completely consistent residue, “.”: residues with weak and similar properties, “:”: residues with very similar properties.
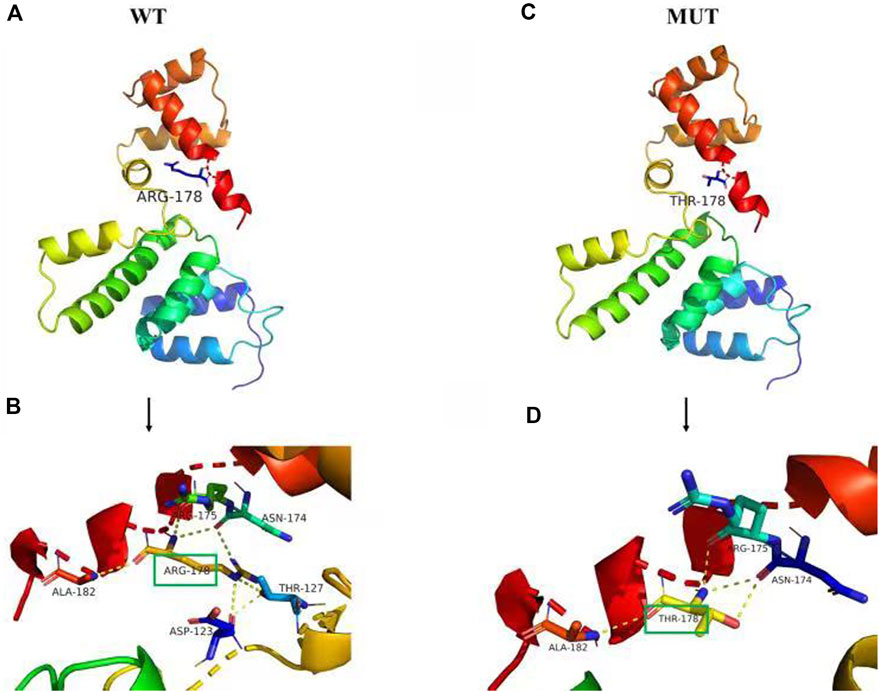
FIGURE 3. Prediction of the tertiary structure of SIX1 protein. The novel variant of the SIX1 gene is c.533 G>C, p.R178T (p.Arg178Thr), (A) is the 3D overall picture of wild-type (WT) SIX1 protein: the 178th amino acid is ARG. (B) is the 3D partial enlarged view of WT SIX1 protein. The 178ARG respectively form a hydrogen bond with the 123ASP, 127THR, 175 ARG, and 182 ALA, and 178ARG form two hydrogen bonds with 174ASN, together to maintain the stability of the protein structure. (C) is the 3D overall picture of mutant (MUT) SIX1 protein: the 178th amino acid is mutated to THR. (D) is the 3D partial enlarged view of mutant SIX1 protein. No hydrogen bond interaction existed between the 178THR and 123ASP, 127THR.
4 Discussion
UHL is often caused by acquired and unnoticed trauma, infection or other factors (van Wieringen et al., 2019). About 35%–60% of UHL cases do not receive an etiological diagnosis, which has stalled treatment for this disease. With the rapid development of next-generation sequencing, genetic alterations have also been identified as a cause of UHL. Previously, UHL frequently inherited as part of Pendred syndrome or Waardenburg syndrome that has been reported continuously. However, NS-UHL is rarely reported. Here, we reported a novel missense mutation in SIX1 gene and provide evidence that this pathogenic mutation may be responsible for dominantly inherited NS-UHL.
The SIX1 gene is located at chromosome 14q23.1 and is composed of an amino terminus, SIX domain (SD), homologous domain (HD) and a carboxyl terminus. The SD and HD domain were highly conserved. The SIX1 gene plays an essential role in the development of several organs, including kidney, muscle and inner ear (Wu et al., 2014; Shah et al., 2020). According to the HGMD, only 24 pathogenic variants of the SIX1 gene have been reported in the literature. Individuals carrying a heterozygous SIX1 mutation were reported to have a very different clinical outcomes, ranging from being non-syndromic bilateral hearing loss to Branchio-otic (BO) syndrome and to Branchio-otic-renal (BOR) syndrome (Salam et al., 2000; Ruf et al., 2004; Mosrati et al., 2011).
Salam et al. demonstrated that heterozygous mutation in the SIX1 gene associated with prelingual bilateral symmetric hearing loss (Salam et al., 2000). Mosrati et al. also proved that dominant mutation in SIX1 result in only auditory defects in humans (Mosrati et al., 2011). Differently, Ruf and Kumar et al. reported SIX1 mutations associated with branchio-otic syndrome or branchio-oto-renal syndrome (Kumar et al., 2000; Ruf et al., 2003; Ruf et al., 2004). Thus, the clinical phenotype associated with SIX1 gene mutation need to be further enriched. In this study, we identified a novel missense mutation, SIX1 c.533G>C in a three-generation Chinese Han pedigree and co-segregate with NS-UHL. Our study added the NS-UHL phenotype associated with SIX1. However, the effect of SIX1 gene on the clinical phenotype has not been fully elucidated, and expanding the hearing loss family sample sizes is essential.
5 Concluding remarks
In conclusion, a novel missense mutation, c.533G>C (p.Arg178Thr), in the SIX1 gene was identified in a Chinese Han family with NS-UHL. To the best of our knowledge, this is the first report on the association of the SIX1 gene with NS-UHL. The mechanisms underlying this unilateral hearing loss is not clearly understood, further functional studies of the SIX1 mutations and the application of in vivo models with genetic deficiency are warranted. This finding not only enriches the spectrum of diseases caused by SIX1 gene, but also provides a new focus for genetic counseling.
Data availability statement
The original contributions presented in the study are publicly available. This data can be found here: https://www.ncbi.nlm.nih.gov/clinvar/ ,SCV002575099
Ethics statement
The studies involving human participants were reviewed and approved by the Ethics Committee of the First Hospital of Jilin University. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
AL drafted the initial manuscript. SL, GL, WG, and YJ were responsible for genetic analysis. SL, PZ, and XH collected clinical information. AL and SL revised the manuscript. YJ designed the study, and critically reviewed the manuscript. All the authors read and approved the final manuscript.
Funding
This research was funded by the National Natural Science Foundation of China (Nos 30972610 and 81273240), National Key Research and Development Program (Nos 2017YFC0910000 and 2017YFD0501300), Jilin Province Science and Technology Agency (Nos JJKH20211210KJ, JJKH20211164KJ, 20200403084SF, JLSWSRCZX 2020-009, 20200901025SF, 20190101022JH, and 2019J026).
Acknowledgments
We are thankful to the patient and her family for their participation in the study. We would like to thank Editage (www.editage.cn) for English language editing.
Conflict of interest
GL and WG were employed by the Chigene (Beijing) Translational Medical Research Center Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.1047230/full#supplementary-material
References
Brookhouser, P. E., Worthington, D. W., and Kelly, W. J. (1991). Unilateral hearing loss in children. Laryngoscope 101 (12), 1264–1272. doi:10.1002/lary.5541011202
Dikkers, F. G., Verheij, J. B., and van Mechelen, M. (2005). Hereditary congenital unilateral deafness: A new disorder? Ann. Otol. Rhinol. Laryngol. 114 (4), 332–337. doi:10.1177/000348940511400414
Dodson, K. M., Georgolios, A., Barr, N., Nguyen, B., Sismanis, A., Arnos, K. S., et al. (2012). Etiology of unilateral hearing loss in a national hereditary deafness repository. Am. J. Otolaryngol. 33 (5), 590–594. doi:10.1016/j.amjoto.2012.03.005
Everberg, G. (1960a). Etiology of unilateral total deafness studied in a series of children and young adults. Ann. Otol. Rhinol. Laryngol. 69, 711–730. doi:10.1177/000348946006900304
Everberg, G. (1960b). Further studies on hereditary unilateral deafness. Acta Otolaryngol. 51, 615–635. doi:10.3109/00016486009124539
Everberg, G. (1957). Hereditary unilateral deafness. Acta Otolaryngol. 47 (4), 303–311. doi:10.3109/00016485709130346
Harrison, S. M., Biesecker, L. G., and Rehm, H. L. (2019). Overview of specifications to the ACMG/AMP variant interpretation guidelines. Curr. Protoc. Hum. Genet. 103 (1), e93. doi:10.1002/cphg.93
Kinney, C. E. (1953). Hearing impairments in children. Laryngoscope 63 (3), 220–226. doi:10.1288/00005537-195303000-00004
Kumar, S., Deffenbacher, K., Marres, H. A., Cremers, C. W., and Kimberling, W. J. (2000). Genomewide search and genetic localization of a second gene associated with autosomal dominant branchio-oto-renal syndrome: Clinical and genetic implications. Am. J. Hum. Genet. 66 (5), 1715–1720. doi:10.1086/302890
Mosrati, M. A., Hammami, B., Rebeh, I. B., Ayadi, L., Dhouib, L., Ben Mahfoudh, K., et al. (2011). A novel dominant mutation in SIX1, affecting a highly conserved residue, result in only auditory defects in humans. Eur. J. Med. Genet. 54 (5), e484–e488. doi:10.1016/j.ejmg.2011.06.001
Prieve, B., Dalzell, L., Berg, A., Bradley, M., Cacace, A., Campbell, D., et al. (2000). The New York state universal newborn hearing screening demonstration project: Outpatient outcome measures. Ear Hear. 21 (2), 104–117. doi:10.1097/00003446-200004000-00005
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Ruf, R. G., Berkman, J., Wolf, M. T., Nurnberg, P., Gattas, M., Ruf, E. M., et al. (2003). A gene locus for branchio-otic syndrome maps to chromosome 14q21.3-q24.3. J. Med. Genet. 40 (7), 515–519. doi:10.1136/jmg.40.7.515
Ruf, R. G., Xu, P. X., Silvius, D., Otto, E. A., Beekmann, F., Muerb, U. T., et al. (2004). SIX1 mutations cause branchio-oto-renal syndrome by disruption of EYA1-SIX1-DNA complexes. Proc. Natl. Acad. Sci. U. S. A. 101 (21), 8090–8095. doi:10.1073/pnas.0308475101
Salam, A. A., Hafner, F. M., Linder, T. E., Spillmann, T., Schinzel, A. A., and Leal, S. M. (2000). A novel locus (DFNA23) for prelingual autosomal dominant nonsyndromic hearing loss maps to 14q21-q22 in a Swiss German kindred. Am. J. Hum. Genet. 66 (6), 1984–1988. doi:10.1086/302931
Shah, A. M., Krohn, P., Baxi, A. B., Tavares, A. L. P., Sullivan, C. H., Chillakuru, Y. R., et al. (2020). Six1 proteins with human branchio-oto-renal mutations differentially affect cranial gene expression and otic development. Dis. Model. Mech. 13 (3), dmm043489. doi:10.1242/dmm.043489
van Wieringen, A., Boudewyns, A., Sangen, A., Wouters, J., and Desloovere, C. (2019). Unilateral congenital hearing loss in children: Challenges and potentials. Hear. Res. 372, 29–41. doi:10.1016/j.heares.2018.01.010
Wu, W., Huang, R., Wu, Q., Li, P., Chen, J., Li, B., et al. (2014). The role of Six1 in the Genesis of muscle cell and skeletal muscle development. Int. J. Biol. Sci. 10 (9), 983–989. doi:10.7150/ijbs.9442
Zazo Seco, C., Serrao de Castro, L., van Nierop, J. W., Morin, M., Jhangiani, S., Verver, E. J., et al. (2015). Allelic mutations of KITLG, encoding KIT ligand, cause asymmetric and unilateral hearing loss and Waardenburg syndrome type 2. Am. J. Hum. Genet. 97 (5), 647–660. doi:10.1016/j.ajhg.2015.09.011
Keywords: non-syndromic unilateral hearing loss, SIX1 gene, whole exome sequencing, genetic counseling, novel mutation
Citation: Li A, Liu S, Zhang P, Hu X, Li G, Gu W and Jiang Y (2022) A novel heterozygous SIX1 missense mutation resulted in non-syndromic unilateral hearing loss. Front. Genet. 13:1047230. doi: 10.3389/fgene.2022.1047230
Received: 17 September 2022; Accepted: 01 November 2022;
Published: 22 November 2022.
Edited by:
Muhammad Naeem, Hebei Normal University, ChinaReviewed by:
Muhammad Sulaiman Saeed, Nishtar Medical University Multan, PakistanAmmar Husami, Cincinnati Children’s Hospital Medical Center, United States
Saba Battelino, University Medical Centre Ljubljana, Slovenia
Copyright © 2022 Li, Liu, Zhang, Hu, Li, Gu and Jiang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yanfang Jiang, eWFuZmFuZ2ppYW5nQGhvdG1haWwuY29t
†These authors have contributed equally to this work