 María Victoria García-Ortiz1,2,3†
María Victoria García-Ortiz1,2,3† María José de la Torre-Aguilar1,4†
María José de la Torre-Aguilar1,4† Teresa Morales-Ruiz1,2,3
Teresa Morales-Ruiz1,2,3 Antonio Gómez-Fernández1,4
Antonio Gómez-Fernández1,4 Katherine Flores-Rojas1,5,6
Katherine Flores-Rojas1,5,6 Mercedes Gil-Campos1,5,6*Pilar Martin-Borreguero1,7
Mercedes Gil-Campos1,5,6*Pilar Martin-Borreguero1,7 Rafael R. Ariza1,2,3
Rafael R. Ariza1,2,3 Teresa Roldán-Arjona1,2,3
Teresa Roldán-Arjona1,2,3 Juan Luis Perez-Navero1,4,8
Juan Luis Perez-Navero1,4,8- 1Maimónides Biomedical Research Institute of Córdoba (IMIBIC), Córdoba, Spain
- 2Department of Genetics, University of Córdoba, Córdoba, Spain
- 3Reina Sofía University Hospital, Córdoba, Spain
- 4Department of Pediatrics, Reina Sofia University Hospital, University of Córdoba, Córdoba, Spain
- 5Pediatric Metabolism Unit, Reina Sofia University Hospital, University of Córdoba, Córdoba, Spain
- 6Physiopathology of Obesity and Nutrition Networking Biomedical Research Center (CIBEROBN), Córdoba, Spain
- 7Department of Child and Adolescent Clinical Psychiatry and Psychology, Reina Sofia University Hospital, Córdoba, Spain
- 8Biomedical Research Center–Rare Diseases (CIBERER), Carlos III Health Institute, Madrid, Spain
The goal of this investigation was to determine whether there are alterations in DNA methylation patterns in children with autism spectrum disorder (ASD).
Material and Methods: Controlled prospective observational case-control study. Within the ASD group, children were sub-classified based on the presence (AMR subgroup) or absence (ANMR subgroup) of neurodevelopmental regression during the first 2 years of life. We analyzed the global levels of DNA methylation, reflected in LINE-1, and the local DNA methylation pattern in two candidate genes, Neural Cell Adhesion Molecule (NCAM1) and Nerve Growth Factor (NGF) that, according to our previous studies, might be associated to an increased risk for ASD. For this purpose, we utilized blood samples from pediatric patients with ASD (n = 53) and their corresponding controls (n = 45).
Results: We observed a slight decrease in methylation levels of LINE-1 in the ASD group, compared to the control group. One of the CpG in LINE-1 (GenBank accession no.X58075, nucleotide position 329) was the main responsible for such reduction, highly significant in the ASD subgroup of children with AMR (p < 0.05). Furthermore, we detected higher NCAM1 methylation levels in ASD children, compared to healthy children (p < 0.001). The data, moreover, showed higher NGF methylation levels in the AMR subgroup, compared to the control group and the ANMR subgroup. These results are consistent with our prior study, in which lower plasma levels of NCAM1 and higher levels of NGF were found in the ANMR subgroup, compared to the subgroup that comprised neurotypically developing children.
Conclusions: We have provided new clues about the epigenetic changes that occur in ASD, and suggest two potential epigenetic biomarkers that would facilitate the diagnosis of the disorder. We similarly present with evidence of a clear differentiation in DNA methylation between the ASD subgroups, with or without mental regression.
Introduction
Autism spectrum disorder (ASD) is a severe neurodevelopmental disorder featuring variable but pronounced deficits in communication and social interaction, and is associated to multiple genetic risk factors (1, 2). Environmental (viral infection, parental age, diet) and epigenetic factors (DNA methylation, histone modification and microRNA) would act on some predisposing genetic factors (3). However, although epigenetic mechanisms, transcriptome profiles, and environmental factors have been suggested to be implicated, no clear pathogenesis mechanisms have been yet identified in ASD (4–12).
One of the best-known epigenetic marks is the 5-methylcytosine (5-mC), generated by the methylation at C5 of cytosine in symmetric CG contexts. A high proportion of regulatory regions, such as promoters and enhancers, contains areas with high density of CpG dinucleotides (called “CpG islands”) that are usually unmethylated, and whose methylation is generally associated to transcriptional silencing (13). Extensive experimental evidence supports that DNA methylation is a crucial step during brain development, and plays a key role in processes including synaptic plasticity, learning, memory, or cognitive decline (14).
Unlike mutations, epigenetic changes are potentially reversible, which has opened new pathways in the study of some diseases. Nowadays, it is accepted that ASD has a strong epigenetic component (14–19). Several genome-wide studies revealed multiple alterations in DNA methylation patterns in the brains of ASD individuals (20, 21). The largest meta-analysis study using peripheral blood samples from about 800 autistic individuals revealed that 55 of examined CpG sites were associated to ASD (22). Nevertheless, the main regions analyzed in these studies are the CpG islands of the protein coding regions, whereas DNA methylation in the genomic non-coding regions, which are often implicated in genetic regulation pathways, has not yet been examined. On the other hand, a significant fraction of the mutations associated to an increased risk of ASD affects transcriptional factors or chromatin-modifying proteins (23). Genetic mutations of the MECP2 gene related to autism spectrum disorders have been recently identified (4). MECP2 is a master epigenetic regulator that exhibits high levels of expression in the brain. However, a reduced expression of MECP2, associated to hypermethylation of its promoter, has been described in the frontal cortex of patients with ASD (8). Understanding the epigenetic changes involved in ASD can also help to identify subgroups of patients, allowing a better stratification for the implementation of pharmacological and/or behavioral therapies.
Long interspersed element-1 (LINE-1) is an autonomous transposable element that makes up roughly 17% of the human genome where it remains active (24). It is possible for retrotransposition of LINE-1 into a new position of the genome to cause duplication, deletion, or insertion at the target site, triggering genomic instability and changes in gene expression (25). Although the process by which LINE-1 transcription and retrotransposition is regulated remains indeterminate, mounting evidence propose that epigenetic pathways, such as DNA methylation and histone modifications, are implicated in the retrotransposition of LINE-1 and could have an impact on the target genes expression (26). There have been reports of altered DNA methylation levels and patterns of CpG residues in LINE-1 promoter regions in many diseases, including ASD (22, 26).
Other authors have posited that both a proinflammatory condition and an alteration in adhesion molecules in the early stages of neurodevelopment may play a role in the pathophysiology of ASD. Our group has recently published a study on the plasma levels of particular adhesion molecules and growth factors in patients with ASD compared to healthy children. When sub-classifying according to the presence or absence of neurodevelopmental regression in children with ASD, lower plasma levels of the Neural Cell Adhesion Molecule (NCAM1) and higher levels of Nerve Growth Factor (NGF) were observed in the subgroup of autistic children without mental regression, compared to the levels obtained in the mental regression subgroup and the control group (27).
Based on evidence of close implication of loci-specific DNA methylation in the pathophysiology of neurological disorders including ASD (20, 21), and previous demonstration of an association between the epigenetic regulation of LINE-1 and its effects in ASD pathobiology (26), the present study aims to compare global methylation levels in blood samples of pediatric patients with ASD and healthy children, and to examine the DNA methylation status of NCAM1 and NGF, two genes that we have previously shown to be deregulated in autistic spectrum disorder. Here, we report the results of a global and local DNA methylation analysis from a pediatric sample with ASD compared to normally developing children.
Materials and Methods
Subjects
This was a controlled prospective observational case-control study. The patients with ASD were recruited from the Department of Child and Adolescent Clinical Psychiatry and Psychology at the Reina Sofia University Hospital. Some of the patients (those aged 2–3) were assimilated into the study at the time of their ASD diagnosis, while the older patients (those aged 3–6) were selected from among children who had already been given diagnoses at the same unit. The diagnosis of ASD was based on the clinical judgment of professionals specializing in identifying the unique developmental profile associated with subjects with ASD. The diagnoses were based on the information obtained from semi-structured clinical development interviews, and psychological and behavioral tests that have been internationally acknowledged to be reliable and valid for this purpose. Two clinical psychologists, a psychiatrist and an occupational therapist with extensive clinical experience and training in diagnostic tests for research in ASD performed the diagnoses, employing the criteria established in DSM-5 (28). In addition, two pediatricians reviewed the subjects' medical histories and conducted an examination of all the children.
Children with ASD presenting with other known neurological, metabolic or genetic diagnoses were excluded from the study, and the same applied to children undergoing medical treatments for autism-related behavioral comorbidities that might interfere with the results, such as sedatives, muscle relaxants or similar. In addition, a control group of normally developing children was recruited, matched to the ASD group in terms of gender and age.
Within the ASD group, children were sub-classified based on the presence or absence of neurodevelopmental regression occurring during the first 2 years of life. This was assessed by means of a five-item questionnaire in accordance with the guide used by the ADI-R clinical interview for the evaluation of this process (29). Children with ASD who obtained a score equal to or greater than three were included in the mental regression subgroup (AMR: Autism Mental Regression), while those with a score inferior to this cut-off value were included in the non-mental regression subgroup (ANMR: Autism Non-Mental Regression) (27).
The study was approved by the Clinical Research and Bioethics Committee at the Reina Sofia University Hospital, conforming to the fundamental principles established in the Declaration of Helsinki (latest version 2013), supplemented by the Declaration of Taipei (2016).
Standardized Diagnostic Measurements and Assessment of ASD Severity
All the cases of ASD were selected after carrying out the Checklist for Autism in Toddlers (M-CHAT). Subsequently, a comprehensive clinical history of each child was taken. The Autism Diagnostic Observation Schedule, Second Edition (ADOS-2), was used in cases of suspected ASD. Each of the following tests was also administered: the Autism Diagnostic Interview-revised (ADI-R), the Pervasive Developmental Disorders Behavior Inventory™ (PDDBI), the Childhood Autism Rating Scale test (CARS), the Battelle Developmental test, and the Strengths and Difficulties Questionnaire (SDQ). These tests enabled the clinical team to diagnose ASD with more certainty.
None of the ASD cases selected for the present study exhibited any other associated pathology (seizures, or other neurological, metabolic or genetic diseases). Each patient was clinically assessed with complementary explorations. The children also underwent a genetic study (karyotype and microarrays) to detect secondary or syndromic ASD. None of the patients required medication to treat behavioral disorders or aggression.
Sample Collection, DNA Extraction and Bisulfite Treatment of the DNA
After overnight fasting, peripheral whole blood samples were collected from participants via antecubital vein into 6-ml blood collection tubes containing EDTA. After centrifugation at 3,500 g for 10 min, plasma was divided into aliquots and processed within 2 h from sampling, and then frozen at −80°C until analysis. Blood count and a general biochemical analysis were performed to confirm the absence of other diseases.
Genomic DNA was extracted from 2 mL of plasma with the QIAamp DNA Blood kit (QIAGEN), following the manufacture's protocol, and quantified in a NanoDrop ND-1,000 spectrophotometer (Nano-Drop Technologies). Bisulfite conversion of isolated DNA (500 ng) was performed with the EZ DNA Methylation Gold Kit (Zymo Research), according to the manufacturer's instructions.
PCR and Pyrosequencing
Utilizing the Immolase DNA Polymerase (Bioline) 50 ng of bisulfite, converted DNA were added to the PCR reaction, following the manufacturer's recommendations. Table 1 shows the forward and reverse primer sequences for each gene. The PCR product was subjected to agarose gel electrophoresis to test for the presence of a single PCR product. 15 μL of the verified biotinylated PCR product was used for each sequencing assay. DNA pyrosequencing was performed in a PyroMark Q24 instrument (Qiagen), following the manufacturer's guidelines, and subsequent methylation analysis was determined with the PyroMark Q24 Software (Qiagen). Sequencing primers for each gene are listed in Table 1. In the light of the literature (30–33) and recent results from this group of children (27), two loci previously linked to ASD (NCAM1 and NGF) were selected for methylation analysis. Primers for NCAM1 were designed to amplify a 3′UTR region (UCSC region chr11:112,965,249–112,965,403) that included CpG positions for which changes in methylation levels have been previously described (34). Primers for NGF were obtained from Qiagen to amplify a region at the 5′UTR, near the non-transcribed first exon of the gene (UCSC region chr1:115,880,563–115,880,774) (Table 1).
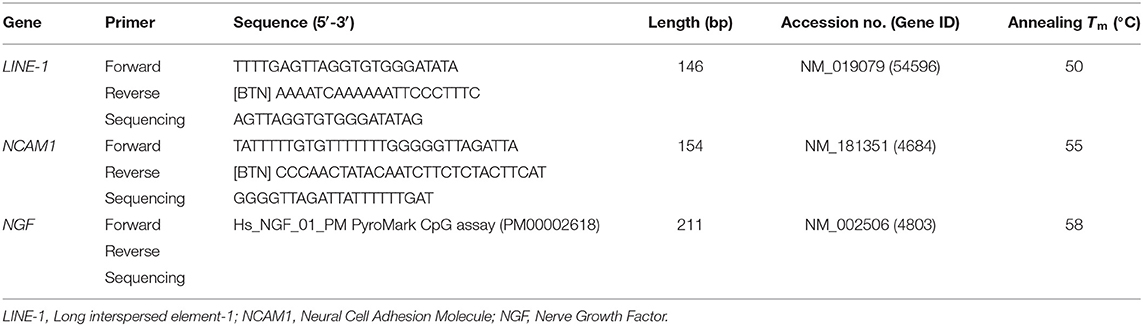
Table 1. List of primers used for pyrosequencing.
Statistical Analysis
The sample size for this study was determined based on the most relevant genome-wide and local DNA methylation studies on ASD using published data results (18, 24, 26). GraphPad Prism 6 Software was used to perform the statistical analyses. The data are expressed as mean ± SD (95% confidence intervals), median (IQR) or absolute (relative frequencies). For data that fit a normal distribution (Shapiro-Wilk normality test) t-tests were used. For data not normally distributed, the non-parametric Mann-Whitney test were applied. Categorical variables were evaluated using the χ2-test or the Fisher exact test. To assess the methylation differences between healthy control group and ASD subgroups, in CpG positions together or individually, unpaired t-test and two-way ANOVA model with Tukey's HSD post-hoc tests were used Correlations between methylation levels and the scores obtained from the various tests performed were carried out using the Spearman's p (rho). Receiver operating characteristics (ROC) analysis was conducted to calculate the area under the curve (AUC). All the tests were two-tailed, and a p < 0.05 was considered statistically significant.
Results
Global Methylation Analysis in Autism Spectrum Disorder
Fifty-four children with ASD and 45 healthy children were included in the study. The principal demographic and test results for the diagnosis are shown in Table 2. Within the ASD group, there were 20 children with AMR and 33 with ANMR; one child could not be classified in these subgroups because he was adopted and there was no previous clinical information.
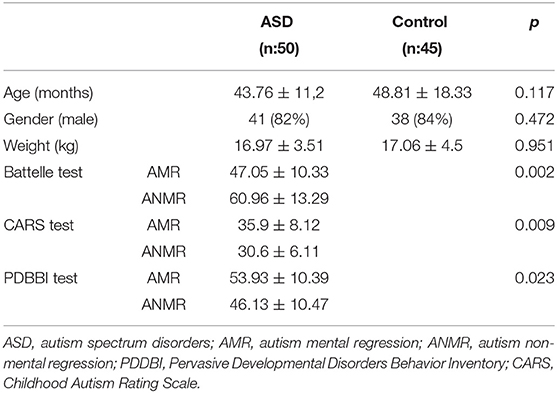
Table 2. Demographic and anthropometric data in children with autism spectrum disorders compared to controls.
Because LINE-1 retrotransposon represents a considerable portion of the human genome, the methylation level of LINE-1 reflects the global DNA methylation status (24). To determine whether global methylation levels were altered in ASD, 53 blood samples from children with ASD and 45 from the control group were drawn to isolate to gDNA. Bisulfite pyrosequencing was performed analyzing 5 CpG positions of the repetitive element LINE-1. Three samples from ASD patients gave inconclusive results, so they were removed from the LINE-1 methylation analysis. Methylation of LINE-1 was slightly reduced in the group of children with ASD compared to the control group (73.45 ± 0.43 vs. 74.56 ± 0.45) (Figures 1A,C). When each studied CpG of the LINE-1 fragment was analyzed separately, position 2 (GenBank accession no.X58075, nucleotide position 329, complementary strand) was the main responsible for the observed decrease in LINE-1 methylation levels in the ASD group (Figure 1B and Table 3). Moreover, when analysis was performed with patients with AMR and AMNR, methylation levels of CpG 2 from LINE-1 in the AMR subgroup was significantly lower (71.16 ± 0.60), compared to the healthy control group (72.98 ± 0.52) (Figure 1D and Table 3). Although methylation levels at position 2 of LINE-1 in the ANMR subgroup were also lower than in the control group, this difference was not statistically significant.
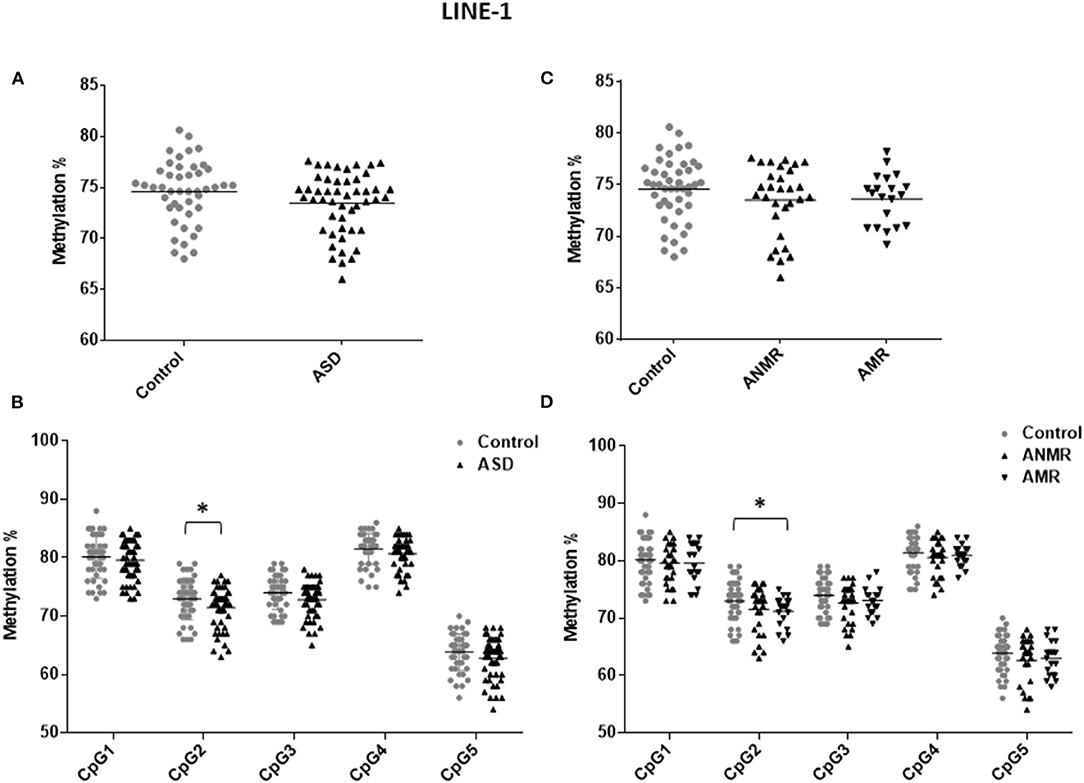
Figure 1. Scatter plot showing comparisons of methylation status of LINE-1 in children with autism spectrum disorders (ASD) compared to controls. (A,B) Methylation levels and differentially methylated sites (CpG1–CpG5) in the LINE-1 sequence in ASD group and healthy control group. (C,D) Methylation levels and differentially methylated sites in the LINE-1 sequence in ASD group subdivided as follows: ANMR (non-mental regression group), AMR (mental regression group), and a healthy control group. The dark bars represent the mean values of each group. Asterisks indicate statistically significant differences (*P ≤ 0.05) compared to control group.
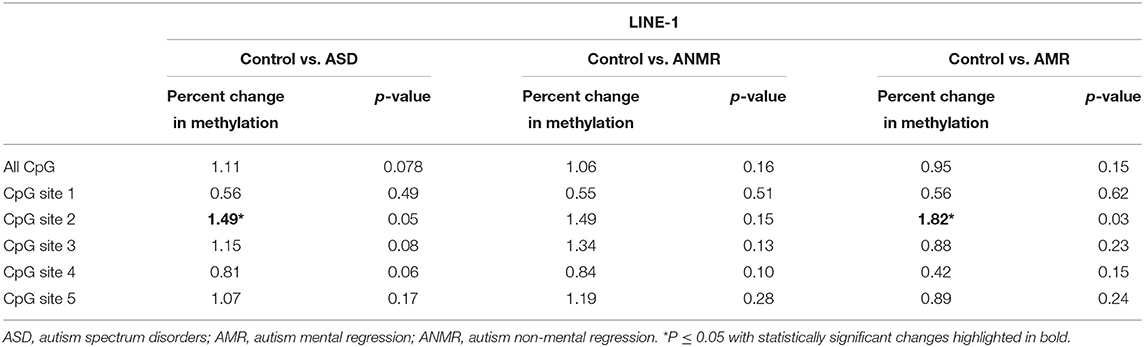
Table 3. Pyrosequencing results of LINE-1 showing percent change in methylation in ASD group, ANMR and AMR subgroups compared to healthy control group.
As expected from slight changes in LINE-1 methylation levels, no clinical correlations were observed between the methylation status of LINE-1 and any of the tests performed to evaluate the diagnosis, severity, and the regressive aspects of the disorder (Supplementary Table 4).
Methylation Status Analysis of NCAM1 and NGF in ASD
DNA methylation status was analyzed for each gene (Figures 2A, 3A), both in the ASD group and in the subgroups ANMR and AMR separately. Pyrosequencing results showed a significant increase in NCAM1 methylation levels in the ASD children and in each subgroup, ANMR and AMR, compared to healthy children (Figures 2B,D and Table 4). Specifically, 70% of ASD individuals presented NCAM1 methylation levels above the mean, with no distinction between AMR and ANMR. In the control group of healthy individuals, only 42% had higher-than-mean NCAM1 methylation levels. In addition, CpG 1 in NCAM1 (UCSC location chr11:112,965,292) was the position that suffered this notable increase in methylation, especially in the AMR subgroup (Figures 2C,E).
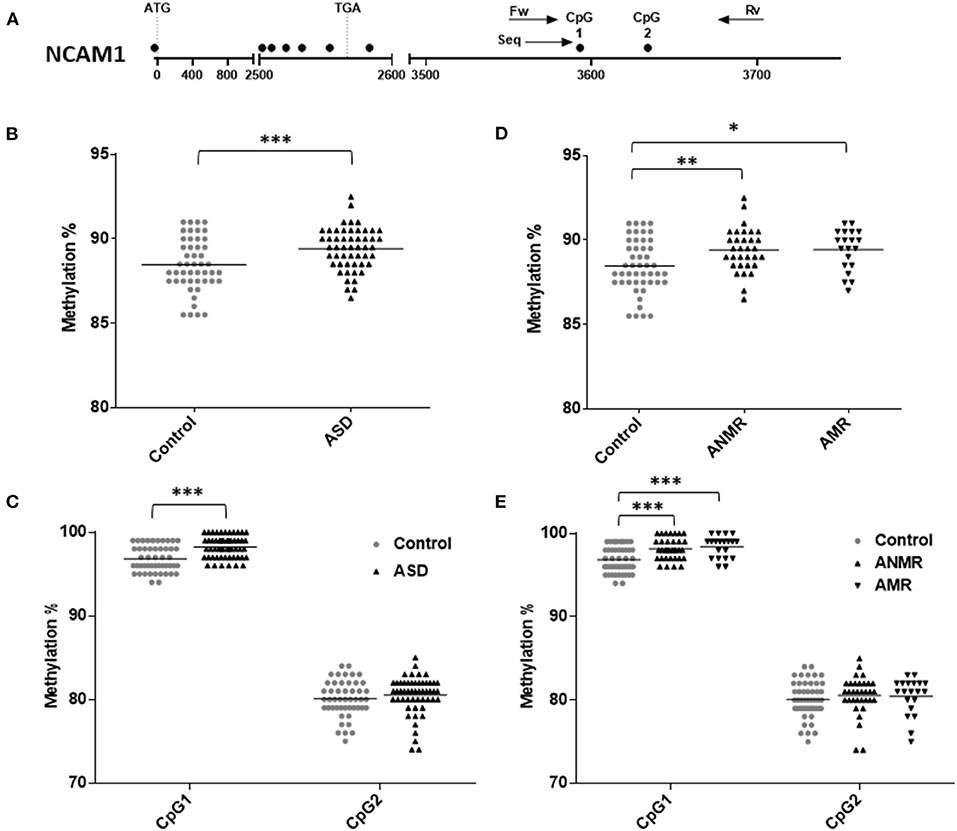
Figure 2. Scatter plot showing comparisons of methylation status of NCAM1 in children with autism spectrum disorders (ASD) compared to controls. (A) Schematic diagram of analyzed gene. Each circle represents a CpG dinucleotide. Position of ATG and TGA codon are indicated. Arrows show the location of pyrosequencing primers. (B,C) Methylation levels and differentially methylated sites (CpG1 and CpG2) in the NCAM1 gene in ASD group and healthy control group. (D,E) Methylation levels and differentially methylated sites in the NCAM1 gene in ASD group subdivided as follows: ANMR (non-mental regression group), AMR (mental regression group), and a healthy control group. The dark bars represent the mean values of each group. Asterisks indicate statistically significant differences (*P < 0.05; **P < 0.01; ***P < 0.001) compared to control group.
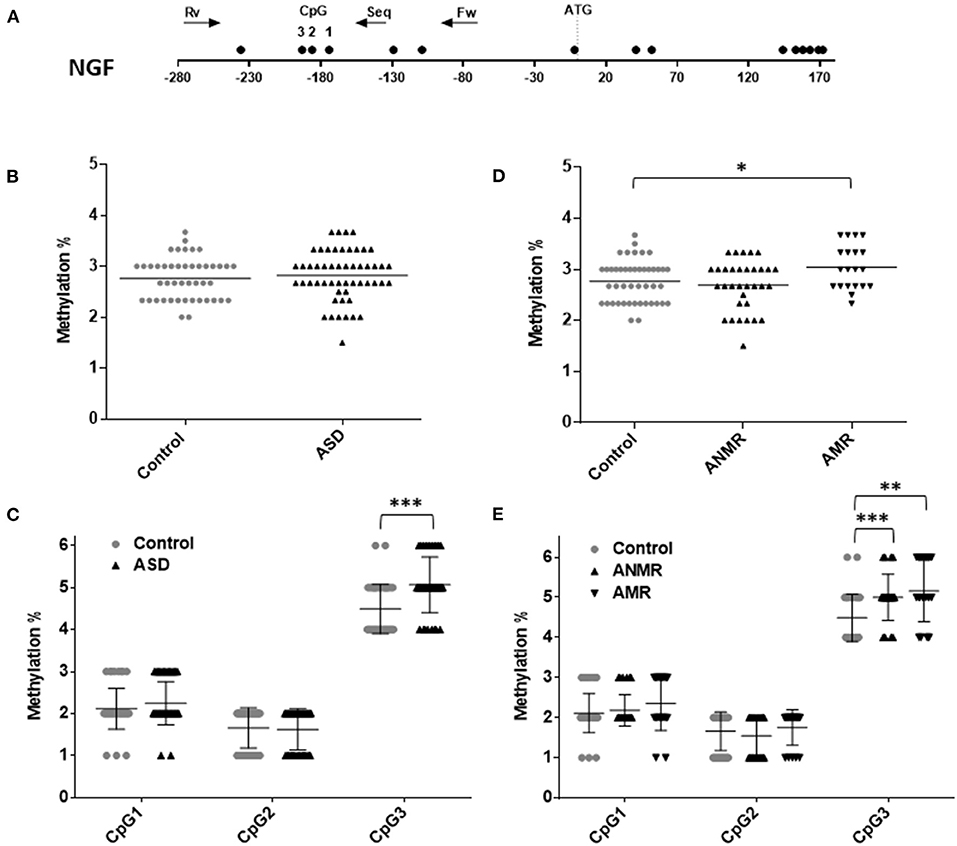
Figure 3. Scatter plot showing comparisons of methylation status of NGF in children with autism spectrum disorders (ASD) compared to controls. (A) Schematic diagram of analyzed gene. Each circle represents a CpG dinucleotide. Position of ATG codon is indicated. Arrows show the location of pyrosequencing primers. (B,C) Methylation levels and differentially methylated sites (CpG 1, 2, and 3) in the NGF gene in ASD group and healthy control group. (D,E) Methylation levels and differentially methylated sites in the NGF gene in ASD group subdivided as follows: ANMR (non-mental regression group), AMR (mental regression group), and a healthy control group. The dark bars represent the mean values of each group. Asterisks indicate statistically significant differences (*P < 0.05; **P < 0.01; ***P < 0.001) compared to control group.
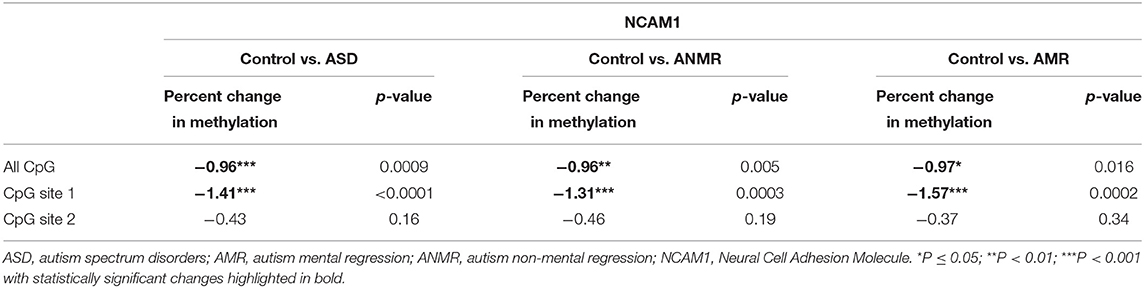
Table 4. Pyrosequencing results of NCAM1 showing percent change in methylation in ASD group, ANMR and AMR subgroups compared to healthy control group.
On the other hand, no differences were observed in the levels of NGF methylation between the ASD group and the healthy group (Figure 3B). Concretely, lower-than-mean levels of NGF methylation were detected in 47% of children diagnosed with ASD and in 53% of neurotypically developing children. Within the ASD group, the AMR subgroup had higher-than-mean NGF methylation levels than both the ANMR subgroup and the normally developing children (Figure 3D). Methylation analysis of each CpG revealed that the CpG at position 3 of the analyzed region (UCSC location chr1: 115,880,705) showed a higher level of methylation in children diagnosed with ASD, with or without mental regression (Figures 3C,E and Table 5).
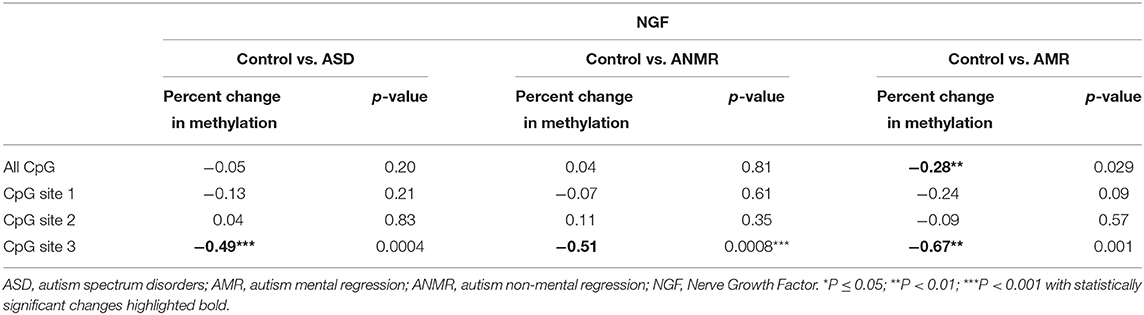
Table 5. Pyrosequencing results of NGF showing percent change in methylation in ASD group, ANMR and AMR subgroups compared to healthy control group.
In the ROC curve, the AUC was calculated to explore differences in LINE-1, NCAM1 and/or NGF methylation between ASD and healthy status (Figure 4). The AUC for NCAM1 was 0.67 (p = 0.02). However, for LINE-1 and NGF, the values resulted not optimal to discriminate between ASD and healthy conditions.
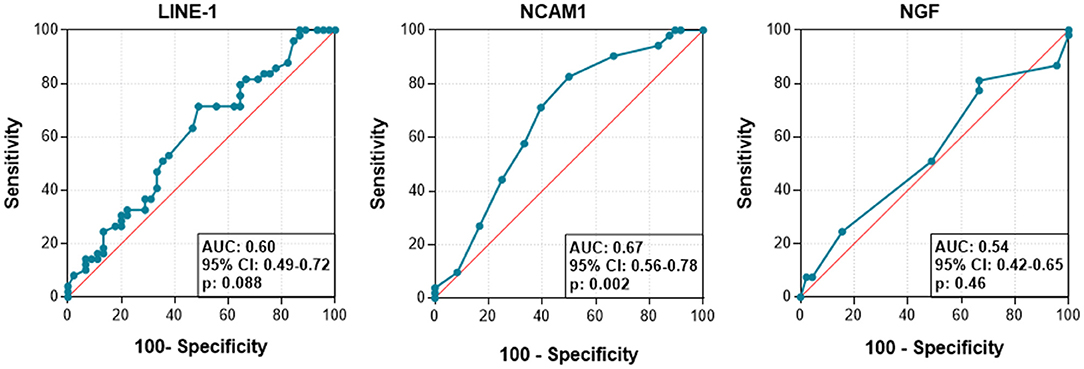
Figure 4. Receiver operator characteristic curves (ROC) and area under the curve (AUC) values to differentiate ASD from healthy condition according to the methylation status of each target.
Discussion
In the present work we report results from a global and local DNA methylation analysis of a pediatric sample with ASD compared to a normally developing children subgroup. ASD is a neurodevelopmental disorder that presumably affects multiple areas of the brain, including the frontal, temporal, and occipital cortex, as well as the cerebellum. Since brain tissue cannot be collected from live individuals, a more accessible tissue sample has been suggested, such as peripheral blood. High correlations have been found between methylation levels of specific genes in the blood and brain regions in children with neurodevelopmental disorders and controls (6, 35, 36).
DNA methylation in non-coding regions, including LINE-1 retrotransposons, which constitutes a substantial portion of the human genome, has been very little investigated in the context of ASD. The first report of an increase in LINE-1 expression in the autism cerebellum was published by Shpyleva et al. (34). These authors carried out a methylated DNA immunoprecipitation (MeDIP) analysis of cytosine-5 methylation in LINE-1, obtaining a downward trend of LINE-1 methylation in ASD, although this difference was not statistically significant, possibly due to heterogeneity within the ASD population. However, they also observed significantly less binding of repressive MeCP2 and histone H3K9me3 to LINE-1 sequences in the autistic cerebellum, suggesting that relaxation of epigenetic repression may have aided in increased expression. Moreover, since the results were obtained from postmortem brain samples, it could not be determined whether these observations were functionally related to the etiology or pathophysiology of autism.
A recent study with 36 ASD subjects and 20 unaffected individuals showed that the LINE-1 methylation levels were significantly reduced in ASD individuals with severe language impairment and were inversely correlated with the transcript level (37). Nevertheless, this study was carried out using lymphoblastoid cell lines derived from peripheral mononuclear cells. Here, we report an analysis of LINE-1 methylation status using blood samples from 50 ASD patients and 45 control individuals. Our results show a slight decrease in the levels of LINE-1 methylation in the ASD group, compared to the normal neurodevelopmental healthy group, with the following mean values: 73.5% in ASD and 74.2% in controls. Although these differences in the pattern of LINE-1 methylation between the ASD group and the control group are not statistically significant, they are higher than those observed in relevant studies also carried out by pyrosequencing in other neurodegenerative diseases. In this regard, Bollati et al. (38) evaluated methylation of ALU, LINE-1 (same CpGs fragment than in the present study), and SAT-α sequences in blood samples from 43 patients with Alzheimer's disease (AD) and 38 healthy donors. LINE-1 methylation increased in AD patients, compared to healthy volunteers, obtaining the following mean methylation values: AD 83.6% and volunteers 83.1% (p-value: 0.05).
Furthermore, our study focused on a LINE-1 fragment containing 5 CpG positions for analysis. Remarkably, we observed a statistically significant decrease in methylation levels at position 2 of the LINE-1 analyzed fragment, which is then reflected in the subgroup of children with mental regression. Thus, while the mean CpG 2 methylation value of LINE-1 in healthy controls is 72.97%, it falls to 71.15% in the subgroup of AMR children (p-value: 0.047). This result should be interpreted as a strong indicator for expanding the LINE-1 study region. Therefore, a more in-depth future study of LINE-1 methylation patterns in patients with ASD and healthy controls with an enlarged analyzed region is needed.
Quantification of DNA methylation in candidate gene promoters using different samples (blood, specific regions in post-mortem brain) from patients with ASD has identified differentially methylated regions (DMR) in OXTR, SHANK3, UBE3A, and MECP2 at CpG sites of specific promoters (5–9). Increasing evidence suggests that growth factors modulate motor, emotional and cognitive functions, which may account for the clinical manifestations of several disorders (39). NGF belongs to the neurotrophin family and is considered a key regulator of the development, differentiation, survival and regeneration of nerve cells. NGF is present mainly in highly functional brain regions. Previous studies from our group of the same cohort of ASD children as this study, as well as other groups of children with this disorder, have shown increased levels of NGF in children with ASD (27). Moreover, in this disorder, different subgroups considering the presence or absence of neurodevelopmental regression have been reported (40, 41). Our results seem to support these notions, since we found different NGF methylation patterns in both subgroups, AMR and ANMR. In this regard, approximately half of both children diagnosed with ASD (47%) and typically developing children (53%) have lower-than-mean levels of NGF methylation.
These results are consistent with those previously observed by our group, in which NGF was detected in < 50% of the children with ASD and typically developing children (27). More specifically, while only 39% of the ANMR subgroup showed increased levels of NGF methylation, this value rose to more than 60% in the AMR subgroup, suggesting a possible higher expression of NGF in the first subgroup. This is precisely what we showed in our previously published results, using samples from the same individuals, where the ANMR subgroup exhibited higher NGF levels than the typically developing children (27), making even more plausible the suggestion that these observations might indicate a disturbance in neuronal development, and pointing to NGF as a possible biomarker in autism disorder.
NCAM1 is a glycoprotein mainly expressed on the surface of nerve cells, in the central and peripheral nervous tissue of vertebrates, and acts as an adhesion molecule between cells and their extracellular environment (14, 41). It plays a critical role in the developmental and plasticity pathways of the nervous system (8). NCAM1 may accumulate in presynaptic and postsynaptic membranes and it has been associated with behavioral phenotypes in ASD children (42). Changes in DNA methylation have not yet been described at the NCAM1 level. In the present study, we reported that when compared to the typically developing children subgroup, higher levels of NCAM1 methylation were observed in children with ASD, without differentiation between the AMR and the ANMR subgroups. Interestingly, and in agreement with these data, our previous studies found lower plasma levels of NCAM1 in the same subgroup of ASD children without neurodevelopmental regression, compared to the levels in the subgroup of typically developing children (27). NCAM1 could be suggested as a possible biomarker, capable of contributing to the diagnostic process of the autistic disorder.
In conclusion, given the high heterogeneity in ASD, it is essential to know the biological factors that trigger this disorder to understand the behavioral variability of the patients. Identification of epigenetic biomarkers based on changes in DNA methylation could help to clarify the pathophysiology of autism, facilitating its diagnosis and prognosis. To date, a consensus has not yet been generated on the gene's specific methylation signature for autism. In this study, we provide new clues about the epigenetic changes that occur in the autistic disorders as well as a clear differentiation in DNA methylation between the ASD subgroups, according to the presence or absence of mental regression. Thus, we have reported a differentially methylated CpG position in the LINE-1 retrotransposon between healthy children and children with ASD. In addition, we have pointed two candidate genes, NGF and NCAM1, as potential epigenetic biomarkers that would facilitate the diagnosis of the disorder. Finally, the findings of this study are in line with previous research and reinforce our understanding of this disorder. However, we consider that further investigation is necessary in this line of research.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The studies involving human participants were reviewed and approved by Ethics Committee of Investigation of Cordoba. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
Author Contributions
MVG-O, TM-R, MT-A, TR-A, RRA, and JP-N were involved in planning and supervised the work. AG-F, KF-R, PM-B, and MT-A processed the clinical data. MVG-O and TM-R processed the experimental data and performed the analysis. MVG-O, TR-A, MG-C, MT-A, and JP-N drafted the manuscript. All authors discussed the results and commented on the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2021.685310/full#supplementary-material
References
1. Menezo YJ, Elder K, Dale B. Link between increased prevalence of autism spectrum disorder syndromes and oxidative stress, DNA methylation, and imprinting: the impact of the environment. JAMA Pediatr. (2015) 169:1066–7. doi: 10.1001/jamapediatrics.2015.2125
2. Fombonne E. Epidemiology of autistic disorder and other pervasive developmental disorders. J Clin Psychiatry. (2005) 66(Suppl. 10):3–8.
3. Yoon SH, Choi J, Lee WJ, Do JT. Genetic and epigenetic etiology underlying autism spectrum disorder. J Clin Med. (2020) 9:966. doi: 10.3390/jcm9040966
4. Wen Z, Cheng TL, Li GZ, Wen Z, Cheng TL, Li GZ, et al. Identification of autism-related MECP2 mutations by whole-exome sequencing and functional validation. Mol Autism. (2017) 8:43. doi: 10.1186/s13229-017-0157-5
5. Elagoz Yuksel M, Yuceturk B, Faruk Karatas O, Ozen M, Dogangun B. The altered promoter methylation of oxytocin receptor gene in autism. J Neurogenet. (2016) 30:280–4. doi: 10.1080/01677063.2016.1202951
6. Gregory SG, Connelly JJ, Towers AJ, Johnson J, Biscocho D, Markunas CA, et al. Genomic and epigenetic evidence for oxytocin receptor deficiency in autism. BMC Med. (2009) 7:62. doi: 10.1186/1741-7015-7-62
7. Jiang YH, Sahoo T, Michaelis RC, Bercovich D, Bressler J, Kashork CD, et al. A mixed epigenetic/genetic model for oligogenic inheritance of autism with a limited role for UBE3A. Am J Med Genet A. (2004) 131:1–10. doi: 10.1002/ajmg.a.30297
8. Nagarajan RP, Hogart AR, Gwye Y, Martin MR, LaSalle JM. Reduced MeCP2 expression is frequent in autism frontal cortex and correlates with aberrant MECP2 promoter methylation. Epigenetics. (2006) 1:e1–11. doi: 10.4161/epi.1.4.3514
9. Zhu L, Wang X, Li XL, Towers A, Cao X, Wang P, et al. Epigenetic dysregulation of SHANK3 in brain tissues from individuals with autism spectrum disorders. Hum Mol Genet. (2014) 23:1563–78. doi: 10.1093/hmg/ddt547
10. Tremblay MW, Jiang YH. DNA methylation and susceptibility to autism spectrum disorder. Annu Rev Med. (2019) 27:151–66. doi: 10.1146/annurev-med-120417-091431
11. Bailey A, Le Couteur A, Gottesman I, Bolton P, Simonoff E, Yuzda E, et al. Autism as a strongly genetic disorder: evidence from a British twin study. Psychol Med. (1995) 25:63–77. doi: 10.1017/S0033291700028099
12. Goldberg AD, Allis CD, Bernstein E. Epigenetics: a landscape takes shape. Cell. (2007) 23:635–8. doi: 10.1016/j.cell.2007.02.006
13. Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. (2012) 29:484–92. doi: 10.1038/nrg3230
14. Vogel Ciernia A, LaSalle J. The landscape of DNA methylation amid a perfect storm of autism aetiologies. Nat Rev Neurosci. (2016) 17:411–23. doi: 10.1038/nrn.2016.41
15. Grayson DR, Guidotti A. Merging data from genetic and epigenetic approaches to better understand autistic spectrum disorder. Epigenomics. (2016) 8:85–104. doi: 10.2217/epi.15.92
16. Keil KP, Lein PJ. DNA methylation: a mechanism linking environmental chemical exposures to risk of autism spectrum disorders. Environ Epigenet. (2016) 2:dvv012. doi: 10.1093/eep/dvv012
17. Loke YJ, Hannan AJ, Craig JM. The role of epigenetic change in autism spectrum disorders. Front Neurol. (2015) 26:107. doi: 10.3389/fneur.2015.00107
18. Siu MT, Weksberg R. Epigenetics of autism spectrum disorder. Adv Exp Med Biol. (2017) 978:63–90. doi: 10.1007/978-3-319-53889-1_4
19. Wiśniowiecka-Kowalnik B, Nowakowska BA. Genetics and epigenetics of autism spectrum disorder-current evidence in the field. J Appl Genet. (2019) 60:37–47. doi: 10.1007/s13353-018-00480-w
20. Ladd-Acosta C, Hansen KD, Briem E, Fallin MD, Kaufmann WE, Feinberg AP. Common DNA methylation alterations in multiple brain regions in autism. Mol Psychiatry. (2014) 19:862–71. doi: 10.1038/mp.2013.114
21. Andrews SV, Ellis SE, Bakulski KM, Sheppard B, Croen LA, Hertz-Picciotto I, et al. Cross-tissue integration of genetic and epigenetic data offers insight into autism spectrum disorder. Nat Commun. (2017) 24:1011. doi: 10.1101/091330
22. Andrews SV, Sheppard B, Windham GC, Schieve LA, Schendel DE, Croen LA, et al. Case-control meta-analysis of blood DNA methylation and autism spectrum disorder. Mol Autism. (2018) 28:40. doi: 10.1101/320622
23. De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. (2014) 13:209–15. doi: 10.1038/nature13772
24. Cordaux R, Batzer MA. The impact of retrotransposons on human genome evolution. Nat Rev Genet. (2009) 10:691–703. doi: 10.1038/nrg2640
25. Han JS, Szak ST, Boeke JD. Transcriptional disruption by the L1 retrotransposon and implications for mammalian transcriptomes. Nature. (2004) 20:268–74. doi: 10.1038/nature02536
26. Kitkumthorn N, Mutirangura A. Long interspersed nuclear element-1 hypomethylation in cancer: biology and clinical applications. Clin Epigenetics. (2011) 2:315–30. doi: 10.1007/s13148-011-0032-8
27. Gomez-Fernandez A, de la Torre-Aguilar MJ, Gil-Campos M, Flores-Rojas K, Cruz-Rico MD, Martin-Borreguero P, et al. Children with autism spectrum disorder with regression exhibit a different profile in plasma cytokines and adhesion molecules compared to children without such regression. Front Pediatr. (2018) 26:264. doi: 10.3389/fped.2018.00264
28. APA. DSM-V. (2013). Available online at: http://www.dsm5.org/Pages/Default.aspx
29. Beck CR, Garcia-Perez JL, Badge RM, Moran JV. LINE-1 elements in structural variation and disease. Annu Rev Genomics Hum Genet. (2011) 12:187–215. doi: 10.1146/annurev-genom-082509-141802
30. Plioplys AV, Hemmens SE, Regan CM. Expression of a neural cell adhesion molecule serum fragment is depressed in autism. J Neuropsychiatry Clin Neurosci. (1990) 2:413–7. doi: 10.1176/jnp.2.4.413
31. Purcell AE, Rocco MM, Lenhart JA, Hyder K, Zimmerman AW, Pevsner J. Assessment of neural cell adhesion molecule (NCAM) in autistic serum and postmortem brain. J Autism Dev Disord. (2001) 31:183–94. doi: 10.1023/A:1010751232295
32. Guney E, Ceylan MF, Kara M, Tekin N, Goker Z, Senses Dinc G, et al. Serum nerve growth factor (NGF) levels in children with attention deficit/hyperactivity disorder (ADHD). Neurosci Lett. (2014) 7:107–11. doi: 10.1016/j.neulet.2013.12.026
33. Anton L, Brown AG, Bartolomei MS, Elovitz MA. Differential methylation of genes associated with cell adhesion in preeclamptic placentas. PLoS ONE. (2014) 25:e100148. doi: 10.1371/journal.pone.0100148
34. Shpyleva S, Melnyk S, Pavliv O, Pogribny I, Jill James S. Overexpression of LINE-1 retrotransposons in autism brain. Mol Neurobiol. (2018) 55:1740–9. doi: 10.1007/s12035-017-0421-x
35. Murphy BC, O'Reilly RL, Singh SM. Site-specific cytosine methylation in S-COMT promoter in 31 brain regions with implications for studies involving schizophrenia. Am J Med Genet B Neuropsychiatr Genet. (2005) 5:37–42. doi: 10.1002/ajmg.b.30134
36. Kaminsky Z, Tochigi M, Jia P, Pal M, Mill J, Kwan A, et al. A multi-tissue analysis identifies HLA complex group 9 gene methylation differences in bipolar disorder. Mol Psychiatry. (2012) 17:728–40. doi: 10.1038/mp.2011.64
37. Tangsuwansri C, Saeliw T, Thongkorn S, Chonchaiya W, Suphapeetiporn K, Mutirangura A, et al. Investigation of epigenetic regulatory networks associated with autism spectrum disorder (ASD) by integrated global LINE-1 methylation and gene expression profiling analyses. PLoS ONE. (2018) 23:13:e0201071. doi: 10.1371/journal.pone.0201071
38. Bollati V, Galimberti D, Pergoli L, Dalla Valle E, Barretta F, Cortini F, et al. DNA methylation in repetitive elements and Alzheimer disease. Brain Behav Immun. (2011) 25:1078–83. doi: 10.1016/j.bbi.2011.01.017
39. Galvez-Contreras AY, Campos-Ordonez T, Gonzalez-Castaneda RE, Gonzalez-Perez O. Alterations of growth factors in autism and attention-deficit/hyperactivity disorder. Front Psychiatry. (2017) 8:126. doi: 10.3389/fpsyt.2017.00126
40. Kern JK, Geier DA, Geier MR. Evaluation of regression in autism spectrum disorder based on parental reports. N Am J Med Sci. (2014) 6:41–7. doi: 10.4103/1947-2714.125867
41. Xi CY, Hua TY, Zhao YJ, Liu XM. Characteristics of developmental regression in autistic children. Zhongguo Dang Dai Er Ke Za Zhi. (2010) 12:781–3.
Keywords: autism spectrum disorder, neurodevelopmental regression, DNA methylation patterns, NCAM, NGF
Citation: García-Ortiz MV, Torre-Aguilar MJ, Morales-Ruiz T, Gómez-Fernández A, Flores-Rojas K, Gil-Campos M, Martin-Borreguero P, Ariza RR, Roldán-Arjona T and Perez-Navero JL (2021) Analysis of Global and Local DNA Methylation Patterns in Blood Samples of Patients With Autism Spectrum Disorder. Front. Pediatr. 9:685310. doi: 10.3389/fped.2021.685310
Received: 24 March 2021; Accepted: 06 September 2021;
Published: 05 October 2021.
Edited by:
Chen Li, Zhejiang University, ChinaReviewed by:
Maud de Dieuleveult, Assistance Publique Hopitaux de Paris, FranceMaria Paola Lombardi, University of Amsterdam, Netherlands
Copyright © 2021 García-Ortiz, Torre-Aguilar, Morales-Ruiz, Gómez-Fernández, Flores-Rojas, Gil-Campos, Martin-Borreguero, Ariza, Roldán-Arjona and Perez-Navero. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mercedes Gil-Campos, bWVyY2VkZXNfZ2lsX2NhbXBvc0B5YWhvby5lcw==
†These authors have contributed equally to this work