 Jiashuo Li
Jiashuo Li Zhenyi Liu2†
Zhenyi Liu2† Gongming Zhang
Gongming Zhang Xue Yin
Xue Yin Xiaoxue Yuan
Xiaoxue Yuan Xiaoyan Ding
Xiaoyan Ding
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol. , 18 March 2025
Sec. Pharmacology of Anti-Cancer Drugs
Volume 15 - 2025 | https://doi.org/10.3389/fonc.2025.1570647
This article is part of the Research Topic Demystifying Fundamental Mechanisms of anticancer drug resistance in Gastrointestinal Oncology View all articles
Background: The tumor microenvironment (TME) plays a critical role in the development, progression, and clinical outcomes of hepatocellular carcinoma (HCC). Despite the critical role of natural killer (NK) cells in tumor immunity, there is limited research on their status within the tumor microenvironment of HCC. In this study, single-cell RNA sequencing (scRNA-seq) analysis of HCC datasets was performed to identify potential biomarkers and investigate the involvement of natural killer (NK) cells in the TME.
Methods: Single-cell RNA sequencing (scRNA-seq) data were extracted from the GSE149614 dataset and processed for quality control using the “Seurat” package. HCC subtypes from the TCGA dataset were classified through consensus clustering based on differentially expressed genes (DEGs). Weighted gene co-expression network analysis (WGCNA) was employed to construct co-expression networks. Furthermore, univariate and multivariate Cox regression analyses were conducted to identify variables linked to overall survival. The single-sample gene set enrichment analysis (ssGSEA) was used to analyze immune cells and the screened genes.
Result: A total of 715 DEGs from GSE149614 and 864 DEGs from TCGA were identified, with 25 overlapping DEGs found between the two datasets. A prognostic risk score model based on two genes was then established. Significant differences in immune cell infiltration were observed between high-risk and low-risk groups. Immunohistochemistry showed that HRG expression was decreased in HCC compared to normal tissues, whereas TUBA1B expression was elevated in HCC.
Conclusion: Our study identified a two-gene prognostic signature based on NK cell markers and highlighted their role in the TME, which may offer novel insights in immunotherapy strategies. Additionally, we developed an accurate and reliable prognostic model, combining clinical factors to aid clinicians in decision-making.
Primary liver cancer is one of the most common malignant tumors, the sixth most prevalent cancer in the world, and the second leading cause of cancer-related deaths (1, 2). Hepatocellular carcinoma is the predominant type of liver cancer (80%-90%). The majority of patients with HCC are diagnosed during the late stages of the disease. Clinical benefit is limited in patients with advanced HCC, with a median overall survival (OS) of only 1-1.5 years (3, 4). In terms of treatment, the primary methods for liver cancer treatment include surgical resection, local ablation, transarterial chemoembolization (TACE), and liver transplantation. In recent years, immunotherapy has also shown potential in the treatment of HCC, providing patients with new treatment options. In addition, immune-related genes play an important role in tumour immunotherapy. Tumor microenvironment (TME) refers to the surrounding environment in which tumor cells exist (5, 6). Nowadays, it is widely accepted that the components of the TME, such as immune cells and inflammatory cells, play a pivotal role in tumor development, progression, and clinical outcomes (7–9). Consequently, alterations in the TME have an impact on patient prognosis, making it critical to identify TME biomarkers to predict patient outcomes (10, 11).
Natural killer (NK) cells are cytotoxic lymphocytes and components of innate immunity, capable of recognizing and eliminating damaged or stressed cells (12). In the liver, NK cells make up 30-50% of intrahepatic lymphocytes (13). Immune cells are able to directly kill tumor cells and promote T-cell immune responses in tumor immunity, thereby inhibiting the occurrence and development of cancer (14, 15). In many types of cancer, the infiltration level of NK cells is reduced, including gastric, esophageal, breast, and colon cancers (16–18). However, studies on NK cells in patients with HCC are scarce. Moreover, high levels of infiltrated NK cells in tumor tissues are associated with better prognosis in patients with cancer (19, 20). Therefore, in-depth characterization of NK cell phenotypes and functionality is crucial for understanding liver cancer and could help determine therapeutic strategies for HCC patients.
Traditional bulk RNA sequencing (bulk RNA-seq) averages the transcriptional profiles of cells in one sample, which is strongly influenced by cell type dominance. In addition, bulk RNA-seq is unable to effectively distinguish between different cell lineages and cellular interactions. The advent of single-cell mRNA sequencing (scRNA-seq) has broadened the understanding of cellular components and gene expression specificities in the TME (21). Single-cell sequencing is a technique that allows the analysis of gene expression at the individual cell level. It involves isolating single cells, amplifying their RNA or DNA, and sequencing the genetic material to uncover cellular heterogeneity and molecular profiles. ScRNA-seq highlight heterogeneity and distinct subpopulations within tumors, allowing for enumeration and quantification of immune infiltration in tumor tissue (22, 23). The heterogeneous immune cell infiltrates are a crucial factor for treatment response and prognosis in HCC and other tumor types (24–26). The majority of scRNA-seq studies on immune cells derived from HCC tissues have predominantly focused on T cell characteristics, with limited research dedicated to NK cells (25, 27). Consequently, there is an urgent need for scRNA-seq analysis of NK cells from both healthy liver and HCC tissues to uncover NK cell-related prognostic genes that contribute to a deeper understanding of HCC prognosis.
In this study, different cell subsets between tumor tissues and normal control tissues were identified from the HCC single-cell dataset of the Gene Expression Omnibus (GEO) database. In conjunction with bulk RNA-seq analysis of The Cancer Genome Atlas (TCGA) cohort and the International Cancer Genome Consortium (ICGC) cohort, NK cell marker gene-related features for predicting HCC prognosis were created, and a model was built with clinical indicators and NK cell risk profiles. Our study expanded the exploration of HCC and contributed novel insights into HCC diagnosis, therapies, and prognosis.
The gene expression dataset (GSE149614) consists of single-cell samples from 10 HCC patients, including ten primary tumor samples and eight adjacent normal samples. The raw data contained a total of 25479 genes and 71915 cells. The Seurat function from the R package was employed to ensure each gene was expressed in a minimum of 3 cells and each cell had at least 250 genes expressed to filter single cells. The percentage of mitochondria and rRNA quantities were calculated using the “PercentageFeatureSet” function. Cells with less than 200 genes, a percentage of mitochondrial reads less than 25%, and a median UMI count of less than 500 were excluded from the downstream analysis. After filtering, 53293 cells were retained.
The RNA-seq transcriptome information and matching clinical data were downloaded from the TCGA database (https://portal.gdc.cancer.gov/) and ICGC database (https://dcc.icgc.org/).
The “SCTransform” method was adopted for data normalization, and the “FindVariableFeatures” function was applied to identify the top 2000 highly variable genes (HVGs) (28). The integrated data were scaled by the “Sclae Data” function, and principal component analysis (PCA) was performed using the “Run PCA” function for 2000 HVGs. The “FindNeighbors” and “FindClusters” functions were implemented to find cell clusters when dim=50 and resolution=0.4 (29, 30). Next, we selected the top 20 principal components in order to further reduce the dimensionality using the UMAP method. UMAP is a method of data dimensionality reduction, which assumes that the available data samples are uniformly distributed in a topological space (Manifold), and these limited data samples can be approximated (Approximation) and mapped (Projection) to a lower dimensional space. Differential genes (DEGs) in different cell types were identified by setting logfc=0.5, minpct=0.25, and adjusting P<0.05 in Seurat’s Findallmarker function. Cell cluster annotation was based on marker genes obtained from the literature and the Cellmarker Database.
DEGs in the dataset GSE149614 were identified and confirmed using the limma package, which applies linear models to detect genes that show significant changes in expression across different experimental conditions. In the TCGA dataset, the limma, DESeq2, and edgeR packages were utilized. Each of these tools employs different statistical approaches for identifying DEGs, such as linear models, data normalization and dispersion estimation, and negative binomial distribution for RNA-seq count data. By using these methods, we ensured a comprehensive and reliable analysis of the differentially expressed genes across the dataset, leading to more accurate results.
The DEGs in the TCGA-LIHC obtained by the three algorithms were taken to be intersected. We determined a total of 356 up-regulated and 168 down-regulated genes. Consistent clustering analysis was performed in TCGA-LIHC samples by using the “ConsensusClusterPlus” R package to identify molecular subtypes. Pam arithmetic and “Spearman” distance were utilized to complete 500 bootstraps, with every bootstrap having specimens (≥80%) of the TCGA-LIHC dataset. Cluster number k was between 2 and 10, and optimal clusters were screened by cumulative distribution function (CDF) curve and consensus CDF. Survival differences among the molecular subtypes were estimated by Kaplan-Meier (K-M) curves using the log-rank test. Additionally, differences in the distribution of clinical characteristics between molecular subtypes were compared with chi-square tests.
The gene expression data profiles of TCGA were constructed for gene co-expression networks using the WCGNA package in R, including module identification, network generation, gene screening, calculation of properties, and data visualization. Correlations between gene pairs were first calculated using gene expression profiling and transformed into a collocation matrix. Then, the soft threshold was set to make network construction among the genes in the network obey scale-free networks, and the adjacency matrix was transformed into a topological overlap matrix (TOM). Branches of the cluster tree and different colors represent different gene modules. The correlation between module eigengenes and clinical traits was assessed using the Pearson correlation test to identify the significant modules.
By analyzing the intersection of prognosis-related genes and DEGs, the death differential genes related to the prognosis of HCC were finally obtained (31). Univariate and multivariate COX analyses were employed to screen for genes with P<0.05 as prognostically relevant genes for HCC and to establish risk regression scores. According to the predictive risk scores, patients were divided into high and low-risk groups to explore the survival difference (32).
After combining risk scores and clinical factors, a nomogram was established. We can accurately predict 1-, 3-, and 5-year survival in HCC patients by calculating cumulative scores based on individual factors. The predictive ability and accuracy of the nomogram were assessed by the ROC curve, calibration curve, and DCA curve.
The relative infiltration of immune cell types in the tumor microenvironment was assessed by Single Sample Gene Set Enrichment Analysis (ssGSEA), including monocyte, central memory CD4 T cell, CD56dim natural killer cell, plasmacytoid dendritic cell, central memory CD8 T cell, immature dendritic cell, natural killer cell, activated dendritic cell, gamma delta T cell, CD56bright natural killer cell, memory B cell, MDSC, T follicular helper cell, activated CD8 T cell, Effector memory CD8 T cell, Type 1 T helper cell, Type T helper cell, natural killer T cell, regulatory T cell, effector memory CD4 T cell, activated CD4 T cell, Type 17 T helper cell, macrophage, immature B cell, mast cell, eosinophil, neutrophil, and activated B cell. Expression data were used to further analyze the correlation between the screened genes and immune cells.
For IHC staining, HCC tissues were fixed in 4% paraformaldehyde and embedded in paraffin blocks, followed by a process of dewaxing and rehydration (33, 34). Peroxidase activity was blocked with 3% hydrogen peroxide. Sections were incubated overnight at four°C with primary antibodies (anti-HRG, anti-TUBAB1) purchased from Abcam. The tissue sections were treated with biotinylated secondary antibody, then stained with diaminobenzidine substrate-chromogen solution, and counterstained with hematoxylin. The images were captured in the XSP-CD204 microscope. It helps validate the expression, localization, and distribution of proteins within tissues, providing insight into the molecular characteristics of diseases, such as cancer, and confirming the presence of specific biomarkers for diagnostic or research purposes.
All statistical analyses were conducted using R software (version 4.1.3). Continuous variables were compared using the Mann-Whitney U or Kruskal-Wallis test, and categorical variables were compared using the Chi-square test or Fisher’s Exact test. Correlations were examined using Spearman rank analysis. Statistical significance was claimed for P<0.05 (two-sided).
By executing the Seurat function and PercentageFeatureSet function, a total of 53293 cells were screened in the scRNA GSE149614 dataset. The quality control before and after filtration was displayed (Supplementary Figure S1). After log-normalization and dimensionality reduction of the data, we obtained the distribution of the nine cell clusters by UMAP visualization (Figures 1A, B). The results of the significant genes with the top20 in rank change values are list in Supplementary Table S1. These cells were manually annotated by cell type based on marker expression, involving NK cells (NKG7, KLRD1), Macrophages (CD68), Epithelial cells, plasma cells (CD138), T cells (CD3D+, CD3E), Endothelial cells (CD31), and B cells (CD79A, MS4A6A), Dendritic Cells (CD141), monocytes (CD14) (Figures 1C–G, Supplementary Figure S2). Comparative the tumor samples from patients HCC01, HCC02, HCC03, HCC04, HCC05, HCC06, HCC07, HCC08, HCC09, HCC10 and adjacent non-tumor tissues from HCC03, HCC04, HCC05, HCC06, HCC07, HCC08, HCC09, HCC 10, it was found that the proportion of NK cells in the tumor tissues was significantly lower than in the adjacent non-tumor tissues (Figure 1H). Figure 1I demonstrated the DEGs between the tumor and para-cancer in NK cells.
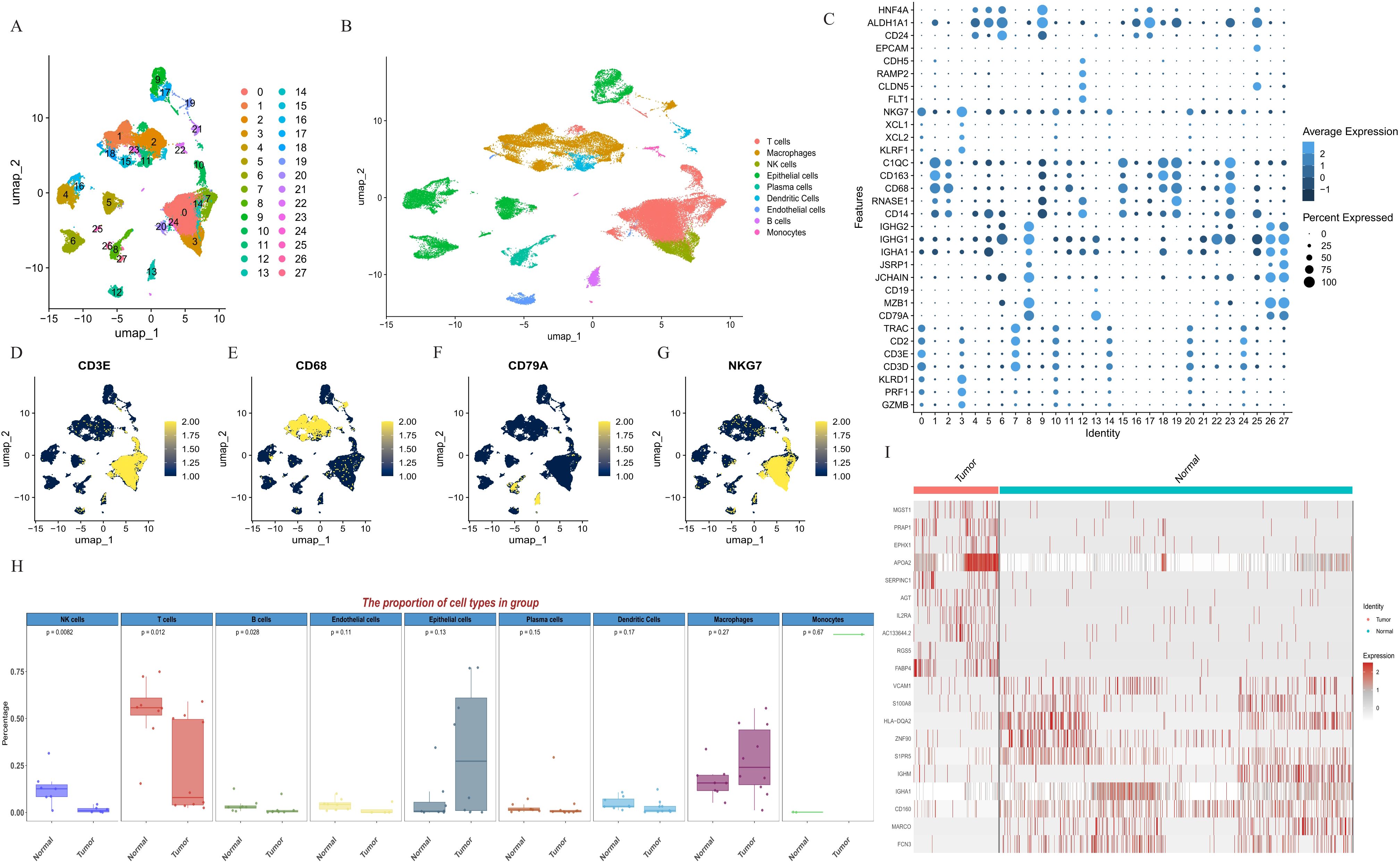
Figure 1. Definition of cell clusters. (A) U-MAP of 27 cell subgroups. (B) U-MAP of nine cell types. (C) The expression of major marker genes in 27 cell clusters. (D) The expression of CD3E in the 27 clusters. (E) The expression of CD68 in the 27 clusters. (F) The expression of CD79A in the 27 clusters. (G) The expression of NKG7 in the 27 clusters. (H) The proportion of cell types in tumor tissues and para-cancer tissues. (I) Differential genes associated with NK cells. NK cells, natural killer cells; U-MAP, uniform manifold approximation and projection.
To investigate the cause of hepatocellular carcinoma development at the molecular level, we performed differential analysis by calling the dataset from the TCGA database and using the Deseq2 package, edgeR package, and limma package (Figure 2A). The common set of differentially expressed genes is represented by the intersection of the three sets of genes, including 356 up-regulated and 168 down-regulated genes (524 genes in total, Figures 2B, C, Supplementary Table S2). Based on the expression of these 524 DEGs, we clustered the 360 samples from TCGA-LIHC using the ConsencusClusterPlus package, and according to the cumulative distribution function (CDF) and incremental area, two clusters (Cluster 1 and Cluster 2) were obtained when K=2 (Figure 2D–F, Supplementary Figure S3). K-M analysis revealed that patients in Cluster 2 had a markedly worse overall survival (OS) than patients in Cluster 1 (Figure 2G).
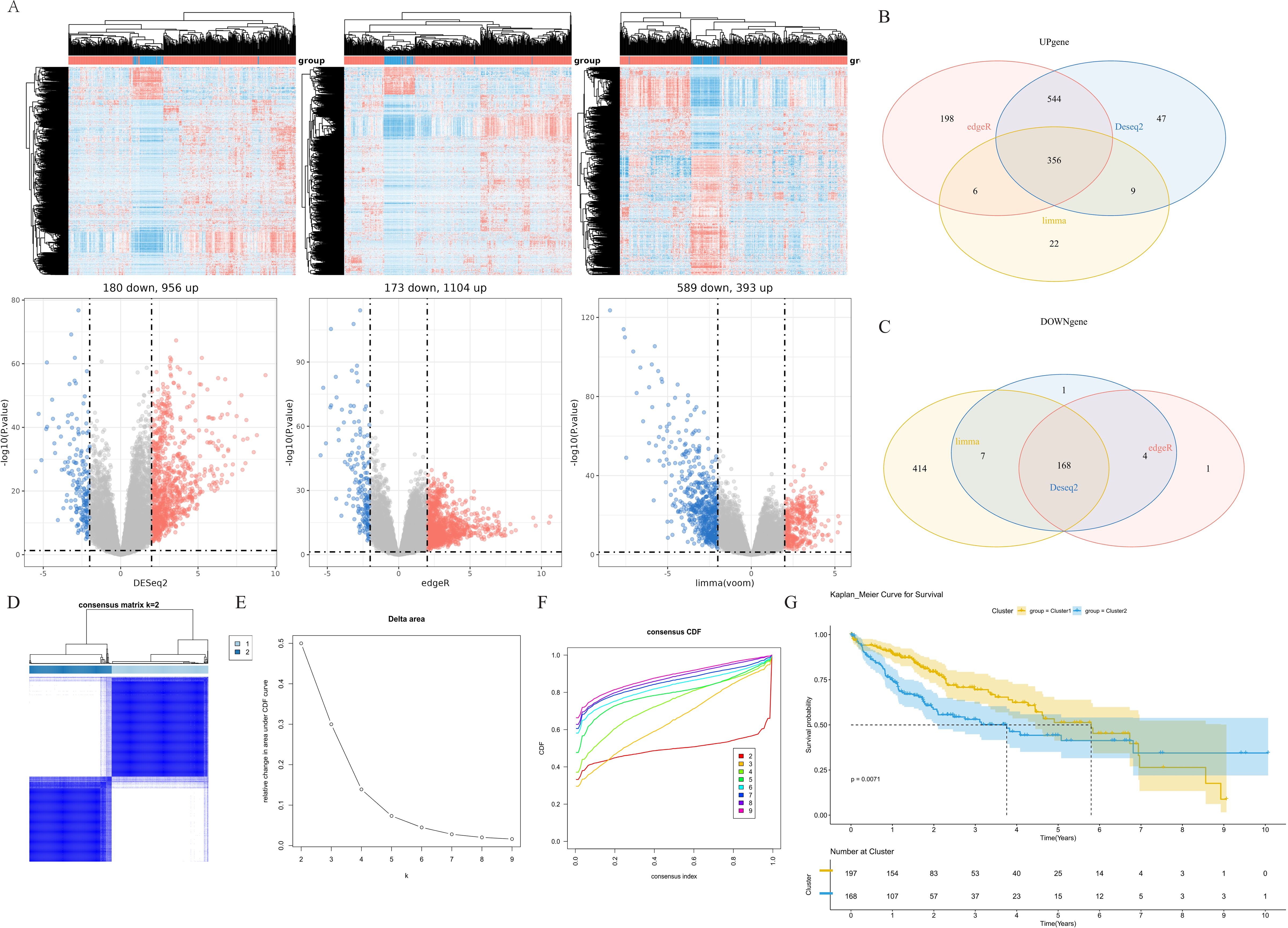
Figure 2. Identification of molecular subtypes. (A) Differential genes associated with different groups in TCGA cohort. (B) Venn diagram of upregulated differential genes. (C) Venn diagram of downregulated differential genes. (D) Heatmap of sample clustering when K=2. (E) Delta area. (F) Cumulative distribution function. (G) K-M survival analysis of cluster 1 and cluster 2 in the TCGA-LIHC dataset. TCGA, The Cancer Genome Atlas; K-M, Kaplan-Meier.
To further analyze the correlation between gene expression patterns and distinct cell subgroups in HCC, we performed the WGCNA method to construct key modules based on the two clusters, and the results of the hierarchical cluster analysis of all samples are presented in Figure 3. We constructed a co-expression network in HCC by calculating the Pearson correlation coefficient in the two clusters based on the WCGNA analysis (Figure 3D). The Pearson correlation test was used to assess the relationship between module eigengenes and clinical traits, helping to identify the significant modules. Modules with a p-value less than 0.05 were considered significant. The hierarchical clustering was applied to cluster genes, and a total of 6 modules were obtained. Among them, the turquoise module was the most relevant module with a prognosis (P<1e-200, Figure 3E). Then, we selected the intersection of highly expressed genes in NK cell lines and genes in the turquoise module in WCGNA analysis for subsequent analysis (Figure 3F). A total of 25 prognosis-related candidate genes were identified.
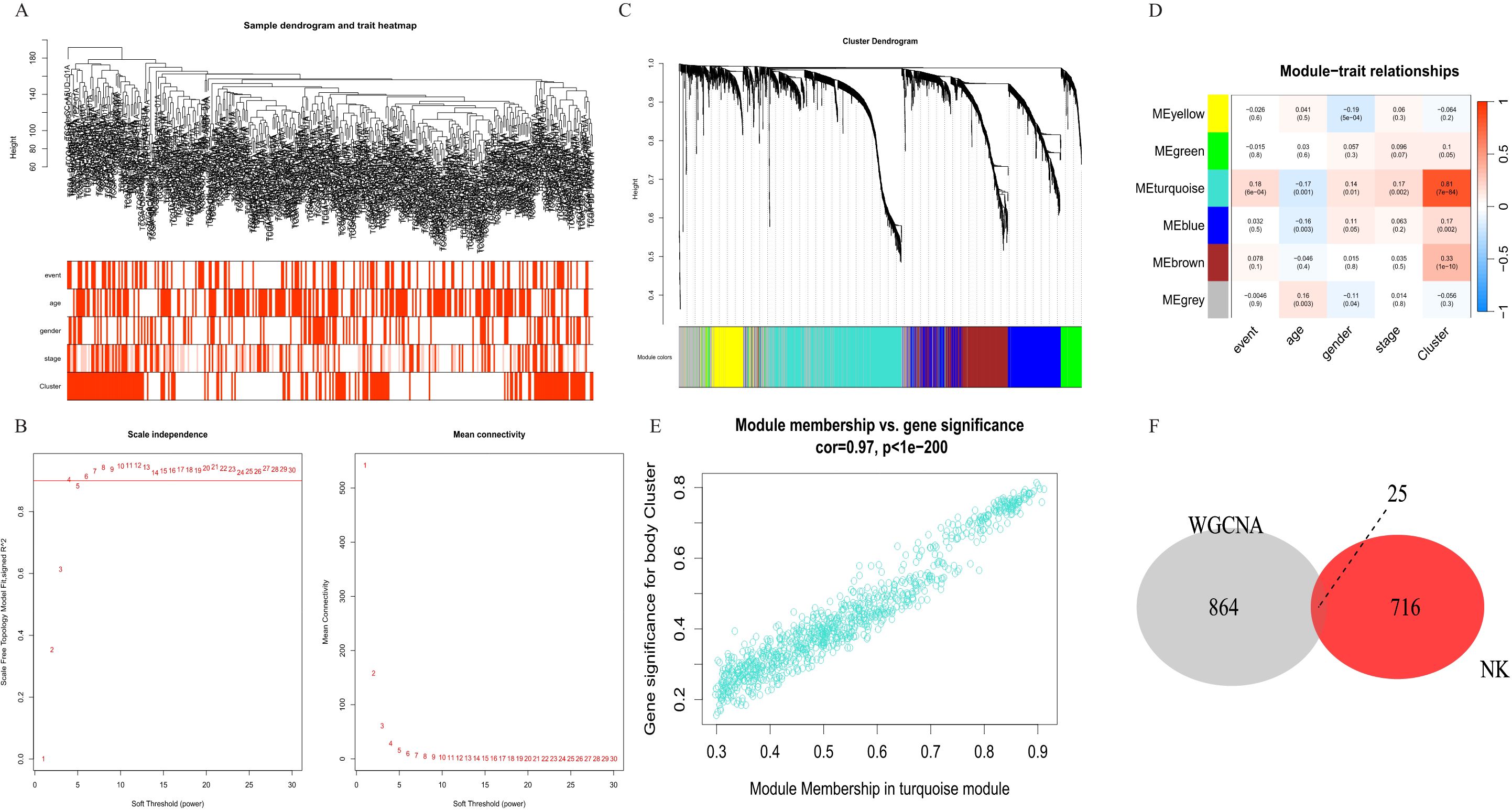
Figure 3. Identification of co-expression modules in HCC. (A) Sample clustering to detect outliers. (B) The scale-free fit index for soft-thresholding powers. (C) Constructing a gene dendrogram based on different metrics. (D) Heatmap of the correlation between 6 modules and clinical characteristics. (E) The correlation between turquoise module and prognostic cluster. (F) Venn diagram of NK cell differential genes and turquoise module. NK cells, natural killer cells; HCC, hepatocellular carcinoma.
HRG and TUBAIB were screened as prognostically relevant genes for HCC by univariate and multivariate COX regression analysis (Figures 4A, B), and the risk score model was established (Figure 4E). Results at both TCGA and ICGC cohorts showed that HRG gene indicated association with better prognosis while TUBA1B indicates association with poor prognosis (Figures 4C, D, Supplementary Figure S4). Patients were assorted into high-risk and low-risk groups in accordance with the median value of risk scores. Survival analysis exhibited significant discrimination in OS among the groups (P<0.001, Figure 4F).
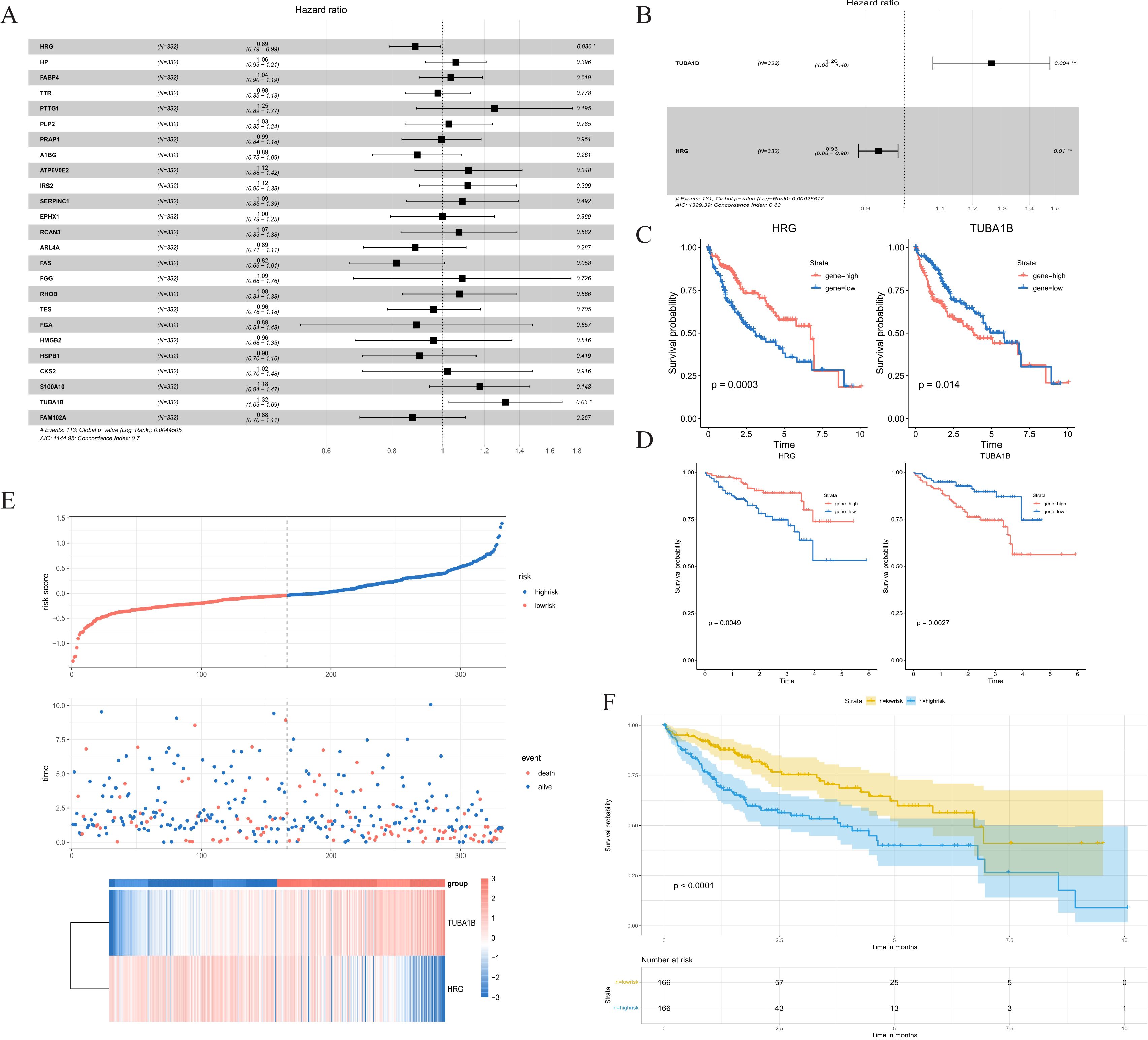
Figure 4. NK cell signature establishment. (A) Univariate Cox analysis of screened genes. (B) Multivariate Cox analysis of screened genes. (C) K-M curve compares the overall HCC patients in the TCGA cohort. (D) K-M curve compares the overall HCC patients in the ICGC cohort. (E) Distribution of risk scores and patient survival between low and high-risk groups in the TCGA cohort. (F) KM curve compares the overall HCC patients between high-risk and low-risk groups in the TCGA cohort. NK cells, natural killer cells; K-M, Kaplan-Meier; TCGA, The Cancer Genome Atlas.
We subsequently examined the association between risk scores and clinical variables, which were statistically different for group, event, and gender. The nomogram was constructed by risk score, age, stage, and gender (Figure 5A). The ROC curves produced the 1-, 3-, and 5-year area under the curve (AUCs) were 0.88, 0.89, and 0.89 (Figure 5B). The calibration curve showed that the predicted value of the nomogram was in good agreement with the actual observed value, and the DCA curve showed that the nomogram had a better clinical net benefit and better clinical applicability (Figures 5C, D). Patients were classified into low-risk and high-risk groups according to a cutoff value of 50% predicted by the nomogram. The KM curves demonstrated significantly higher OS with the low-risk group (Figure 5E).
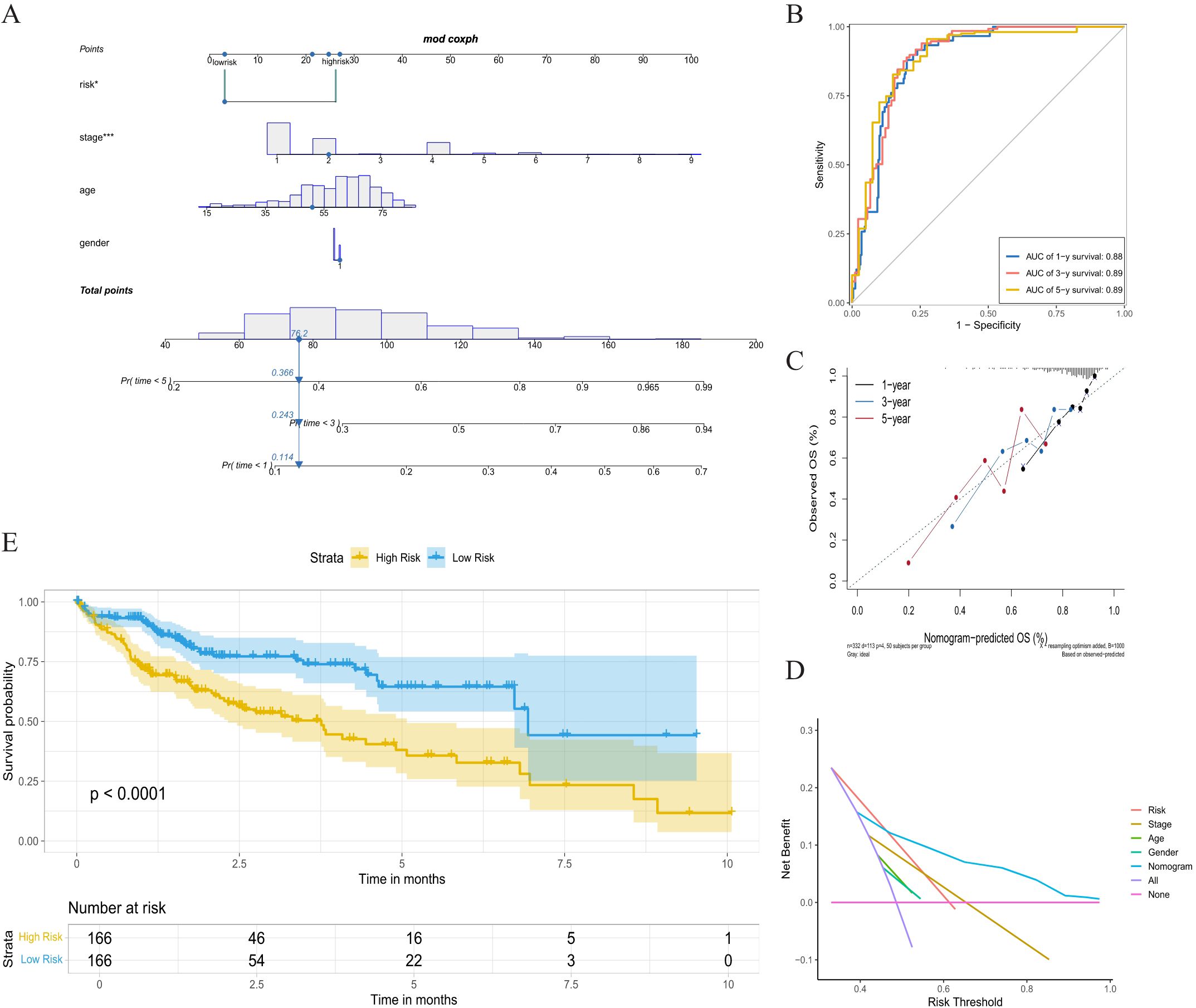
Figure 5. Nomogram analysis based on multivariate Cox regression. (A) Nomogram combing the risk score, stage, age, and gender. (B) Time-dependent ROC curves analysis of 1-year, 3-year, and 5-year survival. (C) The calibration curve of 1-year, 3-year, and 5-year survival. (D) Decision curve analysis. (E) K-M curve between high-risk group and low-risk group. ROC, receiver operating characteristic; K-M, Kaplan-Meier.
We investigated the proportions of various immune cell types in the HCC sample and para-cancer sample (Figure 6A). The considerable variations in immune cell infiltration between the high- and low-risk groups were evident from the single sample GSEA (ssGSEA). The results displayed that the proportion of monocytes, CD56dim NK cells, CD56bright NK cells, and CD8+ T cells was higher in the low-risk group than in the high-risk group (P<0.05). The CD4+ T cell, MDSC, macrophages, and mast cells of the high-risk group were substantially elevated compared with that of the low-risk group (Figure 6B). Correlation analysis showed that HRG was positively correlated with increased NK cells, and TUBA1B was negatively correlated with increased NK cells (Supplementary Figures S5C, D). We further confirmed the results by correlation analyses of immune cells and immune molecules (Figures 6C, D, Supplementary Figure S5).
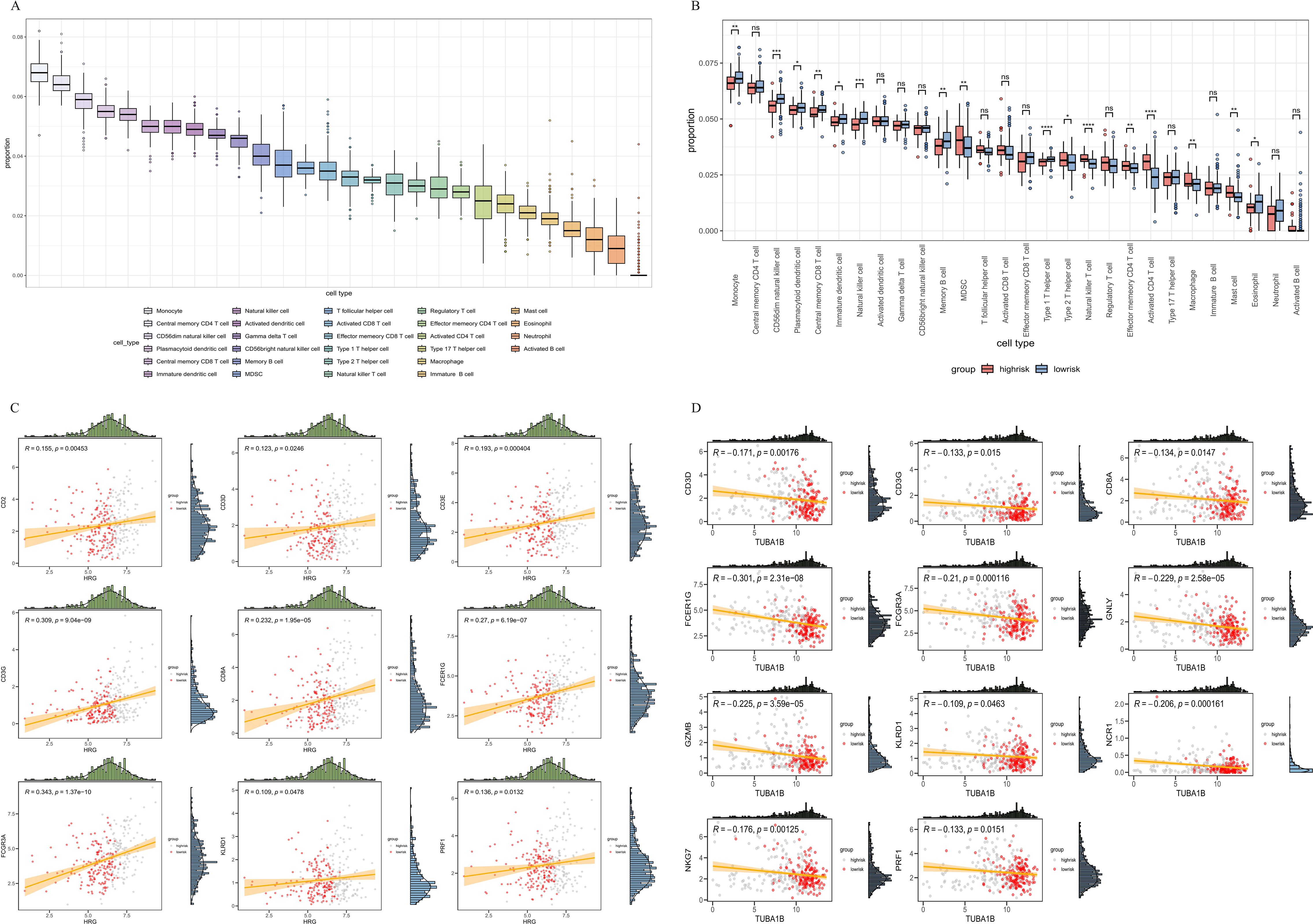
Figure 6. Correlation analysis between the signature and immunity. (A) Immune cell proportion of all tissues in the TCGA-LIHC cohort. (B) Immune cell proportion of high-risk and low-risk groups in the TCGA-LIHC cohort. (C, D) Correlation of immune cell infiltration and risk score. TCGA, The Cancer Genome Atlas.
To validate the prognostic value of the prognostic genes, we used the IHC stain to detect the protein expression of HRG and TUBA1B in tumor tissues and normal tissues. The expression of HRG decreased in HCC compared with normal tissues, whereas TUBA1B expression levels increased in HCC compared with normal tissues (Figure 7).
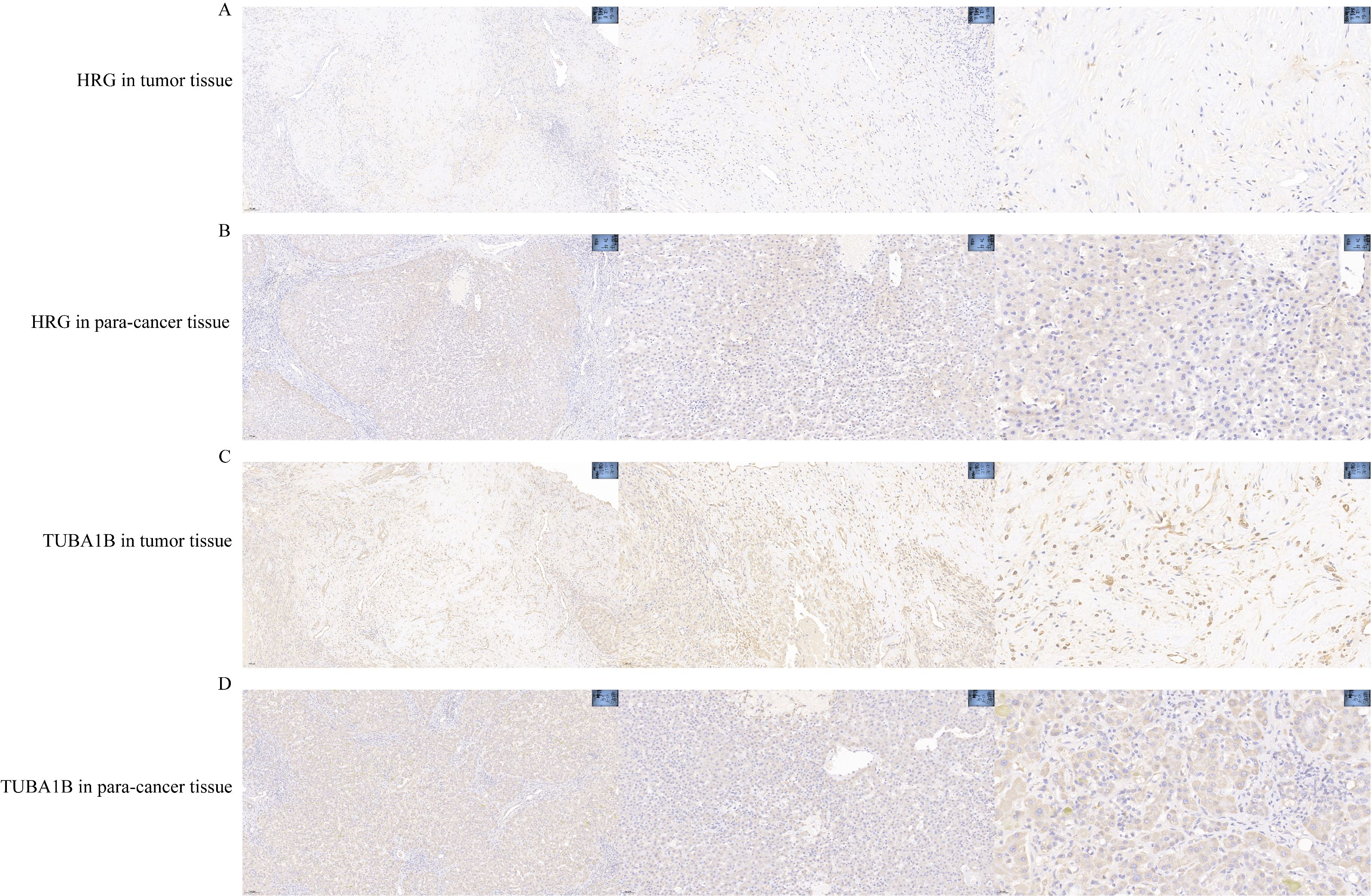
Figure 7. HRG gene and TUBA1B gene expression in HCC. (A) HRG gene in tumor tissue. (B) HRG gene in para-tumor tissue. (C) TUBA1B gene in tumor tissue. (D) TUBA1B gene in para-tumor tissue. HCC, hepatocellular carcinoma.
Hepatocellular carcinoma is highly heterogeneous, characterized by various morphologic features and biologic behaviors (35). Most patients with HCC are diagnosed at an advanced stage, which leads to rapid progress and poor outcomes due to the lack of effective and safe treatment (36, 37). Immune cells and stromal cells in TME are the main cellular components mediating immune tolerance and escape (38–43). Further studies on the heterogeneity of immune cells in the TME are indispensable for understanding their impact on prognosis. We developed a two-gene signature based on GEO (GSE148614) and TCGA-LIHC datasets and constructed a nomogram combining the gene signature and clinical features to predict OS in HCC.
Single-cell sequencing is a booming emerging technology in biomedical research and clinical practice, enabling comprehensive characterization of cell subpopulations, states, and lineages in heterogeneous tissues (44, 45). This is essential for the study of disease progression, tumor metastasis, response to treatment, and assessment of survival probability (25, 46). Therefore, scRNA has great development potential for promoting the diagnosis, targeted therapy, and prognosis prediction of cancers. We found that NK cell subsets were associated with prognosis based on scRNA-seq data from the GSE149614 dataset. NK cell infiltration is positively correlated with good prognosis (47, 48). Many studies have revealed that NK cell regulation of T cell function is an important immunomodulatory component in anticancer immunity (48–50). HCC patients with higher levels of intra-tumoral NK cell infiltration responded better to sorafenib treatment (51). These data strongly suggested that NK cell dysfunction contributes to HCC progression (52). In addition to the ability to kill malignant cells without prior sensitization, NK cells influence the activity of other immune cells by producing cytokines such as IFN-γ (53).
We applied consensus clustering on the TCGA-LIHC dataset, which can be effectively divided into 2 clusters, and the prognosis of the C1 and C2 clusters was significantly different. In WCGNA, we identified six modules and found that the turquoise module had the highest correlation with the prognostic cluster. The intersect was taken between NK cell genes and prognosis-related genes and yielded 25 genes. By univariate and multivariate Cox regression analysis, HRG and TUBA1B were correlated to survival outcomes of HCC. HRG is a secretory glycoprotein that binds to a variety of ligands, thereby regulating immunity, cell adhesion, angiogenesis, and thrombosis (54, 55). Inflammatory factors are pivotal in inflammatory diseases progression (56–60). HRG inhibits the activation of pro-inflammatory signaling (NF-κB) (61). Extensive studies have shown that the NF-κB signaling pathway is related to the development, progression, and invasion of tumors, and targeted regulation of the NF-κB signaling pathway can modulate these processes in various tumors. And NF-κB is bound to the inhibitory protein Farnesoid X (FXR), which retains NF–κB within the cytosol, thus preventing its transcriptional activity (62, 63). Additionally, HRG enhances the interaction between TNFR1 and Caspase8, promoting the formation of TNFR1 complex II, which directly induces apoptosis (64). Microtubules, which consist of a-tubulin and β-tubulin, perform important cellular functions such as protein trafficking, cell cycle, and cell migration (65). HRG also plays a significant role in modulating immune responses and angiogenesis, demonstrating potential in clinical applications for tumor targeting and sepsis-related immune regulation. Its immunomodulatory properties provide a foundation for developing novel therapeutic strategies, with promising applications in personalized treatments.
Microtubulin α1β (TUAB1B), an isoform of α-microtubulin, is associated with the expression of immune-related genes (66, 67). TUBA1B may play crucial roles in promoting tumor progression, including colon adenocarcinoma, osteosarcomas, liver hepatocellular carcinoma, and renal cell carcinoma (68–71). Furthermore, TUBA1B has been shown to mediate the infiltration of several immune cells in hepatocellular carcinoma and colorectal cancer. High TUBA1B expression is reported to be related to high paclitaxel resistance (72). Therefore, TUBA1B could also represent a therapeutic target for overcoming drug resistance, particularly in microtubule-targeting treatments, and holds promise for advancing precision oncology strategies. In our study, we further utilized IHC staining to assess the protein expression of HRG and TUBA1B in tumor and normal tissues. The results indicated that HRG expression was lower in HCC compared to normal tissues, while TUBA1B expression was higher in HCC, thereby confirming the prognostic value of these genes.
The identified genes have significant clinical applications in both immunotherapy and chemotherapy. In immunotherapy, these genes could help predict tumor response to treatment, guide the development of targeted therapies to enhance immune cell function, and enable personalized treatment strategies. As for chemotherapy, these genes can be used to predict drug resistance, assess therapeutic efficacy, and tailor chemotherapy regimens to individual patients, improving treatment outcomes and minimizing side effects. Additionally, the integration of therapies can be optimized by monitoring the expression of these genes, enabling real-time adjustments to treatment plans.
To assess the immune infiltration, ssGSEA analysis was performed. Based on the results of our study, the HRG gene and the TUBA1B gene can predict the prognosis of HCC and accurately respond to the tumor immune microenvironment. The HRG gene was correlated with anti-tumor immune cell infiltration, whereas TUBA1B gene was negatively correlated. The prognosis and immunotherapy outcomes are strongly influenced by the number of tumor-infiltrating lymphocytes (TILs) in the tumor microenvironment (73). Our findings provided new hints and references for the development of the immunotherapeutic approach for hepatocellular carcinoma.
There were limitations of the current study were acknowledged. Firstly, the sample size was small, and a prospective, large-sample, multicenter trial is warranted to confirm these findings. Secondly, functional studies on the molecular and biological functions of key genes will be required to substantiate this hypothesis. Thirdly, this study only identified NK cell-related genes, and further research is needed to explore other potentially related genes. Lastly, external validation using an independent cohort is required to assess the generalizability and reliability of our model.
Our study identified prognostically relevant NK cell signatures, which were further validated in clinical samples by immunohistochemistry. The findings had significant implications for the calculation of prognosis and therapeutic decision-making. Accurately assessing prognostic risk allows clinicians to identify individuals who are more likely to benefit from specific interventions or who require enhanced follow-up. Furthermore, discovering novel NK cell marker genes contributes to our understanding of the role of NK cells in HCC procession and provides a new strategy for precision immunotherapy.
In our study, we found a two-genes prognostic signature based on NK cell marker genes and elaborated on the role of the prognostic genes in the tumor immune microenvironment to provide new ideas for immunotherapy. In addition, we established an efficient and accurate prognostic model combined with clinical indicators to help clinicians make decisions.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
The study had been approved by the Ethics Committee of Beijing Youan Hospital, Capital Medical University. As a minimal-risk study in compliance with the Helsinki protocol, the requirement for informed patient consent was waived by the same ethics committee that approved the study (Beijing Youan Hospital, Capital Medical University), and all methods were performed in accordance with relevant guidelines and regulations. In accordance with state law and institutional requirements, written informed consent for participation was not required in this study.
JL: Data curation, Formal Analysis, Methodology, Software, Writing – original draft. ZL: Data curation, Formal Analysis, Methodology, Software, Writing – original draft. GZ: Data curation, Formal Analysis, Writing – review & editing. XY: Data curation, Formal Analysis, Writing – review & editing. XXY: Conceptualization, Funding acquisition, Supervision, Writing – review & editing. WX: Conceptualization, Funding acquisition, Supervision, Writing – review & editing. XD: Conceptualization, Funding acquisition, Supervision, Writing – review & editing.
The author(s) declare that financial support was received for the research and/or publication of this article. This study was funded by Beijing research center for respiratory infectious diseases project (BJRID2024- 007) and Construction of research-oriented wards in Beijing municipality, Laboratory for Clinical Medicine, Capital Medical University (SYLH2023-06), the grants from the National Natural Science Foundation of China (NSFC, 82202025), the Capital’s Funds for Health Improvement and Research (SF2024-1-2171).
We sincerely thank the researchers and study participants for their contributions to this study. We are also grateful to the data providers of the various public databases.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2025.1570647/full#supplementary-material
Supplementary Figure 1 | Clustering and dimension reduction analysis of single cell data. (A, B) PCA and harmony were performed for dimension reduction analysis. (C, D) Gene numbers (nFeature RNA), sequencing depth (nCount RNA), and mitochondrial gene percentage (pMT) content of samples. (E) Correlation analysis of UMI and number of mRNA.
Supplementary Figure 2 | Cell annotation of clusters was performed by classical markers of immune cells.
Supplementary Figure 3 | Consensus clustering analysis from k=2 to k=9.
Supplementary Figure 4 | Kaplan-Meier survival analysis of selected genes in normal and tumor samples. (A) Kaplan-Meier survival analysis of selected genes in TCGA cohort. (B) Kaplan-Meier survival analysis of selected genes in ICGC cohort. (C) HRG gene expression in relation to different tissues and clinical events. (D) TUBA1B gene expression in relation to different tissues and clinical events.
Supplementary Figure 5 | Correlation analysis between the gene expression and immunity. (A, B) The relationship between HRG and TUBA1B expression with specific immune cell markers. (C–E) Correlation analysis between the gene expression and NK cells.
Supplementary Table 1 | Top 20 marker genes of 27 clusters.
Supplementary Table 2 | The differential genes obtained from TCGA cohort.
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2018) 68:394–424. doi: 10.3322/caac.21492
2. Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA Cancer J Clin. (2023) 73:17–48. doi: 10.3322/caac.21763
3. Liu CY, Chen KF, Chen PJ. Treatment of liver cancer. Cold Spring Harb Perspect Med. (2015) 5:a021535. doi: 10.1101/cshperspect.a021535
4. Oura K, Morishita A, Hamaya S, Fujita K, Masaki T. The roles of epigenetic regulation and the tumor microenvironment in the mechanism of resistance to systemic therapy in hepatocellular carcinoma. Int J Mol Sci. (2023) 24:2805. doi: 10.3390/ijms24032805
5. Xiong J, Chi H, Yang G, Zhao S, Zhang J, Tran LJ, et al. Revolutionizing anti-tumor therapy: unleashing the potential of B cell-derived exosomes. Front Immunol. (2023) 14:1188760. doi: 10.3389/fimmu.2023.1188760
6. Chi H, Gao X, Xia Z, Yu W, Yin X, Pan Y, et al. FAM family gene prediction model reveals heterogeneity, stemness and immune microenvironment of UCEC. Front Mol Biosci. (2023) 10:1200335. doi: 10.3389/fmolb.2023.1200335
7. Lu C, Rong D, Zhang B, Zheng W, Wang X, Chen Z, et al. Current perspectives on the immunosuppressive tumor microenvironment in hepatocellular carcinoma: challenges and opportunities. Mol Cancer. (2019) 18:130. doi: 10.1186/s12943-019-1047-6
8. Wu Q, Zhou L, Lv D, Zhu X, Tang H. Exosome-mediated communication in the tumor microenvironment contributes to hepatocellular carcinoma development and progression. J Hematol Oncol. (2019) 12:53. doi: 10.1186/s13045-019-0739-0
10. Xiang S, Li J, Shen J, Zhao Y, Wu X, Li M, et al. Identification of prognostic genes in the tumor microenvironment of hepatocellular carcinoma. Front Immunol. (2021) 12:653836. doi: 10.3389/fimmu.2021.653836
11. Leonardi GC, Candido S, Cervello M, Nicolosi D, Raiti F, Travali S, et al. The tumor microenvironment in hepatocellular carcinoma (review). Int J Oncol. (2012) 40:1733–47. doi: 10.3892/ijo.2012.1408
12. Guillerey C. NK cells in the tumor microenvironment. Adv Exp Med Biol. (2020) 1273:69–90. doi: 10.1007/978-3-030-49270-0_4
13. Robinson MW, Harmon C, O’Farrelly C. Liver immunology and its role in inflammation and homeostasis. Cell Mol Immunol. (2016) 13:267–76. doi: 10.1038/cmi.2016.3
14. Xie H, Xi X, Lei T, Liu H, Xia Z. CD8(+) T cell exhaustion in the tumor microenvironment of breast cancer. Front Immunol. (2024) 15:1507283. doi: 10.3389/fimmu.2024.1507283
15. Gong X, Chi H, Xia Z, Yang G, Tian G. Advances in HPV-associated tumor management: Therapeutic strategies and emerging insights. J Med Virol. (2023) 95:e28950. doi: 10.1002/jmv.28950
16. Mamessier E, Sylvain A, Thibult ML, Houvenaeghel G, Jacquemier J, Castellano R, et al. Human breast cancer cells enhance self tolerance by promoting evasion from NK cell antitumor immunity. J Clin Invest. (2011) 121:3609–22. doi: 10.1172/JCI45816
17. Izawa S, Kono K, Mimura K, Kawaguchi Y, Watanabe M, Maruyama T, et al. H2O2 production within tumor microenvironment inversely correlated with infiltration of CD56(dim) NK cells in gastric and esophageal cancer: possible mechanisms of NK cell dysfunction. Cancer Immunol Immunother. (2011) 60:1801–10. doi: 10.1007/s00262-011-1082-7
18. Halama N, Braun M, Kahlert C, Spille A, Quack C, Rahbari N, et al. Natural killer cells are scarce in colorectal carcinoma tissue despite high levels of chemokines and cytokines. Clin Cancer Res. (2011) 17:678–89. doi: 10.1158/1078-0432.CCR-10-2173
19. Versluis MAC, Marchal S, Plat A, de Bock GH, van Hall T, de Bruyn M, et al. The prognostic benefit of tumour-infiltrating Natural Killer cells in endometrial cancer is dependent on concurrent overexpression of Human Leucocyte Antigen-E in the tumour microenvironment. Eur J Cancer. (2017) 86:285–95. doi: 10.1016/j.ejca.2017.09.008
20. Feng Q, Huang Z, Song L, Wang L, Lu H, Wu L. Combining bulk and single-cell RNA-sequencing data to develop an NK cell-related prognostic signature for hepatocellular carcinoma based on an integrated machine learning framework. Eur J Med Res. (2023) 28:306. doi: 10.1186/s40001-023-01300-6
21. Grady WM, Yu M, Markowitz SD. Epigenetic alterations in the gastrointestinal tract: current and emerging use for biomarkers of cancer. Gastroenterology. (2021) 160:690–709. doi: 10.1053/j.gastro.2020.09.058
22. Torroja C, Sanchez-Cabo F. Digitaldlsorter: deep-learning on scRNA-seq to deconvolute gene expression data. Front Genet. (2019) 10:978. doi: 10.3389/fgene.2019.00978
23. Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. (2014) 344:1396–401. doi: 10.1126/science.1254257
24. Azizi E, Carr AJ, Plitas G, Cornish AE, Konopacki C, Prabhakaran S, et al. Single-Cell map of diverse immune phenotypes in the breast tumor microenvironment. Cell. (2018) 174:1293–1308.e1236. doi: 10.1016/j.cell.2018.05.060
25. Zhang Q, He Y, Luo N, Patel SJ, Han Y, Gao R, et al. Landscape and dynamics of single immune cells in hepatocellular carcinoma. Cell. (2019) 179:829–845.e820. doi: 10.1016/j.cell.2019.10.003
26. Zhang S, Liu Z, Wu D, Chen L, Xie L. Single-cell RNA-seq analysis reveals microenvironmental infiltration of plasma cells and hepatocytic prognostic markers in HCC with cirrhosis. Front Oncol. (2020) 10:596318. doi: 10.3389/fonc.2020.596318
27. Sun D, Wang J, Han Y, Dong X, Ge J, Zheng R, et al. TISCH: a comprehensive web resource enabling interactive single-cell transcriptome visualization of tumor microenvironment. Nucleic Acids Res. (2021) 49:D1420–30. doi: 10.1093/nar/gkaa1020
28. Zhang P, Pei S, Wu L, Xia Z, Wang Q, Huang X, et al. Integrating multiple machine learning methods to construct glutamine metabolism-related signatures in lung adenocarcinoma. Front Endocrinol (Lausanne). (2023) 14:1196372. doi: 10.3389/fendo.2023.1196372
29. Zhang J, Peng G, Chi H, Yang J, Xie X, Song G, et al. CD8 + T-cell marker genes reveal different immune subtypes of oral lichen planus by integrating single-cell RNA-seq and bulk RNA-sequencing. BMC Oral Health. (2023) 23:464. doi: 10.1186/s12903-023-03138-0
30. Liu J, Zhang P, Yang F, Jiang K, Sun S, Xia Z, et al. Integrating single-cell analysis and machine learning to create glycosylation-based gene signature for prognostic prediction of uveal melanoma. Front Endocrinol (Lausanne). (2023) 14:1163046. doi: 10.3389/fendo.2023.1163046
31. Zhang X, Zhuge J, Liu J, Xia Z, Wang H, Gao Q, et al. Prognostic signatures of sphingolipids: Understanding the immune landscape and predictive role in immunotherapy response and outcomes of hepatocellular carcinoma. Front Immunol. (2023) 14:1153423. doi: 10.3389/fimmu.2023.1153423
32. Wang X, Zhao Y, Strohmer DF, Yang W, Xia Z, Yu C. The prognostic value of MicroRNAs associated with fatty acid metabolism in head and neck squamous cell carcinoma. Front Genet. (2022) 13:983672. doi: 10.3389/fgene.2022.983672
33. Li Z, Zhou H, Xia Z, Xia T, Du G, Franziska SD, et al. HMGA1 augments palbociclib efficacy via PI3K/mTOR signaling in intrahepatic cholangiocarcinoma. biomark Res. (2023) 11:33. doi: 10.1186/s40364-023-00473-w
34. Zhai X, Xia Z, Du G, Zhang X, Xia T, Ma D, et al. LRP1B suppresses HCC progression through the NCSTN/PI3K/AKT signaling axis and affects doxorubicin resistance. Genes Dis. (2023) 10:2082–96. doi: 10.1016/j.gendis.2022.10.021
35. Yang JD, Hainaut P, Gores GJ, Amadou A, Plymoth A, Roberts LR. A global view of hepatocellular carcinoma: trends, risk, prevention and management. Nat Rev Gastroenterol Hepatol. (2019) 16:589–604. doi: 10.1038/s41575-019-0186-y
36. Cabibbo G, Enea M, Attanasio M, Bruix J, Craxì A, Cammà C. A meta-analysis of survival rates of untreated patients in randomized clinical trials of hepatocellular carcinoma. Hepatology. (2010) 51:1274–83. doi: 10.1002/hep.23485
37. Llovet JM, Kelley RK, Villanueva A, Singal AG, Pikarsky E, Roayaie S, et al. Hepatocellular carcinoma. Nat Rev Dis Primers. (2021) 7:6. doi: 10.1038/s41572-020-00240-3
38. Oura K, Morishita A, Tani J, Masaki T. Tumor immune microenvironment and immunosuppressive therapy in hepatocellular carcinoma: A review. Int J Mol Sci. (2021) 22:5801. doi: 10.3390/ijms22115801
39. Wang X, Hassan W, Jabeen Q, Khan GJ, Iqbal F. Interdependent and independent multidimensional role of tumor microenvironment on hepatocellular carcinoma. Cytokine. (2018) 103:150–9. doi: 10.1016/j.cyto.2017.09.026
40. Xia Z, Chen S, He M, Li B, Deng Y, Yi L, et al. Editorial: Targeting metabolism to activate T cells and enhance the efficacy of checkpoint blockade immunotherapy in solid tumors. Front Immunol. (2023) 14:1247178. doi: 10.3389/fimmu.2023.1247178
41. Zhang X, Zhang P, Cong A, Feng Y, Chi H, Xia Z, et al. Unraveling molecular networks in thymic epithelial tumors: deciphering the unique signatures. Front Immunol. (2023) 14:1264325. doi: 10.3389/fimmu.2023.1264325
42. Deng Y, Shi M, Yi L, Naveed Khan M, Xia Z, Li X. Eliminating a barrier: Aiming at VISTA, reversing MDSC-mediated T cell suppression in the tumor microenvironment. Heliyon. (2024) 10:e37060. doi: 10.1016/j.heliyon.2024.e37060
43. Gong X, Chi H, Strohmer DF, Teichmann AT, Xia Z, Wang Q. Exosomes: A potential tool for immunotherapy of ovarian cancer. Front Immunol. (2022) 13:1089410. doi: 10.3389/fimmu.2022.1089410
44. Zhang S, Jiang C, Jiang L, Chen H, Huang J, Gao X, et al. Construction of a diagnostic model for hepatitis B-related hepatocellular carcinoma using machine learning and artificial neural networks and revealing the correlation by immunoassay. Tumour Virus Res. (2023) 16:200271. doi: 10.1016/j.tvr.2023.200271
45. Chi H, Zhao S, Yang J, Gao X, Peng G, Zhang J, et al. T-cell exhaustion signatures characterize the immune landscape and predict HCC prognosis via integrating single-cell RNA-seq and bulk RNA-sequencing. Front Immunol. (2023) 14:1137025. doi: 10.3389/fimmu.2023.1137025
46. Sun D, Guan X, Moran AE, Wu LY, Qian DZ, Schedin P, et al. Identifying phenotype-associated subpopulations by integrating bulk and single-cell sequencing data. Nat Biotechnol. (2022) 40:527–38. doi: 10.1038/s41587-021-01091-3
47. Chew V, Chen J, Lee D, Loh E, Lee J, Lim KH, et al. Chemokine-driven lymphocyte infiltration: an early intratumoural event determining long-term survival in resectable hepatocellular carcinoma. Gut. (2012) 61:427–38. doi: 10.1136/gutjnl-2011-300509
48. Wu M, Mei F, Liu W, Jiang J. Comprehensive characterization of tumor infiltrating natural killer cells and clinical significance in hepatocellular carcinoma based on gene expression profiles. BioMed Pharmacother. (2020) 121:109637. doi: 10.1016/j.biopha.2019.109637
49. Crouse J, Xu HC, Lang PA, Oxenius A. NK cells regulating T cell responses: mechanisms and outcome. Trends Immunol. (2015) 36:49–58. doi: 10.1016/j.it.2014.11.001
50. Mocikat R, Braumüller H, Gumy A, Egeter O, Ziegler H, Reusch U, et al. Natural killer cells activated by MHC class I(low) targets prime dendritic cells to induce protective CD8 T cell responses. Immunity. (2003) 19:561–9. doi: 10.1016/S1074-7613(03)00264-4
51. Pinyol R, Montal R, Bassaganyas L, Sia D, Takayama T, Chau GY, et al. Molecular predictors of prevention of recurrence in HCC with sorafenib as adjuvant treatment and prognostic factors in the phase 3 STORM trial. Gut. (2019) 68:1065–75. doi: 10.1136/gutjnl-2018-316408
52. Sajid M, Liu L, Sun C. The dynamic role of NK cells in liver cancers: role in HCC and HBV associated HCC and its therapeutic implications. Front Immunol. (2022) 13:887186. doi: 10.3389/fimmu.2022.887186
53. Chen Y, Zhou Y, Yan Z, Tong P, Xia Q, He K. Effect of infiltrating immune cells in tumor microenvironment on metastasis of hepatocellular carcinoma. Cell Oncol (Dordr). (2023) 46:1595–604. doi: 10.1007/s13402-023-00841-6
54. Yang LY, Luo Q, Lu L, Zhu WW, Sun HT, Wei R, et al. Increased neutrophil extracellular traps promote metastasis potential of hepatocellular carcinoma via provoking tumorous inflammatory response. J Hematol Oncol. (2020) 13:3. doi: 10.1186/s13045-019-0836-0
55. Johnson LD, Goubran HA, Kotb RR. Histidine rich glycoprotein and cancer: a multi-faceted relationship. Anticancer Res. (2014) 34:593–603.
56. Zhai X, Zhang H, Xia Z, Liu M, Du G, Jiang Z, et al. Oxytocin alleviates liver fibrosis via hepatic macrophages. JHEP Rep. (2024) 6:101032. doi: 10.1016/j.jhepr.2024.101032
57. Xiao J, Lin H, Liu B, Xia Z, Zhang J, Jin J. Decreased S1P and SPHK2 are involved in pancreatic acinar cell injury. biomark Med. (2019) 13:627–37. doi: 10.2217/bmm-2018-0404
58. Xiao J, Huang K, Lin H, Xia Z, Zhang J, Li D, et al. Mogroside II(E) inhibits digestive enzymes via suppression of interleukin 9/interleukin 9 receptor signalling in acute pancreatitis. Front Pharmacol. (2020) 11:859. doi: 10.3389/fphar.2020.00859
59. Zhang H, Xia T, Xia Z, Zhou H, Li Z, Wang W, et al. KIF18A inactivates hepatic stellate cells and alleviates liver fibrosis through the TTC3/Akt/mTOR pathway. Cell Mol Life Sci. (2024) 81:96. doi: 10.1007/s00018-024-05114-5
60. Zhao Y, Wei K, Chi H, Xia Z, Li X. IL-7: A promising adjuvant ensuring effective T cell responses and memory in combination with cancer vaccines? Front Immunol. (2022) 13:1022808. doi: 10.3389/fimmu.2022.1022808
61. Gao S, Wake H, Gao Y, Wang D, Mori S, Liu K, et al. Histidine-rich glycoprotein ameliorates endothelial barrier dysfunction through regulation of NF-κB and MAPK signal pathway. Br J Pharmacol. (2019) 176:2808–24. doi: 10.1111/bph.v176.15
62. Gadaleta RM, Oldenburg B, Willemsen EC, Spit M, Murzilli S, Salvatore L, et al. Activation of bile salt nuclear receptor FXR is repressed by pro-inflammatory cytokines activating NF-κB signaling in the intestine. Biochim Biophys Acta. (2011) 1812:851–8. doi: 10.1016/j.bbadis.2011.04.005
63. Balasubramaniyan N, Ananthanarayanan M, Suchy FJ. Nuclear factor-κB regulates the expression of multiple genes encoding liver transport proteins. Am J Physiol Gastrointest Liver Physiol. (2016) 310:G618–628. doi: 10.1152/ajpgi.00363.2015
64. Zou X, Zhang D, Song Y, Liu S, Long Q, Yao L, et al. HRG switches TNFR1-mediated cell survival to apoptosis in Hepatocellular Carcinoma. Theranostics. (2020) 10:10434–47. doi: 10.7150/thno.47286
65. Kim JY, Han SY, Yoo J, Kim GW, Jeon YH, Lee SW, et al. HDAC8-selective inhibition by PCI-34051 enhances the anticancer effects of ACY-241 in ovarian cancer cells. Int J Mol Sci. (2022) 23:8645. doi: 10.3390/ijms23158645
66. Akhmanova A, Maiato H. Closing the tubulin detyrosination cycle. Science. (2017) 358:1381–2. doi: 10.1126/science.aar3895
67. Snelleksz M, Dean B. Lower levels of tubulin alpha 1b in the frontal pole in schizophrenia supports a role for changed cytoskeletal dynamics in the aetiology of the disorder. Psychiatry Res. (2021) 303:114096. doi: 10.1016/j.psychres.2021.114096
68. Hu J, Han C, Zhong J, Liu H, Liu R, Luo W, et al. Dynamic network biomarker of pre-exhausted CD8(+) T cells contributed to T cell exhaustion in colorectal cancer. Front Immunol. (2021) 12:691142. doi: 10.3389/fimmu.2021.691142
69. Hu X, Zhu H, Chen B, He X, Shen Y, Zhang X, et al. Tubulin alpha 1b is associated with the immune cell infiltration and the response of HCC patients to immunotherapy. Diagn (Basel). (2022) 12:858. doi: 10.3390/diagnostics12040858
70. Dou Y, Zhu K, Sun Z, Geng X, Fang Q. Screening of disorders associated with osteosarcoma by integrated network analysis. Biosci Rep. (2019) 39:BSR20190235. doi: 10.1042/BSR20190235
71. Xu QQ, Qin LT, Liang SW, Chen P, Gu JH, Huang ZG, et al. The expression and potential role of tubulin alpha 1b in wilms’ Tumor. BioMed Res Int. (2020) 2020:9809347. doi: 10.1155/2020/9809347
72. Lu C, Zhang J, He S, Wan C, Shan A, Wang Y, et al. Increased α-tubulin1b expression indicates poor prognosis and resistance to chemotherapy in hepatocellular carcinoma. Dig Dis Sci. (2013) 58:2713–20. doi: 10.1007/s10620-013-2692-z
Keywords: hepatocellular carcinoma, natural kill cell, tumor microenvironment, nomogram, prognosis, TUBA1B, single-cell
Citation: Li J, Liu Z, Zhang G, Yin X, Yuan X, Xie W and Ding X (2025) Uncovering the heterogeneity of NK cells on the prognosis of HCC by integrating bulk and single-cell RNA-seq data. Front. Oncol. 15:1570647. doi: 10.3389/fonc.2025.1570647
Received: 04 February 2025; Accepted: 04 March 2025;
Published: 18 March 2025.
Edited by:
Zhijie Zhao, Shanghai Jiao Tong University, ChinaReviewed by:
Zhijia Xia, Affiliated Hospital of Guilin Medical University, ChinaCopyright © 2025 Li, Liu, Zhang, Yin, Yuan, Xie and Ding. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoxue Yuan, eXVhbnhpYW94dWVfMzJAMTYzLmNvbQ==; Wen Xie, eGlld2VuNjIxOEAxNjMuY29t; Xiaoyan Ding, ZGluZ3hpYW95YW4xOTgxMTFAMTYzLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.