 Louise Evans1†
Louise Evans1† Sarah Trinder2,3†
Sarah Trinder2,3† Andrew Dodgshun4,5
Andrew Dodgshun4,5 David D. Eisenstat6,7,8James R. Whittle9,10,11
David D. Eisenstat6,7,8James R. Whittle9,10,11 Jordan R. Hansford1,12,13*‡
Jordan R. Hansford1,12,13*‡ Santosh Valvi14,15,16‡
Santosh Valvi14,15,16‡- 1Michael Rice Centre for Hematology and Oncology, Women’s and Children’s Hospital, North Adelaide, SA, Australia
- 2Kids Cancer Centre, Sydney Children’s Hospital, Sydney, NSW, Australia
- 3Children’s Cancer Institute, Lowy Cancer Research Centre, University of New South Wales Sydney, Sydney, NSW, Australia
- 4Department of Pediatrics, University of Otago, Christchurch, New Zealand
- 5Children’s Hematology/Oncology Centre, Christchurch Hospital, Christchurch, New Zealand
- 6Children’s Cancer Centre, Royal Children’s Hospital, Melbourne, VIC, Australia
- 7Department of Stem Cell Medicine, Murdoch Children’s Research Institute, Melbourne, VIC, Australia
- 8Department of Pediatrics, University of Melbourne, Melbourne, VIC, Australia
- 9Department of Medical Oncology, Peter MacCallum Cancer Centre, Melbourne, VIC, Australia
- 10Personalized Oncology Division, Walter and Eliza Hall Institute (WEHI), Parkville, VIC, Australia
- 11Department of Medical Biology, University of Melbourne, Parkville, VIC, Australia
- 12Pediatric Neuro-Oncology, Precision Cancer Medicine, South Australia Health and Medical Reseach Institute, Adelaide, SA, Australia
- 13South Australia ImmunoGENomics Cancer Institute, University of Adelaide, Adelaide, SA, Australia
- 14Department of Pediatric and Adolescent Oncology/Hematology, Perth Children’s Hospital, Nedlands, WA, Australia
- 15Brain Tumor Research Program, Telethon Kids Institute, Nedlands, WA, Australia
- 16School of Medicine, Division of Pediatrics, The University of Western Australia, Perth, WA, Australia
Gliomas account for nearly 30% of all primary central nervous system (CNS) tumors in children and adolescents and young adults (AYA), contributing to significant morbidity and mortality. The updated molecular classification of gliomas defines molecularly diverse subtypes with a spectrum of tumors associated with age-distinct incidence. In adults, gliomas are characterized by the presence or absence of mutations in isocitrate dehydrogenase (IDH), with mutated IDH (mIDH) gliomas providing favorable outcomes and avenues for targeted therapy with the emergence of mIDH inhibitors. Despite their rarity, IDH mutations have been reported in 5-15% of pediatric glioma cases. Those with primary mismatch-repair deficient mIDH astrocytomas (PMMRDIA) have a particularly poor prognosis. Here, we describe the biology of mIDH gliomas and review the literature regarding the emergence of mIDH inhibitors, including clinical trials in adults. Given the paucity of clinical trial data from pediatric patients with mIDH glioma, we propose guidelines for the inclusion of pediatric and AYA patients with gliomas onto prospective trials and expanded access programs as well as the potential of combined mIDH inhibition and immunotherapy in the treatment of patients with PMMRDIA at high risk of progression.
1 Introduction
Gliomas are a complex and diverse group of central nervous system (CNS) neoplasms that arise from glial cells, varying in location, grade, and clinical behavior (1–3). These tumors are particularly common in pediatric and adolescent and young adult (AYA) patients, comprising 30% of all CNS tumors and significantly contributing to morbidity and mortality (2, 4–7). The evolution of the integrated histo-molecular classification of gliomas defines tumors according to molecular features that influence patient prognosis and therapeutic decisions. In adults, the identification of isocitrate dehydrogenase (IDH) mutations has resulted in paradigm shift in the classification of tumors, with the recognition that these features play an important role in tumorigenesis and prognosis (7); however, less is understood on the role of mIDH in pediatric and AYA patients.
IDH enzymes are essential in major cellular metabolic processes (8, 9), and exist in three isoforms: IDH1, IDH2, and IDH3 (10, 11) with IDH1 and IDH2 mutations resulting in the accumulation of the oncometabolite D2-hydroxyglutarate (D2-HG) that is implicated in tumorigenesis through a variety of mechanisms. Mutant IDH1 or IHD2 enzyme activity interferes with normal cellular metabolic processes by depleting α-ketoglutarate (α-KG) from the Krebs cycle and disruptions in redox balance (11–14). In addition, D2-HG results in epigenetic modulation and impaired DNA repair, as well as angiogenesis through hydroxylation of hypoxia-inducible factor- α (HIF- α) (11–14).
IDH mutations have been identified in numerous cancers, including CNS tumors, solid tumors, and myeloid malignancies (15–18). In gliomas, these were first described in the context of “secondary glioblastoma (GBM),” before defining lower grade gliomas highlighting prognostic and treatment implications (19–23). The historical standard of care for adults with mIDH glioma involves maximal safe resection followed by radiation and sequential chemotherapy based on the risk of recurrence, although some patients may be suitable for active surveillance (3, 24, 25). Given the toxicity conferred by radiation and chemotherapy, approaches that can delay this intervention are required. Several mIDH inhibitors have been tested in the clinic with the pivotal INDIGO study (26) demonstrating significant improvement in progression free survival (PFS) and time to next treatment intervention in a select group of patients who have not received radiation or chemotherapy. Although patients older than 12 years were eligible for the study, no pediatric patients were enrolled on the treatment arm and thus findings in pediatric patients with mIDH glioma are lacking. Moreover, outstanding questions regarding the biology and prognostic features of these tumors in pediatric and AYA cohorts is yet to be fully elucidated with no consensus for their management (21).
2 The biology of the IDH enzyme
2.1 IDH enzyme
IDH enzymes are crucial in several cellular metabolic processes, including the Krebs cycle, glutamine metabolism, lipogenesis, and regulation of cellular redox status (8, 9). Three IDH isoforms, IDH1, IDH2 and IDH3, encoded by different genes with distinct cellular localization, contribute to the regulation of central metabolic circuits (10–12). IDH1 is located in the cytoplasm and peroxisomes, whereas IDH2 and IDH3 are localized to the mitochondria (10, 22).
IDH enzymes are fundamental to the tricarboxylic acid cycle (TCA, Krebs cycle) (9): a series of metabolic oxidative reactions necessary for the mitochondrial electron transport chain to produce ATP (Figure 1) (6). Nicotinamide adenine dinucleotide phosphate (NADP)-dependent IDH1 and IDH2 catalyze the oxidative decarboxylation of isocitrate into α-ketoglutarate (α-KG), NAPDH, and carbon dioxide (CO2) (3, 9). α-Ketoglutarate is a major modulator of electron transport chain activity and TCA flux (27) and it is a co-factor of numerous important cellular reactions, including fatty acid metabolism (3, 10).
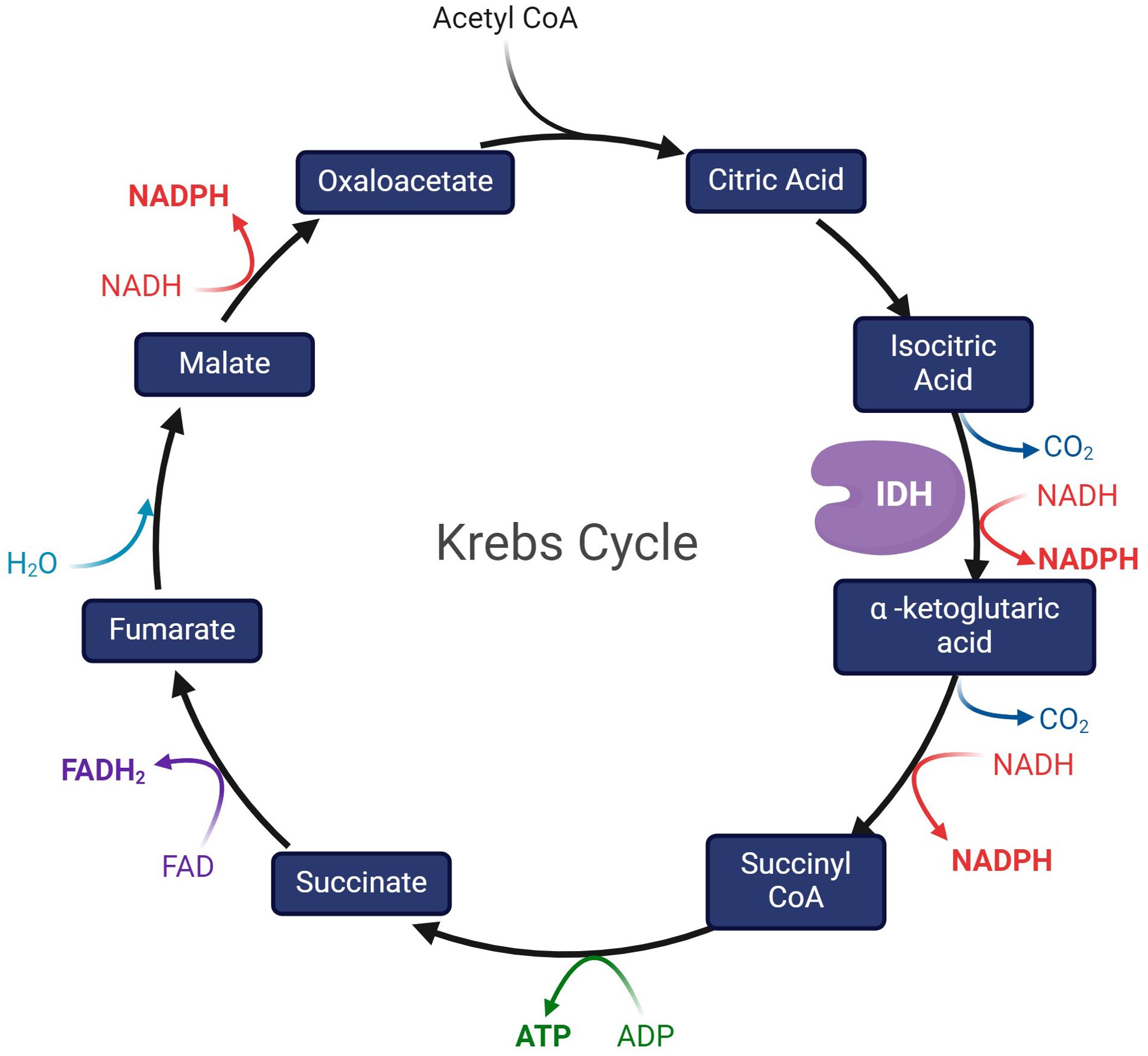
Figure 1. The Krebs cycle, depicting IDH as the enzyme that catalyzes the conversion of isocitric acid to α -ketoglutaric acid.
IDH is critical to maintaining a sufficient pool of reduced glutathione (GSH) and supporting the peroxiredoxin system by generating NADPH (12, 28). The NADPH produced from the reactions catalyzed by IDH1 and IDH2 is critical to maintain redox balance and protect cells from reactive oxygen species (ROS) that cause DNA damage (3, 10, 12, 29, 30). IDH is particularly important in the brain, producing 65% of the brain’s NADPH, which is essential for lipid metabolism (31). IDH1 and IDH2 are also involved in glutamine metabolism (32), which is important in tumorigenesis as glutamine deprivation suppresses cancer growth (28).
IDH3 is a holoenzyme located in the mitochondria and it catalyzes the irreversible nicotinamide adenine dinucleotide (NAD+)-dependent α-KG (6, 10, 11).
2.2 IDH mutations and tumorigenesis
IDH mutations contribute to tumorigenesis in multiple cancer types by interfering with normal cellular metabolism and through the production of the oncometabolite D2-hydroxyglutarate (D2-HG) (Figure 2). Somatic mutations in IDH1 and IDH2 are heterozygous, and primarily consist of missense variants that result in single amino acid substitutions at key arginine residues within the enzyme’s core active sites (10, 11). IDH1 mutations typically occur at Arginine 132, the R132H and R132C variants are the most prevalent (11). IDH1 G97 is mutated in some colon cancers and pediatric astrocytomas (33). IDH2 mutations occur at Arginine 172 and Arginine 140 (34, 35). There are no reports of tumor-associated mutations in the IDH3 gene (10, 36).
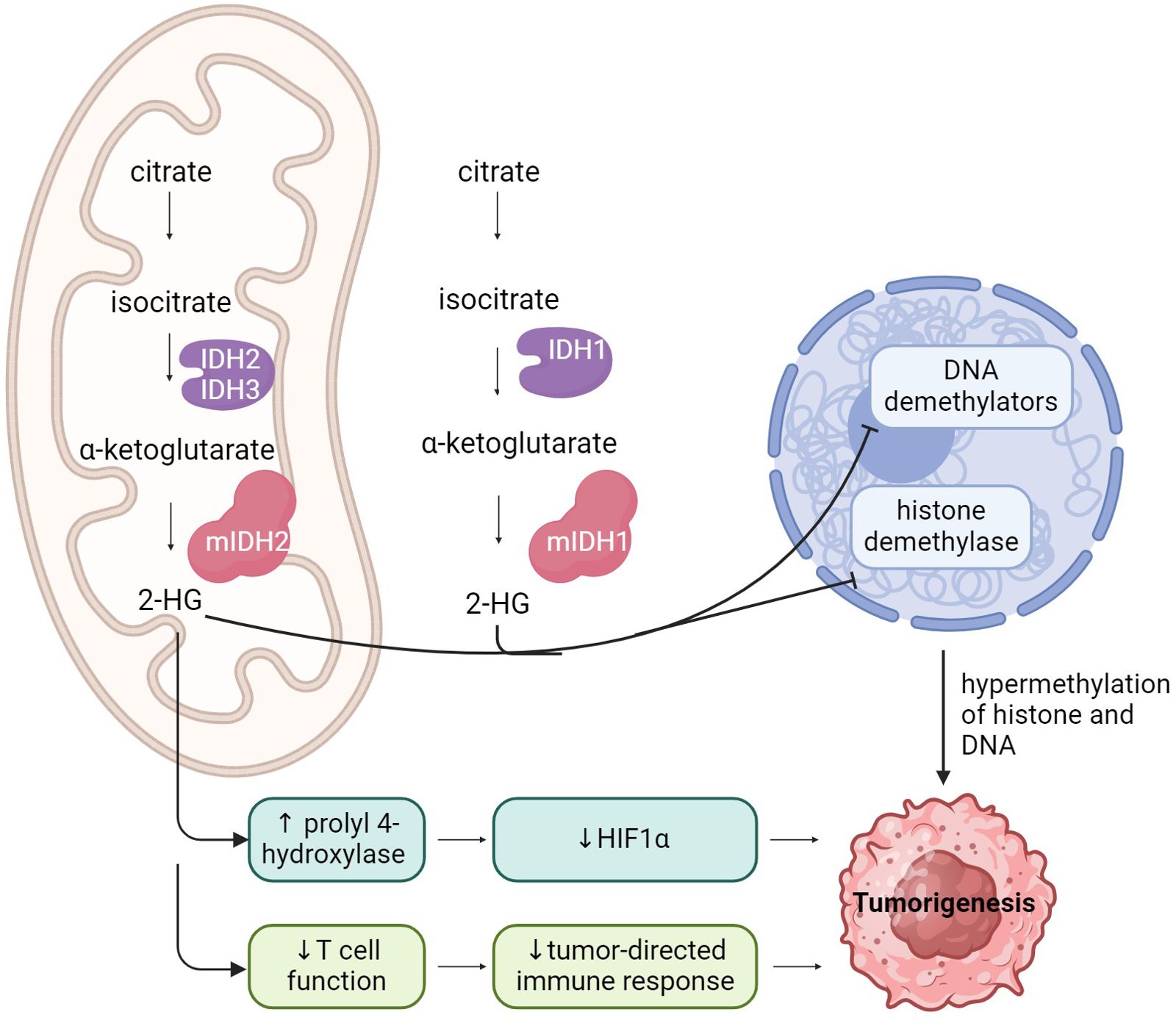
Figure 2. A schematic representation of IDH function, and the impact of IDH mutations on cell function, leading to tumorigenesis.
Uncontrolled cell proliferation associated with cancer often leads to metabolic alterations that support rapid cell growth (37). Mutant IDH enzyme activity interferes with normal cellular metabolism by depleting α-ketoglutarate (α-KG) from the Krebs cycle (2). Mutant IDH enzymes also consume NADPH, reducing its availability for maintaining redox balance and de novo lipogenesis (38). Accumulating oxidative damage is a hallmark of cancer biology for IDH-mutated malignancies, as a direct consequence of disruption to redox balance (39). Under hypoxic conditions, IDH1-mutant cells exhibit increased oxidative TCA metabolism and decreased reductive glutamine metabolism (32, 40). Metabolically reduced glutamine serves as the major carbon source for fatty acid synthesis during hypoxia and impaired cellular respiration, a switch that is crucial for sustaining rapid cell proliferation (37).
In addition to losing its normal catalytic activity, the mutant IDH enzyme catalyzes the reduction of α-KG to its (R)-enantiomer, the oncometabolite D2-HG (9–13, 41–43). Accumulation of D2-HG causes profound metabolic dysregulation, including inhibition of normal cellular differentiation, epigenetic modulation, DNA repair, redox balance as well as alterations to the tumor immune microenvironment (11–14).
D2-HG is a homolog of α-KG, and functions as a competitive inhibitor of α-KG-dependent dioxygenases, vital for DNA and histone demethylation (3, 42, 44). Accumulation of D2-HG has been shown to inhibit several key histone demethylases, including KDM7A (demethylates H3K9me2 and H3K27me2), KDM4A/B (demethylates H3K9 and H3K36), and Jumonji C domain-containing (JmjC) histone demethylases (2, 11, 41, 44). D2-HG also inhibits the DNA modifying enzymes in the ten-eleven translocation (TET) family of 5-methlycytosine (5mC) hydroxylases (44). Interference with the normal activity of dioxygenases disrupts histone and DNA methylation patterns, leading to the signature global DNA hypermethylation phenotype seen in IDH-mutant gliomas, known as the Glioma CpG Island Methylator Phenotype (G-CIMP) (2). This epigenetic reprograming results in a block of cellular differentiation, which in turn causes inappropriate activation of growth-promoting signaling (2).
A high concentration of D2-HG also promotes angiogenesis via VEGFR2 signaling and increased matrix metalloproteinase (MMP2) activity (45). Additionally, D2-HG contributes to gliomagenesis by directly stimulating prolyl 4-hydroxylase activity, thereby decreasing hypoxia-inducible factor- α (HIF1α) activity, which enhances the proliferation of human astrocytes in vitro (2, 46). Furthermore, tumor cell-derived R-2-HG is absorbed by T cells, leading to a disruption of nuclear factor of activated T cells (NFAT) transcriptional activity and polyamine biosynthesis, resulting in the suppression of T cell activity (47). D2-HG drives an immunosuppressive tumor microenvironment, acting directly on CD8+ cells by altering their metabolic and cytotoxic signatures (26, 47, 48).
D2-HG has been shown to accumulate in high levels in glioma cells but is absent in normal brain cells (44). D2-HG also structurally and functionally mimics glutamate, thereby contributing to the genesis of seizures in patients with gliomas (36, 49).
Patients with Ollier disease and Maffucci syndrome, non-hereditary skeletal disorders characterized by multiple enchondromas and spindle cell hemangiomas, are associated with somatic mosaic IDH1 and IDH2 mutations (50). These patients are at increased risk of developing IDH-mutant gliomas (51) and require thorough clinical examination along with a full body MRI every second year, from age 25 years (52).
IDH mutations have been implicated in other cancer types, including AML, myelodysplastic syndrome (15–18), cholangiocarcinoma of intrahepatic origin (53), central and periosteal cartilaginous chondrosarcomas (54), and melanoma (55, 56).
3 Pediatric gliomas
Traditionally, the World Health Organization (WHO) classification divides gliomas into four grades based on histology. Low grade gliomas (LGGs) are well-differentiated, typically slow-growing tumors (grades 1 and 2), whereas high grade gliomas (HGGs) are poorly differentiated or anaplastic, and diffusely infiltrating tumors (grade 3 and 4) (10). Recent advances in the understanding of molecular biology of CNS tumors were incorporated into the fifth edition of the WHO CNS tumor classification (CNS5), incorporating molecular characteristics into the diagnostic criteria in addition to the routine histologic features (57, 58).
Pediatric and AYA LGGs constitute most gliomas in this population, and tend to be more indolent, slow growing lesions. Treatment of LGG is multifaceted and is contingent upon several factors including tumor location, patient age and comorbidities. Whenever feasible, surgical resection is the preferred initial treatment. However, surgical resection is not always possible due to tumor location and surgical morbidity. When surgery is not an option, medical therapy choices include carboplatin as a single agent (59) or in combination with vincristine (60), or single agent vinblastine (61). Despite the intent to cure, treatment interventions can negatively impact neurological, neurocognitive and endocrine function, with long term effects on education, employment and social outcomes (4).
HGGs account for approximately 10% of brain tumors in children and adolescents and are more aggressive, conferring a poor prognosis (62). Standard treatment for HGGs include maximal safe resection, focal radiotherapy, and chemotherapy (62). Temozolomide forms an important part of treatment for adult HGGs concurrent to radiotherapy and as adjuvant therapy. However, in pediatric and AYA patients, the role of temozolomide is less clear given the tumor biology is different to that of adult HGGs.
3.1 IDH mutations in gliomas
IDH1 and IDH2 mutations are important, class-defining mutations in gliomas that carry significant therapeutic and prognostic implications (3, 63). IDH mutations are very early events in gliomagenesis, affecting a common glial precursor cell population in most cases except in patients with replication repair deficiency (RRD) (10, 64, 65), and show almost ubiquitous expression throughout the life-cycle of the disease. Among the three isoforms of IDH, mutations in IDH1 are the most common (66).
The latest WHO Classification of CNS Tumors divides mIDH adult-type diffuse gliomas into astrocytoma grades 2-4 and oligodendroglioma 1p/19q-codeleted. Mutations in IDH1 or IDH2 define WHO grade 2 and 3 diffuse gliomas in adults (5, 34, 66, 67).
Patients with IDH-mutant gliomas tend to be younger than their wild-type counterparts with a median age of 37 years (68). IDH1 gliomas arise from a neural precursor population that is spatially and temporally restricted in the brain (69). Therefore, mIDH tumors tend to be in the frontal lobes compared with other lobes and compared with IDH wild-type tumors (70).
The presence of co-existing mutations also helps subdivide mIDH gliomas and provides prognostic information. Astrocytomas typically harbor ATRX and TP53 mutations (3, 24), whilst oligodendrogliomas are defined by co-deletions in 1p and 19q and may carry CIC and TERT mutations (3). Overall survival (OS) is significantly shorter for astrocytomas harboring CDKN2A/B deletions in both pediatric and adult patients (21, 71), and therefore the CNS5 classification now upgrades astrocytomas with CDKN2A/B homozygous deletion to WHO grade 4 irrespective of high grade histology features with microvascular proliferation or necrosis (58). The role of CDKN2A deletion in patients with oligodendroglioma remains under investigation. The presence of mutations in the PIK3CA, PIK3R1 and amplification of PDGFRA and MYCN also confer a trend toward poorer survival (72). Genetic alterations involving members of the RB1 pathway, including CDK4 amplification and RB1 mutation or homozygous deletion, have also been reported (71).
IDH mutation status is an independent predictor of favorable outcomes among adults with glioma (34, 73). In part, this may be due to increased sensitivity to chemotherapy and radiotherapy, as IDH mutation does not have the same impact on survival in untreated tumors (74).
3.2 Prevalence of IDH-mutant gliomas in pediatric and AYA patients
Given the rarity of IDH mutations in pediatric and AYA tumors, there is less detail on their prevalence and clinical impact (75), with most information found in small cohort studies. The prevalence of IDH mutations in the pediatric and AYA cohort ranges from 1% to over 50%, with IDH1 mutations being more common than IDH2 mutations (Table 1). In a cohort of 43 pediatric patients with newly diagnosed WHO grade 3 or 4 gliomas treated on the Children’s Oncology Group ACNS0423 study, IDH1 mutations were detected in 7 of 43 (16.3%) of children with primary malignant gliomas, and no IDH2 mutations were identified. Older children and AYA are more likely to have mIDH gliomas. In a large AYA case series including patients aged 15-39.9 by Bennet et al. IDH mutations were noted in 57% patients (7) whereas Pollack et al. report IDH mutations in 7 of 20 gliomas (35%) from children ≥14 years and 0 of 23 (0%) in younger children (19). The PRecISion Medicine for Children with Cancer (PRISM), trial has reported two patients with mIDH astrocytoma of the 146 patients enrolled with high-risk pediatric brain tumors who had at least 18 months of follow-up (77).
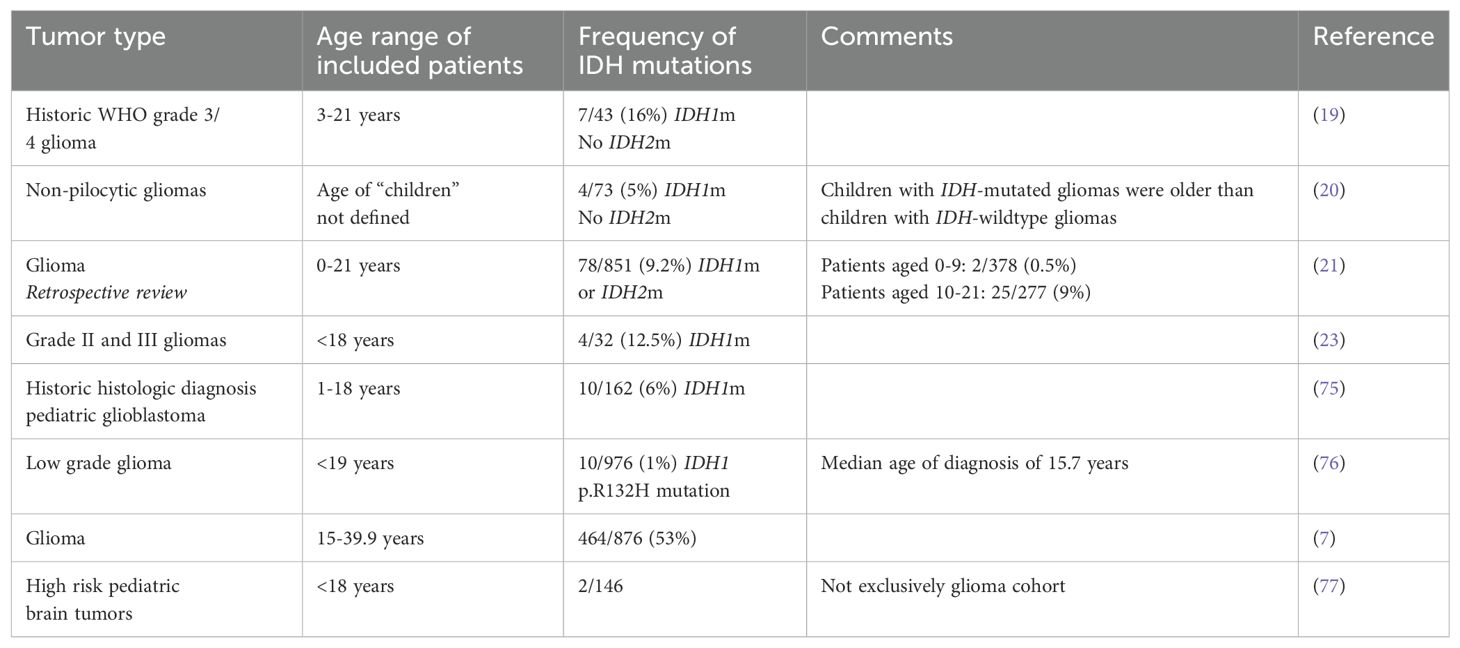
Table 1. Frequency of IDH mutations in pediatric and AYA gliomas.
3.3 Primary mismatch repair-deficient IDH-mutant astrocytomas
Recently, a distinct cohort of patients with IDH-mutant astrocytomas has been recognized with hereditary mismatch repair deficiency (MMR) (78, 79). These patients are unique to those with acquired MMR due to alkylating agents. Instead, primary mismatch repair-deficient IDH-mutant astrocytomas (PMMRDIA) are histologically high grade, often displaying a hypermutant genotype and microsatellite instability (78, 80). These tumors primarily present in younger patients and have worse clinical outcomes compared to other IDH-mutant gliomas (78).
Dodgshun et al. first described six patients with RRD HGG with secondary IDH1 mutations that clustered with other IDH1 mutant gliomas on methylation profiling (79). However, while other mIDH gliomas demonstrated a CpG Island Methylator Phenotype (CIMP), these six tumors with secondary IDH1 mutations displayed the inverse pattern of methylation disturbance; a CpG Island Demethylator Phenotype (CIDP) in keeping with other RRD HGG. These findings suggest that the CIDP phenotype generated by RRD glioma is unable to be overcome by secondary IDH mutations.
In a study by Suwala et al., samples from 32 patients with IDH-mutant gliomas and proven or suspected primary MMR deficiency were sequenced. Patients were clinically diagnosed with Lynch syndrome or Constitutional Mismatch Repair Deficiency Syndrome and/or had germline mutations in DNA mismatch repair genes (MLH1, MSH6, MSH2) in all but one case. Results demonstrated a distinct DNA methylation profile which clustered separately from other IDH-mutant glioma subtypes including those with acquired MMR deficiency on t-distributed stochastic neighbor embedded (t-SNE) plots. Tumors displayed a higher proportion (60%) of unmethylated MGMT promoter compared to other IDH-mutant gliomas. Frequent inactivation of TP53, RB1, ATRX and activation of the RTK/PI3K/AKT pathway was also present. The OS of this cohort of patients was significantly worse, with a mean OS of only 15 months despite treatment with surgery, radiation and chemotherapy. Pediatric and AYA patients diagnosed with a mIDH glioma warrant screening for MMRD.
4 The current standard of care
Recommended management algorithms for mIDH glioma have been defined by several groups including the American Society of Clinical Oncology (ASCO) (24), the Society for Neuro-Oncology (SNO) (2), and the European Association of Neuro-Oncology (EANO) (25). These algorithms incorporate surgery and outcomes from randomized clinical trials on the use of radiotherapy and/or chemotherapy. It is important to consider that most data and recommendations pertain to adult patients with potential for underrepresentation of the AYA group and limited trials conducted in the pediatric setting.
4.1 Role of surgery
The benefits of safe maximal resection in adults with glioma were well-established prior to the era of molecular characterization. These results have been recapitulated in the modern diagnostics era by Wijenga et al. (81) with a retrospective re-analysis of adult tumor subtypes applying new molecular classification. This study demonstrated that for mIDH astrocytomas, each 1cm3 increase in postoperative volume of disease results in poorer OS, hazard ratio (HR) of 1.01 (p<0.0001). However, the impact of extent of upfront resection is less clear in pediatric patients. In a multi-institutional retrospective analysis of 78 pediatric patients with IDH1/2 mutated gliomas including 45 patients with low grade astrocytoma, the 5Y PFS of patients with gross total resection (GTR) was 45.7% with median PFS of 4.4 years compared to 5Y PFS of patients with subtotal resection (STR) of biopsy of 41% with median PFS of 4.7 years (21). There was no statistically significant difference in OS in this cohort. With only 13 patients in the cohort of low-grade oligodendrogliomas in this study, there was no statistically significant differences in PFS or OS for upfront GTR versus STR or biopsy. The discrepancies in outcomes compared to adults may be reflective of small sample size.
In adults, repeat craniotomy and safe resection is feasible for patients with recurrent LGG (82). Re-operation in eloquent or near eloquent brain areas are not associated with higher risk of neurological sequelae compared to initial surgery (83). Up to 50% of patients can achieve a gross total resection at time of recurrence (84). Mounting evidence demonstrates that there is survival benefit to near total or gross resection at time of recurrence (83–85). Thus, standard of care is moving towards aggressive safe early resection at recurrence.
4.2 Adjuvant therapy
In adult IDH-mutant grade 2 gliomas a “watch and wait” approach may be taken for “low-risk” patients, historically defined as younger patients under 40 years with GTR (2, 86). For “high-risk” patients, postoperative combination radiotherapy and sequential chemotherapy is standard of care (2, 24, 25). The most common chemotherapy regimens are procarbazine, lomustine and vincristine (PCV) or temozolomide (TMZ). All cooperative groups recommend radiotherapy and sequential chemotherapy for patients with Grade 2 IDH-mutant gliomas aged over 40 years and with STR (2, 24, 25). Patients with Grade 3 oligodendrogliomas with 1p/19q co-deletion should receive radiotherapy with PCV (2, 24, 25). Those with Grade 3 astrocytomas and no 1p/19q co-deletion receive radiotherapy with adjuvant TMZ (2, 24, 25).
TMZ is an oral alkylating agent that results in the methylation of DNA at the O6-guanine, as well as N7-guanine and N3-adenine residues (87). Methylation of O6-guanine results in the O6-methylgaunine (O6-meg) lesion. O6-meg lesions are repaired by O6-methylguanine DNA methyltransferase (MGMT), a process that is dependent on the number of MGMT molecules per cell and rate of MGMT regeneration (88). Unrepaired O6-meg lesions incorrectly pairs with thymine during DNA replication instead of cytosine, triggering the DNA mismatch repair (MMR) system and repeated cycles of double-stranded breakages in a process called futile cycling (87). The accumulation of these DNA strand breaks eventually results in cell cycle arrest and activation of apoptosis. Thus, cytotoxicity of TMZ is dependent on an intact MMR pathway and low levels of MGMT.
The CATNON trial (89), a phase 3 randomized controlled trial, established the use of adjuvant rather than concurrent TMZ with radiotherapy on 1p/19q non-co-deleted anaplastic gliomas, particularly those with IDH mutations. The CATNON trial used a 2 x 2 factorial design, enrolling 751 patients to receive either radiotherapy alone, or radiotherapy with concurrent and/or adjuvant TMZ. Adjuvant TMZ showed significant improvement in OS (median OS 82.3 months, 5Y OS 56% vs median OS 46.9 months, 5Y OS 44% in control arm). The second interim analysis did not demonstrate a statistically significant benefit of concurrent TMZ compared to radiotherapy alone. TMZ was generally well tolerated with the most common Grade 3 and 4 TRAE being related to hematological side effects, particularly thrombocytopenia and neutropenia.
Importantly, this study holds significant relevance for IDH-mutant gliomas. Mutation status was analyzed in 671 of the 751 patients, with 436 harboring IDH1 or IDH2 mutations, evenly distributed across the four arms of the study. Overall, IDH1/IDH2 mutation status had a significant impact on survival with median OS 19.9 months in the wild-type group vs 98.4 months in the IDH-mutant group (HR 0.14, p<0.0001). Significantly, in patients with IDH1 and IDH2 wild-type tumors, neither concurrent nor adjuvant TMZ improved OS compared with radiotherapy alone, whereas IDH-mutated tumors had significantly improved OS with the addition of adjuvant TMZ. Median PFS in IDH-mutant patients treated with any TMZ was 77.0 months compared to 34.2 months in patients treated with radiotherapy alone (p<0.0001).
The results of the CATNON trial suggest that there is a crucial role for adjuvant TMZ in adult IDH-mutant LGG and this is currently recommended as standard of care in the settings outlined above. However, in the pediatric and AYA population TMZ-induced hypermutation and acquired resistance need to be considered given the potential for prolonged survival and ongoing exposure to alkylating agents. TMZ-resistance may occur through the mutagenic action of TMZ on DNA repair genes resulting in acquired deficiencies in MMR (90). Loss of MMR function leads to ongoing mispairing of guanine with thymine; however, unrepaired DNA damage is no longer detected and cells are unable to activate apoptosis (91). In the absence of MGMT-mediated repair and intact MMR, cells incur a large number of G:C>A:T transitions throughout the genome upon DNA replication, including thousands in coding regions (87). This mutational signature is known as single base substitution (SBS) signature 11 and is commonly seen in hypermutated gliomas after exposure to alkylating agents (92). The acquisition of mutations that inactivate the MMR pathway and continued TMZ exposure results in hypermutation. Despite these considerations, TMZ remains widely used in pediatric HGG.
Another recent trial, the CODEL trial, was initially designed to compare the efficacy of radiotherapy alone, radiotherapy + TMZ, or TMZ monotherapy in patients with 1p/19q codeleted grade 3 oligodendroglioma (93). Initial analysis demonstrated a significantly shorter PFS in patients receiving TMZ monotherapy compared to RT (HR 3.12, p=0.014). Of the patients included that were evaluable for IDH mutation status, 30 (86%) were IDH mutated. Given the results, the trial has been redesigned to compare adjuvant radiotherapy + TMZ and radiotherapy + PCV among patients with grade 2 and 3 oligodendroglioma. The trial is ongoing with results awaited. However, recent results from the French POLA network demonstrate that in a cohort of 306 patients with grade 3 oligodendroglioma, radiotherapy + PCV was associated with significantly improved 5-year and 10-year OS compared to radiotherapy + TMZ (5Y OS 89% PCV vs 75% TMZ, p=0.0014; 10Y OS 73% PCV vs 60% TMZ, p=0.0003) (94).
5 Mutant IDH inhibitors
Surgical resection, chemotherapy and radiation therapy are associated with treatment-related toxicities including long term neurocognitive disorders (1). Targeted therapies are clearly of emerging interest to avoid long-term toxicities particularly in the pediatric and AYA cohort.
IDH inhibitors reduce D2-HG concentrations in tumor xenograft models (95) and in clinical pharmacology studies (26, 96, 97). Inhibition of mutant IDH in tumor cells, and the associated reduction in D2-HG production, can restore normal cellular differentiation and provide therapeutic benefit in cancers harboring IDH mutations (42). There are several IDH inhibitors currently under clinical investigation with biologic activity, good tolerance, and evidence of clinical activity. Studies have demonstrated the safety and feasibility of these agents in patients with glioma, summarized in Table 2.
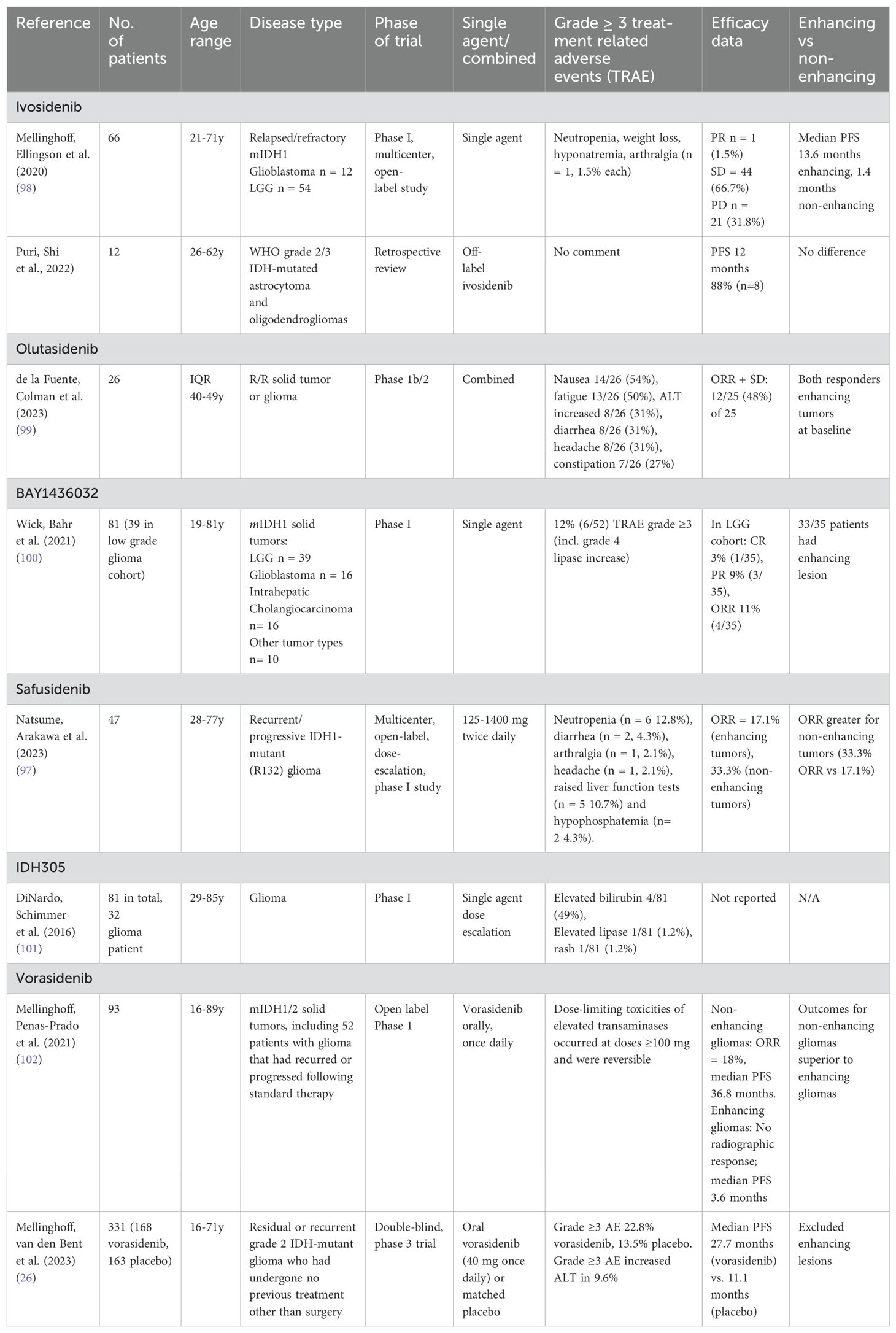
Table 2. Trials of IDH inhibitors in patients with glioma.
The United States Food and Drug Administration (FDA) and the Australian Therapeutic Goods Administration (TGA) recently approved one such agent, vorasidenib, for adult and pediatric patients 12 years and older with grade 2 astrocytoma or oligodendroglioma with a susceptible IDH1 or IDH2 mutation (103).
5.1 IDH1 specific
5.1.1 Ivosidenib (AG120)
Ivosidenib is an oral, potent, highly specific targeted small-molecule inhibitor of mutant IDH1 (1, 13). Preclinical data shows that ivosidenib inhibits invasion and migration of IDH1 mutated chondrosarcoma cell lines (104). Pharmacokinetic analysis showed that ivosidenib was detectable in the brain-tumor tissue, suggesting the molecule is able to cross the blood-brain barrier (105). Mouse xenograft models of human mIDH1-R132H glioma show strong inhibition of D2-HG production in brain tumor samples (105). Furthermore, plasma D2-HG levels decreased in adults treated with ivosidenib (106).
Clinical studies using ivosidenib have been conducted in adults with CNS tumors (98), cholangiocarcinoma (107–109), chondrosarcoma (106), and myeloid malignancies (96, 110–112), either as a single agent or in combination with chemotherapy (Supplementary Table 1). The most common grade ≥ 3 treatment related adverse events (TRAE) include myelosuppression, ascites, and QT prolongation.
5.1.2 Olutasidenib (FT-2102)
Olutasidenib (FT-2102) is a potent, selective, oral, small-molecule inhibitor of mutant IDH1 that entered clinical development in 2016 (17, 42, 113). Olutasidenib is a blood brain barrier penetrant (95, 114) quinolinone-based non-competitive inhibitor of mutant IDH1 that binds to an allosteric site, a hydrophobic pocket near the IDH1 homodimer interface (17, 115).
In adult clinical trials in myeloid malignancies (17, 42, 116) and relapsed/refractory solid tumors (including gliomas) (99), the drug has been well tolerated with most common serious (Grade 3 and above) TRAE include hepatic transaminitis and myelosuppression (Supplementary Table 2). Other common TRAE included nausea, fatigue, diarrhea, constipation, and headache (99). Olutasidenib was granted FDA approval to treat patients with relapsed/refractory IDH1 mutant AML on 1 December 2022 (42, 113).
In a phase 1b/2 trial enrolling 26 adult patients with relapsed/refractory WHO Grade 3 and 4 IDH1 R132X-mutant glioma, olutasidenib resulted in a disease control rate (objective response plus stable disease) of 48% (99). Best response was partial response, achieved in two (8%) patients and eight patients (32%) had stable disease for at least four months as per Response Assessment in Neuro-Oncology (RANO) response criteria. These results suggest olutasidenib demonstrates preliminary evidence of clinical activity in a heavily pretreated population.
5.1.3 BAY1436032
BAY1436032 is an oral small-molecule inhibitor of R132-mutant IDH1 that is active in preclinical models of mIDH1 cancer (117). BAY 1436032 strongly reduces D2-HG levels in cells carrying IDH1-R132H, -R132C, -R132G, -R132S and -R132L mutations, with a median maximal reduction of plasma R-2-hydroxyglutarate levels of 76% (100, 118). BAY 1436032 partially crosses the blood brain barrier; maximal intraparenchymal BAY 1436032 concentration in mouse brain amounted to 38% of that in plasma levels (118).
BAY1436032 has been studied in adults with solid tumors (100) and AML (119) (Supplementary Table 3). The most common serious TRAE include raised lipase levels and myelosuppression.
5.1.4 Safusidenib (DS-1001B/AB-218)
Safusidenib (DS-1001b/AB-218) is an orally available, brain-penetrant, selective inhibitor of R132-mutant IDH1 that has shown efficacy in preclinical models of glioma (49, 120). It has been studied in adults with recurrent or progressive mIDH glioma (Supplementary Table 4) (97). Grade ≥ 3 TRAE related to DS-1001 included neutropenia and hepatotoxicity. Studies of Safusidenib in patients with IDH1-mutated WHO glioma are ongoing (NCT04458272, NCT05303519 and NCT05577416).
5.1.5 IDH305
IDH305 is a potent and selective mutant IDH1 inhibitor that has demonstrated brain exposure in rodents, D2-HG reduction, and efficacy in a patient-derived IDH1 mutant xenograft tumor model (121). It has been studied in adults with CNS tumors and myeloid malignancy, identifying hepatotoxicity, tumor lysis syndrome and differentiation syndrome as the most common serious TRAE (Supplementary Table 5) (101, 122).
5.2 IDH2 specific
5.2.1 Enasidenib (AG221)
Enasidenib is a small molecule inhibitor of the IDH2 enzyme. In preclinical studies, enasidenib decreased total serum D2-HG by more than 90%, reduced abnormal histone hypermethylation, and restored myeloid differentiation (96, 123, 124). In patients with IDH2-mutated AML, enasidenib reduced serum D2-HG levels resulting in increased percentages of mature myeloid cells in the bone marrow (112).
Enasidenib has been studied in adults with AML (Supplementary Table 6) (96, 125, 126). The most common serious adverse reaction was QT prolongation, hepatotoxicity, and myelosuppression.
There is no evidence that enasidenib crosses the blood brain barrier. IDH2 mutations are less prevalent in gliomas, rendering an IDH2 inhibitor less likely to be beneficial in this cohort.
5.3 Combined IDH1 and IDH2 inhibitors
5.3.1 Vorasidenib (AG-881)
Vorasidenib is an oral drug pan inhibitor of both mutant IDH1 and IDH2, shown to penetrate the mouse brain and reduce D2-HG production by over 97% in an orthotopically engrafted patient-derived xenograft (PDX) (127, 128). A recent study revealed that isoform switching from mIDH1 to mIDH2 or vice versa may represent a mechanism of acquired resistance, sparking interest in the use of pan-inhibitors (129, 130).
Vorasedinib has been studied in adults with solid tumors (26) and hematologic malignancy (130) (Supplementary Table 7). Vorasidenib is well tolerated with grade 3 or higher TRAE occurring in less than half of patients (26). The most common adverse Grade 3 or higher TRAE include hepatic transaminitis, diarrhea, and gastrointestinal hemorrhage. A small number of patients require dose reduction or cessation due to adverse effects (26).
Mellinghoff et al. (131) report the use of vorasidenib or ivosidenib for IDH-R132H mutant LGG in a perioperative phase 1 trial. In this study, patients received pre-operative vorasidenib or ivosidenib and continued treatment post-surgery until disease progression or unacceptable toxicity. Tissue analysis was performed from 40/49 patients, demonstrating a reduction in concentration of D-2-hydroxyglutarate (D2-HG), the metabolic product of mIDH enzymes, by 92.6% following vorasidenib and 91.1% following ivosidenib compared to patients receiving placebo. Both drugs were well tolerated without any surgical delays. In the vorasidenib group, 7/24 (29.2%) experienced grade 3 or higher TRAE including brain abscess, tooth infection, aphasia, brain edema, hydrocephalus, alanine aminotransferase (ALT) increase, anemia, hyperglycemia, and hypophosphatemia. Rate of grade 3 TRAE was similar in the ivosidenib group with 6/25 (24%) experiencing hyponatremia, leukopenia, subdural hematoma, invasive ductal breast carcinoma, brain edema, brain injury, hemiparesis, syncope, mental status change and pneumothorax.
Anti-tumor effect was assessed with objective response rate (ORR) in the group treated with 10mg daily vorasidenib 10% (1/10), and 50mg daily vorasidenib 42.9% (6/14). For ivosidenib, those treated with 250mg twice daily had ORR 12.5% (1/8) compared with 35.7% (5/14) treated with 500mg daily. Based on these preliminary antitumor activity and enzyme inhibition findings, vorasidenib 50mg daily was initially carried forward to the global phase 3 INDIGO study in grade 2 mIDH non-enhancing glioma (26). A coated-tablet formulation equivalent to 40mg daily has since been introduced.
The pivotal double-blinded, phase 3 INDIGO study (26) evaluated vorasidenib in adult patients with residual or recurrent grade 2 non-enhancing IDH-mutant gliomas. Of 331 patients, 168 patients were randomized to receive vorasidenib 40mg once daily. Vorasidenib significantly improved imaging-based PFS compared to placebo (median PFS 27.7 months vs 11.1 months, HR 0.39, p<0.001). Additionally, vorasidenib delayed the need for further interventions (HR 0.26, p<0.001). At a median follow-up of 14.0 months, 131 were continuing to receive vorasidenib within the intervention group. The impact on OS will take years to elucidate but will be likely confounded by the crossover design (132). Furthermore, although the eligibility criteria included patients aged 12 years and over, the youngest patient enrolled was 16 years of age and no patients under 18 years of age were randomized to the intervention arm. AYA patients were also underrepresented in this cohort with a mean age of 40.5 years.
5.4 Introducing IDH inhibitors into standard of care treatment
Understanding of the biological impact of IDH inhibitors in glioma is evolving. Preclinical modelling in mIDH glioma is complicated by the indolent nature of the tumors which makes generation of patient-derived cell lines and xenograft models difficult (49, 133). However, using a combination of RNA single-cell analysis, bulk RNA-sequencing analysis and in vitro modelling, Spitzer et al. were able to demonstrate differentiation of oligodendrogliomas to an “astrocytoma-like” phenotype with decreased cell cycle expression, thus resulting in less proliferation, in patients treated with vorasidenib (134).
The specific role of IDH mutations in driving tumor growth and aggressiveness at the time of recurrence is also unclear. Recurrent tumors are more likely to display a hypermutation phenotype and genome-wide loss of DNA methylation (135, 136). Several recent studies also confirm the present of copy number alterations, particularly CDKN2A/B loss and CDK4 amplification, clinically conferring poorer prognosis in IDH-mutant astrocytomas (137–141). However, the effect of CDKN2A/B deletion in oligodendroglioma is less clear (142). These findings may explain the clinical data demonstrating poorer survival outcomes of high-grade mIDH glioma patients treated at recurrence with mIDH inhibitors (143).
The role of IDH inhibitors in standard of care for IDH-mutant gliomas is yet to be determined. Overall, studies suggest superior activity in patients with non-enhancing tumors compared with enhancing tumors (97, 98, 100, 102), with enhancement typically indicating transformation of LGG to higher grades (98). The suggestion based on the above trials would be that these inhibitors may be useful in the early, indolent course of disease (133). However, many questions remain including the efficacy of IDH inhibitors compared to current standard of care radiotherapy and chemotherapy regimens.
5.5 Emerging mechanisms of resistance
There is a lack of current understanding of how prolonged use of IDH inhibitors alters the biology of mIDH gliomas, leading to resistance (133). IDH inhibitor resistance has been reported in adults with AML through trans or cis dimer-interface mutations (144), receptor tyrosine kinase (RTK) pathway mutations (145) or second site mutations (146, 147). In adults with cholangiocarcinoma resistance mechanisms include a second IDH mutation (129, 148). Understanding the mechanism of resistance is important to guide salvage treatment options and to identify other potential targets.
5.6 Combination studies
Several reports and a recently published clinical trial by the International Replication Repair Deficiency Consortium (IRRDC) demonstrating durable responses and prolonged survival in patients with glioma and high mutational burden (149, 150). In contrast, in Suwala’s study of patients with PMMRDIA, limited effect was seen in three patients treated with immune checkpoint inhibitors (ICI) (78). A subsequent IRRDC study of mIDH RRD HGG confirmed lower PFS in mIDH RRD HGG compared to sporadic mIDH HGG treated with ICI monotherapy (151).
The lack of efficacy in this cohort is thought to be attributed to the immune suppressive effects of the IDH mutation on the tumor microenvironment (80). IDH mutations result in down-regulation of genes involved in immune activation of cancer cells likely occurring either by a direct metabolic inhibitory effect of D2-HG or through epigenetic reprogramming by hypermethylation of promotors of immune-related genes (47). Hence, D2-HG strongly represses T-cell activity, acting as a protective mechanism for tumor cells by evading the immune system. Furthermore, hypermethylation has been implicated in the downregulation of immune checkpoints such as PD-L1 and CTLA4 (78, 152).
The combination of IDH inhibitors with ICI is therefore promising, specifically for PMMRDIA where novel therapeutic strategies are required. In this combination, IDH mutation driven immunosuppression could be reversed with IDH inhibitor treatment, enhancing immune checkpoint blockade. In the clinic, Das et al. have reported favorable responses for mIDH-RRD-HGG receiving combination ICI and IDH inhibitor therapy with prolonged survival at 12 months compared to those without IDH mutations (151). To test this further, a number of clinical trials are ongoing including a phase 2 trial combining nivolumab with ivosidenib in patients with advanced IDH-mutant solid tumors (NCT04056910) as well as a phase 1 trial of pembrolizumab and vorasidenib in patients with relapsed/refractory Grade 2 and 3 mIDH glioma (NCT0584622).
6 Need for trials for IDH-mutant LGG in children, adolescents and young adults
As outlined above, pediatric IDH-mutant LGG are rare, although with increased access to advanced genomic testing, these tumors are being identified more readily (21). There remains a paucity of data to describe their natural history, treatment strategies and outcomes. Specifically, the biological characteristics and behavior compared to adult IDH-mutant LGG remains unclear (21). Malignant transformation in the adult cohort is well documented (153) and an aggressive upfront approach is warranted. In comparison to other pediatric/AYA LGG patients, mIDH tumors appear to have a higher risk for malignant transformation (21, 76); however is it unclear whether IDH mutations confer the same prognostic significance as in the adult cohort.
These considerations have critical treatment implications as approaches vary significantly amongst institutions with no established standard of care. Radiotherapy may be delayed or avoided; in the retrospective study by Yeo et al. only 22/76 (28.9%) of patients received upfront radiotherapy (21). In the adult setting, radiotherapy is a cornerstone to the treatment of gliomas and attempts to de-intensify treatment post resection to chemotherapy alone have so far proved unsatisfactory (93, 132). Significant concerns regarding toxicities in children are potential reasons to consider deferral of radiotherapy (132, 154). In particular, CNS directed radiotherapy has neurocognitive sequelae, including decline in cognition, processing speed, fine motor skills, verbal fluency, and delayed attention (154). Endocrinopathies can results in issues with growth and puberty (154). There is an increased risk of late neurovascular events such as stroke (155). However, radiotherapy, particularly in the setting of STR or recurrent/progressive disease, plays an important role in achieving local control and prolonging PFS (154). Therefore, the optimal timing and modality is important to clarify.
The use of novel targeted therapies is an attractive option to prevent long-term toxicities in pediatrics. However, access to IDH inhibitors for pediatric patients with IDH-mutant tumors is difficult. Currently, ivosidenib has available pediatric dosing based on a COG Phase II MATCH trial for IDH1 mutant tumors. However, this trial was closed early prior to meeting study endpoints due to slow accrual (NCT04195555). Although vorasidenib has been approved by the FDA, there is no published dosing for children under 12 years. An upcoming CONNECT trial (NCT06161974) will assess the combination of olutasidenib (FT-2102) and temozolomide for pediatric IDH-mutant HGG. The sparsity of clinical trials leaves many questions left to be answered such as optimal timing and duration of therapy.
Furthermore, the long-term impact of these novel targeted therapies is unknown. Pleasingly, health related quality of life (HrQOL) and neurocognitive outcomes assessed for adult patients enrolled in the INDIGO trial did not demonstrate any clinically meaningful deterioration from baseline at any timepoint compared to placebo (156). While these preliminary data suggest that the neurocognitive effects of these agents are acceptable in the adult population, this needs to be weighed against the impact of potential tumor progression, a key driver of cognitive decline (132). Given the skewed burden of IDH-mutant gliomas to older children and adolescents, fertility is also an important consideration given the unknown impact on fertility, pregnancies, and the fetus (132).
The role of IDH inhibitors in standard of care for IDH-mutant gliomas is yet to be determined. Overall, studies suggest superior activity in patients with non-enhancing tumors compared with enhancing tumors (97, 98, 100, 102), with enhancement typically indicating transformation of LGG to higher grades (98). The suggestion based on the above trials would be that these inhibitors may be useful in the early, indolent course of disease (133). However, many questions remain including the efficacy of IDH inhibitors compared to current standard of care radiotherapy and chemotherapy regimens.
We suggest that by early treatment with these inhibitors we may change the natural history of eventual progression to higher risk disease and eventual death of these patients. By intercepting the physiologic changes to the tumor and its microenvironment, we propose we completely alter the disease course and prevent the eventual development of high grade glioma.
7 Conclusion
While several advances have been made in the field of adult IDH-mutant LGG, much is still left to be elucidated in the pediatric and AYA setting. Increased access to next-generation sequencing has resulted in the increased recognition of these tumors affecting older children and adolescents. Tumor biology in this group is unclear and the impact of available therapies commonly used in the adult setting need to be carefully considered. Temozolomide, used frequently in the adult setting for its favorable toxicity profile, has a clear risk of hypermutation whilst IDH inhibitors have not been tested in the pediatric CNS tumor population and need further study to determine impact on tumor biology and efficacy at recurrence. A small but significant proportion of pediatric patients with mIDH glioma will have PMMRDIA, with poorer survival outcomes and therefore this must be considered at diagnosis in this age group. Current practice remains variable with no standard of care defined, demonstrating an urgent need for pediatric-specific clinical trials.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Author contributions
LE: Writing – original draft, Writing – review & editing. ST: Writing – original draft, Writing – review & editing. AD: Writing – original draft, Writing – review & editing. DE: Writing – original draft, Writing – review & editing. JW: Writing – original draft, Writing – review & editing. JH: Writing – original draft, Writing – review & editing, Conceptualization, Supervision. SV: Writing – original draft, Writing – review & editing, Conceptualization, Supervision.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. JRH receives research support from the McClurg Foundation and the Hospital Research Foundation.
Acknowledgments
Figure 1 was created in BioRender. Evans, L (2024). BioRender.com/e12y091, Figure 2 was created in BioRender. Evans, L (2024). BioRender.com/a88o851.
Conflict of interest
JW reports research funding from AnHeart Therapeutics to institute; received consulting fees from AnHeart Therapeutics and Servier; being on advisory boards for Roche and Merck; is a data safety monitoring member for Telix Pharmaceuticals. JH reports honoraria from Alexion and Bayer Pharmaceuticals. JH sits on advisory board for Servier.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2024.1515538/full#supplementary-material
References
1. Gatto L, Franceschi E, Tosoni A, Di Nunno V, Maggio I, Lodi R, et al. IDH inhibitors and beyond: the cornerstone of targeted glioma treatment. Mol Diagn Ther. (2021) 25:457–73. doi: 10.1007/s40291-021-00537-3
2. Miller JJ, Gonzalez Castro LN, McBrayer S, Weller M, Cloughesy T, Portnow J, et al. Isocitrate dehydrogenase (IDH) mutant gliomas: A Society for Neuro-Oncology (SNO) consensus review on diagnosis, management, and future directions. Neuro Oncol. (2023) 25:4–25. doi: 10.1093/neuonc/noac207
3. Miller JJ, Shih HA, Andronesi OC, Cahill DP. Isocitrate dehydrogenase-mutant glioma: Evolving clinical and therapeutic implications. Cancer. (2017) 123:4535–46. doi: 10.1002/cncr.v123.23
4. Yamasaki F. Adolescent and young adult brain tumors: current topics and review. Int J Clin Oncol. (2022) 27:457–64. doi: 10.1007/s10147-021-02084-7
5. Ostrom QT, Cioffi G, Waite K, Kruchko C, Barnholtz-Sloan JS. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2014-2018. Neuro Oncol. (2021) 23:iii1–iii105. doi: 10.1093/neuonc/noab200
6. Abla H, Sollazzo M, Gasparre G, Iommarini L, Porcelli AM. The multifaceted contribution of α-ketoglutarate to tumor progression: An opportunity to exploit? Semin Cell Dev Biol. (2020) 98:26–33. doi: 10.1016/j.semcdb.2019.05.031
7. Bennett J, Nobre L, Sheth J, Ryall S, Fang K, Johnson M, et al. LGG-41. The clinical and molecular landscape of gliomas in adolescents and young adults. Neuro Oncol. (2022) 24:i97. doi: 10.1093/noajnl/vdad071.008
8. Koh HJ, Lee SM, Son BG, Lee SH, Ryoo ZY, Chang KT, et al. Cytosolic NADP+-dependent isocitrate dehydrogenase plays a key role in lipid metabolism. J Biol Chem. (2004) 279:39968–74. doi: 10.1074/jbc.M402260200
9. Tang F, Pan Z, Wang Y, Lan T, Wang M, Li F, et al. Advances in the immunotherapeutic potential of isocitrate dehydrogenase mutations in glioma. Neurosci Bull. (2022) 38:1069–84. doi: 10.1007/s12264-022-00866-1
10. Kaminska B, Czapski B, Guzik R, Krol SK, Gielniewski B. Consequences of IDH1/2 mutations in gliomas and an assessment of inhibitors targeting mutated IDH proteins. Molecules. (2019) 24:968. doi: 10.3390/molecules24050968
11. Liu Y, Lang F, Chou FJ, Zaghloul KA, Yang C. Isocitrate dehydrogenase mutations in glioma: genetics, biochemistry, and clinical indications. Biomedicines. (2020) 8:294. doi: 10.3390/biomedicines8090294
12. Dang L, Su SM. Isocitrate dehydrogenase mutation and (R)-2-hydroxyglutarate: from basic discovery to therapeutics development. Annu Rev Biochem. (2017) 86:305–31. doi: 10.1146/annurev-biochem-061516-044732
13. Montesinos P, Recher C, Vives S, Zarzycka E, Wang J, Bertani G, et al. Ivosidenib and azacitidine in IDH1-mutated acute myeloid leukemia. N Engl J Med. (2022) 386:1519–31. doi: 10.1056/NEJMoa2117344
14. Prensner JR, Chinnaiyan AM. Metabolism unhinged: IDH mutations in cancer. Nat Med. (2011) 17:291–3. doi: 10.1038/nm0311-291
15. Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. (2009) 361:1058–66. doi: 10.1056/NEJMoa0903840
16. Paschka P, Schlenk RF, Gaidzik VI, Habdank M, Krönke J, Bullinger L, et al. IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with NPM1 mutation without FLT3 internal tandem duplication. J Clin Oncol. (2010) 28:3636–43. doi: 10.1200/JCO.2010.28.3762
17. Watts JM, Baer MR, Yang J, Prebet T, Lee S, Schiller GJ, et al. Olutasidenib alone or with azacitidine in IDH1-mutated acute myeloid leukaemia and myelodysplastic syndrome: phase 1 results of a phase 1/2 trial. Lancet Haematol. (2023) 10:e46–58. doi: 10.1016/S2352-3026(22)00292-7
18. DiNardo CD, Ravandi F, Agresta S, Konopleva M, Takahashi K, Kadia T, et al. Characteristics, clinical outcome, and prognostic significance of IDH mutations in AML. Am J Hematol. (2015) 90:732–6. doi: 10.1002/ajh.24072
19. Pollack IF, Hamilton RL, Sobol RW, Nikiforova MN, Lyons-Weiler MA, LaFramboise WA, et al. IDH1 mutations are common in Malignant gliomas arising in adolescents: a report from the Children’s Oncology Group. Childs Nerv Syst. (2011) 27:87–94. doi: 10.1007/s00381-010-1264-1
20. De Carli E, Wang X, Puget S. IDH1 and IDH2 mutations in gliomas. N Engl J Med. (2009) 360:2248. doi: 10.1056/NEJMc090593
21. Yeo KK, Alexandrescu S, Cotter JA, Vogelzang J, Bhave V, Li MM, et al. Multi-institutional study of the frequency, genomic landscape, and outcome of IDH-mutant glioma in pediatrics. Neuro-oncol. (2023) 25:199–210. doi: 10.1093/neuonc/noac132
22. Reitman ZJ, Yan H. Isocitrate dehydrogenase 1 and 2 mutations in cancer: alterations at a crossroads of cellular metabolism. J Natl Cancer Inst. (2010) 102:932–41. doi: 10.1093/jnci/djq187
23. Hartmann C, Meyer J, Balss J, Capper D, Mueller W, Christians A, et al. Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: a study of 1,010 diffuse gliomas. Acta Neuropathol. (2009) 118:469–74. doi: 10.1007/s00401-009-0561-9
24. Mohile NA, Messersmith H, Gatson NT, Hottinger AF, Lassman A, Morton J, et al. Therapy for diffuse astrocytic and oligodendroglial tumors in adults: ASCO-SNO guideline. J Clin Oncol. (2022) 40:403–26. doi: 10.1200/JCO.21.02036
25. Weller M, van den Bent M, Preusser M, Le Rhun E, Tonn JC, Minniti G, et al. EANO guidelines on the diagnosis and treatment of diffuse gliomas of adulthood. Nat Rev Clin Oncol. (2021) 18:170–86. doi: 10.1038/s41571-020-00447-z
26. Mellinghoff IK, van den Bent MJ, Blumenthal DT, Touat M, Peters KB, Clarke J, et al. Vorasidenib in IDH1- or IDH2-mutant low-grade glioma. N Engl J Med. (2023) 389:589–601. doi: 10.1056/NEJMoa2304194
27. Vatrinet R, Leone G, De Luise M, Girolimetti G, Vidone M, Gasparre G, et al. The α-ketoglutarate dehydrogenase complex in cancer metabolic plasticity. Cancer Metab. (2017) 5:3. doi: 10.1186/s40170-017-0165-0
28. Yoo HC, Yu YC, Sung Y, Han JM. Glutamine reliance in cell metabolism. Exp Mol Med. (2020) 52:1496–516. doi: 10.1038/s12276-020-00504-8
29. Minard KI, McAlister-Henn L. Dependence of peroxisomal beta-oxidation on cytosolic sources of NADPH. J Biol Chem. (1999) 274:3402–6. doi: 10.1074/jbc.274.6.3402
30. Kil IS, Kim SY, Lee SJ, Park JW. Small interfering RNA-mediated silencing of mitochondrial NADP+-dependent isocitrate dehydrogenase enhances the sensitivity of HeLa cells toward tumor necrosis factor-alpha and anticancer drugs. Free Radic Biol Med. (2007) 43:1197–207. doi: 10.1016/j.freeradbiomed.2007.07.009
31. Bleeker FE, Atai NA, Lamba S, Jonker A, Rijkeboer D, Bosch KS, et al. The prognostic IDH1(R132) mutation is associated with reduced NADP+-dependent IDH activity in glioblastoma. Acta Neuropathol. (2010) 119:487–94. doi: 10.1007/s00401-010-0645-6
32. Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature. (2011) 481:380–4. doi: 10.1038/nature10602
33. Ward PS, Cross JR, Lu C, Weigert O, Abel-Wahab O, Levine RL, et al. Identification of additional IDH mutations associated with oncometabolite R(-)-2-hydroxyglutarate production. Oncogene. (2012) 31:2491–8. doi: 10.1038/onc.2011.416
34. Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. (2009) 360:765–73. doi: 10.1056/NEJMoa0808710
35. Pirozzi CJ, Yan H. The implications of IDH mutations for cancer development and therapy. Nat Rev Clin Oncol. (2021) 18:645–61. doi: 10.1038/s41571-021-00521-0
36. Reitman ZJ, Jin G, Karoly ED, Spasojevic I, Yang J, Kinzler KW, et al. Profiling the effects of isocitrate dehydrogenase 1 and 2 mutations on the cellular metabolome. Proc Natl Acad Sci U S A. (2011) 108:3270–5. doi: 10.1073/pnas.1019393108
37. Fendt SM, Bell EL, Keibler MA, Olenchock BA, Mayers JR, Wasylenko TM, et al. Reductive glutamine metabolism is a function of the α-ketoglutarate to citrate ratio in cells. Nat Commun. (2013) 4:2236. doi: 10.1038/ncomms3236
38. Badur MG, Muthusamy T, Parker SJ, Ma S, McBrayer SK, Cordes T, et al. Oncogenic R132 IDH1 mutations limit NADPH for de novo lipogenesis through (D)2-hydroxyglutarate production in fibrosarcoma sells. Cell Rep. (2018) 25:1018–26.e4. doi: 10.1016/j.celrep.2018.09.074
39. Han S, Liu Y, Cai SJ, Qian M, Ding J, Larion M, et al. IDH mutation in glioma: molecular mechanisms and potential therapeutic targets. Br J Cancer. (2020) 122:1580–9. doi: 10.1038/s41416-020-0814-x
40. Grassian AR, Parker SJ, Davidson SM, Divakaruni AS, Green CR, Zhang X, et al. IDH1 mutations alter citric acid cycle metabolism and increase dependence on oxidative mitochondrial metabolism. Cancer Res. (2014) 74:3317–31. doi: 10.1158/0008-5472.CAN-14-0772-T
41. Bhavya B, Anand CR, Madhusoodanan UK, Rajalakshmi P, Krishnakumar K, Easwer HV, et al. To be wild or mutant: role of isocitrate dehydrogenase 1 (IDH1) and 2-hydroxy glutarate (2-HG) in gliomagenesis and treatment outcome in glioma. Cell Mol Neurobiol. (2020) 40:53–63. doi: 10.1007/s10571-019-00730-3
42. de Botton S, Fenaux P, Yee K, Récher C, Wei AH, Montesinos P, et al. Olutasidenib (FT-2102) induces durable complete remissions in patients with relapsed or refractory IDH1-mutated AML. Blood Adv. (2023) 7:3117–27. doi: 10.1182/bloodadvances.2022009411
43. Losman JA, Kaelin WG Jr. What a difference a hydroxyl makes: mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev. (2013) 27:836–52. doi: 10.1101/gad.217406.113
44. Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell. (2011) 19:17–30. doi: 10.1016/j.ccr.2010.12.014
45. Seok J, Yoon SH, Lee SH, Jung JH, Lee YM. The oncometabolite d−2−hydroxyglutarate induces angiogenic activity through the vascular endothelial growth factor receptor 2 signaling pathway. Int J Oncol. (2019) 54:753–63. doi: 10.3892/ijo.2018.4649
46. Koivunen P, Lee S, Duncan CG, Lopez G, Lu G, Ramkissoon S, et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature. (2012) 483:484–8. doi: 10.1038/nature10898
47. Bunse L, Pusch S, Bunse T, Sahm F, Sanghvi K, Friedrich M, et al. Suppression of antitumor T cell immunity by the oncometabolite (R)-2-hydroxyglutarate. Nat Med. (2018) 24:1192–203. doi: 10.1038/s41591-018-0095-6
48. Notarangelo G, Spinelli JB, Perez EM, Baker GJ, Kurmi K, Elia I, et al. Oncometabolite d-2HG alters T cell metabolism to impair CD8(+) T cell function. Science. (2022) 377:1519–29. doi: 10.1126/science.abj5104
49. Ruda R, Horbinski C, van den Bent M, Preusser M, Soffietti R. IDH inhibition in gliomas: from preclinical models to clinical trials. Nat Rev Neurol. (2024). doi: 10.1038/s41582-024-00967-7
50. Pansuriya TC, van Eijk R, d’Adamo P, van Ruler MA, Kuijjer ML, Oosting J, et al. Somatic mosaic IDH1 and IDH2 mutations are associated with enchondroma and spindle cell hemangioma in Ollier disease and Maffucci syndrome. Nat Genet. (2011) 43:1256–61. doi: 10.1038/ng.1004
51. Bonnet C, Thomas L, Psimaras D, Bielle F, Vauléon E, Loiseau H, et al. Characteristics of gliomas in patients with somatic IDH mosaicism. Acta Neuropathol Commun. (2016) 4:31. doi: 10.1186/s40478-016-0302-y
52. El Abiad JM, Robbins SM, Cohen B, Levin AS, Valle DL, Morris CD, et al. Natural history of Ollier disease and Maffucci syndrome: Patient survey and review of clinical literature. Am J Med Genet A. (2020) 182:1093–103. doi: 10.1002/ajmg.a.v182.5
53. Borger DR, Tanabe KK, Fan KC, Lopez HU, Fantin VR, Straley KS, et al. Frequent mutation of isocitrate dehydrogenase (IDH)1 and IDH2 in cholangiocarcinoma identified through broad-based tumor genotyping. Oncologist. (2012) 17:72–9. doi: 10.1634/theoncologist.2011-0386
54. Amary MF, Bacsi K, Maggiani F, Damato S, Halai D, Berisha F, et al. IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J Pathol. (2011) 224:334–43. doi: 10.1002/path.v224.3
55. Shibata T, Kokubu A, Miyamoto M, Sasajima Y, Yamazaki N. Mutant IDH1 confers an in vivo growth in a melanoma cell line with BRAF mutation. Am J Pathol. (2011) 178:1395–402. doi: 10.1016/j.ajpath.2010.12.011
56. Lopez GY, Reitman ZJ, Solomon D, Waldman T, Bigner DD, McLendon RE, et al. IDH1R132 mutation identified in one human melanoma metastasis, but not correlated with metastases to the brain. Biochem Biophys Res Commun. (2010) 398:585–7. doi: 10.1016/j.bbrc.2010.06.125
57. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 world health organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. (2016) 131:803–20. doi: 10.1007/s00401-016-1545-1
58. Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol. (2021) 23:1231–51. doi: 10.1093/neuonc/noab106
59. Dodgshun AJ, Maixner WJ, Heath JA, Sullivan MJ, Hansford JR. Single agent carboplatin for pediatric low-grade glioma: A retrospective analysis shows equivalent efficacy to multiagent chemotherapy. Int J Cancer. (2016) 138:481–8. doi: 10.1002/ijc.v138.2
60. Gnekow AK, Walker DA, Kandels D, Picton S, Giorgio P, Grill J, et al. A European randomised controlled trial of the addition of etoposide to standard vincristine and carboplatin induction as part of an 18-month treatment programme for childhood (≤16 years) low grade glioma - A final report. Eur J Cancer. (2017) 81:206–25. doi: 10.1016/j.ejca.2017.04.019
61. Lassaletta A, Scheinemann K, Zelcer SM, Hukin J, Wilson BA, Jabado N, et al. Phase II weekly vinblastine for chemotherapy-naïve children with progressive low-grade glioma: A canadian pediatric brain tumor consortium study. J Clin Oncol. (2016) 34:3537–43. doi: 10.1200/JCO.2016.68.1585
62. Rallis KS, George AM, Wozniak AM, Bigogno CM, Chow B, Hanrahan JG, et al. Molecular genetics and targeted therapies for paediatric high-grade glioma. Cancer Genomics Proteomics. (2022) 19:390–414. doi: 10.21873/cgp.20328
63. Ahrendsen JT, Torre M, Meredith DM, Hornick JL, Reardon DA, Wen PY, et al. IDH-mutant gliomas with additional class-defining molecular events. Mod Pathol. (2021) 34:1236–44. doi: 10.1038/s41379-021-00795-w
64. Watanabe T, Nobusawa S, Kleihues P, Ohgaki H. IDH1 mutations are early events in the development of astrocytomas and oligodendrogliomas. Am J Pathol. (2009) 174:1149–53. doi: 10.2353/ajpath.2009.080958
65. Johnson BE, Mazor T, Hong C, Barnes M, Aihara K, McLean CY, et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science. (2014) 343:189–93. doi: 10.1126/science.1239947
66. Waitkus MS, Diplas BH, Yan H. Isocitrate dehydrogenase mutations in gliomas. Neuro Oncol. (2016) 18:16–26. doi: 10.1093/neuonc/nov136
67. Gupta R, Flanagan S, Li CC, Lee M, Shivalingham B, Maleki S, et al. Expanding the spectrum of IDH1 mutations in gliomas. Mod Pathol. (2013) 26:619–25. doi: 10.1038/modpathol.2012.210
68. Eckel-Passow JE, Lachance DH, Molinaro AM, Walsh KM, Decker PA, Sicotte H, et al. Glioma groups based on 1p/19q, IDH, and TERT promoter mutations in tumors. N Engl J Med. (2015) 372:2499–508. doi: 10.1056/NEJMoa1407279
69. Lai A, Kharbanda S, Pope WB, Tran A, Solis OE, Peale F, et al. Evidence for sequenced molecular evolution of IDH1 mutant glioblastoma from a distinct cell of origin. J Clin Oncol. (2011) 29:4482–90. doi: 10.1200/JCO.2010.33.8715
70. Brat DJ, Verhaak RG, Aldape KD, Yung WK, Salama SR, Cooper LA, et al. Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N Engl J Med. (2015) 372:2481–98. doi: 10.1056/NEJMoa1402121
71. Brat DJ, Aldape K, Colman H, Figrarella-Branger D, Fuller GN, Giannini C, et al. cIMPACT-NOW update 5: recommended grading criteria and terminologies for IDH-mutant astrocytomas. Acta Neuropathol. (2020) 139:603–8. doi: 10.1007/s00401-020-02127-9
72. Yang RR, Shi ZF, Zhang ZY, Chan AK, Aibaidula A, Wang WW, et al. IDH mutant lower grade (WHO Grades II/III) astrocytomas can be stratified for risk by CDKN2A, CDK4 and PDGFRA copy number alterations. Brain Pathol. (2020) 30:541–53. doi: 10.1111/bpa.12801
73. Sanson M, Marie Y, Paris S, Idbaih A, Laffaire J, Ducray F, et al. Isocitrate dehydrogenase 1 codon 132 mutation is an important prognostic biomarker in gliomas. J Clin Oncol. (2009) 27:4150–4. doi: 10.1200/JCO.2009.21.9832
74. Houillier C, Wang X, Kaloshi G, Mokhtari K, Guillevin R, Laffaire J, et al. IDH1 or IDH2 mutations predict longer survival and response to temozolomide in low-grade gliomas. Neurology. (2010) 75:1560–6. doi: 10.1212/WNL.0b013e3181f96282
75. Korshunov A, Ryzhova M, Hovestadt V, Bender S, Sturm D, Capper D, et al. Integrated analysis of pediatric glioblastoma reveals a subset of biologically favorable tumors with associated molecular prognostic markers. Acta Neuropathol. (2015) 129:669–78. doi: 10.1007/s00401-015-1405-4
76. Ryall S, Zapotocky M, Fukuoka K, Nobre L, Guerreiro Stucklin A, Bennett J, et al. Integrated molecular and clinical analysis of 1,000 pediatric low-grade gliomas. Cancer Cell. (2020) 37:569–83.e5. doi: 10.1016/j.ccell.2020.03.011
77. Lau LMS, Khuong-Quang D-A, Mayoh C, Wong M, Barahona P, Ajuyah P, et al. Precision-guided treatment in high-risk pediatric cancers. Nat Med. (2024) 30:1913–22. doi: 10.1038/s41591-024-03044-0
78. Suwala AK, Stichel D, Schrimpf D, Kloor M, Wefers AK, Reinhardt A, et al. Primary mismatch repair deficient IDH-mutant astrocytoma (PMMRDIA) is a distinct type with a poor prognosis. Acta Neuropathol. (2021) 141:85–100. doi: 10.1007/s00401-020-02243-6
79. Dodgshun AJ, Fukuoka K, Edwards M, Bianchi VJ, Das A, Sexton-Oates A, et al. Germline-driven replication repair-deficient high-grade gliomas exhibit unique hypomethylation patterns. Acta Neuropathol. (2020) 140:765–76. doi: 10.1007/s00401-020-02209-8
80. Ahmad O, Ahmad T, Pfister SM. IDH mutation, glioma immunogenicity, and therapeutic challenge of primary mismatch repair deficient IDH-mutant astrocytoma PMMRDIA: a systematic review. Mol Oncol. (2024). doi: 10.1002/1878-0261.13598
81. Wijnenga MMJ, French PJ, Dubbink HJ, Dinjens WNM, Atmodimedjo PN, Kros JM, et al. The impact of surgery in molecularly defined low-grade glioma: an integrated clinical, radiological, and molecular analysis. Neuro Oncol. (2018) 20:103–12. doi: 10.1093/neuonc/nox176
82. Spitaels J, Devriendt D, Sadeghi N, Luce S, De Witte O, Goldman S, et al. Management of supratentorial recurrent low-grade glioma: A multidisciplinary experience in 35 adult patients. Oncol Lett. (2017) 14:2789–95. doi: 10.3892/ol.2017.6543
83. Kaspera W, Majchrzak K, Bobek-Billewicz B, Hebda A, Stasik-Pres G, Majchrzak H, et al. Reoperations of patients with low-grade gliomas in eloquent or near eloquent brain areas. Neurol Neurochir Pol. (2013) 47:116–25. doi: 10.5114/ninp.2013.34399
84. Ahmadi R, Dictus C, Hartmann C, Zürn O, Edler L, Hartmann M, et al. Long-term outcome and survival of surgically treated supratentorial low-grade glioma in adult patients. Acta Neurochir (Wien). (2009) 151:1359–65. doi: 10.1007/s00701-009-0473-4
85. Ramakrishna R, Hebb A, Barber J, Rostomily R, Silbergeld D. Outcomes in reoperated low-grade gliomas. Neurosurgery. (2015) 77:175–84. doi: 10.1227/NEU.0000000000000753
86. van den Bent MJ, Afra D, de Witte O, Ben Hassel M, Schraub S, Hoang-Xuan K, et al. Long-term efficacy of early versus delayed radiotherapy for low-grade astrocytoma and oligodendroglioma in adults: the EORTC 22845 randomised trial. Lancet. (2005) 366:985–90. doi: 10.1016/S0140-6736(05)67070-5
87. Choi S, Yu Y, Grimmer MR, Wahl M, Chang SM, Costello JF. Temozolomide-associated hypermutation in gliomas. Neuro Oncol. (2018) 20:1300–9. doi: 10.1093/neuonc/noy016
88. Kaina B, Christmann M, Naumann S, Roos WP. MGMT: key node in the battle against genotoxicity, carcinogenicity and apoptosis induced by alkylating agents. DNA Repair (Amst). (2007) 6:1079–99. doi: 10.1016/j.dnarep.2007.03.008
89. van den Bent MJ, Tesileanu CMS, Wick W, Sanson M, Brandes AA, Clement PM, et al. Adjuvant and concurrent temozolomide for 1p/19q non-co-deleted anaplastic glioma (CATNON; EORTC study 26053-22054): second interim analysis of a randomised, open-label, phase 3 study. Lancet Oncol. (2021) 22:813–23. doi: 10.1016/S1470-2045(21)00090-5
90. van Thuijl HF, Mazor T, Johnson BE, Fouse SD, Aihara K, Hong C, et al. Evolution of DNA repair defects during Malignant progression of low-grade gliomas after temozolomide treatment. Acta Neuropathol. (2015) 129:597–607. doi: 10.1007/s00401-015-1403-6
91. Fink D, Aebi S, Howell SB. The role of DNA mismatch repair in drug resistance. Clin Cancer Res. (1998) 4:1–6.
92. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature. (2013) 500:415–21. doi: 10.1038/nature12477
93. Jaeckle KA, Ballman KV, van den Bent M, Giannini C, Galanis E, Brown PD, et al. CODEL: phase III study of RT, RT + TMZ, or TMZ for newly diagnosed 1p/19q codeleted oligodendroglioma. Anal initial study design. Neuro Oncol. (2021) 23:457–67. doi: 10.1093/neuonc/noaa168
94. Kacimi S, Dehais C, Carpentier C, Chinot O, Bonnet C, Vauleon E, et al. Ks02.5.A overall survival associated with first-line pcv or temozolomide in combination with radiotherapy in patients with idh-mutant, 1p/19q codeleted, grade 3 oligodendroglioma: analysis from the pola cohort. Neuro-oncol. (2023) 25:ii4–ii. doi: 10.1093/neuonc/noad137.010
95. Caravella JA, Lin J, Diebold RB, Campbell AM, Ericsson A, Gustafson G, et al. Structure-based design and identification of FT-2102 (Olutasidenib), a potent mutant-selective IDH1 inhibitor. J Med Chem. (2020) 63:1612–23. doi: 10.1021/acs.jmedchem.9b01423
96. Stein EM, DiNardo CD, Fathi AT, Mims AS, Pratz KW, Savona MR, et al. Ivosidenib or enasidenib combined with intensive chemotherapy in patients with newly diagnosed AML: a phase 1 study. Blood. (2021) 137:1792–803. doi: 10.1182/blood.2020007233
97. Natsume A, Arakawa Y, Narita Y, Sugiyama K, Hata N, Muragaki Y, et al. The first-in-human phase I study of a brain-penetrant mutant IDH1 inhibitor DS-1001 in patients with recurrent or progressive IDH1-mutant gliomas. Neuro Oncol. (2023) 25:326–36. doi: 10.1093/neuonc/noac155
98. Mellinghoff IK, Ellingson BM, Touat M, Maher E, de la Fuente MI, Holdhoff M, et al. Ivosidenib in isocitrate dehydrogenase 1–mutated advanced glioma. J Clin Oncol. (2020) 38:3398–406. doi: 10.1200/JCO.19.03327
99. de la Fuente MI, Colman H, Rosenthal M, Van Tine BA, Levacic D, Walbert T, et al. Olutasidenib (FT-2102) in patients with relapsed or refractory IDH1-mutant glioma: A multicenter, open-label, phase Ib/II trial. Neuro Oncol. (2023) 25:146–56. doi: 10.1093/neuonc/noac139
100. Wick A, Bähr O, Schuler M, Rohrberg K, Chawla SP, Janku F, et al. Phase I assessment of safety and therapeutic activity of BAY1436032 in patients with IDH1-mutant solid tumors. Clin Cancer Res. (2021) 27:2723–33. doi: 10.1158/1078-0432.CCR-20-4256
101. DiNardo CD, Schimmer AD, Yee KWL, Hochhaus A, Kraemer A, Carvajal RD, et al. A phase I study of IDH305 in patients with advanced Malignancies including relapsed/refractory AML and MDS that harbor IDH1R132 mutations. Blood. (2016) 128:1073–. doi: 10.1182/blood.V128.22.1073.1073
102. Mellinghoff IK, Penas-Prado M, Peters KB, Burris HA 3rd, Maher EA, Janku F, et al. Vorasidenib, a dual inhibitor of mutant IDH1/2, in recurrent or progressive glioma; results of a first-in-human phase I trial. Clin Cancer Res. (2021) 27:4491–9. doi: 10.1158/1078-0432.CCR-21-0611
103. U.S Food and Drug Administration. FDA approves vorasidenib for Grade 2 astrocytoma or oligodendroglioma with a susceptible IDH1 or IDH2 mutation (2024). Available online at: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-vorasidenib-grade-2-astrocytoma-or-oligodendroglioma-susceptible-idh1-or-idh2-mutation:~:text=On%20August%206%2C%202024%2C%20the,or%20oligodendroglioma%20with%20a%20susceptible (Accessed August 10, 2024).
104. Heredia V, Mendiola M, Ortiz E, Bernabéu D, Pozo-Kreilinger JJ, Miguel M, et al. AG-120, a novel IDH1 targeted molecule, inhibits invasion and migration of chondrosarcoma cells in vitro. Ann Oncol. (2017) 28:v538. doi: 10.1093/annonc/mdx387.049
105. Nicolay B, Narayanaswamy R, Aguado E, Nagaraja R, Murtie J, Liu G, et al. Exth-59. The idh1 mutant inhibitor ag-120 shows strong inhibition of 2-hg production in an orthotopic idh1 mutant glioma model in vivo. Neuro-oncol. (2017) 19:vi86–vi. doi: 10.1093/neuonc/nox168.351
106. Tap WD, Villalobos VM, Cote GM, Burris H, Janku F, Mir O, et al. Phase I study of the mutant IDH1 inhibitor ivosidenib: safety and clinical activity in patients with advanced chondrosarcoma. J Clin Oncol. (2020) 38:1693–701. doi: 10.1200/JCO.19.02492
107. Lowery MA, Burris HA 3rd, Janku F, Shroff RT, Cleary JM, Azad NS, et al. Safety and activity of ivosidenib in patients with IDH1-mutant advanced cholangiocarcinoma: a phase 1 study. Lancet Gastroenterol Hepatol. (2019) 4:711–20. doi: 10.1016/S2468-1253(19)30189-X
108. Abou-Alfa GK, Macarulla T, Javle MM, Kelley RK, Lubner SJ, Adeva J, et al. Ivosidenib in IDH1-mutant, chemotherapy-refractory cholangiocarcinoma (ClarIDHy): a multicentre, randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. (2020) 21:796–807. doi: 10.1016/S1470-2045(20)30157-1
109. Zhu AX, Macarulla T, Javle MM, Kelley RK, Lubner SJ, Adeva J, et al. Final overall survival efficacy results of ivosidenib for patients with advanced cholangiocarcinoma with IDH1 mutation: the phase 3 randomized clinical clarIDHy trial. JAMA Oncol. (2021) 7:1669–77. doi: 10.1001/jamaoncol.2021.3836
110. Roboz GJ, DiNardo CD, Stein EM, de Botton S, Mims AS, Prince GT, et al. Ivosidenib induces deep durable remissions in patients with newly diagnosed IDH1-mutant acute myeloid leukemia. Blood. (2020) 135:463–71. doi: 10.1182/blood.2019002140
111. DiNardo CD, Stein AS, Stein EM, Fathi AT, Frankfurt O, Schuh AC, et al. Mutant isocitrate dehydrogenase 1 inhibitor ivosidenib in combination with azacitidine for newly diagnosed acute myeloid leukemia. J Clin Oncol. (2021) 39:57–65. doi: 10.1200/JCO.20.01632
112. DiNardo CD, Stein EM, Sd B, GJ R, JK A, AS M, et al. Durable remissions with ivosidenib in IDH1-mutated relapsed or refractory AML. N Engl J Med. (2018) 378:2386–98. doi: 10.1056/NEJMoa1716984
113. Venugopal S, Watts J. Olutasidenib: from bench to bedside. Blood Adv. (2023) 7:4358–65. doi: 10.1182/bloodadvances.2023009854
114. Salifu EY, Agoni C, Soliman MES. Highlighting the mechanistic role of Olutasidenib (FT-2102) in the selective inhibition of mutated isocitrate dehydrogenase 1 (mIDH1) in cancer therapy. Inf Med Unlocked. (2022) 28:100829. doi: 10.1016/j.imu.2021.100829
115. Lin J, Lu W, Caravella JA, Campbell AM, Diebold RB, Ericsson A, et al. Discovery and optimization of quinolinone derivatives as potent, selective, and orally bioavailable mutant isocitrate dehydrogenase 1 (mIDH1) inhibitors. J Med Chem. (2019) 62:6575–96. doi: 10.1021/acs.jmedchem.9b00362
116. Cortes J, Jonas BA, Schiller G, Mims A, Roboz GJ, Wei AH, et al. Olutasidenib in post-venetoclax patients with mutant isocitrate dehydrogenase 1 (mIDH1) acute myeloid leukemia (AML). Leuk Lymphoma. (2024), 1–8. doi: 10.1080/10428194.2024.2333451
117. Chaturvedi A, Gupta C, Gabdoulline R, Borchert NM, Goparaju R, Kaulfuss S, et al. Synergistic activity of IDH1 inhibitor BAY1436032 with azacitidine in IDH1 mutant acute myeloid leukemia. Haematologica. (2021) 106:565–73. doi: 10.3324/haematol.2019.236992
118. Pusch S, Krausert S, Fischer V, Balss J, Ott M, Schrimpf D, et al. Pan-mutant IDH1 inhibitor BAY 1436032 for effective treatment of IDH1 mutant astrocytoma in vivo. Acta Neuropathol. (2017) 133:629–44. doi: 10.1007/s00401-017-1677-y
119. Heuser M, Palmisiano N, Mantzaris I, Mims A, DiNardo C, Silverman LR, et al. Safety and efficacy of BAY1436032 in IDH1-mutant AML: phase I study results. Leukemia. (2020) 34:2903–13. doi: 10.1038/s41375-020-0996-5
120. Machida Y, Nakagawa M, Matsunaga H, Yamaguchi M, Ogawara Y, Shima Y, et al. A potent blood-brain barrier-permeable mutant IDH1 inhibitor suppresses the growth of glioblastoma with IDH1 mutation in a patient-derived orthotopic xenograft model. Mol Cancer Ther. (2020) 19:375–83. doi: 10.1158/1535-7163.MCT-18-1349
121. Cho YS, Levell JR, Liu G, Caferro T, Sutton J, Shafer CM, et al. Discovery and evaluation of clinical candidate IDH305, a brain penetrant mutant IDH1 inhibitor. ACS Med Chem Lett. (2017) 8:1116–21. doi: 10.1021/acsmedchemlett.7b00342
122. DiNardo CD, Hochhaus A, Frattini MG, Yee K, Zander T, Krämer A, et al. A phase 1 study of IDH305 in patients with IDH1(R132)-mutant acute myeloid leukemia or myelodysplastic syndrome. J Cancer Res Clin Oncol. (2023) 149:1145–58. doi: 10.1007/s00432-022-03983-6
123. Yen K, Travins J, Wang F, David MD, Artin E, Straley K, et al. AG-221, a first-in-class therapy targeting acute myeloid leukemia harboring oncogenic IDH2 mutations. Cancer Discovery. (2017) 7:478–93. doi: 10.1158/2159-8290.CD-16-1034
124. Shih AH, Shank KR, Meydan C, Intlekofer AM, Ward P, Thompson CB, et al. AG-221, a small molecule mutant IDH2 inhibitor, remodels the epigenetic state of IDH2-mutant cells and induces alterations in self-renewal/differentiation in IDH2-mutant AML model in vivo. Blood. (2014) 124:437. doi: 10.1182/blood.V124.21.437.437
125. de Botton S, Montesinos P, Schuh AC, Papayannidis C, Vyas P, Wei AH, et al. Enasidenib vs conventional care in older patients with late-stage mutant-IDH2 relapsed/refractory AML: a randomized phase 3 trial. Blood. (2023) 141:156–67. doi: 10.1182/blood.2021014901
126. Pollyea DA, Tallman MS, de Botton S, Kantarjian HM, Collins R, Stein AS, et al. Enasidenib, an inhibitor of mutant IDH2 proteins, induces durable remissions in older patients with newly diagnosed acute myeloid leukemia. Leukemia. (2019) 33:2575–84. doi: 10.1038/s41375-019-0472-2
127. Konteatis Z, Artin E, Nicolay B, Straley K, Padyana AK, Jin L, et al. Vorasidenib (AG-881): A first-in-class, brain-penetrant dual inhibitor of mutant IDH1 and 2 for treatment of glioma. ACS Med Chem Lett. (2020) 11:101–7. doi: 10.1021/acsmedchemlett.9b00509
128. Wahid A, Tariq A, Ahsan F, Asif F. Vorasidenib: A promising therapeutic breakthrough for diffuse isocitrate dehydrogenase mutant gliomas. Rare Tumors. (2023) 15:20363613231197991. doi: 10.1177/20363613231197991
129. Harding JJ, Lowery MA, Shih AH, Schvartzman JM, Hou S, Famulare C, et al. Isoform switching as a mechanism of acquired resistance to mutant isocitrate dehydrogenase inhibition. Cancer Discovery. (2018) 8:1540–7. doi: 10.1158/2159-8290.CD-18-0877
130. DiNardo CD, De Botton S, Pollyea DA, Stone RM, Altman JK, Fathi AT, et al. Safety, efficacy, and PK/PD of vorasidenib in previously treated patients with mIDH1/2 hematologic Malignancies: A phase 1 study. Am J Hematol. (2023) 98:E233–e6. doi: 10.1002/ajh.27005
131. Mellinghoff IK, Lu M, Wen PY, Taylor JW, Maher EA, Arrillaga-Romany I, et al. Vorasidenib and ivosidenib in IDH1-mutant low-grade glioma: a randomized, perioperative phase 1 trial. Nat Med. (2023) 29:615–22. doi: 10.1038/s41591-022-02141-2
132. Kotecha R, Schiff D, Chakravarti A, Fleming JL, Brown PD, Puduvalli VK, et al. Multidisciplinary management of isocitrate dehydrogenase-mutated gliomas in a contemporary molecularly defined era. J Clin Oncol. (2024) 42:2588–98. doi: 10.1200/JCO.23.02195
133. Lin MD, Tsai ACY, Abdullah KG, McBrayer SK, Shi DD. Treatment of IDH-mutant glioma in the INDIGO era. NPJ Precis Oncol. (2024) 8:149. doi: 10.1038/s41698-024-00646-2
134. Spitzer A, Gritsch S, Nomura M, Jucht A, Fortin J, Raviram R, et al. Mutant IDH inhibitors induce lineage differentiation in IDH-mutant oligodendroglioma. Cancer Cell. (2024) 42:904–14.e9. doi: 10.1016/j.ccell.2024.03.008
135. Touat M, Li YY, Boynton AN, Spurr LF, Iorgulescu JB, Bohrson CL, et al. Mechanisms and therapeutic implications of hypermutation in gliomas. Nature. (2020) 580:517–23. doi: 10.1038/s41586-020-2209-9
136. Malta TM, Sabedot TS, Morosini NS, Datta I, Garofano L, Vallentgoed W, et al. The epigenetic evolution of glioma is determined by the IDH1 mutation status and treatment regimen. Cancer Res. (2024) 84:741–56. doi: 10.1158/0008-5472.CAN-23-2093
137. Mirchia K, Sathe AA, Walker JM, Fudym Y, Galbraith K, Viapiano MS, et al. Total copy number variation as a prognostic factor in adult astrocytoma subtypes. Acta Neuropathologica Commun. (2019) 7:92. doi: 10.1186/s40478-019-0746-y
138. Tateishi K, Miyake Y, Nakamura T, Iwashita H, Hayashi T, Oshima A, et al. Genetic alterations that deregulate RB and PDGFRA signaling pathways drive tumor progression in IDH2-mutant astrocytoma. Acta Neuropathologica Commun. (2023) 11:186. doi: 10.1186/s40478-023-01683-x
139. Aoki K, Nakamura H, Suzuki H, Matsuo K, Kataoka K, Shimamura T, et al. Prognostic relevance of genetic alterations in diffuse lower-grade gliomas. Neuro-oncol. (2017) 20:66–77. doi: 10.1093/neuonc/nox132
140. Cimino PJ, Holland EC. Targeted copy number analysis outperforms histologic grading in predicting patient survival for WHO grades II/III IDH-mutant astrocytomas. Neuro Oncol. (2019) 21:819–21. doi: 10.1093/neuonc/noz052
141. Appay R, Dehais C, Maurage CA, Alentorn A, Carpentier C, Colin C, et al. CDKN2A homozygous deletion is a strong adverse prognosis factor in diffuse Malignant IDH-mutant gliomas. Neuro Oncol. (2019) 21:1519–28. doi: 10.1093/neuonc/noz126.000
142. Dono A, Ballester LY, Primdahl D, Esquenazi Y, Bhatia A. IDH-mutant low-grade glioma: advances in molecular diagnosis, management, and future directions. Curr Oncol Rep. (2021) 23:20. doi: 10.1007/s11912-020-01006-6
143. Yu Y, Villanueva-Meyer J, Grimmer MR, Hilz S, Solomon DA, Choi S, et al. Temozolomide-induced hypermutation is associated with distant recurrence and reduced survival after high-grade transformation of low-grade IDH-mutant gliomas. Neuro Oncol. (2021) 23:1872–84. doi: 10.1093/neuonc/noab081
144. Intlekofer AM, Shih AH, Wang B, Nazir A, Rustenburg AS, Albanese SK, et al. Acquired resistance to IDH inhibition through trans or cis dimer-interface mutations. Nature. (2018) 559:125–9. doi: 10.1038/s41586-018-0251-7
145. Choe S, Wang H, DiNardo CD, Stein EM, de Botton S, Roboz GJ, et al. Molecular mechanisms mediating relapse following ivosidenib monotherapy in IDH1-mutant relapsed or refractory AML. Blood Adv. (2020) 4:1894–905. doi: 10.1182/bloodadvances.2020001503
146. Reinbold R, Hvinden IC, Rabe P, Herold RA, Finch A, Wood J, et al. Resistance to the isocitrate dehydrogenase 1 mutant inhibitor ivosidenib can be overcome by alternative dimer-interface binding inhibitors. Nat Commun. (2022) 13:4785. doi: 10.1038/s41467-022-32436-4
147. Oltvai ZN, Harley SE, Koes D, Michel S, Warlick ED, Nelson AC, et al. Assessing acquired resistance to IDH1 inhibitor therapy by full-exon IDH1 sequencing and structural modeling. Cold Spring Harb Mol Case Stud. (2021) 7:1–12. doi: 10.1101/mcs.a006007
148. Cleary JM, Rouaisnel B, Daina A, Raghavan S, Roller LA, Huffman BM, et al. Secondary IDH1 resistance mutations and oncogenic IDH2 mutations cause acquired resistance to ivosidenib in cholangiocarcinoma. NPJ Precis Oncol. (2022) 6:61. doi: 10.1038/s41698-022-00304-5
149. Das A, Tabori U, Sambira Nahum LC, Collins NB, Deyell R, Dvir R, et al. Efficacy of nivolumab in pediatric cancers with high mutation burden and mismatch repair deficiency. Clin Cancer Res. (2023) 29:4770–83. doi: 10.1158/1078-0432.CCR-23-0411
150. Das A, Fernandez NR, Levine A, Bianchi V, Stengs LK, Chung J, et al. Combined immunotherapy improves outcome for replication-repair-deficient (RRD) high-grade glioma failing anti-PD-1 monotherapy: A report from the international RRD consortium. Cancer Discovery. (2024) 14:258–73. doi: 10.1158/2159-8290.CD-23-0559
151. Das A, Fernandez NR, Bielamowicz KJ, Abebe-Campino G, Caspi S, Per N, et al. Hgg-11. Trans-species study of idh-mutant replication-repair deficient high-grade gliomas (Rrd-hgg) and response to combined targeted and immunotherapy: an irrdc study. Neuro-oncol. (2024) 26:iv87. doi: 10.1093/neuonc/noae064.295
152. Wang Z, Zhang C, Liu X, Wang Z, Sun L, Li G, et al. Molecular and clinical characterization of PD-L1 expression at transcriptional level via 976 samples of brain glioma. Oncoimmunology. (2016) 5:e1196310. doi: 10.1080/2162402X.2016.1196310
153. Schomas DA, Laack NN, Rao RD, Meyer FB, Shaw EG, O’Neill BP, et al. Intracranial low-grade gliomas in adults: 30-year experience with long-term follow-up at Mayo Clinic. Neuro Oncol. (2009) 11:437–45. doi: 10.1215/15228517-2008-102
154. Phuong C, Qiu B, Mueller S, Braunstein SE. Precision based approach to tailoring radiotherapy in the multidisciplinary management of pediatric central nervous system tumors. J Natl Cancer Center. (2023) 3:141–9. doi: 10.1016/j.jncc.2023.03.001
155. Campen CJ, Kranick SM, Kasner SE, Kessler SK, Zimmerman RA, Lustig R, et al. Cranial irradiation increases risk of stroke in pediatric brain tumor survivors. Stroke. (2012) 43:3035–40. doi: 10.1161/STROKEAHA.112.661561
156. Peters K, Mellinghoff I, van den Bent M, Blumenthal D, Touat M, Clarke J, et al. Qol-26. A randomized, double-blind phase 3 study of vorasidenib vs placebo in patients with mutant idh1/2diffuse glioma (Indigo): analysis of health-related quality of life, neurocognition and seizures. Neuro-oncol. (. 2023) 25:v254–v5. doi: 10.1093/neuonc/noad179.0978
Keywords: IDH mutation, low grade glioma, pediatric, adolescent and young adult, AYA
Citation: Evans L, Trinder S, Dodgshun A, Eisenstat DD, Whittle JR, Hansford JR and Valvi S (2025) IDH-mutant gliomas in children and adolescents - from biology to clinical trials. Front. Oncol. 14:1515538. doi: 10.3389/fonc.2024.1515538
Received: 23 October 2024; Accepted: 10 December 2024;
Published: 06 January 2025.
Edited by:
Ashley Sloane Margol, Children’s Hospital of Los Angeles, United StatesReviewed by:
Joseph Louis Lasky, Cure 4 The Kids, United StatesCopyright © 2025 Evans, Trinder, Dodgshun, Eisenstat, Whittle, Hansford and Valvi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jordan R. Hansford, am9yZGFuLmhhbnNmb3JkQHNhLmdvdi5hdQ==; Santosh Valvi, c2FudG9zaC52YWx2aUBoZWFsdGgud2EuZ292LmF1
†These authors have contributed equally to this work and share first authorship
‡These authors have contributed equally to this work and share senior authorship