 Ira Ekmekciu1*†Doreen Maria Zucha2†Jens Christmann2†Sarah Wisser2Vera Heuer1Buelent Sargin3Stephan Hollerbach4Christof Lamberti5Lothar Müller6Celine Lugnier1Berlinda Verdoodt2Robin Denz7Tobias Terzer1Inke Feder2Anke Reinacher-Schick1Andrea Tannapfel2Iris Tischoff2
Ira Ekmekciu1*†Doreen Maria Zucha2†Jens Christmann2†Sarah Wisser2Vera Heuer1Buelent Sargin3Stephan Hollerbach4Christof Lamberti5Lothar Müller6Celine Lugnier1Berlinda Verdoodt2Robin Denz7Tobias Terzer1Inke Feder2Anke Reinacher-Schick1Andrea Tannapfel2Iris Tischoff2- 1Department of Hematology, Oncology and Palliative Care, St. Josef Hospital, Ruhr University, Bochum, Germany
- 2Institute of Pathology, Ruhr University, Bochum, Germany
- 3Hematology and Medical Oncology, St-Marien-Hospital Lunen, Lunen, Germany
- 4Department of Gastroenterology, Allgemeines Krankenhaus (AKH) Celle, Celle, Germany
- 5Hematology and Oncology, Regiomed Hospital Group, Coburg, Germany
- 6Onkologie UnterEms, Leer, Germany
- 7Department of Medical Informatics, Biometrics and Epidemiology, Ruhr University, Bochum, Germany
Introduction: Understanding the mutational landscape of colon cancer (CC) is crucial for targeted therapy development. Microsatellite instability (MSI-H), rat sarcoma (RAS), and B-Raf proto-oncogene, serine/threonine kinase (BRAF) mutations (MT) are pivotal markers. Further investigation into clinicopathological features of RAS and BRAF MT in microsatellite stable (MSS) and MSI-H tumors is warranted.
Methods: A retrospective analysis of 4883 localized CC patients (pts.) was conducted. Molecular profiling assessed MSI, KRAS, NRAS, and BRAF MT. Correlation with clinicopathological data employed ANOVA and Chi-square tests. Disease-free survival (DFS) and overall survival (OS) were analyzed adjusting for age, gender, sidedness, UICC stage, Charlson Comorbidity Index (CCI). A Cox model incorporated all variables as covariates.
Results: This analysis included 4883 pts. (2302 female/2572 male, 3865 (79.2%) MSS, 1018 (20.8%) MSI-H). MSS pts. had more All-Wild Type (WT), KRAS MT, and NRAS MT tumors vs. MSI-H pts. (42.1% vs. 21.1%; 39.8% vs. 15.4%; 3.6% vs. 0.7%; p<0.001 for each). BRAF MT tumors (95.5% BRAF V600E MT) were more prevalent in MSI-H individuals (62.8% vs. 8.1%, p<0.001). KRAS and BRAF MT tumors were more frequently right-sided, while BRAF MT tumors were associated with female gender, advanced disease stage, lymph node positivity, and poorer differentiation in the MSS subset (p<0.001). Common KRAS mutations included p.G12D (30.44%) and p.G12V (21.3%) in MSS and p.G13D (28.9%) and p.G12D (22.37%) in MSI-H. NRAS MT tumors were dominated by codon 61 mutations (51.7%). Survival analysis revealed worst prognosis in BRAF MT MSS tumors (DFS: HR 1.74 (95% CI 1.15-2.62, p=0.009; OS: HR 1.61 (95% CI 0.99-2.6), p=0.055). The 3-years DFS and 5-years OS rates were lowest in this subset (61.6% and 57.7% respectively).
Discussion: These findings highlight the complex interplay between molecular subtypes, clinicopathological features, and survival outcomes in early CC. Further research is needed to elucidate underlying mechanisms and develop personalized treatment strategies.
1 Introduction
Colon cancer (CC) remains a significant public health concern worldwide, representing a leading cause of cancer-related morbidity and mortality. Despite advancements in diagnosis and treatment, the heterogeneous nature of CC poses challenges for clinicians in predicting patient outcomes and optimizing therapeutic strategies. For stage III and high risk stage II disease adjuvant chemotherapy is recommended using fluopyrimidins +/- oxaliplatin (1, 2). However, the benefit from adjuvant chemotherapy in these patients (pts) appears to be relatively modest, ranging from 5% to 20% (3).
Recent studies have underscored the significant role of molecular changes, particularly mutations in key genes such as those associated with deficient mismatch repair protein (dMMR) leading to microsatellite instability-high status (MSI-H), the rat sarcoma (RAS) family (including KRAS/NRAS/HRAS), and the B-Raf proto-oncogene, serine/threonine kinase (BRAF) in shaping the pathogenesis and clinical behavior of CC. Evaluation of mutational status, especially in metastatic disease, is strongly recommended in all guidelines (4). The development of targeted therapy approaches, particularly for pts with MSI-H and BRAF-V600E mutations, evaluated in clinical trials, has led to more personalized treatment options for these subgroups in metastatic CC (5, 6).
Understanding the interplay between these molecular subtypes and their implications for patient prognosis and treatment response is crucial for advancing personalized medicine approaches in CC management. Activating mutations in KRAS and NRAS are recognized as negative predictors for anti-epidermal growth factor receptor (anti-EGFR) treatment (7, 8). Moreover, targetable KRAS submutations have emerged. Compounds like sotorasib and adagrasib, which selectively inhibit the KRAS p.G12C mutation, are being tested in combination with anti-EGFR antibodies in the context of metastatic CC and have delivered promising results (9, 10).
While these investigations predominantly focus on advanced CC, clinical and molecular data regarding localized disease are less common. In a previous study, we analyzed the AIO Colopredict Plus (CPP) registry, specifically focusing on elderly individuals with CC, and found that adjuvant chemotherapy is an independent positive prognostic factor for overall survival (OS) (11, 12). Furthermore, we identified a subset of elderly pts with fewer comorbidities who may particularly benefit from adjuvant chemotherapy.
In this study, we conducted a comprehensive analysis of the genetic landscape of CC using real-world data from our CPP registry spanning over a decade. Using advanced sequencing methods, we aimed to elucidate the prevalence and clinical significance of MSI, RAS, and BRAF mutations in nonmetastatic CC pts. The primary objective was to investigate the role of RAS and BRAF mutations in correlation with MSI status and associated clinicopathological characteristics. Our findings provide insights into the complex molecular landscape of CC and offer potential implications for pts stratification and treatment decision-making. Through this research, we seek to contribute to the ongoing efforts to improve outcomes for CC pts by uncovering new avenues for targeted therapy and personalized care.
2 Material and methods
In September 2013, the CPP molecular registry study commenced in 20 German community cancer centers, subsequently expanding to encompass over 200 German CC centers. All study participants provided informed consent, and the study protocol was approved by the Ethics Committee of the Ruhr-University Bochum (registration number: 17-6151_13 and 12-4449; DRKS-ID: DRKS00004305). The study gathered demographic data including age, sex, body mass index (BMI), as well as comorbidities, tumor localization and survival status. Newly diagnosed CC cases with histopathological confirmation at Union Internationale Contre le Cancer (UICC) stages II and III were initially enrolled, with the cohort later expanded in 2018 to include UICC stage I cases. Tumors were classified according to the current WHO 2019 guidelines and the TNM system outlined in the American Joint Committee on Cancer (AJCC) Cancer Staging Manual, Eighth Edition. Cases of UICC stage IV CC and rectal cancer were excluded from the study. The data collection cutoff point was February, 2023.
2.1 Tissue analysis by next generation sequencing (NGS)
Molecular analysis was performed centrally at the Institute of Pathology of the Ruhr University Bochum. DNA concentration was measured by QuantiFluor ® ONE dsDNA on the Quantus (Promega Corporation, Madison, USA). All further concentration measurements were performed using QubitTM dsDNA HS. According to the manufacturer’s instructions, the QIAGEN Colorectal Cancer Panel (QIAGEN GmbH, Hilden, GER) was used to amplify target regions of 72 CC related genes (Supplementary Table 1), among others BRAF and RAS (K- and N-RAS). Libraries were prepared with 50 ng of genomic DNA. Sequencing of the final libraries was performed using NextSeq 550 Illumina sequencer (Illumina Inc., San Diego, CA). For data analysis the QIAGEN CLC Genomics Workbench (Version 21) was used. Analysis and assessment of detected variants was performed using IGV, MutationTaster, ClinVar, Cosmic, and Qiagen Clinical Insight. Only disease-causing mutations (Variant Allele Frequency (VAF) ≥ 5%, read coverage ≥100 unique reads mean coverage in min. 80% of sequenced nucleotides, read balance ≥0,2) were reported. In individual cases, hand-curated variants in known driver genes were reported with slightly worse quality parameters if they were classified as true according to expert opinion. Data concerning RAS, BRAF and MSI were extracted from NGS data.
2.2 Detection of MSI status
MSI-testing was performed using both immunohistochemistry for four proteins (MLH1, MLH6, MSH2, PMS2) and PCR-based fragment-length analysis as previously described (11). In cases of inconsistent results, NGS was employed as the gold standard. A discrepancy between immunohistochemistry and PCR-based analysis was observed in 12% of cases (11).
For the detection of MSI status via NGS, eight well-established MSI loci (BAT25, BAT26, D2S123, NR21, NR22, NR24, D5S346, D17S250) were sequenced and the respective reads were compared with those of a validated, non-tumor tissue baseline in the same entity. By analyzing the length distribution histogram, each sample was compared with the baseline to determine the MSI status.
2.3 Statistics
Arithmetic means and standard deviations were computed for continuous variables, while frequencies and percentages were calculated for categorical variables. ANOVA tests and Chi-Square Tests were used to test whether these variables were distributed differently in the relevant subgroups for continuous variables and categorical variables respectively. OS was defined as the time until death from any cause, with pts lost to follow-up or still alive at the end of the study period being censored. Disease-Free Survival (DFS) combined both relapse and death endpoints. Confounder-adjusted survival curves were generated using G-computation based on Cox regression models (13) and hazard ratios (HRs) with 95% confidence intervals (CIs) were calculated. Adjusting factors included age, sex, Charlson Comorbidity Index (CCI), UICC stage, and tumor location (sidedness). Statistical significance was defined as p < 0.05. All statistical analysis were carried out using R (version 4.2.1).
3 Results
3.1 Study population
Between September 2013 and February 2023, a total of 9960 pts. were enrolled in CPP. Following the exclusion of 561 patients who did not meet the inclusion criteria (Figure 1), 9399 patients remained eligible for analysis. Histological analysis and NGS was applied to analyze adequate tissue samples from 5048 randomly selected cases, representing 50.7% of the total cases, up to the cutoff date of February 2023. After excluding 165 cases due to Stage 0, histology indicating appendix carcinoma, missing MSI data, or the presence of double mutations, the final molecular cohort for this analysis comprised 4883 patients. Among these, 3865 individuals (79.2%) were microsatellite stable (MSS), while 1018 (20.8%) were microsatellite instable (MSI-H) (Figure 1). The study population consisted of 2302 females and 2572 males. Baseline characteristics of the entire study population, correlated with the mutational status of KRAS, BRAF, and NRAS, are detailed in Supplementary Table 2. The mean follow-up duration was 29.5 months.
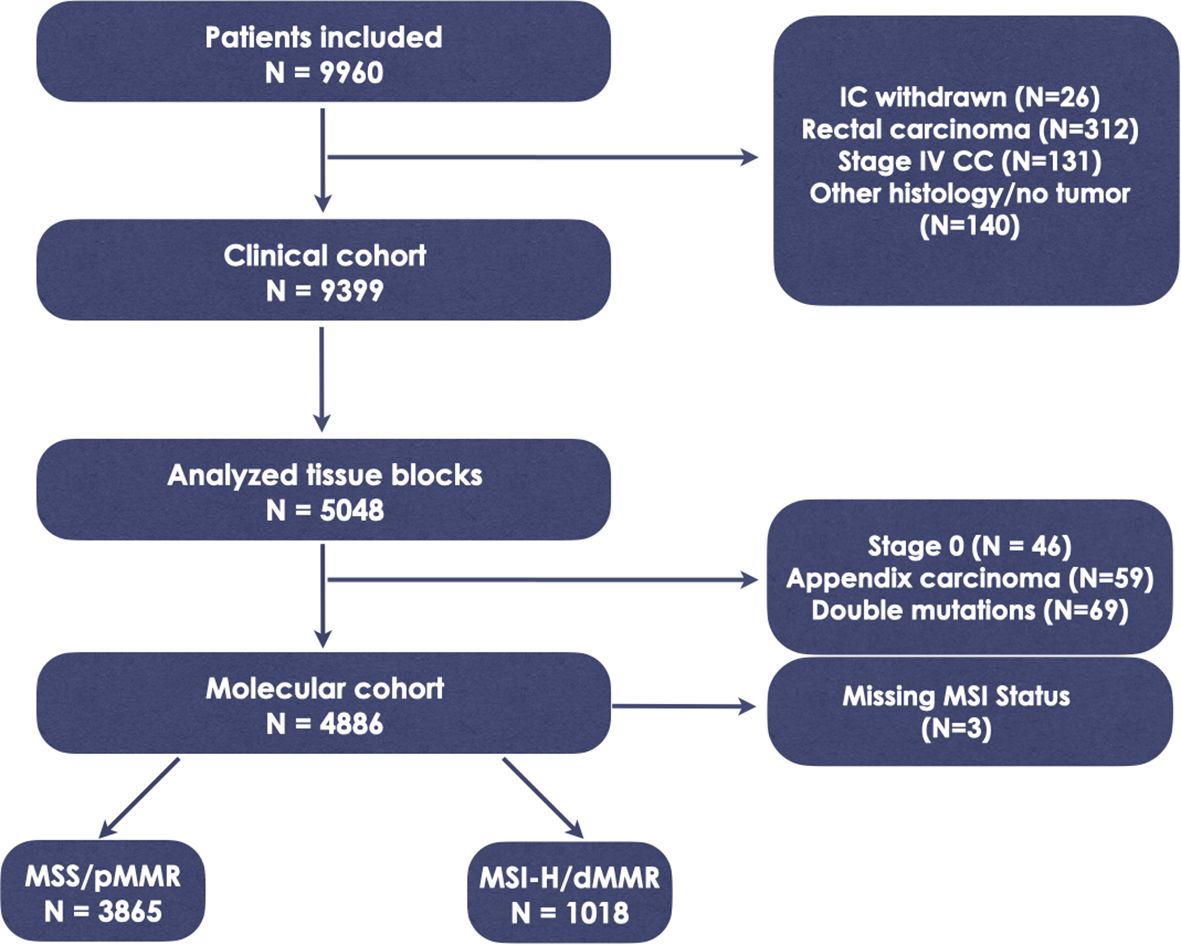
Figure 1. Study flowchart. IC, informed consent; CC, colon cancer; MSS, microsatellite stability; MSI-H, microsatellite instability; dMMR, deficient mismatch repair protein; pMMR, proficient mismatch repair protein.
3.2 Baseline clinical, histopathological and molecular characteristics of MSS and MSI-H cohort
Across the entire study cohort, 37.7% were identified as having All-Wild Type (WT), 39.8% had KRAS mutated (MT) tumors, 3% exhibited NRAS MT, and 19.5% displayed BRAF MT tumors (Supplementary Table 2). In the MSS cohort, the prevalence rate was 42.1% All-WT, 46.2% KRAS MT, 3.6% NRAS MT and 8.1% BRAF MT, respectively. BRAF MT tumors were significantly more prevalent in the MSI-H population, constituting 62.8% of this subgroup (p<0.001), while All-WT, KRAS MT, and NRAS MT cancer specimens were less frequent, accounting for 21.1%, 15.4%, and 0.7%, respectively (Figure 2, p<0.001 for each).
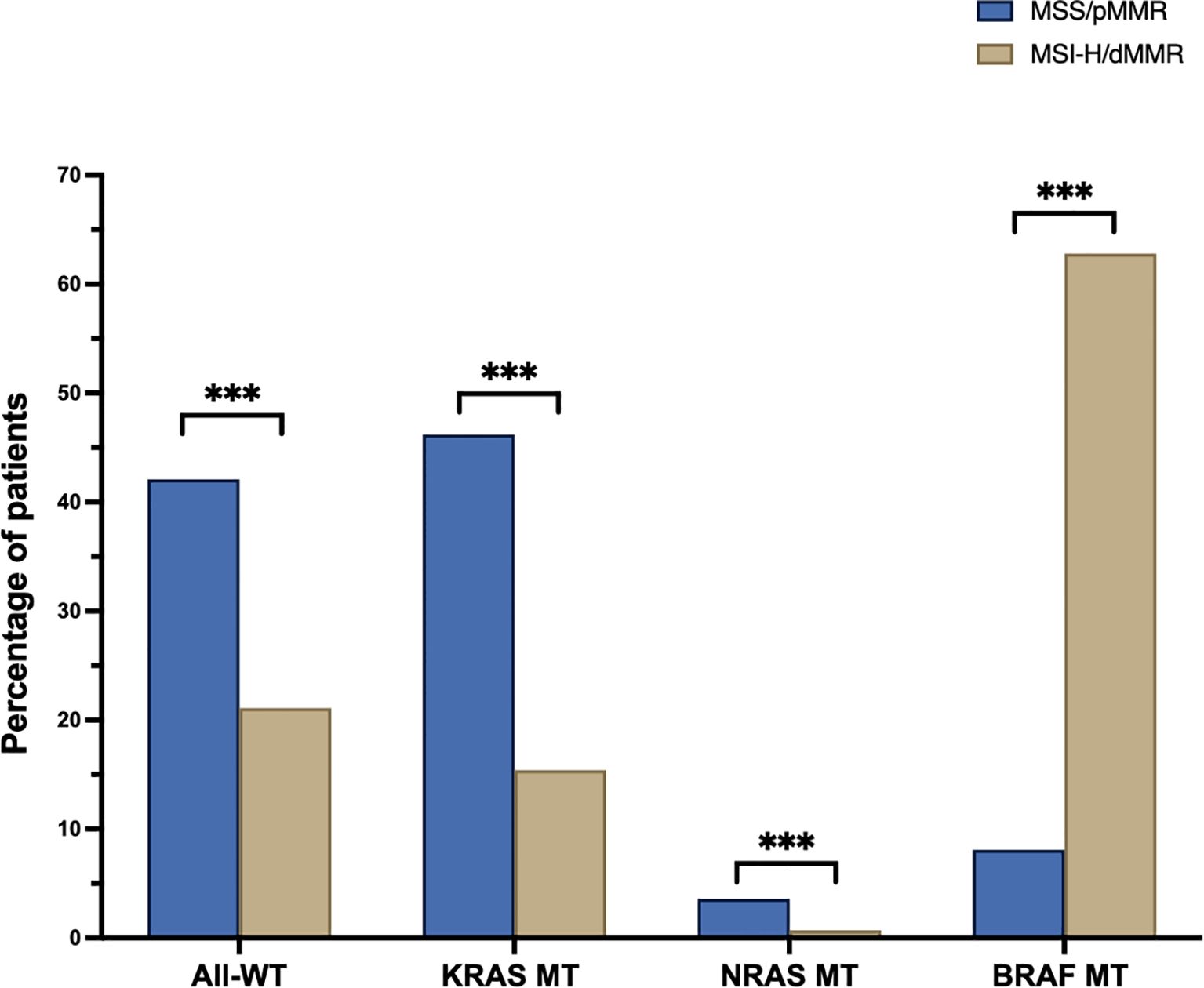
Figure 2. Percentage of wildtype (All-WT) and mutated (MT) pts. in the microsatellite stable (MSS, blue) and instable (MSI-H, sand) cohorts (BRAF, B-Raf proto-oncogene, serine/threonine kinase; KRAS, Kirsten Rat Sarcoma; NRAS, Neuroblastoma ras); ***p < 0.001.
In the MSS population, All-WT as well as KRAS and NRAS MT tumors were more often found in males, while tumors bearing mutations in the BRAF gene were more commonly identified in female pts. All MT tumors exhibited a higher likelihood of presenting with lymph node positivity and were associated with more advanced disease stages at time of diagnosis, particularly in BRAF MT specimens. Notably, both KRAS- and BRAF-MT tumors demonstrated a predilection for localization in the right colon. Furthermore, BRAF mutations correlated with poorer differentiation and the presence of more aggressive histological subtypes, including mucinous adenocarcinoma (Table 1).
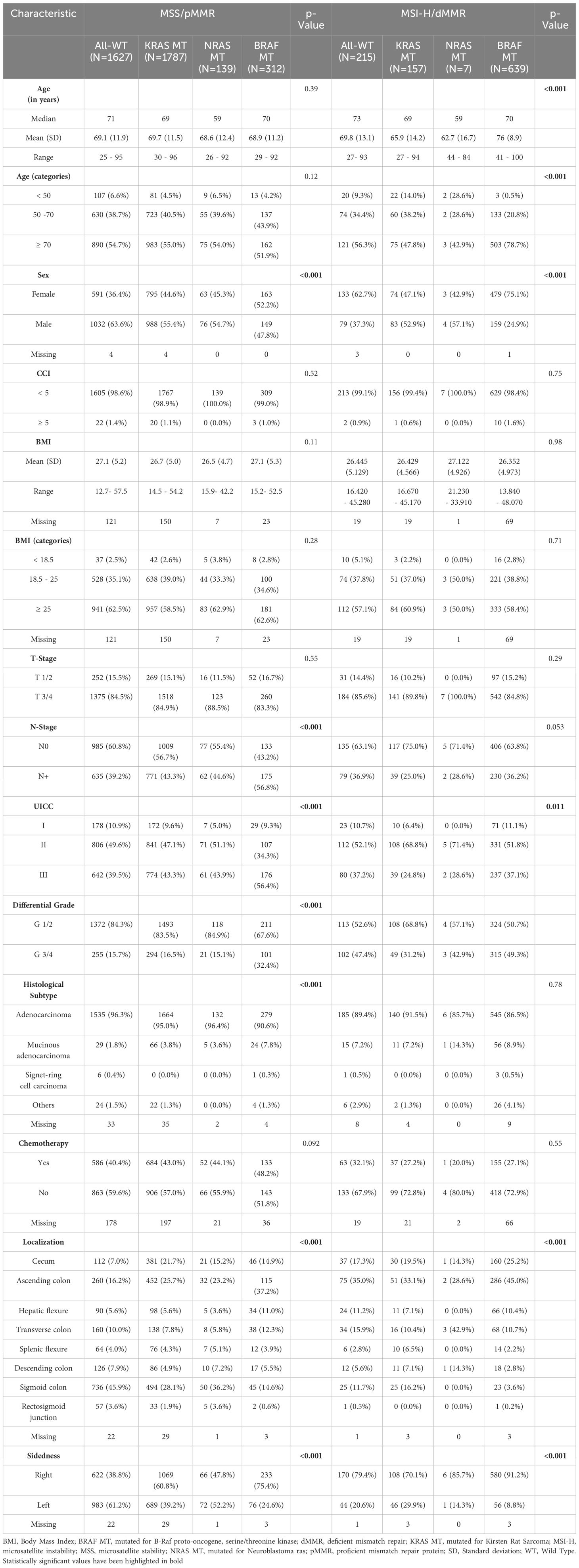
Table 1. Clinical and histopathological baseline characteristics of molecular subgroups in the MSS and MSI- H cohort.
4Among pts. with MSI-H tumors, BRAF mutations were more frequently observed in elderly and female individuals. Additionally, all MSI-H tumors were predominantly localized in the right colon, with this tendency being particularly pronounced in BRAF MT tumors, with 91.2% demonstrating right-sidedness. Conversely, KRAS and NRAS MT MSI-H tumors were more commonly found in younger patients, males, and those with early-stage disease (UICC I-II), often displaying better histological differentiation (Table 1).
In addition, we evaluated the distribution of mutations of the four MMR genes (MLH1, MSH2, PMS2, and MSH6) in MSI-H tumors. In the BRAF wild-type population, the most frequent MMR gene mutation was in MLH1 (22%), followed by MSH6 (21.6%), MSH2 (18.9%), and PMS2 (15.8%). In contrast, the most common MMR gene alteration in the BRAF V600 mutated population was in MSH6 (17.7%), with frequencies of 8.3%, 6.3%, and 9.7% for MLH1, MSH2, and PMS2, respectively (Supplementary Table 4).
3.3 RAS hotspot mutations
In the analysis of individual KRAS hotspot mutations, 62 patients with double mutations in the KRAS gene were excluded and will be described elsewhere. Supplementary Table 3 illustrates the correlations of individual KRAS hotspot mutations with clinical and histopathological characteristics for both the MSS and MSI-H cohorts.
In both cohorts, all KRAS mutations (excluding p.G12D in the MSI-H cohort) were predominantly localized in the right colon. No significant differences were observed regarding age, UICC Stage, or histopathological subtype. In the MSI-H population, mutations such as p.G12D, p.G13D, and p.G12A were more prevalent in males, while p.A146T and p.G12C were more common in females, although the overall numbers were low.
Among the 1751 KRAS-mutated MSS patients, the distribution of mutations was as follows: 30.44% p.G12D, 21.3% p.G12V, 15.31% p.G13D, 5.14% p.G12A, 5.88% p.A146T, and 6.05% p.G12C, with 15.88% classified as other mutations (Figure 3A). The majority of the mutations were located in the known hotspot regions of the RAS gene (Figure 3B). Notably, the most frequent KRAS mutation in the MSI-H cohort was p.G13D, comprising 28.9% of all KRAS mutations. Codon 12 mutations were less prevalent in the MSI-H population (p.G12D 22.37%, p.G12V 6.58%, p.G12A 3.95%, and p.G12C 1.97%), while there were no significant differences in the frequency of the p.A146T mutation between the MSS and MSI-H cohorts (5.88% vs. 5.92%, respectively). The MSI-H population exhibited a greater diversity of KRAS mutations, including rarer and uncommon mutations, categorized as “others” in this analysis (30.26% in the MSI-H vs. 15.88% in the MSS population) (Figure 3A).
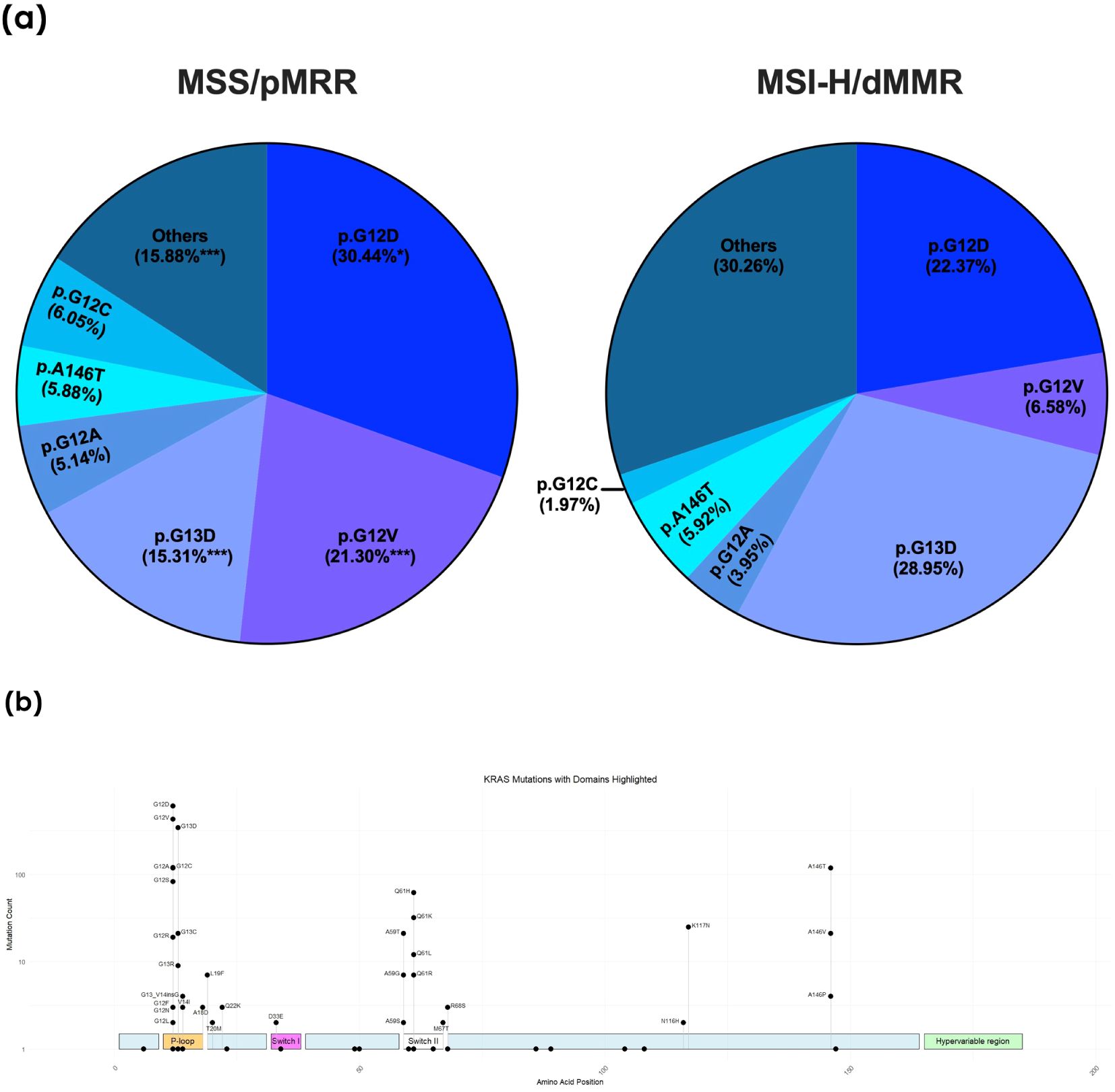
Figure 3. (A) Frequency of individual KRAS hotspot mutations in the microsatellite stable (MSS) and instable (MSI-H) cohorts. dMMR, deficient mismatch repair protein; pMMR, proficient mismatch repair protein: **p < 0.01; ***p < 0.001. (B) Distribution of the individual KRAS mutations with highlighted domains of the KRAS gene. The height of the respective point corresponds to the frequency of the mutation found in the collective.
In the overall population, 146 (3%) NRAS mutations were detected. Three pts. were excluded due to double mutations. The most prevalent NRAS mutations consisted of codon 61 mutations, with p.Q61K observed in 34 out of 143 cases (23.77%), p.Q61R in 22 out of 143 cases (15.38%), and p.Q61L in 14 out of 143 cases (9.79%). Following these, codon 12 mutations were identified, with p.G12D occurring in 30 out of 143 cases (20.98%) and p.G12C in 6 out of 143 cases (6.29%). Of the NRAS-mutated patients, 136 out of 143 (95.1%) belonged to the MSS cohort, indicating a rare co-occurrence of NRAS mutations and MSI-H status (data not shown).
3.4 BRAF mutations
In the cohort of 951 BRAF MT tumors, only 4 possessed double mutations and were thus excluded from this subgroup analysis. Notably, the majority of BRAF MT tumors also demonstrated MSI-H status (636/947, 67.16%). Furthermore, the overwhelming majority of BRAF mutations in both the MSS and MSI subgroups were BRAF V600E mutations, accounting for 89% in the MSS cohort and 98.6% in the MSI-H cohort, respectively (Table 2). BRAF-V600E MT MSS tumors were more frequently associated with patients with a higher BMI and exhibited poorer differentiation compared to Non-V600E mutated tumors. Interestingly, BRAF-V600E mutated MSI-H patients were the oldest, with a median age of 77 years. Moreover, BRAF MT tumors, both in the MSS and MSI-H groups, were predominantly localized in the right colon. Specifically, only 24.1% of BRAF MT MSS tumors and 8.8% of BRAF MT MSI-H tumors were localized in the left colon (Table 2).
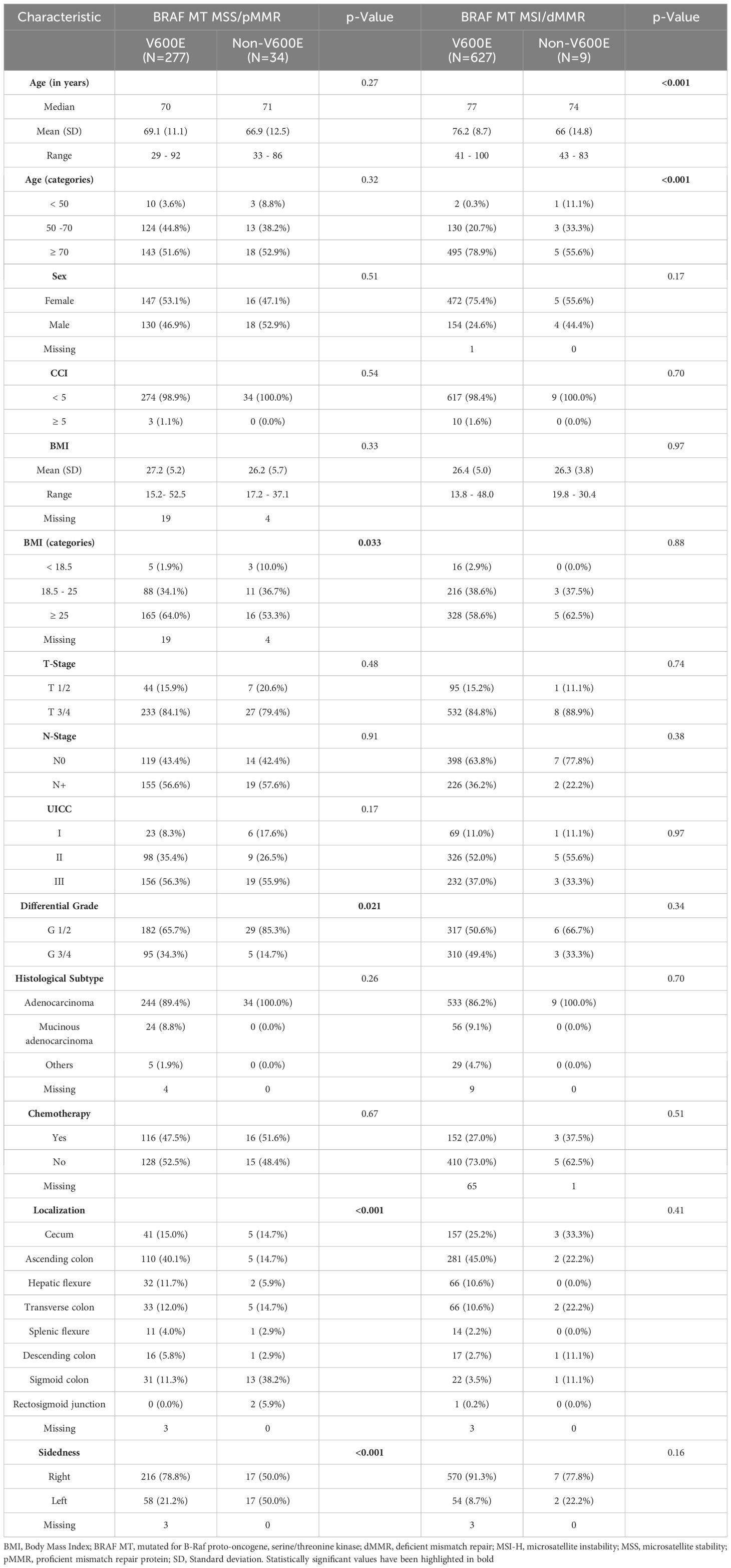
Table 2. Clinical and histopathological baseline characteristics of BRAF V600E and Non-V600E in the MSS and MSI- H cohort.
3.5 Survival
Due to the limited number of NRAS MT cases in the overall population, survival analysis combined KRAS and NRAS MT tumors into the RAS MT category. Cox proportional hazards regressions were conducted to assess both the main effects of MSI status and mutation group, as well as their interaction effects. No significant impact on survival was observed for RAS MT tumors in either MSS or MSI-H subsets. However, in the MSS cohort, BRAF MT tumors exhibited a significantly worse DFS (HR=1.74, 95% CI: 1.15-2.62, p=0.009). Although this subgroup showed similar effects for OS, statistical significance was not reached (HR 1.6, 95% CI: 0.99-2.60, p=0.055). Conversely, the occurrence of BRAF MT did not significantly influence survival in the MSI-H subset.
Adjusted survival probabilities for DFS and OS are presented in Figure 4. In the MSS cohort, the 3-year DFS rates were 73.1% for All-WT (95% CI: 70.8-75.5), 72.2% for RAS MT (95% CI: 70.0-74.3), and 61.4% for BRAF MT (95% CI: 56.1-66.7) pts respectively. The corresponding 5-year OS survival rates were 71% (95% CI: 68.2-73.7), 72.3% (95% CI: 69.8-74.9), and 57.7% (95% CI: 51.6-63.7). For All-WT, RAS MT, and BRAF MT MSI-H tumors the 3-year DFS rate was 75.6% (95% CI: 69.7-81.5), 75.6% (95% CI: 68.2-82.9), and 76.9% (95% CI: 73.6-80.2) with corresponding 5-year OS rates of 76.4% (95% CI: 69.6-83.2), 75% (95% CI: 66.6-83.4), and 74.6% (95% CI: 70.9-78.4).
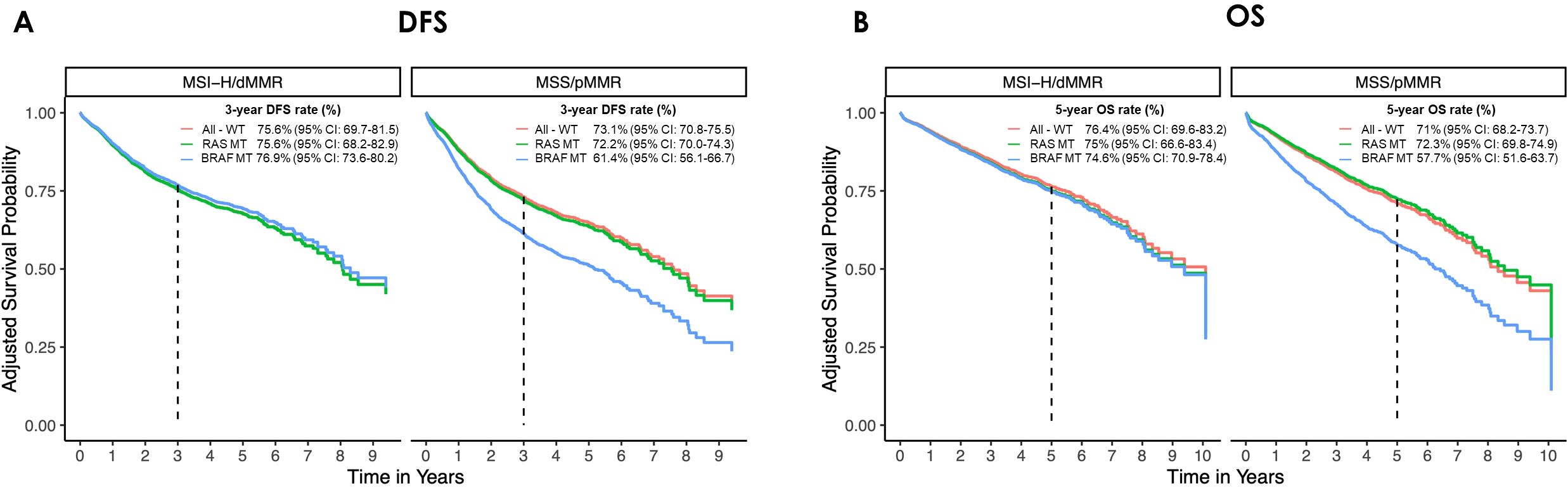
Figure 4. Adjusted survival curves for (A) disease-free survival (DFS) and (B) overall survival (OS) in the microsatellite stable (MSS/pMMR) and instable (MSI-H/dMMR) cohorts by mutational status. The curves have been adjusted for age, gender, UICC stage, sidedness, CCI. (All-WT, wild type tumors; BRAF MT, mutated for B-Raf proto-oncogene, serine/threonine kinase; CI, confidence interval; dMMR: deficient mismatch repair protein; pMMR, proficient mismatch repair protein; RAS MT, mutated for rat sarcoma).
4 Discussion
The findings of this study shed light on the prevalence and clinical implications of KRAS- NRAS- and BRAF mutations stratified by MSI status in localized CC. This dataset represents a big real-world prospective cohort analyzing genetic alterations and their correlations with clinical and histopathological features, as well as prognostic outcomes in this patient population.
In our cohort the prevalence of MSI-H in stage I-III CC was 20.8%. Previous studies have reported MSI-H prevalence in CC ranging from 10% to 20% (14–17). Notably, the distribution of molecular subtypes within our study cohort revealed that 37.7% had All-WT tumors, 39.8% had KRAS mutations, 3% had NRAS mutations, and 19.5% had BRAF mutations. Prior studies have reported varying prevalence rates with KRAS mutations ranging from 28.7% to 49.3%, NRAS mutations from 2.2% to 9%, and BRAF mutations from 4% to 14% (8, 16, 18–28). It is important to acknowledge that previous investigations often focused on specific stages of CC, or included metastatic/recurrent colon and/or rectal cancer, potentially contributing to observed differences. Moreover, some studies narrowly examined specific codon mutations within the KRAS gene, potentially leading to an underrepresentation of mutation frequencies. In contrast, our study identified a broader spectrum of KRAS mutations, with approximately 15% located in codon 146 or other codons, highlighting the complexity of mutational landscapes in CC. In this extensive cohort, we observed a higher prevalence of both All-WT (46.2% vs. 21.1%) and KRAS MT tumors (46.2% vs. 15.4%), and a lower incidence of BRAF MT tumors (8.1% vs. 62.8%) within the MSS population compared to MSI-H CC. Consistent with our findings, a recent study by Taieb et al. demonstrated analogous distinctions between MSS and MSI-H Stage III CC (28). BRAF mutations have been shown to be associated with the MSI-H subtype, as confirmed by our data (29). In terms of clinical and histopathological features, both KRAS and BRAF MT tumors in both MSS and MSI-H subsets exhibited a preference for localization in the right colon, consistent with findings from previous studies (30–32). Supporting this, analysis from the cancer genome atlas dataset revealed that KRAS mutations were present in 45.5% of right-sided and 40.3% of left-sided colorectal cancers, while BRAF mutations occurred in 24.2% and 2.1% of cases, respectively (33). Additionally, BRAF MT tumors in both subsets demonstrated a higher frequency among female pts. Significantly, the vast majority of BRAF mutations were attributed to the well-known BRAF V600E mutation. In the MSS population, both KRAS and BRAF MT tumors were more frequently diagnosed at advanced disease stages, with BRAF mutations being associated with poorer differentiation, findings in line with previous research on CC (20, 24, 28, 30). Notably, BRAF mutations have been linked with more aggressive CC subtypes with a poorer prognosis (18, 34, 35). Conversely, RAS MT tumors in the MSI-H population, although rare, were more prevalent among younger, male pts and exhibited better histological differentiation.
One might speculate whether these differences may be attributed to additional alterations in various genetic pathways. For instance, alterations in the DNA polymerase genes could cause a hypermutated phenotype. In the MSS cohort, the prevalence of POLE and POLD1 mutations shows no correlation with the BRAF/KRAS mutation status. Both KRAS/BRAF mutated and WT cases show a low mutation rate (1-4%) (Supplementary Table 5).
In contrast, the MSI cohort shows a higher frequency of pathogenic POLE and POLD1 mutations. POLE mutations were detected in ~6% of KRAS or BRAF mutated pts. and 13.4% in BRAF/KRAS WT cases. POLD1 mutation were even more frequent (KRAS/BRAF MT 20-25%, WT cases 31.7%) (Supplementary Table 5). Whether these are primary mutations in the polymerase genes or a secondary effect of deficient mismatch repair needs to be subject of further analysis.
These findings underscore the heterogeneity of clinicopathological characteristics associated with different mutation profiles in CC, emphasizing the importance of molecular profiling in patient stratification and the development of personalized treatment approaches. Double mutations involving RAS and BRAF genes or within one gene were rare and therefore excluded from further analysis in this study. Consistent with existing literature, the majority of KRAS mutations were found in codons 12 and 13 (8), constituting ~ 85% of all KRAS mutations in MSS and ~ 70% in MSI-H subsets. In MSS tumors, the most common KRAS mutations were p.G12D, followed by p.G12V, in line with findings from the COSMIC database, suggesting these mutations are the most frequent in CC (36). Conversely, in the MSI-H subset, p.G13D followed by p.G12D were more prevalent, consistent with previous observations (28). Notably, the targetable mutations p.G12C mutation was identified in 5.88% of KRAS-mutated MSS cases, with a lower prevalence in MSI-H cases (1.92%), in accordance with other research as summarized in (37). The MSI-H population exhibited a broader spectrum of KRAS mutations, including rare variants. A study analyzing cancer specimens >13000 CRC pts also found a wider distribution of KRAS mutations in the MSI-H subset, including ~9% p.A146T and ~22% other missense structural variants (38). These differences underscore the biological distinctiveness of MSS and MSI-H CC and support the need to consider them as distinct entities with potentially differing therapeutic strategies in the future. Given the remarkable success of immune checkpoint inhibitors in treating localized MSI-H CC (39), they may become a standard therapeutic option for this patient category. Furthermore, these differences are noteworthy when considering that codon 12, but not codon 13 mutations, have been linked to resistance to anti-EGFR therapy and poorer prognosis in CC (40–43).
The survival analysis conducted in our cohort revealed a distinct pattern regarding the influence of RAS and BRAF mutations on DFS and OS among MSI-H and MSS pts. RAS and BRAF mutations showed no significant impact on survival among MSI-H pts, who generally exhibited longer median DFS and OS compared to MSS pts (although no statistical comparison between MSS and MSI-H groups was conducted in our analysis). In contrast, BRAF MT pts in the MSS cohort displayed a significantly shorter DFS (HR 1.74) and showed a clear tendency towards worse OS (HR 1.6),. Remarkably, RAS mutations did not emerge as a significant factor affecting the survival outcomes of MSS pts within our heterogeneous cohort. KRAS and BRAF mutations have frequently been associated with poorer survival in metastatic disease (44, 45), however, their impact in localized stages has delivered conflicting results. For instance, a pooled analysis of the PETACC-3, EORTC 40993 and SAKK 60-00 trials demonstrated no major prognostic value of KRAS mutations regarding relapse-free survival (RFS) or OS, while BRAF mutation was not prognostic for RFS, but was for OS, particularly in pts with MSS tumors in this analysis (46). Other studies, including post-hoc analysis of large randomized adjuvant trials and meta-analysis have demonstrated detrimental effects of both KRAS (and its subtypes) and BRAF MT on prognostic endpoints not only including DFS and OS but also time to recurrence and survival after recurrence (24, 28, 47).
The conflicting results observed in studies may arise from variations in study populations, methodologies, and treatment modalities. Differences in patient characteristics, such as age, tumor stage, and comorbidities, could confound the interpretation of survival outcomes. Additionally, the inherent biological disparities between MSS and MSI-H tumors may lead to distinct responses to mutational events. MSI-H tumors, characterized by high levels of genomic instability, may trigger immune responses and result in a better prognosis compared to MSS tumors, which are generally more genomically stable (48, 49). This fundamental difference in tumor biology could influence the prognostic significance of RAS and BRAF mutations in each subtype. Importantly, the poor prognosis observed in MSS BRAF MT pts underscores the urgent need for the development of improved therapeutic strategies and tailored treatments for this subset of pts.
However, our study has certain limitations. Firstly, the registry is not based on a random sample, potentially limiting its representativeness for all CC patients. Secondly, the survival analysis could only adjust for a restricted number of factors, leaving room for additional (partially, possibly unknown) factors necessitating adjustment. Furthermore, this study is fundamentally exploratory, thereby precluding definitive confirmatory statements. Nonetheless, these findings highlight the complex interplay between molecular subtypes, clinical and histopathological features, and survival outcomes in CC, emphasizing the necessity for further investigation into the underlying mechanisms driving these associations. Considering these results, stratifying pts according to molecular subtypes may offer advantages in personalized treatment approaches. Given the relatively small numbers of certain mutations and the limited opportunities to analyze pts in randomized trials, real-world data analyzing larger cohorts like ours may play a pivotal role in advancing our understanding of the biological mechanisms driving outcomes for colorectal cancer pts.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by Ethics Committee of the Ruhr-University Bochum (registration number: 17-6151_13 and 12-4449). The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
IE: Conceptualization, Formal analysis, Investigation, Visualization, Writing – original draft, Writing – review & editing. DZ: Conceptualization, Formal analysis, Investigation, Methodology, Resources, Writing – original draft. JC: Conceptualization, Formal analysis, Investigation, Methodology, Resources, Supervision, Validation, Writing – review & editing. SW: Data curation, Methodology, Project administration, Resources, Writing – review & editing. VH: Investigation, Resources, Writing – review & editing. BS: Investigation, Resources, Writing – review & editing. SH: Investigation, Resources, Writing – review & editing. CLa: Investigation, Resources, Writing – review & editing. LM: Investigation, Resources, Writing – review & editing. CLu: Investigation, Project administration, Resources, Supervision, Writing – review & editing. BV: Methodology, Resources, Writing – review & editing. RD: Formal analysis, Visualization, Writing – review & editing. TT: Formal analysis, Visualization, Writing – review & editing. IF: Data curation, Formal analysis, Methodology, Validation, Writing – review & editing. AR-S: Conceptualization, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Writing – review & editing. AT: Conceptualization, Data curation, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Writing – review & editing. IT: Conceptualization, Data curation, Methodology, Project administration, Resources, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was supported by the Protein Research Unit Ruhr within Europe (PURE), funded by the Ministry of Innovation, Science and Research (MIWF) of North-Rhine Westphalia, Germany (grant number: 233-1.08.03.03-031-68079) and by Roche Pharma AG from 2016-2018. The funder was not involved in the study design, collection, analysis, interpretation of data, the writing of this article, or the decision to submit it for publication.
Acknowledgments
The authors thank all CPP study centers, patients and their families, as well as S. Westphal for her work.
Conflict of interest
JC: Advisory Boards: AstraZeneca GmbH, Sanofi-Aventis Deutschland GmbH, Honoraria and Travel Support: AstraZeneca GmbH, Qiagen GmbH, Janssen-Cilag GmbH, MSD Sharp & Dohme GmbH, Merck Healthcare Germany, BS: Trial support institution/study grant institution: BioNTech, SH: Advisory Boards: Fa. Mediglobe ( EUS-KI Project), LM: Travel support: Octapharm, Pierre Fabre, ARS: Honoria: Amgen, Roche, Merck Serono, Bristol-Myers Squibb, MSD, MCI Group, AstraZeneca, Advisory board member: Amgen, AstraZeneca; Bristol-Myers Squibb, Daiichi-Sankyo, Janssen-Cilag, Merck Serono, MSD, Travel support: Roche, Amgen, Pierre Fabre Studies (trial support institution/study grant institution): Roche, Ipsen, Funding for scientific research: Roche, Celgene, Ipsen, Amgen, Alexion Pharmaceuticals, Astra Zeneca, Lilly, Servier, AIO Studien gGmbH, Rafael Pharmaceutics, Erytech Pharma, BioNTech, AT: Trial support institution/study grant institution: BioNTech.
The remaining author(s) declare(s) that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2024.1434791/full#supplementary-material
References
1. Grothey A, Sobrero AF, Shields AF, Yoshino T, Paul J, Taieb J, et al. Duration of adjuvant chemotherapy for stage III colon cancer. N Engl J Med. (2018) 378:1177–88. doi: 10.1056/nejmoa1713709
2. André T, Meyerhardt J, Iveson T, Sobrero A, Yoshino T, Souglakos I, et al. Effect of duration of adjuvant chemotherapy for patients with stage III colon cancer (IDEA collaboration): final results from a prospective, pooled analysis of six randomised, phase 3 trials. Lancet Oncol. (2020) 21:1620–9. doi: 10.1016/s1470-2045(20)30527-1
3. Sargent D, Sobrero A, Grothey A, O'Connell MJ, Buyse M, Andre T, et al. Evidence for cure by adjuvant therapy in colon cancer: observations based on individual patient data from 20,898 patients on 18 randomized trials. J Clin Oncol. (2009) 27:872–7. doi: 10.1200/jco.2008.19.5362
4. Cervantes A, Adam R, Roselló S, Arnold D, Normanno N, Taïeb J, et al. Metastatic colorectal cancer: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann Oncol. (2023) 34:10–32. doi: 10.1016/j.annonc.2022.10.003
5. Kopetz S, Grothey A, Yaeger R, Van Cutsem E, Desai J, Yoshino T, et al. Encorafenib, binimetinib, and cetuximab in BRAF V600E-mutated colorectal cancer. N Engl J Med. (2019) 381:1632–43. doi: 10.1056/nejmoa1908075
6. André T, Shiu KK, Kim TW, Jensen BV, Jensen LH, Punt C, et al. Pembrolizumab in microsatellite-instability-high advanced colorectal cancer. N Engl J Med. (2020) 383:2207–18. doi: 10.1056/NEJMoa2017699
7. Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. (2008) 26:1626–34. doi: 10.1200/jco.2007.14.7116
8. Karapetis CS, Khambata-Ford S, Jonker DJ, O'Callaghan CJ, Tu D, Tebbutt NC, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med. (2008) 359:1757–65. doi: 10.1056/NEJMoa0804385
9. Hallin J, Engstrom LD, Hargis L, Calinisan A, Aranda R, Briere DM, et al. The KRAS(G12C) inhibitor MRTX849 provides insight toward therapeutic susceptibility of KRAS-mutant cancers in mouse models and patients. Cancer Discovery. (2020) 10:54–71. doi: 10.1158/2159-8290.cd-19-1167
10. Fakih MG, Salvatore L, Esaki T, Modest DP, Lopez-Bravo DP, Taieb J, et al. Sotorasib plus panitumumab in refractory colorectal cancer with mutated KRAS G12C. N Engl J Med. (2023) 389:2125–39. doi: 10.1056/nejmoa2308795
11. Noepel-Duennebacke S, Juette H, Feder IS, Kluxen L, Basara N, Hiller W, et al. High microsatellite instability (MSI-H) is associated with distinct clinical and molecular characteristics and an improved survival in early Colon cancer (CC); real world data from the AIO molecular registry Colopredict Plus. Z Gastroenterol. (2020) 58:533–41. doi: 10.1055/a-1156-4433
12. Nöpel-Dünnebacke S, Jütte H, Denz R, Feder IS, Kraeft AL, Lugnier C, et al. Causes of mortality in elderly UICC stage III colon cancer (CC) patients–Tumor-related death and competing risks from the German AIO colorectal study group Colopredict Plus (CPP) registry. Cancer Med. (2022) 11:1735–44. doi: 10.1002/cam4.4540
13. Denz R, Klaaßen-Mielke R, Timmesfeld N. A comparison of different methods to adjust survival curves for confounders. Stat Med. (2023) 42:1461–79. doi: 10.1002/sim.9681
14. Kim JE, Hong YS, Kim HJ, Kim K-P, Lee J-L, Park SJ, et al. Defective mismatch repair status was not associated with DFS and OS in stage II colon cancer treated with adjuvant chemotherapy. Ann Surg Oncol. (2015) 22:630–7. doi: 10.1245/s10434-015-4807-6
15. Hause RJ, Pritchard CC, Shendure J, Salipante SJ. Classification and characterization of microsatellite instability across 18 cancer types. Nat Med. (2016) 22:1342–50. doi: 10.1038/nm.4191
16. Taieb J, Le Malicot K, Shi Q, Penault-Llorca F, Bouché O, Tabernero J, et al. Prognostic value of BRAF and KRAS mutations in MSI and MSS stage III colon cancer. JNCI: J Natl Cancer Instit. (2016) 109(5):djw272. doi: 10.1016/j.annonc.2023.09.1744
17. Bonneville R, Krook MA, Kautto EA, Miya J, Wing MR, Chen HZ, et al. Landscape of microsatellite instability across 39 cancer types. JCO Precis Oncol. (2017) 1:1–15. doi: 10.1200/po.17.00073
18. De Roock W, Claes B, Bernasconi D, De Schutter J, Biesmans B, Fountzilas G, et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol. (2010) 11:753–62. doi: 10.1016/s1470-2045(10)70130-3
19. Peeters M, Price TJ, Cervantes A, Sobrero AF, Ducreux M, Hotko Y, et al. Randomized phase III study of panitumumab with fluorouracil, leucovorin, and irinotecan (FOLFIRI) compared with FOLFIRI alone as second-line treatment in patients with metastatic colorectal cancer. J Clin Oncol. (2010) 28:4706–13. doi: 10.1200/jco.2009.27.6055
20. Hutchins G, Southward K, Handley K, Magill L, Beaumont C, Stahlschmidt J, et al. Value of mismatch repair, KRAS, and BRAF mutations in predicting recurrence and benefits from chemotherapy in colorectal cancer. J Clin Oncol. (2011) 29:1261–70. doi: 10.1200/jco.2010.30.1366
21. Maughan TS, Adams RA, Smith CG, Meade AM, Seymour MT, Wilson RH, et al. Addition of cetuximab to oxaliplatin-based first-line combination chemotherapy for treatment of advanced colorectal cancer: results of the randomised phase 3 MRC COIN trial. Lancet. (2011) 377:2103–14. doi: 10.1016/s0140-6736(11)60613-2
22. Douillard J-Y, Oliner KS, Siena S, Tabernero J, Burkes R, Barugel M, et al. Panitumumab–FOLFOX4 treatment and RAS mutations in colorectal cancer. New Engl J Med. (2013) 369:1023–34. doi: 10.1056/nejmoa1305275
23. Shen Y, Wang J, Han X, Yang H, Wang S, Lin D, et al. Effectors of epidermal growth factor receptor pathway: the genetic profiling ofKRAS, BRAF, PIK3CA, NRAS mutations in colorectal cancer characteristics and personalized medicine. PloS One. (2013) 8:e81628. doi: 10.1371/journal.pone.0081628
24. Sinicrope FA, Shi Q, Smyrk TC, Thibodeau SN, Dienstmann R, Guinney J, et al. Molecular markers identify subtypes of stage III colon cancer associated with patient outcomes. Gastroenterology. (2015) 148:88–99. doi: 10.1053/j.gastro.2014.09.041
25. Gong J, Cho M, Sy M, Salgia R, Fakih M. Molecular profiling of metastatic colorectal tumors using next-generation sequencing: a single-institution experience. Oncotarget. (2017) 8:42198–213. doi: 10.18632/oncotarget.15030
26. Chiu JW, Krzyzanowska MK, Serra S, Knox JJ, Dhani NC, Mackay H, et al. Molecular profiling of patients with advanced colorectal cancer: Princess Margaret cancer centre experience. Clin Colorectal Cancer. (2018) 17:73–9. doi: 10.1016/j.clcc.2017.10.010
27. Sagawa T, Sato Y, Hirakawa M, Hamaguchi K, Fukuya A, Okamoto K, et al. Clinical impact of primary tumour location, early tumour shrinkage, and depth of response in the treatment of metastatic colorectal cancer with first-line chemotherapy plus cetuximab or bevacizumab. Sci Rep. (2020) 10:19815. doi: 10.1038/s41598-020-76756-1
28. Taieb J, Sinicrope FA, Pederson L, Lonardi S, Alberts SR, George TJ, et al. Different prognostic values of KRAS exon 2 submutations and BRAF V600E mutation in microsatellite stable (MSS) and unstable (MSI) stage III colon cancer: an ACCENT/IDEA pooled analysis of seven trials. Ann Oncol. (2023) 34:1025–34. doi: 10.1016/j.annonc.2023.08.006
29. Guinney J, Dienstmann R, Wang X, de Reyniès A, Schlicker A, Soneson C, et al. The consensus molecular subtypes of colorectal cancer. Nat Med. (2015) 21:1350–6. doi: 10.1038/nm.3967
30. Gonsalves WI, Mahoney MR, Sargent DJ, Nelson GD, Alberts SR, Sinicrope FA, et al. Patient and tumor characteristics and BRAF and KRAS mutations in colon cancer, NCCTG/Alliance N0147. J Natl Cancer Inst. (2014) 106(7):dju106. doi: 10.1093/jnci/dju106
31. Lee MS, Menter DG, Kopetz S. Right versus left colon cancer biology: integrating the consensus molecular subtypes. J Natl Compr Canc Netw. (2017) 15:411–9. doi: 10.6004/jnccn.2017.0038
32. Xie M-z, Li J-l, Cai Z-m, Li K-z, Hu B-l. Impact of primary colorectal Cancer location on the KRAS status and its prognostic value. BMC Gastroenterol. (2019) 19:46. doi: 10.1186/s12876-019-0965-5
33. Muzny DM, Bainbridge MN, Chang K, Dinh HH, Drummond JA, Fowler G, et al. Comprehensive molecular characterization of human colon and rectal cancer. Nature. (2012) 487:330–7. doi: 10.1038/nature11252
34. Van Cutsem E, Köhne CH, Láng I, Folprecht G, Nowacki MP, Cascinu S, et al. Cetuximab plus irinotecan, fluorouracil, and leucovorin as first-line treatment for metastatic colorectal cancer: updated analysis of overall survival according to tumor KRAS and BRAF mutation status. J Clin Oncol. (2011) 29:2011–9. doi: 10.1200/jco.2010.33.5091
35. Louisa L, Timothy P, Joanne Y, Amanda T. BRAF Mutation in Colorectal Cancer. In: Luis R, editor. Colorectal Cancer, vol. 4 . IntechOpen, Rijeka (2016).
36. Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. (2011) 39:D945–50. doi: 10.1093/nar/gkq929
37. Meng M, Zhong K, Jiang T, Liu Z, Kwan HY, Su T. The current understanding on the impact of KRAS on colorectal cancer. Biomed Pharmacother. (2021) 140:111717. doi: 10.1016/j.biopha.2021.111717
38. Serebriiskii IG, Connelly C, Frampton G, Newberg J, Cooke M, Miller V, et al. Comprehensive characterization of RAS mutations in colon and rectal cancers in old and young patients. Nat Commun. (2019) 10:3722. doi: 10.1038/s41467-019-11530-0
39. Chalabi M, Fanchi LF, Dijkstra KK, Van den Berg JG, Aalbers AG, Sikorska K, et al. Neoadjuvant immunotherapy leads to pathological responses in MMR-proficient and MMR-deficient early-stage colon cancers. Nat Med. (2020) 26:566–76. doi: 10.1038/s41591-020-0805-8
40. De Roock W, Jonker DJ, Di Nicolantonio F, Sartore-Bianchi A, Tu D, Siena S, et al. Association of KRAS p.G13D mutation with outcome in patients with chemotherapy-refractory metastatic colorectal cancer treated with cetuximab. Jama. (2010) 304:1812–20. doi: 10.1001/jama.2010.1535
41. Imamura Y, Morikawa T, Liao X, Lochhead P, Kuchiba A, Yamauchi M, et al. Specific mutations in KRAS codons 12 and 13, and patient prognosis in 1075 BRAF wild-type colorectal cancers. Clin Cancer Res. (2012) 18:4753–63. doi: 10.1158/1078-0432.ccr-11-3210
42. Tejpar S, Celik I, Schlichting M, Sartorius U, Bokemeyer C, Van Cutsem E. Association of KRAS G13D tumor mutations with outcome in patients with metastatic colorectal cancer treated with first-line chemotherapy with or without cetuximab. J Clin Oncol. (2012) 30:3570–7. doi: 10.1200/jco.2012.42.2592
43. Jones RP, Sutton PA, Evans JP, Clifford R, McAvoy A, Lewis J, et al. Specific mutations in KRAS codon 12 are associated with worse overall survival in patients with advanced and recurrent colorectal cancer. Br J Cancer. (2017) 116:923–9. doi: 10.1038/bjc.2017.37
44. Formica V, Roselli M. Targeted therapy in first line treatment of RAS wild type colorectal cancer. World J Gastroenterol. (2015) 21:2871–4. doi: 10.3748/wjg.v21.i10.2871
45. Modest DP, Ricard I, Heinemann V, Hegewisch-Becker S, Schmiegel W, Porschen R, et al. Outcome according to KRAS-, NRAS- and BRAF-mutation as well as KRAS mutation variants: pooled analysis of five randomized trials in metastatic colorectal cancer by the AIO colorectal cancer study group. Ann Oncol. (2016) 27:1746–53. doi: 10.1093/annonc/mdw261
46. Roth AD, Tejpar S, Delorenzi M, Yan P, Fiocca R, Klingbiel D, et al. Prognostic role of KRAS and BRAF in stage II and III resected colon cancer: results of the translational study on the PETACC-3, EORTC 40993, SAKK 60-00 trial. J Clin Oncol. (2010) 28:466–74. doi: 10.1200/jco.2009.23.3452
47. Formica V, Sera F, Cremolini C, Riondino S, Morelli C, Arkenau HT, et al. KRAS and BRAF mutations in stage II and III colon cancer: A systematic review and meta-analysis. J Natl Cancer Inst. (2022) 114:517–27. doi: 10.1093/jnci/djab190
48. Malesci A, Laghi L, Bianchi P, Delconte G, Randolph A, Torri V, et al. Reduced likelihood of metastases in patients with microsatellite-unstable colorectal cancer. Clin Cancer Res. (2007) 13:3831–9. doi: 10.1158/1078-0432.ccr-07-0366
Keywords: colon cancer, localized, real world data, microsatellite instability, RAS, BRAF
Citation: Ekmekciu I, Zucha DM, Christmann J, Wisser S, Heuer V, Sargin B, Hollerbach S, Lamberti C, Müller L, Lugnier C, Verdoodt B, Denz R, Terzer T, Feder I, Reinacher-Schick A, Tannapfel A and Tischoff I (2024) Exploring the molecular profile of localized colon cancer: insights from the AIO Colopredict Plus registry. Front. Oncol. 14:1434791. doi: 10.3389/fonc.2024.1434791
Received: 18 May 2024; Accepted: 11 September 2024;
Published: 19 November 2024.
Edited by:
Melanie Kucherlapati, Harvard University, United StatesReviewed by:
Elena Tosti, Albert Einstein College of Medicine, United StatesDan Aderka, Sheba Medical Center, Israel
Copyright © 2024 Ekmekciu, Zucha, Christmann, Wisser, Heuer, Sargin, Hollerbach, Lamberti, Müller, Lugnier, Verdoodt, Denz, Terzer, Feder, Reinacher-Schick, Tannapfel and Tischoff. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ira Ekmekciu, ira.ekmekciu@rub.de
†These authors have contributed equally to this work and share first authorship