 Mazharul Haque
Mazharul Haque Ritis K. Shyanti
Ritis K. Shyanti Manoj K. Mishra
Manoj K. Mishra- Cancer Research Center, Department of Biological Sciences, Alabama State University, Montgomery, AL, United States
Triple-negative breast cancer (TNBC) is distinguished by negative expression of estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2), making it an aggressive subtype of breast cancer and contributes to 15-20% of the total incidence. TNBC is a diverse disease with various genetic variations and molecular subtypes. The tumor microenvironment involves multiple cells, including immune cells, fibroblast cells, extracellular matrix (ECM), and blood vessels that constantly interact with tumor cells and influence each other. The ECM undergoes significant structural changes, leading to induced cell proliferation, migration, adhesion, invasion, and epithelial-to-mesenchymal transition (EMT). The involvement of EMT in the occurrence and development of tumors through invasion and metastasis in TNBC has been a matter of concern. Therefore, EMT markers could be prognostic predictors and potential therapeutic targets in TNBC. Chemotherapy has been one of the primary options for treating patients with TNBC, but its efficacy against TNBC is still limited. Targeted therapy is a critical emerging option with enhanced efficacy and less adverse effects on patients. Various targeted therapy approaches have been developed based on the specific molecules and the signaling pathways involved in TNBC. These include inhibitors of signaling pathways such as TGF-β, Wnt/β-catenin, Notch, TNF-α/NF-κB and EGFR, as well as immune checkpoint inhibitors, such as pembrolizumab, 2laparib, and talazoparib have been widely explored. This article reviews recent developments in EMT in TNBC invasion and metastasis and potential targeted therapy strategies.
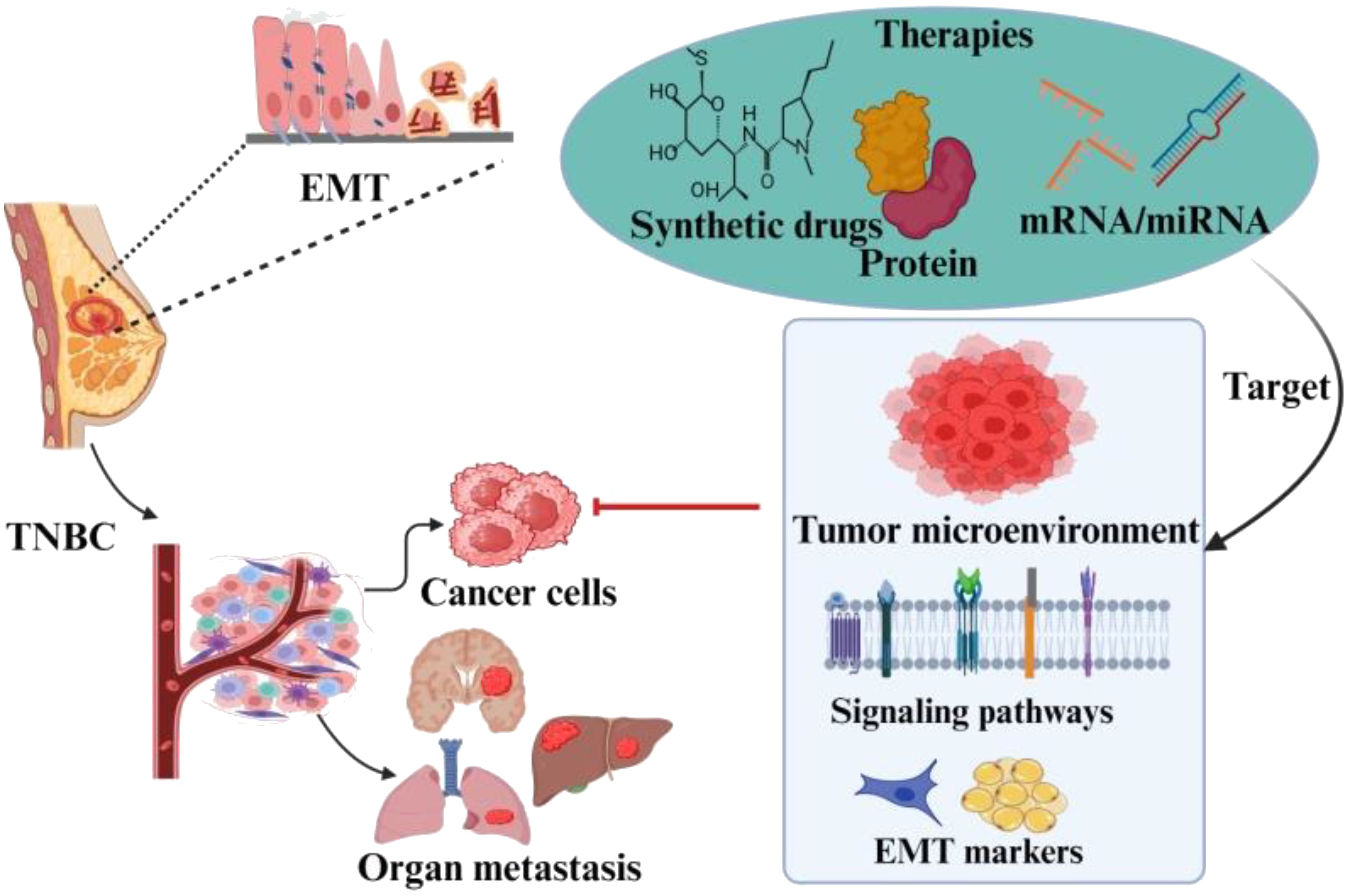
Graphical Abstract. The EMT has profound impact on the TNBC pathogenisity. The factors involved in the metastasis of TNBC due to EMT have been of great concern. The role of these factors in prognosis has enabled researchers to explore it as targeted drug therapies. The aim of this review is to discuss inclusive insights of EMT role in metastasis and the important marker genes and transcription factors to target TNBC and address these challenges.
1 Introduction
Breast cancer (BCa) has become the second most prevalent cause of cancer related mortality among women, consistently increasing the incidence rate annually reported by the American Cancer Society (1, 2). It is the most commonly diagnosed cancer, with approximately 2.3 million cases worldwide annually (3). This unprecedented situation reminds us to investigate and develop improved therapeutic strategies thoroughly. Multiple factors, namely genetic, hormonal, and environmental, as well as other factors associated with lifestyle, are also involved in BCa pathogenesis. Therefore, BCa patients present various clinical, pathological, and molecular abnormalities (4). The expression of estrogen receptors (ER), progesterone receptors (PR), and human epidermal growth factor receptor 2 (HER-2) determines the variability in subtypes of BCa, including luminal A/B, HER-2 overexpression and triple-negative breast cancer (TNBC) (5). TNBC is a subtype, accounting for around 15-20% of all BCa cases, and is characterized by the absence of ER, PR, and HER-2 (6, 7). TNBC is further sub-classified as basal-like, luminal, and mesenchymal based on gene expression profile (8). It is the most heterogeneous among all BCa subtypes and referred as basal A and basal B type. Triple-negative A (basal A) cells are called basal-like as they are enriched with basal markers, including cytokeratins. Phenotypically, triple negative A type cells, highly differentiated subtypes within TNBC subtypes, can exhibit either luminal-like or basal-like morphologies. Thus, triple negative A subtypes closely mimic the core basal tumor subtype. On the other hand, Triple-negative B type (basal B), represents the mesenchymal cluster or normal-like/claudin-low, over-expressed genes and are associated with tumor invasiveness and aggressiveness (9). These subtypes can be utilized for modeling claudin-low or metastatic breast cancer due to their abundance of epithelial-mesenchymal transition (EMT) and stem-cell markers (9).
In comparison to other subtypes, TNBC frequently occurs in young women and is characterized by enormous aggressiveness and mortality rates (10, 11). Around 45% of patients diagnosed with TNBC exhibit distant metastases in the brain or other parts, and the average survival rate ranges from 13.3 to 18 months (12). Several studies have demonstrated that approximately 25% of individuals diagnosed with TNBC can survive for 5 years or more (13). Conventional chemotherapy has shown significant efficacy against TNBC patients. However, its adverse effects become a potential threat, and some patients fail to get any clinical advantages from this treatment. Thus, identifying specific targets for TNBC therapy is a challenging and crucial therapeutic issue that needs to be resolved (14–19). Exploiting whole genome sequencing has provided significant heterogeneity in TNBC and played a critical role in categorizing several subtypes within the TNBC population (20). Different subtypes of TNBC are presented in Figure 1. Due to growing development and advances in bioinformatics tools, the study is gradually moving toward large samples, multi-omics, and refinement of complex data. Potential therapeutic targets derived from genomes, transcriptomics, metabolomics, and proteomics have recently emerged, and many of these research findings have great clinical translational significance and gained enormous importance (21).
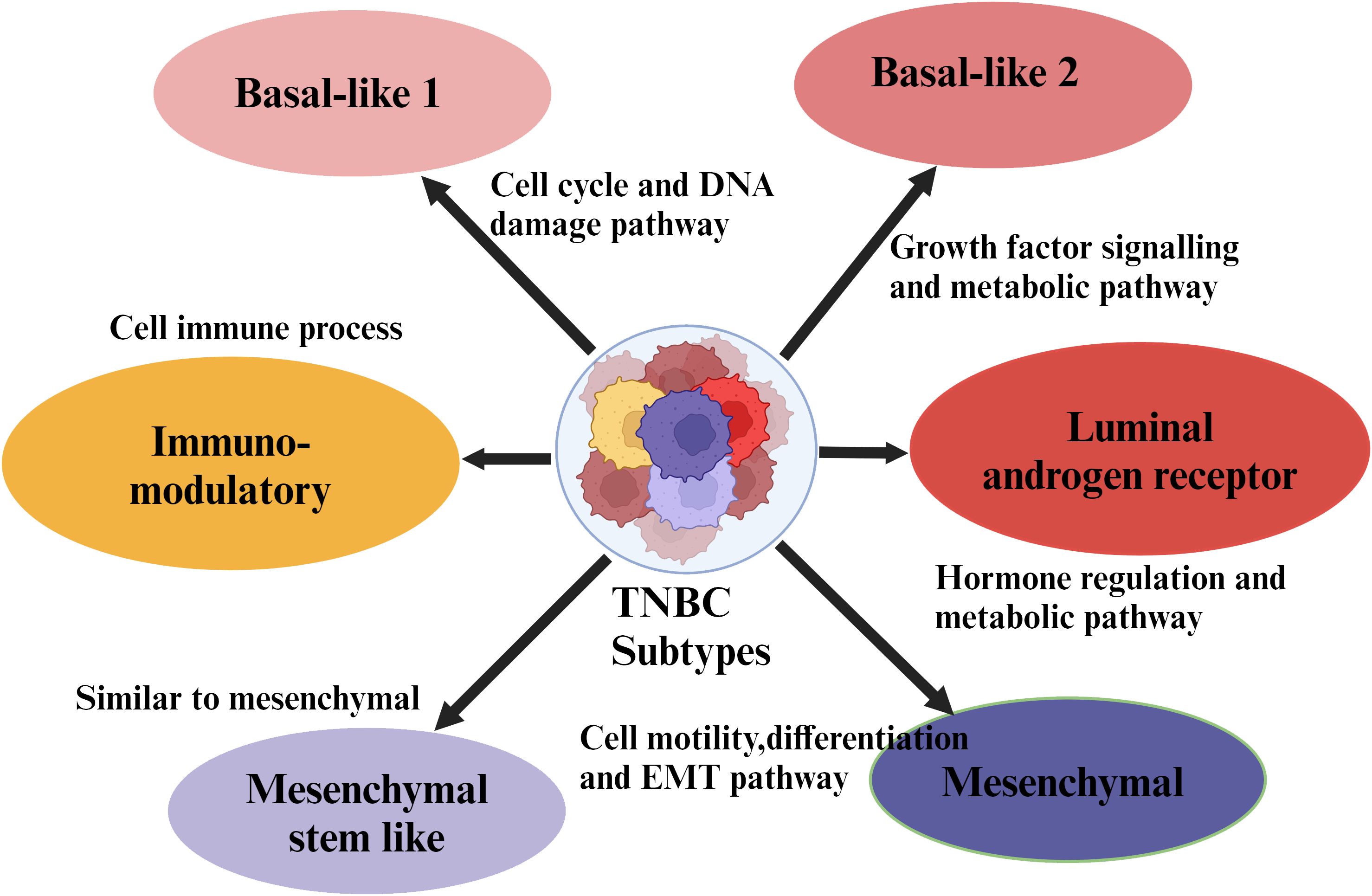
Figure 1. Molecular subtypes of triple negative breast cancer. Major subtypes of breast cancer such as luminal A, luminal B, HER-2 enriched, and TNBC are classified based on the expression of receptors on their cell surface. Further, the TNBC subtypes are sub-classified based on the specific site of the tumor.
1.1 Epithelial-mesenchymal transition
The biological process by which the polarized epithelial cells transform into mesenchymal cell phenotype is called Epithelial to mesenchymal transition (EMT). The significant structural change in extracellular matrix (ECM) induces proliferation, migration, adhesion, invasion, and EMT (22). EMT defines the biological process by which epithelial cells lose their adhesion properties and gain migratory and invasive properties (mesenchymal characteristics), crucial in initiating tumor cell migration and metastasis development (23). This process involves various biochemical changes that result in increased migratory capacity, invasiveness, resistance to apoptosis, and significantly increased production of extracellular matrix (ECM) components (24). Several molecular mechanisms are involved in initiating and facilitating the completion of an EMT (25). These mechanisms involve the activation of transcription factors, expression of particular cell surface proteins, reorganization and expression of cytoskeletal proteins, production of enzymes that degrade ECM, and alterations in the expression of specific microRNA (miRNAs). In numerous instances, the factors involved in the process are utilized as biomarkers to exhibit the progression of a cell undergoing an EMT (Figure 2).
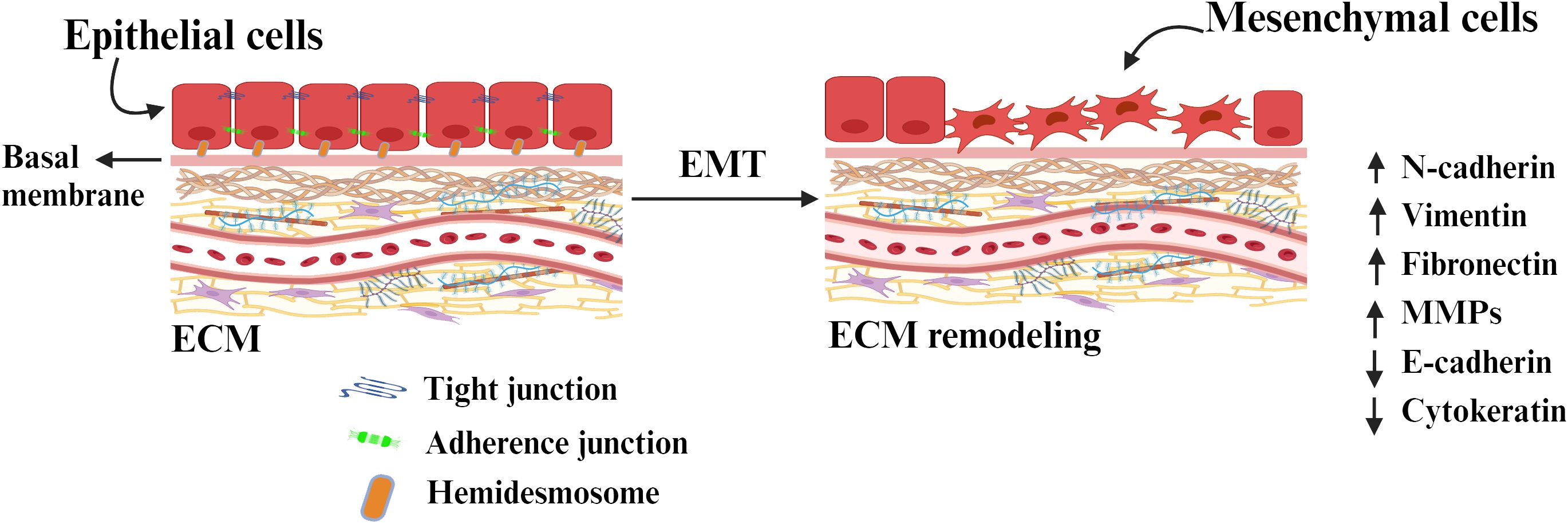
Figure 2. The conversion of epithelial cells to mesenchymal cells through epithelial-mesenchymal transition (EMT) by detaching from the basement membrane. Tight junctions tightly hold together the basal and apical parts of the epithelial cells. The cells are connected to the ECM through adherence junction and hemidesmosome. The epithelial state of the cells is maintained through the expression of a molecule associated with it and retains its polarity. The expression of EMT-inducing molecules and transcription factors remodel the ECM components and induces EMT.
1.2 EMT in TNBC invasion and metastasis
Invasion and metastasis are characteristic features of tumor cells and occur due to transition in intrinsic properties, particularly associated with tumor microenvironment (26). During the progression of a tumor, EMT plays a crucial role and is considered a critical factor in the development and metastasis of TNBC (27). Carcinoma cells in the primary tumor lose cell-cell adhesion due to E-cadherin repression and gain enhanced invasive properties, allowing them to break through the basement membrane and enter the bloodstream via intravasation. Once the tumor approaches a new metastatic site, it may undergo other processes to enhance growth. TGF-β plays a vital role in regulating the morphogenesis and proliferation of normal mammary epithelial cells. However, BCa cells show high resistance towards TGF-β and act as cancer development promoter, which, in turn, modulates angiogenesis, invasion, and resistance against therapeutic interventions (28). Activation of Ras-expressing mammary epithelial cells by TGF-β promotes EMT and inhibits apoptosis (29). It has been discovered that TGF-β plays a crucial role in inducing EMT by regulating through Smad and non-Smad signaling pathways (30). The study revealed that the NF-κB pathway activation leads to EMT induction in TNBC (31). Downregulation of E-cadherin as a result of EMT is indicative of TNBC development. Three distinct core families of transcription factors mediate this regulation. One of the core families belongs to the Snail zinc finger protein family comprises transcription factors Snail 1 and Slug. Another family engaged in regulation is the E-box binding zinc finger protein family, which consists of ZEB 1 and ZEB 2 transcription factors. The basic helix ring helix protein family, consisting of transcription factors TWIST 1, TWIST 2, and E12, is also implicated in the regulation of EMT (25). The overexpression of Snail has been observed in both epithelial and endothelial cells of invasive BCa in comparison to undetectable levels in normal breast tissue (32). The Snail has also been associated with high-grade tumors, metastasis, recurrence, and poor prognosis (33). In addition, the Snail family’s proteins work with other transcription factors to coordinate the combined regulation of EMT. Recent studies have shown a strong association between Slug and TWIST expression in BCa (34). The Snail and TWIST proteins promote ZEB1 expression during EMT (35).
Additionally, ZEB1 has been shown to exhibit stem cell-like properties in TNBC (36). This causes significant risk to cancer patients, as EMT not only allows tumor cells to enter the bloodstream but also enhances their stemness, increasing their potential for tumorigenesis and proliferation (37). The role of EMT and associated factors in the initiation and metastasis of TNBC is presented in Figure 3.
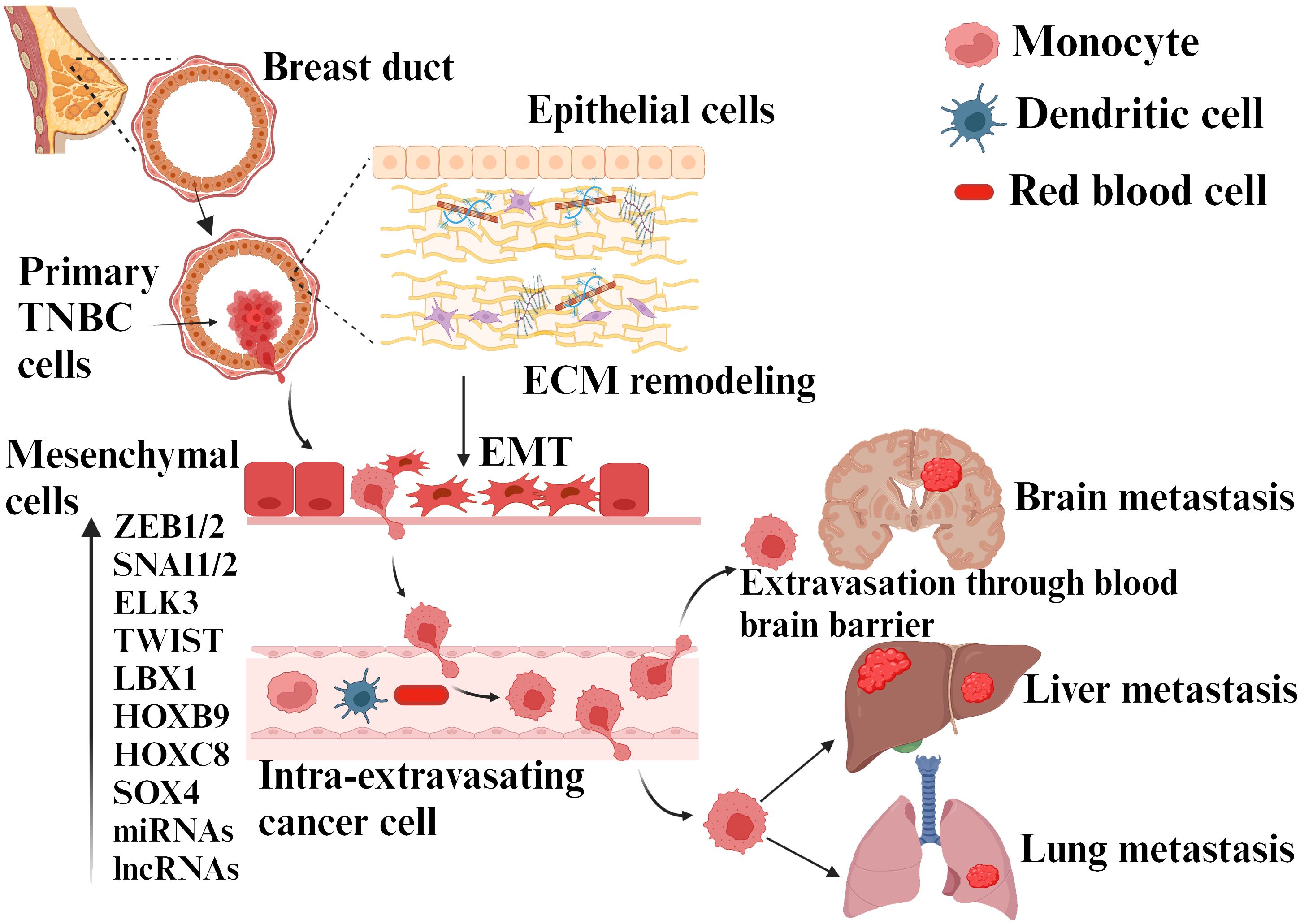
Figure 3. TNBC metastasis due to EMT transition. EMT is induced by various cytokines, molecules secreted by different cells present in the tumor microenvironment, ECM elements, and hypoxia. This process involves different genes, miRNAs/transcription factors, etc. The cancer cells achieve migration, invasion, intravasation, and progression to distant sites by activating EMT and mesenchymal phenotype upon ECM remodeling.
Recent studies also indicate that miRNAs are critical regulators of EMT. miRNAs are short, non-coding, single-stranded RNAs of 20–22 nucleotides that regulate gene expression at the post-transcriptional level (38). The miRNAs are frequently dysregulated in human malignancies and have been linked to the regulation of several cellular processes. According to several reports, specific miRNAs directly target mRNAs of ZEB1 and ZEB2 in cancer cells by upregulating E- cadherin and reducing cell motility (39).
The Protein kinase C iota (PKC-ι) has been also observed to promote TNBC invasion during EMT as well (40). Inhibition of PKC-ι increases the level of E-cadherin and RhoA, while simultaneously decreases vimentin and Par6 (partitioning defective 6 homologs) (41, 42). The role of EMT transcription factors, including SNAIL, TWIST, and ZEB, in BCa invasion mediated via adherens junction has been revealed from several findings (43). Therefore, the effects of EMT and its association with invasion and metastasis appear to be highly dependent on the specific environment. Mesenchymal-to-epithelial transition (MET) is the reversible process of transforming EMT to MET derivatives. It is considered a hallmark of phenotypic plasticity, and the exact mechanism is still poorly understood. However, the MET associated with kidney formation is the most extensively studied example, and it is driven by genes such as bone morphogenetic protein 7 (Bmp7), paired box 2 (Pax2), and Wilms tumor 1 (Wt1) (44). During the MET process, genes associated with epithelium are up-regulated, while mesenchymal-associated genes are downregulated through specific signaling pathways (45).
In addition to these factors related to tumor microenvironments, autophagy has also a role in regulating EMT by activating energy response pathways, initiating EMT-inducing signaling pathways, and managing the breakdown of EMT-related adhesion and cytoskeletal components, as well as EMT-TFs (46). Recent studies have also revealed a shift in the primary effects of EMT, moving away from invasion and metastasis, towards resistance against chemotherapeutic agents.
1.3 TNBC treatment
TNBC has demonstrated sensitivity towards chemotherapy, making it a current standard of care (SOC), though it has limited advantages (47). The Food and Drug Administration (FDA) approved anti-metabolites, paclitaxel, anthracyclines, and neoadjuvant chemotherapy regimens for patients with TNBC (48) presented in Table 1. Patients with TNBC have demonstrated some efficacy with conventional treatment. However, the toxicity of chemotherapy on individuals is a concern, and some may not even experience any therapeutic improvement. Thus, identifying appropriate targets for precise TNBC therapy is a difficult and crucial therapeutic issue that needs to be resolved. Chemotherapeutic drugs such as fluorouracil (5-FU), doxorubicin, and cyclophosphamide have been used for TNBC treatment (49). The current SOC for individuals diagnosed with early TNBC is administered neoadjuvant chemotherapy, along with surgical intervention (50). Currently, no universally accepted chemotherapy treatment option is available for individuals diagnosed with recurrent or resistant TNBC (51). The length of treatment responses is typically short, often followed by rapid relapse, and the occurrence of metastases in visceral organs and the brain is prevalent (52). New therapeutic alternatives have recently become available for patients with advanced TNBC, mainly when surgery is not an option. The therapeutic options currently accessible for individuals diagnosed with advanced TNBC include capecitabine and gemcitabine, eribulin, and DNA cross-linker platinums such as cisplatin and carboplatin (53–55). TNBC has more immunogenic properties than other BCa subtypes with tumor-infiltrating lymphocytes (TILs) in its microenvironment. However, it also exhibits a significant upregulation of programmed cell death ligand 1 (PDL-1) (56, 57). Therefore, the use of immunochemotherapy has been established as a SOC for patients diagnosed with PDL-1 positive, unresectable, locally progressed, or metastatic TNBC.
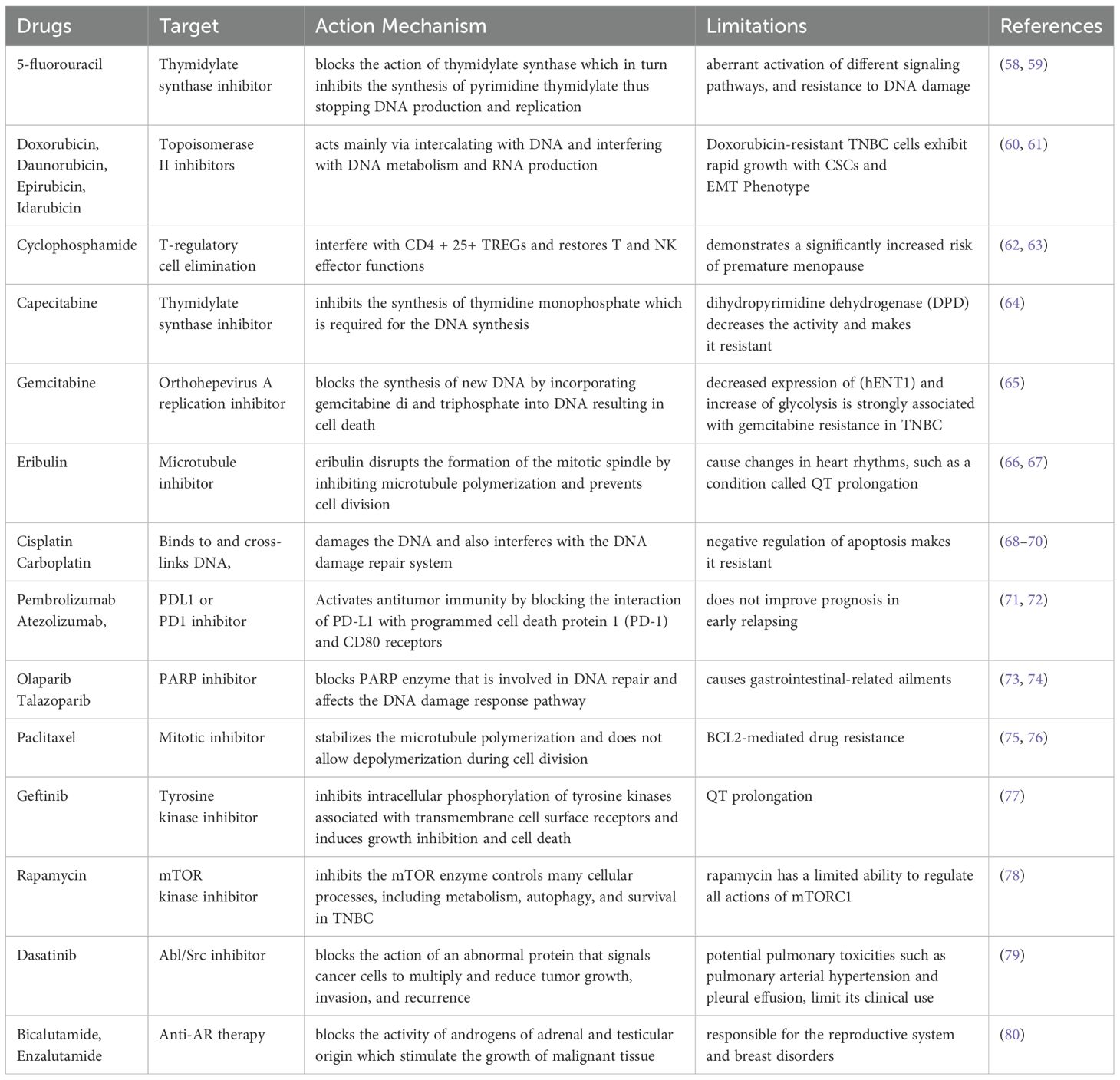
Table 1. List of drugs administered to TNBC patients.
In 2017, pembrolizumab was approved as a histological agonist immunotherapy against tumors involving microsatellite instability and mismatch repair deficiency (81, 82). The FDA approved atezolizumab combined with nanoparticle albumin-bound (nab)-paclitaxel as a first-line therapy against TNBC (83). Tumors with the absence of BRCA1 and BRCA2 show impaired homologous recombination repair and are sensitive to poly(ADP-ribose) polymerase (PARP) inhibitors (84, 85). In 2018, the FDA approved olaparib and talazoparib for treating advanced stage HER2 negative BCa patients with BRCA1/2 mutation. Olaparib was approved upon the significant activity noticed compared to capecitabine or eribulin as chosen by the physician (86). Olaparib demonstrated substantial improvement in median progression-free survival (PFS) in comparison to the control group, with a notable increase of 42% (7 vs. 4 months) (87). Olaparib demonstrated a promising safety profile during prolonged exposure, with no indication of cumulative toxicity. In patients with locally advanced or metastatic BCa with germline BRCA mutation who have previously undergone chemotherapy, talazoparib has been found to increase the average PFS by 46%, with a duration of 8.6 months compared to 5.6 months (88). Another SOC neoadjuvant therapy drug, paclitaxel, for the treatment of patients with TNBC, shows limited benefit for locally advanced or metastatic disease (89). The use of lapatinib and geftinib in combination with capecitabine was approved by FDA as a combination therapy for breast cancer patients (78). The other drug, rapamycin, has been used to target AKT/mTOR pathway and inhibit proliferation of tnbc.
2 Targeted therapy associated with EMT
TNBC is a diverse disease with various genetic variations and molecular subtypes. Due to factors like numerous metastases, extensive organ metastases, and too close to large blood arteries, only a few patients with TNBC metastases are suitable for surgical treatment. To provide patients with personalized therapy and increase their survival chances, investigators studying the metastatic process must bring innovative therapies for patients with advanced BCa. Targeted therapy is one of the critical current options with enhanced efficacy and less adverse effects on patients. The role of different potential targets in EMT is discussed further.
2.1 EMT and genes
Molecular biomarkers are now being studied as possible treatment targets. It has been observed that 11-20% of individuals diagnosed with TNBC, who have not been specifically selected based on their family history, had a hereditary mutation in the BRCA1 or BRCA2 (BRCA1/2) genes. Moreover, current research indicates that deleterious mutations in additional genes associated with cancer susceptibility are also linked to TNBC (90, 91). Other than BRCA1/2 detection, four other established genes (CDH1, PTEN, STK11, TP53) associated with BCa were identified and established as alternate options for diagnosis. The loss of CDH1 gene expression is strongly associated with BCa progression in patients (92). Another critical gene, PTEN expression loss, demonstrates poor prognosis and treatment response. STK11 is a tumor suppressor gene and loss‐of‐function mutations cause tumorigenesis in TNBC (93). Mutation in TP53 is observed in 18%–25% of primary BCa and roughly 80% of TNBCs, which is noticeably more frequent than other BCa subtypes (2, 17).
Later, panels were expanded to include an additional 15-20 candidate genes having similar function to BRCA1/2 in DNA double-strand break repair (ATM, BARD1, CHEK2, PALB2). Furthermore, 25–40 genes (including CDKN2A, MEN1, MLH1, MSH2, MSH6, and MUTYH) that cause cancer risk at different organ sites were identified and added to the existing panel. The number of genes on panels that may be linked to cancer has recently increased to over 100, yet many lack strong enough data to perform patient treatment (94). Before the clinical use of multiple genes panel testing, there was limited understanding of gene alterations in TNBC beyond BRCA1/2. A total of 122 DNA repair genes from germline DNA samples of BCa patients were sequenced, out of which 17 genes (ATM, BRCA1, BRCA2, BARD1, BRIP1, CHEK2, CDH1, MRE11A, NBN, PALB2, PTEN, RAD50, RAD51C, RAD51D, STK11, TP53, and XRCC2) are associated with increased risk of developing BCa were observed (Table 2). A prior report demonstrated that 271 deleterious mutations were detected among patients (95). In this finding, most of these changes (57% or 155 mutations) were observed in BRCA1 mutated tumors, while (18% or 49 mutations) were shown in BRCA2. The remaining (25% or 67 mutations) were distributed among additional susceptibility genes. Notably, PALB2 accounted for 7.7% of these mutations, followed by BARD1 (3.3%), RAD51D (2.5%), RAD50 (2.2%), and RAD51C (2.2%) (90). Of the TNBC patients, 3.7% had these probable harmful mutations in non-BRCA1/2 genes, while 11.2% had BRCA1/2 mutations. Interestingly, no CHEK2 mutations were discovered, which aligns with its link to hormone receptor-positive BCa (96).
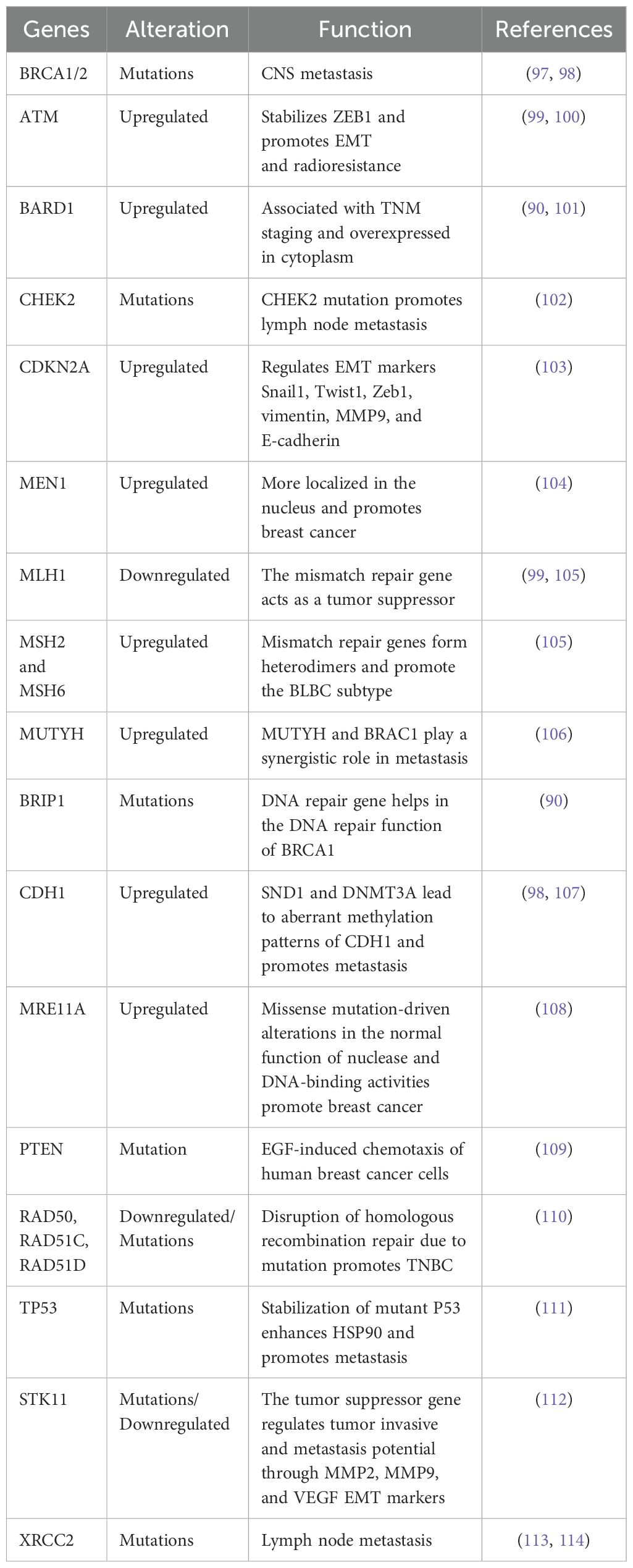
Table 2. List of genes involved in EMT pathogenicity due to altered expression.
2.2 EMT and miRNA
Several studies have evidenced the correlation between different miRNAs and underlying mechanisms of disease progression in TNBC (115–125). These mechanisms involve many processes, such as EMT, cellular migration, invasion, and metastasis (96, 126). Numerous studies suggest that miRNAs have a role in the EMT mechanism. The miR-200 family, which includes miR-200a, miR-200b, miR-200c, miR-141, and miR-429, is thought to contain the majority of miRNAs that negatively regulate EMT. The miR-200a is an EMT inhibitor that targets the E-cadherin repressor ZEB1/2 to maintain the epithelial phenotype (127).
Furthermore, miR-200b inhibits FUT4 expression in BCa cell lines, inhibiting TNBC migration and metastasis via inactivation of EGFR and downstream PI3K/Akt signaling cascade (128). In the meantime, TNBC’s epithelial phenotype is maintained by blocking the genes involved in cell motility, reducing cell growth, and encouraging apoptosis (129). Due to these properties, the miR-200 family is currently considered one of the promising therapeutic targets for the treatment of TNBC (130).
On the other hand, several other miRNAs play significant roles in increasing migration, invasion, and metastasis (131). The upregulation of miR-21 facilitates the proliferation of cancer cells (132). Moreover, 3’ UTR of LZTFL1 (leucine zipper transcription factor-like 1) along with miR-21 activates proliferation and metastasis (133). The upregulation of miR-21 causes increased cell invasion and proliferation in TNBC cells (MDA-MB-468). However, the PTEN gene was observed to be downregulated. This finding highlights the significance of the suppression of miR-21 and the overexpression of PTEN as a prospective therapeutic approach to predicting and assessing individuals diagnosed with TNBC (134). An overview of specific miRNAs linked to the pathophysiology of TNBC and their established targets and associated roles is presented in Table 3.
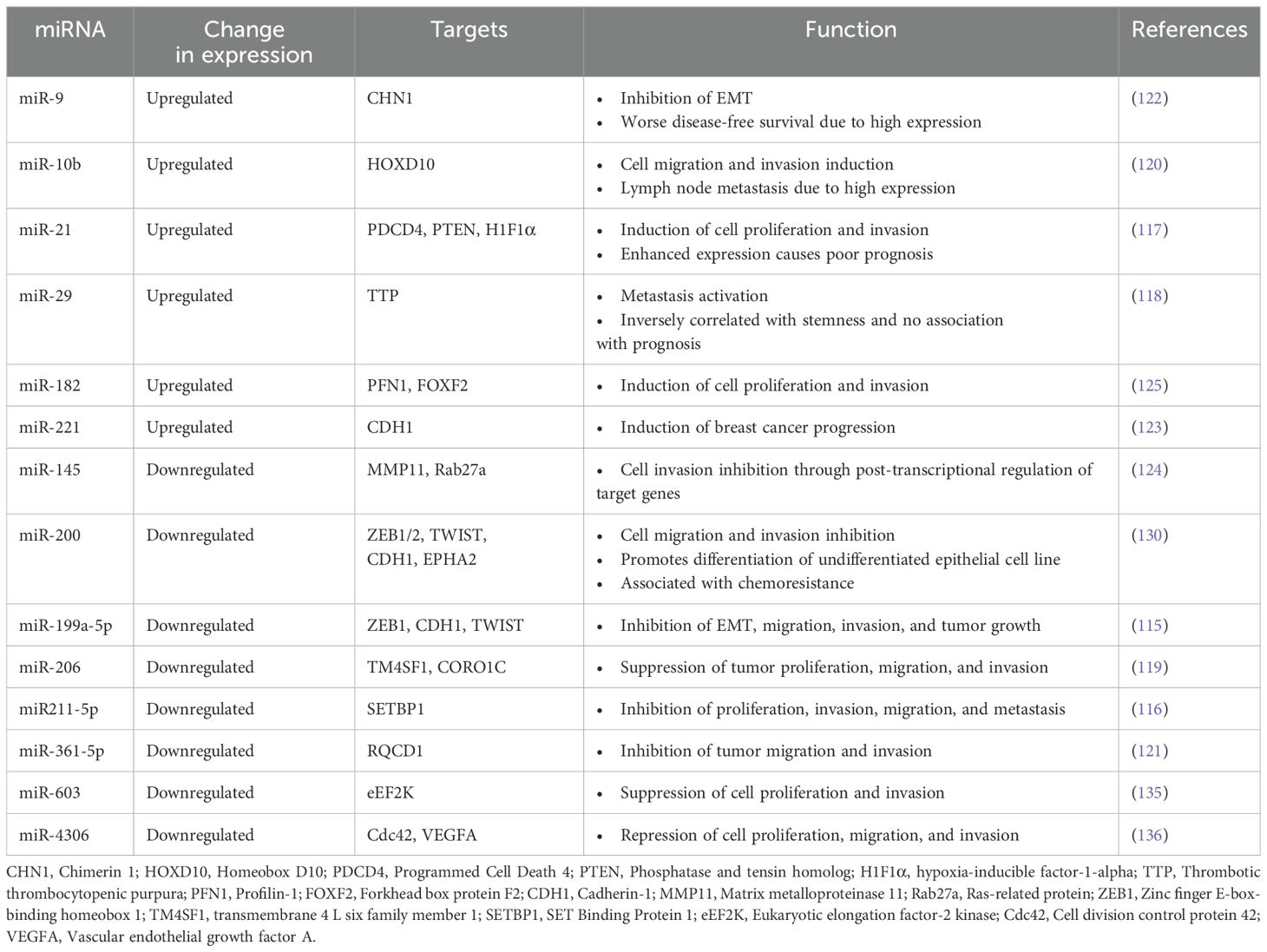
Table 3. Abberated expression of some miRNAs in TNBC and their functions.
2.3 EMT and lncRNA
Non-coding RNAs (ncRNAs) are classified into two groups based on their transcript size. Typically, ncRNAs are comprised of less than 200 nucleotides. These ncRNAs include miRNAs, piwi-interacting RNAs, small nucleolar RNAs (snoRNAs), and short interfering RNAs (siRNA). The second group is long non-coding RNAs (lncRNAs) containing more than 200 nucleotides. lncRNAs include intergenic, intronic, sense, antisense, enhance, and bidirectional (134). The involvement of lncRNAs in several biological processes, including apoptosis, cellular proliferation, cellular differentiation, metastasis, and chromatin remodeling, has been identified. Many tumors, including TNBC, have exhibited abnormal expression patterns of several lncRNAs (137). LncRNAs can bind and compete with miRNA-bound mRNA, resulting in altered regulation of miRNA-mediated genes. They constitute an endogenous RNA network (ceRNA) involving mRNAs and lncRNAs as a post-transcriptional regulatory network in TNBC (138). lncRNA HOTAIR, a well-investigated lncRNA in cancer, has demonstrated its ability to induce alterations in chromatin structure and gene expression, hence facilitating the process of invasion and metastasis, specifically in BCa. Several other lncRNAs have recently been linked to TNBC-related ECM/EMT molecules. The expression of cytoplasmic lncRNA, namely LINK-A (long intergenic non-coding RNA for kinase activation), is involved in growth factor-dependent phosphorylation, stability, and activation of HIF1α and linked with TNBC (139). A summary of different lncRNA and their functions in TNBC is documented in Table 4.
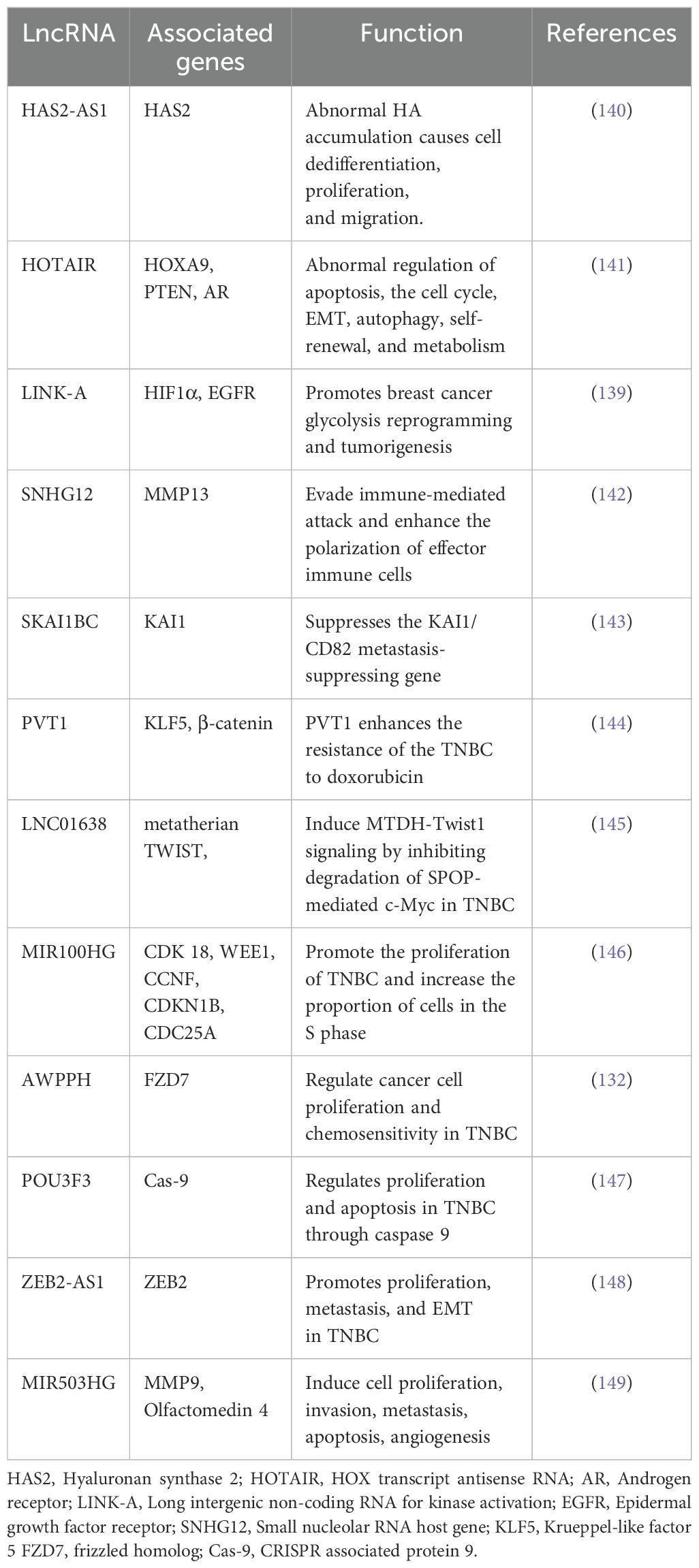
Table 4. LncRNA and associated target genes of TNBC.
The snoRNA host gene 12 (SNHG12), a transcriptional target of c-myc, is highly increased in TNBC. SNHG12 may enhance cell motility through modulating MMP 13 expression (150). Another lncRNA is described as a suppressor of KAI1 in BCa (SKAI1BC) that suppresses the KAI1/CD82 metastasis suppressor gene and promotes TNBC (151). It has been demonstrated that two lncRNAs, Airn and PVT1, control TNBC carcinogenesis by acting opposite to each other on the β-catenin signaling pathway (131). Through TWIST 1 expression, another lncRNA, LINC01638, preserves the ϵμT characteristics of TNBC cells. The tumor growth and metastasis are inhibited through the knockdown of LINC01638 and MIR100HG, which act as oncogenes via controlling p27 (152). The lncRNA AWPPH has been implicated in promoting TNBC growth through the upregulation of frizzled homolog 7 (FZD7) (153) and/or its interaction with miRNA-21 (132). The level of lncRNA POU3F3 in TNBC patient’s plasma has been found to increase as compared to those of normal individuals, and a negative correlation between lncRNA POU3F3 levels and cleaved Caspase 9 was observed. This means that when the level of POU3F3 increases, then caspase 9 decreases, promoting cell proliferation and inhibiting apoptosis in TNBC (147). Many studies have demonstrated that lncRNAs such as HCP, PAPAS, and LUCAT1 have a role in promoting TNBC through the modulation of specific miRNAs (miR-219a-5p, miR-34a, and miR-5702, respectively) (154). The increase of LncRNA-ZEB2-AS1 showed enhanced proliferation and metastasis due to the upregulation of ZEB2 of MDA-MB-231 cells in SCID mice (90). LINC01638 has been found to inhibit the degradation of c-Myc and increase TWIST 1 expression, thereby inducing EMT (145). The lncRNA DLX6-AS1 exhibits an increased expression of EMT markers, promoting cell survival, and enhances resistance to the chemotherapy drug cisplatin in TNBC cells by regulating miR-199b-5p/PXN (155). The inhibition of cell proliferation, invasion, and migration, as well as the enhancement of apoptosis and regulation of the cell cycle, were seen upon the action of lncRNA, RMST (rhabdomyosarcoma 2-associated transcript) (156). The overexpression of another lncRNA, for example, NEF, has been observed to prevent the migration and invasion of TNBC cells (157). The expression of lncRNA PTCSC3 is decreased, but lncRNAH19 has shown increased expression and established an inverse relationship with PTCSC3 levels in TNBC patients. The overexpression of PTCSC3 results in the downregulation of lncRNA H19 in TNBC cells (158). Patients with TNBC who exhibit low expression of lncRNA MIR503HG have shown significantly poorer prognosis than those with high expression. It has also been observed that MIR503HG inhibits the migration and invasion of cells in TNBC by altering the miR-103/OLFM4 axis (159). The LncRNA TCONS_l2_00002973 positively correlates with lower-grade tumors and better survival outcomes. Additionally, it demonstrates inhibitory effects on cancer cell proliferation and promotes apoptosis, specifically in TNBC (160). The in vitro and in vivo studies have shown the inhibitory effects of lncRNA XIST on cell proliferation and EMT in TNBC cells by interfering with the activity of miR-454 (161).
2.4 EMT and proteins
The ECM proteins have a significant role in the tumor microenvironment (Table 5). Many ECM proteins include (BGN, CD44, CD109, DAG1, DCN, ECM1, EFEMP1, FMOD, IGFBP4, IGFBP7, LTBP1, L1CAM, LGALS1, LGALS3BP, LOXL2, LTBP1, NRCAM, P4HB, PLOD1, PPIB, TGF-β I, THBS1, TLN1, and TNC) have found highly expressed in TNBC. However, DCN and TGF-βI are highly expressed in normal cells (162). CD44 is a transmembrane glycoprotein and is highly expressed in TNBC. It plays a pivotal role in mediating cellular adhesion and signaling processes, and its presence in serum is considered a potential prognostic indicator in BCa (163). Recently, significant advancements have been made in developing nanoparticle drug delivery methods, especially targeting CD44 and CEA in colorectal cancer cells and TNBC (164). The insoluble form of CD109 is bound with TGF-β, which acts as a negative regulator and prevents TGF-β signaling (165). The high expression of CD109 in TNBC correlated with a higher histological grade and worse prognosis (166). ECM1, another protein present in the secretome of TNBC, is responsible for inducing angiogenesis and promoting tumor cell proliferation via EGFR signaling (167). Elevated expression of ECM1 in BCa is linked to poor prognosis (168). The glycoprotein Fibulin 3 (FBLN3), also known as EFEMP1, is a protein that interacts with ECM1 and is found in the secretome of TNBC (162). The overexpression of FBLN3 in BCa cases is characterized by low levels of HER2 expression, including TNBC (169). Additionally, it has been observed that FBLN3 has a role in enhancing the invasiveness of tumor cells in xenografts of TNBC (170). The function of FBLN3 in the TNBC secretome is still unknown. The TNBC secretome also contains the closely related protein Fibulin 1 (FBLN1), which interacts with fibronectin (171). The role of FBLN1 in estrogen signaling in BCa is demonstrated in numerous studies. FBLN1 expression is increased by estrogens, particularly that of the spliced variant FBLN1C (172). In an immunohistochemical study of BCa, FBLN1 expression showed an inverse correlation with cathepsin D (173). Interestingly, the TNBC secretome contained both cathepsin D and FBLN1 (174). Importantly, FBLN1 may be involved in BCa cell resistance to doxorubicin therapy (175).
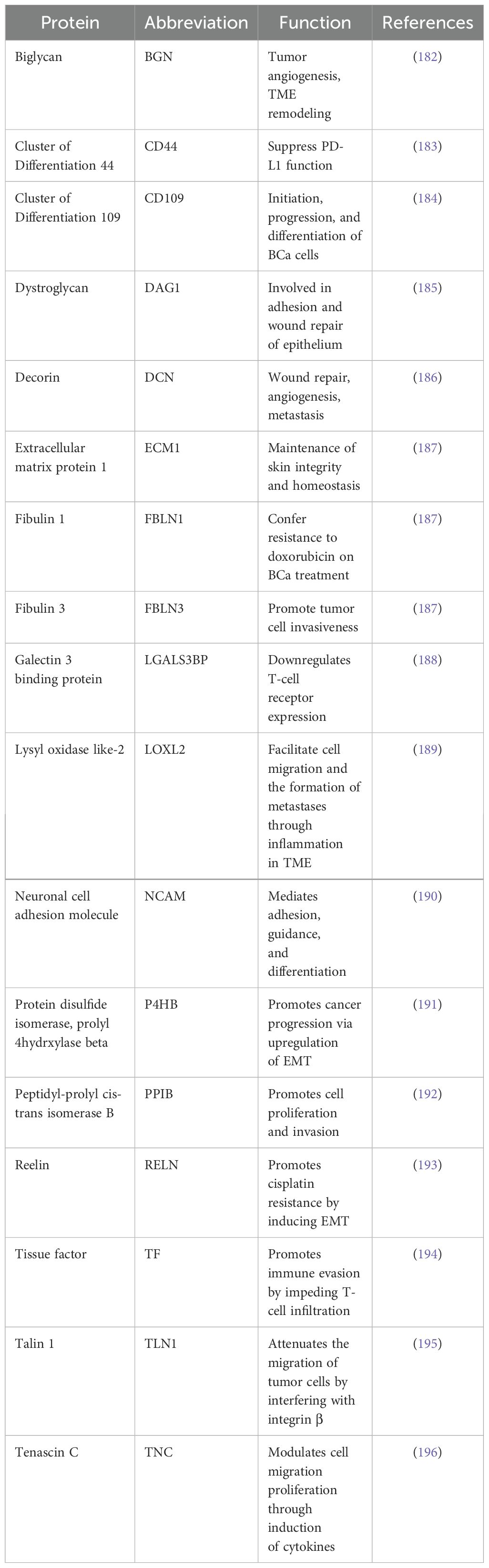
Table 5. ECM proteins in secretome of TNBC.
Several insulin-like growth factor-binding proteins (IGFBPs) have been identified in the secretome of TNBC (176). Proteins belonging to this particular family can bind to IGF and subsequently enhance its half-life (177). The family member Cyr61 (alternatively referred to as CCN1 and IGFBP10) has shown expression in TNBC cells by interacting with the urokinase plasminogen activator receptor (uPAR) (178). It has long been demonstrated that Cyr61 stimulates angiogenesis and tumor development (179). High Cyr61 expression is associated with relapse in TNBC patients, and Cyr61 knockdown decreased TNBC cell line invasiveness, tumor burden, and microvascular density (178). Apart from these ECM proteins, the presence of α2,3-sialylated N-glycoproteins capable of binding lectins in the conditioned media of TNBC cell lines is also reported (180). The expression of polysialic acid in BCa is positively correlated with invasiveness and TNM staging in patients’ tumors. Furthermore, it has also been shown that the suppression of Sialyl transferase X (STX) results in decreased migratory capacity of MDA-MB-231 cells (181). These studies on identifying proteins inside ECM provide us with only a fraction of the overall story since many ECM proteins undergo post-translational modifications, which can also be a target.
3 EMT pathways regulators
Numerous signaling pathways, including TGF-β, Wnt/β-catenin, Notch, TNF-α/NF-κB, Hedgehog (Hh), and receptor tyrosine kinase (RTKs), are involved in EMT regulation (Figure 4) (197). The transcription factors Snail, ZEB1/2, and TWIST, along with miRNA, epigenetic regulators, and alternative splicing, are regulated by these pathways during cancer progression (198). Furthermore, it has been suggested that EMT facilitates the progression of early-stage primary tumors into invasive malignancies and contributes to the development of cancer cells exhibiting stem cell-like properties (36). These properties include enhanced self-renewal capacity, the ability to initiate tumor formation, and resistance to programmed cell death and chemotherapy (199).
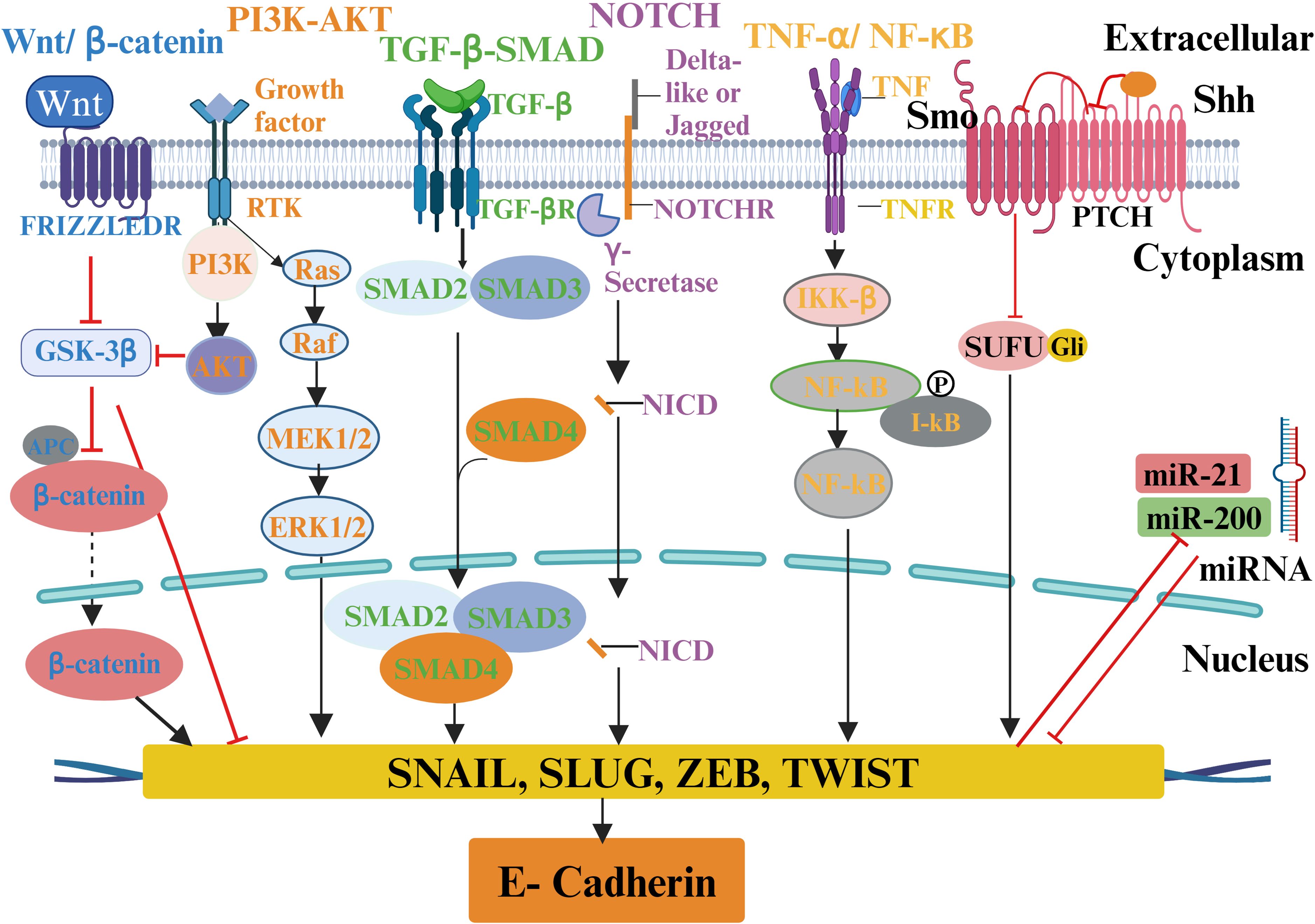
Figure 4. A schematic of various ECM signaling pathways involved in EMT regulation in tumor microenvironment. ECM molecules and proteins interact with the tumor microenvironment to activate multiple biochemical signaling pathways and regulate EMT transcription factors and different EMT inducers to cause EMT and tumor progression. The molecules involved in particular signaling pathways are presented with the same color.
3.1 TGF-β pathway in EMT
TGF-β is a critical cytokine that exhibits a significant role in initiating EMT programming (200). It plays a vital role in activating EMT and interaction with downstream signaling pathways during tumorigenesis (45). The dysregulation of TGF-β expression has been involved in the development of different types of cancer, including breast carcinogenesis (149). In EMT, intracellular Smad2/3 transducer proteins trigger the TGF-β signaling pathway (201). There are three types of TGF-β involved in Smad-dependent signaling: TGF-β1, TGF-β2, and TGF-β3, which are linked to three distinct receptor types: types I, II, and III. When TGF-β binds to TGF-βR-II, TGF-βR-I is activated, triggering the Smad2/3-dependent signaling pathways (202). TGF-β receptors activate Smad2/3, resulting in an active complex of Smad2/3 and Smad4 that regulates the altered expression of different genes associated with EMT (203). The studies found the suppression of invasiveness due to decreased expression of Smad2 and Smad3 (204). On the other hand, upregulation of Smad2 and Smad3 expression is associated with EMT (205). TGF-β activates the AKT/PI3K, Ras/Raf/MEK/ERK, and Wnt/β-catenin signaling pathways, which produce epithelial proteins in non-Smad signaling pathways (206). The regulation of transcription factors, including Snail, Slug, ZEB1/2, and TWIST, is mediated through Smad-dependent and Non-Smad pathways (207). The interaction of TGF-β with several signaling pathways, such as Notch, Wnt/β-catenin, nuclear factor NF-κB, and RTKs, results in the induction of EMT and is essential for preserving the mesenchymal characteristic of invasive and metastatic tumor cells (208). TGF-β signaling during EMT modifies the tight junction formation and triggers the activation of additional signaling pathways, including Wnt, Notch, and Hh MAPK pathways. TGF-β regulates various gene expressions, including core transcription factors Twist (TWIST 1 and TWIST 2), SNAI (SNAI 1 and SNAI 2), ZEB (ZEB 1 and ZEB 2), and Six family of homeobox (Six1) (209). The transcription of E-cadherin, occludin, and claudin is also regulated by TGF-β (208). TGF-β overexpression in BCa is linked to increased EMT (197). Its relationship to BCa stem cells in EMT has recently been discovered (210).
3.2 The Wnt/β-catenin pathway in EMT
The Wnt/β-catenin pathway is essential in EMT regulation in BCa. Many studies have shown the involvement of Wnt signaling in BCa metastasis, immune microenvironment, stemness maintenance, and resistance to therapies (211). The Wnt signaling pathway is controlled by either canonically (β-catenin-dependent expression) or non-canonically (β-catenin-independent expression) (212). The studies revealed that accumulation of β-catenin in the nucleus is the reason for the poor prognosis of BCa (213). The role of glycogen synthase kinase-3 beta (GSK-3β) in regulating β-catenin expression has been elucidated. The increase in the phosphorylation of GSK3β causes the degradation of β-catenin and regulates the Wnt signaling pathway (214). This pathway can regulate the expression of Snail and β-catenin, facilitating EMT and promoting metastasis through inhibition of GSK-3β activity (215). The elevation of SNAI1 expression activates Wnt/β-catenin, resulting in downregulation of E-cadherin and overexpression of vimentin within BCa cells (216). According to reports, the development of several types of BCa is associated with aberrant expression and sub-cellular localization of β-catenin correlated to activation of the Wnt signaling system (217). Even though the Wnt pathway is thought to be linked to the EMT in BCa, β-catenin is not enough to cause EMT on its own (218). β-catenin acts as a molecular bridge in tight junctions of epithelial cells and promotes cell-cell adhesion (219). The process of EMT involves the stabilization of β-catenin and the activation of the Wnt signaling pathway. This activation is closely associated with the involvement of a transcription factor known as T-cell factor/lymphoid enhancer factor (TCF/LEF) and several other components (220).
3.3 Notch pathway in EMT
The Notch pathway is involved in regulation and cell survival during cell development. It plays a vital role in the initiation and progression of cancer (221). Four Notch receptors and five ligands have been reported (222). Unusual or deviating from the norm. The association between Notch signaling and the TNBC subtype has been well established (223). The over-expression of the Notch receptor is associated with the aggressive, metastatic, and therapy-resistant phenotype that is the hallmark of TNBC. Deregulation of the Notch pathway with notch 1, 2, 3, and 4 receptors and ligands (Jagged1, 2 and Delta-like1, 3, 4) are known to be involved in the induction of breast cancer mesenchymal phenotype via interacting with RTKs, MAPK and PI3K signaling (224). The canonical Notch pathway operates through the interaction of the two important ligands delta and Jagged, creating two rounds of dissociation of the Notch receptor at the S2 point (225). The first cleavage is mediated by ADAM10 or ADAM17, followed by the second with γ-secretase generating Notch intracellular domain (NICD) that induces slug-mediated EMT (226) as outlined in Figure 4. The activation of the Notch signaling system induces the NF-κB pathway and regulates TGF-β involved in EMT programming. NUMB is an essential gene that mediates Notch signaling. It has been identified as a suppressor of EMT in human epithelial cells and TNBC cells (225). The downregulation of NUMB has been correlated with the increase in EMT (227). The association between the upregulation of Notch signaling and the overall survival rate of TNBC patients has already been demonstrated (228). The expression of Snail is regulated by Notch signaling via transcriptionally activating either Snail or lysyl oxidase (LOX) (229). Several studies revealed an association between Notch activation and hypoxia. One of the critical factors regulating tumor metastasis is hypoxia. Notch is a crucial bridge connecting the hypoxia response to EMT (230). Notch signaling increases the expression of LOX by triggering a hypoxia-inducible factor 1-α (HIF-1α). This, in turn, stabilizes Snail and leads to the upregulation of EMT programming, which induces the invasion of cancer cells (231). Notch 1- triggers the EMT process in TNBC (228, 232). Notch 2 was found to be involved in TNBC (228). Notch 3 is well known for its anti-metastatic or inhibitor of the EMT pathway via estrogen receptor (ERα) and GATA3 (233). Notch 4 signaling has been found to activate the EMT process in TNBC (234). However, Further evidence suggested that Jagged1-mediated activation of the Notch intracellular domain (NotchIC) through positive regulation of Slug suppresses E-Cadherin, resulting in EMT induction in breast malignancies (235). A report revealed a correlation between TGF-β and Notch activity. Elevated Notch signaling mediated by Smad3 upregulates the expression of Jagged1 and HEY1, thereby inducing upregulation of Slug expression and subsequently inhibiting E-cadherin (229).
3.4 TNF-α/NF-κB signaling pathway in EMT
TNF- α is a transmembrane protein having a molecular weight of 26 kDa. It is a crucial cytokine in inflammation, cellular homeostasis, and tumor progression (236). It promotes angiogenesis, invasion, and metastasis related to EMT reprogramming by activating MMP 9 and preventing E-cadherin. The upregulation of TWIST 1 is associated with the induction of TNF-α in EMT. The upregulation of TNF-α showed an association with enhanced metastatic potential and invasiveness of BCa cells (237). Recent findings suggest that TWIST 1 activity is essential in promoting mouse BCa cell metastasis (238). Recent studies have shown that prolonged exposure to TNF-α activates NF-κB and IKK-β, leading to EMT and the transcriptional repressor TWIST 1 and cancer stemness (239). The direct association between expression of TNF-α by peripheral blood T lymphocytes and EMT markers present in circulating tumor cells is reported (240). The activation of NF-κB is associated with Snail, Slug, TWIST, ZEB1/2, and NF-κB activation (236). The study has revealed the activation of NF-κB leads to Snail stabilization by degrading GSK-3β in the TNF-α/NF-κB activation pathway (236). Similarly, vimentin and MMPs of mesenchymal cell markers are also activated by NF-κB (241).
3.5 Hedgehog pathway in EMT
The hedgehog (Hh) pathway, associated with stem cell renewal, is another signaling system involved in the EMT of BCa. It also requires tissue homeostasis and embryonic development (242). Three glioma-associated oncogenes (GLI) transcription factors, GLI1, GLI2, and GLI3, have a role in either inhibiting or activating the transcription of these components in the Hh pathway (243). The role of the Hh pathway in EMT-derived BCa has already been established. The high expression level of GLI1 in BCa cells attaining EMT has already been reported (244). The role of the Hh pathway in cancer cell stemness and the interplay between NF-κB and GLI1 is also studied. Like the Wnt pathways, it is regulated through canonical or non-canonical signaling. According to a study, it has been revealed that non-canonical activation of GLI1 by hypoxia or other inflammatory cytokines can lead to the induction of EMT, BCa invasiveness, and drug resistance (245). The expression of GLI1 and its role in EMT in BCa via the Hh pathway has been confirmed through in vivo studies (246).
3.6 PI3K-AKT signaling pathway in EMT
Several RTKs have been identified for their role in the EMT of BCa cells (207). There are various factors involved in the activation of RTKs, including hepatocyte growth factor (HGF), epidermal growth factor (EGF), and fibroblast growth factor (FGF). HGF has a role in epithelial differentiation upon downregulating E-cadherin, which is responsible for tumor metastasis. The HGF pathway is also linked to the Snail transcription factor, which induces EMT (207). MAPK and PI3K are two signaling pathways along with TGF-β control invasion and EMT in BCa (197). Ras-activated MAPK stimulates TWIST 1 serine 68 phosphorylation and stabilization of PI3K signaling, which causes EMT and invasion of BCa cells (247). Recent studies indicated a possible connection between RTK, Wnt, and EGFR signaling (248). Though studies revealed the importance of RTK signaling and its role in EMT, various signaling pathways are also involved in EMT. The activation of the RTK pathway alone is not enough to induce EMT; multiple pathways are also involved.
4 Prospects and challenges
The inhibition of EMT inducers is necessary for metastasis and migration suppression (32). The strategies to induce differentiation and target EMT alone may result in adverse effects via the proliferation of metastatic cells. Therefore, targeted therapies combining various EMT marker molecules involved in different cell cycles are the novel approach. Although biomarker genes, miRNA, lncRNA, and multiple proteins of the TNBC secretome have made significant advances as therapeutic targets for particular tumors and as a potential molecular indicator for early clinical detection, there is still much to understand about their proneness of specific drug resistance and new strategies to target the TNBC (249). However, recent studies have shown that these biomarker genes are essential for tumorigenesis and progression of several tumors, including BCa, particularly TNBC, which exhibits challenges to available therapy due to its aggressive behavior (214). A substantial amount of data shows that differential expression of these genes has been significantly associated with BCa subtypes (250). It is well known that the need for effective targeted drugs to improve the survival rate of patients with distant metastasis is discouraging (251). The identification and function of metastasis-related molecular markers such as miRNA and lncRNA and proteins will lead to significant progress.
5 Conclusions
Tumor cell invasion, metastasis, and EMT are responsible for the development of advanced-stage BCa, which needs an effective approach to reduce the tumor burden and improve the patient’s survival. Multiple signaling pathways and factors work together to make these complicated systems operate in BCa patients. This review describes the factors involved and the regulation of signaling pathways Figure 5. Evidence from studies shows that EMT is linked not only to tumor cell invasion and metastasis but also can give tumor stemness properties and induce drug resistance in the cells (36). Consequently, cancer cells with an EMT phenotype show more aggressive behaviors, such as drug resistance, stress, apoptosis, suppression of senescence, immunological evasion, and the acquisition of stem cell-like characteristics, in addition to their mesenchymal characteristics. The association of tumor EMT with autophagy and the surrounding microenvironment has been revealed from the studies. Inflammation, immune cells, tumor-associated fibroblasts, extracellular matrix, signaling chemicals, excessive acidity, and low oxygen are related to the tumor microenvironment. For example, the inflammatory microenvironment within the tumor can result in the EMT phenotype of the tumor cells. Hypoxia induction and inflammatory factors in the tumor microenvironment simultaneously trigger EMT, and the tumor microenvironment is also involved in the process of EMT. Therefore, EMT and the tumor microenvironment interact and affect each other to enhance tumor metastasis.
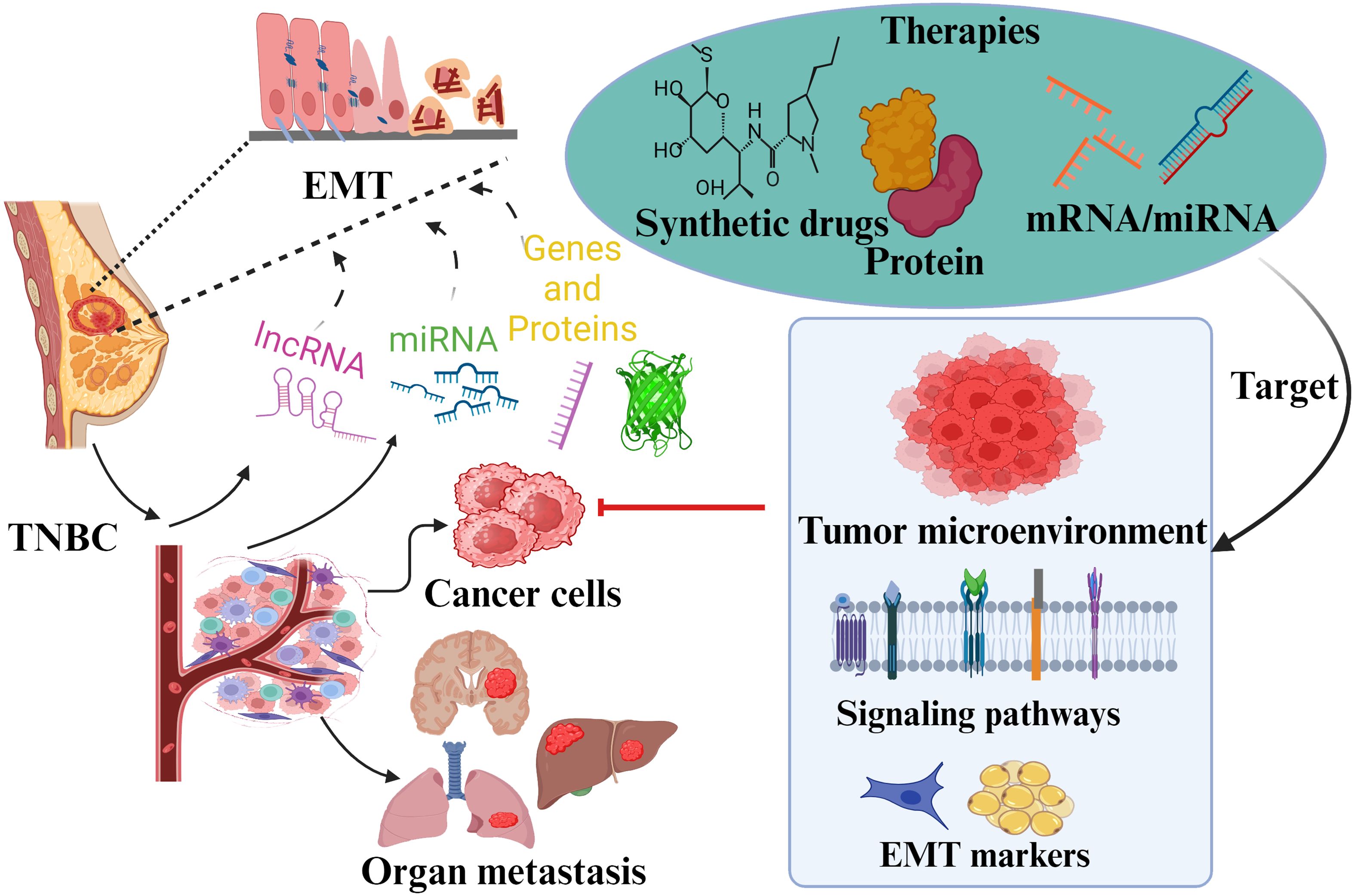
Figure 5. Overview summary shows EMT’s role and associated factors in TNBC pathogenicity. Various transcription factors and markers involved in TNBC metastasis due to EMT are presented. These factors have been discussed as potential approaches for targeted therapy. The role of different signaling pathways in EMT regulation is also reviewed to give insights into specific drug targets against TNBC.
Additionally, since autophagy plays a role in tumor cell invasion and metastasis, a strong correlation has been shown between the emergence of EMT and autophagy. Tumor cells increase the production of autophagosomes, which contributes significantly to the development of EMT and increases tumor resistance against the immune system. Therefore, a strategic approach of combining different agents that can target EMT at multiple levels along with minimum side effects would aid in developing EMT as a successful novel target for tumors and cancer therapy.
Author contributions
MH: Conceptualization, Formal analysis, Investigation, Writing – original draft, Writing – review & editing. RS: Data curation, Formal analysis, Writing – original draft, Writing – review & editing. MM: Conceptualization, Funding acquisition, Project administration, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was partially supported by the National Science Foundation [grant number 1912322; American Cancer Society [grant number DICRIDG-22-1037199-01; and National Institutes of Health grants (R25AG070244 and UO1GM132769) to MM.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA Cancer J Clin. (2023) 73:17–48. doi: 10.3322/caac.21763
2. Yi M, Li T, Niu M, Luo S, Chu Q, Wu K. Epidemiological trends of women’s cancers from 1990 to 2019 at the global, regional, and national levels: a population-based study. biomark Res. (2021) 9:55. doi: 10.1186/s40364-021-00310-y
3. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: Cancer J Clin. (2021) 71:209–49. doi: 10.3322/caac.21660
4. Palomeras S, Ruiz-Martínez S, Puig T. Targeting breast cancer stem cells to overcome treatment resistance. Molecules. (2018) 23:2193. doi: 10.3390/molecules23092193
5. Goldhirsch A, Winer EP, Coates A, Gelber R, Piccart-Gebhart M, Thürlimann B, et al. Personalizing the treatment of women with early breast cancer: highlights of the St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2013. Ann Oncol. (2013) 24:2206–23. doi: 10.1093/annonc/mdt303
6. Borri F, Granaglia A. Pathology of triple negative breast cancer, Seminars in cancer biology. Elsevier. (2021) 72:136–45. doi: 10.1016/j.semcancer.2020.06.005
7. Poteat TC, Adams MA, Malone J, Geffen S, Greene N, Nodzenski M, et al. Delays in breast cancer care by race and sexual orientation: results from a national survey with diverse women in the United States. Cancer. (2021) 127:3514–22. doi: 10.1002/cncr.v127.19
8. Perou CM, Sørlie T, Eisen MB, Van De Rijn M, Jeffrey SS, Rees CA, et al. Molecular portraits of human breast tumours. Nature. (2000) 406:747–52. doi: 10.1038/35021093
9. Dai X, Cheng H, Bai Z, Li J. Breast cancer cell line classification and its relevance with breast tumor subtyping. J Cancer. (2017) 8:3131. doi: 10.7150/jca.18457
10. Dent R, Trudeau M, Pritchard KI, Hanna WM, Kahn HK, Sawka CA, et al. Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res. (2007) 13:4429–34. doi: 10.1158/1078-0432.CCR-06-3045
11. Morris GJ, Naidu S, Topham AK, Guiles F, Xu Y, McCue P, et al. Differences in breast carcinoma characteristics in newly diagnosed African–American and Caucasian patients: A single-institution compilation compared with the National Cancer Institute’s Surveillance, Epidemiology, and end results database. Cancer: Interdiscip Int J Am Cancer Soc. (2007) 110:876–84. doi: 10.1002/cncr.v110:4
12. Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, et al. Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. New Engl J Med. (2018) 379:2108–21. doi: 10.1056/NEJMoa1809615
13. Palmieri C, Brown K, Fraser A, Lough J, Thomas S, Wilson J, et al. A cohort study of the variations in stage of initial presentation of breast cancer by age, deprivation and ethnicity in England 2019–2023. Deprivation Ethnicity Engl. (2019).
14. Dong B, Yi M, Luo S, Li A, Wu K. RDGN-based predictive model for the prognosis of breast cancer. Exp Hematol Oncol. (2020) 9:1–12. doi: 10.1186/s40164-020-00169-z
15. Garrido-Castro AC, Lin NU, Polyak K. Insights into molecular classifications of triple-negative breast cancer: improving patient selection for treatment. Cancer Discovery. (2019) 9:176–98. doi: 10.1158/2159-8290.CD-18-1177
16. Kumar H, Gupta NV, Jain R, Madhunapantula SV, Babu CS, Kesharwani SS, et al. A review of biological targets and therapeutic approaches in the management of triple-negative breast cancer. J Advanced Res. (2023) 54:271–92. doi: 10.1016/j.jare.2023.02.005
17. Leon-Ferre RA, Goetz MP. Advances in systemic therapies for triple negative breast cancer. BMJ. (2023) 381. doi: 10.1136/bmj-2022-071674
18. Spínola-Lasso E, Montero JC, Jiménez-Monzón R, Estévez F, Quintana J, Guerra B, et al. Chemical-proteomics identify peroxiredoxin-1 as an actionable target in triple-negative breast cancer. Int J Biol Sci. (2023) 19:1731. doi: 10.7150/ijbs.78554
19. Yi M, Zhang D, Song B, Zhao B, Niu M, Wu Y, et al. Increased expression of ECT2 predicts the poor prognosis of breast cancer patients. Exp Hematol Oncol. (2022) 11:107. doi: 10.1186/s40164-022-00361-3
20. Guo L, Kong D, Liu J, Zhan L, Luo L, Zheng W, et al. Breast cancer heterogeneity and its implication in personalized precision therapy. Exp Hematol Oncol. (2023) 12:1–27. doi: 10.1186/s40164-022-00363-1
21. Zhu S, Wu Y, Song B, Yi M, Yan Y, Mei Q, et al. Recent advances in targeted strategies for triple-negative breast cancer. J Hematol Oncol. (2023) 16:1–36. doi: 10.1186/s13045-023-01497-3
22. Alibert C, Goud B, Manneville JB. Are cancer cells really softer than normal cells? Biol Cell. (2017) 109:167–89. doi: 10.1016/j.semcancer.2020.06.005
23. Karamanos NK. Translating extracellular matrix: From cancer progression to therapeutics. Semin Cancer Biol. (2019) 62:iii–v. doi: 10.1016/j.semcancer.2019.10.014
24. Kalluri R, Neilson EG. Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Invest. (2003) 112:1776–84. doi: 10.1172/JCI200320530
25. Nieto MA, Huang RY-J, Jackson RA, Thiery JP. EMT: 2016. Cell. (2016) 166:21–45. doi: 10.1016/j.cell.2016.06.028
26. Mani SA, Guo W, Liao M-J, Eaton EN, Ayyanan A, Zhou AY, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. (2008) 133:704–15. doi: 10.1016/j.cell.2008.03.027
27. Grasset EM, Dunworth M, Sharma G, Loth M, Tandurella J, Cimino-Mathews A, et al. Triple-negative breast cancer metastasis involves complex epithelial-mesenchymal transition dynamics and requires vimentin. Sci Trans Med. (2022) 14:eabn7571. doi: 10.1126/scitranslmed.abn7571
28. Moses H, Barcellos-Hoff MH. TGF-β biology in mammary development and breast cancer. Cold Spring Harbor Perspect Biol. (2011) 3:a003277. doi: 10.1101/cshperspect.a003277
30. Faheem MM, ur Rasool R, Ahmad SM, Jamwal VL, Chakraborty S, Katoch A, et al. Par-4 mediated Smad4 induction in PDAC cells restores canonical TGF-β/Smad4 axis driving the cells towards lethal EMT. Eur J Cell Biol. (2020) 99:151076. doi: 10.1016/j.ejcb.2020.151076
31. Sharma P. Biology and management of patients with triple-negative breast cancer. Oncol. (2016) 21:1050–62. doi: 10.1634/theoncologist.2016-0067
32. Martin TA, Goyal A, Watkins G, Jiang WG. Expression of the transcription factors snail, slug, and twist and their clinical significance in human breast cancer. Ann Surg Oncol. (2005) 12:488–96. doi: 10.1245/ASO.2005.04.010
33. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
34. Casas E, Kim J, Bendesky A, Ohno-MaChado L, Wolfe CJ, Yang J. Snail2 is an essential mediator of Twist1-induced epithelial mesenchymal transition and metastasis. Cancer Res. (2011) 71:245–54. doi: 10.1158/0008-5472.CAN-10-2330
35. Dave N, Guaita-Esteruelas S, Gutarra S, Frias A, Beltran M, Peiro S, et al. Functional cooperation between Snail1 and twist in the regulation of ZEB1 expression during epithelial to mesenchymal transition. J Biol Chem. (2011) 286:12024–32. doi: 10.1074/jbc.M110.168625
36. Blick T, Hugo H, Widodo E, Waltham M, Pinto C, Mani SA, et al. Epithelial mesenchymal transition traits in human breast cancer cell lines parallel the CD44 hi/CD24 lo/-stem cell phenotype in human breast cancer. J Mammary Gland Biol Neoplasia. (2010) 15:235–52. doi: 10.1007/s10911-010-9175-z
37. Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. (2010) 29:4741–51. doi: 10.1038/onc.2010.215
38. Stefani G, Slack FJ. Small non-coding RNAs in animal development. Nat Rev Mol Cell Biol. (2008) 9:219–30. doi: 10.1038/nrm2347
39. Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, et al. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol. (2008) 10:593–601. doi: 10.1038/ncb1722
40. Paul A, Gunewardena S, Stecklein S, Saha B, Parelkar N, Danley M, et al. PKCλ/ι signaling promotes triple-negative breast cancer growth and metastasis. Cell Death Differentiation. (2014) 21:1469–81. doi: 10.1038/cdd.2014.62
41. Joberty G, Petersen C, Gao L, Macara IG. The cell-polarity protein Par6 links Par3 and atypical protein kinase C to Cdc42. Nat Cell Biol. (2000) 2:531–9. doi: 10.1038/35019573
42. Paul A, Danley M, Saha B, Tawfik O, Paul S. PKCζ promotes breast cancer invasion by regulating expression of E-cadherin and Zonula Occludens-1 (ZO-1) via NFκB-p65. Sci Rep. (2015) 5:12520. doi: 10.1038/srep12520
43. Shamir ER, Pappalardo E, Jorgens DM, Coutinho K, Tsai W-T, Aziz K, et al. Twist1-induced dissemination preserves epithelial identity and requires E-cadherin. J Cell Biol. (2014) 204:839–56. doi: 10.1083/jcb.201306088
44. Hogan BL, Kolodziej PA. Molecular mechanisms of tubulogenesis. Nat Rev Genet. (2002) 3:513–23. doi: 10.1038/nrg840
45. Sipos F, Galamb O. Epithelial-to-mesenchymal and mesenchymal-to-epithelial transitions in the colon. World J Gastroenterol: WJG. (2012) 18:601. doi: 10.3748/wjg.v18.i7.601
46. Hwang JS, Lai TH, Ahmed M, Pham TM, Elashkar O, Bahar E, et al. Regulation of TGF-β 1-induced EMT by autophagy-dependent energy metabolism in cancer cells. Cancers. (2022) 14:4845. doi: 10.3390/cancers14194845
47. Won KA, Spruck C. Triple−negative breast cancer therapy: Current and future perspectives. Int J Oncol. (2020) 57:1245–61. doi: 10.3892/ijo.2020.5135
48. Wu Q, Qian W, Sun X, Jiang S. Small-molecule inhibitors, immune checkpoint inhibitors, and more: FDA-approved novel therapeutic drugs for solid tumors from 1991 to 2021. J Hematol Oncol. (2022) 15:1–63. doi: 10.1186/s13045-022-01362-9
50. Rubens R, Sexton S, Tong D, Winter P, Knight R, Hayward J. Combined chemotherapy and radiotherapy for locally advanced breast cancer. Eur J Cancer. (1980) 16:351–6. doi: 10.1016/0014-2964(80)90352-7
51. Seltzer S, Corrigan M, O’Reilly S. The clinicomolecular landscape of de novo versus relapsed stage IV metastatic breast cancer. Exp Mol Pathol. (2020) 114:104404. doi: 10.1016/j.yexmp.2020.104404
52. Leone JP, Lee AV, Brufsky AM. Prognostic factors and survival of patients with brain metastasis from breast cancer who underwent craniotomy. Cancer Med. (2015) 4:989–94. doi: 10.1002/cam4.2015.4.issue-7
53. Caswell-Jin JL, Plevritis SK, Tian L, Cadham CJ, Xu C, Stout NK, et al. Change in survival in metastatic breast cancer with treatment advances: meta-analysis and systematic review. JNCI Cancer Spectr. (2018) 2:pky062. doi: 10.1093/jncics/pky062
54. O’Shaughnessy J, Schwartzberg L, Danso MA, Miller KD, Rugo HS, Neubauer M, et al. Phase III study of iniparib plus gemcitabine and carboplatin versus gemcitabine and carboplatin in patients with metastatic triple-negative breast cancer. J Clin Oncol. (2014) 32:3840–7. doi: 10.1200/JCO.2014.55.2984
55. Plevritis SK, Munoz D, Kurian AW, Stout NK, Alagoz O, Near AM, et al. Association of screening and treatment with breast cancer mortality by molecular subtype in US women, 2000-2012. Jama. (2018) 319:154–64. doi: 10.1001/jama.2017.19130
56. Safonov A, Jiang T, Bianchini G, Győrffy B, Karn T, Hatzis C, et al. Immune gene expression is associated with genomic aberrations in breast cancer. Cancer Res. (2017) 77:3317–24. doi: 10.1158/0008-5472.CAN-16-3478
57. Stanton SE, Adams S, Disis ML. Variation in the incidence and magnitude of tumor-infiltrating lymphocytes in breast cancer subtypes: a systematic review. JAMA Oncol. (2016) 2:1354–60. doi: 10.1001/jamaoncol.2016.1061
58. Das PK, Islam F, Lam AK. The roles of cancer stem cells and therapy resistance in colorectal carcinoma. Cells. (2020) 9:1392. doi: 10.3390/cells9061392
59. Longley DB, Harkin DP, Johnston PG. 5-fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer. (2003) 3:330–8. doi: 10.1038/nrc1074
60. Chen N-N, Ma X-D, Miao Z, Zhang X-M, Han B-Y, Almaamari AA, et al. Doxorubicin resistance in breast cancer is mediated via the activation of FABP5/PPARγ and CaMKII signaling pathway. Front Pharmacol. (2023) 14:1150861. doi: 10.3389/fphar.2023.1150861
61. van der Zanden SY, Qiao X, Neefjes J. New insights into the activities and toxicities of the old anticancer drug doxorubicin. FEBS J. (2021) 288:6095–111. doi: 10.1111/febs.v288.21
62. Bernatsky S, Clarke AE, Suissa S. Hematologic Malignant neoplasms after drug exposure in rheumatoid arthritis. Arch Internal Med. (2008) 168:378–81. doi: 10.1001/archinternmed.2007.107
63. Lutsiak MC, Semnani RT, De Pascalis R, Kashmiri SV, Schlom J, Sabzevari H. Inhibition of CD4 + 25+ T regulatory cell function implicated in enhanced immune response by low-dose cyclophosphamide. Blood. (2005) 105:2862–8. doi: 10.1182/blood-2004-06-2410
64. Hammond WA, Swaika A, Mody K. Pharmacologic resistance in colorectal cancer: a review. Ther Adv Med Oncol. (2016) 8:57–84. doi: 10.1177/1758834015614530
65. Airley R. Cancer chemotherapy: basic science to the clinic. Hoboken, NJ: John Wiley & Sons (2009).
66. Maeda S, Saimura M, Minami S, Kurashita K, Nishimura R, Kai Y, et al. Efficacy and safety of eribulin as first-to third-line treatment in patients with advanced or metastatic breast cancer previously treated with anthracyclines and taxanes. Breast. (2017) 32:66–72. doi: 10.1016/j.breast.2016.12.017
67. O’Shaughnessy J, Kaklamani V, Kalinsky K. Perspectives on the mechanism of action and clinical application of eribulin for metastatic breast cancer. Future Oncol. (2019) 15:1641–53. doi: 10.2217/fon-2018-0936
68. Noll DM, Mason TM, Miller PS. Formation and repair of interstrand cross-links in DNA. Chem Rev. (2006) 106:277–301. doi: 10.1021/cr040478b
69. Tchounwou PB, Dasari S, Noubissi FK, Ray P, Kumar S. Advances in our understanding of the molecular mechanisms of action of cisplatin in cancer therapy. J Exp Pharmacol. (2021) 13:303–28. doi: 10.2147/JEP.S267383
70. Yamaguchi A, Kawaguchi K, Kawanishi K, Maeshima Y, Nakakura A, Kataoka TR, et al. Comparison of cisplatin-based versus standard preoperative chemotherapy in patients with operable triple-negative breast cancer: propensity score matching and inverse probability of treatment weighting analysis. Breast Cancer Res Treat. (2024) 204:261–75. doi: 10.1007/s10549-023-07163-z
71. Buqué A, Bloy N, Aranda F, Castoldi F, Eggermont A, Cremer I, et al. Trial Watch: Immunomodulatory monoclonal antibodies for oncological indications. Oncoimmunology. (2015) 4:e1008814. doi: 10.1080/2162402X.2015.1008814
72. Dent R, André F, Gonçalves A, Martin M, Schmid P, Schütz F, et al. IMpassion132 double-blind randomised phase III trial of chemotherapy with or without atezolizumab for early relapsing unresectable locally advanced or metastatic triple-negative breast cancer. Ann Oncol. (2024) 35:630–42. doi: 10.1016/j.esmoop.2024.103202
73. Boussios S, Abson C, Moschetta M, Rassy E, Karathanasi A, Bhat T, et al. Poly (ADP-Ribose) polymerase inhibitors: talazoparib in ovarian cancer and beyond. Drugs R&D. (2020) 20:55–73. doi: 10.1007/s40268-020-00301-8
74. Goulooze SC, Cohen AF, Rissmann R. Olaparib. Br J Clin Pharmacol. (2016) 81:171–3. doi: 10.1111/bcp.12761
75. Ben-Hamo R, Zilberberg A, Cohen H, Bahar-Shany K, Wachtel C, Korach J, et al. Resistance to paclitaxel is associated with a variant of the gene BCL2 in multiple tumor types. NPJ Precis Oncol. (2019) 3:12. doi: 10.1038/s41698-019-0084-3
76. Kampan NC, Madondo MT, McNally OM, Quinn M, Plebanski M. Paclitaxel and its evolving role in the management of ovarian cancer. BioMed Res Int. (2015) 2015:413076. doi: 10.1155/2015/413076
77. Masuda H, Zhang D, Bartholomeusz C, Doihara H, Hortobagyi GN, Ueno NT. Role of epidermal growth factor receptor in breast cancer. Breast Cancer Res Treat. (2012) 136:331–45. doi: 10.1007/s10549-012-2289-9
78. Higa GM, Abraham J. Lapatinib in the treatment of breast cancer. Expert Rev Anticancer Ther. (2007) 7:1183–92. doi: 10.1586/14737140.7.9.1183
79. Chang AY, Wang M. Molecular mechanisms of action and potential biomarkers of growth inhibition of dasatinib (BMS-354825) on hepatocellular carcinoma cells. BMC Cancer. (2013) 13:1–12. doi: 10.1186/1471-2407-13-267
80. Osguthorpe D, Hagler A. Mechanism of androgen receptor antagonism by bicalutamide in the treatment of prostate cancer. Biochemistry. (2011) 50:4105–13. doi: 10.1021/bi102059z
81. Dudley JC, Lin M-T, Le DT, Eshleman JR. Microsatellite instability as a biomarker for PD-1 blockade. Clin Cancer Res. (2016) 22:813–20. doi: 10.1158/1078-0432.CCR-15-1678
82. Kurata K, Kubo M, Mori H, Kawaji H, Motoyama Y, Kuroki L, et al. Abstract P1-06-11: microsatellite instability in triple negative breast cancers. Cancer Res. (2019) 79. doi: 10.1158/1538-7445.SABCS18-P1-06-11
83. Emens LA, Adams S, Barrios C, Diéras V, Iwata H, Loi S, et al. First-line atezolizumab plus nab-paclitaxel for unresectable, locally advanced, or metastatic triple-negative breast cancer: IMpassion130 final overall survival analysis. Ann Oncol. (2021) 32:983–93. doi: 10.1016/j.annonc.2021.05.355
84. Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly (ADP-ribose) polymerase. Nature. (2005) 434:913–7. doi: 10.1038/nature03443
85. Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. (2005) 434:917–21. doi: 10.1038/nature03445
86. Robson M, Im S-A, Senkus E, Xu B, Domchek SM, Masuda N, et al. Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. New Engl J Med. (2017) 377:523–33. doi: 10.1056/NEJMoa1706450
87. Robson M, Tung N, Conte P, Im S-A, Senkus E, Xu B, et al. OlympiAD final overall survival and tolerability results: Olaparib versus chemotherapy treatment of physician’s choice in patients with a germline BRCA mutation and HER2-negative metastatic breast cancer. Ann Oncol. (2019) 30:558–66. doi: 10.1093/annonc/mdz012
88. Litton JK, Rugo HS, Ettl J, Hurvitz SA, Gonçalves A, Lee K-H, et al. Talazoparib in patients with advanced breast cancer and a germline BRCA mutation. New Engl J Med. (2018) 379:753–63. doi: 10.1056/NEJMoa1802905
89. Mustacchi G, De Laurentiis M. The role of taxanes in triple-negative breast cancer: literature review. Drug Design Dev Ther. (2015) 9:4303–18. doi: 10.2147/DDDT.S86105
90. Couch FJ, Hart SN, Sharma P, Toland AE, Wang X, Miron P, et al. Inherited mutations in 17 breast cancer susceptibility genes among a large triple-negative breast cancer cohort unselected for family history of breast cancer. J Clin Oncol. (2015) 33:304. doi: 10.1200/JCO.2014.57.1414
91. Gonzalez-Angulo AM, Timms KM, Liu S, Chen H, Litton JK, Potter J, et al. Incidence and outcome of BRCA mutations in unselected patients with triple receptor-negative breast cancer. Clin Cancer Res. (2011) 17:1082–9. doi: 10.1158/1078-0432.CCR-10-2560
92. Aftimos P, Oliveira M, Irrthum A, Fumagalli D, Sotiriou C, Gal-Yam EN, et al. Genomic and transcriptomic analyses of breast cancer primaries and matched metastases in AURORA, the Breast International Group (BIG) molecular screening initiative. Cancer Discovery. (2021) 11:2796–811. doi: 10.1158/2159-8290.CD-20-1647
93. Laderian B, Mundi P, Fojo T, E. Bates S. Emerging therapeutic implications of STK11 mutation: case series. Oncol. (2020) 25:733–7. doi: 10.1634/theoncologist.2019-0846
94. Kurian AW, Ford JM. Multigene panel testing in oncology practice: how should we respond? JAMA Oncol. (2015) 1:277–8. doi: 10.1001/jamaoncol.2015.28
95. Gervas P, Molokov A, Ivanova A, Panferova Y, Kiselev A, Chernyshova A, et al. New germline mutations in BRCA1, ATM, MUTYH, and RAD51D genes in Tuvans early-onset breast cancer patients. Exp Oncol. (2021) 43:52–5. doi: 10.32471/exp-oncology.2312-8852.vol-43-no-1.15587
96. The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. (2012) 490:61–70. doi: 10.1038/nature11412
97. Song Y, Barry WT, Seah DS, Tung NM, Garber JE, Lin NU. Patterns of recurrence and metastasis in BRCA1/BRCA2-associated breast cancers. Cancer. (2020) 126:271–80. doi: 10.1002/cncr.v126.2
98. Zhang H, Gao M, Zhao W, Yu L. The chromatin architectural regulator SND1 mediates metastasis in triple-negative breast cancer by promoting CDH1 gene methylation. Breast Cancer Res. (2023) 25:129. doi: 10.1186/s13058-023-01731-3
99. Li A, Schleicher SM, Andre F, Mitri ZI. Genomic alteration in metastatic breast cancer and its treatment. Am Soc Clin Oncol Educ Book. (2020) 40:30–43. doi: 10.1200/EDBK_280463
100. Zhang P, Wei Y, Wang L, Debeb BG, Yuan Y, Zhang J, et al. ATM-mediated stabilization of ZEB1 promotes DNA damage response and radioresistance through CHK1. Nat Cell Biol. (2014) 16:864–75. doi: 10.1038/ncb3013
101. Śniadecki M, Brzeziński M, Darecka K, Klasa-Mazurkiewicz D, Poniewierza P, Krzeszowiec M, et al. BARD1 and breast cancer: the possibility of creating screening tests and new preventive and therapeutic pathways for predisposed women. Genes. (2020) 11:1251. doi: 10.3390/genes11111251
102. Cybulski C, Wokołorczyk D, Jakubowska A, Huzarski T, Byrski T, Gronwald J, et al. Risk of breast cancer in women with a CHEK2 mutation with and without a family history of breast cancer. J Clin Oncol. (2011) 29:3747–52. doi: 10.1200/JCO.2010.34.0778
103. Wan H, Yang X, Sang G, Ruan Z, Ling Z, Zhang M, et al. CDKN2A was a cuproptosis-related gene in regulating chemotherapy resistance by the MAGE-A family in breast cancer: based on artificial intelligence (AI)-constructed pan-cancer risk model. Aging. (2023) 15:11244–67. doi: 10.18632/aging.205125
104. Massey S, Khan MA, Rab SO, Mustafa S, Khan A, Malik Z, et al. Evaluating the role of MEN1 gene expression and its clinical significance in breast cancer patients. PloS One. (2023) 18:e0288482. doi: 10.1371/journal.pone.0288482
105. Mo J, Borcherding N, Jo S, Tithi TI, Cho E, Cash KE, et al. Contrasting roles of different mismatch repair proteins in basal-like breast cancer. bioRxiv. (2023). doi: 10.1101/2023.07.20.549745
106. Chen C, Lin C-J, Pei Y-C, Ma D, Liao L, Li S-Y, et al. Comprehensive genomic profiling of breast cancers characterizes germline-somatic mutation interactions mediating therapeutic vulnerabilities. Cell Discovery. (2023) 9:125. doi: 10.1038/s41421-023-00614-3
107. Xie D, Chen Y, Wan X, Li J, Pei Q, Luo Y, et al. The potential role of CDH1 as an oncogene combined with related miRNAs and their diagnostic value in breast cancer. Front Endocrinol. (2022) 13:916469. doi: 10.3389/fendo.2022.916469
108. Wang Y-Y, Hung AC, Lo S, Hsieh Y-C, Yuan S-SF. MRE11 as a molecular signature and therapeutic target for cancer treatment with radiotherapy. Cancer Lett. (2021) 514:1–11. doi: 10.1016/j.canlet.2021.05.013
109. Wan W, Zou H, Sun R, Liu Y, Wang J, Ma D, et al. Investigate the role of PTEN in chemotaxis of human breast cancer cells. Cell Signalling. (2007) 19:2227–36. doi: 10.1016/j.cellsig.2007.06.007
110. Granado-Martínez P, Garcia-Ortega S, González-Sánchez E, McGrail K, Selgas R, Grueso J, et al. STK11 (LKB1) missense somatic mutant isoforms promote tumor growth, motility and inflammation. Commun Biol. (2020) 3:366. doi: 10.1038/s42003-020-1092-0
111. Marvalim C, Datta A, Lee SC. Role of p53 in breast cancer progression: An insight into p53 targeted therapy. Theranostics. (2023) 13:1421. doi: 10.7150/thno.81847
112. Zhuang Z-G, Di G-H, Shen Z-Z, Ding J, Shao Z-M. Enhanced expression of LKB1 in breast cancer cells attenuates angiogenesis, invasion, and metastatic potential. Mol Cancer Res. (2006) 4:843–9. doi: 10.1158/1541-7786.MCR-06-0118
113. Bashir N, Sana S, Mahjabeen I, Kayani MA. Association of reduced XRCC2 expression with lymph node metastasis in breast cancer tissues. Familial Cancer. (2014) 13:611–7. doi: 10.1007/s10689-014-9745-0
114. Shi Y, Shen M, Xu M, Tao M, Chen K, Zhu Q. Comprehensive analysis of the expression and prognosis for RAD51 family in human breast cancer. Int J Gen Med. (2022) 15:4925–36. doi: 10.2147/IJGM.S350971
115. Chen J, Shin VY, Siu MT, Ho JC, Cheuk I, Kwong A. miR-199a-5p confers tumor-suppressive role in triple-negative breast cancer. BMC Cancer. (2016) 16:1–12. doi: 10.1186/s12885-016-2916-7
116. Chen L-l, Zhang Z-j, Yi Z-b, Li J-j. MicroRNA-211-5p suppresses tumour cell proliferation, invasion, migration and metastasis in triple-negative breast cancer by directly targeting SETBP1. Br J Cancer. (2017) 117:78–88. doi: 10.1038/bjc.2017.150
117. De Mattos-Arruda L, Bottai G, Nuciforo PG, Di Tommaso L, Giovannetti E, Peg V, et al. MicroRNA-21 links epithelial-to-mesenchymal transition and inflammatory signals to confer resistance to neoadjuvant trastuzumab and chemotherapy in HER2-positive breast cancer patients. Oncotarget. (2015) 6:37269. doi: 10.18632/oncotarget.v6i35
118. Drago-Ferrante R, Pentimalli F, Carlisi D, De Blasio A, Saliba C, Baldacchino S, et al. Suppressive role exerted by microRNA-29b-1-5p in triple negative breast cancer through SPIN1 regulation. Oncotarget. (2017) 8:28939. doi: 10.18632/oncotarget.15960
119. Fan C, Liu N, Zheng D, Du J, Wang K. MicroRNA-206 inhibits metastasis of triple-negative breast cancer by targeting transmembrane 4 L6 family member 1. Cancer Manage Res. (2019) 11:6755–64. doi: 10.2147/CMAR.S199027
120. Fkih M’hamed I, Privat M, Trimeche M, Penault-Llorca F, Bignon Y-J, Kenani A. miR-10b, miR-26a, miR-146a And miR-153 expression in triple negative vs non triple negative breast cancer: potential biomarkers. Pathol Oncol Res. (2017) 23:815–27. doi: 10.1007/s12253-017-0188-4
121. Han C, Li X, Fan Q, Liu G, Yin J. CCAT1 promotes triple-negative breast cancer progression by suppressing miR-218/ZFX signaling. Aging (Albany NY). (2019) 11:4858. doi: 10.18632/aging.102080
122. Jang MH, Kim HJ, Gwak JM, Chung YR, Park SY. Prognostic value of microRNA-9 and microRNA-155 expression in triple-negative breast cancer. Hum Pathol. (2017) 68:69–78. doi: 10.1016/j.humpath.2017.08.026
123. Piasecka D, Braun M, Kordek R, Sadej R, Romanska H. MicroRNAs in regulation of triple-negative breast cancer progression. J Cancer Res Clin Oncol. (2018) 144:1401–11. doi: 10.1007/s00432-018-2689-2
124. Tang L, Wei D, Yan F. MicroRNA-145 functions as a tumor suppressor by targeting matrix metalloproteinase 11 and Rab GTPase family 27a in triple-negative breast cancer. Cancer Gene Ther. (2016) 23:258–65. doi: 10.1038/cgt.2016.27
125. Yu J, Shen W, Gao B, Zhao H, Xu J, Gong B. MicroRNA-182 targets FOXF2 to promote the development of triple-negative breast cancer. Neoplasma. (2017) 64:209–15. doi: 10.4149/neo_2017_206
126. Sabit H, Cevik E, Tombuloglu H, Abdel-Ghany S, Tombuloglu G, Esteller M. Triple negative breast cancer in the era of miRNA. Crit Rev Oncol/hematol. (2021) 157:103196. doi: 10.1016/j.critrevonc.2020.103196
127. Tsouko E, Wang J, Frigo DE, Aydoğdu E, Williams C. miR-200a inhibits migration of triple-negative breast cancer cells through direct repression of the EPHA2 oncogene. Carcinogenesis. (2015) 36:1051–60. doi: 10.1093/carcin/bgv087
128. Zheng Q, Cui X, Zhang D, Yang Y, Yan X, Liu M, et al. miR-200b inhibits proliferation and metastasis of breast cancer by targeting fucosyltransferase IV and α1, 3-fucosylated glycans. Oncogenesis. (2017) 6:e358–8. doi: 10.1038/oncsis.2017.58
129. Li D, Wang H, Song H, Xu H, Zhao B, Wu C, et al. The microRNAs miR-200b-3p and miR-429-5p target the LIMK1/CFL1 pathway to inhibit growth and motility of breast cancer cells. Oncotarget. (2017) 8:85276. doi: 10.18632/oncotarget.19205
130. Mekala JR, Naushad SM, Ponnusamy L, Arivazhagan G, Sakthiprasad V, Pal-Bhadra M. Epigenetic regulation of miR-200 as the potential strategy for the therapy against triple-negative breast cancer. Gene. (2018) 641:248–58. doi: 10.1016/j.gene.2017.10.018
131. Tang J, Li Y, Sang Y, Yu B, Lv D, Zhang W, et al. LncRNA PVT1 regulates triple-negative breast cancer through KLF5/beta-catenin signaling. Oncogene. (2018) 37:4723–34. doi: 10.1038/s41388-018-0310-4
132. Liu AN, Qu HJ, Gong WJ, Xiang JY, Yang MM, Zhang W. LncRNA AWPPH and miRNA-21 regulates cancer cell proliferation and chemosensitivity in triple-negative breast cancer by interacting with each other. J Cell Biochem. (2019) 120:14860–6. doi: 10.1002/jcb.v120.9
133. Wang H, Tan Z, Hu H, Liu H, Wu T, Zheng C, et al. microRNA-21 promotes breast cancer proliferation and metastasis by targeting LZTFL1. BMC Cancer. (2019) 19:1–13. doi: 10.1186/s12885-019-5951-3
134. Fang H, Xie J, Zhang M, Zhao Z, Wan Y, Yao Y. miRNA-21 promotes proliferation and invasion of triple-negative breast cancer cells through targeting PTEN. Am J Trans Res. (2017) 9:953.
135. Bayraktar R, Pichler M, Kanlikilicer P, Ivan C, Bayraktar E, Kahraman N, et al. MicroRNA 603 acts as a tumor suppressor and inhibits triple-negative breast cancer tumorigenesis by targeting elongation factor 2 kinase. Oncotarget. (2017) 8:11641. doi: 10.18632/oncotarget.14264
136. Zhao Z, Li L, Du P, Ma L, Zhang W, Zheng L, et al. Transcriptional Downregulation of miR-4306 serves as a New Therapeutic Target for Triple Negative Breast Cancer: Erratum. Theranostics. (2023) 13:1287. doi: 10.7150/thno.82636
137. Zhang W, Guan X, Tang J. The long non-coding RNA landscape in triple-negative breast cancer. Cell Proliferation. (2021) 54:e12966. doi: 10.1111/cpr.12966
138. Yuan N, Zhang G, Bie F, Ma M, Ma Y, Jiang X, et al. Integrative analysis of lncRNAs and miRNAs with coding RNAs associated with ceRNA crosstalk network in triple negative breast cancer. OncoTargets Ther. (2017) 10:5883–97. doi: 10.2147/OTT.S149308
139. Lin A, Li C, Xing Z, Hu Q, Liang K, Han L, et al. The LINK-A lncRNA activates normoxic HIF1α signalling in triple-negative breast cancer. Nat Cell Biol. (2016) 18:213–24. doi: 10.1038/ncb3295
140. Parnigoni A, Caon I, Moretto P, Viola M, Karousou E, Passi A, et al. The role of the multifaceted long non-coding RNAs: A nuclear-cytosolic interplay to regulate hyaluronan metabolism. Matrix Biol. (2021) Plus 11:100060. doi: 10.1016/j.mbplus.2021.100060
141. Zhu C, Wang X, Wang Y, Wang K. Functions and underlying mechanisms of lncRNA HOTAIR in cancer chemotherapy resistance. Cell Death Discovery. (2022) 8:383. doi: 10.1038/s41420-022-01174-3
142. Tamang S, Acharya V, Roy D, Sharma R, Aryaa A, Sharma U, et al. SNHG12: an LncRNA as a potential therapeutic target and biomarker for human cancer. Front Oncol. (2019) 9:901. doi: 10.3389/fonc.2019.00901
143. Komlosh PG, Chen JL, Childs-Disney J, Disney MD, Canaani D. Broad-spectrum metastasis suppressing compounds and therapeutic uses thereof in human tumors. Sci Rep. (2023) 13:20420. doi: 10.1038/s41598-023-47478-x
144. Luo Y, Zhang W, Xu L, Chen Y, Xu Y, Yuan L. Long non-coding RNA PVT1 regulates the resistance of the breast cancer cell line MDA-MB-231 to doxorubicin via Nrf2. Technol Cancer Res Treat. (2020) 19:1533033820980763. doi: 10.1177/1533033820980763
145. Luo L, Tang H, Ling L, Li N, Jia X, Zhang Z, et al. LINC01638 lncRNA activates MTDH-Twist1 signaling by preventing SPOP-mediated c-Myc degradation in triple-negative breast cancer. Oncogene. (2018) 37:6166–79. doi: 10.1038/s41388-018-0396-8
146. Wu Y, Wang Z, Yu S, Liu D, Sun L. LncmiRHG-MIR100HG: A new budding star in cancer. Front Oncol. (2022) 12:997532. doi: 10.3389/fonc.2022.997532
147. Yang J, Meng X, Yu Y, Pan L, Zheng Q, Lin W. LncRNA POU3F3 promotes proliferation and inhibits apoptosis of cancer cells in triple-negative breast cancer by inactivating caspase 9. Biosci Biotechnol Biochem. (2019) 83:1117–23. doi: 10.1080/09168451.2019.1588097
148. Zhang G, Li H, Sun R, Li P, Yang Z, Liu Y, et al. Long non-coding RNA ZEB2-AS1 promotes the proliferation, metastasis and epithelial mesenchymal transition in triple-negative breast cancer by epigenetically activating ZEB2. J Cell Mol Med. (2019) 23:3271–9. doi: 10.1111/jcmm.2019.23.issue-5
149. Han X, Li B, Zhang S. MIR503HG: A potential diagnostic and therapeutic target in human diseases. Biomed Pharmacother. (2023) 160:114314. doi: 10.1016/j.biopha.2023.114314
150. Wang O, Yang F, Liu Y, Lv L, Ma R, Chen C, et al. C-MYC-induced upregulation of lncRNA SNHG12 regulates cell proliferation, apoptosis and migration in triple-negative breast cancer. Am J Trans Res. (2017) 9:533.
151. Aram R, Dotan I, Hotz-Wagenblatt A, Canaani D. Identification of a novel metastasis inducing lncRNA which suppresses the KAI1/CD82 metastasis suppressor gene and is upregulated in triple-negative breast cancer. Oncotarget. (2017) 8:67538. doi: 10.18632/oncotarget.18733
152. Wang S, Ke H, Zhang H, Ma Y, Ao L, Zou L, et al. LncRNA MIR100HG promotes cell proliferation in triple-negative breast cancer through triplex formation with p27 loci. Cell Death Dis. (2018) 9:805. doi: 10.1038/s41419-018-0869-2
153. Wang K, Li X, Song C, Li M. LncRNA AWPPH promotes the growth of triple-negative breast cancer by up-regulating frizzled homolog 7 (FZD7). Biosci Rep. (2018) 38:BSR20181223. doi: 10.1042/BSR20181223
154. Mou E, Wang H. LncRNA LUCAT1 facilitates tumorigenesis and metastasis of triple-negative breast cancer through modulating miR-5702. Biosci Rep. (2019) 39:BSR20190489. doi: 10.1042/BSR20190489
155. Du C, Wang Y, Zhang Y, Zhang J, Zhang L, Li J. LncRNA DLX6-AS1 contributes to epithelial–mesenchymal transition and cisplatin resistance in triple-negative breast cancer via modulating Mir-199b-5p/paxillin axis. Cell Transplant. (2020) 29:0963689720929983. doi: 10.1177/0963689720929983
156. Wang L, Liu D, Wu X, Zeng Y, Li L, Hou Y, et al. Long non-coding RNA (LncRNA) RMST in triple-negative breast cancer (TNBC): expression analysis and biological roles research. J Cell Physiol. (2018) 233:6603–12. doi: 10.1002/jcp.v233.10
157. Song X, Liu Z, Yu Z. LncRNA NEF is downregulated in triple negative breast cancer and correlated with poor prognosis. Acta Biochim Biophys Sin. (2019) 51:386–92. doi: 10.1093/abbs/gmz021
158. Wang N, Hou M, Zhan Y, Sheng X. LncRNA PTCSC3 inhibits triple-negative breast cancer cell proliferation by downregulating lncRNA H19. J Cell Biochem. (2019) 120:15083–8. doi: 10.1002/jcb.v120.9
159. Fu J, Dong G, Shi H, Zhang J, Ning Z, Bao X, et al. LncRNA MIR503HG inhibits cell migration and invasion via miR-103/OLFM4 axis in triple negative breast cancer. J Cell Mol Med. (2019) 23:4738–45. doi: 10.1111/jcmm.2019.23.issue-7
160. Yan J, Wang R, Wu Z. LncRNA TCONS_l2_00002973 correlates with less advanced tumor stage and favorable survival, and also inhibits cancer cells proliferation while enhancing apoptosis in triple-negative breast cancer. J Buon. (2019) 24:535–42.
161. Li X, Hou L, Yin L, Zhao S. LncRNA XIST interacts with miR-454 to inhibit cells proliferation, epithelial mesenchymal transition and induces apoptosis in triple-negative breast cancer. J Biosci. (2020) 45:45. doi: 10.1007/s12038-020-9999-7
162. Ziegler YS, Moresco JJ, Yates JR III, Nardulli AM. Integration of breast cancer secretomes with clinical data elucidates potential serum markers for disease detection, diagnosis, and prognosis. PloS One. (2016) 11:e0158296. doi: 10.1371/journal.pone.0158296
163. Kong Y, Lyu N, Wu J, Tang H, Xie X, Yang L, et al. Breast cancer stem cell markers CD44 and ALDH1A1 in serum: distribution and prognostic value in patients with primary breast cancer. J Cancer. (2018) 9:3728. doi: 10.7150/jca.28032
164. Kalyane D, Polaka S, Vasdev N, Tekade RK. CD44-receptor targeted gold-doxorubicin nanocomposite for pulsatile chemo-photothermal therapy of triple-negative breast cancer cells. Pharmaceutics. (2022) 14:2734. doi: 10.3390/pharmaceutics14122734
165. Li C, Hancock MA, Sehgal P, Zhou S, Reinhardt DP, Philip A. Soluble CD109 binds TGF-β and antagonizes TGF-β signalling and responses. Biochem J. (2016) 473:537–7. doi: 10.1042/BJ20141488
166. Tao J, Li H, Li Q, Yang Y. CD109 is a potential target for triple-negative breast cancer. Tumor Biol. (2014) 35:12083–90. doi: 10.1007/s13277-014-2509-5
167. Han Z, Ni J, Smits P, Underhill CB, Xie B, Chen Y, et al. Extracellular matrix protein 1 (ECM1) has angiogenic properties and is expressed by breast tumor cells. FASEB J. (2001) 15:988–94. doi: 10.1096/fsb2fj990934com
168. Lal G, Hashimi S, Smith BJ, Lynch CF, Zhang L, Robinson RA, et al. Extracellular matrix 1 (ECM1) expression is a novel prognostic marker for poor long-term survival in breast cancer: a Hospital-based Cohort Study in Iowa. Ann Surg Oncol. (2009) 16:2280–7. doi: 10.1245/s10434-009-0533-2
169. Fararjeh AS, Kaddumi E, Al Khader A, Aloliqi AA. The diagnostic and prognostic significance of EFEMP1 in breast cancer: an immunohistochemistry study. Int J Surg Pathol. (2023) 31:1057–66. doi: 10.1177/10668969221126122
170. Noonan MM, Dragan M, Mehta MM, Hess DA, Brackstone M, Tuck AB, et al. The matrix protein Fibulin-3 promotes KISS1R induced triple negative breast cancer cell invasion. Oncotarget. (2018) 9:30034. doi: 10.18632/oncotarget.25682
171. Jabbari K, Winkelmaier G, Andersen C, Yaswen P, Quilici D, Furuta S, et al. Protein ligands in the secretome of CD36+ fibroblasts induce growth suppression in a subset of breast cancer cell lines. Cancers. (2021) 13:4521. doi: 10.3390/cancers13184521
172. Marano F, Zunino V, Frairia R, Arvat E, Castellano I, Bosco O, et al. Fibulin-1 interacts with Sex Hormone Binding Globulin and is linked to less aggressive estrogen-dependent breast cancers. Life Sci. (2018) 207:372–80. doi: 10.1016/j.lfs.2018.06.024
173. Pupa SM, Argraves SW, Forti S, Casalini P, Berno V, Agresti R, et al. Immunological and pathobiological roles of fibulin-1 in breast cancer. Oncogene. (2004) 23:2153–60. doi: 10.1038/sj.onc.1207323
174. Ramdas P, Radhakrishnan AK, Abdu Sani AA, Abdul-Rahman PS. Tocotrienols modulate breast cancer secretomes and affect cancer-signaling pathways in MDA-MB-231 cells: A label-free quantitative proteomic analysis. Nutr Cancer. (2019) 71:1263–71. doi: 10.1080/01635581.2019.1607407
175. Pupa SM, Giuffré S, Castiglioni F, Bertola L, Cantú M, Bongarzone I, et al. Regulation of breast cancer response to chemotherapy by fibulin-1. Cancer Res. (2007) 67:4271–7. doi: 10.1158/0008-5472.CAN-06-4162
176. Winiarski BK, Wolanska KI, Rai S, Ahmed T, Acheson N, Gutowski NJ, et al. Epithelial ovarian cancer-induced angiogenic phenotype of human omental microvascular endothelial cells may occur independently of VEGF signaling. Trans Oncol. (2013) 6:703–23. doi: 10.1593/tlo.13529
177. Allard JB, Duan C. IGF-binding proteins: why do they exist and why are there so many? Front Endocrinol. (2018) 9:117. doi: 10.3389/fendo.2018.00117
178. Huber MC, Falkenberg N, Hauck SM, Priller M, Braselmann H, Feuchtinger A, et al. Cyr61 and YB-1 are novel interacting partners of uPAR and elevate the Malignancy of triple-negative breast cancer. Oncotarget. (2016) 7:44062. doi: 10.18632/oncotarget.v7i28
179. Babic AM, Kireeva ML, Kolesnikova TV, Lau LF. CYR61, a product of a growth factor-inducible immediate early gene, promotes angiogenesis and tumor growth. Proc Natl Acad Sci. (1998) 95:6355–60. doi: 10.1073/pnas.95.11.6355
180. Lee LY, Thaysen-Andersen M, Baker MS, Packer NH, Hancock WS, Fanayan S. Comprehensive N-glycome profiling of cultured human epithelial breast cells identifies unique secretome N-glycosylation signatures enabling tumorigenic subtype classification. J Proteome Res. (2014) 13:4783–95. doi: 10.1021/pr500331m
181. Wang X, Li X, Zeng Y-N, He F, Yang X-M, Guan F. Enhanced expression of polysialic acid correlates with Malignant phenotype in breast cancer cell lines and clinical tissue samples. Int J Mol Med. (2016) 37:197–206. doi: 10.3892/ijmm.2015.2395
182. Zheng S, Zou Y, Tang Y, Yang A, Liang J-Y, Wu L, et al. Landscape of cancer-associated fibroblasts identifies the secreted biglycan as a protumor and immunosuppressive factor in triple-negative breast cancer. Oncoimmunology. (2022) 11:2020984. doi: 10.1080/2162402X.2021.2020984
183. Verma A, Kapoor R, Mittal RD. Cluster of differentiation 44 (CD44) gene variants: a putative cancer stem cell marker in risk prediction of bladder cancer in North Indian population. Indian J Clin Biochem. (2017) 32:74–83. doi: 10.1007/s12291-016-0580-y
184. Lee KY, Shueng PW, Chou CM, Lin BX, Lin MH, Kuo DY, et al. Elevation of CD109 promotes metastasis and drug resistance in lung cancer via activation of EGFR-AKT-mTOR signaling. Cancer Sci. (2020) 111:1652–62. doi: 10.1111/cas.v111.5
185. Quereda C, Pastor À, Martín-Nieto J. Involvement of abnormal dystroglycan expression and matriglycan levels in cancer pathogenesis. Cancer Cell Int. (2022) 22:1–30. doi: 10.1186/s12935-022-02812-7
186. Neill T, Schaefer L, Iozzo RV. Decorin as a multivalent therapeutic agent against cancer. Advanced Drug Delivery Rev. (2016) 97:174–85. doi: 10.1016/j.addr.2015.10.016
187. McHenry PR, Prosperi JR. Proteins found in the triple-negative breast cancer secretome and their therapeutic potential. Int J Mol Sci. (2023) 24:2100. doi: 10.3390/ijms24032100
188. Raiter A, Lipovetsky J, Stenbac A, Lubin I, Yerushalmi R. TNBC-derived Gal3BP/Gal3 complex induces immunosuppression through CD45 receptor. OncoImmunology. (2023) 12:2246322. doi: 10.1080/2162402X.2023.2246322
189. Ramos S, Ferreira S, Fernandes AS, Saraiva N. Lysyl oxidases expression and breast cancer progression: A bioinformatic analysis. Front Pharmacol. (2022) 13:883998. doi: 10.3389/fphar.2022.883998
190. Weledji EP, Assob JC. The ubiquitous neural cell adhesion molecule (N-CAM). Ann Med Surg. (2014) 3:77–81. doi: 10.1016/j.amsu.2014.06.014
191. Ma X, Wang J, Zhuang J, Ma X, Zheng N, Song Y, et al. P4HB modulates epithelial−mesenchymal transition and the β−catenin/snail pathway influencing chemoresistance in liver cancer cells. Oncol Lett. (2020) 20:257–65. doi: 10.3892/ol.2020.11569
192. Zheng F, Wang Y, Zhang Q, Chen Q, Liang C-L, Liu H, et al. Polyphyllin I suppresses the gastric cancer growth by promoting cancer cell ferroptosis. Front Pharmacol. (2023) 14:1145407. doi: 10.3389/fphar.2023.1145407
193. Li J-M, Yang F, Li J, Yuan W-Q, Wang H, Luo Y-Q. Reelin promotes cisplatin resistance by induction of epithelial-mesenchymal transition via p38/gsk3β/snail signaling in non-small cell lung cancer. Med Sci Monitor: Int Med J Exp Clin Res. (2020) 26:e925298–925291. doi: 10.12659/MSM.925298
194. Ren Z, Xue Y, Liu L, Zhang X, Pei J, Zhang Y, et al. Tissue factor overexpression in triple-negative breast cancer promotes immune evasion by impeding T-cell infiltration and effector function. Cancer Lett. (2023) 565:216221. doi: 10.1016/j.canlet.2023.216221
195. Zhang Y, Sun L, Li H, Ai L, Ma Q, Qiao X, et al. Binding blockade between TLN1 and integrin β1 represses triple-negative breast cancer. Elife. (2022) 11:e68481. doi: 10.7554/eLife.68481.sa2
196. Li Z-L, Zhang H-L, Huang Y, Huang J-H, Sun P, Zhou N-N, et al. Autophagy deficiency promotes triple-negative breast cancer resistance to T cell-mediated cytotoxicity by blocking tenascin-C degradation. Nat Commun. (2020) 11:3806. doi: 10.1038/s41467-020-17395-y
197. Hao Y, Baker D, Ten Dijke P. TGF-β-mediated epithelial-mesenchymal transition and cancer metastasis. Int J Mol Sci. (2019) 20:2767. doi: 10.3390/ijms20112767
198. Moyret-Lalle C, Ruiz E, Puisieux A. Epithelial-mesenchymal transition transcription factors and miRNAs:“Plastic surgeons” of breast cancer. World J Clin Oncol. (2014) 5:311. doi: 10.5306/wjco.v5.i3.311
199. Scimeca M, Antonacci C, Colombo D, Bonfiglio R, Buonomo OC, Bonanno E. Emerging prognostic markers related to mesenchymal characteristics of poorly differentiated breast cancers. Tumor Biol. (2016) 37:5427–35. doi: 10.1007/s13277-015-4361-7
200. Wendt MK, Allington TM, Schiemann WP. Mechanisms of the epithelial–mesenchymal transition by TGF-β. Future Oncol. (2009) 5:1145–68. doi: 10.2217/fon.09.90
201. Hua W, Ten Dijke P, Kostidis S, Giera M, Hornsveld M. TGFβ-induced metabolic reprogramming during epithelial-to-mesenchymal transition in cancer. Cell Mol Life Sci. (2020) 77:2103–23. doi: 10.1007/s00018-019-03398-6
202. Taylor MA, Parvani JG, Schiemann WP. The pathophysiology of epithelial-mesenchymal transition induced by transforming growth factor-β in normal and Malignant mammary epithelial cells. J Mammary Gland Biol Neoplasia. (2010) 15:169–90. doi: 10.1007/s10911-010-9181-1
203. Xu P, Liu J, Derynck R. Post-translational regulation of TGF-β receptor and Smad signaling. FEBS Lett. (2012) 586:1871–84. doi: 10.1016/j.febslet.2012.05.010
204. Wu Y, Li Q, Zhou X, Yu J, Mu Y, Munker S, et al. Decreased levels of active SMAD2 correlate with poor prognosis in gastric cancer. PloS One. (2012) 7:e35684. doi: 10.1371/journal.pone.0035684
205. Xu J, Lamouille S, Derynck R. TGF-β-induced epithelial to mesenchymal transition. Cell Res. (2009) 19:156–72. doi: 10.1038/cr.2009.5
206. Zhang YE. Non-Smad signaling pathways of the TGF-β family. Cold Spring Harbor Perspect Biol. (2017) 9:a022129. doi: 10.1101/cshperspect.a022129
207. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial–mesenchymal transition. Nat Rev Mol Cell Biol. (2014) 15:178–96. doi: 10.1038/nrm3758
208. Pelullo M, Zema S, Nardozza F, Checquolo S, Screpanti I, Bellavia D. Wnt, Notch, and TGF-β pathways impinge on hedgehog signaling complexity: an open window on cancer. Front Genet. (2019) 10:711. doi: 10.3389/fgene.2019.00711
209. Singha PK, Pandeswara S, Geng H, Lan R, Venkatachalam MA, Dobi A, et al. Increased Smad3 and reduced Smad2 levels mediate the functional switch of TGF-β from growth suppressor to growth and metastasis promoter through TMEPAI/PMEPA1 in triple negative breast cancer. Genes Cancer. (2019) 10:134. doi: 10.18632/genesandcancer.v10i5-6
210. Sun Q, Wang Y, Officer A, Pecknold B, Lee G, Harismendy O, et al. Stem-like breast cancer cells in the activated state resist genetic stress via TGFBI-ZEB1. NPJ Breast Cancer. (2022) 8:5. doi: 10.1038/s41523-021-00375-w
211. Xu X, Zhang M, Xu F, Jiang S. Wnt signaling in breast cancer: biological mechanisms, challenges and opportunities. Mol Cancer. (2020) 19:1–35. doi: 10.1186/s12943-020-01276-5
212. Komiya Y, Habas R. Wnt signal transduction pathways. Organogenesis. (2008) 4:68–75. doi: 10.4161/org.4.2.5851
213. MacDonald BT, Tamai K, He X. Wnt/β-catenin signaling: components, mechanisms, and diseases. Dev Cell. (2009) 17:9–26. doi: 10.1016/j.devcel.2009.06.016
214. Beurel E, Grieco SF, Jope RS. Glycogen synthase kinase-3 (GSK3): regulation, actions, and diseases. Pharmacol Ther. (2015) 148:114–31. doi: 10.1016/j.pharmthera.2014.11.016
215. Zheng H, Yang Z, Xin Z, Yang Y, Yu Y, Cui J, et al. Glycogen synthase kinase-3β: a promising candidate in the fight against fibrosis. Theranostics. (2020) 10:11737. doi: 10.7150/thno.47717
216. Katz E, Dubois-Marshall S, Sims AH, Gautier P, Caldwell H, Meehan RR, et al. An in vitro model that recapitulates the epithelial to mesenchymal transition (EMT) in human breast cancer. PloS One. (2011) 6:e17083. doi: 10.1371/journal.pone.0017083
217. Geyer FC, Lacroix-Triki M, Savage K, Arnedos M, Lambros MB, MacKay A, et al. [amp]]beta;-Catenin pathway activation in breast cancer is associated with triple-negative phenotype but not with CTNNB1 mutation. Modern Pathol. (2011) 24:209–31. doi: 10.1038/modpathol.2010.205
218. Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of Malignant and stem cell traits. Nat Rev Cancer. (2009) 9:265–73. doi: 10.1038/nrc2620
219. Valenta T, Hausmann G, Basler K. The many faces and functions of β-catenin. EMBO J. (2012) 31:2714–36. doi: 10.1038/emboj.2012.150
220. Sun Y, Zhang J, Ma L. [amp]]alpha;-catenin: A tumor suppressor beyond adherens junctions. Cell Cycle. (2014) 13:2334–9. doi: 10.4161/cc.29765
221. Li Y, Ma J, Qian X, Wu Q, Xia J, Miele L, et al. Regulation of EMT by Notch signaling pathway in tumor progression. Curr Cancer Drug Targets. (2013) 13:957–62. doi: 10.2174/15680096113136660101
222. Wang MM. Notch signaling and Notch signaling modifiers. Int J Biochem Cell Biol. (2011) 43:1550–62. doi: 10.1016/j.biocel.2011.08.005
223. Giuli M, Giuliani E, Screpanti I, Bellavia D, Checquolo S. Notch signaling activation as a hallmark for triple-negative breast cancer subtype. J Oncol. (2019) 2019:8707053. doi: 10.1155/2019/8707053
224. Al-Hussaini H, Subramanyam D, Reedijk M, Sridhar SS. Notch signaling pathway as a therapeutic target in breast cancer. Mol Cancer Ther. (2011) 10:9–15. doi: 10.1158/1535-7163.MCT-10-0677
225. Bellavia D, Checquolo S, Campese AF, Felli M, Gulino A, Screpanti I. Notch3: from subtle structural differences to functional diversity. Oncogene. (2008) 27:5092–8. doi: 10.1038/onc.2008.230
226. Kar R, Jha NK, Jha SK, Sharma A, Dholpuria S, Asthana N, et al. A “NOTCH” deeper into the epithelial-to-mesenchymal transition (EMT) program in breast cancer. Genes. (2019) 10:961. doi: 10.3390/genes10120961
227. Zhang J, Shao X, Sun H, Liu K, Ding Z, Chen J, et al. NUMB negatively regulates the epithelial-mesenchymal transition of triple-negative breast cancer by antagonizing Notch signaling. Oncotarget. (2016) 7:61036. doi: 10.18632/oncotarget.11062
228. Shah H, Mistry M, Patel N, Vora H. Clinical significance of Notch receptors in triple negative breast cancer. Breast Dis. (2023) 42:85–100. doi: 10.3233/BD-220041
229. Wang Z, Li Y, Kong D, H Sarkar F. The role of Notch signaling pathway in epithelial-mesenchymal transition (EMT) during development and tumor aggressiveness. Curr Drug Targets. (2010) 11:745–51. doi: 10.2174/138945010791170860
230. Ishida T, Hijioka H, Kume K, Miyawaki A, Nakamura N. Notch signaling induces EMT in OSCC cell lines in a hypoxic environment. Oncol Lett. (2013) 6:1201–6. doi: 10.3892/ol.2013.1549
231. Sahlgren C, Gustafsson MV, Jin S, Poellinger L, Lendahl U. Notch signaling mediates hypoxia-induced tumor cell migration and invasion. Proc Natl Acad Sci. (2008) 105:6392–7. doi: 10.1073/pnas.0802047105
232. Shi Q, Xue C, Zeng Y, Yuan X, Chu Q, Jiang S, et al. Notch signaling pathway in cancer: from mechanistic insights to targeted therapies. Signal Transduction Targeted Ther. (2024) 9:128. doi: 10.1038/s41392-024-01828-x
233. Chen W, Zhang Y, Li R, Huang W, Wei X, Zeng D, et al. Notch3 transactivates glycogen synthase kinase-3-beta and inhibits epithelial-to-mesenchymal transition in breast cancer cells. Cells. (2022) 11:2872. doi: 10.3390/cells11182872
234. Tian Y, Zhang P, Mou Y, Yang W, Zhang J, Li Q, et al. Silencing Notch4 promotes tumorigenesis and inhibits metastasis of triple-negative breast cancer via Nanog and Cdc42. Cell Death Discovery. (2023) 9:148. doi: 10.1038/s41420-023-01450-w
235. Kontomanolis EN, Kalagasidou S, Pouliliou S, Anthoulaki X, Georgiou N, Papamanolis V, et al. The notch pathway in breast cancer progression. Sci World J. (2018) 2018. doi: 10.1155/2018/2415489
236. Li C-W, Xia W, Huo L, Lim S-O, Wu Y, Hsu JL, et al. Epithelial–mesenchymal transition induced by TNF-α requires NF-κB–mediated transcriptional upregulation of Twist1. Cancer Res. (2012) 72:1290–300. doi: 10.1158/0008-5472.CAN-11-3123
237. Mercogliano MF, Bruni S, Elizalde PV, Schillaci R. Tumor necrosis factor α blockade: an opportunity to tackle breast cancer. Front Oncol. (2020) 10:584. doi: 10.3389/fonc.2020.00584
238. Xu Y, Qin L, Sun T, Wu H, He T, Yang Z, et al. Twist1 promotes breast cancer invasion and metastasis by silencing Foxa1 expression. Oncogene. (2017) 36:1157–66. doi: 10.1038/onc.2016.286
239. Wang W, Nag SA, Zhang R. Targeting the NFκB signaling pathways for breast cancer prevention and therapy. Curr Medicinal Chem. (2015) 22:264–89.
240. Leone K, Poggiana C, Zamarchi R. The interplay between circulating tumor cells and the immune system: from immune escape to cancer immunotherapy. Diagnostics. (2018) 8:59. doi: 10.3390/diagnostics8030059
241. Rinkenbaugh AL, Baldwin AS. The NF-κB pathway and cancer stem cells. Cells. (2016) 5:16. doi: 10.3390/cells5020016
242. Armas-López L, Zúñiga J, Arrieta O, Ávila-Moreno F. The Hedgehog-GLI pathway in embryonic development and cancer: implications for pulmonary oncology therapy. Oncotarget. (2017) 8:60684. doi: 10.18632/oncotarget.19527
243. Niewiadomski P, Niedziółka SM, Markiewicz Ł, Uśpieński T, Baran B, Chojnowska K. Gli proteins: regulation in development and cancer. Cells. (2019) 8:147. doi: 10.3390/cells8020147
244. Colavito SA, Zou MR, Yan Q, Nguyen DX, Stern DF. Significance of glioma-associated oncogene homolog 1 (GLI1) expression in claudin-low breast cancer and crosstalk with the nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) pathway. Breast Cancer Res. (2014) 16:1–18. doi: 10.1186/s13058-014-0444-4
245. Jeng K-S, Sheen I-S, Leu C-M, Tseng P-H, Chang C-F. The role of smoothened in cancer. Int J Mol Sci. (2020) 21:6863. doi: 10.3390/ijms21186863
246. Kasper M, Jaks V, Fiaschi M, Toftgård R. Hedgehog signalling in breast cancer. Carcinogenesis. (2009) 30:903–11. doi: 10.1093/carcin/bgp048
247. Tan E-J, Olsson A-K, Moustakas A. Reprogramming during epithelial to mesenchymal transition under the control of TGFβ. Cell Adhesion Migration. (2015) 9:233–46. doi: 10.4161/19336918.2014.983794
248. Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. (2010) 141:1117–34. doi: 10.1016/j.cell.2010.06.011
249. Feng Y, Spezia M, Huang S, Yuan C, Zeng Z, Zhang L, et al. Breast cancer development and progression: Risk factors, cancer stem cells, signaling pathways, genomics, and molecular pathogenesis. Genes Dis. (2018) 5:77–106. doi: 10.1016/j.gendis.2018.05.001
250. Peinado H, Ballestar E, Esteller M, Cano A. Snail mediates E-cadherin repression by the recruitment of the Sin3A/histone deacetylase 1 (HDAC1)/HDAC2 complex. Mol Cell Biol. (2004) 24:306–19. doi: 10.1128/MCB.24.1.306-319.2004
Keywords: breast cancer, triple-negative breast cancer, tumor microenvironment, epithelial-mesenchymal transition, regulatory pathway, targeted therapy
Citation: Haque M, Shyanti RK and Mishra MK (2024) Targeted therapy approaches for epithelial-mesenchymal transition in triple negative breast cancer. Front. Oncol. 14:1431418. doi: 10.3389/fonc.2024.1431418
Received: 11 May 2024; Accepted: 24 September 2024;
Published: 10 October 2024.
Edited by:
Cristian Scatena, University of Pisa, ItalyReviewed by:
Jia Li, University of North Carolina at Charlotte, United StatesCarla Van Den Berg, The University of Texas at Austin, United States
Copyright © 2024 Haque, Shyanti and Mishra. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Manoj K. Mishra, bW1pc2hyYUBhbGFzdS5lZHU=