 Laura Klement
Laura Klement- Institut für Molekulare Zellbiologie, CMB - Center for Molecular Biomedicine, Universitätsklinikum Jena, Friedrich-Schiller-Universität Jena, Jena, Germany
FLT3 mutations are very frequent in AML and utilization of FLT3 inhibitors as approved treatment options are very common. Despite the initial success of inhibitor treatment, the development of resistances against this treatment is a major challenge in AML therapy. One of the mechanisms causing resistance is the homing of the leukemic cells in the protective niche of the bone marrow microenvironment (BMM). A pathway mediating homing to the BMM and leukemic cell survival is the CXCL12/CXCR4 axis. The analysis of patient samples in several independent studies indicated that FLT3-ITD expression led to higher CXCR4 surface expression. However, several in vitro studies reported contradictory findings, suggesting that FLT3-ITD signaling negatively influenced CXCR4 expression. In this commentary, we provide an overview summarizing the studies dealing with the relationship of FLT3 and CXCR4. Taken together, the current research status is not sufficient to answer the question whether FLT3 and CXCR4 act together or independently in leukemia progression. Systematic analyses in model cell systems are needed to understand the interplay between FLT3 and CXCR4, since this knowledge could lead to the development of more effective treatment strategies for AML patients.
1 The role of FLT3 in AML
Acute myeloid leukemia (AML) is a heterogeneous and malignant clonal disorder of the hematopoietic system. It is characterized by the uncontrolled proliferation of abnormal myeloid progenitors which are impaired in their differentiation into mature blood cells (1). Despite significant advances in therapy, AML remains a challenging disease to treat, with high relapse rates and poor overall survival.
The FMS-like tyrosine kinase 3 (FLT3, CD135) is expressed on the surface of hematopoietic stem and progenitor cells and is involved in the regulation of cell survival, proliferation, and differentiation (2–4). FLT3 plays a crucial role in the development and progression of AML as the FLT3 gene is mutated in approximately 25 to 35% of AML patients (5). The mutations are classified into two major types: internal tandem duplications (ITD) of varying length mostly in the juxtamembrane domain and point mutations of e.g., D385 or I836 in the tyrosine kinase domain (TKD) (4–6). Both types of mutations lead to ligand independent constitutive activation of the receptor, but they differ in their signal transduction: in contrast to TKD and wild type (WT) FLT3, FLT3-ITD strongly activates Signal transducer and activator of transcription 5 (STAT5) (7–9).
The ITD mutations are the most common mutations of FLT3 found in AML patients and are associated with a poor prognosis, increased relapse rates, and decreased overall survival (10, 11). The prognostic relevance of FLT3-TKD is not verified yet (6). Targeting FLT3 signaling with specific inhibitors in patients with a FLT3 mutation is a commonly used treatment strategy in AML patients (6, 12, 13). Several FLT3 inhibitors have been developed and tested in clinical trials for the treatment of AML, including quizartinib (AC220), sorafenib, or the FDA-approved drugs gilteritinib and midostaurin (PKC-412). However, despite their initial success in inducing remission and improving outcomes, the development of resistance to FLT3 inhibitors remains a major challenge (5, 12, 13).
2 CXCR4 as a relevant target for BMM modulation
One of the mechanisms causing resistance to inhibitor treatment is the association of leukemic cells with the bone marrow microenvironment (BMM). The bone marrow provides a protective niche for AML cells impeding the effective elimination by FLT3 inhibitors.
The stromal cells of the bone marrow produce and secrete CXCL12 [previously known as stromal cell-derived factor-1 (SDF-1)], the ligand for the C-X-C chemokine receptor type 4 (CXCR4, SDF-1 receptor, CD184). The G protein-coupled receptor CXCR4 is expressed on the surface of hematopoietic cells, where it plays a crucial role in chemotactic migration to the bone marrow, as well as cellular proliferation and survival (14). Moreover, dysregulation of CXCR4 signaling has been implicated in the pathogenesis and progression of hematopoietic malignancies and CXCR4 is widely discussed as a promising therapeutic target (14–21). The surface expression of CXCR4 was found to be of prognostic relevance in leukemia, as either low expression of CXCR4 was found to be favorable for the treatment outcome, or high expression was associated with a poor prognosis (22–26). CXCR4-positive cells can migrate to and reside in the BMM and are shielded from chemotherapeutic agents. The leukemic cells can bypass the inhibitor therapy by survival signals coming from the CXCL12/CXCR4 axis and may contribute to the development of recurrences (20, 21, 27–29). Consequently, targeting CXCR4 signaling via e.g., CXCR4 antagonists, is a potent strategy for disrupting the influence of the protective microenvironment thus enhancing the efficacy of chemotherapy in AML (29–31).
3 Combinatorial targeting of FLT3 and CXCR4 in AML
Since CXCR4 contributes to the survival of leukemic cells in the BMM, the combinatorial treatment of CXCR4 antagonists and FLT3 inhibitors may lead to improved therapeutic outcomes in AML. For example, Kim et al. have demonstrated that the combination of the CXCR4 antagonist LY2510924 and the FLT3 inhibitor quizartinib reversed the stroma-mediated resistance against the FLT3 inhibitor and induced the mobilization and differentiation of AML cells in mice, resulting in enhanced anti-leukemia effects (32). In addition, Borthakur et al. conducted a phase I clinical trial utilizing granulocyte colony stimulating factor (G-CSF which regulates the expression of CXCL12 in the bone marrow), the CXCR4 inhibitor plerixafor and the FLT3 inhibitor sorafenib in relapsed/refractory FLT3-ITD AML patients (NCT00943943). The combinatorial treatment provoked a mobilization of AML cells from the bone marrow to the peripheral circulation and exhibited encouraging response rates in relapsed AML patients (33). In the same line, an RNA oligonucleotide that inhibits CXCL12, showed synergistic activity in combinatorial treatment with midostaurin against FLT3-ITD-dependent leukemia cells (34). We summarized these and additional studies targeting FLT3 and CXCL12/CXCR4 simultaneously in Table 1.
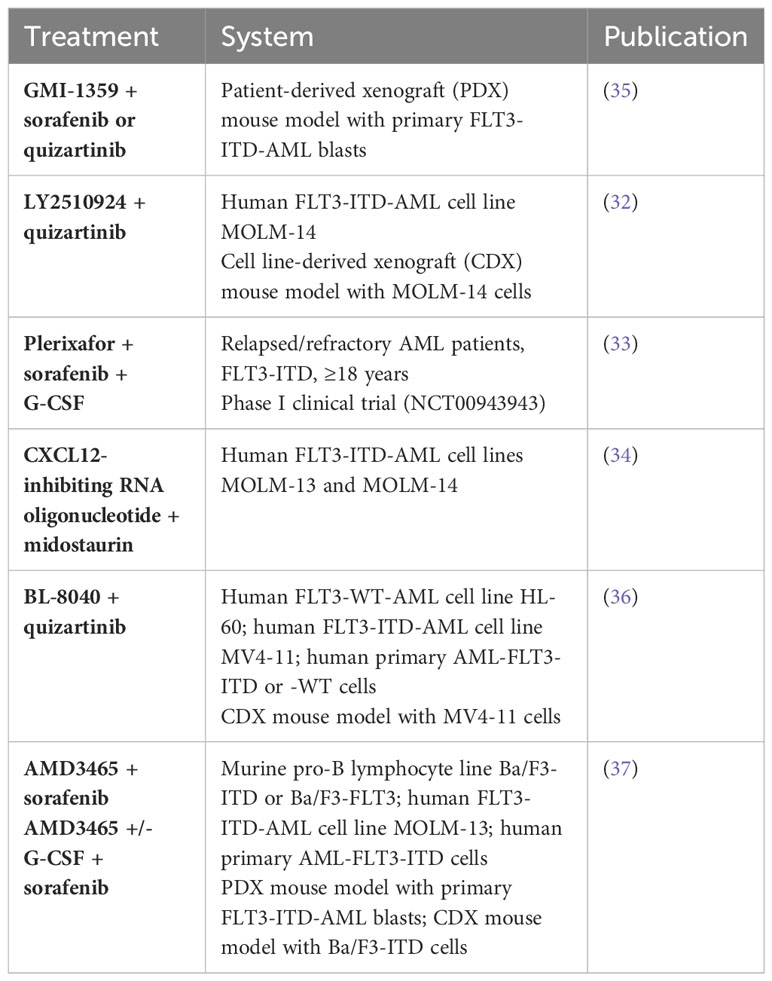
Table 1 Summary of studies combining FLT3 and CXCL12/CXCR4 treatments.
A different approach, using a CXCL12 knock out mouse model also resulted in enhanced targeting of AML cells by FLT3 inhibitors and chemotherapy treatment as compared to mice with normal CXCL12 expression (38). This study describes the mitogen-activated protein kinase p38 as a possible key player in this regulation. Taken together, although targeting both FLT3 and CXCR4 simultaneously has shown promising results in several studies, the specific mechanism underlying their interplay and potential crosstalk is not yet fully understood.
4 Contradictory findings describing the relationship between FLT3 and CXCR4
Until now, several studies have examined the interplay of FLT3 and CXCR4. For instance, Rombouts et al. found that AML patient samples with FLT3-ITD mutation have a higher expression of surface CXCR4 (39). Similar findings were reported in a study based on the analysis of 122 patient samples in which significantly higher CXCR4 surface expression was found on cells from patients expressing FLT3-ITD than in samples expressing FLT3-WT (26). In addition, Cao et al. analyzed 466 samples from newly diagnosed AML patients as well as from healthy donors and found that cells of the healthy control group had the least, AML patients expressing FLT3-WT had medium, and AML patients with FLT3-ITD expression showed the highest CXCR4 expression (40).
In addition to the analysis of patient samples, multiple in vitro experiments have been conducted, but they reveal contrasting findings. Moreover, the used cell models and experimental setups are very diverse. To elucidate the current research status, the findings of several publications will be recited in the following.
In 2005, Fukuda et al. presented data where the effects of CXCL12 and FLT3 ligand (FL) co-stimulation were analyzed (41). They report that FL and CXCL12 act synergistically to mediate short term migration, and in FLT3-ITD expressing cells, the migration towards CXCL12 is strongly enhanced. Most interestingly though, long-term stimulation of FLT3-WT receptor with FL leads to a downregulation of CXCR4. Moreover, CXCR4 was downregulated in Ba/F3 or 32D cells if expressing FLT3-ITD as compared to FLT3-WT expressing cells, indicating a negative influence of FLT3 signaling on the CXCR4 surface expression (41).
In 2009, the PIM1 kinase was introduced as a possible factor influencing the CXCR4 surface expression, but not the mRNA level of CXCR4 (42). PIM1 is one of the first target genes of STAT5, which is abnormally activated in cells expressing FLT3-ITD. This leads to the assumption that FLT3-ITD could increase CXCR4 surface expression via induction of PIM1 expression. However, this study does not comment on the possible influence of FLT3-WT on the CXCR4 expression.
Shorty after, Jacobi et al. present their results: They found a reduction of CXCR4 protein and mRNA levels if FLT3-ITD was overexpressed (43). Interestingly, a co-culture of FLT3-ITD expressing cells with stromal cells led to an increased CXCR4 surface expression, higher cell growth rate, and an increase in migration. Taking this into account, the high expression of CXCR4 might lead to an overgrowth of these cells compared to cells with lower CXCR4 expression.
Cao et al. addressed the question of interplay by analyzing the CXCR4 levels in two different hematopoietic cell lines representing FLT3-ITD expression (MV4-11 cells) or FLT3-WT expression (HL-60, an acute promyelocytic cell line). In contrast to the above-mentioned studies, they report that FLT3-WT expressing cells do not express CXCR4 whereas the FLT3-ITD expressing cell line does (40). Nevertheless, an exogenous expression of FLT3-ITD in the HL-60 cells or inhibition of FLT3-ITD in the MV4-11 cells that might show a direct influence on CXCR4 were not conducted.
A recently published study by Jia et al. suggests that FLT3-ITD signaling could inhibit CXCR4 expression as it reports that treatment of FLT3-ITD expressing MOLM-14 cells with a FLT3 inhibitor resulted in an upregulation of CXCR4 on the cell surface, as well as on the mRNA level (35). If this mechanism proves true, the CXCR4 surface expression would increase during FLT3 inhibitor treatment, which might then lead to enhanced residing of the AML cells in the BMM. A combinatorial treatment with CXCR4 antagonists would therefore be mandatory to overcome the resistance to inhibitor treatment and avoid leukemia relapse.
5 Conclusions
It has been clearly shown that the level of CXCR4 expression in AML patient samples does have prognostic value and that targeting CXCR4 in AML could be a promising strategy to eliminate leukemic cells hiding in the bone marrow niche.
However, contrasting findings on the influence of FLT3-ITD on the surface expression of CXCR4 were reported and simplified results are summarized in Figure 1. The analysis of patient data indicates that the presence of FLT3-ITD leads to a higher CXCR4 expression, which was also supported by a cell model study (Figure 1A), but several reports of FLT3 overexpression experiments or FLT3 inhibitor usage point in the opposite direction (Figure 1B). All the above-mentioned studies show that the situation found in patients cannot easily be reproduced in cell culture experiments. Obviously, the complicated interplay of different stimuli and tissues in AML patients leads to the development of a specific cell phenotype that is perfectly adapted to evade the AML treatments.
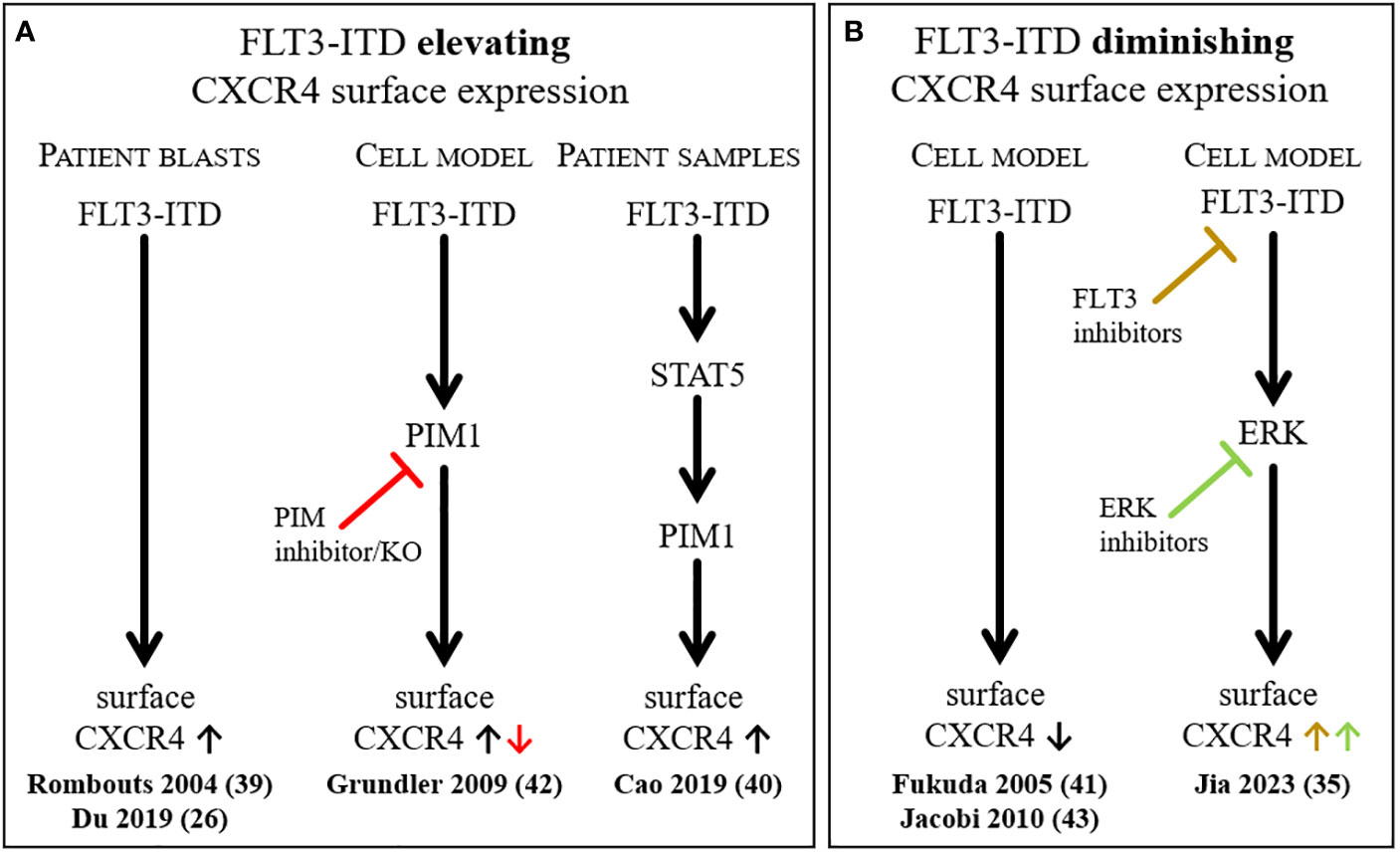
Figure 1 Summary of selected findings describing the influence of FLT3-ITD on the surface expression of CXCR4. Simplified depiction of literature reports on FLT3-ITD elevating (A) or diminishing (B) CXCR4 surface expression.
In any way, the more detailed information would be available, the better the treatment strategies could be optimized to improve the prognosis of patients suffering from AML.
Author contributions
JD: Conceptualization, Funding acquisition, Writing – original draft, Writing – review & editing. LK: Writing – original draft, Writing – review & editing.
Funding
The authors declare financial support was received for the research, authorship, and/or publication of this article. This research was funded by the Interdisciplinary Center of Clinical Research of the Medical Faculty Jena, (IZKF), grant number MSP10 to JD. LK is supported by the Deutsche Forschungsgemeinschaft (Polytarget; SFB1278: 316213987, project D02). We additionally acknowledge support by the German Research Foundation Projekt-Nr. 512648189 and the Open Access Publication Fund of the Thueringer Universitaets- und Landesbibliothek Jena.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Marcucci G, Haferlach T, Dohner H. Molecular genetics of adult acute myeloid leukemia: prognostic and therapeutic implications. J Clin Oncol (2011) 29(5):475–86. doi: 10.1200/JCO.2010.30.2554
2. Brasel K, Escobar S, Anderberg R, de Vries P, Gruss HJ, Lyman SD. Expression of the flt3 receptor and its ligand on hematopoietic cells. Leukemia (1995) 9(7):1212–8.
3. Turner AM, Lin NL, Issarachai S, Lyman SD, Broudy VC. FLT3 receptor expression on the surface of normal and Malignant human hematopoietic cells. Blood (1996) 88(9):3383–90. doi: 10.1182/blood.V88.9.3383.bloodjournal8893383
4. Stirewalt DL, Radich JP. The role of FLT3 in haematopoietic Malignancies. Nat Rev Cancer (2003) 3(9):650–65. doi: 10.1038/nrc1169
5. Kazi JU, Ronnstrand L. FMS-like tyrosine kinase 3/FLT3: from basic science to clinical implications. Physiol Rev (2019) 99(3):1433–66. doi: 10.1152/physrev.00029.2018
6. Daver N, Schlenk RF, Russell NH, Levis MJ. Targeting FLT3 mutations in AML: review of current knowledge and evidence. Leukemia (2019) 33(2):299–312. doi: 10.1038/s41375-018-0357-9
7. Hayakawa F, Towatari M, Kiyoi H, Tanimoto M, Kitamura T, Saito H, et al. Tandem-duplicated Flt3 constitutively activates STAT5 and MAP kinase and introduces autonomous cell growth in IL-3-dependent cell lines. Oncogene (2000) 19(5):624–31. doi: 10.1038/sj.onc.1203354
8. Mizuki M, Fenski R, Halfter H, Matsumura I, Schmidt R, Muller C, et al. Flt3 mutations from patients with acute myeloid leukemia induce transformation of 32D cells mediated by the Ras and STAT5 pathways. Blood (2000) 96(12):3907–14. doi: 10.1182/blood.V96.12.3907
9. Grundler R, Miething C, Thiede C, Peschel C, Duyster J. FLT3-ITD and tyrosine kinase domain mutants induce 2 distinct phenotypes in a murine bone marrow transplantation model. Blood (2005) 105(12):4792–9. doi: 10.1182/blood-2004-11-4430
10. Wu X, Feng X, Zhao X, Ma F, Liu N, Guo H, et al. Prognostic significance of FLT3-ITD in pediatric acute myeloid leukemia: a meta-analysis of cohort studies. Mol Cell Biochem (2016) 420(1-2):121–8. doi: 10.1007/s11010-016-2775-1
11. Lagunas-Rangel FA, Chavez-Valencia V. FLT3-ITD and its current role in acute myeloid leukaemia. Med Oncol (2017) 34(6):114. doi: 10.1007/s12032-017-0970-x
12. Antar AI, Otrock ZK, Jabbour E, Mohty M, Bazarbachi A. FLT3 inhibitors in acute myeloid leukemia: ten frequently asked questions. Leukemia (2020) 34(3):682–96. doi: 10.1038/s41375-019-0694-3
13. Muller JP, Schmidt-Arras D. Novel approaches to target mutant FLT3 leukaemia. Cancers (Basel) (2020) 12(10). doi: 10.3390/cancers12102806
14. Pozzobon T, Goldoni G, Viola A, Molon B. CXCR4 signaling in health and disease. Immunol Lett (2016) 177:6–15. doi: 10.1016/j.imlet.2016.06.006
15. Burger JA. CXCR4 in acute myelogenous leukemia (AML): when too much attraction is bad for you. Leuk Res (2009) 33(6):747–8. doi: 10.1016/j.leukres.2008.11.007
16. Burger JA, Peled A. CXCR4 antagonists: targeting the microenvironment in leukemia and other cancers. Leukemia (2009) 23(1):43–52. doi: 10.1038/leu.2008.299
17. Peled A, Tavor S. Role of CXCR4 in the pathogenesis of acute myeloid leukemia. Theranostics (2013) 3(1):34–9. doi: 10.7150/thno.5150
18. Ladikou EE, Chevassut T, Pepper CJ, Pepper AG. Dissecting the role of the CXCL12/CXCR4 axis in acute myeloid leukaemia. Br J Haematol (2020) 189(5):815–25. doi: 10.1111/bjh.16456
19. Meng J, Ge Y, Xing H, Wei H, Xu S, Liu J, et al. Synthetic CXCR4 antagonistic peptide assembling with nanoscaled micelles combat acute myeloid leukemia. Small (2020) 16(31):e2001890. doi: 10.1002/smll.202001890
20. Su L, Hu Z, Yang YG. Role of CXCR4 in the progression and therapy of acute leukaemia. Cell Prolif (2021) 54(7):e13076. doi: 10.1111/cpr.13076
21. Mehrpouri M. The contributory roles of the CXCL12/CXCR4/CXCR7 axis in normal and Malignant hematopoiesis: a possible therapeutic target in hematologic Malignancies. Eur J Pharmacol (2022) 920:174831. doi: 10.1016/j.ejphar.2022.174831
22. Konoplev S, Rassidakis GZ, Estey E, Kantarjian H, Liakou CI, Huang X, et al. Overexpression of CXCR4 predicts adverse overall and event-free survival in patients with unmutated FLT3 acute myeloid leukemia with normal karyotype. Cancer (2007) 109(6):1152–6. doi: 10.1002/cncr.22510
23. Spoo AC, Lubbert M, Wierda WG, Burger JA. CXCR4 is a prognostic marker in acute myelogenous leukemia. Blood (2007) 109(2):786–91. doi: 10.1182/blood-2006-05-024844
24. Tavernier-Tardy E, Cornillon J, Campos L, Flandrin P, Duval A, Nadal N, et al. Prognostic value of CXCR4 and FAK expression in acute myelogenous leukemia. Leuk Res (2009) 33(6):764–8. doi: 10.1016/j.leukres.2008.10.014
25. Ahn JY, Seo K, Weinberg OK, Arber DA. The prognostic value of CXCR4 in acute myeloid leukemia. Appl Immunohistochem Mol Morphol (2013) 21(1):79–84. doi: 10.1097/PAI.0b013e3182606f4d
26. Du W, Lu C, Zhu X, Hu D, Chen X, Li J, et al. Prognostic significance of CXCR4 expression in acute myeloid leukemia. Cancer Med (2019) 8(15):6595–603. doi: 10.1002/cam4.2535
27. Tavor S, Petit I, Porozov S, Avigdor A, Dar A, Leider-Trejo L, et al. CXCR4 regulates migration and development of human acute myelogenous leukemia stem cells in transplanted NOD/SCID mice. Cancer Res (2004) 64(8):2817–24. doi: 10.1158/0008-5472.CAN-03-3693
28. Wysoczynski M, Reca R, Ratajczak J, Kucia M, Shirvaikar N, Honczarenko M, et al. Incorporation of CXCR4 into membrane lipid rafts primes homing-related responses of hematopoietic stem/progenitor cells to an SDF-1 gradient. Blood (2005) 105(1):40–8. doi: 10.1182/blood-2004-04-1430
29. Mousavi A. CXCL12/CXCR4 signal transduction in diseases and its molecular approaches in targeted-therapy. Immunol Lett (2020) 217:91–115. doi: 10.1016/j.imlet.2019.11.007
30. Yazdani Z, Mousavi Z, Moradabadi A, Hassanshahi G. Significance of CXCL12/CXCR4 ligand/receptor axis in various aspects of acute myeloid leukemia. Cancer Manag Res (2020) 12:2155–65. doi: 10.2147/CMAR.S234883
31. Yao Y, Li F, Huang J, Jin J, Wang H. Leukemia stem cell-bone marrow microenvironment interplay in acute myeloid leukemia development. Exp Hematol Oncol (2021) 10(1):39. doi: 10.1186/s40164-021-00233-2
32. Kim BR, Jung SH, Han AR, Park G, Kim HJ, Yuan B, et al. CXCR4 inhibition enhances efficacy of FLT3 inhibitors in FLT3-mutated AML augmented by suppressed TGF-b signaling. Cancers (Basel) (2020) 12(7). doi: 10.3390/cancers12071737
33. Borthakur G, Zeng Z, Cortes JE, Chen HC, Huang X, Konopleva M, et al. Phase 1 study of combinatorial sorafenib, G-CSF, and plerixafor treatment in relapsed/refractory, FLT3-ITD-mutated acute myelogenous leukemia patients. Am J Hematol (2020) 95(11):1296–303. doi: 10.1002/ajh.25943
34. Weisberg EL, Sattler M, Azab AK, Eulberg D, Kruschinski A, Manley PW, et al. Inhibition of SDF-1-induced migration of oncogene-driven myeloid leukemia by the L-RNA aptamer (Spiegelmer), NOX-A12, and potentiation of tyrosine kinase inhibition. Oncotarget (2017) 8(66):109973–84. doi: 10.18632/oncotarget.22409
35. Jia Y, Zhang W, Basyal M, Chang KH, Ostermann L, Burks JK, et al. FLT3 inhibitors upregulate CXCR4 and E-selectin ligands via ERK suppression in AML cells and CXCR4/E-selectin inhibition enhances anti-leukemia efficacy of FLT3-targeted therapy in AML. Leukemia (2023) 37, 1379–1383. doi: 10.21203/rs.3.rs-2277452/v1
36. Abraham M, Klein S, Bulvik B, Wald H, Weiss ID, Olam D, et al. The CXCR4 inhibitor BL-8040 induces the apoptosis of AML blasts by downregulating ERK, BCL-2, MCL-1 and cyclin-D1 via altered miR-15a/16-1 expression. Leukemia (2017) 31(11):2336–46. doi: 10.1038/leu.2017.82
37. Zeng Z, Shi YX, Samudio IJ, Wang RY, Ling X, Frolova O, et al. Targeting the leukemia microenvironment by CXCR4 inhibition overcomes resistance to kinase inhibitors and chemotherapy in AML. Blood (2009) 113(24):6215–24. doi: 10.1182/blood-2008-05-158311
38. Anderson NR, Sheth V, Li H, Harris MW, Qiu S, Crossman DK, et al. Microenvironmental CXCL12 deletion enhances Flt3-ITD acute myeloid leukemia stem cell response to therapy by reducing p38 MAPK signaling. Leukemia (2023) 37, 560–570. doi: 10.1038/s41375-022-01798-5
39. Rombouts EJ, Pavic B, Lowenberg B, Ploemacher RE. Relation between CXCR-4 expression, Flt3 mutations, and unfavorable prognosis of adult acute myeloid leukemia. Blood (2004) 104(2):550–7. doi: 10.1182/blood-2004-02-0566
40. Cao T, Jiang N, Liao H, Shuai X, Su J, Zheng Q. The FLT3-ITD mutation and the expression of its downstream signaling intermediates STAT5 and Pim-1 are positively correlated with CXCR4 expression in patients with acute myeloid leukemia. Sci Rep (2019) 9(1):12209. doi: 10.1038/s41598-019-48687-z
41. Fukuda S, Broxmeyer HE, Pelus LM. Flt3 ligand and the Flt3 receptor regulate hematopoietic cell migration by modulating the SDF-1alpha(CXCL12)/CXCR4 axis. Blood (2005) 105(8):3117–26. doi: 10.1182/blood-2004-04-1440
42. Grundler R, Brault L, Gasser C, Bullock AN, Dechow T, Woetzel S, et al. Dissection of PIM serine/threonine kinases in FLT3-ITD-induced leukemogenesis reveals PIM1 as regulator of CXCL12-CXCR4-mediated homing and migration. J Exp Med (2009) 206(9):1957–70. doi: 10.1084/jem.20082074
Keywords: acute myeloid leukemia, cancer, FLT3-ITD, stem cell niche, treatment options, CXCR4, SDF-1, CXCL12
Citation: Klement L and Drube J (2023) The interplay of FLT3 and CXCR4 in acute myeloid leukemia: an ongoing debate. Front. Oncol. 13:1258679. doi: 10.3389/fonc.2023.1258679
Received: 14 July 2023; Accepted: 08 September 2023;
Published: 02 October 2023.
Edited by:
Spiros Vlahopoulos, University of Athens, GreeceReviewed by:
Anna M. Eiring, Texas Tech University Health Sciences Center El Paso, United StatesCopyright © 2023 Klement and Drube. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Julia Drube, Julia.Drube@med.uni-jena.de