 Swapnil Parashram Bhavsar
Swapnil Parashram Bhavsar
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Oncol., 18 May 2023
Sec. Molecular and Cellular Oncology
Volume 13 - 2023 | https://doi.org/10.3389/fonc.2023.1196861
Oncogenic drivers like MYCN in neuroblastoma subsets continues to present a significant challenge owing to its strong correlation with high-risk metastatic disease and poor prognosis. However, only a limited number of MYCN-regulatory proteins associated with tumor initiation and progression have been elucidated. In this minireview, I summarize the recent progress in understanding the functional role of MYCN and its regulatory partners in neuroblastoma metastasis.
Genetic changes are acquired by rare cells in the primary tumor mass which confer them dominant phenotypes like the ability to resist growth inhibiting signals, avoiding apoptosis, acquisition of constitutive mitogenic signals and induction of angiogenesis. A subset of these rare cells harbors yet more mutations that increases the propensity to metastasize to distant organs and seed new colonies (1). This process termed - tumor metastasis, is the main cause of cancer related deaths and is the most important obstacle in the treatment of cancer patients. Despite decades of research and improvements in treatment approaches thereof, the survival rates of patients with metastatic disease are very low. For example, the overall survival of high-risk neuroblastoma patients remains around 50 - 60% (2). Therefore, there is an immediate need to better understand the molecular mechanisms underlying tumor metastasis to develop novel and effective therapeutics to improve the quality of cure in this vulnerable population.
The most common pediatric cancer of the developing sympathetic nervous system – Neuroblastoma, as coined by James Wright in 1910 (3), after a century of research is still enigmatic due to its extremely heterogenous clinical nature. It has been demonstrated to range from an aggressive metastatic disease with poor prognosis to spontaneous regression or differentiation into benign histological variants (4). This explains the highly variable clinical behavior of neuroblastoma tumors, some of which are easily treatable while majority of them are aggressive (5).
Despite technological advances, the understanding of the genomic features of neuroblastoma is still modest. Numerical and segmental chromosomal abnormalities, transcriptomics, amplifications and/or overexpression of specific genes, variations in ploidy and epigenetics have all proven to play a critical role in neuroblastoma pathogenesis (6, 7). The most frequently harbored alterations of specific genes in neuroblastoma include heritable mutations in the ALK (6) and PHOX2B (8) observed in familial neuroblastoma, chromatin remodeling genes like ATRX (6), ARID1A and ARID1B (9) and other important genes like PTPN11, MYCN, and NRAS (6). In addition, cytogenetic abnormalities like gain of the long arm of chromosome 17 (17q gain) (10), 11q loss of heterozygosity (LOH) and 1p deletion (11) are also observed in neuroblastoma. Interestingly, all these aberrations are predictor of poor outcome in neuroblastoma (12).
The MYCN gene in neuroblastoma subsets continues to present a significant challenge. It is deregulated in both the pediatric and adult cancers (13). Multiple studies have demonstrated MYCN to function as an oncogenic driver in neuroblastoma. This is mainly proven by the guided ectopic expression of MYCN (alone or in combination of LMO1 or ALK) in specific cell lineages of zebrafish models (14, 15) or genetically engineered mouse models (GEMM) (16, 17) which results in the development of neuroblastoma. MYCN as a transcription factor, activate and/or repress various genes involved in cell proliferation, survival, and apoptosis ultimately leading to increased cellular self-renewal capacity, apoptotic resistance, and metabolic flexibility (18). Amplification of MYCN indicates aggressive or high-risk metastatic disease and poor patient prognosis (19). Nearly half of all the neuroblastoma patients at diagnosis show metastasis (20). The most frequent sites of metastasis are in bone marrow (70.5%), bone (55.7%), lymph nodes (30.9%), liver (29.6%), and intracranial and orbital sites (18.2%) (21). Abnormal MYCN expression is present at diagnosis and is never acquired during later tumorigenesis of MYCN-non-amplified neuroblastoma (18) which might be the reason behind observation of metastasis in neuroblastoma patients at diagnosis.
Discovered in 1983, MYCN (22, 23), belongs to a Myc family of genes which consists of c-myc (MYC), l-myc (MYCL) and n-myc (MYCN) (24). It is located on chromosome 2p24.3 (25) and its normal function include regulation of the cell cycle and apoptosis (26). The MYCN gene in turn is regulated by different mechanisms including and not limited to epigenetic regulation, miRNAs, and formation of G-quadraplexes (27). It is commonly amplified and overexpressed in a cancer setting (28). And it contributes to multiple facets of metastasis like adhesion, motility, invasion, and degradation of surrounding matrices (19). Molecular processes central to the MYCN’s oncogenic activity include the hallmarks of cancer influencing the cell cycle, apoptosis or cell death and cellular metabolism reviewed elsewhere (29). It is thus, an important genetic marker for prognosis, diagnosis, and therapeutics.
MYCN is extensively studied in neuroblastoma. It is amplified in 25% of all neuroblastomas and nearly 50% in high-risk neuroblastoma cases (30). It is the best characterized negative prognostic indicator that not only stratifies risk in neuroblastoma but also predict poor clinical outcome. Therefore, overexpression of MYCN is associated with poor prognosis, advanced stage of disease, rapid tumor growth and metastasis (31). MYCN thus plays a significant role in neuroblastoma biology and is the most important target for therapy.
The interplay of the host genetic factors, somatic mutations, chromosomal abnormalities, and epigenetic alterations lead to highly aggressive metastatic neuroblastoma (6). But the fundamental molecular mechanisms remain to be elucidated. Very few studies have demonstrated a crucial role of MYCN and its regulatory proteins in neuroblastoma metastasis (Table 1). Investigating these thoroughly, I define here, the ‘Metastatic wheel of neuroblastoma’ driven by MYCN oncogene (Figure 1). I explored eight different MYCN regulatory networks and sought to determine the mechanisms by which this oncogene influences neuroblastoma metastasis.
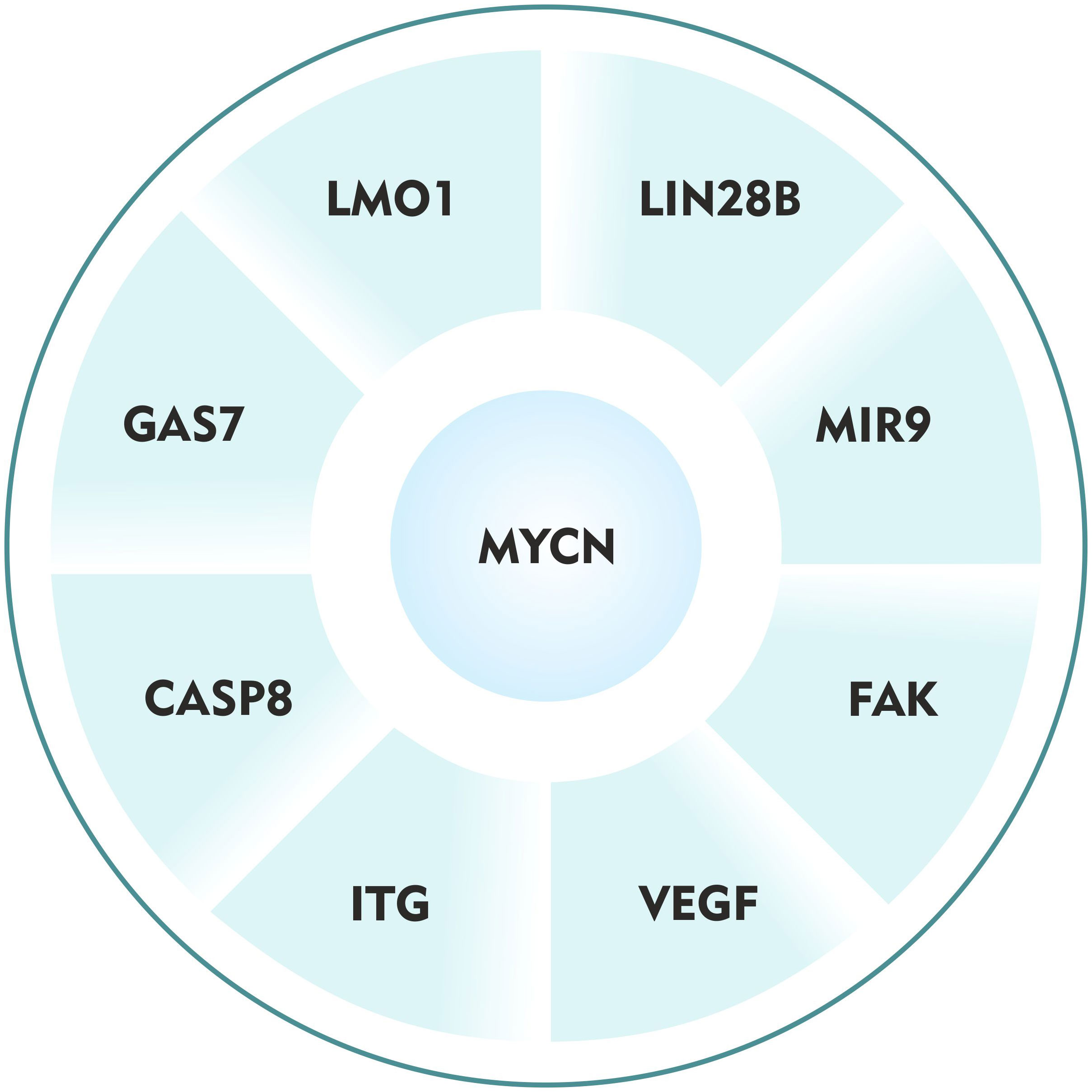
Figure 1 The ‘Metastatic wheel of neuroblastoma’ driven by MYCN oncogene.
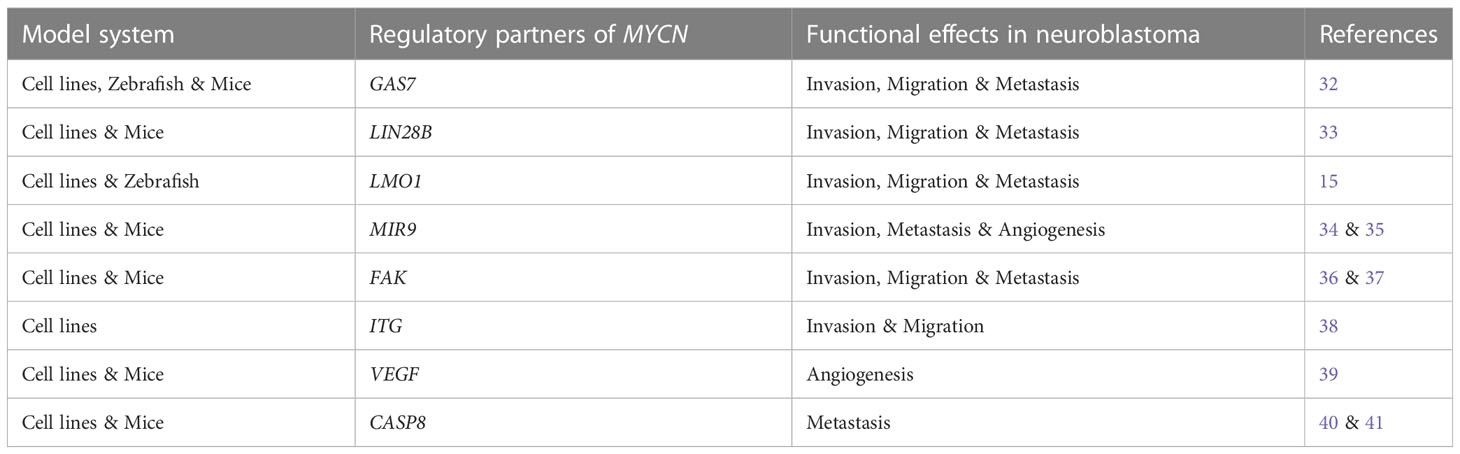
Table 1 The articles investigating the role of MYCN and its regulatory genes in neuroblastoma.
GAS7 is an evolutionary conserved gene which play key role in cell structure and motility. It has been associated with multiple development pathways and recently characterized as a metastasis suppressor gene (42). Dong and colleagues have reported the first genetic evidence linking GAS7 deficiency and metastasis in MYCN-driven neuroblastoma. Chromosome 17p13.1 harbors GAS7 and TP53 genes. And they observed a heterozygous deletion of 17p arm in high-risk cases. Bioinformatics analyses revealed association of 17p deletion with poor clinical outcome. Moreover, they also observed significantly lower level of GAS7 expression in MYCN-amplified vs. MYCN-non-amplified cases. This prompted them to consider if MYCN binds to GAS7 promoter directly. But they did not observe any such binding. However, by dual cross-linking ChIP-PCR assay they showed that MYCN interacts with SP1 transcription factor, by forming a transcription repression complex at the GAS7 promoter region to regulate GAS7 activity indirectly. Next, using both zebrafish and mammalian model systems, they show that reduced expression of the GAS7 promotes metastasis of MYCN-amplified neuroblastoma cells (32). Thus, loss of GAS7 is one of the main events driving metastasis in neuroblastoma.
LIN28 and LIN28B are highly conserved RNA-binding proteins associated with advanced human malignancies (43). Activation of these proteins is correlated with poor clinical prognosis (44). Molenaar et al., has demonstrated an oncogenic role of LIN28B in neuroblastoma. They reported that LIN28B is genomically altered and overexpressed in high-risk neuroblastoma and it’s correlated with adverse clinical outcome (45). Interestingly, the role of LIN28 and LIN28B in blocking the maturation of the tumor suppressor microRNA let-7 family is well established (46). Since, the 3’ UTRs of MYC and MYCN harbor let-7 binding sites, repression of let-7 results in higher expression of MYC and MYCN. Thus, the genetic loss of LIN28B in MYCN-driven neuroblastoma results in loss of metastatic potential both in vitro using MYCN-amplified neuroblastoma cell lines and/or PDX samples and in vivo using immunocompromised mice (33). Therefore, the role of LIN28B in neuroblastoma initiation and its correlation with MYCN expression makes it an important therapeutic target for intervention. Taken together, these studies highlight the significant role of LIN28B and MYCN in metastasis.
LMO1 gene is a member of LMO protein family, which includes LMO1, LMO2, LMO3 and LMO4. These proteins have been shown to play an oncogenic role in types of cancer including neuroblastoma (47). Disease-associated single nucleotide polymorphisms (SNPs) are often located within the super-enhancer elements (48). Oldridge and colleagues identified SNP within a super-enhancer element in the intronic region of LMO1 which led to the high expression of LMO1 and neuroblastoma pathogenesis (49). Further to this study, Zhu et al., using a novel zebrafish model, demonstrate how LMO1 and MYCN genes cooperate to first initiate neuroblastoma and further contribute to metastatic disease progression. They generated a stable transgenic zebrafish model that overexpress LMO1 gene in the peripheral sympathetic nervous system (PSNS). They observed that overexpressing LMO1 alone do not develop neuroblastoma but its overexpression along with increased MYCN expression led to enhanced neuroblastoma initiation and disease penetrance. Interestingly, they also observed distant metastasis in the transgenic fish overexpressing both MYCN and LMO1. This observation was supported by the increase in expression of panel of genes involved in tumor cell-extracellular matrix interactions like LOXL3 and integrins - ITGA2B, ITGA3, and ITGA5 (15, 50). Thus, these findings confirm the important role of MYCN and LMO1 in promoting neuroblastoma initiation, progression, and metastasis.
The ubiquitous involvement of microRNAs in shaping the cellular properties have paved way to speculate its role in influencing metastasis. Thus, multiple microRNAs have demonstrated its role either in promoting or repressing metastasis in different cancers. However, the relation between miRNA and MYCN in inducing metastasis is few explored in neuroblastoma. Interestingly, in breast cancer, studies by Ma et al., showed that miR-9, a microRNA induced by MYC/MYCN, targets E-cadherin, and not only primes cancer cells for epithelial to mesenchymal transition (EMT) and invasion, but also contribute to promoting angiogenesis. Thus, leading to growth, proliferation, invasion, and migration of cancer cells and hence metastasis. However, this miR-9 effect is not observed in neuroblastoma since the tumors do not express E-cadherin (34, 51). Nonetheless, studies by Zhang et al., showed that miR-9 expression was downregulated in neuroblastoma cell lines and primary tissues. They also observed an inverse correlation between MMP14, a matrix metalloproteinase playing a critical role in tumor metastasis and angiogenesis, and endogenous miR-9 expression. Further bioinformatic and functional assays confirmed MMP14 as a target of miR-9. The effect of targeting MMP14 resulted in suppression of invasion, metastasis, and angiogenesis of neuroblastoma cells both in vitro and in vivo (35). This study was performed using MYCN-non-amplified neuroblastoma cell lines SH-SY5Y and SK-N-SH. Therefore, the role of MYCN in inducing miR-9 expression was not addressed.
FAK is a cytoplasmic protein tyrosine kinase, a crucial signaling component activated by numerous stimuli, which play a key role in normal and tumor cell migration (52). FAK is shown to be overexpressed and activated in several advanced-stage solid cancers and promotes tumor progression and metastasis (53). Beierle and colleagues observed upregulation of FAK in MYCN-amplified and overexpressed neuroblastoma cell lines and in the advanced-stage human neuroblastoma tumor specimens. This observation prompted them to investigate if MYCN regulates FAK expression. Interestingly, bioinformatic analysis revealed two MYCN binding sites in the FAK promoter region. Moreover, series of in vitro and in vivo assays confirmed the binding of MYCN to sites in the FAK promoter. Conditional expression of MYCN or inhibition of FAK expression resulted in decreased cell viability and increase in apoptosis. Thus, this study clearly demonstrated MYCN regulation of FAK expression (36, 54). Furthermore, similar group set out to determine if the abrogation of FAK would result in alteration of metastatic potential of neuroblastoma cells. As expected, RNA interference-mediated silencing and/or small molecule inhibitors inhibition of FAK, decreased the metastatic properties like invasion and migration in vitro and decrease the ability of neuroblastoma cells to form metastasis in a nude mouse model in vivo (37).
Integrin signaling is crucial in regulating multiple functions in normal and transformed cells. Increased expression of integrins (e.g., αvβ3, αvβ5, α5β1, α6β4, α4β1 and αvβ6) is correlated with proliferation survival, invasion, and metastasis in various cancers (55). Study by Wu and colleagues showed that α4β1 and α5β1 integrins were expressed in the late stage of neuroblastoma tumors and cell lines. To find out how integrins initiated neuroblastoma motility, they performed knockdown and reconstitution experiments. Whereas knockdown of α5β1 led to FAK/Src/p130Cas dependent neuroblastoma motility, knockdown of α4β1 led to Src/p130Cas dependent neuroblastoma motility but not FAK (56). In another study, Tanaka et al., observed that MYCN-non-amplified cell lines (SK-N-SH and NB69) had higher expression of integrin α1 whereas MYCN-amplified cell lines (IMR-32, NB1, NB9, and NB19) had its lower expression. Therefore, knockdown of MYCN in NB1 and NB19 led to increased expression of integrin α1 and reduced migration. And overexpression of MYCN in SK-N-SH and NB69 led to decreased expression of integrin α1 and enhanced migration (38). This observation thus suggests that MYCN promotes neuroblastoma metastasis by downregulating integrin α1.
VEGF is a key regulator of the physiological and pathological angiogenesis, a highly complex and coordinated process for the formation of the new blood vessel growth and maturation (57). VEGF inhibition is a strategy for the prevention of angiogenesis and its inhibitors are undergoing clinical trial in several malignancies (58, 59). Kang et al., showed that tumor angiogenesis correlates with increased expression of the VEGF and poor clinical outcome in neuroblastoma. And they proposed that MYCN plays a significant role as a novel effector of PI3K-mediated regulation of VEGF and hence tumor angiogenesis, in a highly vascularized, malignant neuroblastoma (39). The observation that PI3K inhibition reduce tumor growth of murine neuroblastoma model (60) prompted Kang and colleagues to check its effect on angiogenic capacity of human neuroblastoma in mice bearing neuroblastoma xenografts. Selective inhibition of PI3K in this regard led to reduction of VEGF expression and secretion and reduced growth of established neuroblastoma tumors. Next, they investigated if MYCN inhibition could also regulate the VEGF expression in neuroblastoma cells. Interestingly, siRNA knockdown of MYCN in MYCN-amplified neuroblastoma cells led to significant decrease in VEGF expression and no difference in MYCN-nonamplified cells. This observation indicated the role of MYCN in the regulation of VEGF expression (39). Taken together, these findings indicate the important role of MYCN in the underlying molecular mechanisms of PI3K/MYCN/VEGF regulation of critical angiogenic pathways in neuroblastoma.
In 2000, Teitz et al., in a seminal paper proposed that Caspase-8 is deleted or preferentially silenced through epigenetic mechanisms in MYCN-amplified neuroblastomas. This observation indicated that Caspase-8 act as a tumor suppressor in neuroblastoma (40, 61). CASP8 gene is mapped to chromosome 2q33. Takita and colleagues demonstrated a common region of allelic imbalance on chromosome 2q and an alteration of CASP8 in neuroblastoma. They concluded that epigenetic silencing and allelic imbalance are two important mechanisms for the inactivation of Caspase-8 in neuroblastoma (62). Further studies in this regard led Cui and colleagues to establish a link between MYCN and the death receptor apoptotic pathways (involves Caspase-8) in neuroblastoma (63).
In 2006, Stupack and colleagues, were successfully able to demonstrate for the first time the role of Caspase-8 in metastases. They provided evidence that Caspase-8 act as a as a metastasis suppressor gene and regulated the survival and invasive capacity of neuroblastoma cells (64). This study was further complimented with yet another publication from Teitz et al., establishing the role of Caspase-8 in metastasis. In this study, they established an immunocompetent mouse model for metastatic neuroblastoma which recapitulated not only overexpression of MYCN but also loss of Caspase-8 expression. Microarray expression studies from the mouse primary tumors revealed genes involved in epithelial to mesenchymal transition (EMT) and Extracellular Matrix (ECM) (41). Thus, given the necessity of animal models in testing therapies for metastatic neuroblastoma, Th-MYCN/caspase-8–deleted mouse could serve as an important model system.
Due to the clinical and biological heterogeneity of neuroblastoma tumors, the treatment approaches tailored are mainly based on low, intermediate, and high-risk stage groups. Children with low and intermediate-risk get surgical tumor resection with or without chemotherapy. On the other hand, high-risk patients require intensive multimodal treatment regimens, which is complex and contains multiple consecutive phases (65). However, despite intensive modalities, 50-60% patients with high-risk disease will ultimately relapse with no curative treatment available for these patients (5). Therefore, new alternate approaches are being tested to combat this deadly disease. Recent novel immunotherapy approaches to target neuroblastoma have demonstrated that anti-GD2 monoclonal antibody therapy and CAR T cell therapy for patients with high-risk neuroblastoma improves the event-free and overall survival (66, 67). Given the significant role of MYCN in neuroblastoma biology and poor clinical outcomes, novel treatments targeting MYCN should be developed for patients with neuroblastoma.
The focus of this review is to explore the novel mechanisms underlying tumor metastasis in MYCN-driven neuroblastoma. The fact that around 50% of the neuroblastoma patients show metastasis with MYCN amplifications at diagnosis indicate that this oncogene might have a significant role in promoting metastasis. Decades of research in this regard has led to discoveries of only a couple of regulatory genes of MYCN (GAS7, LIN28B, LMO1, MIR9, FAK, ITG, VEGF and CASP8) having direct or indirect role in facets of metastasis as illustrated in this review. Though some of these studies imply the influence of MYCN and its regulatory proteins in metastatic neuroblastoma, very few studies successfully demonstrate its role both in vitro and in vivo (33, 35, 36). Therefore, advanced bioinformatic techniques and/or high throughput technologies are necessitated to find new candidate genes and its association with MYCN in promoting metastasis. Studies in types of cancers have implicated effectors of the EMT (68, 69), which are poorly investigated in neuroblastoma. Thus, either independent or co-operative role of such genes with MYCN requires further investigation both in vitro and in vivo.
Modeling of the pediatric cancer in diverse animal models is necessary to understand the role of MYCN in metastasis. Models like the widely used TH-MYCN transgenic mouse model and the orthotopic xenografts of primary human neuroblastomas or cell lines may be of valuable resource for evaluating therapeutics (15, 61, 64, 70–72).
Taken together, considering the metastatic role of MYCN, it is an ideal and the most wanted target for cancer therapy. Though MYCN is thought ‘undruggable’, due to multiple reasons like its MYC similarity, few known MYCN-interacting proteins, lack of in vivo testing of MYCN-targets, lack of structural information on MYCN-protein complexes, and the challenges of using traditional small -molecule inhibitors of protein-DNA and/or protein-protein interactions’ (73), novel alternate approaches to target MYCN are warranted to combat this deadly pediatric malignancy. I believe that insights provided in this minireview could help develop new ideas and strategies to counter tumor metastasis in MYCN-driven high-risk neuroblastoma.
The author confirms being the sole contributor of this work and has approved it for publication.
This study was supported by research grant from the Barnekreftforeningen (Project number: 210009). The publication charges for this article have been funded by a grant from the publication fund of UiT - The Arctic University of Norway. The funding organizations have no role in the design of the study, analysis, and interpretation of the data and in writing the manuscript.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
2. Irwin MS, Naranjo A, Zhang FF, Cohn SL, London WB, Gastier-Foster JM, et al. Revised neuroblastoma risk classification system: a report from the children's oncology group. J Clin Oncol (2021) 39(29):3229–41. doi: 10.1200/JCO.21.00278
3. Wright JH. Neurocytoma or neuroblastoma, a kind of tumor not generally recognized. J Exp Med (1910) 12(4):556–61. doi: 10.1084/jem.12.4.556
4. Brodeur GM. Spontaneous regression of neuroblastoma. Cell Tissue Res (2018) 372(2):277–86. doi: 10.1007/s00441-017-2761-2
5. Maris JM. Recent advances in neuroblastoma. N Engl J Med (2010) 362(23):2202–11. doi: 10.1056/NEJMra0804577
6. Pugh TJ, Morozova O, Attiyeh EF, Asgharzadeh S, Wei JS, Auclair D, et al. The genetic landscape of high-risk neuroblastoma. Nat Genet (2013) 45(3):279–84. doi: 10.1038/ng.2529
7. Barr EK, Applebaum MA. Genetic predisposition to neuroblastoma. Children (Basel) (2018) 5(9):119. doi: 10.3390/children5090119
8. Trochet D, Bourdeaut F, Janoueix-Lerosey I, Deville A, de Pontual L, Schleiermacher G, et al. Germline mutations of the paired-like homeobox 2B (PHOX2B) gene in neuroblastoma. Am J Hum Genet (2004) 74(4):761–4. doi: 10.1086/383253
9. Sausen M, Leary RJ, Jones S, Wu J, Reynolds CP, Liu X, et al. Integrated genomic analyses identify ARID1A and ARID1B alterations in the childhood cancer neuroblastoma. Nat Genet (2013) 45(1):12–7. doi: 10.1038/ng.2493
10. Bown N, Cotterill S, Lastowska M, O'Neill S, Pearson AD, Plantaz D, et al. Gain of chromosome arm 17q and adverse outcome in patients with neuroblastoma. N Engl J Med (1999) 340(25):1954–61. doi: 10.1056/NEJM199906243402504
11. Attiyeh EF, London WB, Mosse YP, Wang Q, Winter C, Khazi D, et al. Chromosome 1p and 11q deletions and outcome in neuroblastoma. N Engl J Med (2005) 353(21):2243–53. doi: 10.1056/NEJMoa052399
12. Morgenstern DA, Irwin MS. Cancer genomics : chapter 21. Neuroblastoma. London: Elsevier/Academic Press (2014). doi: 10.1016/B978-0-12-396967-5.00021-9
13. Liu Z, Chen SS, Clarke S, Veschi V, Thiele CJ. Targeting MYCN in pediatric and adult cancers. Front Oncol (2020) 10:623679. doi: 10.3389/fonc.2020.623679
14. Zhu S, Lee JS, Guo F, Shin J, Perez-Atayde AR, Kutok JL, et al. Activated ALK collaborates with MYCN in neuroblastoma pathogenesis. Cancer Cell (2012) 21(3):362–73. doi: 10.1016/j.ccr.2012.02.010
15. Zhu S, Zhang X, Weichert-Leahey N, Dong Z, Zhang C, Lopez G, et al. LMO1 synergizes with MYCN to promote neuroblastoma initiation and metastasis. Cancer Cell (2017) 32(3):310–323.e315. doi: 10.1016/j.ccell.2017.08.002
16. Weiss WA, Aldape K, Mohapatra G, Feuerstein BG, Bishop JM. Targeted expression of MYCN causes neuroblastoma in transgenic mice. EMBO J (1997) 16(11):2985–95. doi: 10.1093/emboj/16.11.2985
17. Chesler L, Weiss WA. Genetically engineered murine models–contribution to our understanding of the genetics, molecular pathology and therapeutic targeting of neuroblastoma. Semin Cancer Biol (2011) 21(4):245–55. doi: 10.1016/j.semcancer.2011.09.011
18. Otte J, Dyberg C, Pepich A, Johnsen JI. MYCN function in neuroblastoma development. Front Oncol (2020) 10:624079. doi: 10.3389/fonc.2020.624079
19. Huang M, Weiss WA. Neuroblastoma and MYCN. Cold Spring Harb Perspect Med (2013) 3(10):a014415. doi: 10.1101/cshperspect.a014415
20. Maris JM, Hogarty MD, Bagatell R, Cohn SL. Neuroblastoma. Lancet (2007) 369(9579):2106–20. doi: 10.1016/S0140-6736(07)60983-0
21. DuBois SG, Kalika Y, Lukens JN, Brodeur GM, Seeger RC, Atkinson JB, et al. Metastatic sites in stage IV and IVS neuroblastoma correlate with age, tumor biology, and survival. J Pediatr Hematol Oncol (1999) 21(3):181–9. doi: 10.1097/00043426-199905000-00005
22. Kohl NE, Kanda N, Schreck RR, Bruns G, Latt SA, Gilbert F, et al. Transposition and amplification of oncogene-related sequences in human neuroblastomas. Cell (1983) 35(2 Pt 1):359–67. doi: 10.1016/0092-8674(83)90169-1
23. Schwab M, Alitalo K, Klempnauer KH, Varmus HE, Bishop JM, Gilbert F, et al. Amplified DNA with limited homology to myc cellular oncogene is shared by human neuroblastoma cell lines and a neuroblastoma tumour. Nature (1983) 305(5931):245–8. doi: 10.1038/305245a0
24. Nau MM, Brooks BJ, Battey J, Sausville E, Gazdar AF, Kirsch IR, et al. L-myc, a new myc-related gene amplified and expressed in human small cell lung cancer. Nature (1985) 318(6041):69–73. doi: 10.1038/318069a0
25. Schwab M, Ellison J, Busch M, Rosenau W, Varmus HE, Bishop JM. Enhanced expression of the human gene n-myc consequent to amplification of DNA may contribute to malignant progression of neuroblastoma. Proc Natl Acad Sci U.S.A. (1984) 81(15):4940–4. doi: 10.1073/pnas.81.15.4940
26. Henriksson M, Luscher B. Proteins of the myc network: essential regulators of cell growth and differentiation. Adv Cancer Res (1996) 68:109–82. doi: 10.1016/s0065-230x(08)60353-x
27. Braoudaki M, Hatziagapiou K, Zaravinos A, Lambrou GI. MYCN in neuroblastoma: "Old wine into new wineskins". Diseases (2021) 9(4):78. doi: 10.3390/diseases9040078
28. Beltran H. The n-myc oncogene: maximizing its targets, regulation, and therapeutic potential. Mol Cancer Res (2014) 12(6):815–22. doi: 10.1158/1541-7786.MCR-13-0536
29. Ruiz-Perez MV, Henley AB, Arsenian-Henriksson M. The MYCN protein in health and disease. Genes (Basel) (2017) 8(4):113. doi: 10.3390/genes8040113
30. Strieder V, Lutz W. Regulation of n-myc expression in development and disease. Cancer Lett (2002) 180(2):107–19. doi: 10.1016/s0304-3835(02)00020-4
31. Seeger RC, Brodeur GM, Sather H, Dalton A, Siegel SE, Wong KY, et al. Association of multiple copies of the n-myc oncogene with rapid progression of neuroblastomas. N Engl J Med (1985) 313(18):1111–6. doi: 10.1056/NEJM198510313131802
32. Dong Z, Yeo KS, Lopez G, Zhang C, Dankert Eggum EN, Rokita JL, et al. GAS7 deficiency promotes metastasis in MYCN-driven neuroblastoma. Cancer Res (2021) 81(11):2995–3007. doi: 10.1158/0008-5472.CAN-20-1890
33. Missios P, da Rocha EL, Pearson DS, Philipp J, Aleman MM, Pirouz M, et al. LIN28B alters ribosomal dynamics to promote metastasis in MYCN-driven malignancy. J Clin Invest (2021) 131(22):e145142. doi: 10.1172/JCI145142
34. Ma L, Young J, Prabhala H, Pan E, Mestdagh P, Muth D, et al. miR-9, a MYC/MYCN-activated microRNA, regulates e-cadherin and cancer metastasis. Nat Cell Biol (2010) 12(3):247–56. doi: 10.1038/ncb2024
35. Zhang H, Qi M, Li S, Qi T, Mei H, Huang K, et al. microRNA-9 targets matrix metalloproteinase 14 to inhibit invasion, metastasis, and angiogenesis of neuroblastoma cells. Mol Cancer Ther (2012) 11(7):1454–66. doi: 10.1158/1535-7163.MCT-12-0001
36. Beierle EA, Trujillo A, Nagaram A, Kurenova EV, Finch R, Ma X, et al. N-MYC regulates focal adhesion kinase expression in human neuroblastoma. J Biol Chem (2007) 282(17):12503–16. doi: 10.1074/jbc.M701450200
37. Megison ML, Stewart JE, Nabers HC, Gillory LA, Beierle EA. FAK inhibition decreases cell invasion, migration and metastasis in MYCN amplified neuroblastoma. Clin Exp Metastasis (2013) 30(5):555–68. doi: 10.1007/s10585-012-9560-7
38. Tanaka N, Fukuzawa M. MYCN downregulates integrin alpha1 to promote invasion of human neuroblastoma cells. Int J Oncol (2008) 33(4):815–21. doi: 10.3892/ijo00000069
39. Kang J, Rychahou PG, Ishola TA, Mourot JM, Evers BM, Chung DH. N-myc is a novel regulator of PI3K-mediated VEGF expression in neuroblastoma. Oncogene (2008) 27(28):3999–4007. doi: 10.1038/onc.2008.15
40. Teitz T, Wei T, Valentine MB, Vanin EF, Grenet J, Valentine VA, et al. Caspase 8 is deleted or silenced preferentially in childhood neuroblastomas with amplification of MYCN. Nat Med (2000) 6(5):529–35. doi: 10.1038/75007
41. Teitz T, Inoue M, Valentine MB, Zhu K, Rehg JE, Zhao W, et al. Th-MYCN mice with caspase-8 deficiency develop advanced neuroblastoma with bone marrow metastasis. Cancer Res (2013) 73(13):4086–97. doi: 10.1158/0008-5472.CAN-12-2681
42. Menard MJ. Loss of Gas7 is a key metastatic switch in neuroblastoma. Cancer Res (2021) 81(11):2815–6. doi: 10.1158/0008-5472.CAN-21-0783
43. Viswanathan SR, Powers JT, Einhorn W, Hoshida Y, Ng TL, Toffanin S, et al. Lin28 promotes transformation and is associated with advanced human malignancies. Nat Genet (2009) 41(7):843–8. doi: 10.1038/ng.392
44. Diskin SJ, Capasso M, Schnepp RW, Cole KA, Attiyeh EF, Hou C, et al. Common variation at 6q16 within HACE1 and LIN28B influences susceptibility to neuroblastoma. Nat Genet (2012) 44(10):1126–30. doi: 10.1038/ng.2387
45. Molenaar JJ, Domingo-Fernandez R, Ebus ME, Lindner S, Koster J, Drabek K, et al. LIN28B induces neuroblastoma and enhances MYCN levels via let-7 suppression. Nat Genet (2012) 44(11):1199–206. doi: 10.1038/ng.2436
46. Balzeau J, Menezes MR, Cao S, Hagan JP. The LIN28/let-7 pathway in cancer. Front Genet (2017) 8:31. doi: 10.3389/fgene.2017.00031
47. Wang K, Diskin SJ, Zhang H, Attiyeh EF, Winter C, Hou C, et al. Integrative genomics identifies LMO1 as a neuroblastoma oncogene. Nature (2011) 469(7329):216–20. doi: 10.1038/nature09609
48. Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-Andre V, Sigova AA, et al. Super-enhancers in the control of cell identity and disease. Cell (2013) 155(4):934–47. doi: 10.1016/j.cell.2013.09.053
49. Oldridge DA, Wood AC, Weichert-Leahey N, Crimmins I, Sussman R, Winter C, et al. Genetic predisposition to neuroblastoma mediated by a LMO1 super-enhancer polymorphism. Nature (2015) 528(7582):418–21. doi: 10.1038/nature15540
50. Liu Z, Thiele CJ. When LMO1 meets MYCN, neuroblastoma is metastatic. Cancer Cell (2017) 32(3):273–5. doi: 10.1016/j.ccell.2017.08.014
51. Khew-Goodall Y, Goodall GJ. Myc-modulated miR-9 makes more metastases. Nat Cell Biol (2010) 12(3):209–11. doi: 10.1038/ncb0310-209
52. Mitra SK, Hanson DA, Schlaepfer DD. Focal adhesion kinase: in command and control of cell motility. Nat Rev Mol Cell Biol (2005) 6(1):56–68. doi: 10.1038/nrm1549
53. Sulzmaier FJ, Jean C, Schlaepfer DD. FAK in cancer: mechanistic findings and clinical applications. Nat Rev Cancer (2014) 14(9):598–610. doi: 10.1038/nrc3792
54. Beierle EA, Massoll NA, Hartwich J, Kurenova EV, Golubovskaya VM, Cance WG, et al. Focal adhesion kinase expression in human neuroblastoma: immunohistochemical and real-time PCR analyses. Clin Cancer Res (2008) 14(11):3299–305. doi: 10.1158/1078-0432.CCR-07-1511
55. Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer (2010) 10(1):9–22. doi: 10.1038/nrc2748
56. Wu L, Bernard-Trifilo JA, Lim Y, Lim ST, Mitra SK, Uryu S, et al. Distinct FAK-src activation events promote alpha5beta1 and alpha4beta1 integrin-stimulated neuroblastoma cell motility. Oncogene (2008) 27(10):1439–48. doi: 10.1038/sj.onc.1210770
57. Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med (2003) 9(6):669–76. doi: 10.1038/nm0603-669
58. Holash J, Davis S, Papadopoulos N, Croll SD, Ho L, Russell M, et al. VEGF-trap: a VEGF blocker with potent antitumor effects. Proc Natl Acad Sci U.S.A. (2002) 99(17):11393–8. doi: 10.1073/pnas.172398299
59. Kabbinavar F, Hurwitz HI, Fehrenbacher L, Meropol NJ, Novotny WF, Lieberman G, et al. Phase II, randomized trial comparing bevacizumab plus fluorouracil (FU)/leucovorin (LV) with FU/LV alone in patients with metastatic colorectal cancer. J Clin Oncol (2003) 21(1):60–5. doi: 10.1200/JCO.2003.10.066
60. Chesler L, Goldenberg DD, Seales IT, Satchi-Fainaro R, Grimmer M, Collins R, et al. Malignant progression and blockade of angiogenesis in a murine transgenic model of neuroblastoma. Cancer Res (2007) 67(19):9435–42. doi: 10.1158/0008-5472.CAN-07-1316
61. Teitz T, Lahti JM, Kidd VJ. Aggressive childhood neuroblastomas do not express caspase-8: an important component of programmed cell death. J Mol Med (Berl) (2001) 79(8):428–36. doi: 10.1007/s001090100233
62. Takita J, Yang HW, Chen YY, Hanada R, Yamamoto K, Teitz T, et al. Allelic imbalance on chromosome 2q and alterations of the caspase 8 gene in neuroblastoma. Oncogene (2001) 20(32):4424–32. doi: 10.1038/sj.onc.1204521
63. Cui H, Li T, Ding HF. Linking of n-myc to death receptor machinery in neuroblastoma cells. J Biol Chem (2005) 280(10):9474–81. doi: 10.1074/jbc.M410450200
64. Stupack DG, Teitz T, Potter MD, Mikolon D, Houghton PJ, Kidd VJ, et al. Potentiation of neuroblastoma metastasis by loss of caspase-8. Nature (2006) 439(7072):95–9. doi: 10.1038/nature04323
65. Pinto NR, Applebaum MA, Volchenboum SL, Matthay KK, London WB, Ambros PF, et al. Advances in risk classification and treatment strategies for neuroblastoma. J Clin Oncol (2015) 33(27):3008–17. doi: 10.1200/JCO.2014.59.4648
66. Yu AL, Gilman AL, Ozkaynak MF, London WB, Kreissman SG, Chen HX, et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J Med (2010) 363(14):1324–34. doi: 10.1056/NEJMoa0911123
67. Richards RM, Sotillo E, Majzner RG. CAR T cell therapy for neuroblastoma. Front Immunol (2018) 9:2380. doi: 10.3389/fimmu.2018.02380
68. Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer (2002) 2(6):442–54. doi: 10.1038/nrc822
69. Banyard J, Bielenberg DR. The role of EMT and MET in cancer dissemination. Connect Tissue Res (2015) 56(5):403–13. doi: 10.3109/03008207.2015.1060970
70. Teitz T, Stanke JJ, Federico S, Bradley CL, Brennan R, Zhang J, et al. Preclinical models for neuroblastoma: establishing a baseline for treatment. PloS One (2011) 6(4):e19133. doi: 10.1371/journal.pone.0019133
71. Delloye-Bourgeois C, Bertin L, Thoinet K, Jarrosson L, Kindbeiter K, Buffet T, et al. Microenvironment-driven shift of Cohesion/Detachment balance within tumors induces a switch toward metastasis in neuroblastoma. Cancer Cell (2017) 32(4):427–443.e428. doi: 10.1016/j.ccell.2017.09.006
72. Dankert EN, Look AT, Zhu S. Unraveling neuroblastoma pathogenesis with the zebrafish. Cell Cycle (2018) 17(4):395–6. doi: 10.1080/15384101.2017.1414683
Keywords: MYCN, oncogene, poor prognosis, metastasis, neuroblastoma
Citation: Bhavsar SP (2023) Metastasis in neuroblastoma: the MYCN question. Front. Oncol. 13:1196861. doi: 10.3389/fonc.2023.1196861
Received: 30 March 2023; Accepted: 08 May 2023;
Published: 18 May 2023.
Edited by:
Eugenia Broude, University of South Carolina, United StatesReviewed by:
Luz Jubierre Zapater, Memorial Sloan Kettering Cancer Center, United StatesCopyright © 2023 Bhavsar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Swapnil Parashram Bhavsar, swapnil.p.bhavsar@uit.no
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.