 Paloma Tejera Nevado
Paloma Tejera Nevado André Fehr
André Fehr Mattias K. Andersson
Mattias K. Andersson- Sahlgrenska Center for Cancer Research, Department of Pathology, University of Gothenburg, Gothenburg, Sweden
T-cell acute lymphoblastic leukemia (T-ALL) is an aggressive hematologic malignancy with poor prognosis. The MYB oncogene encodes a master transcription factor that is activated in the majority of human T-ALLs. In the present study, we have performed a large-scale screening with small-molecule drugs to find clinically useful inhibitors of MYB gene expression in T-ALL. We identified several pharmacological agents that potentially could be used to treat MYB-driven malignancies. In particular, treatment with the synthetic oleanane triterpenoids (OTs) bardoxolone methyl and omaveloxolone decreased MYB gene activity and expression of MYB downstream target genes in T-ALL cells with constitutive MYB gene activation. Notably, treatment with bardoxolone methyl and omaveloxolone led to a dose-dependent reduction in cell viability and induction of apoptosis at low nanomolar concentrations. In contrast, normal bone marrow-derived cells were unaffected at these concentrations. Bardoxolone methyl and omaveloxolone treatment downregulated the expression of DNA repair genes and sensitized T-ALL cells to doxorubicin, a drug that is part of the standard therapy of T-ALL. OT treatment may thus potentiate DNA-damaging chemotherapy through attenuation of DNA repair. Taken together, our results indicate that synthetic OTs may be useful in the treatment of T-ALL and potentially also in other MYB-driven malignancies.
1 Introduction
T-cell acute lymphoblastic leukemia (T-ALL) is an aggressive hematologic malignancy characterized by abnormal expansion of transformed lymphoid progenitor cells in the bone marrow, blood, and extramedullary sites (1, 2). It accounts for approximately 25% of ALL cases in adults and 10-15% of cases in children. Even though risk-based stratification and dose-intensification therapy have led to marked improvements in survival of pediatric patients, the prognosis for adults remains poor, particularly for older patients (2). Less than half of adults diagnosed with T-ALL survive more than five years (3). Thus, development of improved therapies for this disease is needed.
The MYB oncogene is activated in the majority of T-ALLs (4, 5). It encodes a pioneer master transcription factor that binds to super-enhancers and regulates chromatin accessibility and genes involved in cell division and maturation (5, 6). In T-ALL, MYB gene activation leads to blocked differentiation and increased proliferation of neoplastic lymphoid progenitor cells, suggesting that the gene is a potential therapeutic target in this disease (4, 7). The MYB gene is activated by various molecular mechanisms in T-ALL. MYB is activated by the TAL1 complex that is overexpressed in up to 60% of T-ALL (8) and is a downstream target of MLL-fusions (9). In 10-15% of T-ALLs, MYB is activated by gene duplication or gene fusion (7, 10, 11). Furthermore, potentially activating hot spot mutations in the 5´-part of MYB were recently described in T-ALL (11). We and others have previously shown that the MYB protein and its downstream effectors can be successfully targeted in acute myeloid leukemia (AML) (12–24). However, very few MYB inhibitors with therapeutic effects in T-ALL have been identified (25). Given that MYB is an oncogenic driver in the majority of T-ALLs and primarily regulated at the transcriptional level (26), we decided to perform a large-scale drug screening with small-molecule agents to identify inhibitors of MYB gene expression. We hypothesized that suppression of MYB might improve the treatment of patients with this aggressive disease that are refractory to the conventional therapies currently used. We now show that synthetic oleanane triterpenoids both inhibit MYB expression and sensitize T-ALL cells to chemotherapy.
2 Materials and methods
2.1 Cell culture
The T-ALL cell lines (27–29) MOLT-4 (ATCC no. CRL-1582), CCRF-CEM (ATCC no. CCL-119), P12-ICHIKAWA (DSMZ no. ACC-34), and RPMI-8402 (DSMZ no. ACC-290) were obtained from ATCC (Manassas, VA, USA) and DSMZ (Braunschweig, Germany). The cell lines have MYB-activation by gene duplication (7, 10). Cell identity was confirmed by STR-analysis and all cell lines were shown to be mycoplasma free prior to experiments. T-ALL cells were maintained in RPMI-1640 medium with GlutaMAX, 10% or 20% FBS, and 1% penicillin-streptomycin (Thermo Fisher Scientific, Waltham, MA, USA). Human mononuclear cells (Merck, Darmstadt, Germany) isolated from a healthy donor (HMCs) were maintained in Mononuclear cell medium (Merck) according to the instructions of the supplier. All cells were kept in a humidified incubator at 37°C and 5% CO2.
2.2 Chemical library, drug screening, and validation experiments
The L1100 Inhibitor Library, bardoxolone methyl (CDDO-Me, RTA 402), omaveloxolone (RTA 408), and doxorubicin were purchased from Selleck Chemicals (Houston, TX, USA). The IKK inhibitor VII and IKK-2 inhibitor IV were obtained from Merck. The KI696 peptide (30) was a kind gift from Dr. Volkan Sayin. Drugs were kept at -80°C or -20°C in stock solutions of 10 µM in DMSO and diluted to working concentrations in growth medium prior to experiments. For drug screening, 4,000 MOLT-4 or CCRF-CEM cells were seeded in V-bottom 96-well plates (Greiner Bio-One, Kremsmünster, Austria) and after 24 h treated with 1 µM of 768 small-molecule inhibitors from the Inhibitor Library (Supplementary Table S1). Cells were incubated with drugs, or DMSO as control, for 48 h whereafter they were washed with PBS and harvested using direct lysis (31) in 100 µl of 1 mg/ml BSA (Thermo Fisher Scientific). The screening was performed three times and cell plates were stored at -80°C until further analysis. For validation experiments, 2-2.5x105 T-ALL cells were seeded in 24-well plates (RNA analysis) and 4x105 cells were seeded in 6-well plates (protein analysis) and on the next day treated for 24 h with drugs or equal volumes of DMSO.
2.3 RNA isolation and cDNA synthesis
Total RNA was isolated using the RNeasy Micro-kit (Qiagen, Hilden, Germany) following the protocol of the manufacturer. RNA purity and concentration was measured with the NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific). Reverse transcription was performed with the iScript cDNA synthesis kit (Bio-Rad, Hercules, CA, USA) according to instructions of the manufacturer. For drug screening, 2 µl of cell lysate were used in 10 µl cDNA reactions.
2.4 Western blot
Total protein was isolated using RIPA buffer supplemented with the Halt protease and phosphatase inhibitor (Thermo Fisher Scientific). Protein concentrations were measured with the DC protein assay (Bio-Rad). Protein expression was analyzed by Western blotting using the NuPAGE system (Thermo Fisher Scientific) with monoclonal antibodies to MYB (clone 1-1; Merck Millipore) and beta-actin (ab8226, Abcam, Cambridge, UK). Bands were visualized with horseradish peroxidase-conjugated secondary antibodies and chemiluminescent detection with the Supersignal West Femto Max Sensitivity Substrate (Thermo Fisher Scientific). Blots were scanned with an Amersham ImageQuant 800 imaging system (Cytiva, Marlborough, MA, USA). MYB protein expression was quantified using ImageJ v2.0.0-rc-43 and normalized with beta-actin expression.
2.5 Quantitative real-time PCR (qPCR)
qPCR was done with the AB 7500 Fast Real-Time PCR system using TaqMan gene expression assays (Thermo Fisher Scientific) for MYB (Hs00920556_m1), CD34 (Hs00156373_m1), CCNB1 (Hs00259126_m1), MCM4 (Hs00381533_m1), CDK1 (Hs00364293_m1), and the reference genes UBC (Hs01871556_s1) and 18S (Hs99999901_s1). Relative gene expression was calculated by the ddCT method (32).
2.6 RNA-sequencing (RNA-seq)
RNA-seq was performed at Eurofins Genomics (Luxembourg City, Luxembourg) using the INVIEW Transcriptome Discover service. Library preparation was done with poly A enrichment, random primed strand-specific cDNA synthesis, adapter ligation, and adapter-specific PCR amplification. Libraries were subjected to Illumina paired-end sequencing (2x150 bps) with at least 30 million read pairs generated. Gene counts were extracted from FastQ files on the Uppsala Multidisciplinary Center for Advanced Computational Science (UPPMAX) cluster with FastQC/0.11.5, STAR v2.5.3a, Samtools v1.5, and FeatureCounts (Subread v1.5.2). Samples were analyzed with unsupervised hierarchical clustering using the hclust function in R and heatmaps were generated using R or Morpheus (Broad Institute). Differential gene expression was analyzed with DESeq2 v1.22.2. Gene ontology analysis was performed with ToppGene (33) and Gene set enrichment analysis (GSEA) (34) was done with GSEA v4.2.2 (Broad Institute).
2.7 Cell viability, cell cycle and apoptosis assays
For cell viability assays, 2,000 T-ALL cells were seeded in black 96-well plates (BD, Franklin Lakes, NJ, USA) and after 24 h treated with single or combinations of drugs. The AlamarBlue reagent (Thermo Fisher Scientific) was added to wells after 72 h and plates were analyzed with the VICTOR3 multilabel plate reader (PerkinElmer, Waltham, MA, USA). For apoptosis assays, 2,000 cells were seeded in Corning white 96-well plates (Corning, New York, USA) and treated with drugs after 24 h. The Caspase-Glo 3/7 Assay (Promega, Madison, WI, USA) was used to estimate apoptosis and luminescent signals were quantified on the VICTOR3. For cell cycle analysis, 8x105 MOLT-4 cells were seeded in 6-well plates and on the next day treated for 24 h with drugs. Cells were supplemented with 10 μM BrdU during the last 4 h and then fixed and stained using the FITC BrdU Flow kit (BD). Stained cells were analyzed with an Accuri C6 flow cytometer (BD). All experiments were performed three times.
2.8 siRNA experiments
MYB siRNA-mediated knockdown was done with the Cell Line Nucleofector Kit L on the Amaxa Nucleofector II device (Cologne, Germany) using 2x106 MOLT-4 cells according to the instructions provided by the manufacturer. Cells were electroporated with 1 µM Silencer Select MYB (s9109, s9110) or negative control siRNAs (Thermo Fisher Scientific). Total RNA and protein were isolated from transfected cells after 48 h. All experiments were done three times.
2.9 Statistical analysis
Unsupervised hierarchical clustering was done with R and overlap of differentially expressed genes was visualized with BioVenn (https://www.biovenn.nl) and analyzed with chi-square tests. Drug kernel density estimation plots were generated with the density function of the stats package of R (https://www.r-project.org). Correlation of treatment effects between T-ALL cell lines was estimated by Pearson correlation. Dose-response curves were generated and analyzed by nonlinear regression in Prism 9 (GraphPad Software, San Diego, CA, USA); IC25 and IC50 were defined as drug concentrations that reduced cell viability by 25% and 50%, respectively. For combination treatments, the definition of drug additivity and synergy was according to Bliss (35). Differences between experimentally measured effects of drug combinations and the expected effects calculated from single drug treatments were analyzed using independent samples t-test. Bar graph values are presented as mean ± SEM. Differences between groups in qPCR, cell cycle, and apoptosis analyses were evaluated with one-way ANOVA or independent samples t-test. All statistical tests were two-sided. A P-value of less than 0.05 was considered statistically significant.
3 Results
3.1 Large-scale drug screening identifies synthetic oleanane triterpenoids as inhibitors of MYB gene expression in T-ALL cells
We performed a large-scale in vitro screening of two T-ALL cell lines, CCRF-CEM and MOLT-4, with MYB-activation using 768 small-molecule drugs from the L1100 Inhibitor Library (Figure 1, Supplementary Table S1). Cells were treated with 1 μM of inhibitors for 48 h after which relative MYB mRNA levels were analyzed by qPCR. Density plots of MYB gene expression following drug treatments showed similar distributions in three independent experiments for each cell line (Figure 1A). There was a significant (P < 0.0001) correlation between drug effects in the two cell lines (Figure 1B). A larger number of drugs reduced MYB mRNA levels in CCRF-CEM cells compared with MOLT-4 cells (Figure 1A). Further analysis showed that 46 drugs caused a greater than 50% decrease in MYB gene expression in both cases (Figure 1C). These agents included histone-deacetylase inhibitors (HDACi), intracellular kinase (IK) inhibitors, receptor tyrosine kinase (RTK) inhibitors, and other drugs with diverse targets (Figure 1D, Supplementary Table S2). Among these were drugs that were previously identified as inhibitors of MYB gene expression (AT7519, flavopiridol, givinostat, and vorinostat) (36–39) as well as an inhibitor of MYB protein activity (celastrol) (12). To validate our drug screen, we treated MOLT-4 cells with two of the identified HDACi (givinostat and panobinostat) and two of the cyclin-dependent kinase inhibitors (CDKi) (AT7519 and SNS-032) from the screen. All tested drugs significantly decreased MYB mRNA levels at nanomolar concentrations (Supplementary Figure S1). We next used hierarchical clustering and heat map analysis of MYB mRNA levels to investigate the potency of the inhibitors of MYB gene expression identified in our screen (Figure 1E). These analyses identified bardoxolone methyl and omaveloxolone, two related oleanane triterpenoids (OTs), as top hits in the screen.
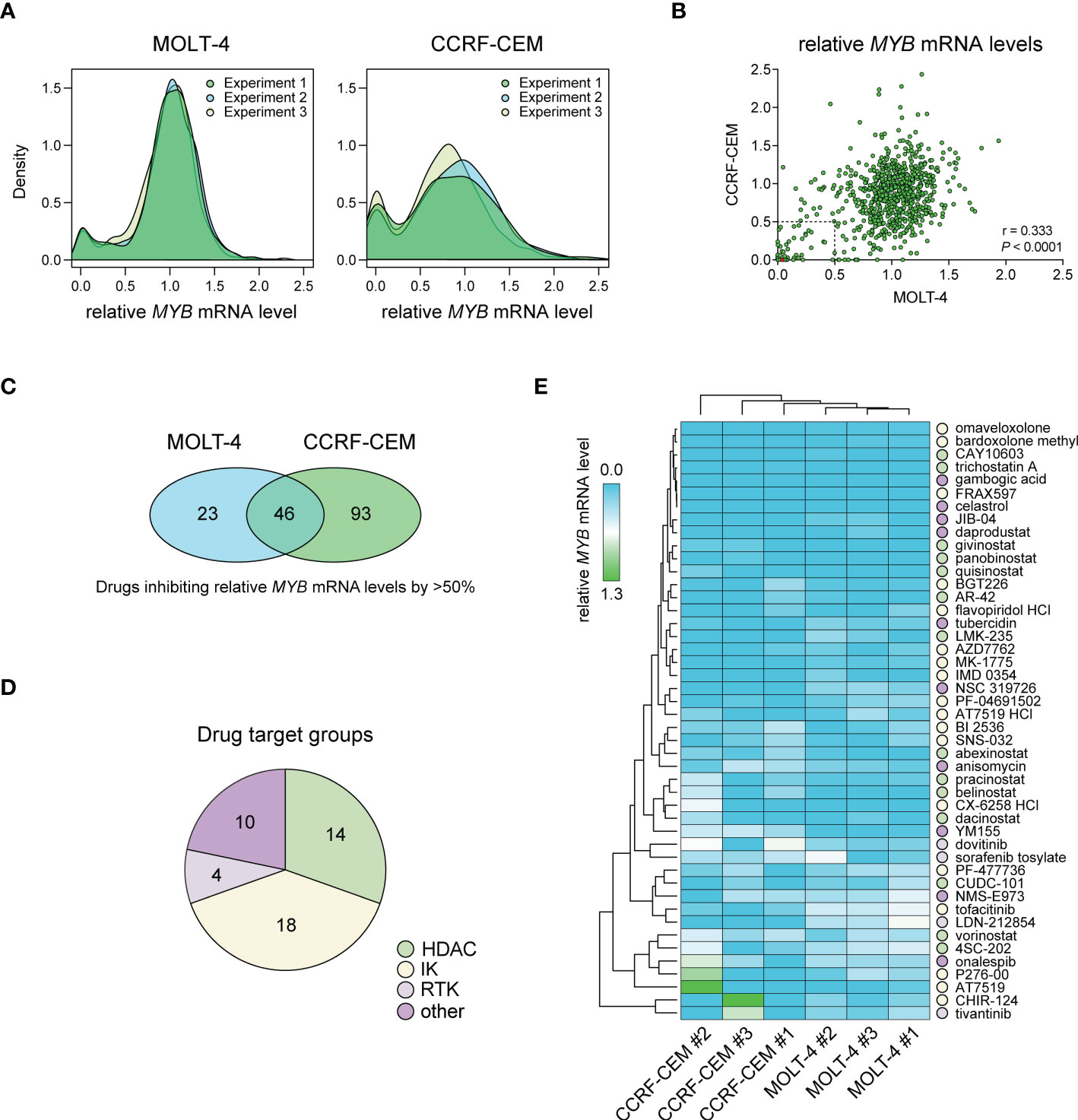
Figure 1 Drug screening for inhibitors of MYB gene expression in T-ALL cells with MYB gene activation. (A) Density plots showing treatment effects of 768 small-molecule inhibitors on MYB gene expression in two T-ALL cell lines. Data is derived from three independent experiments. (B) Pearson correlation analysis of treatment response for 768 inhibitors in two T-ALL cell lines. Dashed lines indicate thresholds for 50% MYB mRNA expression compared with DMSO treated control cells. The red dot shows bardoxolone methyl and omaveloxolone that had near identical effects on MYB expression in the screen. (C) Venn diagram showing the number of drugs reducing MYB gene expression with more than 50% in T-ALL cells. (D) Drug target groups of 46 inhibitors reducing MYB mRNA levels with more than 50% in two T-ALL cell lines. (E) Heat map and hierarchical clustering analyses showing the effects of 46 inhibitors on MYB gene expression. Drug targets with grouping as in D are indicated. IK, intracellular kinase; RTK, receptor tyrosine kinase; HDAC, histone deacetylase.
3.2 Treatment of T-ALL cells with OTs leads to downregulation of MYB expression and downstream target genes
To confirm the effects of OT treatment on MYB gene expression in T-ALL cells, we treated MOLT-4, CCRF-CEM, P12-ICHIKAWA, and RPMI-8402 cells with nanomolar concentrations of bardoxolone methyl or omaveloxolone for 24 h. This treatment led to a dose-dependent decrease in MYB mRNA levels in all T-ALL cell lines tested (Figure 2A, Supplementary Figure S2). A corresponding reduction in MYB protein levels after OT treatment was confirmed by immunoblotting (Figure 2B, Supplementary Figure S3A). To investigate the biological consequences of MYB suppression in OT treated cells, we analyzed the expression of known MYB target genes involved in cell cycle regulation and leukemic stem cell function. The expression of CCNB1, CDK1, MCM4, and CD34 were significantly downregulated in MOLT-4 cells with OT treatment (Figure 2C). To verify that these genes are regulated by MYB in T-ALL cells, we knocked down MYB mRNA and protein levels in MOLT-4 cells using siRNAs (Figures 2D, E, Supplementary Figure S3B). Analogous to treatment with OTs, transfection with MYB siRNAs resulted in downregulation of CCNB1, CDK1, MCM4, and CD34 (Figure 2F). Our results indicate that these genes are MYB targets in T-ALL that are downregulated following OT treatment.
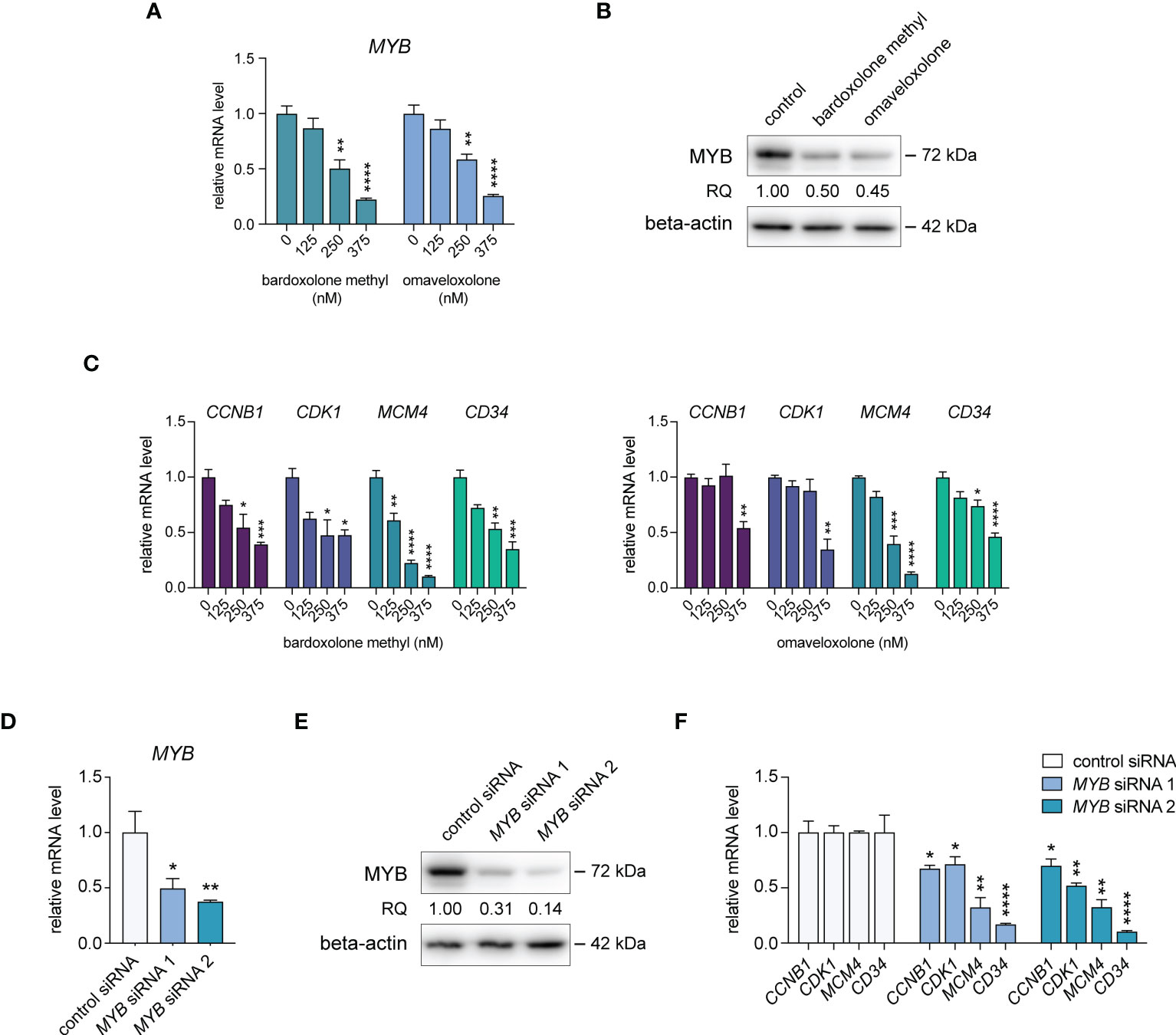
Figure 2 Treatment with oleanane triterpenoids (OTs) leads to decreased expression of MYB and its downstream target genes. (A) MYB mRNA expression in MOLT-4 cells treated with bardoxolone methyl and omaveloxolone for 24 h. (B) MYB protein expression in MOLT-4 cells treated for 24 h with 250 nM OTs. (C) Expression of MYB downstream target genes in MOLT-4 cells treated as in (A, D) Knockdown of MYB mRNA in MOLT-4 cells after treatment with 1 µM of MYB siRNAs for 48 h. (E) MYB protein expression in MOLT-4 cells treated with MYB siRNAs as in (D, F) Expression of MYB downstream target genes after MYB knockdown as in D and (E) Error bars indicate standard error of the mean. One-way ANOVA test (* P < 0.05; ** P < 0.01; *** P < 0.001; **** P < 0.0001). Data shown represents one of three independent experiments. RQ, relative quantification of MYB protein levels normalized with beta-actin expression.
Both bardoxolone methyl and omaveloxolone inhibit NF-κB (40, 41), a signaling pathway that has been shown to positively regulate MYB transcription in murine erythroleukemia cells (42, 43). To test whether NF-κB signaling is involved in MYB gene regulation also in human T-ALL cells, we treated MOLT-4 cells with two IKK-1 and IKK-2 inhibitors and measured MYB mRNA levels by qPCR (Supplementary Figure S4A). Neither of these inhibitors decreased MYB gene expression, which may indicate that OTs downregulate MYB gene activity independently of NF-κB signaling in T-ALL cells. Similarly, treatment with the cell-penetrating NRF2-activating peptide KI696 (30, 44) did not affect MYB gene expression in T-ALL cells (Supplementary Figure S4B), suggesting that OTs target other pathways than NF-κB and NRF2 to regulate the MYB gene in T-ALL cells.
3.3 RNA-seq reveals molecular signatures associated with a reversed MYB-driven transcriptional program and induction of apoptosis in OT treated T-ALL cells
To study the effects of OT treatment on critical cellular processes in T-ALL cells, we performed RNA-seq analysis of MOLT-4 cells treated with 375 nM bardoxolone methyl or omaveloxolone for 24 h. Global gene expression analysis showed that OT treated cells clustered separately from control cells (Figure 3A). There was a significant overlap of differentially regulated genes between the two drugs (Figures 3A–C). To investigate if OT treatment affects the MYB-driven transcriptional program in T-ALL cells, we used GSEA to compare our RNA-seq data with a previously published dataset (45) of direct MYB target genes in hematopoietic progenitor cells (Figures 3D, E). OT treatment downregulated genes that are normally activated by MYB (e.g. MYC, TCF7, MAT2A) and upregulated known MYB-repressed genes (e.g. CEBPB, IL17RA, BCL6). Thus, our findings indicate that treatment with bardoxolone methyl and omaveloxolone can reverse the MYB-driven transcriptional program in T-ALL cells. GSEA also revealed activation of the p53 and apoptosis pathways in OT treated cells as well as decreased expression of genes regulating cell cycle progression, metabolism, and DNA repair (Figures 4A, B). Moreover, gene ontology analysis showed that genes upregulated by OT treatment are involved in cell differentiation and cell morphogenesis (Figure 4C) whereas genes downregulated are involved in ribosome biogenesis, RNA processing/metabolism, and nucleotide synthesis (Figure 4D). These results imply impaired synthesis of nucleic acids, RNA maturation, and translation in OT treated cells. Taken together, our RNA-seq data indicate that OT treatment reverses a MYB-driven gene expression program, inhibits cell proliferation, and induces differentiation and apoptosis in T-ALL cells.
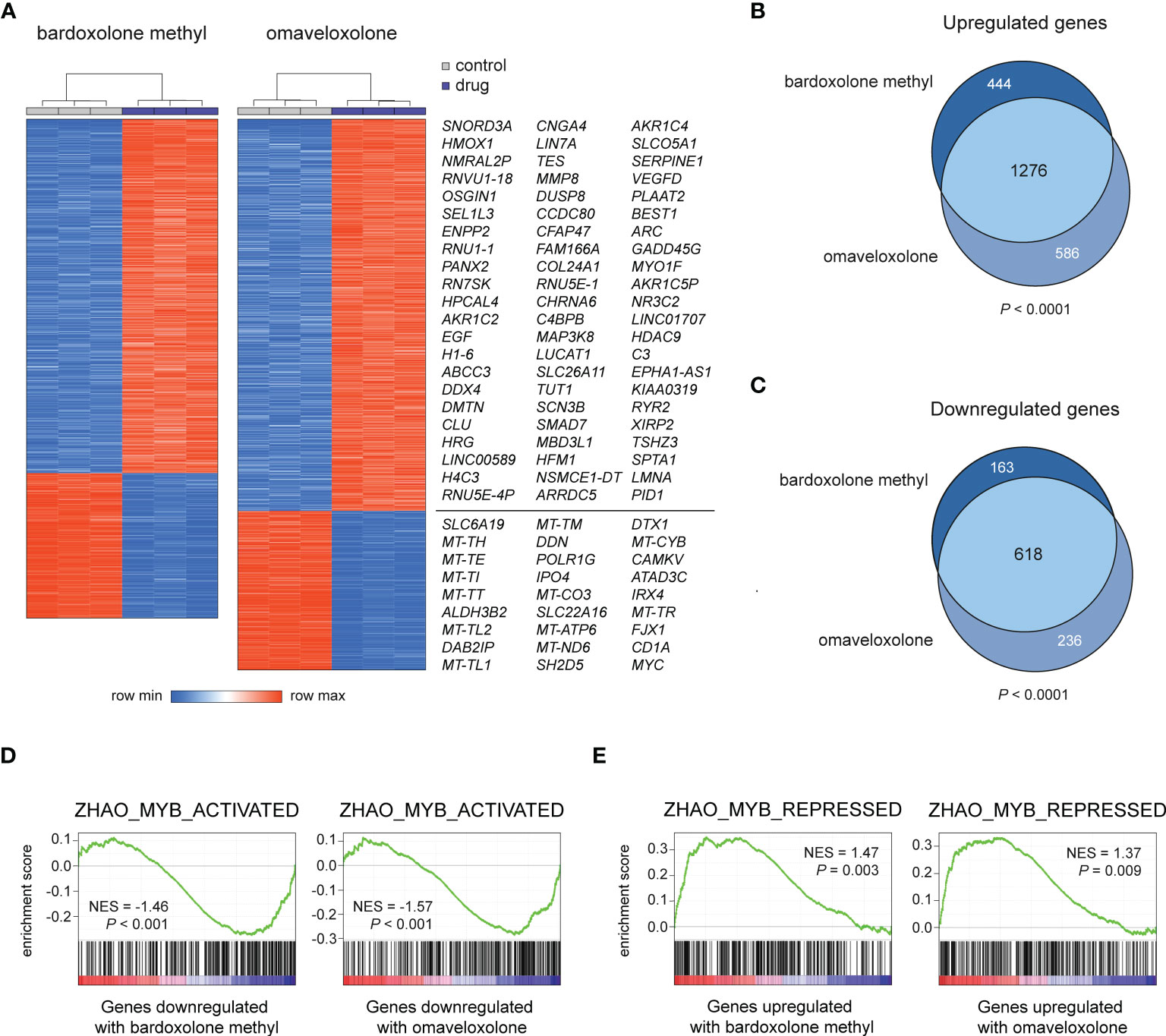
Figure 3 RNA-seq analysis of OT treated T-ALL cells. (A) Hierarchical clustering and heatmap analysis of global gene expression data from MOLT-4 cells treated for 24 h with 375 nM OTs. The most up- and downregulated genes by both drugs are shown to the right. (B, C) Venn diagrams of up- and downregulated genes in OT treated T-ALL cells. The significance of overlap was estimated by chi-square tests. (D, E) Gene set enrichment analysis of MYB-activated and repressed genes in OT treated T-ALL cells. NES, normalized enrichment score.
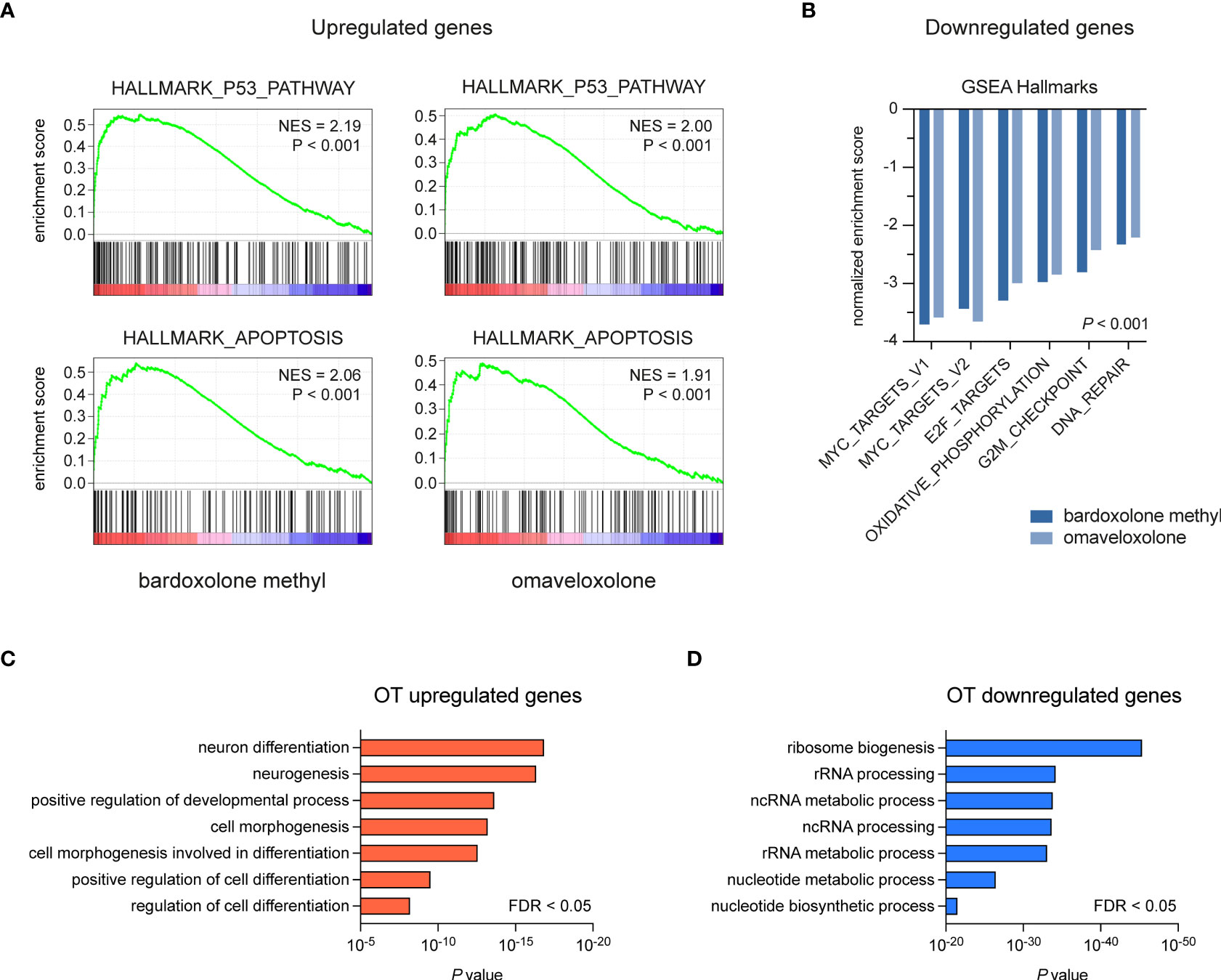
Figure 4 Gene set enrichment and gene ontology analysis of OT treated T-ALL cells. (A, B) Gene set enrichment analysis of pathways and gene sets affected by OT treatment in MOLT-4 cells. (C, D) Gene ontology analysis of up- and downregulated genes following OT treatment. NES, normalized enrichment score. FDR, false-discovery rate.
3.4 Treatment of MYB-activated T-ALL cells with OTs results in apoptosis and decreased cell proliferation
To investigate if T-ALL cell viability is affected by treatment with OTs, we treated T-ALL cell lines derived from four cases with 50-1000 nM of bardoxolone methyl or omaveloxolone for 72 h (Figure 5A). The treatments resulted in a dose-dependent decrease in cell viability at nanomolar concentrations. There was also a significant induction of apoptosis in T-ALL cells after treatment with both drugs (Figure 5B). Importantly, HMC control cells were unaffected at low nanomolar concentrations and showed markedly less apoptosis at 500 nM compared with T-ALL cells. Cell cycle analysis showed that OT treated T-ALL cells displayed a marked G2/M arrest (Figure 5C). This was in agreement with both OT treatment and MYB knockdown causing a decreased expression of the G2 regulating genes CCNB1 and CDK1 (Figures 2C, F). OT treated cells also displayed a reduced entry into the S phase and an increased accumulation of apoptotic cells in sub G1 (Figure 5C). These results, which corroborate our findings from the global gene expression analysis (Figures 3, 4), show that T-ALL cells are sensitive to OT treatment.
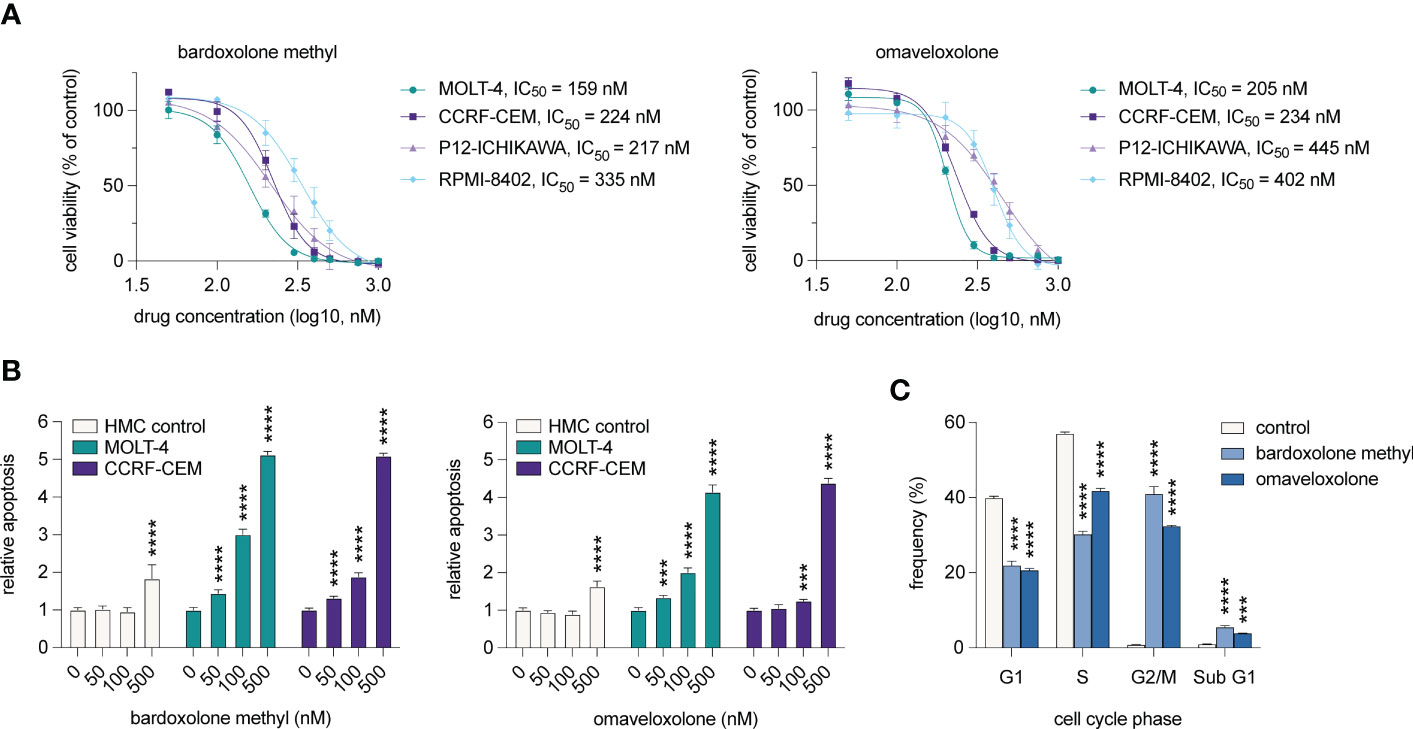
Figure 5 OT treatment leads to G2/M arrest, decreased cell viability, and induction of apoptosis in T-ALL cells. (A) Cell viability of T-ALL cells treated with OTs for 72 h. Data points represent the average of three independent experiments. Error bars indicate standard error of the mean. IC50 is defined as the drug concentration reducing cell viability by 50%. (B) Apoptosis as measured by caspase 3/7 activity after 48 h of treatment with OTs. (C) Cell cycle distribution of MOLT-4 cells after 24 h treatment with 250 nM OTs. Error bars indicate standard error of the mean. One-way ANOVA test (*** P < 0.001; **** P < 0.0001). Data shown represents one of three independent experiments. HMC, human mononuclear cells isolated from a healthy donor.
3.5 OT treatment sensitizes T-ALL cells to chemotherapy
Since genes involved in DNA repair were downregulated following OT treatment (Figure 4B), we tested whether the OTs could synergize with the DNA-damaging agent doxorubicin that is part of the standard therapy of T-ALL. Treatments with bardoxolone methyl or omaveloxolone combined with doxorubicin at IC25 and IC50 concentrations (Figure 5A, Supplementary Figure S5) resulted in a significant synergistic decrease in cell viability at both concentrations in MOLT-4 cells (Figure 6). These results suggest that synthetic OTs may sensitize T-ALL cells to chemotherapy.
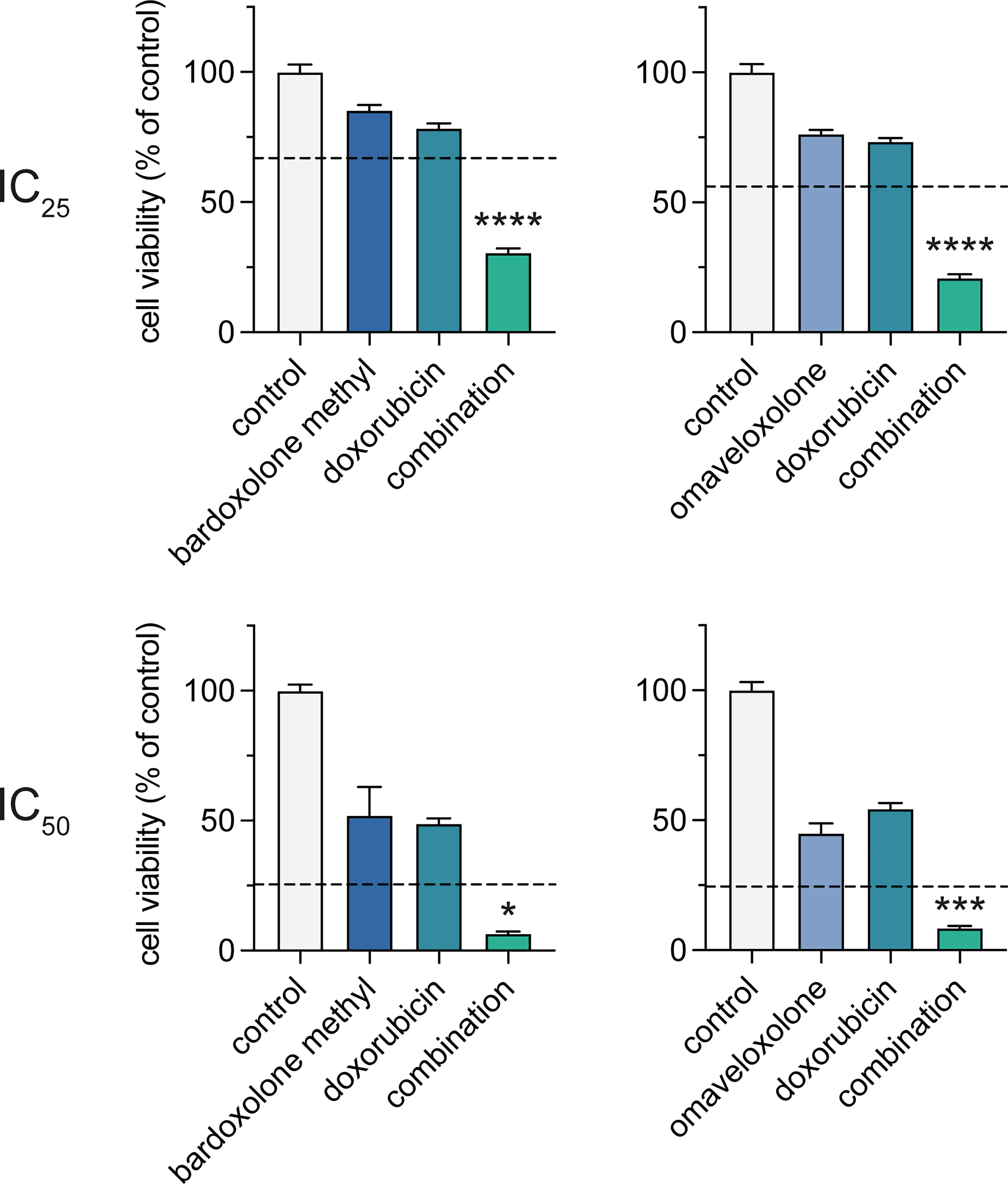
Figure 6 Combined treatment with OTs and doxorubicin leads to a synergistic reduction in cell viability of T-ALL cells. Cell viability of MOLT-4 cells after treatment with IC25 and IC50 concentrations of bardoxolone methyl or omaveloxolone combined with doxorubicin for 72 h. The expected additive effects of combination treatments, as predicated by Bliss interaction, are indicated with dashed lines. Independent samples t-test (* P < 0.05; *** P < 0.001; **** P < 0.0001). Error bars indicate standard error of the mean. Data shown represents one of three independent experiments.
4 Discussion
The MYB oncogene is an oncogenic driver in the majority of leukemias and encodes a master transcription factor with critical roles in cell proliferation, cell survival, and leukemic stem cell maintenance (4). To find novel inhibitors of MYB gene expression, we have now performed a large-scale screening to identify drugs with inhibitory effects on MYB mRNA expression in T-ALL cells with constitutive MYB gene activation.
Notably, the most potent inhibitors of MYB gene expression in our drug screen were bardoxolone methyl and omaveloxolone, two structurally related synthetic OTs that inhibit NF-κB and activate NRF2 signaling (46, 47). Treatment with bardoxolone methyl and omaveloxolone led to downregulation of MYB mRNA and protein levels and decreased expression of MYB downstream target genes in a dose-dependent manner. The OTs also negatively affected cell proliferation and viability of T-ALL cells through induction of a marked G2/M arrest, apoptosis, and differentiation. Since both NF-κB pathway inhibition and NRF2 activation failed to downregulate MYB gene expression in T-ALL cells, this suggests that the two OT drugs act through alternative mechanisms to inhibit MYB activity. Our results indicate that the OTs inhibit one or more positive regulators of MYB gene expression in T-ALL cells, possibly independent of NF-κB and NRF2-signaling.
OTs are natural compounds present in a large number of dietary and medicinal plants (48). Synthetic OTs such as bardoxolone methyl and omaveloxolone are generated by chemical modification of oleanolic acid and have anti-inflammatory and anticarcinogenic properties. We have now, for the first time, identified members of this group of compounds as potent inhibitors of MYB gene expression. OTs mediate their pharmacological effects in part by interaction with sulfhydryl groups of cysteine residues through Michael addition, yet do not bind non-discriminately to all cysteine-containing proteins (49). Instead, OT binding is markedly influenced by the accessibility of the drug to a specific cysteine residue and the redox potential of the cell. Thus, individual OTs are expected to have different target proteins depending on the cellular context. In addition to targeting the NF-κB and NRF2 pathways, synthetic OTs negatively affect proliferation by inhibiting specific proteins involved in the JAK/STAT and PI3K/AKT/mTOR signaling pathways (49). These pathways may regulate MYB expression in T-ALL cells as several JAK/STAT and PI3K/AKT/mTOR inhibitors were among the top hits in our drug screen. Moreover, mTOR signaling has previously been shown to regulate MYB expression in normal human T-cells (50). Previous studies have shown that OTs also may induce cell differentiation (49). Similarly, we found that genes involved in positive regulation of differentiation were upregulated in T-ALL cells following treatment with bardoxolone methyl or omaveloxolone. Since the hematopoietic progenitor marker CD34 (51) was downregulated by both OTs and MYB knockdown, this suggests that the differentiation seen following OT treatment is, at least partly, mediated by decreased MYB expression in T-ALL cells. This conclusion is further supported by studies showing that MYB expression is high in immature and progenitor-like cells and suppress differentiation of both normal and leukemic cells (7, 26).
Bardoxolone methyl is in late clinical stage development for treatment of chronic kidney disease and omaveloxolone recently received FDA approval for treatment of Friedreich’s ataxia. These drugs are currently used in several active and completed clinical trials in which they have demonstrated acceptable adverse effects (https://clinicaltrials.gov). In the present study, the two OTs were active in the low nanomolar range in T-ALL cells whereas normal bone marrow-derived cells were unaffected at these concentrations. Thus, there may be a therapeutic window for these drugs to be repurposed for use in T-ALL patients with acceptable side-effects. Notably, both bardoxolone methyl and omaveloxolone showed a synergistic negative effect on T-ALL cell viability when used in combination with the cytotoxic DNA-damaging agent doxorubicin, which is part of the standard therapy of T-ALL. Since the expression of DNA repair genes was attenuated following treatment, suppression of DNA repair may be part of the underlying mechanism that sensitizes T-ALL cells to combined treatment with OTs and doxorubicin. However, further studies are needed to determine the association between OT treatment, DNA repair, and sensitivity to chemotherapy in T-ALL. It would also be interesting to study the effects of OTs in combination with other drugs such as for example angiogenesis or NOTCH inhibitors (52, 53). Taken together, our studies suggest that bardoxolone methyl and omaveloxolone are new potential drugs for treatment of T-ALL.
Other drugs that we identified with potent inhibitory effects on MYB gene expression included several HDACi (e.g. givinostat, panobinostat, quisinostat) and CDKi (e.g. AT7519, SNS-032, p276-00). Previous studies have shown that HDACi (e.g. givinostat and vorinostat, also identified in our screen) downregulate MYB gene expression in myeloproliferative neoplasms and myeloid leukemias (36, 39). Here, we identified HDACi as potent inhibitors of MYB gene expression also in lymphoid leukemia cells, suggesting a conserved mechanism of MYB gene regulation in neoplasms of both the myeloid and lymphoid lineages. Our findings, and those of others (36, 39, 54), thus support the involvement of chromatin remodeling in the regulation of the MYB gene. Notably, several CDK9 inhibitors had also inhibitory effects on MYB gene expression in our drug screen. The inhibitory effect on MYB gene activity by these inhibitors is most likely a result of blocked transcription, since MYB gene expression is known to be positively regulated by the transcriptional elongation factor P-TEFb (which includes CDK9) and is inhibited by CDKi (e.g. AT7519 and flavopiridol, also identified in our screen) in breast cancer cells (37, 38). Notably, the plant-derived triterpenoid celastrol was one of the top hits in our screen. Celastrol was previously reported as an inhibitor of MYB transcriptional activity in AML cells (12) and, like the OTs identified here, forms covalent Michael adducts with target proteins (55). This suggests similar mechanisms of MYB inhibition by celastrol, bardoxolone methyl, and omaveloxolone. In contrast to the synthetic OTs identified in our screen, celastrol is a natural compound that cause severe side effects, has low bioavailability and poor water solubility, which have hindered its clinical application (56). A number of other MYB inhibitors have been identified with therapeutic effects on AML cells (13–24). It would be interesting to test if these inhibitors also have anti-leukemic effects in MYB-driven T-ALL cells. Only one of these, mebendazole, has to our knowledge been tested and shown effects in T-ALL preclinical models (25). However, many of these inhibitors, unlike the OTs found in our study, are early-stage experimental drugs that have not been tested in humans or show poor pharmacokinetics and/or toxicity. Future studies will evaluate the potential of these inhibitors as new therapies for T-ALL.
Targeting the MYB oncogene in a clinical setting will require information about the MYB activation status. Since the mechanism of activation of MYB varies in T-ALL, analysis of the expression of MYB by RT-PCR, RNA-seq or immunohistochemistry are the preferable methods to determine whether the gene is activated or not (57). T-ALL patients with overexpression of MYB are thus candidates for MYB inhibitory treatments, whereas those with low or undetectable MYB levels are not. MYB expression analysis will be crucial for stratification of patients in clinical trials using MYB inhibitors.
A limitation of this study is the lack of testing of bardoxolone methyl and omaveloxolone in in vivo T-ALL models and patient primary cells. However, we could demonstrate that normal bone marrow-derived cells are largely unaffected by the drugs compared with T-ALL cells. This is in line with the fact that the drugs show low toxicity in clinical trials (49). Furthermore, the mechanism by which the OTs negatively affect MYB expression is unknown. We believe that the drugs may act by similar mechanisms as celastrol, which blocks MYB activity in AML (12), but they may also regulate MYB expression through inhibition of for example JAK/STAT and/or PI3K/AKT/mTOR signaling. OTs do not fit the single-target paradigm and has multiple molecular targets, yet they show low cytotoxicity in normal cells (49). This multifunctionality may be an advantage in order to potentiate treatment and to avoid drug resistance. In T-ALL, multiple driver events, in addition to MYB activation, is involved in the pathogenesis of the disease, including NOTCH1 mutations and loss of CDK2NA (11). This strongly implies that inhibition of MYB must be combined with targeting of additional pathways for successful treatment.
In conclusion, we have identified several novel suppressors of MYB gene expression by large-scale drug screening of cultured T-ALL cells with constitutive MYB gene activation. In particular, the structurally related OTs bardoxolone methyl and omaveloxolone demonstrated potent inhibitory effects on MYB gene expression and MYB downstream targets, and induced apoptosis in MYB gene activated T-ALL cells. Notably, these drugs sensitized T-ALL cells in culture to DNA-damaging chemotherapy. Our results indicate that both bardoxolone methyl and omaveloxolone may synergize with the standard treatment of T-ALL but also have the potential to improve current therapies of other MYB-driven malignancies.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/sra/PRJNA811771.
Author contributions
MA conceptualized, designed, and supervised the study. PN, TT, AA, AF, and MA performed experiments. PN, TT, AA, AF, GS, and MA analyzed data. GS and MA provided project resources. MA drafted the manuscript. All authors reviewed and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by the Swedish Childhood Cancer Fund (PR2020-0015), the Swedish Cancer Society (CAN 21 1773), Mary Béves Foundation for Child Cancer Research (2018-04), the Magnus Bergvall Foundation (2017-02214), and the AG-Fond (FB 17-93).
Acknowledgments
Resources for RNA-seq analysis were provided by SNIC through Uppsala Multidisciplinary Center for Advanced Computational Science (UPPMAX) under project SNIC 2021/22-661. The authors thank Dr. Volkan Sayin for the KI696 peptide.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2023.1126354/full#supplementary-material
References
1. Hunger SP, Mullighan CG. Acute lymphoblastic leukemia in children. N Engl J Med (2015) 373(16):1541–52. doi: 10.1056/NEJMra1400972
2. Terwilliger T, Abdul-Hay M. Acute lymphoblastic leukemia: A comprehensive review and 2017 update. Blood Cancer J (2017) 7(6):e577. doi: 10.1038/bcj.2017.53
3. Marks DI, Rowntree C. Management of adults with T-cell lymphoblastic leukemia. Blood (2017) 129(9):1134–42. doi: 10.1182/blood-2016-07-692608
4. Pattabiraman DR, Gonda TJ. Role and potential for therapeutic targeting of myb in leukemia. Leukemia (2013) 27(2):269–77. doi: 10.1038/leu.2012.225
5. Fuglerud BM, Lemma RB, Wanichawan P, Sundaram AYM, Eskeland R, Gabrielsen OS. A c-myb mutant causes deregulated differentiation due to impaired histone binding and abrogated pioneer factor function. Nucleic Acids Res (2017) 45(13):7681–96. doi: 10.1093/nar/gkx364
6. Mansour MR, Abraham BJ, Anders L, Berezovskaya A, Gutierrez A, Durbin AD, et al. Oncogene regulation. an oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science (2014) 346(6215):1373–7. doi: 10.1126/science.1259037
7. Lahortiga I, De Keersmaecker K, Van Vlierberghe P, Graux C, Cauwelier B, Lambert F, et al. Duplication of the myb oncogene in T cell acute lymphoblastic leukemia. Nat Genet (2007) 39(5):593–5. doi: 10.1038/ng2025
8. Sanda T, Lawton LN, Barrasa MI, Fan ZP, Kohlhammer H, Gutierrez A, et al. Core transcriptional regulatory circuit controlled by the Tal1 complex in human T cell acute lymphoblastic leukemia. Cancer Cell (2012) 22(2):209–21. doi: 10.1016/j.ccr.2012.06.007
9. Zuber J, Rappaport AR, Luo W, Wang E, Chen C, Vaseva AV, et al. An integrated approach to dissecting oncogene addiction implicates a myb-coordinated self-renewal program as essential for leukemia maintenance. Genes Dev (2011) 25(15):1628–40. doi: 10.1101/gad.17269211
10. Clappier E, Cuccuini W, Kalota A, Crinquette A, Cayuela JM, Dik WA, et al. The c-myb locus is involved in chromosomal translocation and genomic duplications in human T-cell acute leukemia (T-all), the translocation defining a new T-all subtype in very young children. Blood (2007) 110(4):1251–61. doi: 10.1182/blood-2006-12-064683
11. Liu Y, Easton J, Shao Y, Maciaszek J, Wang Z, Wilkinson MR, et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat Genet (2017) 49(8):1211–8. doi: 10.1038/ng.3909
12. Uttarkar S, Dasse E, Coulibaly A, Steinmann S, Jakobs A, Schomburg C, et al. Targeting acute myeloid leukemia with a small molecule inhibitor of the Myb/P300 interaction. Blood (2016) 127(9):1173–82. doi: 10.1182/blood-2015-09-668632
13. Ramaswamy K, Forbes L, Minuesa G, Gindin T, Brown F, Kharas MG, et al. Peptidomimetic blockade of myb in acute myeloid leukemia. Nat Commun (2018) 9(1):110. doi: 10.1038/s41467-017-02618-6
14. Walf-Vorderwulbecke V, Pearce K, Brooks T, Hubank M, van den Heuvel-Eibrink MM, Zwaan CM, et al. Targeting acute myeloid leukemia by drug-induced c-myb degradation. Leukemia (2018) 32(4):882–9. doi: 10.1038/leu.2017.317
15. Xu Y, Milazzo JP, Somerville TDD, Tarumoto Y, Huang YH, Ostrander EL, et al. A tfiid-saga perturbation that targets myb and suppresses acute myeloid leukemia. Cancer Cell (2018) 33(1):13–28 e8. doi: 10.1016/j.ccell.2017.12.002
16. Yusenko M, Jakobs A, Klempnauer KH. A novel cell-based screening assay for small-molecule myb inhibitors identifies podophyllotoxins teniposide and etoposide as inhibitors of myb activity. Sci Rep (2018) 8(1):13159. doi: 10.1038/s41598-018-31620-1
17. Yusenko MV, Trentmann A, Andersson MK, Ghani LA, Jakobs A, Arteaga Paz MF, et al. Monensin, a novel potent myb inhibitor, suppresses proliferation of acute myeloid leukemia and adenoid cystic carcinoma cells. Cancer Lett (2020) 479:61–70. doi: 10.1016/j.canlet.2020.01.039
18. Joy ST, Henley MJ, De Salle SN, Beyersdorf MS, Vock IW, Huldin AJL, et al. A dual-site inhibitor of Cbp/P300 kix is a selective and effective modulator of myb. J Am Chem Soc (2021) 143(37):15056–62. doi: 10.1021/jacs.1c04432
19. Yusenko MV, Biyanee A, Andersson MK, Radetzki S, von Kries JP, Stenman G, et al. Proteasome inhibitors suppress myb oncogenic activity in a P300-dependent manner. Cancer Lett (2021) 520:132–42. doi: 10.1016/j.canlet.2021.07.010
20. Yusenko MV, Biyanee A, Frank D, Kohler LHF, Andersson MK, Khandanpour C, et al. Bcr-tmp, a novel nanomolar-active compound that exhibits both myb- and microtubule-inhibitory activity. Cancers (Basel) (2021) 14(1):43. doi: 10.3390/cancers14010043
21. Yusenko MV, Trentmann A, Casolari DA, Abdel Ghani L, Lenz M, Horn M, et al. C/Ebpbeta is a myb- and P300-cooperating pro-leukemogenic factor and promising drug target in acute myeloid leukemia. Oncogene (2021) 40(29):4746–58. doi: 10.1038/s41388-021-01800-x
22. Biyanee A, Yusenko MV, Klempnauer KH. Src-family protein kinase inhibitors suppress myb activity in a P300-dependent manner. Cells (2022) 11(7):1162. doi: 10.3390/cells11071162
23. Clesham K, Walf-Vorderwulbecke V, Gasparoli L, Virely C, Cantilena S, Tsakaneli A, et al. Identification of a c-Myb-Directed therapeutic for acute myeloid leukemia. Leukemia (2022) 36(6):1541–9. doi: 10.1038/s41375-022-01554-9
24. Suetaka S, Oka Y, Kunihara T, Hayashi Y, Arai M. Rational design of a helical peptide inhibitor targeting c-Myb-Kix interaction. Sci Rep (2022) 12(1):816. doi: 10.1038/s41598-021-04497-w
25. Smith C, Touzart A, Simonin M, Tran-Quang C, Hypolite G, Latiri M, et al. Harnessing the myb-dependent Tal1 5'super-enhancer for targeted therapy in T-all. Mol Cancer (2023) 22(1):12. doi: 10.1186/s12943-022-01701-x
26. Ramsay RG, Gonda TJ. Myb function in normal and cancer cells. Nat Rev Cancer (2008) 8(7):523–34. doi: 10.1038/nrc2439
27. Foley GE, Lazarus H, Farber S, Uzman BG, Boone BA, McCarthy RE. Continuous culture of human lymphoblasts from peripheral blood of a child with acute leukemia. Cancer (1965) 18:522–9. doi: 10.1002/1097-0142(196504)18:4<522::aid-cncr2820180418>3.0.co;2-j
28. Huang CC, Hou Y, Woods LK, Moore GE, Minowada J. Cytogenetic study of human lymphoid T-cell lines derived from lymphocytic leukemia. J Natl Cancer Inst (1974) 53(3):655–60. doi: 10.1093/jnci/53.3.655
29. Watanabe S, Shimosato Y, Kameya T, Kuroki M, Kitahara T, Minato K, et al. Leukemic distribution of a human acute lymphocytic leukemia cell line (Ichikawa strain) in nude mice conditioned with whole-body irradiation. Cancer Res (1978) 38(10):3494–8.
30. Davies TG, Wixted WE, Coyle JE, Griffiths-Jones C, Hearn K, McMenamin R, et al. Monoacidic inhibitors of the kelch-like ech-associated protein 1: Nuclear factor erythroid 2-related factor 2 (Keap1:Nrf2) protein-protein interaction with high cell potency identified by fragment-based discovery. J Med Chem (2016) 59(8):3991–4006. doi: 10.1021/acs.jmedchem.6b00228
31. Svec D, Dolatabadi S, Thomsen C, Cordes N, Shannon M, Fitzpatrick P, et al. Identification of inhibitors regulating cell proliferation and fus-Ddit3 expression in myxoid liposarcoma using combined DNA, mrna, and protein analyses. Lab Invest (2018) 98(7):957–67. doi: 10.1038/s41374-018-0046-3
32. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative pcr and the 2(-delta delta C(T)) method. Methods (2001) 25(4):402–8. doi: 10.1006/meth.2001.1262
33. Chen J, Bardes EE, Aronow BJ, Jegga AG. Toppgene suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res (2009) 37(Web Server issue):W305–11. doi: 10.1093/nar/gkp427
34. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U.S.A. (2005) 102(43):15545–50. doi: 10.1073/pnas.0506580102
35. Bliss CI. The toxicity of poisons applied jointly. Ann Appl Biol (1939) 26(3):585–615. doi: 10.1111/j.1744-7348.1939.tb06990.x
36. Amaru Calzada A, Todoerti K, Donadoni L, Pellicioli A, Tuana G, Gatta R, et al. The hdac inhibitor givinostat modulates the hematopoietic transcription factors Nfe2 and c-myb in Jak2(V617f) myeloproliferative neoplasm cells. Exp Hematol (2012) 40(8):634–45 e10. doi: 10.1016/j.exphem.2012.04.007
37. Mitra P, Pereira LA, Drabsch Y, Ramsay RG, Gonda TJ. Estrogen receptor-alpha recruits p-tefb to overcome transcriptional pausing in intron 1 of the myb gene. Nucleic Acids Res (2012) 40(13):5988–6000. doi: 10.1093/nar/gks286
38. Mitra P, Yang RM, Sutton J, Ramsay RG, Gonda TJ. Cdk9 inhibitors selectively target estrogen receptor-positive breast cancer cells through combined inhibition of myb and mcl-1 expression. Oncotarget (2016) 7(8):9069–83. doi: 10.18632/oncotarget.6997
39. Slaughter MJ, Shanle EK, Khan A, Chua KF, Hong T, Boxer LD, et al. Hdac inhibition results in widespread alteration of the histone acetylation landscape and Brd4 targeting to gene bodies. Cell Rep (2021) 34(3):108638. doi: 10.1016/j.celrep.2020.108638
40. Ahmad R, Raina D, Meyer C, Kharbanda S, Kufe D. Triterpenoid cddo-me blocks the nf-kappab pathway by direct inhibition of ikkbeta on cys-179. J Biol Chem (2006) 281(47):35764–9. doi: 10.1074/jbc.M607160200
41. Probst BL, Trevino I, McCauley L, Bumeister R, Dulubova I, Wigley WC, et al. Rta 408, a novel synthetic triterpenoid with broad anticancer and anti-inflammatory activity. PloS One (2015) 10(4):e0122942. doi: 10.1371/journal.pone.0122942
42. Suhasini M, Reddy CD, Reddy EP, DiDonato JA, Pilz RB. Camp-induced nf-kappab (P50/Relb) binding to a c-myb intronic enhancer correlates with c-myb up-regulation and inhibition of erythroleukemia cell differentiation. Oncogene (1997) 15(15):1859–70. doi: 10.1038/sj.onc.1201530
43. Suhasini M, Pilz RB. Transcriptional elongation of c-myb is regulated by nf-kappab (P50/Relb). Oncogene (1999) 18(51):7360–9. doi: 10.1038/sj.onc.1203158
44. Sayin VI, LeBoeuf SE, Singh SX, Davidson SM, Biancur D, Guzelhan BS, et al. Activation of the Nrf2 antioxidant program generates an imbalance in central carbon metabolism in cancer. Elife (2017) 6:e28083 . doi: 10.7554/eLife.28083
45. Zhao L, Glazov EA, Pattabiraman DR, Al-Owaidi F, Zhang P, Brown MA, et al. Integrated genome-wide chromatin occupancy and expression analyses identify key myeloid pro-differentiation transcription factors repressed by myb. Nucleic Acids Res (2011) 39(11):4664–79. doi: 10.1093/nar/gkr024
46. Honda T, Rounds BV, Bore L, Favaloro FG Jr., Gribble GW, Suh N, et al. Novel synthetic oleanane triterpenoids: A series of highly active inhibitors of nitric oxide production in mouse macrophages. Bioorg Med Chem Lett (1999) 9(24):3429–34. doi: 10.1016/s0960-894x(99)00623-x
47. Reisman SA, Lee CY, Meyer CJ, Proksch JW, Sonis ST, Ward KW. Topical application of the synthetic triterpenoid rta 408 protects mice from radiation-induced dermatitis. Radiat Res (2014) 181(5):512–20. doi: 10.1667/RR13578.1
48. Parikh NR, Mandal A, Bhatia D, Siveen KS, Sethi G, Bishayee A. Oleanane triterpenoids in the prevention and therapy of breast cancer: Current evidence and future perspectives. Phytochem Rev (2014) 13(4):793–810. doi: 10.1007/s11101-014-9337-5
49. Liby KT, Sporn MB. Synthetic oleanane triterpenoids: Multifunctional drugs with a broad range of applications for prevention and treatment of chronic disease. Pharmacol Rev (2012) 64(4):972–1003. doi: 10.1124/pr.111.004846
50. Rohwer F, Todd S, McGuire KL. The effect of il-2 treatment on transcriptional attenuation in proto-oncogenes pim-1 and c-myb in human thymic blast cells. J Immunol (1996) 157(2):643–9. doi: 10.4049/jimmunol.157.2.643
51. Civin CI, Strauss LC, Brovall C, Fackler MJ, Schwartz JF, Shaper JH. Antigenic analysis of hematopoiesis. iii. a hematopoietic progenitor cell surface antigen defined by a monoclonal antibody raised against kg-1a cells. J Immunol (1984) 133(1):157–65.
52. Ribatti D, Solimando AG, Pezzella F. The anti-Vegf(R) drug discovery legacy: Improving attrition rates by breaking the vicious cycle of angiogenesis in cancer. Cancers (Basel) (2021) 13(14):3433. doi: 10.3390/cancers13143433
53. Habets RA, de Bock CE, Serneels L, Lodewijckx I, Verbeke D, Nittner D, et al. Safe targeting of T cell acute lymphoblastic leukemia by pathology-specific notch inhibition. Sci Transl Med (2019) 11(494):eaau6246. doi: 10.1126/scitranslmed.aau6246
54. Stadhouders R, Thongjuea S, Andrieu-Soler C, Palstra RJ, Bryne JC, van den Heuvel A, et al. Dynamic long-range chromatin interactions control myb proto-oncogene transcription during erythroid development. EMBO J (2012) 31(4):986–99. doi: 10.1038/emboj.2011.450
55. Salminen A, Lehtonen M, Paimela T, Kaarniranta K. Celastrol: Molecular targets of thunder God vine. Biochem Biophys Res Commun (2010) 394(3):439–42. doi: 10.1016/j.bbrc.2010.03.050
56. Shi J, Li J, Xu Z, Chen L, Luo R, Zhang C, et al. Celastrol: A review of useful strategies overcoming its limitation in anticancer application. Front Pharmacol (2020) 11:558741. doi: 10.3389/fphar.2020.558741
Keywords: MYB, acute lymphoblastic leukemia, oleanane triterpenoid, bardoxolone methyl, omaveloxolone
Citation: Tejera Nevado P, Tešan Tomić T, Atefyekta A, Fehr A, Stenman G and Andersson MK (2023) Synthetic oleanane triterpenoids suppress MYB oncogene activity and sensitize T-cell acute lymphoblastic leukemia cells to chemotherapy. Front. Oncol. 13:1126354. doi: 10.3389/fonc.2023.1126354
Received: 17 December 2022; Accepted: 24 March 2023;
Published: 03 April 2023.
Edited by:
Daekyu Sun, University of Arizona, United StatesReviewed by:
Daniela Sanchez Bassères, University of São Paulo, BrazilAntonio Giovanni Solimando, University of Bari Aldo Moro, Italy
Claudia Scotti, University of Pavia, Italy
Copyright © 2023 Tejera Nevado, Tešan Tomić, Atefyekta, Fehr, Stenman and Andersson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mattias K. Andersson, bWF0dGlhcy5hbmRlcnNzb25AbGxjci5tZWQuZ3Uuc2U=