 Wenyue Shen1,2†
Wenyue Shen1,2† Quanwei Zhou1,2†
Quanwei Zhou1,2† Chenxi Peng1,2
Chenxi Peng1,2 Jiaheng Li1,2Qizhi Yuan1,2Hecheng Zhu3,4Ming Zhao4Xingjun Jiang1,2*Weidong Liu3,5*
Jiaheng Li1,2Qizhi Yuan1,2Hecheng Zhu3,4Ming Zhao4Xingjun Jiang1,2*Weidong Liu3,5* Caiping Ren1,2,3,5*
Caiping Ren1,2,3,5*- 1Department of Neurosurgery, Xiangya Hospital, Central South University, Changsha, China
- 2National Clinical Research Center for Geriatric Disorders, Xiangya Hospital, Central South University, Changsha, China
- 3Cancer Research Institute, School of Basic Medical Science, Central South University, Changsha, China
- 4Changsha Kexin Cancer Hospital, Changsha, China
- 5The Key Laboratory of Carcinogenesis of the Chinese Ministry of Health and the Key Laboratory of Carcinogenesis and Cancer Invasion of the Chinese Ministry of Education, School of Basic Medicine, Central South University, Changsha, China
FBXW7, a member of the F-box protein family within the ubiquitin–proteasome system, performs an indispensable role in orchestrating cellular processes through ubiquitination and degradation of its substrates, such as c-MYC, mTOR, MCL-1, Notch, and cyclin E. Mainly functioning as a tumor suppressor, inactivation of FBXW7 induces the aberrations of its downstream pathway, resulting in the occurrence of diseases especially tumorigenesis. Here, we decipher the relationship between FBXW7 and the hallmarks of cancer and discuss the underlying mechanisms. Considering the interplay of cancer hallmarks, we propose several prospective strategies for circumventing the deficits of therapeutic resistance and complete cure of cancer patients.
1 Introduction
Over the past few decades, accumulated knowledge in cancer research has gradually sketched the outline of cancer hallmarks in reference to molecular, biochemical, and cellular characteristics. In 2000, Hanahan and Weinberg generalized the hallmarks of cancers into 6 major biological capabilities, which comprises self-sufficiency in growth signals, limitless replicative potential, evading apoptosis, insensitivity to anti-growth signals, sustained angiogenesis, and invasion and metastasis (1). A decade later, this notion was further refined by introducing two emerging hallmarks: deregulating cellular energetics and avoiding immune destruction. The aforementioned eight hallmarks can be explained by two enabling characteristics: genome instability and mutation as well as tumor-promoting inflammation (2). Recently, this concept of cancer hallmarks has been updated again owing to the in-depth mining of cancer mechanisms, incorporating novel emerging hallmarks as well as enabling characteristics including unlocking phenotypic plasticity, non-mutational epigenetic reprogramming, polymorphic microbiomes, and senescent cells (3).
Rapid advances have gradually deepened our understanding of cancer biological behaviors. During these years, unremitting efforts in the fields of cancer biology have also shed light on its link with the main degradation mechanism of eukaryotes, that is, the ubiquitin–proteasome system (UPS). Through the degradation of proteins involved in a broad spectrum of cellular processes, such as cell cycle progression, signal transduction, and endocytosis, UPS is of great significance to regulate immune response, development, and apoptosis to maintain homeostasis, the abnormalities of which contribute to uncontrolled development, genomic instability, and even transformation of malignancy. The UPS-based degradation, termed ubiquitination, is a multi-level cascade associated with the covalent binding of ubiquitin throughout the course, mainly consisting of the following components: a ubiquitin-activating enzyme (E1), a ubiquitin-conjugating enzyme (E2), a ubiquitin ligase (E3), and a 26S proteasome. The initiation of ubiquitin-mediated degradation involves sequential reactions. After the ATP-dependent activation triggered by E1, the 76-amino-acid-residue protein ubiquitin (Ub) binds to E1 in a thiolester linkage. Then, Ub is transferred to E2, and subsequently transferred through E3 to substrate proteins (4, 5). Afterwards, a polyubiquitin chain can be formed for further degradation by the 26S proteasome complex.
The S-phase kinase-associated protein 1 (SKP1)-cullin-1 (CUL1)-F-box-protein (SCF) complex belongs to the really interesting new gene (RING) family of E3 ubiquitin ligases, mediating the degradation of ~20% proteins related to cell-cycle regulation, transcription, oncogenesis, and tumor suppression (6). The SCF complex contains the invariable subunits CULl, RING-box 1 (RBX1), and SKP1, as well as the variable component F-box proteins (7), which is characterized by an approximately 40-amino-acid motif and determines the substrate specificity of the SCF complex.
F-box and WD repeat domain containing 7 (FBXW7, also known as AGO, hCdc4, FBW6, FBW7, SEL10 or FBX30) is a member of F-box proteins encoded by FBXW7 gene. The FBXW7 gene (Gene ID: 55294) maps to chromosome region 4q31.3 including 18 exons, where mutations are frequently detected in multiple human cancers, such as colorectal cancer (CRC) (8, 9), ovarian cancer (10), breast cancer (BC) (11), endometrial cancer (12), and human T-cell acute lymphoblastic leukemia (T-ALL) (13). Based on the statistics of COSMIC Cancer Gene Census list (Tier 1), the calculated population-level mutation proportion of FBXW7 is approximately 4.17% in Americans, ranking 28th among all genes counted (14). FBXW7 is composed of a tandem WD40-repeat domain, an F-box domain, a 5-residue tail, and an α-helical linker domain (15). Additionally, FBXW7 can be subdivided into three isoforms: FBXW7α localized in the nucleoplasm, FBXW7β residing in the cytoplasm, and FBXW7γ within the nucleolus (16), all of which recognize substrates through a conserved CDC4 phosphodegron (CPD) motif, requiring phosphorylation of substrates at particular sites by protein kinases such as the glycogen synthase kinase-3 (GSK3) to accelerate the ubiquitination and proteolysis process of substrates (17). As a consequence, the process of UPS-mediated proteolysis involved in FBXW7 is shown in Figure 1.
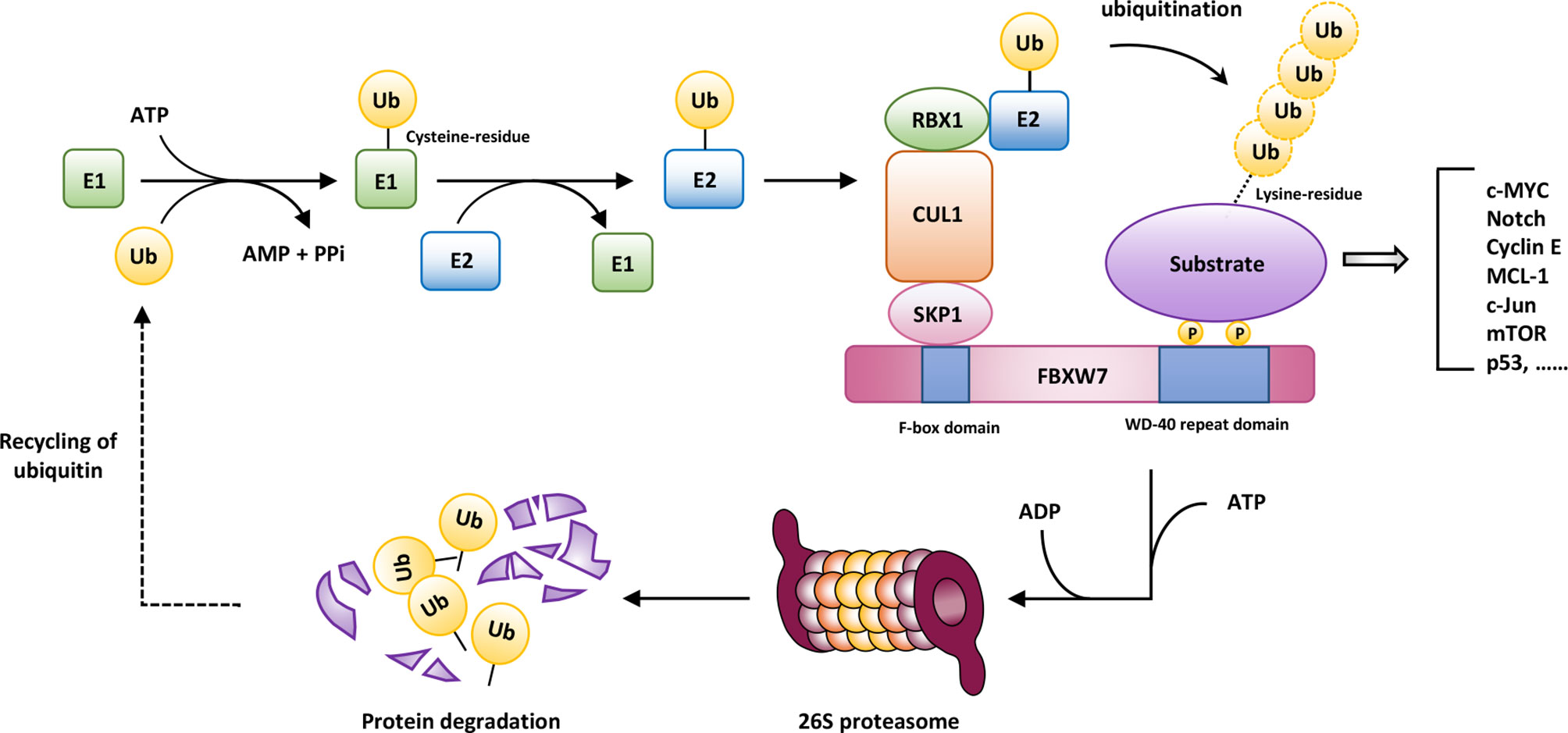
Figure 1 The ubiquitin-mediated degradation involved in FBXW7. After the ATP-dependent activation triggered by the ubiquitin-activating enzyme (E1), the 76-amino-acid-residue protein ubiquitin binds to E1 in a thiolester linkage. Then, ubiquitin (Ub) is transferred to the ubiquitin-conjugating enzyme (E2), and subsequently transferred through the ubiquitin ligase (E3) to substrate proteins. Afterwards, a polyubiquitin chain can be formed for further degradation by 26S proteasome complex, conferring the recycling of ubiquitin. AMP, adenosine monophosphate; ATP, adenosine triphosphate; CUL1, cullin 1; RBX1, RING-box 1; SKP1, S-phase kinase-associated protein 1.
The mRNA and protein expression of FBXW7 can be detected almost in all human normal organs and tissues (18). FBXW7 protein expression level can be high, seen in lung, stomach, colon, kidney, breast, skin, etc.; it also can be medium, detected in rectum, liver, endometrium, cervix, etc. In addition, FBXW7 mRNA expression has organ specificity, such as brain (18). However, in malignant organs and tissues, FBXW7 protein expression is decreased in glioma, lung cancer, liver cancer, urothelial cancer, ovarian cancer, melanoma, etc. (18), and the expression levels of aforementioned cancers are positively correlated with prognosis in the majority (besides liver cancer and melanoma).
It is universally acknowledged that FBXW7 exerts its major function as a tumor suppressor (19–21), orchestrating cellular processes by virtue of interacting with its substrates, such as c-MYC (22), Notch (23), cyclin E (11), myeloid cell leukemia-1 (MCL-1) (24), c-Jun (25), p53 (15, 26), and mechanistic target of rapamycin (mTOR) (27). As a consequence, in the following sections, we will mainly elaborate on the relationship between FBXW7 and cancer hallmarks and unravel the underlying mechanisms (Figure 2). In light of the interplay of cancer hallmarks, we highlight the prospects to battle against therapeutic resistance, one of the thorniest obstacles in the course of completely curing the patients.
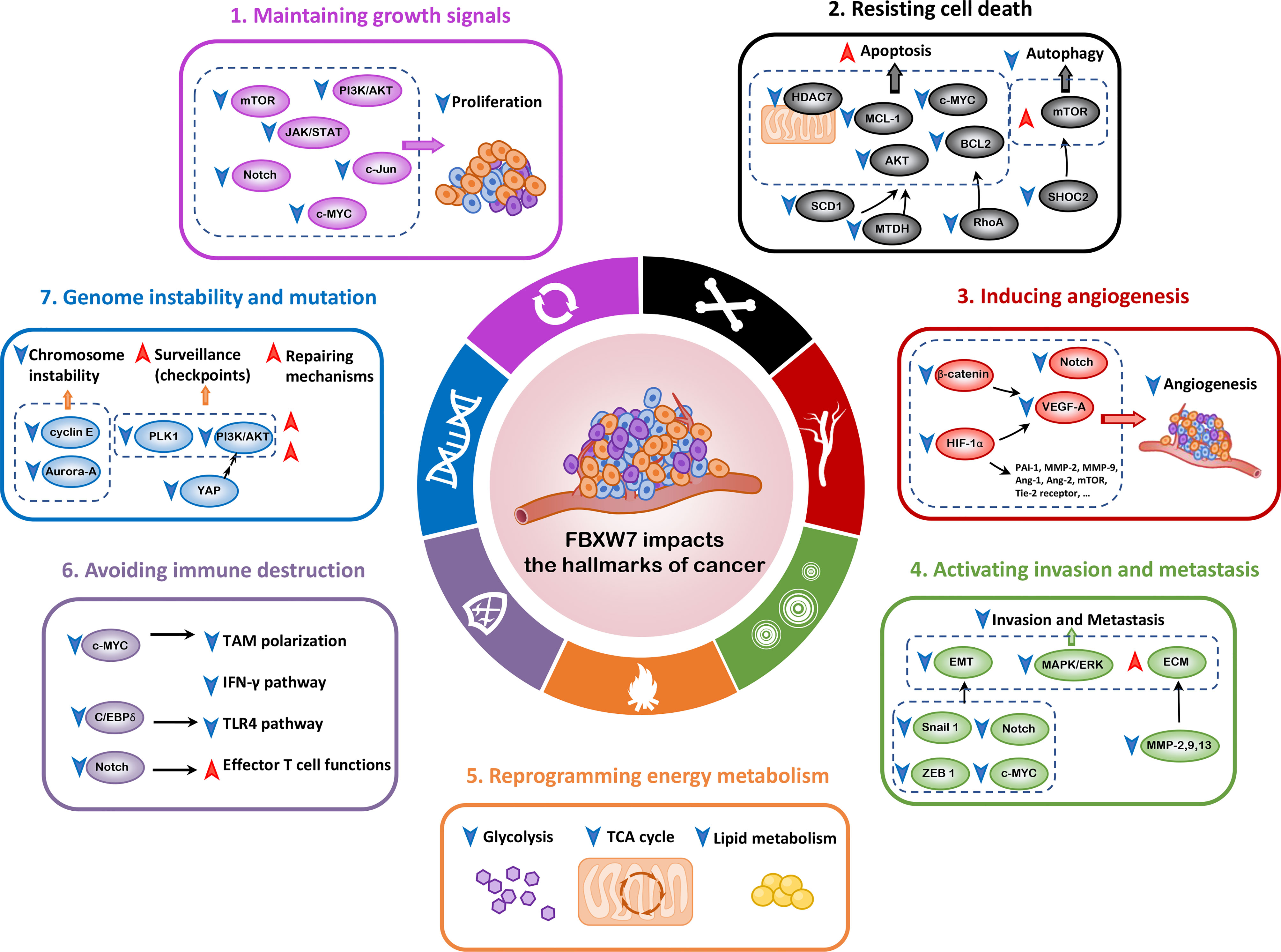
Figure 2 FBXW7 and the hallmarks of cancer. In this review, we mainly elaborate on seven hallmarks of cancer influenced by FBXW7, including maintaining growth signals, resisting cell death, inducing angiogenesis, activating invasion and metastasis, reprogramming energy metabolism, avoiding immune destruction, and genome instability and mutation. Ang-1, Angioprotein-1; BCL2, B cell lymphoma 2; C/EBPδ, CCAAT/enhancer binding protein δ; ECM, extracellular matrix; EMT, epithelial–mesenchymal transition; HDAC7, histone deacetylase 7; HIF-1α, hypoxia-inducible factor-1α; ICL, interstrand cross-link; MAPK/ERK, mitogen-activated protein kinase/extracellular signal–regulated kinase; MCL-1, myeloid cell leukemia-1; MMP, matrix metalloproteinase; MTDH, metadherin; NHEJ, nonhomologous end-joining; PAI-1, plasminogen activator inhibitor-1; PLK1, polo-like kinase 1; SCD1, stearoyl-CoA desaturase 1; VEGF-A, vascular endothelial growth factor-A; YAP, Yes-associated protein; ZEB1, zinc-finger E-box-binding homeobox 1.
2 FBXW7 and the Hallmarks of Cancer
2.1 Maintaining Growth Signals
Tumor cells are independent of exogenous growth signal stimulation and generate their own proliferative signals, which serves as one of the six hallmarks of cancer. FBXW7 reshapes the proliferative niches of tumor cells through ubiquitinating and degrading several crucial signal molecules, such as c-MYC, c-Jun, phosphatidylinositol 3-kinase (PI3K)/AKT, mTOR, Notch, as well as JAK/STAT signaling pathway.
c-MYC is a critical transcription factor in control of various biological processes such as proliferation, differentiation, and apoptosis (28, 29). Accumulated evidence has indicated that FBXW7 strictly regulates c-MYC turnover at the post-translational level following phosphorylation of its specific sites, where the sequential steps is associated with feedback mechanisms (30, 31). In adult T-cell leukemia/lymphoma (ATLL) cell lines, the mRNA and protein level of c-MYC is higher than normal due to the aberrations of FBXW7 expression, which is correlated with ATLL proliferation and poor prognosis of patients (32). Another study has disclosed that the oncogene Ecotropic viral integration site 5 (Evi5) accelerates laryngeal cancer cell proliferation by counteracting FBXW7, thus facilitating the accumulation of its substrate c-MYC (33).
c-Jun, a crucial member of the AP-1 family, participates in cellular proliferation, apoptosis, survival, and tumorigenesis. FBXW7 exerts its role as a component of c-Jun’s E3 ligase, and knockdown of FBXW7 confers the accumulation of c-Jun (25). In colon cancer cells, the lysine demethylase KDM5c could attenuate FBXW7-mediated degradation of c-Jun by epigenetically modifying FBXW7 and decreasing FBXW7 transcription, further promoting cell proliferation (34).
The PI3K/AKT signaling pathway regulates the functions of cellular growth, survival, and cell cycle, the dysregulation of which is frequently detected in human cancers (35). For example, microRNA-27a (miR-27a) downregulates FBXW7 expression by binding to its 3’-untranslated region (3’-UTR), and promotes OSCC cell proliferation via the PI3K/AKT axis (36). The serine/threonine kinase mTOR is capable of interacting with both upstream and downstream proteins of the PI3K/AKT pathway, which performs an indispensable role in orchestrating metabolism, cellular proliferation, and survival (37). FBXW7 deficiency results in elevated mTOR and phosphorylated mTOR (p-mTOR) protein levels, supporting that the turnover of mTOR is controlled by FBXW7 via targeted ubiquitination and degradation (38). Ectopic expression of the oncogene family with sequence similarity 83, member D (FAM83D) facilitates breast cancer cell proliferation partly through the accumulation of mTOR by reducing the expression level of FBXW7 (39).
As a substrate of FBXW7-dependent ubiquitination and proteolysis, the Notch signaling pathway has significant influences on cellular differentiation, proliferation, and apoptosis (40), while the dysregulated Notch signaling is also linked to oncogenesis of both solid tumors and leukemias (41, 42). Liu et al. have revealed that the serine/threonine/tyrosine interacting protein (STYX) accelerates endometrial cancer cell proliferation and suppresses apoptosis partly through the Notch/mTOR pathway by downregulating the expression of FBXW7 (43). The relationship between Notch and mTOR has been explored, presumably that the HES1 protein activated by Notch suppresses PTEN transcription, subsequently promoting the phosphorylation of AKT and mTOR activation (44).
In gastric cancer (GC), the downregulated level of FBXW7 attenuated its impact on growth arrest and apoptosis (45). One possible mechanism has elucidated that the transcription factor growth factor independence 1 (GFI1) promotes cell proliferation partly by means of suppressing transcription of Gastrokine-2 (GKN2) (46). Overexpression of GKN2 in GC cell lines suppresses GC cell proliferation through the downregulation of the JAK/STAT signaling pathway. Noteworthily, GFI1 mutant on S94A/S98A inhibits its phosphorylation mediated by GSK3β and ubiquitination by FBXW7, eventually facilitating tumor progression.
2.2 Resisting Cell Death
2.2.1 Apoptosis
The apoptotic machinery comprises both upstream regulators and downstream effectors, the latter eliciting a series of cascades pointed to the death program. The signals between regulators and effectors are transmitted by the so-called “apoptotic trigger” in the control of the B cell lymphoma 2 (BCL2) protein family including both pro- and anti-apoptotic regulatory proteins (1, 2).
By virtue of flow cytometry, apoptosis assays, and caspase 3/7 activity assays, researchers found that overexpression of FBXW7 accelerates GC tissue apoptosis through inducing the ubiquitination and degradation of RhoA (45), which is known to promote survival by triggering the expression of BCL2 gene (47). MCL-1, a member of BCL2 protein family, functions as a crucial anti-apoptotic regulator in cellular differentiation and apoptosis (48), which is overexpressed in multiple malignancies giving rise to chemotherapeutic resistance (49–51). It has been reported that FBXW7 regulates apoptosis cascades through ubiquitination and degradation of MCL-1. In T-ALL cell lines, FBXW7 deficiency confers resistance to chemotherapy, which can be reversed by FBXW7 restoration or MCL-1 knockout, implicating that MCL-1 promotes FBXW7-deficient cells to escape from apoptosis (52, 53). Likewise, FBXW7 mutations also mediates chemoresistance to some solid tumors such as CRC (24, 54), squamous cell carcinoma (55), and BC (56), which attributes to the downregulation of MCL-1. Meanwhile, favorable response to chemoradiotherapy is correlated with a high level of FBXW7, which is in parallel with a low level of MCL-1 (57).
In terms of targeting FBXW7 itself, the downregulation of prolyl isomerase Pin1 contributes to upregulation of FBXW7 and ensuing destruction of MCL-1, which potentiates the toxicity of sorafenib to prevent cell proliferation and induce apoptosis in hepatocellular carcinoma (HCC) (58). Similarly, the alkaloid lycorine hydrochloride (LH) has been reported to upregulate the level of FBXW7 as well as destabilize MCL-1, facilitating BCL2-drug-resistant GC cell apoptosis and inhibiting proliferation (59). Moreover, tyrosine kinase inhibitors (TKIs) give rise to GSK3β-dependent phosphorylation of MCL-1, translocation of MCL-1 to nucleus, as well as FBXW7-mediated degradation through targeting PI3K/AKT signaling, which overcome the resistance to targeted therapy in non-small cell lung cancer (NSCLC) (60). In addition, directly targeting MCL-1 with small molecular MCL-1 inhibitors exhibits potent efficacy such as S63845, AZD5991, and AMG176, sensitizing CRC cells to treatment of the targeted drug regorafenib by virtue of restoring apoptotic response (61). Collectively, accumulated evidence has deciphered MCL-1’s role in therapeutic resistance, in combination with novel strategies to dampen therapeutic resistance by targeting MCL-1, FBXW7, and their upstream proteins.
The mitochondria integrate an array of proapoptotic signals, further releasing proapoptotic signaling proteins such as cytochrome c to trigger the progression of a cascade of apoptotic caspases. FBXW7 facilitates apoptosis of glioblastoma cells through the ubiquitination and proteolysis of histone deacetylase 7 (HDAC7) (62), which localizes to the inner membrane of mitochondria and will be released into and sequestered within the cytoplasm in response to the initiation of apoptotic cascades (63).
c-MYC has been reported to have fundamental impacts on cell cycle progression as well as programmed cell death (64–66). The Aurora B kinase (AUKRB) impedes the phosphorylation of GSK3-3β and subsequent FBXW7-mediated degradation, rendering the accumulation of c-MYC and further promoting the progression of T-ALL (67). Nevertheless, the AUKRB inhibitor can reverse leukemogenesis and induce apoptosis due to the destabilization of c-MYC. Furthermore, c-MYC influences apoptosis partly through the p53 signaling pathway (68). The FBXW7 deficiency in the T-cell lineage of mice results in the accumulation of c-MYC, which promotes p53 expression, and further cell cycle arrest and apoptosis. The abnormalities can be reversed by the suppression of p53 (69). The development of glioblastoma can be ascribed to p53 mutations, which promotes the accumulation of c-MYC through the depression of FBXW7 and prevents apoptosis (70).
In pancreatic cancer tissues, FBXW7 induces apoptosis by inhibiting the binding of nuclear receptor subfamily 4 group A member 1 (NR4A1) to stearoyl-CoA desaturase 1 (SCD1) promoter, thus suppressing the transcription of SCD1 (71). SCD1 inhibition has been reported before to inactivate the AKT signaling, seen in higher rates of cell apoptosis in human lung adenocarcinoma (72). FBXW7 promotes apoptosis of BC cells through the ubiquitination and proteolysis of the oncoprotein metadherin (MTDH) (73), whose expression is related to various pathways, for example, the AKT signaling pathway (74). Furthermore, the inhibition of MTDH gives rise to apoptosis of cancer cells probably due to the downregulation of the AKT signaling (75, 76).
2.2.2 Autophagy
Autophagy is recognized as an evolutionarily conserved, intracellular degradation process, capable of delivering cytoplasmic materials to lysosomes (77) and regulating energy homeostasis as well as cellular renovation (78). Based on the differences of physiological functions and delivering modes, autophagy can be subdivided into mainly three types: macroautophagy that is thought to be the major type, microautophagy, and chaperone-mediated autophagy (CMA) (77). Dysregulated autophagy is involved in the development of multiple cancers, exerting a bifunctional role in suppressing benign tumor development and inducing cancer progression (79). In addition, autophagy can be induced during treatment of drug-sensitive cancers, resulting in drug resistance and cancer relapse (80). In general, inhibition of autophagy has been proposed as an effective therapeutic intervention (81).
The central metabolism modulator mTOR could be directly regulated by FBXW7 through ubiquitination and subsequent degradation (38, 82). mTOR is an essential inhibitory regulator of autophagy in response to growth factors as well as abundant nutrients (77), especially mTOR complex 1 (mTORC1), which regulates autophagy both in the process of initiation and transcription (83). Given the pivotal suppressive functions of mTOR in modulating autophagy, we propose the possible metabolic crosstalk between FBXW7 and autophagy. Perifosine is an oral alkylphospholipid with antitumor activity, whereas the pharmacological efficacy can be partly counteracted by inhibiting the mTOR axis dependent on GSK3/FBXW7 and inducing prosurvival autophagy (84). SHOC2, a RAS/ERK activator and a substrate of FBXW7, selectively combines with Raptor to impede Raptor-mTOR binding and subsequent mTORC1 activity, contributing to the stimulation of autophagy and acceleration of cancer proliferation (27, 85). Furthermore, there is a negative feedback loop between mTORC1 and RAS/ERK pathways, both under the regulation of FBXW7 targeting SHOC2 (85).
In addition to tumorigenesis, autophagy is also integral to therapeutic effects, capable of mediating resistance or sensitivity in response to chemotherapy agents (86). Wang et al. demonstrated that the microRNA-223 (miR-223) overexpression downregulates the level of FBXW7 protein, resulting in the activation of autophagy and cisplatin resistance of non-small lung cancer cells (NSLCCs) (87). Furthermore, this result indicates that targeting microRNAs can be of great value to inhibit autophagy and attenuate chemoresistance, which will be discussed in detail in later sections.
2.3 Inducing Angiogenesis
Growing malignant tumors are consistently endowed with the identity of neovascularization, referred to the vigorous and persistent growth of new capillaries (88). Angiogenesis plays a pivotal part in supplying essential oxygen and nutrients for solid tumor to satisfy ever-growing metabolic demands. Hypoxia-inducible factor (HIF) is a heterodimeric transcription factor, with the ability to trigger a large scale of pro-angiogenic factor expressions containing vascular endothelial growth factor (VEGF), VEGF-R1, and VEGF-R2, plasminogen activator inhibitor‐1 (PAI-1), matrix metalloproteinase-2 (MMP-2) and MMP-9, Angioprotein-1 (Ang-1) and Ang-2, as well as Tie-2 receptor (89, 90). In particular, the HIF-1α pathway is acknowledged to mainly regulate vasculature formation (89). GSK-3β/FBXW7-mediated ubiquitination and degradation is shown to influence angiogenesis and cell metastasis by decreasing the level of HIF-1α (91), in contrast to ubiquitin-specific protease 28 (USP28) which abrogates FBXW7-dependent degradation (92). miR-182, as previously mentioned, figures as an oncogenic miRNA by targeting its downstream gene FBXW7 in BC, capable of accelerating HIF-1α/VEGF-A-induced angiogenesis under hypoxia condition (93). miR-144, enriched in tumor-derived extracellular vesicles (EVs) from nasopharyngeal carcinoma (NPC), is demonstrated in vitro and in vivo to promote angiogenesis by downregulating FBXW7 and promoting HIF-1α-induced VEGF-A expression (94). Notably, FBXW7 can inactivate β-catenin pathway to influence the expression of VEGF-A, ultimately suppressing angiogenesis of ovarian cancer cells (95).
The Notch signaling pathway is crucial to vascular construction reflected in regulation of sprouting tip cells, endothelial cell development, as well as arterial differentiation (96). In melanoma, loss of FBXW7 is relevant to enrichment of Notch1, enhanced expression of Notch1 target genes, as well as acceleration of angiogenesis, which vividly elucidates FBXW7 as a critical suppressor of angiogenesis partly through degrading its substrate Notch1 and influencing its downstream signaling (97). In view of the pivotal role of FBXW7 in regulating angiogenesis, it is ponderable to investigate the underlying efficacy of dampening angiogenesis by targeting FBXW7 and its downstream factors.
2.4 Activating Invasion and Metastasis
Epithelial–mesenchymal transition (EMT) refers to epithelial cells’ acquisition of mesenchymal phenotypes, and is a highly conserved cellular program that is crucial to embryonic development and malignant progression such as migration, invasion, stemness, and therapeutic resistance (98–101). FBXW7 expression abundance is relevant to tumor clinicopathologic features, and FBXW7 is capable of dampening EMT process partly through downregulating EMT upstream transcription factors such as Snail 1 (45, 102) and zinc-finger E-box-binding homeobox 1 (ZEB1) (45), whereas the downregulation of FBXW7 expression reverses its inhibitory role. Research reveals that FBXW7 is involved in renal cell carcinoma (RCC) cell invasion and metastasis by virtue of suppressing EMT, which has great potential for future therapeutic targets (103, 104). Moreover, non-coding RNAs regulate tumor properties of invasion and metastasis via EMT in part through interacting with FBXW7. MiR-27a overexpression in human BC induces cell migration and EMT dependent on its target gene FBXW7, while ectopic expression of FBXW7 attenuates the properties of invasion and metastasis partly associated with EMT (105). Cancer susceptibility candidate 2 (CASC2), a long non-coding RNA (lncRNA) downregulated in HCC cell lines, can inhibit tumorigenesis by means of functioning as a competing endogenous RNA (ceRNA) for miR-367 and weakening its pro-metastatic effects through targeting FBXW7 (106).
Consequently, this pathway provides insights into potential targeted therapies. It is well-known that FBXW7 controls various cellular processes by regulating its substrates, of which c-MYC and Notch participate in the promotion of EMT process (107, 108). The ATPase Thyroid hormone receptor interactor 13 (TRIP13), usually overexpressed in a myriad of human cancers, promotes glioblastoma migration and invasion by decreasing the transcription of FBXW7 and attenuating its inhibitory effects on c-MYC (109). Dysregulated FBXW7 appears to be a prognostic hallmark of HCC, exhibiting that lower expression corresponds to worse progression and survival. Moreover, FBXW7 is capable of modulating HCC invasion and metastasis via FBXW7/Notch1 axis (23).
In addition to EMT process, there are multiple underlying mechanisms accounting for the invasion, migration and metastasis of cancer cells. For example, mitogen-activated protein kinase/extracellular signal–regulated kinase (MAPK/ERK) pathway appears to be involved in melanoma tumorigenesis, for the treatment of MAPK/ERK kinase (MEK) inhibitors significantly reverses FBXW7 knockdown-induced cell migration (110). Enhancer of zeste homolog 2 (EZH2), a substrate of FBXW7, figures as an oncogenic protein facilitating invasion and metastasis of pancreatic cancer cells, which can be abrogated by CDK5/FBXW7-dependent degradation (111). Cai et al. reported that FBXW7 participates in regulating RCC metastasis partly through modulating expression of MMP-2, MMP-9, and MMP-13 (103), which have been disclosed to degrade extracellular matrix (ECM) protein to accelerate cancer metastasis (112).
2.5 Reprogramming Energy Metabolism
Under the condition of hypoxia or even adequate oxygen supply, cancer cells choose inefficient aerobic glycolysis rather than oxidative phosphorylation (113), giving rise to increased glucose consumption, high-speed ATP production, as well as transformation of glycolytic pyruvate to lactate, widely known as “the Warburg effect” (114). The rewired metabolic network produces intermediates, for example, figuring in the process of glycolysis or tricarboxylic acid (TCA) cycle, not only favoring cancer cells to meet essential energy, and anabolic and redox demands on acquired nutrients during early stages of cancer development (115), but also supporting cancer malignant progression especially metastasis and therapeutic resistance in later stages (116, 117) based on the resistance to oxidative stress. The circuit of energy reprogramming involves a series of signaling pathways (118), where FBXW7 participates in the regulation of crucial metabolic molecules (Figure 3).
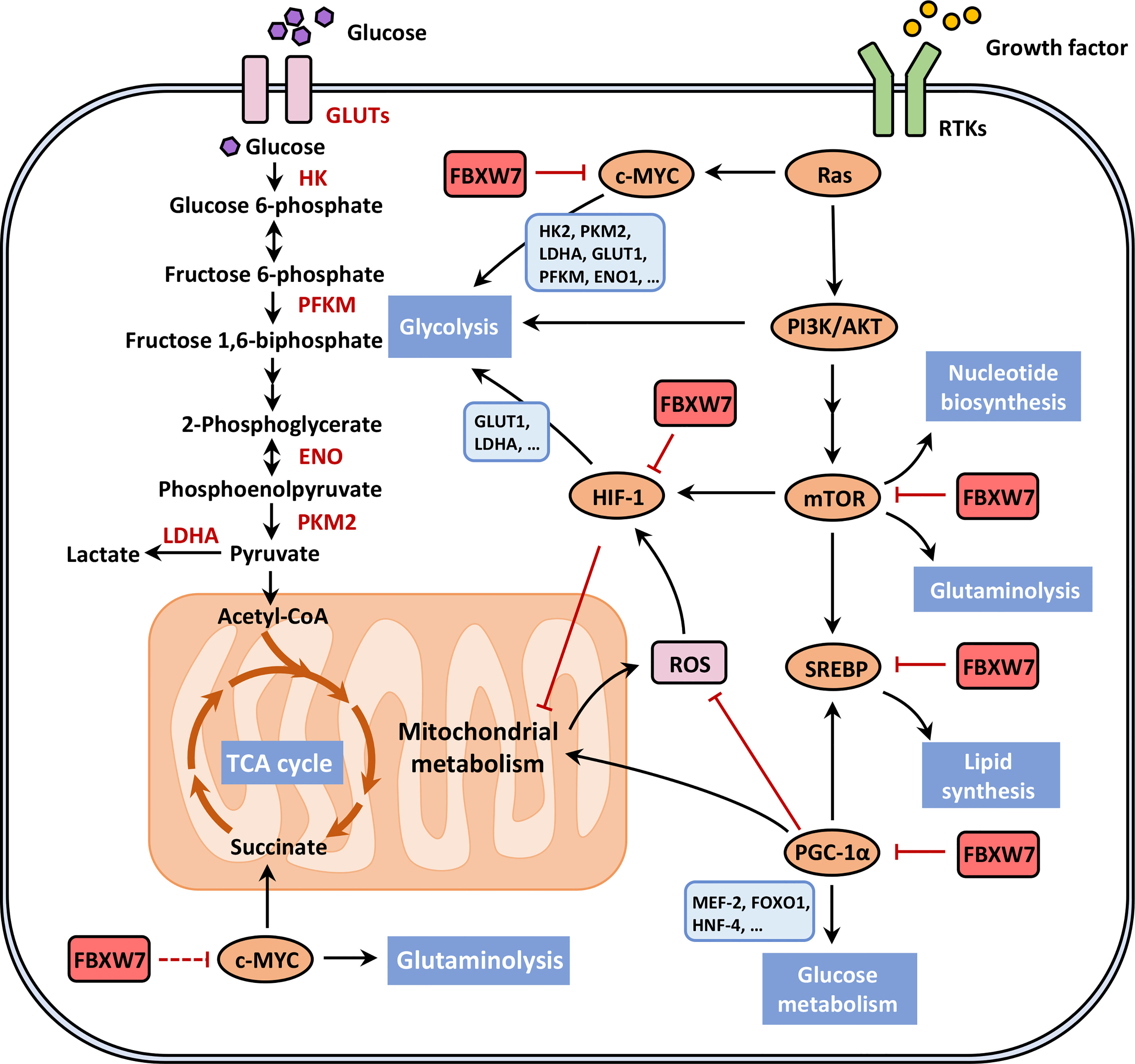
Figure 3 FBXW7 participates in the reprogramming energy metabolism. The rewired metabolic network favors cancer cells to meet essential energy, anabolic and redox demands on acquired nutrients during early stages of cancer development, where FBXW7 modulates metabolic reprogramming through ubiquitin-dependent degradation of crucial metabolic molecules, such as mTORC1, SREBP, HIF-1, c-MYC and PGC-1α, their relationship seen in red solid blocking arrows. We are awaiting the potential roles of FBXW7 in regulating tricarboxylic acid (TCA) cycle and glutamine metabolism through interacting with c-MYC, so the red dotted line blocking arrow is shown in the lower left part of the figure. This figure was adapted from DeBerardinis and Chandel (118). ENO, enolase; FOXO1, fork head box O1; GLUT, glucose transporter; HK, hexokinase; HNF-4, hepatic nuclear factor-4; LDHA, lactate dehydrogenase A; MEF-2, myocyte enhancer factor-2; PFKM, phosphofructokinase; PKM2, pyruvate kinase M2; ROS, reactive oxygen species; RTK, receptor tyrosine kinase.
Growth factors trigger the PI3K/AKT axis as well as the downstream mTOR pathway, inducing the glycolytic flux and fatty acids (FAs) production due to activation of HIF and sterol regulatory element binding protein (SREBP), respectively. The mTOR pathway performs an indispensable role in nutrient metabolism, energetic regulation, and promotion of cancer cell survival (119). mTORC1 enhances the transcription of glycolysis-related genes (120), and mTOR was proven to be a crucial activator of Warburg effect (121). Evidence has shown that the activation of mTORC1 promotes nucleotide synthesis as well as glutaminolysis (118). Various studies report that FBXW7 targets and degrades mTOR through ubiquitin–proteasome pathway (38, 122, 123). Fructose-1,6-bisphosphatase (FBP1) was reported to promote the autoubiquitination of FBXW7, then resulting in lower levels of mTOR and subsequently deregulated glycolysis in NPC cells (122).
In addition to the functions of HIF-1 in angiogenesis, HIF-1 exhibits a pivotal role in the aerobic glycolysis under hypoxic conditions, including the induction of glucose transporter 1 (GLUT1) and critical glycolytic enzymes such as lactate dehydrogenase A (LDH-A) (124). Furthermore, HIF-1 suppresses the functions of mitochondria to regulate the Warburg effect, comprising the inactivation of TCA cycle, enzymes, and microRNAs associated with the mitochondrial process, as well as the decrease of activated mitochondria. Under the circumstance of hypoxia, HIF-1α is negatively regulated by FBXW7-mediated ubiquitination and proteolysis in human ovarian cancer cells, proposing an underlying direction for energy reprogramming and angiogenesis (91). The oncoprotein TAR (HIV-1) RNA binding protein 2 (TARBP2) can elevate the stability of HIF-1α by decreasing its ubiquitination level through decreasing the E3 ligases activity including FBXW7 in BC cells (125).
SREBPs, a family of membrane-bound transcription factors (126), are able to induce the expression of genes associated with lipid metabolism (127–129) and multiple cellular processes (130), which can be subdivided into three major members encoded by two distinct genes (126). SREBPs translocate to the nucleus in an active form and regulate corresponding gene expressions, and are degraded rapidly by the ubiquitin–proteasome pathway (131). In recent years, researchers have deciphered the role of FBXW7 in lipogenesis related with SREBPs, whereas the precise regulatory mechanisms remain to be elucidated. Dependent on the phosphorylation of Cdc4 phosphodegron (CPD) motifs, FBXW7 interacts with and degrades SREBP1 and SREBP2 in control of lipid metabolism (132, 133), reflected in the elevated cholesterol and FAs synthesis as well as the enhanced uptake of LDL in FBXW7-/- cells (132). miR-182, originated from a single primary transcript (134), is emerging as an oncogenic role in various malignancies (135, 136). miR-182 was reported to target FBXW7 and indirectly enhance the accumulation of nuclear SREBPs; subsequently, lipid synthesis was accelerated by stimulation of SREBP-targeted genes (137). To meet the demand for rapidly excessive proliferation, cancer cells rely on glucose and glutamine for de novo lipogenesis responsive to dysregulated SREBP pathways (138). In an experiment, different cancer cells were treated with inhibitors of mTOR complex 2 (mTORC2) and then detected to express decreased mature SREBP1 (mSREBP1). It was unraveled that mTOR2 suppresses the GSK3/FBXW7-mediated ubiquitination of SREBP1 and serves as a positive regulator of SREBP1-related genes and lipogenesis (139). Likewise, protein arginine methyltransferase 5 (PRMT5) facilitates de novo lipogenesis by virtue of epigenetic modification of SREBP1a and blockade of GSK3/FBXW7-mediated ubiquitin–proteasome pathway (140).
c-MYC exerts its roles in various biological processes such as proliferation, metabolism, differentiation, and transformation as mentioned before (28). c-MYC modulates almost all glucose metabolism genes, including hexokinase 2 (HK2), pyruvate kinase M2 (PKM2), LDHA, GLUT1, phosphofructokinase (PFKM), and enolase 1 (ENO1) (141, 142), promoting the development of glycolysis. Additionally, c-MYC plays a significant part in FA biosynthesis, glutaminolysis, serine metabolism, and mitochondrial metabolism. Several studies have demonstrated that FBXW7 interacts with and ubiquitylates c-MYC, and phosphorylation of c-MYC on Thr-58 and Ser-62 is essential for proteasome-dependent degradation (31). FBXW7 dramatically suppresses glucose metabolism and reduces the 18F-FDG uptake of pancreatic cancer cells through regulating the expression of thioredoxin binding protein (TXNIP) in a c-MYC-dependent manner, whose regulatory mode can be summarized into the FBXW7/c-MYC/TXNIP axis (114). PRMT5 epigenetically regulated the expression of FBXW7, contributing to elevated c-MYC levels and subsequently enhanced glycolysis and proliferation of pancreatic cancer cells (22). Reversely, dioscin treatment enhanced the interaction between FBXW7 and c-MYC, which leads to inhibition of HK2, a crucial enzyme catalyzing glucose phosphorylation and hence the suppression of glycolysis (143). Likewise, Tanshinone IIA (Tan IIA) suppresses HK-mediated glycolysis of oral squamous cell carcinoma (OSCC) cells by promoting FBXW7-mediated degradation of c-MYC and inhibiting the AKT-c-MYC signaling, exhibiting its natural antitumor activity (144).
In addition, FBXW7 also ubiquitinates and degrades PPARγ coactivator-1α (PGC-1α), which serves as a critical energy metabolism regulator, coordinating mitochondrial biogenesis and oxidative phosphorylation (145). Outside the mitochondrion, PGC-1α also modulates lipid and glucose metabolism through the interaction with nuclear factors, such as SREBP1, myocyte enhancer factor-2 (MEF-2), fork head box O1 (FOXO1), and hepatic nuclear factor-4 (HNF-4) (146). In human melanoma cell lines, the microphthalmia-associated transcription factor (MITF) directly controls PGC-1α expression; hence, the MITF/PGC-1α axis can regulate mitochondrial oxidative phosphorylation and rescue the oxidative stress of reactive oxygen species (ROS) (147, 148). FBXW7 expression was detected downregulated, which is correlated with elevated expression of mitochondrial functional genes as well as poor prognosis of melanoma patients, dependent on its downstream MITF/PGC-1α pathway satisfying the metabolic needs of tumor cells (149). In contrast to the aforementioned tumorigenic functions of PGC-1α, the diminution of PGC-1α indirectly by HIF renders metabolic reprogramming, accelerating tumorigenesis and the resistance to chemotherapies in RCC cells (150). The dual roles of PGC-1α in metabolic reprogramming remain elusive and require to be explored. In addition, FBXW7 mutations in CRC cells enhance the mitochondrial gene expression, in which the so-called “metabolic reprogramming” points to oxidative phosphorylation and is possessed with metabolic vulnerabilities (151).
2.6 Avoiding Immune Destruction
The consistently alerted immune system has been monitoring and eliminating cells and tissues from forming and progressing early-stage neoplasms, terminal tumors, and micrometastases. According to this theory of immune surveillance, solid tumors avoid immune eradication through evading immune detection or curtailing the damage of immunological killing (2), both of which can be realized through presenting tumor antigens resistant to immune effectors (also called “immunoediting”) and the progressive formation of immunosuppressive microenvironment (152).
Tumor-associated macrophages (TAM), the macrophages that are characterized as immunosuppressive infiltrating tumors, promote tumor progression through subversion of genetic stability, nurturing cancer stem cells (CSCs), promoting metastasis, and restraining adaptive immune response (153). Driven by cytokines derived from tumors and T cells, macrophages polarize into classically activated macrophages (also called M1) and alternatively activated macrophages (also called M2), in which TAM mainly belong to the latter, showing anti-inflammatory and pro-tumorigenic functions (154). FBXW7 is capable of restricting cancer progression by haltering immunosuppressive niche and immune evasion. Myeloid cell-specific ablation of FBXW7 enhances tumor proliferation by decreasing its ubiquitination on c-MYC, which promotes TAM polarization (155).
Moreover, in a triple-negative BC (TNBC) model, low-expressed E74-like transcription factor (ELF5) downregulates the level of FBXW7, attenuating its degradation on interferon-γ receptor 1 (IFNGR1). Afterwards, the triggered intrinsic interferon-γ (IFN-γ) pathway facilitates immunosuppressive neutrophil aggregation, accompanied by programmed death ligand 1 (PD-L1) upregulation, which not only promotes tumor proliferation and metastasis but also appears to accelerate CD8+ T cell exhaustion favoring immune evasion. Therapeutic drugs targeting IFNGR1 and PD-L1 potentiate antitumor efficacy in TNBC patients, providing new insights into underlying targets for immunotherapies (156). FBXW7 inhibits the transcription of Toll-like receptor 4 (TLR4) signaling by degrading CCAAT/enhancer binding protein delta (C/EBPδ) phosphorylated by GSK3β, detected as transcriptional activator of Tlr4 genes (157). Reversely, C/EBPδ also represses the expression of FBXW7 (158), thus forming a negative feedback cycle through which FBXW7 inhibits inflammatory responses and TLR4-associated immune evasion (159). In addition, high expression of miR-101 and miR-26a are detected in ovarian cells, which downregulate EZH2 and confer functional inactivation of effector T cells in the absence of glucose. In mechanism, the histone methyltransferase EZH2 triggers notch signaling by repressing its suppressor Numb and FBXW7, maintaining effector T-cell survival and poly-functions (160).
2.7 Genome Instability and Mutation
Genomic alterations promote the acquisition of other hallmarks of cancer cells, prioritizing subclone of cells with superior mutant genotype to expand progressively and play a leading role within the local environment. The maintenance of genome stability requires delicate modulation of DNA replication in the cell cycle process, precise detection, and repairing any defects of DNA.
As a substrate of FBXW7, cyclin E, a nuclear protein that interacts with and activates cyclin-dependent kinase 2 (CDK2) to phosphorylate proteins, facilitates G1/S phase transition and is degraded at the boundary of the S/G2 phase, whereas the overexpression of cyclin E has been detected in human cancer cells associated with cell cycle deregulation and chromosome instability (CIN) (161, 162). FBXW7 mutations lead to the aberrant accumulation of its substrate cyclin E in CRC samples, conferring abnormality of chromosome congression during metaphase as well as ensuing chromosome transmission, implicating the role of FBXW7/cyclin E axis in regulating CIN (163). An in vivo study generated a mouse model to disturb the regulation of cyclin E relying on FBXW7, which was found fragile in response to hematologic stress and induced CIN, ultimately pointing to fatal T-cell malignancies (164). Additionally, cyclin E/CDK2 overactivation due to FBXW7 deficiency could induce hyper-phosphorylation of the centromere protein CENtromere Protein A (CENP-A) at the N-terminal Ser18 site, thus promoting chromosome missegregation and micronucleus formation, demonstrating that the FBXW7-dependent cyclin E is related with CIN via centromere dysfunction (165, 166).
Several essential proteins and microRNAs existing in or interacting with the FBXW7/cyclin E pathway may be underlying targets to overturn genomic instability and disrupt tumorigenesis. Both functioning as the upstream proteins of FBXW7, the integrator protein TRIP-Br coordinates with the transcription regulator early 2 factor (E2F) to regulate S phase execution and maintain genomic stability partly via the FBXW7/cyclin E axis (167). MiR-223 targets the 3’-untranslated region of FBXW7 to regulate cyclin E activity and cyclin E-mediated genomic stability, whereas miR-223 expression can be responsive to the acute changes of cyclin E expression, thus forming a negative feedback circuit among miR-223, FBXW7, and cyclin E (168). Meanwhile, Aurora-A amplification was detected frequently in CRC tissues and significantly correlated with a fractional allelic loss (FAL) score, so targeting Aurora-A kinase in the FBXW7/cyclin E pathway can be a potential CRC therapy associated with CIN (169). The excessive activation of the oncogenic ERK1/2 MAP kinase (MAPK) pathway results in the downregulation of FBXW7β both in epithelial cell lines and in the intestine tissue of a transgenic mouse model, conferring the stabilization of Aurora-A, abnormal cell division, and polyploidization. Consequently, the ERK1/2/FBXW7/Aurora-A axis is associated with aneuploidy and epithelial malignancies, and the MAPK can be a good candidate for target therapy in the not-too-distant future (170). Under the governance of p53-dependent transcription regulation, FBXW7 protects epithelial cells from DNA damage and malignant progression presumably through its substrate such as cyclin E, Aurora A, or c-MYC, and p53 heterozygosity combined with FBXW7 loss can confer aneuploidy as well as epithelial cancers (171). Another in vitro study generated adenocarcinomas exhibiting aneuploidy, which were derived from FBXW7-/-; p53-/- cell lines, implicating that ablation of FBXW7 and p53 synergistically promotes the occurrence of CIN and CRC tumorigenesis (172).
As major components of surveillance mechanism, cell cycle checkpoints ensure the order, integrity, and fidelity of crucial events throughout the cell cycle, monitoring cell size, the responses to DNA replication, and DNA damage (173). Under the stress of UV irradiation, FBXW7α induces proliferation arrest in the S phase through interacting with and ubiquitinating polo-like kinase 1 (PLK1) that functions in eukaryotic cytokinesis, showing the role of the FBXW7/PLK1 axis in the regulation of the intra-S-phase DNA-damage checkpoint (174). The mice bearing P48-Cre; LSL-KRASG12D; FBXW7fl/fl (KFCfl/fl) mutations exhibited progressive pancreatic tumorigenesis with elevated numbers of micronuclei, as a result of Yes-associated protein (YAP) overexpression due to FBXW7 deficiency (175). The role of oncogenic protein YAP in driving genomic instability has also been established in a medulloblastoma mouse model. Mechanically, YAP triggers the expression of insulin-like growth factor 2 (IGF2) and ensuing activation of the PI3K/AKT pathway after radiation, thus avoiding DNA repair and inactivating the checkpoint regulators of ATM and CHK2, the latter inhibiting the G1/S and G2/M checkpoints (176).
In terms of repairing machinery, FBXW7 participates in the Fanconi anemia (FA) pathway that deals with DNA interstrand cross-link (ICL) repair through ubiquitinating and degrading the crucial component of the FA pathway, FAAP20 (177). Additionally, FBXW7 promotes nonhomologous end-joining (NHEJ) that resolves DNA double-strand breaks (DSBs) in mammalian cells via FBXW7α-dependent XRCC4 polyubiquitylation (178).
3 Perspectives: Potential Therapeutic Strategies
Therapeutic resistance remains one of the thorniest obstacles in the course of cancer treatment, which are associated with relapse and poor prognosis of patients. Tumors can be resistant due to a single or a combination of biological determining factors, involving intrinsic factors such as tumor burden and growth kinetics, tumor heterogeneity, physical barriers, tumor microenvironment, and undruggable genomic drivers (e.g., c-MYC and TP53), and extrinsic factors such as selective therapeutic pressure. The discussed hallmarks of cancer associated with FBXW7 present necessary insights to better understand the relationship between FBXW7 and therapeutic resistance, emphasizing the urgency of establishing novel targets for precise therapy. We propose three potential strategies against therapeutic resistance as follows (seen in Figure 4).
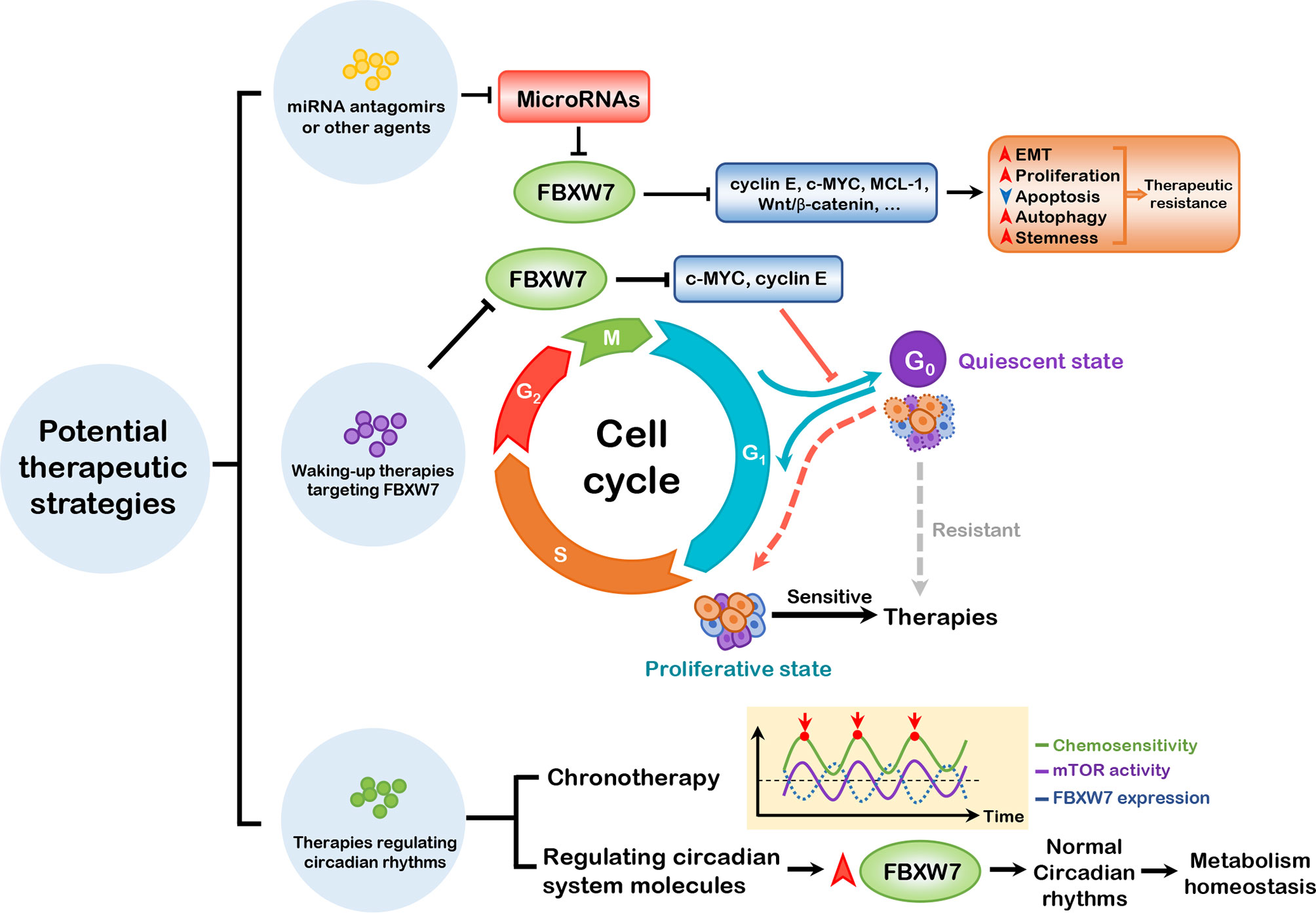
Figure 4 Three potential therapeutic strategies. miRNA antagomirs or other agents can be administered to block the suppression of FBXW7 expression by miRNAs, thus inhibiting the substrates of FBXW7, which promote EMT, proliferation, autophagy, and stemness and dampen apoptosis to induce therapeutic resistance. Waking-up therapies can be applied to prevent exit of cells from cell cycle regulated by FBXW7, thus waking up cancer stem cells from quiescent to proliferative state. Based on the role of FBXW7 in regulating circadian rhythms, it is ponderable to introduce chronotherapy and elevating FBXW7 expression to attain better therapeutic efficacy. EMT, epithelial–mesenchymal transition; MCL-1, myeloid cell leukemia-1.
3.1 Targeting MicroRNAs to Enhance Therapeutic Sensitivity of Cancers
miRNAs refer to short endogenous non-coding RNAs with a length of ∼22 nucleotides, targeting at the complementary site of mRNAs to mediate the degradation or repression of different genes that modulate development, apoptosis, differentiation, and even tumorigenesis (179). Frequently altered miRNAs in diseases make it possible to target these molecules in the form of miRNA mimics or anti-miRs for cancer therapy, combined with the effective application of RNA-delivering constructs (180). An increasing number of in vitro and in vivo studies has implicated that microRNAs can regulate cancer progression and therapeutic resistance by virtue of interacting with FBXW7, such as miR-223, miR-363, miR-27b-3p, and miR-92a-3p (181–184).
In addition to functioning in the hematopoietic system, miR-223 is in close relationship with multiple malignancies, given that miR-223 targets (e.g., IGF-1 receptor and stathmin 1) have been disclosed to impact the hallmarks of cancer, involving proliferation, invasion, metastasis, etc. (185). In vitro and in vivo studies have substantiated that the upregulated expression of miR-223 dampens therapeutic sensitivity in several cancer types, such as T-ALL, GC, NSCLC, HCC, and CRC (seen in Table 1). For example, miR-223 was detected upregulated in cisplatin-resistant GC cell lines, and the elevated expression of miR-223 inhibits cisplatin sensitivity of GC cells in vitro; these results can be explained by miR-223 target protein FBXW7, which regulates cell cycle and apoptosis presumably through ubiquitin-mediated proteolysis of MCL-1 (187).

Table 1 MicroRNAs regulate therapeutic resistance via targeting FBXW7 and the possible mechanisms.
Furthermore, several therapeutic agents or signaling molecules are involved in the regulation of miRNAs and its downstream FBXW7, so that miRNAs or its upstream proteins can be critical targets to enhance the sensitivity of chemo-, immuno-, and radiotherapies. We find that inhibiting the upstream regulators of miRNAs can be of great value to combat therapeutic resistance. For example, serving as negative regulators of miR-223, the Notch and Akt inhibitors have shown efficacy in increasing drug sensitivity of T-ALL and NSCLC, respectively. Another example is that the bioactive natural product honokiol downregulates miR-188-5p and further attenuates the inhibition of the FBXW7/c-MYC axis, thus increasing doxorubicin sensitivity in BC cells (195). Nonetheless, the in vivo combined with in vitro studies on miRNA upstream regulators are relatively rare, the mechanisms of miRNAs targeting FBXW7 to confer therapeutic resistance of cancers remain elusive, and the effective delivery of miRNA mimics or anti-miRNAs to realize significant clinical values is awaiting to be elucidated.
3.2 Waking-up Therapy
CSCs, which consistently dwell within the tumor microenvironment, is defined as a subpopulation of cancer cells with the ability of self-renewing and differentiating into multiple types of heterogeneous cancers (196) like normal stem cells. The stemness properties maintain CSCs in a quiescent and non-proliferative state (also called dormancy), persistently staying in the G0 stage and infrequently entering the cell cycle, which is associated with therapeutic resistance, maintenance, as well as anti-apoptosis (197).
Serving as a suppressor protein, FBXW7 attenuates tumor stemness by orchestrating different pathways. EMT, a critical cellular process as mentioned before, is associated with acquisition of stem cell traits of normal as well as cancer cells (198, 199), and functions as a crucial regulator of CSCs (101, 200). The mTOR complex (composed of mTORC1 and mTORC2) has been reported to play a key role in activating the EMT process (201), which is one of FBXW7’s substrates. In colon cancer cells, FBXW7 deficiency results in accumulation of mTOR, thus in turn favoring EMT-related stem-like properties (202). The ZEB family includes ZEB1 and ZEB2, both of which are crucial transcription factors subordinated to EMT-inducing transcription factors (EMT-TFs), which are usually endowed with stemness features (203). ZEB1, serving as a transcription factor promoting the EMT process (204), is associated with the stem-like properties of cholangiocarcinoma (CCA) cells, which can be explained by the fact that FBXW7 dampens EMT and stemness of CCA cells via the mTOR/ZEB1 signaling pathway (205). ZEB2 can be directly bound and degraded by FBXW7, and the FBXW/ZEB2 axis modulates stemness/differentiation, therapeutic resistance, as well as metastasis in CRC cells (206).
Similar to the EMT process, the Wnt/β-catenin signaling exerts its pivotal role in regulating functions of normal stem cells as well as CSCs (207, 208). Cancer-associated fibroblasts (CAFs), the stromal cells dwelling in tumor microenvironment, is involved in tumor progression. CAFs can deliver exosomal miR-92a-3p to CRC cells, which in turn activates Wnt/β-catenin pathway by suppressing FBXW7 and Modulator of apoptosis 1 (MOAP1) and finally promotes cell stemness (184). Moreover, the exosomal miR-19b derived from CRC cells promotes stemness by inhibiting FBXW7 expression, which subsequently induces Wnt/β-catenin signaling pathway (193). In perihilar cholangiocarcinoma (pCCA), the high mobility group A1 (HMGA1) interacts with thyroid hormone receptor interactor 13 (TRIP13), and the HMGA1-TRIP13 axis promotes cell stemness by downregulating FBXW7 expression and thus stabilizing c-MYC, the latter in interplay with Wnt/β-catenin signaling (209).
Contrary to the aforementioned onco-suppressive role, FBXW7 is capable of promoting cancer progression by maintaining stemness of cancer cells. FBXW7 targets positive regulators of cell cycle, such as cyclin E (210) and c-MYC (211) for ubiquitination and subsequent degradation, hence inducing exit from cell cycle and maintaining cells within the quiescent state (212, 213). Among these substrates, c-MYC is pivotal to reverse the quiescent state of CSCs and thus induce them into cell cycle. For example, FBXW7 deficiency gives rise to premature loss of normal HSC, which largely boils down to c-Myc-triggered active cell cycling, showing that FBXW7 is significant for hematopoietic stem cell (HSC) maintenance (214). In chronic myeloid leukemia (CML), FBXW7 expression is integral to initiation, progression, and maintenance of leukemia stem cells (LSCs) associated with downregulation of c-MYC (215), and ablation of FBXW7 results in increased c-MYC and activated p53 pathway, further promoting LSC apoptosis (216). In addition to non-solid tumors, FBXW7 also exerts a functional role in CSCs of solid tumors. Under the condition of cycle arrest due to anticancer agent therapy, c-MYC expression is reduced due to the elevated level of FBXW7, and upregulated FBXW7 confers chemoresistance on colorectal CSCs (217). Depletion of FBXW7 dominantly reduces gefitinib-resistant CSC population in NSCLCs and alters cell cycling in the G0/G1 phase (218). Similarly, FBXW7 diminution enhances the proliferative identity of breast CSCs and the sensitivity to paclitaxel therapy in vitro, and FBXW7 ablation combined with chemotherapy exhibits longer survival period and more favorable prognosis of mice in vivo (219).
Based on this, waking up CSCs from a quiescent to a proliferative state by targeting its internal molecules and cues can be a potential strategy to break the vital bottleneck. Waking-up therapy has been applied for CML patients in several clinical trials, with no distinct risk detected in combination with standard regiments (219). Nonetheless, there remain several barriers to finally realize efficacy of the waking-up therapy targeting FBXW7. First, we wonder how to leverage the bi-functions of FBXW7 in stemness maintenance in order to countervail therapeutic resistance. Just as mentioned before, on the one hand, FBXW7 has been reported to dampen stemness properties of CSCs through the EMT and Wnt/β-catenin signaling pathways; on the other hand, the genetic silencing of FBXW7 confers accumulation of c-MYC, finally repressing the state of dormancy and sensitizing cancer cells to therapies, which has been substantiated in vitro and in vivo studies. Second, the delivery of drugs may bring out side effects on cancer cells aside from CSCs, such as alterations of proliferation, apoptosis, invasion, and metastasis based on our general concept of FBXW7 as a tumor suppressor. Third, although a part of these thorniest puzzles can be handled out by virtue of combined therapies, there is still a long way to go for real clinical applications on cancer patients not just mice models.
3.3 Therapies Based on Regulating Circadian Rhythms
Circadian rhythms can govern a large array of biological processes linked to body homeostasis, characterized by periodic oscillations of 24 h, which is modulated by an endogenous clock system (220, 221). The autonomous, organized circadian system is composed of three critical components including input pathways, central pacemaker, and output pathways, among which the central pacemaker is located in suprachiasmatic nucleus (SCN) of anterior hypothalamus (220). Circadian rhythms are orchestrated by intricate transcriptional–translational system feedback loops, which are governed by clock genes. Circadian locomotor output cycles kaput (CLOCK) and brain and muscle ARNT-like 1 (BMAL1) heterodimerize as a complex and bind to E-boxes of promoters on clock-controlled genes, which triggers their transcriptions containing Period (Per) and Cryptochrome (Cry) families during the day; proteins encoded by Per and Cry families suppress CLOCK/BMAL1 transcription activity during the night (222). In addition, core clock proteins are under the control of the orphan nuclear receptors REV-ERBs (including REV-ERBα and REV-ERBβ) and retinoic acid receptor-related orphan receptors (RORs), respectively (223). Dysregulation of circadian rhythms can contribute to metabolic disorders, carcinogenesis, immune inefficiency, poorer cancer prognosis, and attenuation of drug efficacy on cancer patients (221, 224).
The circadian clock protein cryptochrome 2 (CRY2) is overexpressed in specimen of CRC patients, correlated with chemoresistance and poor prognosis. Furthermore, FBXW7 negatively regulates CRY2 via a ubiquitin-dependent pathway and potentiates chemosensitivity of CRC cells, which proposes the method to enhance the antitumor efficacy targeting clock proteins (225). After being phosphorylated by cyclin-dependent kinase 1 (CDK1), the negative regulator of clock transcription REV-ERBα can be degraded by FBXW7, favoring amplitude of clock transcription. Meanwhile, rhythm disruption induced by FBXW7-specific deficiency of liver perturbs the circadian homeostasis of lipid and glucose metabolism (226), recognized as a critical impediment to tumorigenesis. In addition, as an integral part of regulating cell metabolism, mTOR signaling pathway is detected to show an oscillated 24-h rhythm in RCC cells in part caused by fluctuated expression of FBXW7 protein, whose pattern shows anti-phase of rhythm of mTOR protein. Experiment results indicate that D-site binding protein (DBP) binds to D-site element in the FBXW7 promoter and also exhibits a circadian rhythm of protein levels, thus regulating the transcriptional activity of FBXW7. The higher activity of mTOR in tumor-bearing mice is correlated to better efficacy of everolimus, underpinning the significant impacts of circadian rhythm of mTOR signaling on the antitumor therapies (82).
Based on the association of FBXW7 and circadian rhythms, we outline this potential therapy in the bud, which elevates therapeutic responses through regulation of circadian rhythms involving FBXW7. Dysregulated FBXW7 expression renders tumorigenesis by conferring disrupted circadian rhythms as well as metabolism, thus providing a potential target for curbing tumorigenesis and elevating therapeutic responses. In addition, FBXW7 modulates the activity of critical molecules residing in the circadian system (such as mTOR), and the activity of mTOR in tumor-bearing mice is correlated to better efficacy of mTOR inhibitor everolimus, so leveraging the oscillated therapeutic sensitivity is critical to our therapy. As a consequence, FBXW7 expression is upregulated to maintain circadian rhythm and metabolism homeostasis, and chronotherapy is provided when the activity of molecules within the circadian system regulated by FBXW7 is the highest, both of which can be taken advantage of in order to attain better efficacy. Nevertheless, the potential therapy we have proposed is in the bud, which urgently requires in-depth exploration of critical molecules within the clock system as well as their oscillated rhythms and fluctuated sensitivity at different times of the day.
4 Concluding Remarks
UPS has been heralded as the main degradation mechanism of eukaryotes, located at the crossroads of multiple cellular processes. Considering the relationship between abnormalities of UPS and carcinogenesis, we focus on FBXW7, a component of the SCF ubiquitin ligase complexes, which mainly serves as a tumor suppressor through ubiquitin-mediated degradation of its substrates, such as c-MYC, mTOR, MCL-1, Notch, c-Jun, and cyclin E. Being devoid of FBXW7 results in the accumulation of target proteins, demonstrated in in vitro and in vivo studies to impact the initiation, development, relapse, and therapeutic responses of multiple cancers. The expression levels of FBXW7 in most malignancies are downregulated compared to normal tissues, such as glioma, lung cancer, liver cancer, urothelial cancer, ovarian cancer, and melanoma (18), which have a positive correlation with prognosis of patients, aside from liver cancer and melanoma. Other studies have also shown that expression pattern alterations of FBXW7 are correlated with malignancy progression of cancer cells, as well as clinicopathological classification and prognosis of patients who are predisposed to relapse, so FBXW7 may figure as a predictor of initiation, development, and prognosis of cancers (213, 227, 228).
Here, we discuss how FBXW7 influences the hallmarks of cancer that are subdivided into 7 parts. We infer that the pathways underlying cancer hallmarks can be intricate and interwoven with each other, for example, that the crucial transcriptional factor c-MYC regulated by FBXW7 participates in proliferation, apoptosis, invasion and metastasis, energy reprogramming, and immune evasion of cancers. In light of c-MYC as one of the undruggable genomic drivers, attenuating oncogenic c-MYC-induced cellular processes by targeting FBXW7 can be new therapeutic targets, which have been elaborated in previous sections. FBXW7’s role as a tumor suppressor has been pervasively explored. In practice, FBXW7 figures as a double-edged sword impacting tumorigenesis, especially its role in maintenance of cancer cell stemness.
Tumor burden and growth kinetics, tumor microenvironment, cell death inhibition, and EMT contribute to therapeutic resistance (229), which can be leveraged by targeting FBXW7 to elevate therapeutic sensitivity and the prognosis of patients. We provide three prospective strategies to deal with this problem. First, we take advantage of miRNAs interacting with FBXW7 to regulate therapeutic sensitivity, among which miR-223 has gained great attention of many researchers. miR-223 is an evolutionarily conserved microRNA mainly expressed in hematopoietic cells, which comes from endogenous expression or releasing of exosomes and EVs. miR-223 has been detected to be upregulated in T-ALL, GC, NSCLC, and HCC in vitro and in vivo, mediating therapeutic resistance presumably through FBXW7-regulated proliferation, apoptosis, EMT, and autophagy. More in vivo studies should substantiate the exact roles of microRNAs in tumorigenesis, and the effective delivery of miRNA mimics or anti-miRNAs remains to be elucidated, emerging as a novel target therapy in cancers over the years (180). Second, given FBXW7’s role in the maintenance of CSCs, we introduce the waking-up therapy regulating cell cycle by targeting the switch between quiescent and proliferative states, and propose several potential risks for application of this therapy. Through the genetic ablation of FBXW7, LSCs are predisposed to enter the cell cycle and are sensitive to chemotherapy. FBXW7 diminution combined with other drug therapies demonstrates great efficacy in patients of CML chronic phase. Last but not least, we unravel the relationship between FBXW7 and circadian rhythms, the latter in close interaction with metabolism, immune functions, and carcinogenesis. In order to maximize the therapeutic efficacy, taking advantage of chronotherapy or targeting critical molecules of the circadian machinery emerges as a new treatment paradigm in cancers in the foreseeable future. In addition, we are looking forward to incorporating additional hallmarks, continuing to explore FBXW7’s role in other facets of cancer.
Author Contributions
WS, QZ, CR, WL, and XJ conceived and designed the study and analyzed the results. Other authors performed analysis procedures. WS wrote the manuscript. QZ and WL revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the National Natural Science Foundation of China [Grant Nos. 81773179 and 81272972 (CR); Grant No. 81472355 (XJ)] and the Hunan Provincial Science and Technology Department [Grant No. 2016JC2049 (CR); Grant No. 2014FJ6006 (XJ)]. This work was also supported by the Students Innovations in Central South University of China (No. 2021105330188).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Hanahan D, Weinberg RA. The Hallmarks of Cancer. Cell (2000) 100(1):57–70. doi: 10.1016/s0092-8674(00)81683-9
2. Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
3. Hanahan D. Hallmarks of Cancer: New Dimensions. Cancer Discovery (2022) 12(1):31–46. doi: 10.1158/2159-8290.Cd-21-1059
4. Hershko A, Ciechanover A. The Ubiquitin System. Annu Rev Biochem (1998) 67:425–79. doi: 10.1146/annurev.biochem.67.1.425
5. Morreale FE, Walden H. Types of Ubiquitin Ligases. Cell (2016) 165(1):248–.e1. doi: 10.1016/j.cell.2016.03.003
6. Jia L, Sun Y. Scf E3 Ubiquitin Ligases as Anticancer Targets. Curr Cancer Drug Targets (2011) 11(3):347–56. doi: 10.2174/156800911794519734
7. Satija YK, Bhardwaj A, Das S. A Portrayal of E3 Ubiquitin Ligases and Deubiquitylases in Cancer. Int J Cancer (2013) 133(12):2759–68. doi: 10.1002/ijc.28129
8. Miyaki M, Yamaguchi T, Iijima T, Takahashi K, Matsumoto H, Mori T. Somatic Mutations of the Cdc4 (Fbxw7) Gene in Hereditary Colorectal Tumors. Oncology (2009) 76(6):430–4. doi: 10.1159/000217811
9. Kemp Z, Rowan A, Chambers W, Wortham N, Halford S, Sieber O, et al. Cdc4 Mutations Occur in a Subset of Colorectal Cancers But Are Not Predicted to Cause Loss of Function and Are Not Associated With Chromosomal Instability. Cancer Res (2005) 65(24):11361–6. doi: 10.1158/0008-5472.Can-05-2565
10. Kwak EL, Moberg KH, Wahrer DC, Quinn JE, Gilmore PM, Graham CA, et al. Infrequent Mutations of Archipelago (Hago, Hcdc4, Fbw7) in Primary Ovarian Cancer. Gynecol Oncol (2005) 98(1):124–8. doi: 10.1016/j.ygyno.2005.04.007
11. Strohmaier H, Spruck CH, Kaiser P, Won KA, Sangfelt O, Reed SI. Human F-Box Protein Hcdc4 Targets Cyclin E for Proteolysis and Is Mutated in a Breast Cancer Cell Line. Nature (2001) 413(6853):316–22. doi: 10.1038/35095076
12. Spruck CH, Strohmaier H, Sangfelt O, Müller HM, Hubalek M, Müller-Holzner E, et al. Hcdc4 Gene Mutations in Endometrial Cancer. Cancer Res (2002) 62(16):4535–9.
13. Malyukova A, Dohda T, von der Lehr N, Akhoondi S, Corcoran M, Heyman M, et al. The Tumor Suppressor Gene Hcdc4 Is Frequently Mutated in Human T-Cell Acute Lymphoblastic Leukemia With Functional Consequences for Notch Signaling. Cancer Res (2007) 67(12):5611–6. doi: 10.1158/0008-5472.Can-06-4381
14. Mendiratta G, Ke E, Aziz M, Liarakos D, Tong M, Stites EC. Cancer Gene Mutation Frequencies for the U.S. Population. Nat Commun (2021) 12(1):5961. doi: 10.1038/s41467-021-26213-y
15. Cao J, Ge MH, Ling ZQ. Fbxw7 Tumor Suppressor: A Vital Regulator Contributes to Human Tumorigenesis. Med (Baltimore) (2016) 95(7):e2496. doi: 10.1097/md.0000000000002496
16. Welcker M, Orian A, Grim JE, Eisenman RN, Clurman BE. A Nucleolar Isoform of the Fbw7 Ubiquitin Ligase Regulates C-Myc and Cell Size. Curr Biol (2004) 14(20):1852–7. doi: 10.1016/j.cub.2004.09.083
17. Xu C, Kim NG, Gumbiner BM. Regulation of Protein Stability by Gsk3 Mediated Phosphorylation. Cell Cycle (2009) 8(24):4032–9. doi: 10.4161/cc.8.24.10111
18. Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Proteomics. Tissue-Based Map Hum Proteome Sci (2015) 347(6220):1260419. doi: 10.1126/science.1260419
19. Koepp DM, Schaefer LK, Ye X, Keyomarsi K, Chu C, Harper JW, et al. Phosphorylation-Dependent Ubiquitination of Cyclin E by the Scffbw7 Ubiquitin Ligase. Science (2001) 294(5540):173–7. doi: 10.1126/science.1065203
20. Yeh CH, Bellon M, Nicot C. Fbxw7: A Critical Tumor Suppressor of Human Cancers. Mol Cancer (2018) 17(1):115. doi: 10.1186/s12943-018-0857-2
21. Xu W, Taranets L, Popov N. Regulating Fbw7 on the Road to Cancer. Semin Cancer Biol (2016) 36:62–70. doi: 10.1016/j.semcancer.2015.09.005
22. Qin Y, Hu Q, Xu J, Ji S, Dai W, Liu W, et al. Prmt5 Enhances Tumorigenicity and Glycolysis in Pancreatic Cancer Via the Fbw7/Cmyc Axis. Cell Commun Signal (2019) 17(1):30. doi: 10.1186/s12964-019-0344-4
23. Wang X, Zhang J, Zhou L, Sun W, Zheng ZG, Lu P, et al. Fbxw7 Regulates Hepatocellular Carcinoma Migration and Invasion Via Notch1 Signaling Pathway. Int J Oncol (2015) 47(1):231–43. doi: 10.3892/ijo.2015.2981
24. Tong J, Tan S, Zou F, Yu J, Zhang L. Fbw7 Mutations Mediate Resistance of Colorectal Cancer to Targeted Therapies by Blocking Mcl-1 Degradation. Oncogene (2017) 36(6):787–96. doi: 10.1038/onc.2016.247
25. Anzi S, Finkin S, Shaulian E. Transcriptional Repression of C-Jun’s E3 Ubiquitin Ligases Contributes to C-Jun Induction by Uv. Cell Signal (2008) 20(5):862–71. doi: 10.1016/j.cellsig.2007.12.020
26. Mao JH, Perez-Losada J, Wu D, Delrosario R, Tsunematsu R, Nakayama KI, et al. Fbxw7/Cdc4 Is a P53-Dependent, Haploinsufficient Tumour Suppressor Gene. Nature (2004) 432(7018):775–9. doi: 10.1038/nature03155
27. Xie CM, Sun Y. The Mtorc1-Mediated Autophagy Is Regulated by the Fbxw7-Shoc2-Rptor Axis. Autophagy (2019) 15(8):1470–2. doi: 10.1080/15548627.2019.1609864
28. Meyer N, Penn LZ. Reflecting on 25 Years With Myc. Nat Rev Cancer (2008) 8(12):976–90. doi: 10.1038/nrc2231
29. Lemaitre JM, Buckle RS, Méchali M. C-Myc in the Control of Cell Proliferation and Embryonic Development. Adv Cancer Res (1996) 70:95–144. doi: 10.1016/s0065-230x(08)60873-8
30. Yeh E, Cunningham M, Arnold H, Chasse D, Monteith T, Ivaldi G, et al. A Signalling Pathway Controlling C-Myc Degradation That Impacts Oncogenic Transformation of Human Cells. Nat Cell Biol (2004) 6(4):308–18. doi: 10.1038/ncb1110
31. Welcker M, Orian A, Jin J, Grim JE, Harper JW, Eisenman RN, et al. The Fbw7 Tumor Suppressor Regulates Glycogen Synthase Kinase 3 Phosphorylation-Dependent C-Myc Protein Degradation. Proc Natl Acad Sci USA (2004) 101(24):9085–90. doi: 10.1073/pnas.0402770101
32. Mihashi Y, Mizoguchi M, Takamatsu Y, Ishitsuka K, Iwasaki H, Koga M, et al. C-Myc and Its Main Ubiquitin Ligase, Fbxw7, Influence Cell Proliferation and Prognosis in Adult T-Cell Leukemia/Lymphoma. Am J Surg Pathol (2017) 41(8):1139–49. doi: 10.1097/pas.0000000000000871
33. Mao CG, Zhou XC, Jiang YD, Wan LJ, Tao ZZ, Guo J. The Evi5 Oncogene Promotes Laryngeal Cancer Cells Proliferation by Stabilizing C-Myc Protein. Cancer Cell Int (2020) 20:44. doi: 10.1186/s12935-020-1127-0
34. Lin H, Ma N, Zhao L, Yang G, Cao B. Kdm5c Promotes Colon Cancer Cell Proliferation Through the Fbxw7-C-Jun Regulatory Axis. Front Oncol (2020) 10:535449. doi: 10.3389/fonc.2020.535449
35. Fresno Vara JA, Casado E, de Castro J, Cejas P, Belda-Iniesta C, González-Barón M. Pi3k/Akt Signalling Pathway and Cancer. Cancer Treat Rev (2004) 30(2):193–204. doi: 10.1016/j.ctrv.2003.07.007
36. Li C, Lin XF, Wang JN, Ren XS. Fbxw7 Inhibited Cell Proliferation and Invasion Regulated by miR-27a Through Pi3k/Akt Signaling Pathway and Epithelial-To-Mesenchymal Transition in Oral Squamous Cell Carcinoma. Eur Rev Med Pharmacol Sci (2020) 24(7):3701–9. doi: 10.26355/eurrev_202004_20833
37. Hua H, Kong Q, Zhang H, Wang J, Luo T, Jiang Y. Targeting Mtor for Cancer Therapy. J Hematol Oncol (2019) 12(1):71. doi: 10.1186/s13045-019-0754-1
38. Mao JH, Kim IJ, Wu D, Climent J, Kang HC, DelRosario R, et al. Fbxw7 Targets Mtor for Degradation and Cooperates With Pten in Tumor Suppression. Science (2008) 321(5895):1499–502. doi: 10.1126/science.1162981
39. Wang Z, Liu Y, Zhang P, Zhang W, Wang W, Curr K, et al. Fam83d Promotes Cell Proliferation and Motility by Downregulating Tumor Suppressor Gene Fbxw7. Oncotarget (2013) 4(12):2476–86. doi: 10.18632/oncotarget.1581
40. Hori K, Sen A, Artavanis-Tsakonas S. Notch Signaling at a Glance. J Cell Sci (2013) 126(Pt 10):2135–40. doi: 10.1242/jcs.127308
41. Aster JC, Pear WS, Blacklow SC. The Varied Roles of Notch in Cancer. Annu Rev Pathol (2017) 12:245–75. doi: 10.1146/annurev-pathol-052016-100127
42. Aster JC, Pear WS, Blacklow SC. Notch Signaling in Leukemia. Annu Rev Pathol (2008) 3:587–613. doi: 10.1146/annurev.pathmechdis.3.121806.154300
43. Liu L, Jiang H, Wang X, Wang X, Zou L. Styx/Fbxw7 Axis Participates in the Development of Endometrial Cancer Cell Via Notch-Mtor Signaling Pathway. Biosci Rep (2020) 40(4):BSR20200057. doi: 10.1042/bsr20200057
44. Palomero T, Sulis ML, Cortina M, Real PJ, Barnes K, Ciofani M, et al. Mutational Loss of Pten Induces Resistance to Notch1 Inhibition in T-Cell Leukemia. Nat Med (2007) 13(10):1203–10. doi: 10.1038/nm1636
45. Li H, Wang Z, Zhang W, Qian K, Xu W, Zhang S. Fbxw7 Regulates Tumor Apoptosis, Growth Arrest and the Epithelial-To-Mesenchymal Transition in Part Through the Rhoa Signaling Pathway in Gastric Cancer. Cancer Lett (2016) 370(1):39–55. doi: 10.1016/j.canlet.2015.10.006
46. Kuai X, Li L, Chen R, Wang K, Chen M, Cui B, et al. Scf(Fbxw7)/Gsk3β-Mediated Gfi1 Degradation Suppresses Proliferation of Gastric Cancer Cells. Cancer Res (2019) 79(17):4387–98. doi: 10.1158/0008-5472.Can-18-4032
47. Gómez J, Martínez C, Giry M, García A, Rebollo A. Rho Prevents Apoptosis Through Bcl-2 Expression: Implications for Interleukin-2 Receptor Signal Transduction. Eur J Immunol (1997) 27(11):2793–9. doi: 10.1002/eji.1830271108
48. Michels J, Johnson PW, Packham G. Mcl-1. Int J Biochem Cell Biol (2005) 37(2):267–71. doi: 10.1016/j.biocel.2004.04.007
49. Warr MR, Shore GC. Unique Biology of Mcl-1: Therapeutic Opportunities in Cancer. Curr Mol Med (2008) 8(2):138–47. doi: 10.2174/156652408783769580
50. Akgul C. Mcl-1 Is a Potential Therapeutic Target in Multiple Types of Cancer. Cell Mol Life Sci (2009) 66(8):1326–36. doi: 10.1007/s00018-008-8637-6
51. Quinn BA, Dash R, Azab B, Sarkar S, Das SK, Kumar S, et al. Targeting Mcl-1 for the Therapy of Cancer. Expert Opin Investig Drugs (2011) 20(10):1397–411. doi: 10.1517/13543784.2011.609167
52. Inuzuka H, Shaik S, Onoyama I, Gao D, Tseng A, Maser RS, et al. Scf(Fbw7) Regulates Cellular Apoptosis by Targeting Mcl1 for Ubiquitylation and Destruction. Nature (2011) 471(7336):104–9. doi: 10.1038/nature09732
53. Inuzuka H, Fukushima H, Shaik S, Liu P, Lau AW, Wei W. Mcl-1 Ubiquitination and Destruction. Oncotarget (2011) 2(3):239–44. doi: 10.18632/oncotarget.242
54. Tong J, Tan S, Nikolovska-Coleska Z, Yu J, Zou F, Zhang L. Fbw7-Dependent Mcl-1 Degradation Mediates the Anticancer Effect of Hsp90 Inhibitors. Mol Cancer Ther (2017) 16(9):1979–88. doi: 10.1158/1535-7163.Mct-17-0032
55. He L, Torres-Lockhart K, Forster N, Ramakrishnan S, Greninger P, Garnett MJ, et al. Mcl-1 and Fbw7 Control a Dominant Survival Pathway Underlying Hdac and Bcl-2 Inhibitor Synergy in Squamous Cell Carcinoma. Cancer Discovery (2013) 3(3):324–37. doi: 10.1158/2159-8290.Cd-12-0417
56. Gasca J, Flores ML, Giráldez S, Ruiz-Borrego M, Tortolero M, Romero F, et al. Loss of Fbxw7 and Accumulation of Mcl1 and Plk1 Promote Paclitaxel Resistance in Breast Cancer. Oncotarget (2016) 7(33):52751–65. doi: 10.18632/oncotarget.10481
57. Gombodorj N, Yokobori T, Tanaka N, Suzuki S, Kuriyama K, Kumakura Y, et al. Correlation Between High Fbxw7 Expression in Pretreatment Biopsy Specimens and Good Response to Chemoradiation Therapy in Patients With Locally Advanced Esophageal Cancer: A Retrospective Study. J Surg Oncol (2018) 118(1):101–8. doi: 10.1002/jso.25127
58. Zheng M, Xu H, Liao XH, Chen CP, Zhang AL, Lu W, et al. Inhibition of the Prolyl Isomerase Pin1 Enhances the Ability of Sorafenib to Induce Cell Death and Inhibit Tumor Growth in Hepatocellular Carcinoma. Oncotarget (2017) 8(18):29771–84. doi: 10.18632/oncotarget.15967
59. Li C, Deng C, Pan G, Wang X, Zhang K, Dong Z, et al. Lycorine Hydrochloride Inhibits Cell Proliferation and Induces Apoptosis Through Promoting Fbxw7-Mcl1 Axis in Gastric Cancer. J Exp Clin Cancer Res (2020) 39(1):230. doi: 10.1186/s13046-020-01743-3
60. Ye M, Zhang Y, Zhang X, Zhang J, Jing P, Cao L, et al. Targeting Fbw7 as a Strategy to Overcome Resistance to Targeted Therapy in Non-Small Cell Lung Cancer. Cancer Res (2017) 77(13):3527–39. doi: 10.1158/0008-5472.Can-16-3470
61. Song X, Shen L, Tong J, Kuang C, Zeng S, Schoen RE, et al. Mcl-1 Inhibition Overcomes Intrinsic and Acquired Regorafenib Resistance in Colorectal Cancer. Theranostics (2020) 10(18):8098–110. doi: 10.7150/thno.45363
62. Nie X, Jia W, Li X, Pan X, Yin R, Liu N, et al. Fbxw7 Induces Apoptosis in Glioblastoma Cells by Regulating Hdac7. Cell Biol Int (2021) 45(10):2150–8. doi: 10.1002/cbin.11668
63. Bakin RE, Jung MO. Cytoplasmic Sequestration of Hdac7 From Mitochondrial and Nuclear Compartments Upon Initiation of Apoptosis. J Biol Chem (2004) 279(49):51218–25. doi: 10.1074/jbc.M409271200
64. Eilers M, Schirm S, Bishop JM. The Myc Protein Activates Transcription of the Alpha-Prothymosin Gene. EMBO J (1991) 10(1):133–41. doi: 10.1002/j.1460-2075.1991.tb07929.x
65. Evan GI, Wyllie AH, Gilbert CS, Littlewood TD, Land H, Brooks M, et al. Induction of Apoptosis in Fibroblasts by C-Myc Protein. Cell (1992) 69(1):119–28. doi: 10.1016/0092-8674(92)90123-t
66. Shi Y, Glynn JM, Guilbert LJ, Cotter TG, Bissonnette RP, Green DR. Role for C-Myc in Activation-Induced Apoptotic Cell Death in T Cell Hybridomas. Science (1992) 257(5067):212–4. doi: 10.1126/science.1378649
67. Jiang J, Wang J, Yue M, Cai X, Wang T, Wu C, et al. Direct Phosphorylation and Stabilization of Myc by Aurora B Kinase Promote T-Cell Leukemogenesis. Cancer Cell (2020) 37(2):200–15.e5. doi: 10.1016/j.ccell.2020.01.001
68. Zindy F, Eischen CM, Randle DH, Kamijo T, Cleveland JL, Sherr CJ, et al. Myc Signaling Via the Arf Tumor Suppressor Regulates P53-Dependent Apoptosis and Immortalization. Genes Dev (1998) 12(15):2424–33. doi: 10.1101/gad.12.15.2424
69. Onoyama I, Tsunematsu R, Matsumoto A, Kimura T, de Alborán IM, Nakayama K, et al. Conditional Inactivation of Fbxw7 Impairs Cell-Cycle Exit During T Cell Differentiation and Results in Lymphomatogenesis. J Exp Med (2007) 204(12):2875–88. doi: 10.1084/jem.20062299
70. Kim HS, Woolard K, Lai C, Bauer PO, Maric D, Song H, et al. Gliomagenesis Arising From Pten- and Ink4a/Arf-Deficient Neural Progenitor Cells Is Mediated by the P53-Fbxw7/Cdc4 Pathway, Which Controls C-Myc. Cancer Res (2012) 72(22):6065–75. doi: 10.1158/0008-5472.Can-12-2594
71. Ye Z, Zhuo Q, Hu Q, Xu X, Mengqi L, Zhang Z, et al. Fbw7-Nra41-Scd1 Axis Synchronously Regulates Apoptosis and Ferroptosis in Pancreatic Cancer Cells. Redox Biol (2021) 38:101807. doi: 10.1016/j.redox.2020.101807
72. Scaglia N, Igal RA. Inhibition of Stearoyl-Coa Desaturase 1 Expression in Human Lung Adenocarcinoma Cells Impairs Tumorigenesis. Int J Oncol (2008) 33(4):839–50.
73. Chen X, Li XY, Long M, Wang X, Gao ZW, Cui Y, et al. The Fbxw7 Tumor Suppressor Inhibits Breast Cancer Proliferation and Promotes Apoptosis by Targeting Mtdh for Degradation. Neoplasma (2018) 65(2):201–9. doi: 10.4149/neo_2018_170228N149
74. Tian W, Hao S, Gao B, Jiang Y, Zhang X, Zhang S, et al. Lobaplatin Inhibits Breast Cancer Progression, Cell Proliferation While It Induces Cell Apoptosis by Downregulating Mtdh Expression. Drug Des Devel Ther (2018) 12:3563–71. doi: 10.2147/dddt.S163157
75. Hu G, Wei Y, Kang Y. The Multifaceted Role of Mtdh/Aeg-1 in Cancer Progression. Clin Cancer Res (2009) 15(18):5615–20. doi: 10.1158/1078-0432.Ccr-09-0049
76. Li WF, Dai H, Ou Q, Zuo GQ, Liu CA. Overexpression of MicroRNA-30a-5p Inhibits Liver Cancer Cell Proliferation and Induces Apoptosis by Targeting Mtdh/Pten/Akt Pathway. Tumour Biol (2016) 37(5):5885–95. doi: 10.1007/s13277-015-4456-1
77. Levine B, Kroemer G. Autophagy in the Pathogenesis of Disease. Cell (2008) 132(1):27–42. doi: 10.1016/j.cell.2007.12.018
78. Mizushima N, Komatsu M. Autophagy: Renovation of Cells and Tissues. Cell (2011) 147(4):728–41. doi: 10.1016/j.cell.2011.10.026
79. Onorati AV, Dyczynski M, Ojha R, Amaravadi RK. Targeting Autophagy in Cancer. Cancer (2018) 124(16):3307–18. doi: 10.1002/cncr.31335
80. Li YJ, Lei YH, Yao N, Wang CR, Hu N, Ye WC, et al. Autophagy and Multidrug Resistance in Cancer. Chin J Cancer (2017) 36(1):52. doi: 10.1186/s40880-017-0219-2
81. Amaravadi RK, Kimmelman AC, Debnath J. Targeting Autophagy in Cancer: Recent Advances and Future Directions. Cancer Discovery (2019) 9(9):1167–81. doi: 10.1158/2159-8290.Cd-19-0292
82. Okazaki H, Matsunaga N, Fujioka T, Okazaki F, Akagawa Y, Tsurudome Y, et al. Circadian Regulation of Mtor by the Ubiquitin Pathway in Renal Cell Carcinoma. Cancer Res (2014) 74(2):543–51. doi: 10.1158/0008-5472.Can-12-3241
83. Kim YC, Guan KL. Mtor: A Pharmacologic Target for Autophagy Regulation. J Clin Invest (2015) 125(1):25–32. doi: 10.1172/jci73939
84. Fu L, Kim YA, Wang X, Wu X, Yue P, Lonial S, et al. Perifosine Inhibits Mammalian Target of Rapamycin Signaling Through Facilitating Degradation of Major Components in the Mtor Axis and Induces Autophagy. Cancer Res (2009) 69(23):8967–76. doi: 10.1158/0008-5472.Can-09-2190
85. Xie CM, Tan M, Lin XT, Wu D, Jiang Y, Tan Y, et al. The Fbxw7-Shoc2-Raptor Axis Controls the Cross-Talks Between the Ras-Erk and Mtorc1 Signaling Pathways. Cell Rep (2019) 26(11):3037–50.e4. doi: 10.1016/j.celrep.2019.02.052
86. Jing Y, Liang W, Liu J, Zhang L, Wei J, Yang J, et al. Autophagy-Mediating MicroRNAs in Cancer Chemoresistance. Cell Biol Toxicol (2020) 36(6):517–36. doi: 10.1007/s10565-020-09553-1
87. Wang H, Chen J, Zhang S, Zheng X, Xie S, Mao J, et al. miR-223 Regulates Autophagy Associated With Cisplatin Resistance by Targeting Fbxw7 in Human Non-Small Cell Lung Cancer. Cancer Cell Int (2020) 20:258. doi: 10.1186/s12935-020-01284-x
88. Folkman J. Tumor Angiogenesis: Therapeutic Implications. N Engl J Med (1971) 285(21):1182–6. doi: 10.1056/nejm197111182852108
89. Lv X, Li J, Zhang C, Hu T, Li S, He S, et al. The Role of Hypoxia-Inducible Factors in Tumor Angiogenesis and Cell Metabolism. Genes Dis (2017) 4(1):19–24. doi: 10.1016/j.gendis.2016.11.003
90. Hickey MM, Simon MC. Regulation of Angiogenesis by Hypoxia and Hypoxia-Inducible Factors. Curr Top Dev Biol (2006) 76:217–57. doi: 10.1016/s0070-2153(06)76007-0
91. Cassavaugh JM, Hale SA, Wellman TL, Howe AK, Wong C, Lounsbury KM. Negative Regulation of Hif-1α by an Fbw7-Mediated Degradation Pathway During Hypoxia. J Cell Biochem (2011) 112(12):3882–90. doi: 10.1002/jcb.23321
92. Flügel D, Görlach A, Kietzmann T. Gsk-3β Regulates Cell Growth, Migration, and Angiogenesis Via Fbw7 and Usp28-Dependent Degradation of Hif-1α. Blood (2012) 119(5):1292–301. doi: 10.1182/blood-2011-08-375014
93. Chiang CH, Chu PY, Hou MF, Hung WC. miR-182 Promotes Proliferation and Invasion and Elevates the Hif-1α-Vegf-A Axis in Breast Cancer Cells by Targeting Fbxw7. Am J Cancer Res (2016) 6(8):1785–98.
94. Tian X, Liu Y, Wang Z, Wu S. miR-144 Delivered by Nasopharyngeal Carcinoma-Derived Evs Stimulates Angiogenesis Through the Fbxw7/Hif-1α/Vegf-A Axis. Mol Ther Nucleic Acids (2021) 24:1000–11. doi: 10.1016/j.omtn.2021.03.016
95. Zhong L, Pan Y, Shen J. Fbxw7 Inhibits Invasion, Migration and Angiogenesis in Ovarian Cancer Cells by Suppressing Vegf Expression Through Inactivation of β-Catenin Signaling. Exp Ther Med (2021) 21(5):514. doi: 10.3892/etm.2021.9945
96. Izumi N, Helker C, Ehling M, Behrens A, Herzog W, Adams RH. Fbxw7 Controls Angiogenesis by Regulating Endothelial Notch Activity. PloS One (2012) 7(7):e41116. doi: 10.1371/journal.pone.0041116
97. Aydin IT, Melamed RD, Adams SJ, Castillo-Martin M, Demir A, Bryk D, et al. Fbxw7 Mutations in Melanoma and a New Therapeutic Paradigm. J Natl Cancer Inst (2014) 106(6):dju107. doi: 10.1093/jnci/dju107
98. Thiery JP. Epithelial-Mesenchymal Transitions in Tumour Progression. Nat Rev Cancer (2002) 2(6):442–54. doi: 10.1038/nrc822
99. Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-Mesenchymal Transitions in Development and Disease. Cell (2009) 139(5):871–90. doi: 10.1016/j.cell.2009.11.007
100. Dongre A, Weinberg RA. New Insights Into the Mechanisms of Epithelial-Mesenchymal Transition and Implications for Cancer. Nat Rev Mol Cell Biol (2019) 20(2):69–84. doi: 10.1038/s41580-018-0080-4
101. Lu W, Kang Y. Epithelial-Mesenchymal Plasticity in Cancer Progression and Metastasis. Dev Cell (2019) 49(3):361–74. doi: 10.1016/j.devcel.2019.04.010
102. Zhang Y, Zhang X, Ye M, Jing P, Xiong J, Han Z, et al. Fbw7 Loss Promotes Epithelial-To-Mesenchymal Transition in Non-Small Cell Lung Cancer Through the Stabilization of Snail Protein. Cancer Lett (2018) 419:75–83. doi: 10.1016/j.canlet.2018.01.047
103. Cai Y, Zhang M, Qiu X, Wang B, Fu Y, Zeng J, et al. Upregulation of Fbxw7 Suppresses Renal Cancer Metastasis and Epithelial Mesenchymal Transition. Dis Markers (2017) 2017:8276939. doi: 10.1155/2017/8276939
104. He H, Dai J, Xu Z, He W, Wang X, Zhu Y, et al. Fbxw7 Regulates Renal Cell Carcinoma Migration and Invasion Via Suppression of the Epithelial-Mesenchymal Transition. Oncol Lett (2018) 15(3):3694–702. doi: 10.3892/ol.2018.7744
105. Jiang G, Shi W, Fang H, Zhang X. miR−27a Promotes Human Breast Cancer Cell Migration by Inducing Emt in a Fbxw7−Dependent Manner. Mol Med Rep (2018) 18(6):5417–26. doi: 10.3892/mmr.2018.9587
106. Wang Y, Liu Z, Yao B, Li Q, Wang L, Wang C, et al. Long Non-Coding RNA Casc2 Suppresses Epithelial-Mesenchymal Transition of Hepatocellular Carcinoma Cells Through Casc2/miR-367/Fbxw7 Axis. Mol Cancer (2017) 16(1):123. doi: 10.1186/s12943-017-0702-z
107. Cowling VH, Cole MD. E-Cadherin Repression Contributes to C-Myc-Induced Epithelial Cell Transformation. Oncogene (2007) 26(24):3582–6. doi: 10.1038/sj.onc.1210132
108. Timmerman LA, Grego-Bessa J, Raya A, Bertrán E, Pérez-Pomares JM, Díez J, et al. Notch Promotes Epithelial-Mesenchymal Transition During Cardiac Development and Oncogenic Transformation. Genes Dev (2004) 18(1):99–115. doi: 10.1101/gad.276304
109. Zhang G, Zhu Q, Fu G, Hou J, Hu X, Cao J, et al. Trip13 Promotes the Cell Proliferation, Migration and Invasion of Glioblastoma Through the Fbxw7/C-Myc Axis. Br J Cancer (2019) 121(12):1069–78. doi: 10.1038/s41416-019-0633-0
110. Cheng Y, Chen G, Martinka M, Ho V, Li G. Prognostic Significance of Fbw7 in Human Melanoma and Its Role in Cell Migration. J Invest Dermatol (2013) 133(7):1794–802. doi: 10.1038/jid.2013.58
111. Jin X, Yang C, Fan P, Xiao J, Zhang W, Zhan S, et al. Cdk5/Fbw7-Dependent Ubiquitination and Degradation of Ezh2 Inhibits Pancreatic Cancer Cell Migration and Invasion. J Biol Chem (2017) 292(15):6269–80. doi: 10.1074/jbc.M116.764407
112. Fata JE, Ho AT, Leco KJ, Moorehead RA, Khokha R. Cellular Turnover and Extracellular Matrix Remodeling in Female Reproductive Tissues: Functions of Metalloproteinases and Their Inhibitors. Cell Mol Life Sci (2000) 57(1):77–95. doi: 10.1007/s000180050500
113. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science (2009) 324(5930):1029–33. doi: 10.1126/science.1160809
114. Ji S, Qin Y, Liang C, Huang R, Shi S, Liu J, et al. Fbw7 (F-Box and Wd Repeat Domain-Containing 7) Negatively Regulates Glucose Metabolism by Targeting the C-Myc/Txnip (Thioredoxin-Binding Protein) Axis in Pancreatic Cancer. Clin Cancer Res (2016) 22(15):3950–60. doi: 10.1158/1078-0432.Ccr-15-2380
115. Pavlova NN, Thompson CB. The Emerging Hallmarks of Cancer Metabolism. Cell Metab (2016) 23(1):27–47. doi: 10.1016/j.cmet.2015.12.006
116. Vaupel P, Schmidberger H, Mayer A. The Warburg Effect: Essential Part of Metabolic Reprogramming and Central Contributor to Cancer Progression. Int J Radiat Biol (2019) 95(7):912–9. doi: 10.1080/09553002.2019.1589653
117. Faubert B, Solmonson A, DeBerardinis RJ. Metabolic Reprogramming and Cancer Progression. Science (2020) 368(6487):eaaw5473. doi: 10.1126/science.aaw5473
118. DeBerardinis RJ, Chandel NS. Fundamentals of Cancer Metabolism. Sci Adv (2016) 2(5):e1600200. doi: 10.1126/sciadv.1600200
119. Xu RH, Pelicano H, Zhang H, Giles FJ, Keating MJ, Huang P. Synergistic Effect of Targeting Mtor by Rapamycin and Depleting Atp by Inhibition of Glycolysis in Lymphoma and Leukemia Cells. Leukemia (2005) 19(12):2153–8. doi: 10.1038/sj.leu.2403968
120. Perl A. Mtor Activation Is a Biomarker and a Central Pathway to Autoimmune Disorders, Cancer, Obesity, and Aging. Ann N Y Acad Sci (2015) 1346(1):33–44. doi: 10.1111/nyas.12756
121. Sun Q, Chen X, Ma J, Peng H, Wang F, Zha X, et al. Mammalian Target of Rapamycin Up-Regulation of Pyruvate Kinase Isoenzyme Type M2 Is Critical for Aerobic Glycolysis and Tumor Growth. Proc Natl Acad Sci USA (2011) 108(10):4129–34. doi: 10.1073/pnas.1014769108
122. Zhang P, Shao Y, Quan F, Liu L, Yang J. Fbp1 Enhances the Radiosensitivity by Suppressing Glycolysis Via the Fbxw7/Mtor Axis in Nasopharyngeal Carcinoma Cells. Life Sci (2021) 283:119840. doi: 10.1016/j.lfs.2021.119840
123. Xu Y, Tian C, Sun J, Zhang J, Ren K, Fan XY, et al. Fbxw7-Induced Mtor Degradation Forces Autophagy to Counteract Persistent Prion Infection. Mol Neurobiol (2016) 53(1):706–19. doi: 10.1007/s12035-014-9028-7
124. Nagao A, Kobayashi M, Koyasu S, Chow CCT, Harada H. Hif-1-Dependent Reprogramming of Glucose Metabolic Pathway of Cancer Cells and Its Therapeutic Significance. Int J Mol Sci (2019) 20(2):238. doi: 10.3390/ijms20020238
125. Li JN, Chen PS, Chiu CF, Lyu YJ, Lo C, Tsai LW, et al. Tarbp2 Suppresses Ubiquitin-Proteasomal Degradation of Hif-1α in Breast Cancer. Int J Mol Sci (2021) 23(1):208. doi: 10.3390/ijms23010208
126. Brown MS, Goldstein JL. The Srebp Pathway: Regulation of Cholesterol Metabolism by Proteolysis of a Membrane-Bound Transcription Factor. Cell (1997) 89(3):331–40. doi: 10.1016/s0092-8674(00)80213-5
127. Shimano H, Horton JD, Hammer RE, Shimomura I, Brown MS, Goldstein JL. Overproduction of Cholesterol and Fatty Acids Causes Massive Liver Enlargement in Transgenic Mice Expressing Truncated Srebp-1a. J Clin Invest (1996) 98(7):1575–84. doi: 10.1172/jci118951
128. Edwards PA, Tabor D, Kast HR, Venkateswaran A. Regulation of Gene Expression by Srebp and Scap. Biochim Biophys Acta (2000) 1529(1-3):103–13. doi: 10.1016/s1388-1981(00)00140-2
129. Horton JD, Shah NA, Warrington JA, Anderson NN, Park SW, Brown MS, et al. Combined Analysis of Oligonucleotide Microarray Data From Transgenic and Knockout Mice Identifies Direct Srebp Target Genes. Proc Natl Acad Sci USA (2003) 100(21):12027–32. doi: 10.1073/pnas.1534923100
130. Jeon TI, Osborne TF. Srebps: Metabolic Integrators in Physiology and Metabolism. Trends Endocrinol Metab (2012) 23(2):65–72. doi: 10.1016/j.tem.2011.10.004
131. Hirano Y, Yoshida M, Shimizu M, Sato R. Direct Demonstration of Rapid Degradation of Nuclear Sterol Regulatory Element-Binding Proteins by the Ubiquitin-Proteasome Pathway. J Biol Chem (2001) 276(39):36431–7. doi: 10.1074/jbc.M105200200
132. Sundqvist A, Bengoechea-Alonso MT, Ye X, Lukiyanchuk V, Jin J, Harper JW, et al. Control of Lipid Metabolism by Phosphorylation-Dependent Degradation of the Srebp Family of Transcription Factors by Scf(Fbw7). Cell Metab (2005) 1(6):379–91. doi: 10.1016/j.cmet.2005.04.010
133. Bengoechea-Alonso MT, Ericsson J. A Phosphorylation Cascade Controls the Degradation of Active Srebp1. J Biol Chem (2009) 284(9):5885–95. doi: 10.1074/jbc.M807906200
134. Xu S, Witmer PD, Lumayag S, Kovacs B, Valle D. MicroRNA (miRNA) Transcriptome of Mouse Retina and Identification of a Sensory Organ-Specific miRNA Cluster. J Biol Chem (2007) 282(34):25053–66. doi: 10.1074/jbc.M700501200
135. Huynh C, Segura MF, Gaziel-Sovran A, Menendez S, Darvishian F, Chiriboga L, et al. Efficient in Vivo MicroRNA Targeting of Liver Metastasis. Oncogene (2011) 30(12):1481–8. doi: 10.1038/onc.2010.523
136. Wei Q, Lei R, Hu G. Roles of miR-182 in Sensory Organ Development and Cancer. Thorac Cancer (2015) 6(1):2–9. doi: 10.1111/1759-7714.12164
137. Jeon TI, Esquejo RM, Roqueta-Rivera M, Phelan PE, Moon YA, Govindarajan SS, et al. An Srebp-Responsive MicroRNA Operon Contributes to a Regulatory Loop for Intracellular Lipid Homeostasis. Cell Metab (2013) 18(1):51–61. doi: 10.1016/j.cmet.2013.06.010
138. Shimano H, Sato R. Srebp-Regulated Lipid Metabolism: Convergent Physiology - Divergent Pathophysiology. Nat Rev Endocrinol (2017) 13(12):710–30. doi: 10.1038/nrendo.2017.91
139. Li S, Oh YT, Yue P, Khuri FR, Sun SY. Inhibition of Mtor Complex 2 Induces Gsk3/Fbxw7-Dependent Degradation of Sterol Regulatory Element-Binding Protein 1 (Srebp1) and Suppresses Lipogenesis in Cancer Cells. Oncogene (2016) 35(5):642–50. doi: 10.1038/onc.2015.123
140. Liu L, Zhao X, Zhao L, Li J, Yang H, Zhu Z, et al. Arginine Methylation of Srebp1a Via Prmt5 Promotes De Novo Lipogenesis and Tumor Growth. Cancer Res (2016) 76(5):1260–72. doi: 10.1158/0008-5472.Can-15-1766
141. Miller DM, Thomas SD, Islam A, Muench D, Sedoris K. C-Myc and Cancer Metabolism. Clin Cancer Res (2012) 18(20):5546–53. doi: 10.1158/1078-0432.Ccr-12-0977
142. Dang CV, Le A, Gao P. Myc-Induced Cancer Cell Energy Metabolism and Therapeutic Opportunities. Clin Cancer Res (2009) 15(21):6479–83. doi: 10.1158/1078-0432.Ccr-09-0889
143. Wu Z, Han X, Tan G, Zhu Q, Chen H, Xia Y, et al. Dioscin Inhibited Glycolysis and Induced Cell Apoptosis in Colorectal Cancer Via Promoting C-Myc Ubiquitination and Subsequent Hexokinase-2 Suppression. Onco Targets Ther (2020) 13:31–44. doi: 10.2147/ott.S224062
144. Li M, Gao F, Zhao Q, Zuo H, Liu W, Li W. Tanshinone Iia Inhibits Oral Squamous Cell Carcinoma Via Reducing Akt-C-Myc Signaling-Mediated Aerobic Glycolysis. Cell Death Dis (2020) 11(5):381. doi: 10.1038/s41419-020-2579-9
145. Olson BL, Hock MB, Ekholm-Reed S, Wohlschlegel JA, Dev KK, Kralli A, et al. Scfcdc4 Acts Antagonistically to the Pgc-1alpha Transcriptional Coactivator by Targeting It for Ubiquitin-Mediated Proteolysis. Genes Dev (2008) 22(2):252–64. doi: 10.1101/gad.1624208
146. Fontecha-Barriuso M, Martin-Sanchez D, Martinez-Moreno JM, Monsalve M, Ramos AM, Sanchez-Niño MD, et al. The Role of Pgc-1α and Mitochondrial Biogenesis in Kidney Diseases. Biomolecules (2020) 10(2):347. doi: 10.3390/biom10020347
147. Vazquez F, Lim JH, Chim H, Bhalla K, Girnun G, Pierce K, et al. Pgc1α Expression Defines a Subset of Human Melanoma Tumors With Increased Mitochondrial Capacity and Resistance to Oxidative Stress. Cancer Cell (2013) 23(3):287–301. doi: 10.1016/j.ccr.2012.11.020
148. Haq R, Shoag J, Andreu-Perez P, Yokoyama S, Edelman H, Rowe GC, et al. Oncogenic Braf Regulates Oxidative Metabolism Via Pgc1α and Mitf. Cancer Cell (2013) 23(3):302–15. doi: 10.1016/j.ccr.2013.02.003
149. Abbate F, Badal B, Mendelson K, Aydin IT, Serasinghe MN, Iqbal R, et al. Fbxw7 Regulates a Mitochondrial Transcription Program by Modulating Mitf. Pigment Cell Melanoma Res (2018) 31(5):636–40. doi: 10.1111/pcmr.12704
150. LaGory EL, Wu C, Taniguchi CM, Ding CC, Chi JT, von Eyben R, et al. Suppression of Pgc-1α Is Critical for Reprogramming Oxidative Metabolism in Renal Cell Carcinoma. Cell Rep (2015) 12(1):116–27. doi: 10.1016/j.celrep.2015.06.006
151. Davis RJ, Gönen M, Margineantu DH, Handeli S, Swanger J, Hoellerbauer P, et al. Pan-Cancer Transcriptional Signatures Predictive of Oncogenic Mutations Reveal That Fbw7 Regulates Cancer Cell Oxidative Metabolism. Proc Natl Acad Sci USA (2018) 115(21):5462–7. doi: 10.1073/pnas.1718338115
152. Vinay DS, Ryan EP, Pawelec G, Talib WH, Stagg J, Elkord E, et al. Immune Evasion in Cancer: Mechanistic Basis and Therapeutic Strategies. Semin Cancer Biol (2015) 35 Suppl:S185–s98. doi: 10.1016/j.semcancer.2015.03.004
153. Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-Associated Macrophages as Treatment Targets in Oncology. Nat Rev Clin Oncol (2017) 14(7):399–416. doi: 10.1038/nrclinonc.2016.217
154. Najafi M, Hashemi Goradel N, Farhood B, Salehi E, Nashtaei MS, Khanlarkhani N, et al. Macrophage Polarity in Cancer: A Review. J Cell Biochem (2019) 120(3):2756–65. doi: 10.1002/jcb.27646
155. Zhong L, Zhang Y, Li M, Song Y, Liu D, Yang X, et al. E3 Ligase Fbxw7 Restricts M2-Like Tumor-Associated Macrophage Polarization by Targeting C-Myc. Aging (Albany NY) (2020) 12(23):24394–423. doi: 10.18632/aging.202293
156. Singh S, Kumar S, Srivastava RK, Nandi A, Thacker G, Murali H, et al. Loss of Elf5-Fbxw7 Stabilizes Ifngr1 to Promote the Growth and Metastasis of Triple-Negative Breast Cancer Through Interferon-Γ Signalling. Nat Cell Biol (2020) 22(5):591–602. doi: 10.1038/s41556-020-0495-y
157. Balamurugan K, Sharan S, Klarmann KD, Zhang Y, Coppola V, Summers GH, et al. Fbxw7α Attenuates Inflammatory Signalling by Downregulating C/Ebpδ and Its Target Gene Tlr4. Nat Commun (2013) 4:1662. doi: 10.1038/ncomms2677
158. Balamurugan K, Wang JM, Tsai HH, Sharan S, Anver M, Leighty R, et al. The Tumour Suppressor C/Ebpδ Inhibits Fbxw7 Expression and Promotes Mammary Tumour Metastasis. EMBO J (2010) 29(24):4106–17. doi: 10.1038/emboj.2010.280
159. Huang B, Zhao J, Li H, He KL, Chen Y, Chen SH, et al. Toll-Like Receptors on Tumor Cells Facilitate Evasion of Immune Surveillance. Cancer Res (2005) 65(12):5009–14. doi: 10.1158/0008-5472.Can-05-0784
160. Zhao E, Maj T, Kryczek I, Li W, Wu K, Zhao L, et al. Cancer Mediates Effector T Cell Dysfunction by Targeting MicroRNAs and Ezh2 Via Glycolysis Restriction. Nat Immunol (2016) 17(1):95–103. doi: 10.1038/ni.3313
161. Spruck CH, Won KA, Reed SI. Deregulated Cyclin E Induces Chromosome Instability. Nature (1999) 401(6750):297–300. doi: 10.1038/45836
162. Fagundes R, Teixeira LK. Cyclin E/Cdk2: DNA Replication, Replication Stress and Genomic Instability. Front Cell Dev Biol (2021) 9:774845. doi: 10.3389/fcell.2021.774845
163. Rajagopalan H, Jallepalli PV, Rago C, Velculescu VE, Kinzler KW, Vogelstein B, et al. Inactivation of Hcdc4 Can Cause Chromosomal Instability. Nature (2004) 428(6978):77–81. doi: 10.1038/nature02313
164. Siu KT, Xu Y, Swartz KL, Bhattacharyya M, Gurbuxani S, Hua Y, et al. Chromosome Instability Underlies Hematopoietic Stem Cell Dysfunction and Lymphoid Neoplasia Associated With Impaired Fbw7-Mediated Cyclin E Regulation. Mol Cell Biol (2014) 34(17):3244–58. doi: 10.1128/mcb.01528-13
165. Zhang W, Karpen GH, Zhang Q. Exploring the Role of Cenp-A Ser18 Phosphorylation in Cin and Tumorigenesis. Cell Cycle (2017) 16(24):2323–5. doi: 10.1080/15384101.2017.1387698
166. Takada M, Zhang W, Suzuki A, Kuroda TS, Yu Z, Inuzuka H, et al. Fbw7 Loss Promotes Chromosomal Instability and Tumorigenesis Via Cyclin E1/Cdk2-Mediated Phosphorylation of Cenp-A. Cancer Res (2017) 77(18):4881–93. doi: 10.1158/0008-5472.Can-17-1240
167. Sim KG, Zang Z, Yang CM, Bonventre JV, Hsu SI. Trip-Br Links E2f to Novel Functions in the Regulation of Cyclin E Expression During Cell Cycle Progression and in the Maintenance of Genomic Stability. Cell Cycle (2004) 3(10):1296–304. doi: 10.4161/cc.3.10.1157
168. Xu Y, Sengupta T, Kukreja L, Minella AC. MicroRNA-223 Regulates Cyclin E Activity by Modulating Expression of F-Box and Wd-40 Domain Protein 7. J Biol Chem (2010) 285(45):34439–46. doi: 10.1074/jbc.M110.152306
169. Nishida N, Nagasaka T, Kashiwagi K, Boland CR, Goel A. High Copy Amplification of the Aurora-A Gene Is Associated With Chromosomal Instability Phenotype in Human Colorectal Cancers. Cancer Biol Ther (2007) 6(4):525–33. doi: 10.4161/cbt.6.4.3817
170. Duhamel S, Girondel C, Dorn JF, Tanguay PL, Voisin L, Smits R, et al. Deregulated Erk1/2 Map Kinase Signaling Promotes Aneuploidy by a Fbxw7β-Aurora a Pathway. Cell Cycle (2016) 15(12):1631–42. doi: 10.1080/15384101.2016.1183851
171. Perez-Losada J, Mao JH, Balmain A. Control of Genomic Instability and Epithelial Tumor Development by the P53-Fbxw7/Cdc4 Pathway. Cancer Res (2005) 65(15):6488–92. doi: 10.1158/0008-5472.Can-05-1294
172. Grim JE, Knoblaugh SE, Guthrie KA, Hagar A, Swanger J, Hespelt J, et al. Fbw7 and P53 Cooperatively Suppress Advanced and Chromosomally Unstable Intestinal Cancer. Mol Cell Biol (2012) 32(11):2160–7. doi: 10.1128/mcb.00305-12
173. Barnum KJ, O’Connell MJ. Cell Cycle Regulation by Checkpoints. Methods Mol Biol (2014) 1170:29–40. doi: 10.1007/978-1-4939-0888-2_2
174. Giráldez S, Herrero-Ruiz J, Mora-Santos M, Japón M, Tortolero M, Romero F. Scf(Fbxw7α) Modulates the Intra-S-Phase DNA-Damage Checkpoint by Regulating Polo Like Kinase-1 Stability. Oncotarget (2014) 5(12):4370–83. doi: 10.18632/oncotarget.2021
175. Zhang Q, Zhang Y, Parsels JD, Lohse I, Lawrence TS, Pasca di Magliano M, et al. Fbxw7 Deletion Accelerates Kras(G12d)-Driven Pancreatic Tumorigenesis Via Yap Accumulation. Neoplasia (2016) 18(11):666–73. doi: 10.1016/j.neo.2016.08.009
176. Fernandez LA, Squatrito M, Northcott P, Awan A, Holland EC, Taylor MD, et al. Oncogenic Yap Promotes Radioresistance and Genomic Instability in Medulloblastoma Through Igf2-Mediated Akt Activation. Oncogene (2012) 31(15):1923–37. doi: 10.1038/onc.2011.379
177. Wang J, Jo U, Joo SY, Kim H. Fbw7 Regulates DNA Interstrand Cross-Link Repair by Modulating Faap20 Degradation. Oncotarget (2016) 7(24):35724–40. doi: 10.18632/oncotarget.9595
178. Zhang Q, Karnak D, Tan M, Lawrence TS, Morgan MA, Sun Y. Fbxw7 Facilitates Nonhomologous End-Joining Via K63-Linked Polyubiquitylation of Xrcc4. Mol Cell (2016) 61(3):419–33. doi: 10.1016/j.molcel.2015.12.010
179. Bartel DP. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell (2004) 116(2):281–97. doi: 10.1016/s0092-8674(04)00045-5
180. Rupaimoole R, Slack FJ. MicroRNA Therapeutics: Towards a New Era for the Management of Cancer and Other Diseases. Nat Rev Drug Discovery (2017) 16(3):203–22. doi: 10.1038/nrd.2016.246
181. Kumar V, Palermo R, Talora C, Campese AF, Checquolo S, Bellavia D, et al. Notch and Nf-Kb Signaling Pathways Regulate miR-223/Fbxw7 Axis in T-Cell Acute Lymphoblastic Leukemia. Leukemia (2014) 28(12):2324–35. doi: 10.1038/leu.2014.133
182. Zhang PF, Sheng LL, Wang G, Tian M, Zhu LY, Zhang R, et al. miR-363 Promotes Proliferation and Chemo-Resistance of Human Gastric Cancer Via Targeting of Fbw7 Ubiquitin Ligase Expression. Oncotarget (2016) 7(23):35284–92. doi: 10.18632/oncotarget.9169
183. Frassanito MA, Desantis V, Di Marzo L, Craparotta I, Beltrame L, Marchini S, et al. Bone Marrow Fibroblasts Overexpress miR-27b and miR-214 in Step With Multiple Myeloma Progression, Dependent on Tumour Cell-Derived Exosomes. J Pathol (2019) 247(2):241–53. doi: 10.1002/path.5187
184. Hu JL, Wang W, Lan XL, Zeng ZC, Liang YS, Yan YR, et al. Cafs Secreted Exosomes Promote Metastasis and Chemotherapy Resistance by Enhancing Cell Stemness and Epithelial-Mesenchymal Transition in Colorectal Cancer. Mol Cancer (2019) 18(1):91. doi: 10.1186/s12943-019-1019-x
185. Aziz F, Chakraborty A, Khan I, Monts J. Relevance of miR-223 as Potential Diagnostic and Prognostic Markers in Cancer. Biol (Basel) (2022) 11(2):249. doi: 10.3390/biology11020249
186. Eto K, Iwatsuki M, Watanabe M, Ishimoto T, Ida S, Imamura Y, et al. The Sensitivity of Gastric Cancer to Trastuzumab Is Regulated by the miR-223/Fbxw7 Pathway. Int J Cancer (2015) 136(7):1537–45. doi: 10.1002/ijc.29168
187. Zhou X, Jin W, Jia H, Yan J, Zhang G. miR-223 Promotes the Cisplatin Resistance of Human Gastric Cancer Cells Via Regulating Cell Cycle by Targeting Fbxw7. J Exp Clin Cancer Res (2015) 34(1):28. doi: 10.1186/s13046-015-0145-6
188. Gao H, Ma J, Cheng Y, Zheng P. Exosomal Transfer of Macrophage-Derived miR-223 Confers Doxorubicin Resistance in Gastric Cancer. Onco Targets Ther (2020) 13:12169–79. doi: 10.2147/ott.S283542
189. Zhang H, Chen F, He Y, Yi L, Ge C, Shi X, et al. Sensitivity of Non-Small Cell Lung Cancer to Erlotinib Is Regulated by the Notch/miR-223/Fbxw7 Pathway. Biosci Rep (2017) 37(3):BSR20160478. doi: 10.1042/bsr20160478
190. Tang X, Yang W, Shu Z, Shen X, Zhang W, Cen C, et al. MicroRNA−223 Promotes Hepatocellular Carcinoma Cell Resistance to Sorafenib by Targeting Fbw7. Oncol Rep (2019) 41(2):1231–7. doi: 10.3892/or.2018.6908
191. Ding J, Zhao Z, Song J, Luo B, Huang L. miR-223 Promotes the Doxorubicin Resistance of Colorectal Cancer Cells Via Regulating Epithelial-Mesenchymal Transition by Targeting Fbxw7. Acta Biochim Biophys Sin (Shanghai) (2018) 50(6):597–604. doi: 10.1093/abbs/gmy040
192. Lin H, Zhang L, Zhang C, Liu P. Exosomal miR-500a-3p Promotes Cisplatin Resistance and Stemness Via Negatively Regulating Fbxw7 in Gastric Cancer. J Cell Mol Med (2020) 24(16):8930–41. doi: 10.1111/jcmm.15524
193. Sun T, Yin YF, Jin HG, Liu HR, Tian WC. Exosomal MicroRNA-19b Targets Fbxw7 to Promote Colorectal Cancer Stem Cell Stemness and Induce Resistance to Radiotherapy. Kaohsiung J Med Sci (2022) 38(2):108–19. doi: 10.1002/kjm2.12449
194. Wang J, Li T, Wang B. Exosomal Transfer of miR−25−3p Promotes the Proliferation and Temozolomide Resistance of Glioblastoma Cells by Targeting Fbxw7. Int J Oncol (2021) 59(2):64. doi: 10.3892/ijo.2021.5244
195. Yi X, Lou L, Wang J, Xiong J, Zhou S. Honokiol Antagonizes Doxorubicin Resistance in Human Breast Cancer Via miR-188-5p/Fbxw7/C-Myc Pathway. Cancer Chemother Pharmacol (2021) 87(5):647–56. doi: 10.1007/s00280-021-04238-w
196. Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, Jones DL, et al. Cancer Stem Cells–Perspectives on Current Status and Future Directions: Aacr Workshop on Cancer Stem Cells. Cancer Res (2006) 66(19):9339–44. doi: 10.1158/0008-5472.Can-06-3126
197. Essers MA, Trumpp A. Targeting Leukemic Stem Cells by Breaking Their Dormancy. Mol Oncol (2010) 4(5):443–50. doi: 10.1016/j.molonc.2010.06.001
198. Polyak K, Weinberg RA. Transitions Between Epithelial and Mesenchymal States: Acquisition of Malignant and Stem Cell Traits. Nat Rev Cancer (2009) 9(4):265–73. doi: 10.1038/nrc2620
199. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, et al. The Epithelial-Mesenchymal Transition Generates Cells With Properties of Stem Cells. Cell (2008) 133(4):704–15. doi: 10.1016/j.cell.2008.03.027
200. Nieto MA, Huang RY, Jackson RA, Thiery JP. Emt: 2016. Cell (2016) 166(1):21–45. doi: 10.1016/j.cell.2016.06.028
201. Karimi Roshan M, Soltani A, Soleimani A, Rezaie Kahkhaie K, Afshari AR, Soukhtanloo M. Role of Akt and Mtor Signaling Pathways in the Induction of Epithelial-Mesenchymal Transition (Emt) Process. Biochimie (2019) 165:229–34. doi: 10.1016/j.biochi.2019.08.003
202. Wang Y, Liu Y, Lu J, Zhang P, Wang Y, Xu Y, et al. Rapamycin Inhibits Fbxw7 Loss-Induced Epithelial-Mesenchymal Transition and Cancer Stem Cell-Like Characteristics in Colorectal Cancer Cells. Biochem Biophys Res Commun (2013) 434(2):352–6. doi: 10.1016/j.bbrc.2013.03.077
203. Bruneel K, Verstappe J, Vandamme N, Berx G. Intrinsic Balance Between Zeb Family Members Is Important for Melanocyte Homeostasis and Melanoma. Progression Cancers (Basel) (2020) 12(8):2248. doi: 10.3390/cancers12082248
204. Zhang P, Sun Y, Ma L. Zeb1: At the Crossroads of Epithelial-Mesenchymal Transition, Metastasis and Therapy Resistance. Cell Cycle (2015) 14(4):481–7. doi: 10.1080/15384101.2015.1006048
205. Yang H, Lu X, Liu Z, Chen L, Xu Y, Wang Y, et al. Fbxw7 Suppresses Epithelial-Mesenchymal Transition, Stemness and Metastatic Potential of Cholangiocarcinoma Cells. Oncotarget (2015) 6(8):6310–25. doi: 10.18632/oncotarget.3355
206. Li N, Babaei-Jadidi R, Lorenzi F, Spencer-Dene B, Clarke P, Domingo E, et al. An Fbxw7-Zeb2 Axis Links Emt and Tumour Microenvironment to Promote Colorectal Cancer Stem Cells and Chemoresistance. Oncogenesis (2019) 8(3):13. doi: 10.1038/s41389-019-0125-3
207. Zhan T, Rindtorff N, Boutros M. Wnt Signaling in Cancer. Oncogene (2017) 36(11):1461–73. doi: 10.1038/onc.2016.304
208. Fodde R, Brabletz T. Wnt/Beta-Catenin Signaling in Cancer Stemness and Malignant Behavior. Curr Opin Cell Biol (2007) 19(2):150–8. doi: 10.1016/j.ceb.2007.02.007
209. Li Z, Liu J, Chen T, Sun R, Liu Z, Qiu B, et al. Hmga1-Trip13 Axis Promotes Stemness and Epithelial Mesenchymal Transition of Perihilar Cholangiocarcinoma in a Positive Feedback Loop Dependent on C-Myc. J Exp Clin Cancer Res (2021) 40(1):86. doi: 10.1186/s13046-021-01890-1
210. Klotz K, Cepeda D, Tan Y, Sun D, Sangfelt O, Spruck C. Scf(Fbxw7/Hcdc4) Targets Cyclin E2 for Ubiquitin-Dependent Proteolysis. Exp Cell Res (2009) 315(11):1832–9. doi: 10.1016/j.yexcr.2008.11.017
211. Hydbring P, Castell A, Larsson LG. Myc Modulation Around the Cdk2/P27/Skp2 Axis. Genes (Basel) (2017) 8(7):174. doi: 10.3390/genes8070174
212. Onoyama I, Nakayama KI. Fbxw7 in Cell Cycle Exit and Stem Cell Maintenance: Insight From Gene-Targeted Mice. Cell Cycle (2008) 7(21):3307–13. doi: 10.4161/cc.7.21.6931
213. Iwatsuki M, Mimori K, Ishii H, Yokobori T, Takatsuno Y, Sato T, et al. Loss of Fbxw7, a Cell Cycle Regulating Gene, in Colorectal Cancer: Clinical Significance. Int J Cancer (2010) 126(8):1828–37. doi: 10.1002/ijc.24879
214. Matsuoka S, Oike Y, Onoyama I, Iwama A, Arai F, Takubo K, et al. Fbxw7 Acts as a Critical Fail-Safe Against Premature Loss of Hematopoietic Stem Cells and Development of T-All. Genes Dev (2008) 22(8):986–91. doi: 10.1101/gad.1621808
215. Takeishi S, Matsumoto A, Onoyama I, Naka K, Hirao A, Nakayama KI. Ablation of Fbxw7 Eliminates Leukemia-Initiating Cells by Preventing Quiescence. Cancer Cell (2013) 23(3):347–61. doi: 10.1016/j.ccr.2013.01.026
216. Reavie L, Buckley SM, Loizou E, Takeishi S, Aranda-Orgilles B, Ndiaye-Lobry D, et al. Regulation of C-Myc Ubiquitination Controls Chronic Myelogenous Leukemia Initiation and Progression. Cancer Cell (2013) 23(3):362–75. doi: 10.1016/j.ccr.2013.01.025
217. Izumi D, Ishimoto T, Miyake K, Eto T, Arima K, Kiyozumi Y, et al. Colorectal Cancer Stem Cells Acquire Chemoresistance Through the Upregulation of F-Box/Wd Repeat-Containing Protein 7 and the Consequent Degradation of C-Myc. Stem Cells (2017) 35(9):2027–36. doi: 10.1002/stem.2668
218. Hidayat M, Mitsuishi Y, Takahashi F, Tajima K, Yae T, Miyahara K, et al. Role of Fbxw7 in the Quiescence of Gefitinib-Resistant Lung Cancer Stem Cells in Egfr-Mutant Non-Small Cell Lung Cancer. Bosn J Basic Med Sci (2019) 19(4):355–67. doi: 10.17305/bjbms.2019.4227
219. Shimizu H, Takeishi S, Nakatsumi H, Nakayama KI. Prevention of Cancer Dormancy by Fbxw7 Ablation Eradicates Disseminated Tumor Cells. JCI Insight (2019) 4(4):e125138. doi: 10.1172/jci.insight.125138
220. Fu L, Lee CC. The Circadian Clock: Pacemaker and Tumour Suppressor. Nat Rev Cancer (2003) 3(5):350–61. doi: 10.1038/nrc1072
221. Sahar S, Sassone-Corsi P. Metabolism and Cancer: The Circadian Clock Connection. Nat Rev Cancer (2009) 9(12):886–96. doi: 10.1038/nrc2747
222. Kinouchi K, Sassone-Corsi P. Metabolic Rivalry: Circadian Homeostasis and Tumorigenesis. Nat Rev Cancer (2020) 20(11):645–61. doi: 10.1038/s41568-020-0291-9
223. Kojetin DJ, Burris TP. Rev-Erb and Ror Nuclear Receptors as Drug Targets. Nat Rev Drug Discovery (2014) 13(3):197–216. doi: 10.1038/nrd4100
224. Lévi F, Okyar A, Dulong S, Innominato PF, Clairambault J. Circadian Timing in Cancer Treatments. Annu Rev Pharmacol Toxicol (2010) 50:377–421. doi: 10.1146/annurev.pharmtox.48.113006.094626
225. Fang L, Yang Z, Zhou J, Tung JY, Hsiao CD, Wang L, et al. Circadian Clock Gene Cry2 Degradation Is Involved in Chemoresistance of Colorectal Cancer. Mol Cancer Ther (2015) 14(6):1476–87. doi: 10.1158/1535-7163.Mct-15-0030
226. Zhao X, Hirota T, Han X, Cho H, Chong LW, Lamia K, et al. Circadian Amplitude Regulation Via Fbxw7-Targeted Rev-Erbα Degradation. Cell (2016) 165(7):1644–57. doi: 10.1016/j.cell.2016.05.012
227. Kawashita Y, Morine Y, Ikemoto T, Saito Y, Iwahashi S, Yamada S, et al. Loss of Fbxw7 Expression Is a Predictor of Recurrence in Colorectal Liver Metastasis. J Hepatobiliary Pancreat Sci (2017) 24(10):576–83. doi: 10.1002/jhbp.500
228. Li MR, Zhu CC, Ling TL, Zhang YQ, Xu J, Zhao EH, et al. Fbxw7 Expression Is Associated With Prognosis and Chemotherapeutic Outcome in Chinese Patients With Gastric Adenocarcinoma. BMC Gastroenterol (2017) 17(1):60. doi: 10.1186/s12876-017-0616-7
Keywords: FBXW7, hallmarks of cancer, metabolic reprogramming, therapeutic resistance, c-MYC, mTOR
Citation: Shen W, Zhou Q, Peng C, Li J, Yuan Q, Zhu H, Zhao M, Jiang X, Liu W and Ren C (2022) FBXW7 and the Hallmarks of Cancer: Underlying Mechanisms and Prospective Strategies. Front. Oncol. 12:880077. doi: 10.3389/fonc.2022.880077
Received: 21 February 2022; Accepted: 15 March 2022;
Published: 19 April 2022.
Edited by:
Carmela De Marco, Magna Græcia University of Catanzaro, ItalyReviewed by:
Lisa Crawford, Queen’s University Belfast, United KingdomJie Li, Chinese Academy of Medical Sciences and Peking Union Medical College, China
Copyright © 2022 Shen, Zhou, Peng, Li, Yuan, Zhu, Zhao, Jiang, Liu and Ren. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xingjun Jiang, amlhbmd4akBjc3UuZWR1LmNu; Weidong Liu, TGl1d2Q2ODhAMTI2LmNvbQ==; Caiping Ren, cmVuY2FpcGluZ0Bjc3UuZWR1LmNu
†These authors have contributed equally to this work and share first authorship