 Xiaoye Duan
Xiaoye Duan Weihao Wang
Weihao Wang Qi Pan
Qi Pan Lixin Guo1,2*
Lixin Guo1,2*
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 16 August 2021
Sec. Gastrointestinal Cancers
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.730038
This article is part of the Research Topic Hard but Hopeful: the Clinical and Translational Research Progress in Pancreatic Cancer View all 13 articles
The relationship between type 2 diabetes mellitus (T2DM) and pancreatic cancer (PC) is complex. Diabetes is a known risk factor for PC, and new-onset diabetes (NOD) could be an early manifestation of PC that may be facilitate the early diagnosis of PC. Metformin offers a clear benefit of inhibiting PC, whereas insulin therapy may increase the risk of PC development. No evidence has shown that novel hypoglycemic drugs help or prevent PC. In this review, the effects of T2DM on PC development are summarized, and novel strategies for the prevention and treatment of T2DM and PC are discussed.
In recent years, the incidence of pancreatic cancer (PC) in the world has increased annually. PC has become the third leading cause of cancer-related death in the United States and the fourth in Japan (1). Despite considerable efforts in diagnosis and treatment, the 5-year survival rate has increased to only 10% (1). Because of nonspecific symptoms and a lack of screening recommendations, the vast majority of patients with PC are diagnosed at a late stage, and there is no opportunity for surgical intervention (2). According to data from Chinese Pancreatic Surgery Association, the 5-year overall survival rate of pancreatic cancer was is only 7.2% and the incidence of pancreatic cancer is expected to soar to the second place by 2030 in China (3). Unfortunately, early diagnosis rate of pancreatic cancer is only 5%. The proportion of estimated new cases of pancreatic cancer in China showed obvious regional characteristics, which is consistent with the result that the incidence and mortality increased from low to high urbanization areas in China, and the prevalence of diabetes increased from underdeveloped to developed region (4).
Type 2 diabetes mellitus (T2DM) is considered a risk factor for various malignant tumors, such as hepatocellular cancer, breast cancer, ovarian cancer, endometrial cancer, and gastrointestinal cancer. The incidence of cancer in patients with T2DM has increased by 10%, comparing the public population (5–7). Approximately 50% of patients with PC develop T2DM or impaired glucose tolerance at the very beginning (8). T2DM is a known risk factor for PC, and new-onset diabetes (NOD) may be an early manifestation of PC (9–11). Therefore, T2DM, especially NOD, may be a clue to early detection of PC and may improve the prognosis of this intractable malignant tumor.
However, the incidence of T2DM is too high to justify screening all patients with the condition for PC: the cost-benefit ratio does not justify such widespread use of medical resources. Additional risk stratification is needed in patients with T2DM. In this review, we discuss the mechanism of the relationship between T2DM and PC, update the literature about risk factors and biomarkers of PC in patients with T2DM, and summarize PC prevention and treatment strategies.
The mechanisms connecting T2DM with the formation and development of PC are multilayered and complex. Hyperglycemia, hyperinsulinemia, insulin resistance, chronic inflammation, and genetic factors all contribute to the association between these conditions (12).
In T2DM, hyperglycemia is caused by long-term excessive hepatic gluconeogenesis, decreased insulin activity, low peripheral glucose uptake, and changes in insulin signaling (13, 14). These events can cause cancer, especially PC (5–8) (Figure 1). In fact, patients can remain asymptomatic for many years, with undiscovered glucose intolerance and transient hyperglycemia. This time of prediabetes greatly increases the likelihood of developing PC (15, 16). One possible mechanism is the activation of the transforming growth factor-β1 (TGF-β1) pathway by glucose, which results in a decrease in the level of E-cadherin in pancreatic ductal cells and a significant mesenchymal phenotype that promotes tumor growth and metastasis (17). Hyperglycemia may also increase genetic instability and lead to KRAS mutations by activating O-GlcN acetylation and nucleotide deficiency (17, 18). Finally, the mTOR pathway controls protein synthesis and autophagy, and its deregulation is associated with diabetes and PC (19–23). Interestingly, inhibition of mTOR can reduce tumorigenesis in KRAS-dependent PC.
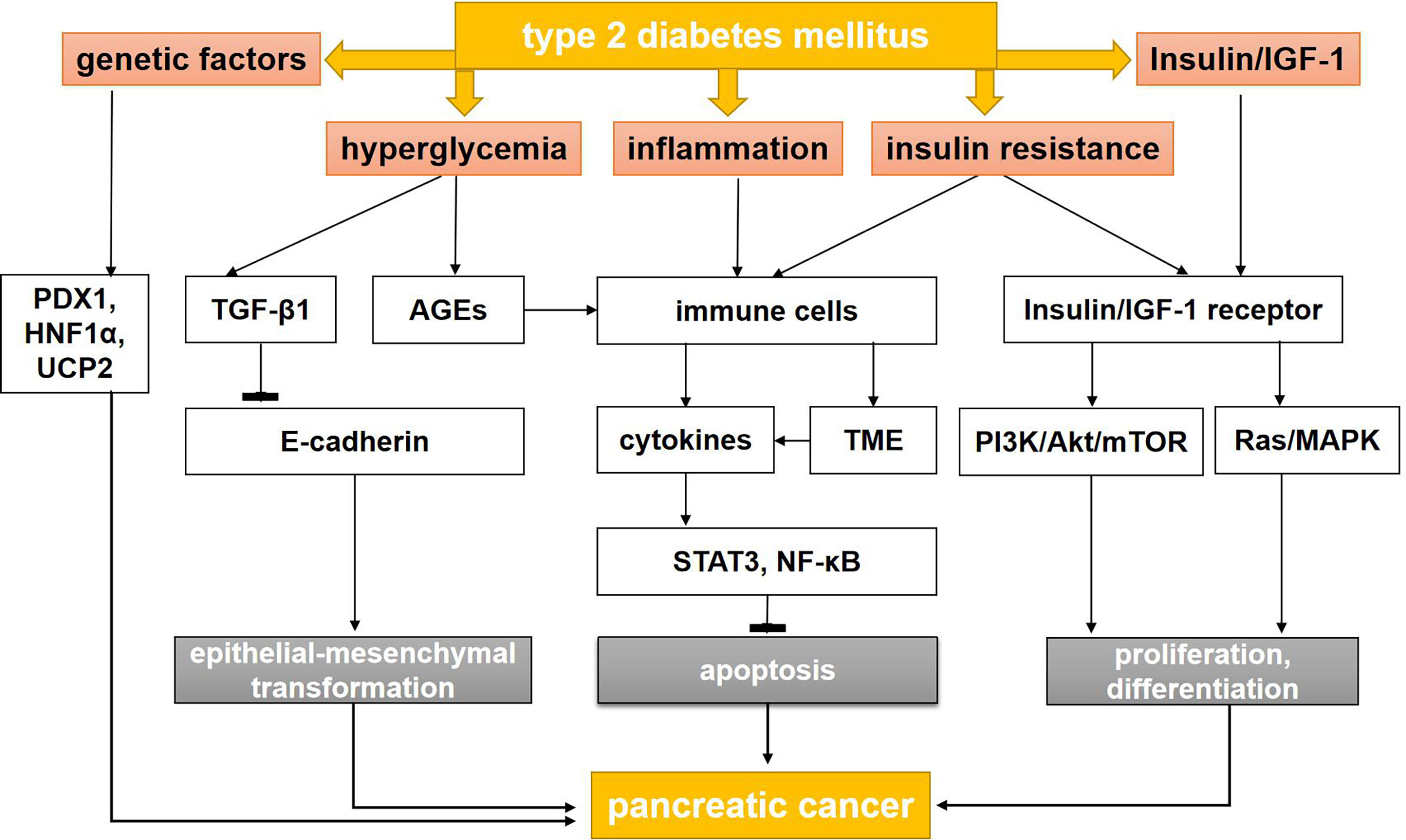
Figure 1 The mechanisms between type 2 diabetes mellitus and pancreatic cancer. AGEs, advanced glycation end products; AMPK, adenosine monophosphate protein-activated kinase; IGF-1, insulin-like growth factor-1; LKB, liver kinase B; MAPK, mitogen-activated protein kinase; mTOR, mammalian target of rapamycin; NF-κB, nuclear factor kappa B; PI3K, phosphatidyl inositol-3 kinase; STAT3, signal transducer and activator of transcription 3; TGF-β1, transforming growth factor-β1; TME, tumor microenvironment; PDX1, pancreatic and duodenal homeobox-1; HNF1A, HNF1 Homeobox A; UCP2, uncoupling protein 2.
The tumor-promoting effect of N-carboxymethyllysine was found. N-carboxymethyllysine is a RAGE ligand and a major AGE in pancreatic cancer cell lines. The researchers found that PC was observed in eight (72.7%) of the 11 mice treated with N-carboxymethyllysine but in only one mouse (9.1%) in the control group (25). N-carboxymethyllysine upregulated the expression of RAGE in a concentration- and time-dependent manner, activated leukocyte cell adhesion molecule, and promoted the growth of PC cells (25). Reducing AGEs may be a good way to prevent PC.
Prospective cohort and case-control studies have shown that hyperglycemia is associated with increased free radical formation and may lead to the development of advanced glycosylation end product (AGEs), which may increase inflammation (24). The use of exogenous AGE in PC-susceptible mice can upregulate the expression of the AGE receptor (RAGE) in pancreatic intraepithelial neoplasia and greatly stimulate the development of invasive PC (25).
In 2018, Rahn et al. (17) explored the role of hyperglycemia in the malignant transformation of pancreatic ductal epithelial cell (PDEC), the occurrence and maintenance of cancer stem cells (CSCs), and the promotion of cancer-related epithelial-mesenchymal transformation (EMT). Hyperglycemia did not affect the mesenchymal phenotype of Panc-1 cells but did increase the characteristics of CSCs. In addition, in another study using H6c7-KRAS cells, high glucose stimulated the expression of a TGF-β1 signal and decreased the expression of E-cadherin, increased the expression of nestin, and increased the number of polyclonal cells in a TGF-β1–dependent manner (26). This study also found that decreased E-cadherin was detected in the pancreatic duct of hyperglycemic, but not normoglycemic, mice. These findings suggest that hyperglycemia promotes the acquisition of PDEC mesenchymal and vascular stem cell characteristics by activating TGF-β1 signaling, which may explain how T2DM promotes PC (Figure 1).
In addition, hyperglycemia also produce a large number of reactive oxygen species (ROS) (27–29) and reduce the activity of antioxidant enzymes (30, 31) to promote mitosis and stimulate cell proliferation. Luo et al. (28) found that the inactivation of the JNK pathway caused by the increase in ROS levels has a pivotal role on high glucose-induced cell proliferation. Their findings indicated that ROS stimulates proliferation of pancreatic cancer cells under high glucose conditions via inactivating the JNK pathway.
T2DM is characterized by insulin resistance (IR) with hyperinsulinemia and high levels of insulin-like growth factor (IGF)-1 (32–35). In patients with T2DM, IR can lead to hyperinsulinemia through serine phosphorylation of insulin receptor substrate proteins, thus activating protein kinase C and the mTOR complex/S6K and so participating in the downregulation of the insulin signal (36, 37). Insulin can reduce the production of IGF binding proteins 1 and 2 in the liver, both of which have high affinity for IGF-1 and IGF-2, thus increasing the levels of free IGF-1 in circulating blood (36–39).
Most cancer cells highly express insulin and IGF-1, because they are important members of the tyrosine kinase class of membrane receptors and are highly homologous to tyrosine kinase oncogenes (39–44). When insulin and IGF-1 bind to their receptors, they can mediate signal transduction, activate important intracellular signaling pathways—including Ras/Raf/MAPK and PI3K/Akt/mTOR pathways—and lead to the development of PC (45, 46).
Many studies have shown that IGF-1 has a stronger mitotic and anti-apoptotic effect than insulin (47–49). In addition, studies have indicated that cancer cell proliferation increases in a dose-dependent manner with increasing concentrations of IGF-1. Activation of the IGF-1 signal pathway leads to increased PC cell proliferation, invasion, and angiogenesis and to decreased apoptosis (50–53).
Inflammation may increase the risk of PC in patients with T2DM. In patients with T2DM, insulin resistance and hyperinsulinemia often occur, accompanied by abundant adipocytes and a large amount of inflammatory cell infiltrating the pancreas tissue (54–56). The high glucose and fat diet may accelerate the inflammatory response by increasing oxidative stress and activating transcription factors, such as nuclear factor kappa B (NF-κB) and activator protein 1, which leads to the development of genomic aberration and carcinogenesis (57–59).
Inflammatory cytokines, ROS, and mediators of inflammatory pathways, such as cyclooxygenase-2 and NF-κB, are closely related to the STAT3 pathways. STAT3 and NF-κB signaling pathways are proven inhibitors of apoptosis and promoters of cell cycle progression. They also downregulate the expression of E-cadherin to induce EMT. During the inflammatory response, immune cells may directly promote the growth and progression of PC by releasing a large number of cytokines and growth factors into the microenvironment. The environment around the tumor is called as tumor microenvironment (TME), including Carcinoma-associated fibroblasts, endothelial cells and immune cells, which plays an significant role in growth, invasion and metastasis of pancreatic cancer. Inflammation change the TME and break balance of cancer cells in growth and apoptosis (60–63) (Figure 1).
A genome-wide association study has identified the relationship between diabetes and PC. Some pancreatic developmental genes, such as NR5A2, PDX1, and HNF1A, have been identified as susceptibility factors for PC in T2DM patients. Heterozygous mutations in some of these genes, such as PDX1 and HNF1A, also lead to different types of monogenic diabetes in young people (types 4 and 5). Some variants in PDX1 and HNF1A are also associated with an increased risk of T2DM (64, 65), obesity, or hyperglycemia (66).
The antioxidant mitochondrial uncoupling protein 2 (UCP2) controls pancreatic development and insulin secretion (67). UCP2 is overexpressed in PC tumors compared with normal adjacent tissues, indicating that its overexpression is a biomarker of poor prognosis. However, other recent studies using the PC cell line MiaPACA2 have shown that UCP2 can inhibit cancer cell proliferation and tumorigenesis (68). This effect is mediated by the retrograde mitochondrial signal on Warburg, which redirects mitochondrial function to oxidative phosphorylation rather than to glycolysis (69). Additional analysis is needed to clarify the differences between these two studies involving UCP2. Taken together, these data suggest a link between genes that control DM and PC.
NOD may be an early sign of PC, and a sudden increase in blood glucose in patients with previously well-controlled T2DM may also be a sign of PC (70). However, universal screening of PC in all elderly patients with NOD is difficult to achieve and not cost effective. In recent years, many studies have proposed different strategies to stratify T2DM groups and facilitate targeted screening (71–74).
A prospective observation cohort study initiated by the Consortium for the Study of Chronic Pancreatitis, Diabetes, and Pancreatic Cancer proposed a new approach (define, enrich, find) to clarify the population at high risk of PC and to detect lesions in the high-risk groups (75). Patients older than age 50 years were divided into high-, medium-, and low-risk groups using the Enriched New-Onset Diabetes Score for Pancreatic Cancer (END-PAC). This scoring model provides a reference for early clinical screening of PC (76). Elderly patients with weight loss [low body mass index (BMI)] and rapidly rising blood glucose levels in a short period may be the target population for early screening of pancreatic cancer (77). Other indicators for screening PC in patients with T2DM include BMI; age of T2DM onset; hepatitis B virus infection; and total bilirubin, alanine aminotransferase, creatinine, apolipoprotein A1, and leukocyte (WBC) levels (78). Fatigue and depression caused by elevated interleukin-6, combined with severe weight loss (>10%) and NOD, may represent paraneoplastic syndrome and be early manifestations of PC (79).
In addition to contributing to risk stratification, the development of biomarkers to distinguish PC-associated DM is expected to be an important aspect of PC screening. Studies have shown that the numerous molecules, described in the following sections, may be effective biomarkers for an early diagnosis of PC (Table 1).
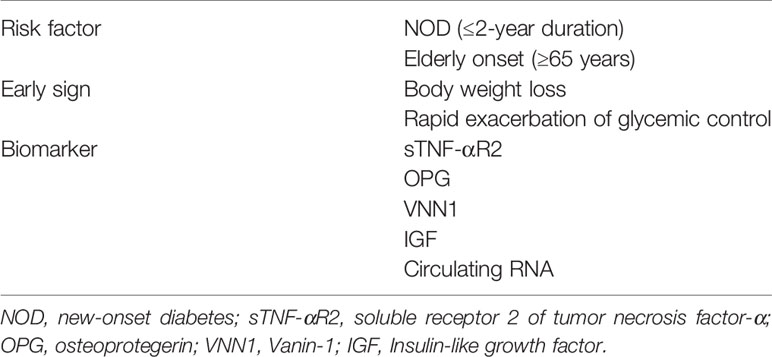
Table 1 Risk factors, early signs, and biomarkers for pancreatic cancer in patients with type 2 diabetes mellitus.
The level of carbohydrate antigen 19-9 (CA19-9) secreted by cancer cells in patients with NOD may be a reliable indicator for predicting PC. However, CA19-9 has a high false-positive rate; any condition that causes inflammation of the pancreas increases the CA 19-9 level. One study has shown that the CA19-9 level is of little significance in screening PC in NOD, because the positive predictive value and sensitivity are zero, and the false-positive rate is 9% (80). However, the sample size of this study was small, so the conclusions cannot be applied to the entire population. Other studies have shown that, in the first 2 years of NOD, CA19-9 can be used as a cost-effective approach to detect small PC lesions that cannot be detected on imaging (81, 82).
During the systemic inflammatory response to PC, C-reactive protein (CRP) levels can increase. Tumor necrosis factor-α (TNF-α) is the upstream regulator of CRP. Grote et al. (83) found that an increase in soluble TNF receptor 2 (sTNF-R2), significantly increases the risk of PC in patients with diabetes. In the diabetes arm of the study, the odds ratio of PC when the sTNF-R2 doubled was 4.76 (95% CI, 1.11–20.37); in the arm without diabetes, the odds ratio was only 1.12 (95% CI, 0.73–1.72) (83).
Osteoprotegerin (OPG) is a soluble decoy receptor of TNF-related apoptosis-inducing ligand, which belongs to the TNF receptor superfamily. Shi et al. (84) found that serum OPG was significantly increased in patients with PC-related DM. The sensitivity of serum OPG in identifying PC in patients with NOD was 68%; the specificity was 73.9%; and the area under the curve (AUC) was 73.7%.
The enzyme vascular non-inflammatory molecule-1 (vanin-1) is highly expressed at gene and protein level in many organs. Recently, many researches have elucidated the role of vanin-1 under physiological conditions in relation to oxidative stress and inflammation, which is important in the pancreatic microenvironment (85). Huang et al. (86) identified vanin-1 (VNN1) as a potential biomarker for PC, using microarray analysis of the peripheral blood in patients with PC-associated DM compared with T2DM (84). Kang et al. (87) also explored the functional mechanism of VNN1 in PC-associated DM and found that overexpression of VNN1 in tumor tissues can decrease glutathione concentration and increase ROS, thus aggravating paraneoplastic islet dysfunction.
Recently, circulating RNAs have become research hotspots as noninvasive biomarkers for the early detection of PC (88). PC cells release a large amount of RNA into the bloodstream. These RNAs can effectively resist the RNA enzyme, thus increasing the expression level in the serum. Dai et al. (89) reported a microRNA panel (miR-483-5p, miR-19a, miR-29a, miR-20a, miR-24, miR-25) that distinguished PC-related DM from T2DM with an AUC of 0.887.
Although a number of studies have reported biomarkers for PC-related DM, most of them are case-control studies with limited sample sizes. Future studies must verify the role of these discussed biomarkers in distinguishing T2DM with PC versus without PC in larger samples.
Some antidiabetic medications may have an impact on PC development, progression, and outcome because of their direct effects on the key factors mediating the association between T2DM and PC. The safety of antidiabetic medications with regard to PC risk is discussed in the following sections.
Insulin therapy is usually necessary to treat T2DM in the long term. However, abundant research has shown that insulin therapy may increase the incidence of PC (90, 91).
To explore the risk relationship between insulin therapy and PC, Bosetti et al. (92) analyzed 15 case-control studies, which included 8,305 patient cases and 13,987 controls. Studies indicated that short-term insulin use (<5 years) was independently associated with a higher risk of PC (odds ratio [OR] = 5.6, 95% CI, 3.75–8.35), whereas long-term insulin use (≥15 years) was not (OR = 0.95, 95% CI, 0.53–1.70). At the same time, studies also showed that long-term oral antidiabetic use (≥15 years) in patients with T2DM might reduce the risk of PC (OR = 0.31, 95% CI, 0.14–0.69).
In 2018, Lee et al. (93) conducted a population-based study comparing PC risk in patients expose to antidiabetic drugs versus no drug exposure. The study concluded that, among several kinds of antidiabetic drugs, insulin alone was associated with an increased risk of PC (hazard ratio [HR] = 2.86, 95% CI, 1.43–5.74). The conclusion is similar with the research by Liu et al. (94), a case–control study using 12 years of data from Taiwan’s National Health Insurance Research Database. The association between insulin use and high pancreatic cancer risk is significant.
Wang et al. (95) also found that insulin can promote the proliferation and glucose utilization of PC cells by activating ERK and PI3K and by increasing the expression of MMP-2. Insulin promotes migration and invasion in PC by activating the MMP-2 signal pathway. In addition, insulin induces phosphorylation of ERK and PI3K/Akt, which indicates that insulin can stimulate the Ras/Raf/MAPK and PI3K/Akt pathways and accelerate tumorigenesis and development (Figure 1). In summary, insulin use is associated with an increased risk of PC, so patients with T2DM who have a high risk of PC may not be candidates for insulin treatment. While insulin treatment was imperative for the patients with insulin secretion absolutely insufficient. For clinical physicians, we should pay attention to the risk of PC during long-term treatment with insulin and screen early PC in islet β-cell dysfunction patients with long-term treatment with insulin. We need more evidence for PC risk for patients with long-term treatment with insulin in the further research.
Metformin is the cornerstone treatment of diabetes. Retrospective studies have shown that metformin can improve the survival of patients with T2DM and PC. During the past 5 years, numerous studies have suggested that metformin can reduce the risk of PC (96–99).
In the analysis of case-control studies by Bosetti et al. (100), long-term oral metformin use (≥15 years) reduced the risk of PC in patients with T2DM (OR = 0.31, 95% CI, 0.14–0.69). In 2018, Lee et al. (101) conducted a population-based study to assess the effects of T2DM and antidiabetic drugs on PC risk. That study identified metformin, among the antidiabetic drugs studied, as an independent risk factor for PC (HR = 0.86, 95% CI, 0.77–0.96). However, patients who received metformin combined with a thiazolidinedione or with dipeptidyl peptidase-4 inhibitors had lower risks of PC than patients receiving metformin alone. There are several clinical trials about metformin on PC treatment, such as metformin combined With Chemotherapy. Although addition of metformin does not improve outcome in patients with advanced PC treated with gemcitabine and erlotinib, future research should include studies of more potent biguanides, and should focus on patients with tumors showing markers of sensitivity to energetic stress, such as a lack of function of AMP kinase (102).
Currently, most scholars believe that metformin can reduce the risk of PC, because metformin can activate the liver kinase B1 (LKB1)–adenosine monophosphate protein-activated kinase (AMPK) pathway, which can not only promote cell energy production and inhibit liver glucose production but also inhibit the signal pathway of cancer cell proliferation (103). As a tumor suppressor, LKB1 can activate AMPK, which is a potent inhibitor of mTOR complex 1, and disrupt cross-talk between insulin/IGF-1 receptor and G protein–coupled receptors, thus regulating protein synthesis and replication. More importantly, metformin may play a role in the development of PC stem cells through the mTOR pathway (23, 104–107).
In a study evaluating the effect of metformin on PC, cancer stem cells (Alk4, nodal, activin, and Smad2) and pluripotency-related RNA proteins (Nanog, Oct4, and Sox2) changed significantly after metformin treatment. These changes may be due to the inhibition of nicotinamide-adenine dinucleotide dehydrogenase and the production of free ROS, which would directly increase the damage to PC stem cells (108). Ma M et al. found that metformin significantly inhibited proliferation and viability, induced apoptosis of pancreatic cancer cells, which was more pronounced in low-glucose than in high-glucose group, and metformin may play protective effect by suppressing glycolysis and inducing energy stress via up-regulation of miR-210-5p (109).
These studies have shown that metformin can reduce the risk of PC and activate the LKB1/AMPK pathway, thus inhibiting cell proliferation by mTOR. Therefore, metformin is expected to become part of the standard treatment for patients with PC.
The use of incretin-based medicines—glucagon-like peptide 1 receptor agonists (GLP1-RAs) and dipeptidyl peptidase-4 inhibitors (DPP4is)—is increasingly popular. Some studies in animal models have been speculated that the chronic overstimulation of GLP1 receptors in exocrine pancreatic cells may induce pancreatitis, ultimately increasing the risk of PC (110, 111). However, this hypothesis has not been supported by evidence from clinical trials (112, 113).
Monami et al. (112) analyzed 113 trials, and 15 studies that reported at least one event. Those 15 studies enrolled 14,866 and 12,849 patients in GLP1-RA and comparator groups, respectively, and the number of reported PCs was 24 for GLP1-RAs and was 23 for comparators (Mantel-Haenszel odds ratio [MH-OR] for PC with GLP1-RA treatment = 0.94, 95% CI, 0.52–1.70, P = 0.84). Similar results were obtained in a post-hoc analysis excluding comparisons with DPP4is (MH-OR = 0.93, 95% CI, 0.51–1.69, P = 0.80).
In 2020, Nreu et al. (113) analyzed 43 randomized, controlled trials that met the following inclusion criteria: at least 52 weeks in duration, and comparison of a GLP1-RA versus any non–GLP1-RA treatment in patients with T2DM and PC. They found that GLP1-RA use showed no association with PC (MH-OR = 1.28, 95% CI, 0.87–1.89, P = 0.20) (107).
Currently, no clear evidence of risk for PC has been observed with the use of incretin-based medications. Data about the relationship between incretin-based medicines and PC may be too scarce to draw any conclusion.
Sodium-glucose cotransporter 2 (SGLT2) inhibitors represent a novel class of oral antidiabetic drugs that help maintain glycemic control by decreasing the reabsorption of glucose and increasing the excretion of urinary glucose (114). In addition to substantial cardiovascular benefits, anti-tumor benefits or the safety of SGLT2 inhibitors have been considered by the public. Scafoglio et al. (115) found that SGLT2 was functionally expressed in pancreatic carcinomas and that SGLT2 inhibitors blocked glucose uptake and reduced tumor growth and survival in a xenograft model of PC. These findings suggest that SGLT2 inhibitors may be useful for cancer therapy.
In 2019, Tang et al. (116) undertook a study to systematically evaluate the association between SGLT2 inhibitors and pancreatic safety in patients with T2DM. Of the 35 trials, involving 44,912 patients with T2DM, 40 PC events (in 18 trials and 27,806 patients) were reported during a median follow-up of 52 weeks. SGLT2 inhibitors were not associated with PC (OR=1.34; 95% CI, 0.71–2.54; very-low-quality evidence) (116).
PC is highly aggressive and lethal malignancy, and T2DM is the most common metabolic disease. T2DM is a risk factor for PC. Conversely, NOD may be a sign and consequence of PC. Screening in patients with NOD combined with assessment of risk factors and biomarkers may be an important way to improve the early diagnosis of PC. The mechanisms that contribute to the relationship between PC and diabetes include insulin resistance, hyperinsulinemia, hyperglycemia, and chronic inflammation. Metformin, insulin, GLP1-RAs, DPP4is, and SGLT2 inhibitors are common drugs that treat T2DM. Studies have shown that metformin can reduce the risk of PC, whereas insulin therapy is associated with a higher risk of PC. Therefore, metformin may be used to prevent the development of malignant lesions and is expected to become an anticancer agent. T2DM-related studies will likely be crucial to improve the morbidity and mortality associated with PC.
XD consulted literatures and wrote the manuscript. LG designed the review. WW and QP assisted with writing and revised the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (grants 81670763 and 81471050).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor and the reviewer (YL) declared a shared affiliation with the authors at time of review.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We sincerely thank editors at Charlesworth Author Services for editing the language of this article.
1. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics, 2021. CA: A Cancer J Clin (2021) 71(1):7–33. doi: 10.3322/caac.21654
2. Klein AP. Pancreatic Cancer Epidemiology: Understanding the Role of Lifestyle and Inherited Risk Factors. Nat Rev Gastro Hepat (2021) 18(7):493–502. doi: 10.1038/s41575-021-00457-x
3. Chinese Pancreatic Surgery Association, Chinese Society of Surgery, Chinese Medical Association. Guidelines for the Diagnosis and Treatment of Pancreatic Cancer in China (2021). Zhonghua wai ke za zhi [Chinese J Surgery] (2021) 59(7):561–77. doi: 10.3760/cma.j.cn112139-20210416-00171
4. Zhao C, Gao F, Li Q, Liu Q, Lin X. The Distributional Characteristic and Growing Trend of Pancreatic Cancer in China. Pancreas (2019) 48(3):309–14. doi: 10.1097/MPA.0000000000001222
5. Cheng F, Carroll L, Joglekar MV, Januszewski AS, Wong KK, Hardikar AA, et al. Diabetes, Metabolic Disease, and Telomere Length. Lancet Diabetes Endocrinol (2021) 9(2):117–26. doi: 10.1016/S2213-8587(20)30365-X
6. Gallagher EJ, LeRoith D. Hyperinsulinaemia in Cancer. Nat Rev Cancer (2020) 20(11):629–44. doi: 10.1038/s41568-020-0295-5
7. Mizrahi JD, Surana R, Valle JW, Shroff RT. Pancreatic Cancer. Lancet (2020) 395(10242):2008–20. doi: 10.1016/S0140-6736(20)30974-0
8. Toledo FGS, Chari S, Yadav D. Understanding the Contribution of Insulin Resistance to the Risk of Pancreatic Cancer. Am J Gastroenterol (2021) 116(4):669–70. doi: 10.14309/ajg.0000000000001104
9. Gallo M, Adinolfi V, Morviducci L, Acquati S, Tuveri E, Ferrari P, et al. Early Prediction of Pancreatic Cancer From New-Onset Diabetes: An Associazione Italiana Oncologia Medica (AIOM)/Associazione Medici Diabetologi (AMD)/Società Italiana Endocrinologia (SIE)/Società Italiana Farmacologia (SIF) Multidisciplinary Consensus Position Paper. ESMO Open (2021) 6(3):100155. doi: 10.1016/j.esmoop.2021.100155
10. Takikawa T, Kikuta K, Kume K, Hamada S, Miura S, Yoshida N, et al. New-Onset or Exacerbation of Diabetes Mellitus Is a Clue to the Early Diagnosis of Pancreatic Cancer. Tohoku J Exp Med (2020) 252(4):353–64. doi: 10.1620/tjem.252.353
11. Khan S, Safarudin RF, Kupec JT. Validation of the ENDPAC Model: Identifying New-Onset Diabetics at Risk of Pancreatic Cancer. Pancreatology (2021) 21(3):550–5. doi: 10.1016/j.pan.2021.02.001
12. Quoc Lam B, Shrivastava SK, Shrivastava A, Shankar S, Srivastava RK. The Impact of Obesity and Diabetes Mellitus on Pancreatic Cancer: Molecular Mechanisms and Clinical Perspectives. J Cell Mol Med (2020) 24(14):7706–16. doi: 10.1111/jcmm.15413
13. Perreault L, Skyler JS, Rosenstock J. Novel Therapies With Precision Mechanisms for Type 2 Diabetes Mellitus. Nat Rev Endocrinol (2021) 17(6):364–77. doi: 10.1038/s41574-021-00489-y
14. Targher G, Corey KE, Byrne CD, Roden M. The Complex Link Between NAFLD and Type 2 Diabetes Mellitus - Mechanisms and Treatments. Nat Rev Gastroenterol Hepatol (2021) in press. doi: 10.1038/s41575-021-00448-y
15. Alpertunga I, Sadiq R, Pandya D, Lo T, Dulgher M, Evans S, et al. Glycemic Control as an Early Prognostic Marker in Advanced Pancreatic Cancer. Front Oncol (2021) 11:571855. doi: 10.3389/fonc.2021.571855
16. Yu Q, Zhang Z, Zhang H. Effect of Glucose Variability on Pancreatic Cancer Through Regulation of COL6A1. Cancer Manag Res (2021) 13:1291–8. doi: 10.2147/CMAR.S293473
17. Rahn S, Zimmermann V, Viol F, Knaack H, Stemmer K, Peters L, et al. Diabetes as Risk Factor for Pancreatic Cancer: Hyperglycemia Promotes Epithelial-Mesenchymal-Transition and Stem Cell Properties in Pancreatic Ductal Epithelial Cells. Cancer Lett (2018) 415:129–50. doi: 10.1016/j.canlet.2017.12.004
18. Sato K, Hikita H, Myojin Y, Fukumoto K, Murai K, Sakane S, et al. Hyperglycemia Enhances Pancreatic Cancer Progression Accompanied by Elevations in Phosphorylated STAT3 and MYC Levels. PloS One (2020) 15(7):e235573. doi: 10.1371/journal.pone.0235573
19. Milton CK, Self AJ, Clarke PA, Banerji U, Piccioni F, Root DE, et al. A Genome-Scale CRISPR Screen Identifies the ERBB and mTOR Signaling Networks as Key Determinants of Response to PI3K Inhibition in Pancreatic Cancer. Mol Cancer Ther (2020) 19:1423–35. doi: 10.1158/1535-7163.MCT-19-1131
20. Javle MM, Shroff RT, Xiong H, Varadhachary GA, Fogelman D, Reddy SA, et al. Inhibition of the Mammalian Target of Rapamycin (mTOR) in Advanced Pancreatic Cancer: Results of Two Phase II Studies. BMC Cancer (2010) 10(1):368. doi: 10.1186/1471-2407-10-368
21. Wolpin BM, Hezel AF, Abrams T, Blaszkowsky LS, Meyerhardt JA, Chan JA, et al. Oral mTOR Inhibitor Everolimus in Patients With Gemcitabine-Refractory Metastatic Pancreatic Cancer. J Clin Oncol (2009) 27(2):193–8. doi: 10.1200/JCO.2008.18.9514
22. Kordes S, Klümpen HJ, Weterman MJ, Schellens JHM, Richel DJ, Wilmink JW. Phase II Study of Capecitabine and the Oral mTOR Inhibitor Everolimus in Patients With Advanced Pancreatic Cancer. Cancer Chemoth Pharm (2015) 75(6):1135–41. doi: 10.1007/s00280-015-2730-y
23. Chen YH, Huang YC, Yang SF, Yen HH, Tsai HD, Hsieh MC, et al. Pitavastatin and Metformin Synergistically Activate Apoptosis and Autophagy in Pancreatic Cancer Cells. Environ Toxicol (2021) 36(8):1491–503. doi: 10.1002/tox.23146
24. Grote VA, Rohrmann S, Nieters A, Dossus L, Tjønneland A, Halkjær J, et al. Diabetes Mellitus, Glycated Haemoglobin and C-Peptide Levels in Relation to Pancreatic Cancer Risk: A Study Within the European Prospective Investigation Into Cancer and Nutrition (EPIC) Cohort. Diabetologia (2011) 54(12):3037–46. doi: 10.1007/s00125-011-2316-0
25. Menini S, Iacobini C, de Latouliere L, Manni I, Ionta V, Blasetti Fantauzzi C, et al. The Advanced Glycation End-Product N-Carboxymethyllysine Promotes Progression of Pancreatic Cancer: Implications for Diabetes-Associated Risk and Its Prevention. J Pathol (2018) 245(2):197–208. doi: 10.1002/path.5072
26. Kiss K, Baghy K, Spisák S, Szanyi S, Tulassay Z, Zalatnai A, et al. Chronic Hyperglycemia Induces Trans-Differentiation of Human Pancreatic Stellate Cells and Enhances the Malignant Molecular Communication With Human Pancreatic Cancer Cells. PloS One (2015) 10(5):e128059. doi: 10.1371/journal.pone.0128059
27. Martinez-Useros J, Li W, Cabeza-Morales M, Garcia-Foncillas J. Oxidative Stress: A New Target for Pancreatic Cancer Prognosis and Treatment. J Clin Med (2017) 6. doi: 10.3390/jcm6030029
28. Luo J, Xiang Y, Xu X, Fang D, Li D, Ni F, et al. High Glucose-Induced ROS Production Stimulates Proliferation of Pancreatic Cancer via Inactivating the JNK Pathway. Oxid Med Cell Longev (2018) 2018:1–10. doi: 10.1155/2018/6917206
29. Cao L, Chen X, Xiao X, Ma Q, Li W. Resveratrol Inhibits Hyperglycemia-Driven ROS-Induced Invasion and Migration of Pancreatic Cancer Cells via Suppression of the ERK and P38 MAPK Signaling Pathways. Int J Oncol (2016) 49(2):735–43. doi: 10.3892/ijo.2016.3559
30. Li W. Hyperglycemia as a Mechanism of Pancreatic Cancer Metastasis. Front Biosci (2012) 17(1):1761. doi: 10.2741/4017
31. Li W, Wu Z, Ma Q, Liu J, Xu Q, Han L, et al. Hyperglycemia Regulates TXNIP/TRX/ROS Axis via P38 MAPK and ERK Pathways in Pancreatic Cancer. Curr Cancer Drug Tar (2014) 14(4):348–56. doi: 10.2174/1568009614666140331231658
32. Shlomai G, Neel B, LeRoith D, Gallagher EJ. Type 2 Diabetes Mellitus and Cancer: The Role of Pharmacotherapy. J Clin Oncol (2016) 34(35):4261–9. doi: 10.1200/JCO.2016.67.4044
33. Gallagher EJ, LeRoith D. The Proliferating Role of Insulin and Insulin-Like Growth Factors in Cancer. Trends Endocrinol Metab (2010) 21(10):610–8. doi: 10.1016/j.tem.2010.06.007
34. Sathishkumar C, Prabu P, Balakumar M, Lenin R, Prabhu D, Anjana RM, et al. Augmentation of Histone Deacetylase 3 (HDAC3) Epigenetic Signature at the Interface of Proinflammation and Insulin Resistance in Patients With Type 2 Diabetes. Clin Epigenet (2016) 8(1):125. doi: 10.1186/s13148-016-0293-3
35. Sharma S, Taliyan R. Histone Deacetylase Inhibitors: Future Therapeutics for Insulin Resistance and Type 2 Diabetes. Pharmacol Res (2016) 113:320–6. doi: 10.1016/j.phrs.2016.09.009
36. Paneni F, Costantino S, Cosentino F. Insulin Resistance, Diabetes, and Cardiovascular Risk. Curr Atheroscler Rep (2014) 16(7):419. doi: 10.1007/s11883-014-0419-z
37. Ross JS, Russo SB, Chavis GC, Cowart LA. Sphingolipid Regulators of Cellular Dysfunction in Type 2 Diabetes Mellitus: A Systems Overview. Clin Lipidol (2014) 9(5):553–69. doi: 10.2217/clp.14.37
38. Rojas-Rodriguez R, Ziegler R, DeSouza T, Majid S, Madore AS, Amir N, et al. PAPPA-Mediated Adipose Tissue Remodeling Mitigates Insulin Resistance and Protects Against Gestational Diabetes in Mice and Humans. Sci Transl Med (2020) 12(571):y4145. doi: 10.1126/scitranslmed.aay4145
39. Thomas RJ, Kenfield SA, Jimenez A. Exercise-Induced Biochemical Changes and Their Potential Influence on Cancer: A Scientific Review. Brit J Sport Med (2017) 51(8):640–4. doi: 10.1136/bjsports-2016-096343
40. Belfiore A, Malaguarnera R, Vella V, Lawrence MC, Sciacca L, Frasca F, et al. Insulin Receptor Isoforms in Physiology and Disease: An Updated View. Endocr Rev (2017) 38(5):379–431. doi: 10.1210/er.2017-00073
41. Bassil F, Canron M, Vital A, Bezard E, Li Y, Greig NH, et al. Insulin Resistance and Exendin-4 Treatment for Multiple System Atrophy. Brain (2017) 140(5):1420–36. doi: 10.1093/brain/awx044
42. Matsushita M, Fujita K, Hayashi T, Kayama H, Motooka D, Hase H, et al. Gut Microbiota-Derived Short-Chain Fatty Acids Promote Prostate Cancer Growth via IGF-1 Signaling. Cancer Res (2021) 81(15):4014–26. doi: 10.1158/0008-5472.CAN-20-4090
43. Baxter RC. IGF Binding Proteins in Cancer: Mechanistic and Clinical Insights. Nat Rev Cancer (2014) 14(5):329–41. doi: 10.1038/nrc3720
44. Werner H, Sarfstein R, Laron Z. The Role of Nuclear Insulin and IGF1 Receptors in Metabolism and Cancer. Biomolecules (2021) 11:531. doi: 10.3390/biom11040531
45. Badarni M, Prasad M, Golden A, Bhattacharya B, Levin L, Yegodayev KM, et al. IGF2 Mediates Resistance to Isoform-Selective-Inhibitors of the PI3K in HPV Positive Head and Neck Cancer. Cancers (2021) 13:2250. doi: 10.3390/cancers13092250
46. Pollak M. The Insulin and Insulin-Like Growth Factor Receptor Family in Neoplasia: An Update. Nat Rev Cancer (2012) 12(3):159–69. doi: 10.1038/nrc3215
47. Hua H, Kong Q, Yin J, Zhang J, Jiang Y. Insulin-Like Growth Factor Receptor Signaling in Tumorigenesis and Drug Resistance: A Challenge for Cancer Therapy. J Hematol Oncol (2020) 13(1):64. doi: 10.1186/s13045-020-00904-3
48. LeRoith D, Holly JMP, Forbes BE. Insulin-Like Growth Factors: Ligands, Binding Proteins, and Receptors. Mol Metab (2021), in press. doi: 10.1016/j.molmet.2021.101245
49. Hua H, Kong Q, Zhang H, Wang J, Luo T, Jiang Y. Targeting mTOR for Cancer Therapy. J Hematol Oncol (2019) 12(1):71. doi: 10.1186/s13045-019-0754-1
50. Geleta B, Park KC, Jansson PJ, Sahni S, Maleki S, Xu Z, et al. Breaking the Cycle: Targeting of NDRG1 to Inhibit Bi-Directional Oncogenic Cross-Talk Between Pancreatic Cancer and Stroma. FASEB J (2021) 35(2):e21347. doi: 10.1096/fj.202002279R
51. Włodarczyk B, Borkowska A, Włodarczyk P, Małecka-Panas E, Gąsiorowska A. Insulin-Like Growth Factor 1 and Insulin-Like Growth Factor Binding Protein 2 Serum Levels as Potential Biomarkers in Differential Diagnosis Between Chronic Pancreatitis and Pancreatic Adenocarcinoma in Reference to Pancreatic Diabetes. Gastroenterol Rev (2021) 16(1):36–42. doi: 10.5114/pg.2020.95091
52. Zheng Y, Wu C, Yang J, Zhao Y, Jia H, Xue M, et al. Insulin-Like Growth Factor 1-Induced Enolase 2 Deacetylation by HDAC3 Promotes Metastasis of Pancreatic Cancer. Signal Transduct Target Ther (2020) 5(1):53. doi: 10.1038/s41392-020-0146-6
53. Wlodarczyk B, Gasiorowska A, Malecka-Panas E. The Role of Insulin-Like Growth Factor (IGF) Axis in Early Diagnosis of Pancreatic Adenocarcinoma (PDAC). J Clin Gastroenterol (2018) 52(7):569–72. doi: 10.1097/MCG.0000000000001073
54. Jin Q, Hart PA, Shi N, Joseph JJ, Donneyong M, Conwell DL, et al. Dietary Patterns of Insulinemia, Inflammation and Glycemia, and Pancreatic Cancer Risk: Findings From the Women’s Health Initiative. Cancer Epidem Biomar (2021) 30(6):1229–40. doi: 10.1158/1055-9965.EPI-20-1478
55. Desai V, Patel K, Sheth R, Barlass U, Chan Y, Sclamberg J, et al. Pancreatic Fat Infiltration Is Associated With a Higher Risk of Pancreatic Ductal Adenocarcinoma. Visceral Med (2020) 36(3):220–6. doi: 10.1159/000507457
56. Liao WC, Chen PR, Huang CC, Chang YT, Huang BS, Chang CC, et al. Relationship Between Pancreatic Cancer-Associated Diabetes and Cachexia. J Cachexia Sarcopenia Muscle (2020) 11(4):899–908. doi: 10.1002/jcsm.12553
57. Spyrou N, Avgerinos KI, Mantzoros CS, Dalamaga M. Classic and Novel Adipocytokines at the Intersection of Obesity and Cancer: Diagnostic and Therapeutic Strategies. Curr Obes Rep (2018) 7(4):260–75. doi: 10.1007/s13679-018-0318-7
58. Mannelli M, Gamberi T, Magherini F, Fiaschi T. The Adipokines in Cancer Cachexia. Int J Mol Sci (2020) 21(14):4860. doi: 10.3390/ijms21144860
59. Garikapati KK, Ammu VVVR, Krishnamurthy PT, Chintamaneni PK, Pindiprolu SKSS. Type-II Endometrial Cancer: Role of Adipokines. Arch Gynecol Obstet (2019) 300(2):239–49. doi: 10.1007/s00404-019-05181-1
60. Takahashi M, Mutoh M, Ishigamori R, Fujii G, Imai T, Tanaka T, et al. Involvement of Inflammatory Factors in Pancreatic Carcinogenesis and Preventive Effects of Anti-Inflammatory Agents. Semin Immunopathol (2013) 35(2):203–27. doi: 10.1007/s00281-012-0340-x
61. Pothuraju R, Rachagani S, Junker WM, Chaudhary S, Saraswathi V, Kaur S, et al. Pancreatic Cancer Associated With Obesity and Diabetes: An Alternative Approach for Its Targeting. J Exp Clin Canc Res (2018) 37(1):319. doi: 10.1186/s13046-018-0963-4
62. Yadav RK, Gautam DK, Muj C, Madhubabu GB, Paddibhatla I. Methotrexate Negatively Acts on Inflammatory Responses Triggered in Drosophila Larva With Hyperactive JAK/STAT Pathway. Dev Comp Immunol (2021), in press. doi: 10.1016/j.dci.2021.104161
63. Paternoster S, Falasca M. The Intricate Relationship Between Diabetes, Obesity and Pancreatic Cancer. Biochim Biophys Acta (BBA) - Rev Cancer (2020) 1873(1):188326. doi: 10.1016/j.bbcan.2019.188326
64. Menini S, Iacobini C, de Latouliere L, Manni I, Vitale M, Pilozzi E, et al. Diabetes Promotes Invasive Pancreatic Cancer by Increasing Systemic and Tumour Carbonyl Stress in KrasG12D/+ Mice. J Exp Clin Canc Res (2020) 39(1):1–152. doi: 10.1186/s13046-020-01665-0
65. Tang Z, Chu Y, Tan Y, Li J, Gao S. Pancreatic and Duodenal Homeobox-1 in Pancreatic Ductal Adenocarcinoma and Diabetes Mellitus. Chin Med J Peking (2020) 133(3):344–50. doi: 10.1097/CM9.0000000000000628
66. Španinger E, Potočnik U, Bren U. Molecular Dynamics Simulations Predict That rSNP Located in the HNF-1α Gene Promotor Region Linked With MODY3 and Hepatocellular Carcinoma Promotes Stronger Binding of the HNF-4α Transcription Factor. Biomolecules (2020) 10(12):1700. doi: 10.3390/biom10121700
67. Broche B, Ben Fradj S, Aguilar E, Sancerni T, Bénard M, Makaci F, et al. Mitochondrial Protein UCP2 Controls Pancreas Development. Diabetes (2017) 67(1):78–84. doi: 10.2337/db17-0118
68. Esteves P, Pecqueur C, Ransy C, Esnous C, Lenoir V, Bouillaud F, et al. Mitochondrial Retrograde Signaling Mediated by UCP2 Inhibits Cancer Cell Proliferation and Tumorigenesis. Cancer Res (2014) 74(14):3971–82. doi: 10.1158/0008-5472.CAN-13-3383
69. Brandi J, Cecconi D, Cordani M, Torrens-Mas M, Pacchiana R, Dalla Pozza E, et al. The Antioxidant Uncoupling Protein 2 Stimulates Hnrnpa2/B1, GLUT1 and PKM2 Expression and Sensitizes Pancreas Cancer Cells to Glycolysis Inhibition. Free Radical Bio Med (2016) 101:305–16. doi: 10.1016/j.freeradbiomed.2016.10.499
70. Henrikson NB, Aiello Bowles EJ, Blasi PR, Morrison CC, Nguyen M, Pillarisetty VG, et al. Screening for Pancreatic Cancer. JAMA (2019) 322(5):445. doi: 10.1001/jama.2019.6190
71. Pereira SP, Oldfield L, Ney A, Hart PA, Keane MG, Pandol SJ, et al. Early Detection of Pancreatic Cancer. Lancet Gastroenterol Hepatol (2020) 5(7):698–710. doi: 10.1016/S2468-1253(19)30416-9
72. Singhi AD, Koay EJ, Chari ST, Maitra A. Early Detection of Pancreatic Cancer: Opportunities and Challenges. Gastroenterology (2019) 156(7):2024–40. doi: 10.1053/j.gastro.2019.01.259
73. Liao W, Huang B, Yu Y, Yang H, Chen P, Huang C, et al. Galectin-3 and S100A9: Novel Diabetogenic Factors Mediating Pancreatic Cancer–Associated Diabetes. Diabetes Care (2019) 42(9):1752–9. doi: 10.2337/dc19-0217
74. Petrusel L, Bilibou M, Drug V, Leucuta DC, Seicean R, Cainap C, et al. Risk Factors in Pancreatic Adenocarcinoma: The Interrelation With Familial History and Predictive Role on Survival. J Gastrointest Liver Diseases: JGLD (2020) 29:391–8. doi: 10.15403/jgld-2529
75. Maitra A, Sharma A, Brand RE, Van Den Eeden SK, Fisher WE, Hart PA, et al. A Prospective Study to Establish a New-Onset Diabetes Cohort. Pancreas (2018) 47(10):1244–8. doi: 10.1097/MPA.0000000000001169
76. Sharma A, Kandlakunta H, Nagpal SJS, Feng Z, Hoos W, Petersen GM, et al. Model to Determine Risk of Pancreatic Cancer in Patients With New-Onset Diabetes. Gastroenterology (2018) 155(3):730–9. doi: 10.1053/j.gastro.2018.05.023
77. Mueller AM, Meier CR, Jick SS, Schneider C. Weight Change and Blood Glucose Concentration as Markers for Pancreatic Cancer in Subjects With New-Onset Diabetes Mellitus: A Matched Case-Control Study. Pancreatology (2019) 19(4):578–86. doi: 10.1016/j.pan.2019.03.006
78. Dong X, Lou YB, Mu YC, Kang MX, Wu YL. Predictive Factors for Differentiating Pancreatic Cancer-Associated Diabetes Mellitus From Common Type 2 Diabetes Mellitus for the Early Detection of Pancreatic Cancer. Digestion (2018) 98(4):209–16. doi: 10.1159/000489169
79. Walter FM, Mills K, Mendonça SC, Abel GA, Basu B, Carroll N, et al. Symptoms and Patient Factors Associated With Diagnostic Intervals for Pancreatic Cancer (SYMPTOM Pancreatic Study): A Prospective Cohort Study. Lancet Gastroenterol Hepatol (2016) 1(4):298–306. doi: 10.1016/S2468-1253(16)30079-6
80. Illés D, Terzin V, Holzinger G, Kosár K, Róka R, Zsóri G, et al. New-Onset Type 2 Diabetes Mellitus – A High-Risk Group Suitable for the Screening of Pancreatic Cancer? Pancreatology (2016) 16(2):266–71. doi: 10.1016/j.pan.2015.12.005
81. Choe JW, Kim JS, Kim HJ, Hwang SY, Joo MK, Lee BJ, et al. Value of Early Check-Up of Carbohydrate Antigen 19-9 Levels for Pancreatic Cancer Screening in Asymptomatic New-Onset Diabetic Patients. Pancreas (2016) 45(5):730–4. doi: 10.1097/MPA.0000000000000538
82. Choe JW, Kim HJ, Kim JS, Cha J, Joo MK, Lee BJ, et al. Usefulness of CA 19–9 for Pancreatic Cancer Screening in Patients With New-Onset Diabetes. Hepatob Pancreat Dis (2018) 17(3):263–8. doi: 10.1016/j.hbpd.2018.04.001
83. Grote VA, Kaaks R, Nieters A, Tjønneland A, Halkjær J, Overvad K, et al. Inflammation Marker and Risk of Pancreatic Cancer: A Nested Case–Control Study Within the EPIC Cohort. Brit J Cancer (2012) 106(11):1866–74. doi: 10.1038/bjc.2012.172
84. Shi W, Qiu W, Wang W, Zhou X, Zhong X, Tian G, et al. Osteoprotegerin Is Up-Regulated in Pancreatic Cancers and Correlates With Cancer-Associated New-Onset Diabetes. Biosci Trends (2014) 8(6):322–6. doi: 10.5582/bst.2014.01092
85. Kang M, Qin W, Buya M, Dong X, Zheng W, Lu W, et al. VNN1, a Potential Biomarker for Pancreatic Cancer-Associated New-Onset Diabetes, Aggravates Paraneoplastic Islet Dysfunction by Increasing Oxidative Stress. Cancer Lett (2016) 373(2):241–50. doi: 10.1016/j.canlet.2015.12.031
86. Huang H, Dong X, Kang MX, Xu B, Chen Y, Zhang B, et al. Novel Blood Biomarkers of Pancreatic Cancer–Associated Diabetes Mellitus Identified by Peripheral Blood–Based Gene Expression Profiles. Am J Gastroenterol (2010) 105(7):1661–9. doi: 10.1038/ajg.2010.32
87. Kang M, Qin W, Buya M, Dong X, Zheng W, Lu W, et al. VNN1, a Potential Biomarker for Pancreatic Cancer-Associated New-Onset Diabetes, Aggravates Paraneoplastic Islet Dysfunction by Increasing Oxidative Stress. Cancer Lett (2016) 373(2):241–50. doi: 10.1016/j.canlet.2015.12.031
88. Iovanna J. Implementing Biological Markers as a Tool to Guide Clinical Care of Patients With Pancreatic Cancer. Transl Oncol (2021) 14(1):100965. doi: 10.1016/j.tranon.2020.100965
89. Dai X, Pang W, Zhou Y, Yao W, Xia L, Wang C, et al. Altered Profile of Serum microRNAs in Pancreatic Cancer-Associated New-Onset Diabetes Mellitus. J Diabetes (2016) 8(3):422–33. doi: 10.1111/1753-0407.12313
90. Tan J, You Y, Guo F, Xu J, Dai H, Bie P. Association of Elevated Risk of Pancreatic Cancer in Diabetic Patients: A Systematic Review and Meta-Analysis. Oncol Lett (2017) 13(3):1247–55. doi: 10.3892/ol.2017.5586
91. Kautzky-Willer A, Thurner S, Klimek P. Use of Statins Offsets Insulin-Related Cancer Risk. J Intern Med (2017) 281(2):206–16. doi: 10.1111/joim.12567
92. Bosetti C, Rosato V, Li D, Silverman D, Petersen GM, Bracci PM, et al. Diabetes, Antidiabetic Medications, and Pancreatic Cancer Risk: An Analysis From the International Pancreatic Cancer Case-Control Consortium. Ann Oncol (2014) 25(10):2065–72. doi: 10.1093/annonc/mdu276
93. Lee DY, Yu JH, Park S, Han K, Kim NH, Yoo HJ, et al. The Influence of Diabetes and Antidiabetic Medications on the Risk of Pancreatic Cancer: A Nationwide Population-Based Study in Korea. Sci Rep Uk (2018) 8(1):9719. doi: 10.1038/s41598-018-27965-2
94. Liu YC, Nguyen PA, Humayun A, Chien SC, Yang HC, Asdary RN, et al. Does Long-Term Use of Antidiabetic Drugs Changes Cancer Risk? Medicine (2019) 98(40):e17461. doi: 10.1097/MD.0000000000017461
95. Wang G, Yin L, Peng Y, Gao Y, Gao H, Zhang J, et al. Insulin Promotes Invasion and Migration of KRASG12D Mutant HPNE Cells by Upregulating MMP-2 Gelatinolytic Activity via ERK- and PI3K-Dependent Signalling. Cell Proliferat (2019) 52(3):e12575. doi: 10.1111/cpr.12575
96. Cho J, Scragg R, Pandol SJ, Goodarzi MO, Petrov MS. Antidiabetic Medications and Mortality Risk in Individuals With Pancreatic Cancer–Related Diabetes and Postpancreatitis Diabetes: A Nationwide Cohort Study. Diabetes Care (2019) 42(9):1675–83. doi: 10.2337/dc19-0145
97. Wang C, Zhang T, Liao Q, Dai M, Guo J, Yang X, et al. Metformin Inhibits Pancreatic Cancer Metastasis Caused by SMAD4 Deficiency and Consequent HNF4G Upregulation. Protein Cell (2021) 12(2):128–44. doi: 10.1007/s13238-020-00760-4
98. Chen K, Qian W, Jiang Z, Cheng L, Li J, Sun L, et al. Metformin Suppresses Cancer Initiation and Progression in Genetic Mouse Models of Pancreatic Cancer. Mol Cancer (2017) 16(1):131. doi: 10.1186/s12943-017-0701-0
99. Han H, Hou Y, Chen X, Zhang P, Kang M, Jin Q, et al. Metformin-Induced Stromal Depletion to Enhance the Penetration of Gemcitabine-Loaded Magnetic Nanoparticles for Pancreatic Cancer Targeted Therapy. J Am Chem Soc (2020) 142(10):4944–54. doi: 10.1021/jacs.0c00650
100. Bosetti C, Rosato V, Li D, Silverman D, Petersen GM, Bracci PM, et al. Diabetes, Antidiabetic Medications, and Pancreatic Cancer Risk: An Analysis From the International Pancreatic Cancer Case-Control Consortium. Ann Oncol (2014) 25(10):2065–72. doi: 10.1093/annonc/mdu276
101. Lee DY, Yu JH, Park S, Han K, Kim NH, Yoo HJ, et al. The Influence of Diabetes and Antidiabetic Medications on the Risk of Pancreatic Cancer: A Nationwide Population-Based Study in Korea. Sci Rep Uk (2018) 8(1):9719. doi: 10.1038/s41598-018-27965-2
102. Kordes S, Pollak MN, Zwinderman AH, Mathôt RA, Weterman MJ, Beeker A, et al. Metformin in Patients With Advanced Pancreatic Cancer: A Double-Blind, Randomised, Placebo-Controlled Phase 2 Trial. Lancet Oncol (2015) 16:839–47. doi: 10.1016/S1470-2045(15)00027-3
103. De Souza A, Khawaja KI, Masud F, Saif MW. Metformin and Pancreatic Cancer: Is There a Role? Cancer Chemoth Pharm (2016) 77(2):235–42. doi: 10.1007/s00280-015-2948-8
104. Yoshida J, Ishikawa T, Endo Y, Matsumura S, Ota T, Mizushima K, et al. Metformin Inhibits TGF−β1−Induced Epithelial−Mesenchymal Transition and Liver Metastasis of Pancreatic Cancer Cells. Oncol Rep (2020) 44(1):371–81. doi: 10.3892/or.2020.7595
105. Zhao H, Zhou N, Jin F, Wang R, Zhao J. Metformin Reduces Pancreatic Cancer Cell Proliferation and Increases Apoptosis Through MTOR Signaling Pathway and its Dose-Effect Relationship. Eur Rev Med Pharmaco (2020) 24(10):5336. doi: 10.26355/eurrev_202005_21316
106. Fitzgerald TL, Lertpiriyapong K, Cocco L, Martelli AM, Libra M, Candido S, et al. Roles of EGFR and KRAS and Their Downstream Signaling Pathways in Pancreatic Cancer and Pancreatic Cancer Stem Cells. Adv Biol Regul (2015) 59:65–81. doi: 10.1016/j.jbior.2015.06.003
107. Soliman GA, Shukla SK, Etekpo A, Gunda V, Steenson SM, Gautam N, et al. The Synergistic Effect of an ATP-Competitive Inhibitor of mTOR and Metformin on Pancreatic Tumor Growth. Curr Developments Nutr (2020) 4(9):a131. doi: 10.1093/cdn/nzaa131
108. Lonardo E, Cioffi M, Sancho P, Sanchez-Ripoll Y, Trabulo SM, Dorado J, et al. Metformin Targets the Metabolic Achilles Heel of Human Pancreatic Cancer Stem Cells. PloS One (2013) 8(10):e76518. doi: 10.1371/journal.pone.0076518
109. Ma M, Ma C, Li P, Ma C, Ping F, Li W, et al. Low Glucose Enhanced Metformin’s Inhibitory Effect on Pancreatic Cancer Cells by Suppressing Glycolysis and Inducing Energy Stress via Up-Regulation of miR-210-5p. Cell Cycle (Georgetown Tex) (2020) 19(17):2168–81. doi: 10.1080/15384101.2020.1796036
110. Gale EAM. Response to Comment on: Butler Et al. A Critical Analysis of the Clinical Use of Incretin-Based Therapies: Are the GLP-1 Therapies Safe? Diabetes Care (2013) 36:2118–25. doi: 10.2337/dc13-1542
111. Gier B, Matveyenko AV, Kirakossian D, Dawson D, Dry SM, Butler PC. Chronic GLP-1 Receptor Activation by Exendin-4 Induces Expansion of Pancreatic Duct Glands in Rats and Accelerates Formation of Dysplastic Lesions and Chronic Pancreatitis in the KrasG12D Mouse Model. Diabetes (2012) 61(5):1250–62. doi: 10.2337/db11-1109
112. Monami M, Nreu B, Scatena A, Cresci B, Andreozzi F, Sesti G, et al. Safety Issues With Glucagon-Like Peptide-1 Receptor Agonists (Pancreatitis, Pancreatic Cancer and Cholelithiasis): Data From Randomized Controlled Trials. Diabetes Obes Metab (2017) 19(9):1233–41. doi: 10.1111/dom.12926
113. Nreu B, Dicembrini I, Tinti F, Mannucci E, Monami M. Pancreatitis and Pancreatic Cancer in Patients With Type 2 Diabetes Treated With Glucagon-Like Peptide-1 Receptor Agonists: An Updated Meta-Analysis of Randomized Controlled Trials. Minerva Endocrinol (2020) in press. doi: 10.23736/S0391-1977.20.03219-8
114. Ferrannini E, Solini A. SGLT2 Inhibition in Diabetes Mellitus: Rationale and Clinical Prospects. Nat Rev Endocrinol (2012) 8(8):495–502. doi: 10.1038/nrendo.2011.243
115. Scafoglio C, Hirayama BA, Kepe V, Liu J, Ghezzi C, Satyamurthy N, et al. Functional Expression of Sodium-Glucose Transporters in Cancer. Proc Natl Acad Sci (2015) 112(30):E4111–9. doi: 10.1073/pnas.1511698112
Keywords: type 2 diabetes mellitus, pancreatic cancer, hyperglycemia, insulin resistance, screening strategy, hypoglycemic therapy
Citation: Duan X, Wang W, Pan Q and Guo L (2021) Type 2 Diabetes Mellitus Intersects With Pancreatic Cancer Diagnosis and Development. Front. Oncol. 11:730038. doi: 10.3389/fonc.2021.730038
Received: 24 June 2021; Accepted: 30 July 2021;
Published: 16 August 2021.
Edited by:
Taiping Zhang, Peking Union Medical College Hospital (CAMS), ChinaReviewed by:
Bei Li, Sichuan University, ChinaCopyright © 2021 Duan, Wang, Pan and Guo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lixin Guo, Z2x4MTIxOEAxNjMuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.