 Yangheng Xu
Yangheng Xu Zhisheng Hong
Zhisheng Hong Sheng Yu1
Sheng Yu1 Lvyun Zhu
Lvyun Zhu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Oncol. Rev., 30 April 2024
Sec. Oncology Reviews: Reviews
Volume 18 - 2024 | https://doi.org/10.3389/or.2024.1379323
SLC25A26 is the only known human mitochondrial S-adenosylmethionine carrier encoding gene. Recent studies have shown that SLC25A26 is abnormally expressed in some cancers, such as cervical cancer, low-grade glioma, non-small cell lung cancer, and liver cancer, which suggests SLC25A26 can affect the occurrence and development of some cancers. This article in brief briefly reviewed mitochondrial S-adenosylmethionine carrier in different species and its encoding gene, focused on the association of SLC25A26 aberrant expression and some cancers as well as potential mechanisms, summarized its potential for cancer prognosis, and characteristics of mitochondrial diseases caused by SLC25A26 mutation. Finally, we provide a brief expectation that needs to be further investigated. We speculate that SLC25A26 will be a potential new therapeutic target for some cancers.
The mitochondrial S-adenosylmethionine carrier (mSAMC), a member of the mitochondrial carrier family (MCF), widely exists in all eukaryotes and is located in the mitochondrial inner membrane [1–3]. The mSAMC catalyzes the import of S-adenosylmethionine (SAM) from the cytosol into the mitochondria and the export of S-adenosylhomocysteine (SAH) from the mitochondria into the cytosol [4–11]. In the cytoplasm, SAM participates in the methionine cycle and polyamine biosynthesis (Figure 1) [12–18]. In the mitochondria, SAM provides the methyl for methylation of DNA, RNA, proteins, and amino acids (Figure 1). Therefore, SAM has a great influence on sustained epigenetic modifications and has been implicated in several diseases and potential pathogenesis of cancer development [19]. For example, high or extremely high activity of methionine cycle metabolism was observed in cancer cells, especially tumor-initiating cells or tumor stem cells. Moreover, the proliferation of tumor cells is significantly inhibited by drug intervention of methionine cycle metabolism [20]. Schober et al. reported that mSAMC is the only carrier for transporting SAM from cytoplasm to mitochondria, and mitochondrial SAM levels in flies, mice, and humans are directly dependent on cytosolic SAM generated in the methionine cycle. Therefore, SLC25A26 abnormal expression or mSAMC dysfunction can be associated with multiple disease states, including cancer, nutrient deficiencies, and cardiovascular diseases [21]. The mitochondrial carrier family in humans, called solute carrier family 25 (SLC25), has been identified for 53 members which are widely distributed in eukaryotes with similar structure and are mainly embedded in the inner membrane of mitochondria. They are involved in the transmembrane transport of various substrates including amino acids and their derivatives, cofactors, inorganic ions, nucleotides, and so on, which is further associated with mitochondrial metabolism and cell function [3, 22, 23]. Human mSAMC was encoded by solute carrier family 25 member 26 (SLC25A26) gene [9]. Growing studies showed that aberrant expression of SLC25A26 may be involved in the occurrence and development of some cancers [24–32].
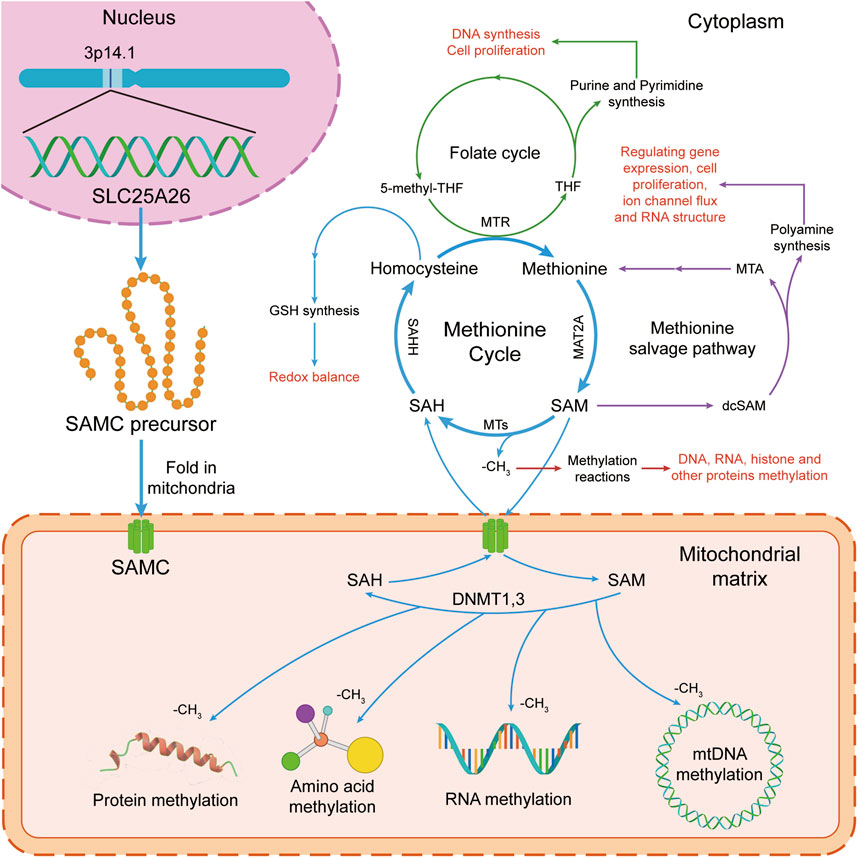
Figure 1. The S-adenosylmethionine (SAM) transport and function in human cells. In the cytoplasm, SAM takes part in the methionine cycle and polyamine synthesis. In mitochondria, SAM donates a methyl group for methylation reactions. The methionine cycle is interconnected with the transsulfuration pathway, the folate cycle, the methionine salvage pathway, and polyamine synthesis, all of which maintain important cellular functions. Firstly, SAM is converted by methionine via methionine adenosyltransferase 2A (MAT2A). Secondly, after donating a methyl group for methylation reactions, SAM is converted into S-adenosylhomocysteine (SAH) by methyltransferases (MTs). Then, SAH is then hydrolyzed by S-adenosyl-L-homocysteine hydrolase (SAHH) to generate homocysteine. Finally, homocysteine becomes methionine by receiving a methyl group from the folate cycle, which is mediated by 5-methyltetrahydrofolate: homocysteine methyltransferase (MTR). At the same time, SAM is transported to mitochondria for methylation reactions of mitochondrial DNA, RNA, protein and amino acid by mitochondrial S-adenosylmethionine carrier (mSAMC) which is encoded by SLC25A26 nuclear gene on chromosome 3p14.1. GSH, glutathione; MTR, 5-methyltetrahydrofolate: homocysteine methyltransferase; SAHH, S-adenosyl-L-homocysteine hydrolase; MTs, methyltransferases; MAT2A, methionine adenosyltransferase 2A; SAM, S-adenosylmethionine; SAMC, mitochondrial S-adenosylmethionine carrier; SAH, S-adenosylhomocysteine. THF, tetrahydrofolate; 5-methyl-THF, 5-methyltetrahydrofolate.
Therefore, we will briefly review mSAMC in different species and its encoding gene, focusing on the association of SLC25A26 gene aberrant expression and some cancers as well as potential mechanisms, and characteristics of mitochondrial diseases caused by SLC25A26 mutation. Finally, we provide a brief expectation that needs to be further investigated.
To date, mSAMC was identified in saccharomyces cerevisiae in 2003, human in 2004 and Arabidopsis in 2006 [5, 9, 10]. In Saccharomyces cerevisiae, it was named Sam5p (Pet8p is used now) and encoded by PET8 gene [5, 33–37]. In human, it was named SAMC and encoded by SLC25A26 gene which is located on chromosome 3p14.1 as an autosomal recessive hereditary double allele gene with a highly conserved exon similar to the other mitochondrial carrier genes [38]. In Arabidopsis, it has two isoforms, named SAMC1 and SAMC2 which were encoded by At4g39460 and At1g34065 gene, respectively. Sam5p (Pet8p), SAMC, SAMC1, and SAMC2 have the characteristics of the MCF and are located at mitochondria [3, 39]. However, later research proved that SAMC1 also exists on the chloroplast envelope membrane [24, 40–42]. SAMC1, SAMC2, and Sam5p (Pet8p) catalyze countertransport between SAM and mitochondrial SAH (mainly), S-adenosylcysteine (SAC), or adenosylornithine [5]. SAMC catalyzes countertransport between SAM and mitochondrial SAH [9]. SAMC1 and SAMC2 catalyze countertransport between SAM and mitochondrial SAH (mainly) or SAC [10]. Table 1 showed the differences and function of the mitochondrial S-adenosylmethionine carrier in yeast, human, and Arabidopsis, respectively.

Table 1. Comparison of mSAMC in yeast, human and Arabidopsis.
Additionally, the 2004 in vitro study on human recombinant SAMC identified several potent inhibitors [9]. These include pyridoxal 5′-phosphate, p-hydroxymercuribenzoate, mersalyl, and mercuric chloride, which are general inhibitors of mitochondrial carriers, and tannic acid and Bromocresol Purple, which specifically inhibit glutamate carriers. Notably, inhibitors characteristic of other mitochondrial carriers showed minimal impact on the activity of recombinant SAMC.
Regarding the regulation of the SLC25A26 gene, FOXD3 has been identified as a repressor in the CaSki cell line [25], while CTB enhances senescence in HCC cells by inducing the upregulation of SLC25A26 expression, potentially influencing its activation [28].
In some cancers, SLC25A26 expression is aberrant (Table 2). Patients with invasive cervical cancer have recurrent chromosome 3p12-p14 loss. In 2013, Lando et al. reported that 8 genes including SLC25A26 were highly downregulated in invasive cervical cancer patients with recurrent chromosome 3p12-p14 loss, which may be associated with cancer invasiveness [43]. In cervical cancer line CaSki and HeLa cells, SLC25A26 gene is also downregulated [26]. A large-scale genome-wide association study of low-grade glioma revealed 2 causal single nucleotide polymorphisms at rs11706832 site. One is rs11706832 reference allele: A, which may generate a binding site for a transcriptional repressor named LEF1 to inhibit SLC25A26 expression. The other is rs11706832 alternative allele: C, a risk alternative allele, which promotes SLC25A26 expression [27]. In human liver cancer tissues, SLC25A26 expression was low compared to adjacent tissues. A novel copper complex [Cu(ttpy-tpp)Br2]Br (Referred to as CTB) exerted an anti-hepatocellular carcinoma effect by up-regulating SLC25A26 expression level in mice [28]. In non-small cell lung cancer, expression of SLC25A26 and the 10-year survival rate of the patients were negatively correlated [31]. In most colorectal cancer patients, SLC25A26 gene was highly expressed. Moreover, in a subcutaneous transplanted tumor model with MC38 cells, a mouse colon cancer cell lines, knockdown of SLC25A26 caused tumor volume to begin to decrease by day 9 and completely disappear by day 30, whereas control tumor volume continued to increase [32]. The expression of SLC25A26 in different cancers is shown in Table 2. These studies suggest that aberrant expression of SLC25A26 may be associated with the occurrence and development of some cancers.
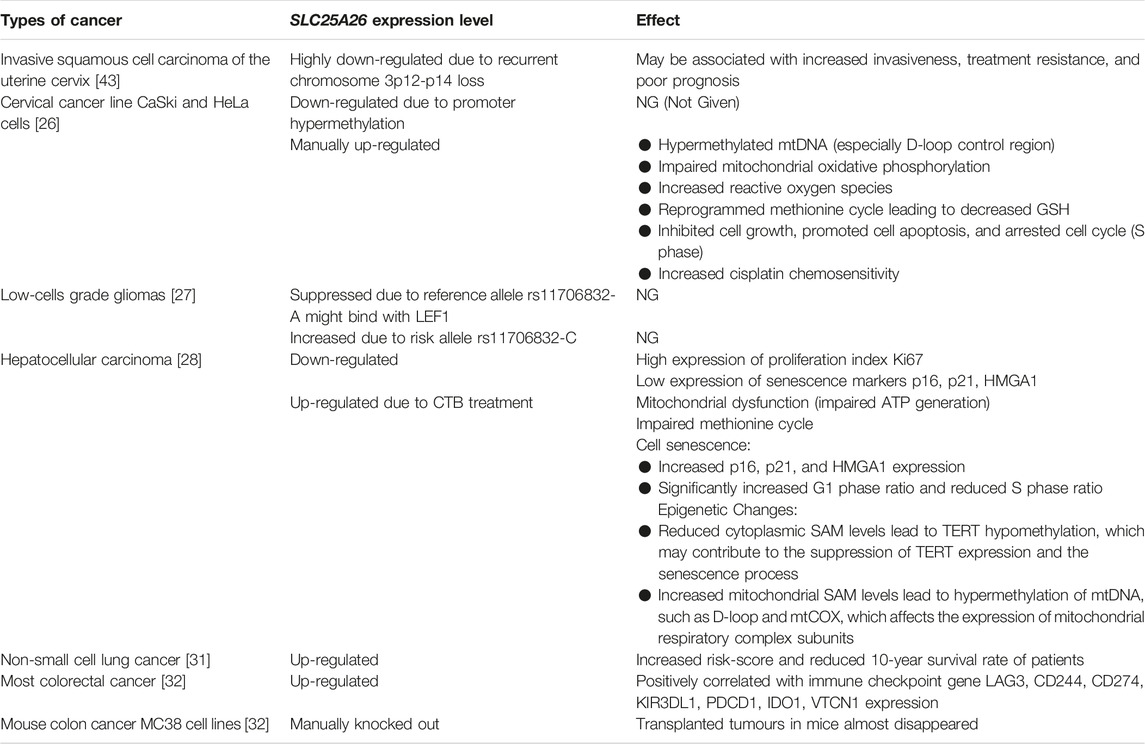
Table 2. Altered SLC25A26 expression and its effects on some cancers.
Studies from clinical trials, and animal and cell models indicate that SLC25A26 abnormal expression can regulate the occurrence and development of cancers. Its potential mechanisms are as follows (Figure 2).
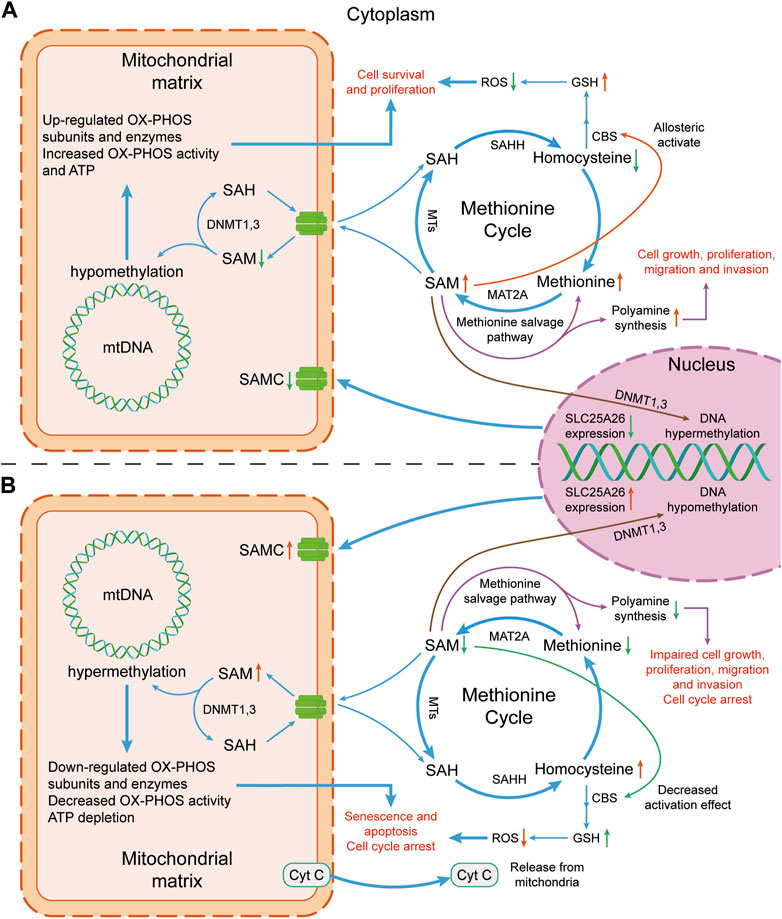
Figure 2. Mechanism diagram of the effect of altered SLC25A26 expression level on cancer cells. (A) Lower expression level of SLC25A26 may be beneficial to cancer cells. Lower expression level of SLC25A26 reduces mitochondrial SAMC levels, thereby reducing mitochondrial uptake of cytosolic SAM, resulting in a decrease in mitochondrial SAM concentration and an increase in cytosolic SAM concentration. Such changes in SAM level will eventually lead to the growth, survival, proliferation, migration and invasion of cancer cells by promoting GSH synthesis and polyamine synthesis, as well as up-regulating mitochondrial respiratory enzyme levels. (B) Higher expression levels of SLC25A26 may be detrimental to cancer cells. In contrast to panel (A), higher SLC25A26 expression level increases mitochondrial SAMC levels and therefore increases mitochondrial uptake of cytosolic SAM, resulting in an increase in mitochondrial SAM concentration and a decrease in cytosolic SAM concentration. Such changes in SAM level will eventually lead to senescence, apoptosis, cell cycle arrest of cancer cells, and inhibition of its growth, proliferation, migration and invasion by reducing GSH synthesis and polyamine synthesis, as well as down-regulating mitochondrial respiratory enzymes. SAH, S-adenosylhomocysteine; SAHH, S-adenosyl-L-homocysteine hydrolase; SAM, S-adenosylmethionine; SAMC, S-adenosylmethionine carrier; ROS, reactive oxygen species; GSH, glutathione; CBS, cystathione-β synthase; mtDNA, mitochondrial DNA; MTs, Methyltransferases; MAT2A, methionine adenosyltransferase 2A; DNMT, DNA methyltransferase; OX-PHOS, oxidative phosphorylation; Cyt C, cytochrome c.
The methionine cycle consists of a series of reactions that break down and regenerate methionine which is essential for many cellular functions, such as DNA and polyamine synthesis, histone and DNA methylation, and redox balance. Reports showed that the proliferation process of cancer cells is highly dependent on the methionine cycle [44]. Moreover, lung tumor-initiating cells showed highly elevated methionine cycle activity and transmethylation rates driven by MAT2A. High methionine cycling activity results in methionine consumption far exceeding its regeneration, leading to extrinsic methionine addiction. When the methionine cycle was, even transiently, inhibited by drug, the tumor-initiating ability of tumor-initiating cells was sufficiently decreased. At the same time, methionine cycling flux specifically affects the epigenetic state of cancer cells and drives cancer initiation [20]. Therefore, interference with methionine cycle is one of the targets of tumor therapy.
The novel copper complex CTB, a candidate anti-tumor compound, significantly inhibits the methionine cycle and promotes hepatocellular carcinoma cell senescence via inducing the accumulation of SAMC. siSLC25A26 treatment has opposite effects [28]. Therefore, the occurrence and development of cancers can be controlled by regulating the expression of SLC25A26 to affect the activity of the methionine cycle metabolism.
Cancer is an outcome of abnormal genetic and epigenetic changes. Epigenetic mechanisms, responsible for regulating gene expression without altering the DNA sequence, mainly include DNA methylation, histone modification, and chromatin remodelling. DNA methylation is the most studied mechanism [45]. The process of DNA methylation refers to the enzymatic addition of methyl groups to the cytosine residues by DNA methyltransferases, which relies on the availability of methyl groups and the function of the corresponding methyl donors and acceptors [11]. DNA methylation is a normal mechanism by which cells regulate gene expression in the mammalian genome. However, in 1983, Feinberg and Vogelstein first reported that there was extensive global loss of 5′-methylcytosine (the main form of DNA methylation) in colon cancers compared with the normal colon [46]. Subsequent studies confirmed that aberrant DNA methylation is a nearly universal finding in cancers and a major early contributor to cancer development [47, 48]. Therefore, regulating DNA methylation is one of the therapeutic targets for cancers.
No methylated sites were found in a CpG island in non-tumoral SLC25A26 promoter, while in CaSki and HeLa cancer cells, 13 and 15 methylated sites of DNA were found, respectively. In CaSki cells (a cervical cancer cell), SLC25A26 gene promoter hypermethylation results in downregulation of SLC25A26 expression, which decreased SAM levels in mitochondria, induced hypomethylation of mitochondrial DNA, enhanced expression of key respiratory complex subunits, generation of mitochondrial ATP. At the same time, increased SAM in cytoplasm enhanced the methionine cycle, which reduces homocysteine and ROS but increases glutathione (GSH) that enhances cellular antioxidant defenses. All these events occur almost simultaneously to promote cancer cell survival and proliferation. On the contrary, overexpression of SLC25A26 increased mitochondrial SAM level and enhanced hypermethylation of mitochondrial DNA, which inhibited expression of key respiratory complex subunits, and generation of mitochondrial ATP but enhanced release of cytochrome C (a trigger of type II apoptosis when released from the mitochondria). At the same time, methionine cycle was disrupted because SAM was in large quantities transported into mitochondria, which increases homocysteine and ROS and reduces GSH that reduces cellular antioxidant defenses. All these events occur almost simultaneously to arrest the cell cycle in the S phase and enhance cisplatin chemosensitivity [26].
Subsequently, Cianciulli et al. found that FOXD3, a member of the forkhead protein family, was a repressor of SLC25A26 gene expression. Moreover, after CaSki cells were treated with folate, the repressive role of FOXD3 was completely abrogated via affecting methylation of FOXD3 gene promoter [25]. Additionally, studies on CTB have shown that the suppression of extra-mitochondrial methylation by CTB-induced SLC25A26 overexpression can inhibit cancer cell genesis, progression, and proliferation by inhibiting the methylation of the telomerase reverse transcriptase promoter [28]. These studies showed that SLC25A26 may regulate the occurrence and development of cancers via affecting DNA methylation.
Cellular senescence, a stable cell cycle arrest that limits cell proliferation, is pivotal in various stages of tumorigenesis, including initiation, formation, and escape [49–51]. This process can also inhibit cancer development through autocrine and paracrine manners [49–52]. Consequently, therapies aimed at selectively enhancing senescence in cancer cells, known as “prosenescence” therapies, can be instrumental in anti-cancer treatment strategies [51].
In the human clinical liver cancer tissues with high expression of SLC25A26, proliferation index Ki67 had low expression and the senescence markers p16, p21, and HMGA1 had high expression, and the tissues with low expression of SLC25A26 had opposite effects [28]. Moreover, during novel copper complex CTB-induced hepatocellular carcinoma cell senescence, the accumulation of SAMC is essential [28]. These results indicated that SLC25A26 may regulate cancer cell senescence.
Apoptosis is considered to be the most vital form of cell death, which plays a crucial role for all multicellular organisms to regulate cell proliferation and maintain tissue homeostasis as well as get rid of unnecessary or harmful cells from an organism [53, 54]. Defects of apoptotic physiological mechanisms may lead to different human diseases, such as cancer. Cancer cells can alter apoptotic pathways through transcription, translation, and post-translation as well as escape apoptosis via several different strategies. Therefore, apoptosis has become one of the main molecular targets for drug discovery and development, especially for diseases such as cancer [55–57]. In CaSki cells, overexpression of SLC25A26 promotes apoptosis, inhibits cell growth, and enhances chemosensitivity to cisplatin by reducing cytoplasmic SAM levels and increasing mitochondrial SAM, thereby suppressing methionine metabolism and methylation [26]. Moreover, several studies have demonstrated that restricting exogenous methionine intake in cancer cells can enhance control over various cancers and improve interventions with radiotherapy and chemotherapy for cancer [58–61]. Therefore, upregulating SLC25A26 and limiting methionine intake in cancer cells may be able to further affect cellular methionine metabolism, potentially enhancing apoptosis and enhancing the effect of chemotherapy drugs.
Cancer, a genomic disease, prompts the immune system to recognize tumor antigens as foreign and triggers cellular immune responses [62, 63], inducing immune cells to infiltrate into the tumor microenvironment and regulate tumor progression [64–66]. Therefore, immunotherapy provides a more effective and less toxic alternative to chemotherapy for cancer patients [66–68], which is revolutionizing cancer treatment.
In patients with non-small cell lung cancer, immune infiltration analysis showed that SLC25A26 expression was negatively correlated with 10-year survival [31]. SAM and histone methylation were supported by one-carbon metabolism to drive inflammatory macrophage infiltration [21]. Immuno-infiltration analysis also showed that SLC25A26 expression in several tumors has a positive or negative correlation with the abundance of immune cell subsets around the tumor tissue [32]. Expression of SLC25A26 in lower-grade glioma (GBMLGG) and glioblastoma (GBM), melanoma (SKCM), uveal melanoma (UVM), and colon and rectal cancer (COADREAD) has a significant negative correlation with the abundance of dendritic cells, M1 macrophages, and T helper 2 cells around them. On the contrary, expression of SLC25A26 in cholangiocarcinoma (CHOL), esophageal cancer (ESCA), and sarcoma (SARC) has a significant positive correlation with the abundance of dendritic cells and M1 macrophages around them [32]. Moreover, since DC, M1 macrophages, and Th2 have different mechanisms to directly or indirectly inhibit or kill cancer cells, increased SLC25A26 expression may promote the development of GBMLGG, SKCM, UVM, and COADREAD, whereas decreased SLC25A26 expression is beneficial to inhibit or kill them. In contrast, low expression of SLC25A26 may promote the development of CHOL, ESCA, and SARC, while high expression of SLC25A26 is beneficial to inhibit or kill them [32].
These results indicated that SLC25A26 may regulate cancer cell development via affecting the immune system, which may be a research hotspot of tumor treatment in the future and provide a new direction for tumor immunotherapy.
The low expression levels of eight genes located on chromosome 3p12-14, including SLC25A26, have been verified to be associated with poor prognosis in patients with cervical squamous cell carcinomas (SCC). In addition, SCC is more likely to have chromosome 3p12-14 loss and is associated with aggressive tumour growth compared to precancerous lesions. Therefore, these eight genes may contribute to evaluating cancer aggressiveness in the early invasive stage. Based on the low intratumor heterogeneity of these genes, the gene expression level can be obtained by FISH for clinical diagnostic testing [43].
SLC25A26 expression is detected as low in HCC tissues, which is correlated with a high level of proliferation index Ki67 and low expression of senescence markers p16, p21 and HMGA1 (28). Therefore, we speculate that the low expression of SLC25A26 in HCC tissues may be related to the poor prognosis of patients.
For NSCLC, a total of 5 genes including SLC25A26 were selected and made as a risk assessment model. In the model, high expression of SLC25A26 in NSCLC patients is positively correlated with the risk score and predicts a poor 10-year survival rate [31].
In brief, these studies suggest that SLC25A26 expression level may be differently correlated with the prognosis in different cancers. However, the relationship between SLC25A26 expression and the prognosis of other cancers as well as the specific predictive indicators remain to be investigated. In addition, it should be explored whether SLC25A26 expression is relevant for predicting, detecting, or diagnosing certain cancers.
It has also been reported that SLC25A26 mutation may lead to mitochondrial defects and then induce a mitochondrial disease called combined oxidative phosphorylation deficiency 28 (COXPD28).
COXPD28 is an autosomal recessive genetic disease first identified by Kishita et al. in 2015 in three unrelated severe infant cases [69]. Subsequently, two new infants with COXPD28 were reported in 2022 [70, 71]. The five patients presented with varying degrees of clinical symptoms, ranging from mild muscle weakness, cardiopulmonary insufficiency, and developmental delay to respiratory and circulatory/multiple organ failure and death, accompanied by hyperlactatemia. Muscle biopsy revealed varying degrees of decrease in mitochondrial complexes I, III, and IV [69]. Further research on infants with COXPD28 indicated that the SLC25A26 mutation severely impairs the function of SAMC in transporting SAM into the mitochondria [69], which may lead to a more severe early-onset phenotype. This mutation results in a deficiency of mitochondrial SAM input, leading to a lack of methylation substrates and impaired mitochondrial biosynthesis. Specifically, mtRNA methylation is impaired, affecting rRNA stability and tRNA maturation, further impacting ribosomal assembly and de novo translation processes in mitochondria. The steady-state levels of COXII, a subunit of complex IV, was reduced, and the methylation of ADP/ATP translocase genes ANT1 and ANT2, as well as the electron transfer flavoprotein ETFβ, were found to be hypomethylated. The biosynthesis of LA [72], which relies on SAM for methylation, is impaired, leading to reduced levels of the pyruvate dehydrogenase complex E2 (PDHC-E2) and the α-ketoglutarate dehydrogenase E2 (α-KGDH-E2), of which LA is a subunit, and further affecting the activity of PDH and α-KDGH. The biosynthesis of CoQ10, an electron carrier in the mitochondrial oxidative respiratory chain, is also impaired, leading to a significant decrease in mitochondrial ATP production [69].
Additionally, three adult female patients with milder phenotypes of COXPD28 have been reported [73, 74]. The common symptoms of these three patients were muscle weakness, gastrointestinal discomfort, and hyperlactacidemia. Independent symptoms include respiratory and multi-organ failure, exercise intolerance with muscle pain and disability. Studies in adult cases of COXPD28 suggest that the SLC25A26 mutation primarily affects the function of SAMC in transporting SAH into the cytoplasm, which may result in a milder late-onset phenotype. In mouse embryonic fibroblast models, cells with the SLC25A26 mutation also showed reduced mitochondrial LA levels and PDH activity [73]. In Drosophila Melanogaster larvae with the SLC25A26 mutation, a decrease in the pentose phosphate pathway and bile acid synthesis was found, while iron-sulfur clusters and ubiquinone biosynthesis proteins were upregulated, and OXPHOS components UQCR-11 and COX7A proteins both showed increased expression, along with biomarkers of mitochondrial methylation defects [21], including the absence of glycine-N-methyltransferase (GNMT) and an increase in serine biosynthetic enzyme phosphoserine phosphatase (PSPH) [73].
In conclusion, SLC25A26 mutations impair mitochondrial RNA stability, protein synthesis, LA, and CoQ10 biosynthesis, which further affect TCA and the mitochondrial oxidative respiratory chain, resulting in the intractable mitochondrial disease COXPD28. This also emphasizes the importance of SLC25A26 for mitochondrial function. By the way, in patients with major depression disorder, the plasma SAMC level was found to be significantly increased [75].
In conclusion, we briefly reviewed the essential roles of SAMC and its encoding gene, further empathizes association of SLC25A26 aberrant expression and some cancers, as well as potential mechanisms for SLC25A26 regulating the occurrence and development of cancers including regulating the activity of methionine cycle metabolism, DNA methylation, senescence, apoptosis and immune cell infiltration. Next, we summarized its potential in cancer prognosis and finally emphasized its importance in mitochondria function by reviewing an intractable mitochondrial disease named COXPD28 caused by SLC25A26 mutation.
However, the role of SLC25A26 in cancer regulation remains to be fully elucidated. Key questions include how varying expression levels of SLC25A26 affect the methylation of genes in the nucleus or mitochondria, and how these changes regulate various aspects of cancer through specific pathways. Additionally, the impact of SLC25A26 expression on the infiltration of immune cell subsets adjacent to different cancer cells and the abundance of cancer cells, and its subsequent influence on cancer progression through immune modulation, is an area that needs further investigation. The potential of SLC25A26 as a target for cancer therapy is yet to be fully exploited, either. For instance, the discovery of potent inhibitors of the expression product of SLC25A26, SAMC, has been made [9], but their effects on cancer have not yet been studied. Moreover, the downregulation of SLC25A26 in invasive cervical SCC may enhance invasiveness and resistance to treatment [43]. This finding prompts a deeper exploration into the mechanisms linking SLC25A26 downregulation with cancer cell invasiveness and treatment resistance, potentially revealing new strategies to reduce cancer lethality and improve therapeutic efficacy. Furthermore, the novel copper complex CTB has been shown to inhibit tumorigenesis in vivo by up-regulating SLC25A26 expression [28], while MC38 cells with SLC25A26 knockout exhibited massive cell death [32], and SLC25A26 knockout mice were embryonically lethal [21]. These observations suggest that both extremely high and low levels of SLC25A26 expression could lead to the inhibition or death of cancer cells via distinct pathways, indicating that SLC25A26 inhibitors or activators may represent novel cancer gene therapies awaiting further research. Besides, further investigation is needed to determine the role of SLC25A26 in predicting, diagnosing, and prognosing cancer.
Overall, SLC25A26 may regulate the occurrence and development of cancers via many pathways, which suggests that SLC25A26 will be a potential new therapeutic target for cancers.
LZ and SX designed the manuscript and approved the final manuscript for publication; YX wrote the manuscript and prepared all the figures and tables; and ZH, SY, KL, RH, and ML collected the references and modified the manuscript. No Large Language Models are used for this review. All authors contributed to the article and approved the submitted version.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by grants from the National Natural Science Foundation of China (32171429), the “Huxiang Young Talents Plan” Project of Hunan Province (2019RS2030), the Natural Science Foundation of Hunan Province (2022JJ30672 and 2020JJ5657), Fund for NUDT Young Innovator Awards (20190104), and Postgraduate Scientific Research Innovation Project of Hunan Province.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Picault, N, Hodges, M, Palmieri, L, and Palmieri, F. The Growing Family of Mitochondrial Carriers in Arabidopsis. Trends Plant Sci (2004) 9(3):138–46. doi:10.1016/j.tplants.2004.01.007
2. Palmieri, F, Agrimi, G, Blanco, E, Castegna, A, Di Noia, MA, Iacobazzi, V, et al. Identification of Mitochondrial Carriers in Saccharomyces Cerevisiae by Transport Assay of Reconstituted Recombinant Proteins. Biochim Biophys Acta (Bba) - Bioenerg (2006) 1757(9-10):1249–62. doi:10.1016/j.bbabio.2006.05.023
3. Palmieri, F. The Mitochondrial Transporter Family SLC25: Identification, Properties and Physiopathology. Mol Aspects Med (2013) 34(2-3):465–84. doi:10.1016/j.mam.2012.05.005
4. Kumar, A, Agarwal, S, Heyman, JA, Matson, S, Heidtman, M, Piccirillo, S, et al. Subcellular Localization of the Yeast Proteome. Genes Dev (2002) 16(6):707–19. doi:10.1101/gad.970902
5. Marobbio, CM, Agrimi, G, Lasorsa, FM, and Palmieri, F. Identification and Functional Reconstitution of Yeast Mitochondrial Carrier for S-Adenosylmethionine. EMBO J (2003) 22(22):5975–82. doi:10.1093/emboj/cdg574
6. Schröder, G, Eichel, J, Breinig, S, and Schröder, J. Three Differentially Expressed S-Adenosylmethionine Synthetases From Catharanthus Roseus: Molecular and Functional Characterization. Plant Mol Biol (1997) 33(2):211–22. doi:10.1023/a:1005711720930
7. Ravanel, S, Gakière, B, Job, D, and Douce, R. The Specific Features of Methionine Biosynthesis and Metabolism in Plants. Proc Natl Acad Sci USA (1998) 95(13):7805–12. doi:10.1073/pnas.95.13.7805
8. Hanson, AD, and Roje, S. One-Carbon Metabolism in Higher Plants. Annu Rev Plant Physiol Plant Mol Biol (2001) 52:119–37. doi:10.1146/annurev.arplant.52.1.119
9. Agrimi, G, Di Noia, MA, Marobbio, CM, Fiermonte, G, Lasorsa, FM, and Palmieri, F. Identification of the Human Mitochondrial S-Adenosylmethionine Transporter: Bacterial Expression, Reconstitution, Functional Characterization and Tissue Distribution. Biochem J (2004) 379(Pt 1):183–90. doi:10.1042/BJ20031664
10. Palmieri, L, Arrigoni, R, Blanco, E, Carrari, F, Zanor, MI, Studart-Guimaraes, C, et al. Molecular Identification of an Arabidopsis S-Adenosylmethionine Transporter. Analysis of Organ Distribution, Bacterial Expression, Reconstitution Into Liposomes, and Functional Characterization. Plant Physiol (2006) 142(3):855–65. doi:10.1104/pp.106.086975
11. Ducker, GS, and Rabinowitz, JD. One-Carbon Metabolism in Health and Disease. Cell Metab (2017) 25(1):27–42. doi:10.1016/j.cmet.2016.08.009
12. Lauinger, L, and Kaiser, P. Sensing and Signaling of Methionine Metabolism. Metabolites (2021) 11(2):83. doi:10.3390/metabo11020083
13. Wang, H, Wu, Y, and Tang, W. Methionine Cycle in Nonalcoholic Fatty Liver Disease and Its Potential Applications. Biochem Pharmacol (2022) 200:115033. doi:10.1016/j.bcp.2022.115033
14. Mäkinen, K, and De, S. The Significance of Methionine Cycle Enzymes in Plant Virus Infections. Curr Opin Plant Biol (2019) 50:67–75. doi:10.1016/j.pbi.2019.03.002
15. Casero, RA, Murray Stewart, T, and Pegg, AE. Polyamine Metabolism and Cancer: Treatments, Challenges and Opportunities. Nat Rev Cancer (2018) 18(11):681–95. doi:10.1038/s41568-018-0050-3
16. Madeo, F, Eisenberg, T, Pietrocola, F, and Kroemer, G. Spermidine in Health and Disease. Science (2018) 359(6374):eaan2788. doi:10.1126/science.aan2788
17. Greenberg, MVC, and Bourc'his, D. The Diverse Roles of DNA Methylation in Mammalian Development and Disease. Nat Rev Mol Cell Biol. (2019) 20(10):590–607. doi:10.1038/s41580-019-0159-6
18. Parkhitko, AA, Jouandin, P, Mohr, SE, and Perrimon, N. Methionine Metabolism and Methyltransferases in the Regulation of Aging and Lifespan Extension Across Species. Aging Cell (2019) 18:e13034. doi:10.1111/acel.13034
19. Kaelin, WG, and McKnight, SL. Influence of Metabolism on Epigenetics and Disease. Cell (2013) 153(1):56–69. doi:10.1016/j.cell.2013.03.004
20. Wang, Z, Yip, LY, Lee, J, Wu, Z, Chew, HY, Chong, P, et al. Methionine Is a Metabolic Dependency of Tumor-Initiating Cells. Nat Med (2019) 25(5):825–37. doi:10.1038/s41591-019-0423-5
21. Rosenberger, FA, Moore, D, Atanassov, I, Moedas, MF, Clemente, P, Végvári, Á, et al. The One-Carbon Pool Controls Mitochondrial Energy Metabolism Via Complex I and Iron-Sulfur Clusters. Sci Adv (2021) 7(8):eabf0717. doi:10.1126/sciadv.abf0717
22. Ruprecht, JJ, and Kunji, E. The SLC25 Mitochondrial Carrier Family: Structure and Mechanism. Trends Biochem Sci (2020) 45(3):244–58. doi:10.1016/j.tibs.2019.11.001
23. Kunji, E, King, MS, Ruprecht, JJ, and Thangaratnarajah, C. The SLC25 Carrier Family: Important Transport Proteins in Mitochondrial Physiology and Pathology. Physiology (Bethesda) (2020) 35(5):302–27. doi:10.1152/physiol.00009.2020
24. Monné, M, Marobbio, C, Agrimi, G, Palmieri, L, and Palmieri, F. Mitochondrial Transport and Metabolism of the Major Methyl Donor and Versatile Cofactor S-Adenosylmethionine, and Related Diseases: A Review†. IUBMB life (2022) 74(7):573–91. doi:10.1002/iub.2658
25. Cianciulli, A, Menga, A, Ferdinando, P, and Iacobazzi, V. FOXD3 Acts as a Repressor of the Mitochondrial S-Adenosylmethionine Carrier (SLC25A26) Gene Expression in Cancer Cells. Biochimie (2018) 154:25–34. doi:10.1016/j.biochi.2018.07.025
26. Menga, A, Palmieri, EM, Cianciulli, A, Infantino, V, Mazzone, M, Scilimati, A, et al. SLC25A26 Overexpression Impairs Cell Function Via mtDNA Hypermethylation and Rewiring of Methyl Metabolism. FEBS J (2017) 284(6):967–84. doi:10.1111/febs.14028
27. Manjunath, M, Yan, J, Youn, Y, Drucker, KL, Kollmeyer, TM, McKinney, AM, et al. Functional Analysis of Low-Grade Glioma Genetic Variants Predicts Key Target Genes and Transcription Factors. Neuro-Oncology (2021) 23(4):638–49. doi:10.1093/neuonc/noaa248
28. Jin, C, Li, Y, Su, Y, Guo, Z, Wang, X, Wang, S, et al. Novel Copper Complex CTB Regulates Methionine Cycle Induced TERT Hypomethylation to Promote HCC Cells Senescence Via Mitochondrial SLC25A26. Cell Death Dis (2020) 11(10):844. doi:10.1038/s41419-020-03048-x
29. Lee, DD, Komosa, M, Nunes, NM, and Tabori, U. DNA Methylation of the TERT Promoter and Its Impact on Human Cancer. Curr Opin Genet Dev (2020) 60:17–24. doi:10.1016/j.gde.2020.02.003
30. Yu, J, Yuan, X, Sjöholm, L, Liu, T, Kong, F, Ekström, TJ, et al. Telomerase Reverse Transcriptase Regulates DNMT3B Expression/aberrant DNA Methylation Phenotype and AKT Activation in Hepatocellular Carcinoma. Cancer Lett (2018) 434:33–41. doi:10.1016/j.canlet.2018.07.013
31. Ye, Q, Singh, S, Qian, PR, and Guo, NL. Immune-Omics Networks of CD27, PD1, and PDL1 in Non-Small Cell Lung Cancer. Cancers (Basel) (2021) 13(17):4296. doi:10.3390/cancers13174296
32. Yu, S, Xu, Y, Zhao, Y, Zhao, Y, Zhang, Y, Sisi Xie, S, et al. Immune Infiltration of Colorectal Cancer Based on SLC25A26 Gene Expression. China Science and Technology Papers Online. Preprint (2022). Available from: www.paper.edu.cn (Accessed July 16, 2023).
33. Roussel, D, Harding, M, Runswick, MJ, Walker, JE, and Brand, MD. Does Any Yeast Mitochondrial Carrier Have a Native Uncoupling Protein Function? J Bioenerg Biomembranes (2002) 34(3):165–76. doi:10.1023/a:1016027302232
34. Steinmetz, LM, Scharfe, C, Deutschbauer, AM, Mokranjac, D, Herman, ZS, Jones, T, et al. Systematic Screen for Human Disease Genes in Yeast. Nat Genet (2002) 31(4):400–4. doi:10.1038/ng929
35. Mortimer, RK, Schild, D, Contopoulou, CR, and Kans, JA. Genetic and Physical Maps of Saccharomyces Cerevisiae. Methods Enzymol (1991) 194:827–63. doi:10.1016/0076-6879(91)94060-p
36. Cherry, JM, Ball, C, Weng, S, Juvik, G, Schmidt, R, Adler, C, et al. Genetic and Physical Maps of Saccharomyces Cerevisiae. Nature (1997) 387(6632):67–73. doi:10.1038/387s067
37. Lalo, D, Stettler, S, Mariotte, S, Gendreau, E, and Thuriaux, P. XIV. Yeast Sequencing Reports. Organization of the Centromeric Region of Chromosome XIV in Saccharomyces Cerevisiae. Yeast (Chichester, England) (1994) 10(4):523–33. doi:10.1002/yea.320100412
38. Cianciulli, A, Calvello, R, and Panaro, MA. Determinism and Randomness in the Evolution of Introns and Sine Inserts in Mouse and Human Mitochondrial Solute Carrier and Cytokine Receptor Genes. Comput Biol Chem (2015) 55:49–59. doi:10.1016/j.compbiolchem.2015.02.012
39. Millar, AH, and Heazlewood, JL. Genomic and Proteomic Analysis of Mitochondrial Carrier Proteins in Arabidopsis. Plant Physiol (2003) 131(2):443–53. doi:10.1104/pp.009985
40. Zybailov, B, Rutschow, H, Friso, G, Rudella, A, Emanuelsson, O, Sun, Q, et al. Sorting Signals, N-Terminal Modifications and Abundance of the Chloroplast Proteome. PloS one (2008) 3(4):e1994. doi:10.1371/journal.pone.0001994
41. Ferro, M, Salvi, D, Brugière, S, Miras, S, Kowalski, S, Louwagie, M, et al. Proteomics of the Chloroplast Envelope Membranes From Arabidopsis Thaliana. Mol Cell Proteomics (2003) 2(5):325–45. doi:10.1074/mcp.M300030-MCP200
42. Bryant, N, Lloyd, J, Sweeney, C, Myouga, F, and Meinke, D. Identification of Nuclear Genes Encoding Chloroplast-Localized Proteins Required for Embryo Development in Arabidopsis. Plant Physiol (2011) 155(4):1678–89. doi:10.1104/pp.110.168120
43. Lando, M, Wilting, SM, Snipstad, K, Clancy, T, Bierkens, M, Aarnes, EK, et al. Identification of Eight Candidate Target Genes of the Recurrent 3p12-P14 Loss in Cervical Cancer by Integrative Genomic Profiling. J Pathol (2013) 230(1):59–69. doi:10.1002/path.4168
44. Pavillard, V, Nicolaou, A, Double, JA, and Phillips, RM. Methionine Dependence of Tumours: A Biochemical Strategy for Optimizing Paclitaxel Chemosensitivity In Vitro. Biochem Pharmacol (2006) 71:772–8. doi:10.1016/j.bcp.2005.12.014
45. Biswas, S, and Rao, CM. Epigenetics in Cancer: Fundamentals and Beyond. Pharmacol Ther (2017) 173:118–34. doi:10.1016/j.pharmthera.2017.02.011
46. Feinberg, A, and Vogelstein, B. Hypomethylation Distinguishes Genes of Some Human Cancers From Their normal Counterparts. Nature (1983) 301(89):89–92. doi:10.1038/301089a0
47. Klutstein, M, Nejman, D, Greenfield, R, and Cedar, H. DNA Methylation in Cancer and Aging. Cancer Res (2016) 76(12):3446–50. doi:10.1158/0008-5472.CAN-15-3278
48. Morgan, AE, Davies, TJ, and Mc Auley, MT. The Role of DNA Methylation in Ageing and Cancer. Proc Nutr Soc (2018) 77(4):412–22. doi:10.1017/S0029665118000150
49. He, S, and Sharpless, NE. Senescence in Health and Disease. Cell (2017) 169(6):1000–11. doi:10.1016/j.cell.2017.05.015
50. Calcinotto, A, Kohli, J, Zagato, E, Pellegrini, L, Demaria, M, and Alimonti, A. Cellular Senescence: Aging, Cancer, and Injury. Physiol Rev (2019) 99(2):1047–78. doi:10.1152/physrev.00020.2018
51. Wang, L, Lankhorst, L, and Bernards, R. Exploiting Senescence for the Treatment of Cancer. Nat Rev Cancer (2022) 22(6):340–55. doi:10.1038/s41568-022-00450-9
52. Radiloff, DR, Wakeman, TP, Feng, J, Schilling, S, Seto, E, and Wang, XF. Trefoil Factor 1 Acts to Suppress Senescence Induced by Oncogene Activation During the Cellular Transformation Process. Proc Natl Acad Sci USA (2011) 108(16):6591–6. doi:10.1073/pnas.1017269108
53. Wyllie, AH. Glucocorticoid-Induced Thymocyte Apoptosis Is Associated With Endogenous Endonuclease Activation. Nature (1980) 284:555–6. doi:10.1038/284555a0
54. Kaufmann, SH, Desnoyers, S, Ottaviano, Y, Davidson, NE, and Poirier, GG. Specific Proteolytic Cleavage of Poly (ADP-Ribose) Polymerase: An Early Marker of Chemotherapy-Induced Apoptosis. Cancer Res (1993) 53:3976–85.
55. Fulda, S. Tumor Resistance to Apoptosis. Int J Cancer (2009) 124(3):511–5. doi:10.1002/ijc.24064
56. Carneiro, BA, and El-Deiry, WS. Targeting Apoptosis in Cancer Therapy. Nat Rev Clin Oncol (2020) 17(7):395–417. doi:10.1038/s41571-020-0341-y
57. Goldar, S, Khaniani, MS, Derakhshan, SM, and Baradaran, B. Molecular Mechanisms of Apoptosis and Roles in Cancer Development and Treatment. Asian Pac J Cancer Prev (2015) 16(6):2129–44. doi:10.7314/apjcp.2015.16.6.2129
58. Gao, X, Sanderson, SM, Dai, Z, Reid, MA, Cooper, DE, Lu, M, et al. Dietary Methionine Influences Therapy in Mouse Cancer Models and Alters Human Metabolism. Nature (2019) 572(7769):397–401. doi:10.1038/s41586-019-1437-3
59. Kokkinakis, DM. Methionine-Stress: A Pleiotropic Approach in Enhancing the Efficacy of Chemotherapy. Cancer Lett (2006) 233(2):195–207. doi:10.1016/j.canlet.2005.02.034
60. Lu, WC, Saha, A, Yan, W, Garrison, K, Lamb, C, Pandey, R, et al. Enzyme-Mediated Depletion of Serum L-Met Abrogates Prostate Cancer Growth Via Multiple Mechanisms Without Evidence of Systemic Toxicity. Proc Natl Acad Sci (2020) 117(23):13000–11. doi:10.1073/pnas.1917362117
61. Xu, Q, Li, Y, Gao, X, Kang, K, Williams, JG, Tong, L, et al. HNF4α Regulates Sulfur Amino Acid Metabolism and Confers Sensitivity to Methionine Restriction in Liver Cancer. Nat Commun (2020) 11(1):3978. doi:10.1038/s41467-020-17818-w
62. Matsushita, H, Vesely, MD, Koboldt, DC, Rickert, CG, Uppaluri, R, Magrini, VJ, et al. Cancer Exome Analysis Reveals a T-cell-Dependent Mechanism of Cancer Immunoediting. Nature (2012) 482(7385):400–4. doi:10.1038/nature10755
63. Schreiber, RD, Old, LJ, and Smyth, MJ. Cancer Immunoediting: Integrating Immunity’s Roles in Cancer Suppression and Promotion. Science (2011) 331:1565–70. doi:10.1126/science.1203486
64. Demaria, O, Cornen, S, Daëron, M, Morel, Y, Medzhitov, R, and Vivier, E. Harnessing Innate Immunity in Cancer Therapy. Nature (2019) 574:45–56. doi:10.1038/s41586-019-1593-5
65. Grivennikov, SI, Greten, FR, and Karin, M. Immunity, Inflammation, and Cancer. Cell (2010) 140:883–99. doi:10.1016/j.cell.2010.01.025
66. Zhang, Y, and Zhang, Z. The History and Advances in Cancer Immunotherapy: Understanding the Characteristics of Tumor-Infiltrating Immune Cells and Their Therapeutic Implications. Cell Mol. Immunol. (2020) 17(8):807–21. doi:10.1038/s41423-020-0488-6
67. Cm, JS, Dammeijer, F, Aerts, J, and Cornelissen, R. Immunotherapeutic Strategies in Non-Small-Cell Lung Cancer: The Present and the Future. Immunotherapy (2016) 9:507–20. doi:10.2217/imt-2016-0151
68. Kaufman, HL. Rational Combination Immunotherapy: Understand the Biology. Cancer Immunol Res (2017) 5:355–6. doi:10.1158/2326-6066.cir-17-0128
69. Kishita, Y, Pajak, A, Bolar, NA, Marobbio, CM, Maffezzini, C, Miniero, DV, et al. Intra-Mitochondrial Methylation Deficiency Due to Mutations in SLC25A26. Am J Hum Genet (2015) 97(5):761–8. doi:10.1016/j.ajhg.2015.09.013
70. Toyama, Y, Fujii, Y, Hori, SI, Yoshimura, K, and Kaneko, K. L-Carnitine Rescue for Neonatal Intractable Mitochondrial Cardiomyopathy. Pediatr Int official J Jpn Pediatr Soc (2022) 64(1):e15143. doi:10.1111/ped.15143
71. Wang, J, Xu, F, Shang, L, Li, W, and Zhang, X. Combined Oxidative Phosphorylation Deficiency Type 28: A Case Report and Literature Review (Article in Chinese). Chin J Appl Clin Pediatr (2022) 37(8):631–3.
72. Booker, SJ, Cicchillo, RM, and Grove, TL. Self-Sacrifice in Radical S-Adenosylmethionine Proteins. Curr Opin Chem Biol (2007) 11(5):543–52. doi:10.1016/j.cbpa.2007.08.028
73. Rosenberger, FA, Tang, JX, Sergeant, K, Moedas, MF, Zierz, CM, Moore, D, et al. Pathogenic SLC25A26 Variants Impair SAH Transport Activity Causing Mitochondrial Disease. Hum Mol Genet (2022) 31(12):2049–62. doi:10.1093/hmg/ddac002
74. Ji, Y, Wang, S, Cheng, Y, Fang, L, Zhao, J, Gao, L, et al. Identification and Characterization of Novel Compound Variants in SLC25A26 Associated With Combined Oxidative Phosphorylation Deficiency 28. Gene (2021) 804:145891. doi:10.1016/j.gene.2021.145891
Keywords: SLC25A26, S-adenosylmethionine carrier protein, cancer, gene expression regulation, methylation
Citation: Xu Y, Hong Z, Yu S, Huang R, Li K, Li M, Xie S and Zhu L (2024) Fresh Insights Into SLC25A26: Potential New Therapeutic Target for Cancers: A Review. Oncol. Rev. 18:1379323. doi: 10.3389/or.2024.1379323
Received: 31 January 2024; Accepted: 02 April 2024;
Published: 30 April 2024.
Edited by:
Deepa Kushwaha, Rare Genomics Institute, United StatesReviewed by:
Jiarui Chen, University of Wisconsin-Madison, United StatesCopyright © 2024 Xu, Hong, Yu, Huang, Li, Li, Xie and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lvyun Zhu, emh1bHZ5dW5AbnVkdC5lZHUuY24=; Sisi Xie, eGllc2lzaUBudWR0LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.