 Wei Gu
Wei Gu Guohuo Wu1,2†
Guohuo Wu1,2† Guijie Chen
Guijie Chen Zhongwen Xie
Zhongwen Xie- 1State Key Laboratory of Tea Plant Biology and Utilization, School of Tea and Food Sciences and Technology, Anhui Agricultural University, Hefei, Anhui, China
- 2Joint Research Center for Food Nutrition and Health of IHM, Anhui Agricultural University, Hefei, Anhui, China
- 3The First Affiliated Hospital of University of Science and Technology of China, Hefei, Anhui, China
Metabolic disorders include obesity, nonalcoholic fatty liver disease, insulin resistance and type 2 diabetes. It has become a major health issue around the world. Ubiquitin-proteasome system (UPS) is essential for nearly all cellular processes, functions as a primary pathway for intracellular protein degradation. Recent researches indicated that dysfunctions in the UPS may result in the accumulation of toxic proteins, lipotoxicity, oxidative stress, inflammation, and insulin resistance, all of which contribute to the development and progression of metabolic disorders. An increasing body of evidence indicates that specific dietary polyphenols ameliorate metabolic disorders by preventing lipid synthesis and transport, excessive inflammation, hyperglycemia and insulin resistance, and oxidative stress, through regulation of the UPS. This review summarized the latest research progress of natural polyphenols improving metabolic disorders by regulating lipid accumulation, inflammation, oxidative stress, and insulin resistance through the UPS. In addition, the possible mechanisms of UPS-mediated prevention of metabolic disorders are comprehensively proposed. We aim to provide new angle to the development and utilization of polyphenols in improving metabolic disorders.
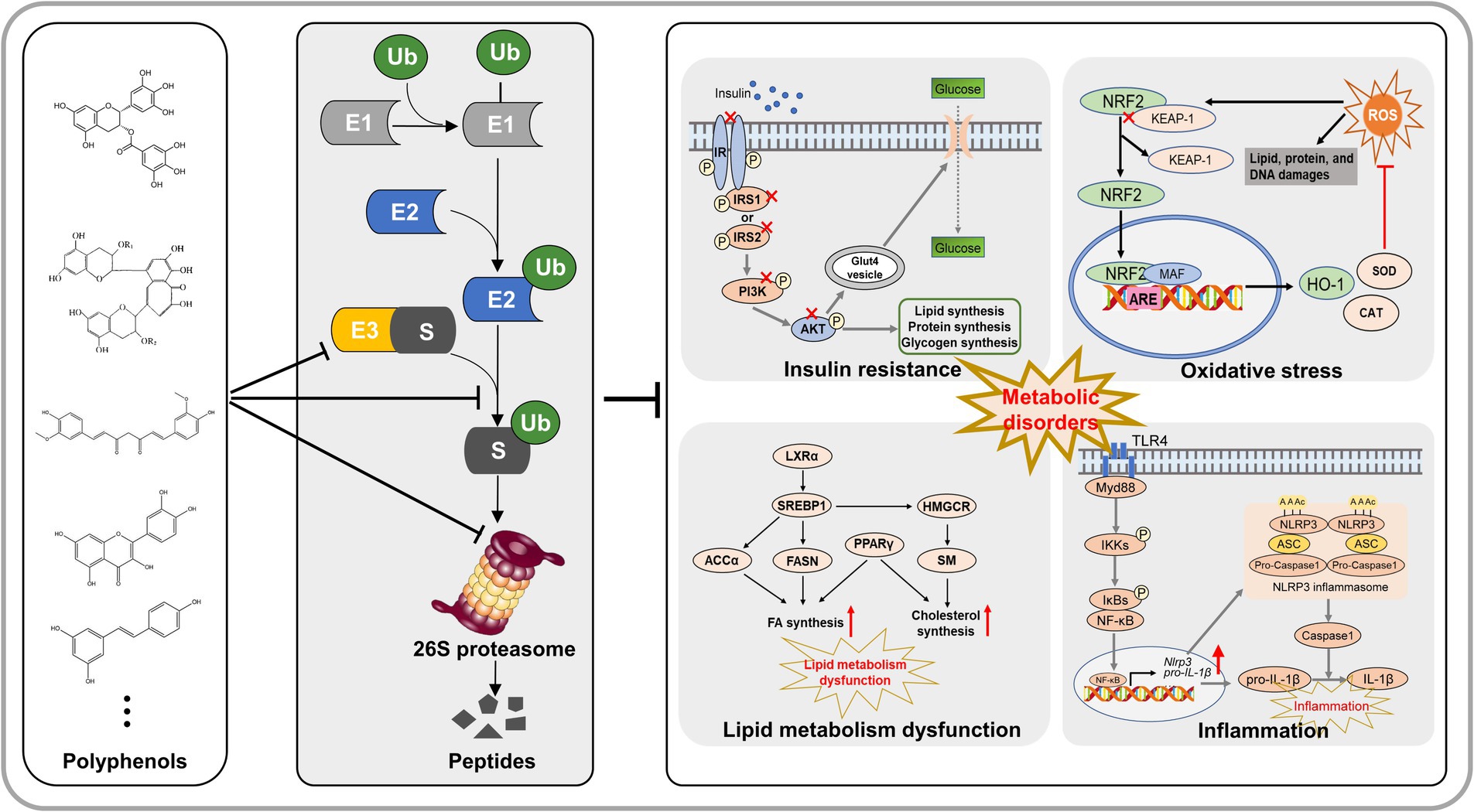
Graphical Abstract.
1 Introduction
Metabolic disorders are a set of syndrome, which include obesity, nonalcoholic fatty liver disease, insulin resistance, and type 2 diabetes. During recent years, metabolic disorders have been recognized as a heavy public health burden (1). Studies have indicated that the morbidity rates are associated with metabolic disorders in the general population, while reaching as high as 75% among individuals with obesity or type 2 diabetes mellitus (T2DM) (2, 3). Nonalcoholic fatty liver disease (NAFLD) is a typical syndrome of metabolic disorders. The constellation of NAFLD symptoms encompasses a spectrum of liver pathology, spanning from beginning hepatic steatosis to more advanced stages including steatohepatitis (NASH), fibrosis, cirrhosis, and ultimately hepatocellular carcinoma (HCC) development (4). The “Two-Hit Hypothesis” was initially proposed as a mechanistic framework to elucidate the initiation and progression of NAFLD (5, 6). Recent researches posits that various factors, including oxidative stress, insulin resistance, lipotoxicity, adipokine secretion by adipocytes, gut microbiota-derived endotoxins, and endoplasmic reticulum (ER) stress, collectively contribute to the progressive development of metabolic disorders (3, 7). Dyslipidemia, inflammation, oxidative stress, and insulin resistance are important factors that contribute to its development (3, 5). Numerous studies have demonstrated that dietary polyphenols can improve dyslipidemia and insulin resistance, possess anti-inflammatory and antioxidant properties, and show significant effectiveness in improving metabolic diseases (8, 9). However, the exact mechanism for the development of metabolic disorders remains incompletely to be understood. Therefore, summarizing the effects of polyphenols on improving metabolic diseases is great significance for exploring the mechanisms of metabolic disorders.
The ubiquitin-proteasome system (UPS) is a crucial cellular pathway accountable for the degradation of undesired or misfolded proteins, and it plays a pivotal function in governing a multitude of cellular processes, encompassing cell differentiation, proliferation, apoptosis, gene expression regulation, DNA repair, and stress response (10, 11). Abnormal UPS activity has been observed in animal models and human patients with metabolic disorders, suggesting its substantial contribution to the development of the diseases (12–14). Dysregulation of the UPS can lead to the buildup of misfolded or damaged proteins, triggering stress pathways, inflammation, and cell death, ultimately contributing to the initiation and progression of metabolic disorders. Therefore, targeting UPS may be a promising therapeutic approach for treating metabolic disorders. Wang et al. (15) demonstrated that Trim16 exerts a beneficial effect in alleviating nonalcoholic steatohepatitis by facilitating the degradation of phospho-TAK1. Yang et al. (16) revealed that RNF5, an E3 ubiquitin ligase, inhibits metabolic disorders progression by targeting HRD1 in the ubiquitin-proteasome pathway. Currently, bortezomib and carfilzomib, two UPS inhibitors, have been approved for clinical use of treating NAFLD (17, 18). Bortezomib has been shown to improve insulin resistance and reduce liver fat accumulation in animal models of NAFLD (19). However, the use of proteasome inhibitors is limited by their potential side effects and systemic toxicity. Therefore, screening nature products targeting UPS emerge as effective and safety therapies for metabolic disorders.
Recently, prevention of metabolic disorders by using dietary regimens and nutraceuticals has attracted much attention. This strategy is driven by the desire to find dietary compounds that can prevent the onset, progression, and severity of metabolic disorders. There is a growing body of evidence that supports the beneficial effects of natural phytochemicals, especially polyphenolic compounds, on suppressing metabolic disorders via protein clearance mechanisms (8, 9). Therefore, this review first summarizes how polyphenols ameliorate metabolic disorders by regulating the UPS, indicating that targeting UPS with polyphenols might have the potential to prevent the pathologies of metabolic disorders. In summarizing the mechanisms by which polyphenols targeting the UPS to ameliorate metabolic disorders, the four possible pathways are proposed: lipid metabolism, inflammation, insulin resistance, and oxidative stress. Hence, the relationship between UPS and these four metabolic pathways is comprehensively summarized. Therefore, this review aims to provide novel theoretical data for the prevention of metabolic disorders through polyphenol targeting UPS.
2 Polyphenols protect metabolic disorders by regulating UPS activity
Polyphenols are a large family of organic compounds found abundantly in natural products. It can be classified into flavonoids, phenolic acids, lignans, and stilbenes based on their phenol ring numbers and structural elements (20, 21). Various phenolic compounds, including EGCG, curcumin, quercetin, resveratrol, theaflavin, and rutin, have been identified for their proteasome-inhibitory activity. Currently, EGCG, quercetin, curcumin, and resveratrol have received the highest research attention, either as standalone agents or in conjunction with other conventional drugs. At present, a substantial body of experimental evidence indicates the remarkable efficacy of these secondary metabolites in combating the progression of NAFLD and metabolic syndrome (22, 23). In addition, the researchers suggested that various polyphenolic compounds targeting different components of UPS can alleviate metabolic disorder. The effect of phenolic compounds on the UPS may be through the following two ways. Firstly, it facilitates the degradation of related proteins indirectly by engaging the UPS. Secondly, it serves as a proteasome regulator, directly controlling the intracellular proteasome levels. In this review, various polyphenols and their influence on UPS activity are summarized and illustrated in Figure 1.
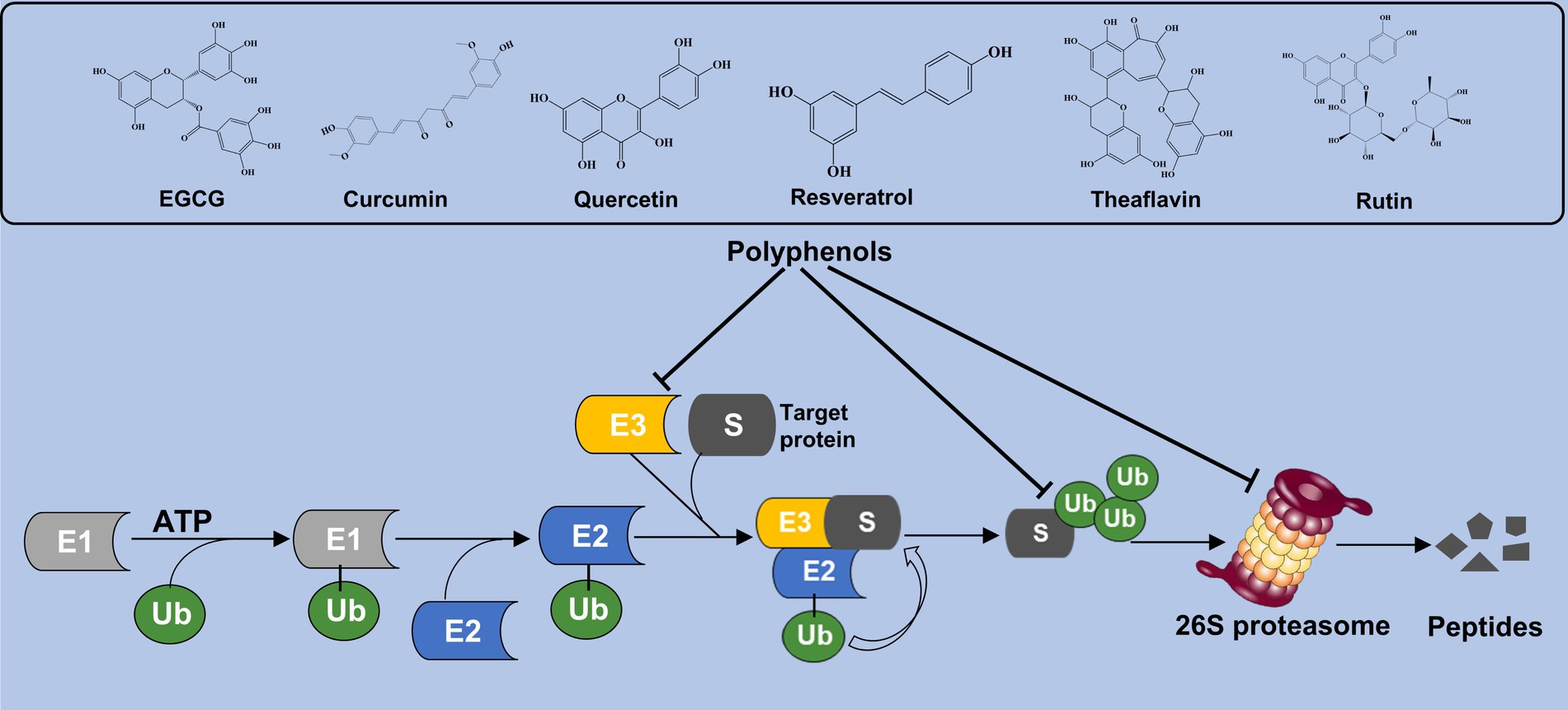
Figure 1. Polyphenols interact the UPS activity. Polyphenolic compounds such as EGCG, curcumin, quercetin, and resveratrol modulate UPS function by either promoting the degradation of proteins indirectly through UPS interactions or directly regulating intracellular proteasome levels.
2.1 EGCG modulates the UPS process to protect against metabolic disorders
Green tea contains high concentration of polyphenols, and epigallocatechin-3-gallate (EGCG) comprises 50–75% of the total catechin content of green tea (24). Studies have demonstrated that EGCG has potential in the treatment of metabolic disorders such as obesity, insulin resistance, and dyslipidemia, although the underlying mechanisms are not fully understood (25, 26). The recent researches have reported that EGCG can modulate E3 ligase and 26S proteasome to regulate lipid metabolism and exhibit antioxidative and anti-inflammatory effect.
2.1.1 EGCG modulates the UPS process to regulate lipid metabolism
Epigallocatechin-3-gallate has been shown to inhibit the ubiquitin/proteasome-mediated degradation of active SREBP-2, leading to the upregulation of LDLR, which potentially contributed to its fatty liver-improving, cholesterol-lowering, and heart disease-preventive effects (27). A research has reported that EGCG (50 μM) inhibits the secretion of apolipoprotein B-100 (apoB) in HepG2 cells through a proteasome-independent pathway, thereby reducing lipid synthesis (28). Kumazoe et al. demonstrated that EGCG3'”Me served as a natural agonist for the 67-kDa laminin receptor (67LR), effectively attenuated Toll-like receptor 4 (TLR4) expression through the upregulation of E3 ubiquitin-protein ring finger protein 216 (RNF216), thereby mitigating high insulinemia and hypertriglyceridemia induced by high-fat/high-sugar conditions. The study confirmed that EGCG3”Me acts as a natural agonist of 67LR, showing a dose-dependent effect, and this was validated in the HF/HS mouse model. Therefore, 67LR serves as an attractive target, providing strong support for developing EGCG3”Me into a therapeutic agent for hyperinsulinemia and hypertriglyceridemia. EGCG restores AKT activity in skeletal muscle through the myo-statin/miRNAs/ubiquitin-proteasome (miRNA-486-5p) signaling pathway, alleviates age-related muscle mass loss and insulin resistance (29). DNA damage-inducible alpha (GADD45A) is a key regulatory factor in intramuscular fat (IMAT) infiltration, induces ubiquitination and degradation of ATP synthase F1 subunit alpha (ATP5A1) through recruiting the E3 ubiquitin ligase TRIM25, inhibits the cAMP/PKA/LKB1 signaling pathway, upregulates lipogenic gene expression, and promotes adipogenesis in intramuscular preadipocytes. And EGCG effectively reduces lipogenic gene expression by inhibiting GADD45A (30). Hence, EGCG holds promising potential for ameliorating metabolic disorders by addressing lipid metabolism imbalance and insulin resistance through modulation of the UPS system (Figure 2). However, the regulation of lipid disorders and insulin resistance by EGCG through the UPS lacks further in-depth research, limits its development as a drug.
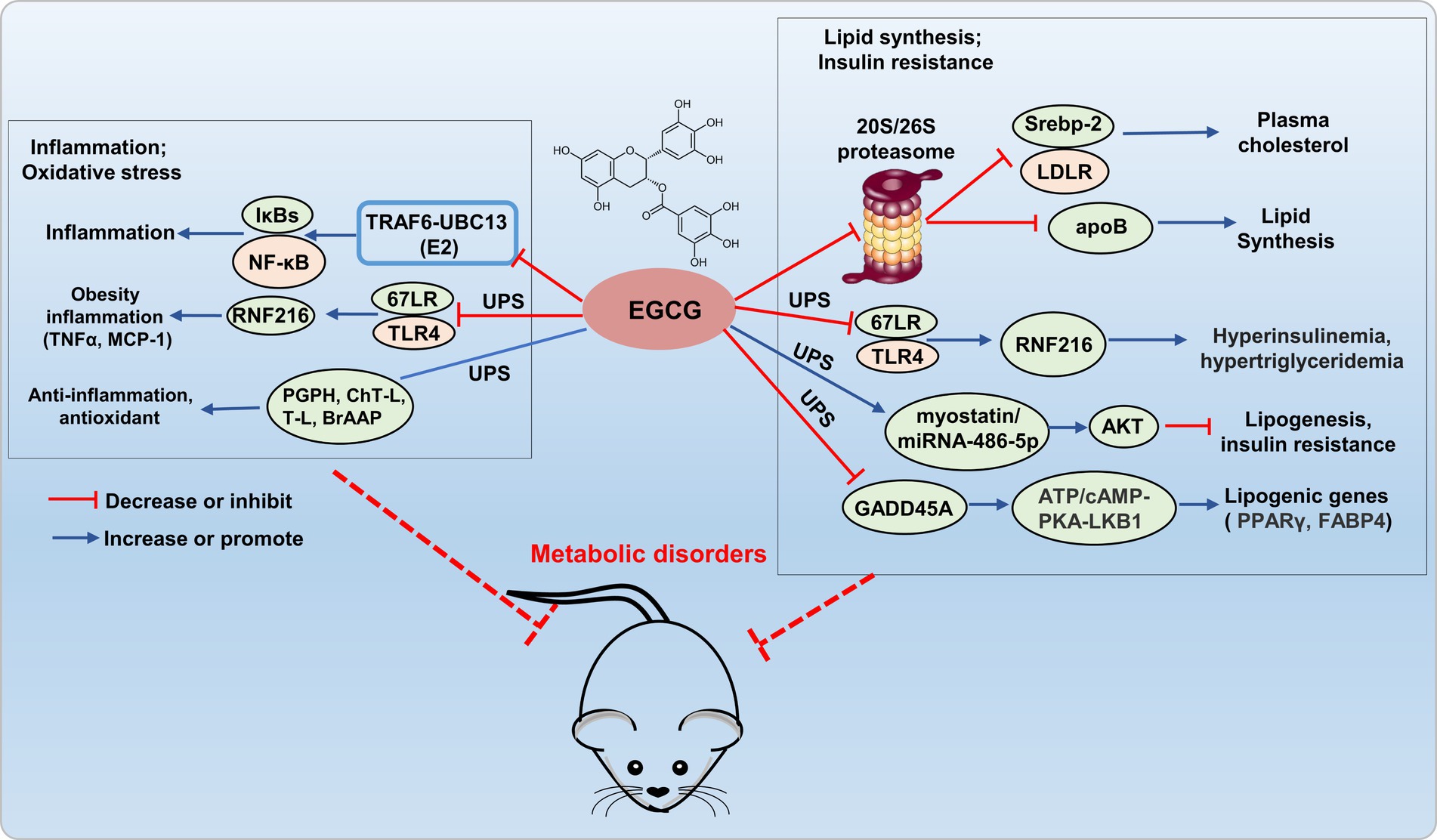
Figure 2. Potential mechanism of EGCG targets UPS to prevent metabolic disorder. EGCG exerts anti-inflammatory and antioxidant effects by regulating E2 (TRAF6-UBC13), E3 (67LR/TLR4), and the 20S proteasome (PGPH, ChT-L, T-L, and BrAAP) within the UPS. It inhibits 20S/26S proteasome, E3 (67LR/TLR4), and GADD45A (interacting with E3 ligase TRIM25), reducing adipogenic gene expression, thus improving lipid metabolism disorders and insulin resistance.
2.1.2 EGCG modulates the UPS process to exert antioxidative and anti-inflammatory effect
Epigallocatechin-3-gallate can also directly interact with E3-TRAF6 ubiquitin ligases, which leading to the attenuation of TRAF6-UBC13 (E2) interaction and inhibition of p65 and p50 translocation, consequently deactivating the NF-κB pathway (31). Kumazoe et al. re-ported that 3″ methlated EGCG inhibited the expression of TLR4 by upregulating RNF216, and further inhibited the expression of tumor necrosis factor α (TNFα) in adipose tissue, and increased serum monocyte chemotactic protein-1 (MCP-1), thus alleviating adipocyte inflammation (32). EGCG exhibited diverse biological activities, including anti-inflammatory, antioxidant, antiviral, and antibacterial effects, achieved through interactions with the PGPH, ChT-L, T-L, and BrAAP subunits of constitutive and immuno-proteasomes, modulated the functionality of the 20S proteasome, thereby conferred antioxidant, anti-inflammatory, and related functionalities (33). Therefore, EGCG exerted antioxidative and anti-inflammatory effects by modulating the UPS system, which demonstrating promising potential as a novel therapeutic agent for the treatment of metabolic disorders (Figure 2).
2.2 Curcumin modulates UPS process to alleviate metabolic disorders
Curcumin, a diaryl-heptanoid and a member of curcuminoids, naturally presents in turmeric. Due to its plentiful antioxidant and anti-inflammatory properties, there is considerable interest in exploring the therapeutic potential of curcumin for various metabolic disorders (34). Previous studies have demonstrated that curcumin can improve lipid metabolism abnormalities and insulin resistance, and exhibit significant anti-inflammatory effects by regulating E3 ligase and the 26S proteasome.
2.2.1 Curcumin modulates the UPS process to improve lipid metabolism and insulin resistance
Recent research indicates that curcumin prevents obesity complication by inhibiting TRAF4-induced ubiquitylation. TRAF4, functioning as an E3 RING ubiquitin ligase, facilitates the degradation of the adipocyte differentiation regulator PPARγ through the ubiquitin-proteasome pathway, thereby impeding adipogenesis (35). Curcumin inhibited the UPS function in 3 T3-L1 adipocytes, activated autophagy, reduced protein kinase B (Akt) protein levels, and concurrently inhibited insulin-stimulated Akt phosphorylation and membrane translocation of glucose transporter type 4 (GLUT4), which leading to the amelioration of insulin resistance (36). In the TGF-β (2 ng/mL) induced BNL CL.2 model, curcumin (20 μM) mitigated hepatic oxidative stress through autophagy activation by inhibiting TGF-β/Smad (Smad2 and Smad3) signaling transmission, inducing ubiquitination and proteasomal degradation of Smad ubiquitin regulatory factor 2 (SMURF2), thereby alleviated hepatic fibrosis by increasing hepatic cell autophagy (37). This discovery confirms that curcumin can mediate hepatocyte autophagy through the UPS pathway to alleviate liver fibrosis, as demonstrated in both animal and cell studies. This provides theoretical support for the development and use of curcumin in liver-protective drugs. In addition, curcumin inhibited scavenger receptor class A (SR-A)-mediated oxidized low-density lipoprotein (oxLDL) uptake through the ubiquitin-proteasome-calpain pathway, and promoted ATP-binding cassette transporter (ABC) A1-dependent cholesterol efflux, thereby retarding cholesterol accumulation in apoE−/− mice (38). Therefore, curcumin shows greatly potential in improving metabolic disorders by addressing the imbalance of lipid metabolism and insulin resistance through the modulation of the UPS (Figure 3).
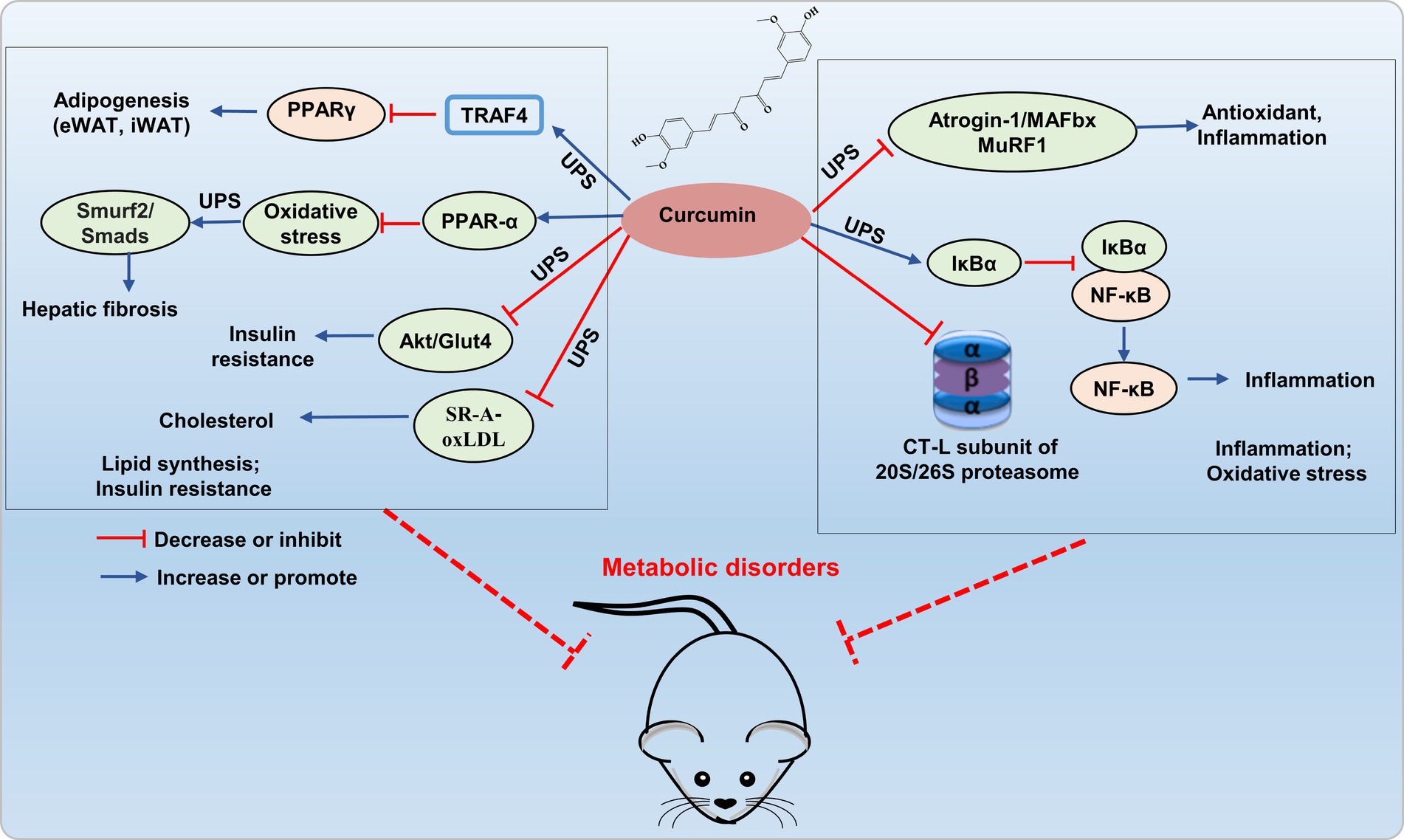
Figure 3. Potential mechanism of curcumin interferes UPS to alleviating metabolic disorder. Curcumin improves lipid accumulation by inhibiting E3 ubiquitin ligases (TRAF4, Smurf2/Smad) and reducing Akt/GLUT4 and SR-A/oxLDL expression via UPS inhibition, thereby enhancing insulin resistance and reducing cholesterol accumulation. It also hinders 26S proteasomal degradation of IκBα, thereby inhibiting NF-κB activation, and inhibits E3 ligases atrogin-1/MAFbx and MuRF1 to decrease inflammatory factor expression.
2.2.2 Curcumin regulates the UPS process to exert anti-inflammatory effects
Curcumin exerts inhibitory effects on the activation of the proinflammatory transcription factor NF-κB by hindering the 26S proteasomal degradation of IκBα. Curcumin improved skeletal muscle atrophy in type 1 diabetic mice by inhibiting the muscle-specific ubiquitin E3 ligases atrogin-1/MAFbx and MuRF1, oxidative stress, and inflammatory (NF-κB, TNF-α, and IL-1β) (39). In addition, curcumin is suggested to regulate UPS by inhibiting the proteasome protease activity (40, 41). Curcumin’s carbonyl carbons directly interact with the hydroxyl group of the amino-terminal threonine residue within the proteasomal CT-L subunit found both in 20S proteasome and cellular 26S proteasome (42). Interestingly, curcumin exhibits a dose-dependent effect on the 26S proteasome. In HeLa cells, low doses (1 μM) enhance its activity, while high doses (10 μM) inhibited proteasome activity (43). Research also suggested that curcumin is a powerful inhibitor for COP9 and its associated kinases, such as protein kinase D and casein kinase 2, all of which are closely related to the UPS (44). Additionally, curcumin directly hinder ubiquitin isopeptidases, which are part of the DUB family that responsible for recycling ubiquitin and making it available again for the 26S proteasome system. The regulation of the UPS pathway by curcumin is associated with transcription factors (NF-κB, AP-1, STAT3, HIF-1α, and GADD153/CHOP), which play crucial roles in oxidative stress and inflammation (37, 45). Therefore, curcumin, as an effective inhibitor of proteasome, plays an important role in regulating inflammation and oxidative stress, and has potential value in the prevention and treatment of metabolic disorders (Figure 3). However, most researches on curcumin improving metabolic imbalance through the UPS pathway are based on cell experiments, with fewer studies on animal models.
2.3 Quercetin modulates UPS process to alleviate metabolic disorders
Quercetin is a polyphenol distinguished by 3-hydroxyl flavone structure. It exists in several foods, including tea, nuts, apples, berries, onions, shallots, and grapes (46). Extensive researches have consistently demonstrated that quercetin has potent anti-inflammatory and antioxidative properties, and inhibits hepatocyte apoptosis and NAFLD development (22, 47). Current literature has demonstrated that quercetin can improve lipid metabolism and show promising anti-inflammatory effects by modulating the proteasome.
2.3.1 Quercetin modulates the UPS process to improve lipid metabolism
The increased NF-κB activation induced by the ubiquitin-proteasome system may be implicated in the pathogenesis of proteinuria in diabetic nephropathy. Quercetin reduces NF-κB p65 ubiquitination, and significantly lowers fasting insulin, fasting blood glucose, TG, TC, and LDL-C levels, exerts a protective effect against various complications in diabetic rats (48). Liu’s research revealed that quercetin alleviates NAFLD induced by high-fat diet feeding in C57BL/6 J mice by enhancing frataxin-mediated PINK1/Parkin-dependent mitochondrial autophagy (49).
2.3.2 Quercetin regulates the UPS process to exert anti-inflammatory effects
Quercetin also inhibits the LPS-induced enhancement of TNFα and the production of the C-C chemokine superfamily member RANTES in Raw264.7 cells, mediated by ubiquitin-like protein MNSFβ and HSC70 siRNA (50). Furthermore, quercetin inhibited the activity of all three catalytic subunits of the proteasome, namely CT-L, T-L, and C-L/PGPH, with the CT-L subunit displaying the most potent inhibitory effect (51). Additionally, Sagrario et al. (52) reported that quercetin enhanced proteasome activities and promoted Nrf2 transcription and expression. Quercetin, aside from its modulation of proteasomes to suppress lipid synthesis and anti-inflammatory responses, may constitute an alternative prospective strategy for treating metabolic dis-orders through targeting UPS (Figure 4). Currently, there is limited research on quercetin improving metabolic imbalance through the UPS, and more theoretical studies are needed to enrich the foundational knowledge of quercetin to prevent and treat metabolic disorders.
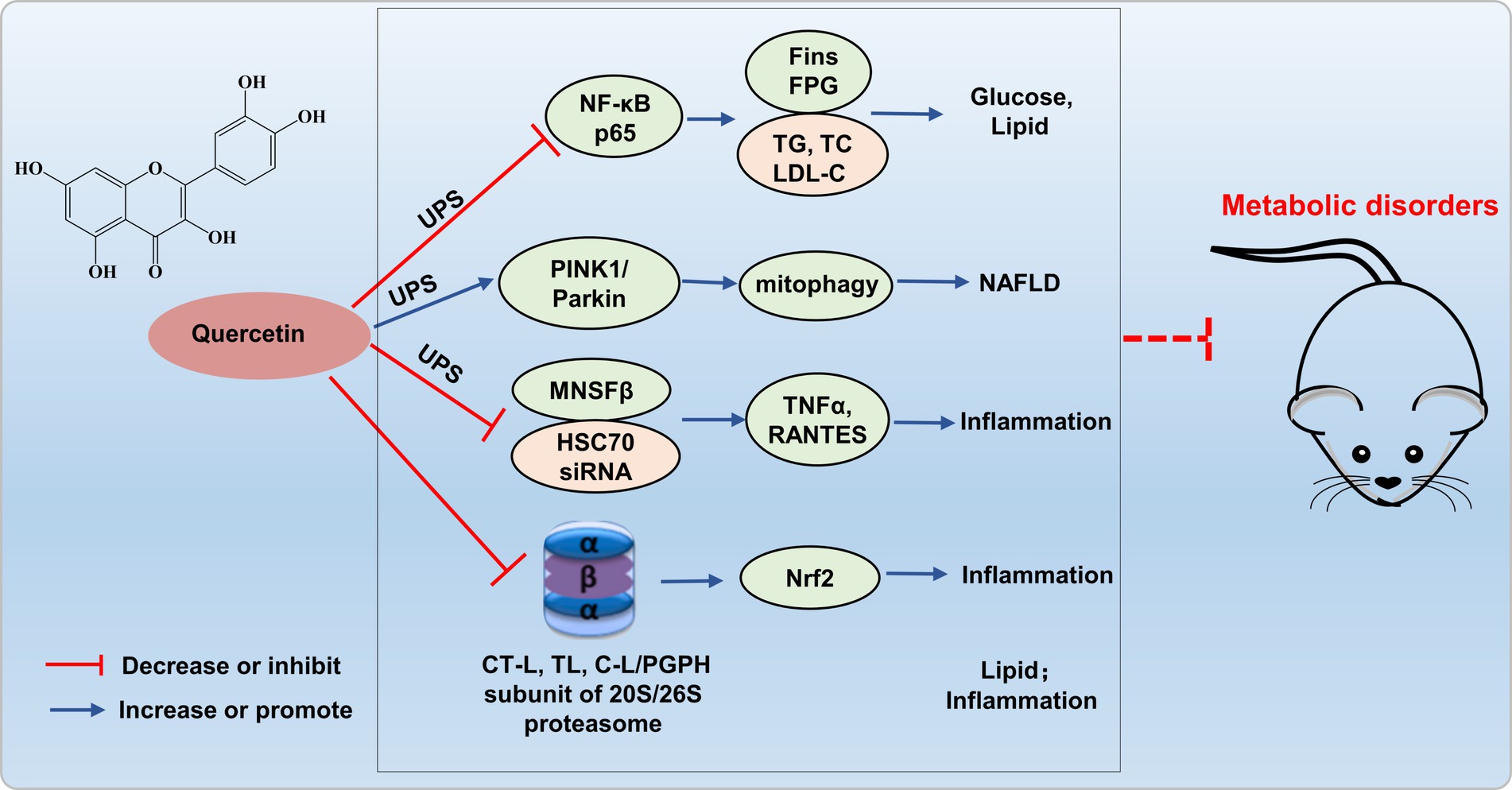
Figure 4. Possible mechanism of quercetin targets UPS to alleviate metabolic disorders. Quercetin reduces NF-κB p65 ubiquitination, enhances frataxin-mediated PINK1/Parkin-dependent mitochondrial autophagy, and inhibits ubiquitin-like protein MNSFβ and HSC70 siRNA, thereby inhibiting inflammation and improving glucose and lipid metabolism disorders.
2.4 Resveratrol modulates UPS process to prevent metabolic disorders
Resveratrol, a polyphenol found in grapes and berries, has various biological and pharmacological properties (53). Currently, some studies have reported that resveratrol can improve lipid metabolism imbalances, demonstrates strong anti-inflammatory and antioxidative properties by regulating the UPS, providing protective effects against metabolic disorders.
2.4.1 Resveratrol modulates the UPS process to improve lipid metabolism
Resveratrol treated of hepatic cells efficiently activated SIRT1, thereby reducing PPARα protein levels through deacetylation, ubiquitination, and degradation, ultimately improved liver and metabolic dysfunction caused by nutritional deficiencies (54). Resveratrol (100 μM) also exerted a critical role in hepatic metabolic regulation by modulating the triiodothyronine (T3) response through dual target genes TRβ3/SIRT1 in HepG2 cells, facilitating the SIRT1-TRβ1 interaction, promoting TRβ1 deacetylation in the presence of T3, and enhancing ubiquitin-dependent turnover of TRβ1 (55). Floyd’s research revealed that resveratrol modulates PPARγ protein levels in 3 T3-L1 adipocytes by inhibiting PPARγ gene expression and increasing ubiquitin-proteasome-dependent degradation of PPARγ protein, thereby inhibiting lipid synthesis and regulating insulin sensitivity (56). These results suggest that resveratrol may exert a protective effect on the metabolic disorders by modulating lipid metabolism through the UPS (Figure 5).
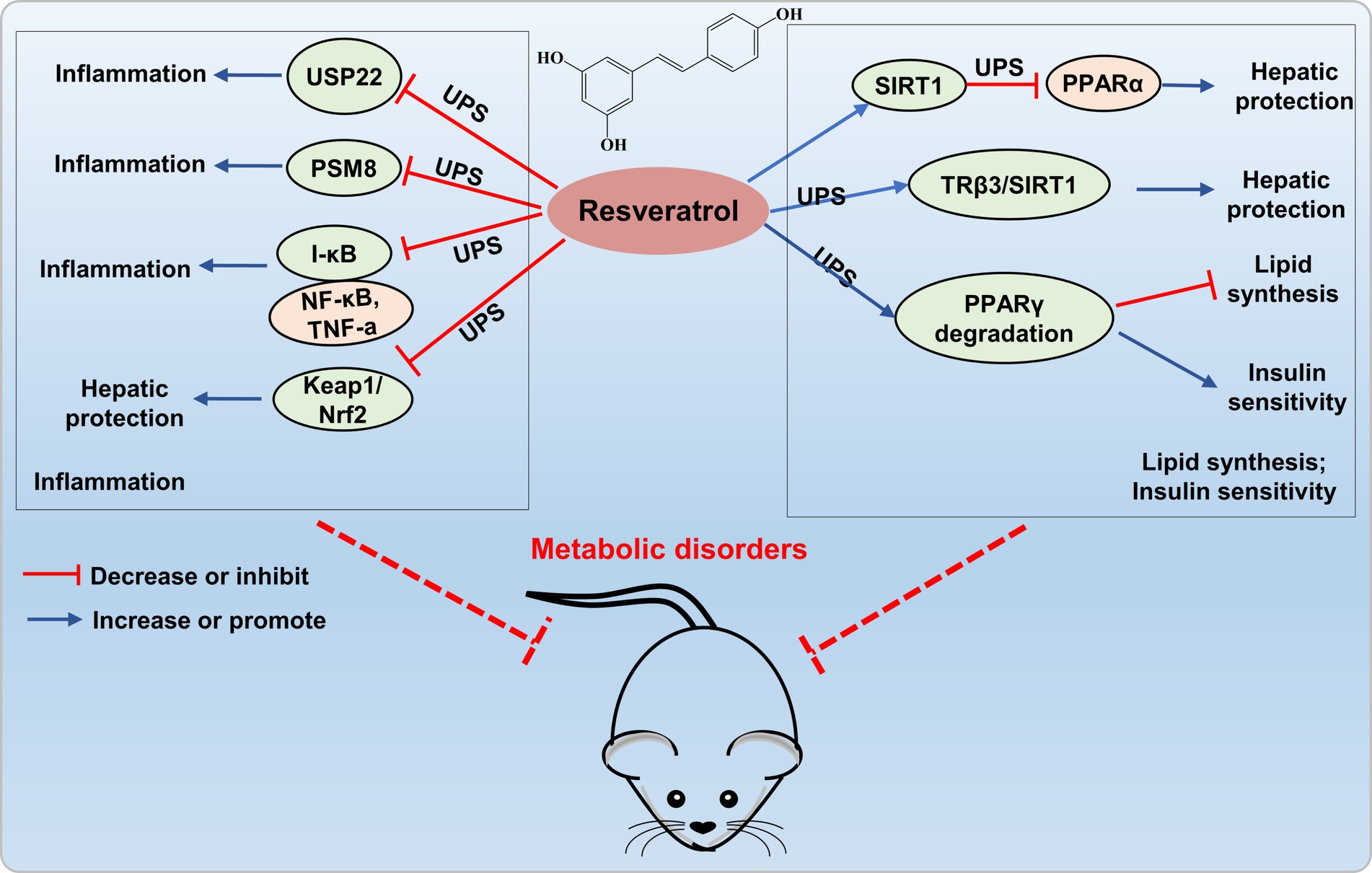
Figure 5. Resveratrol targets UPS to prevent metabolic disorders. Resveratrol inhibits the expression of ubiquitin-specific protease 22 (USP22), proteasome subunit PSMB8, and the Nrf2-Keap1 signaling pathway, thereby suppressing the occurrence of inflammation. Resveratrol activates SIRT1 to decrease PPARα protein levels through ubiquitination, enhances SIRT1 interaction with TRβ1 to facilitate ubiquitin-dependent degradation of TRβ1, and increases ubiquitin-proteasome-dependent degradation of PPARγ protein levels, thereby providing liver protection and improving metabolic disorders.
Researches have shown that resveratrol is a potent proteasome inhibitor and can also function by regulating the UPS (57, 58). Seungkyu et al. (59) reported that resveratrol dose-dependently inhibits HIF-1α and VEGF through the blockade of the PI3K/Akt/mTOR signaling pathway while facilitating proteasomal degradation of HIF-1α. Moreover, He’s research demonstrated a novel synergistic interaction of curcumin and resveratrol (150 mg/kg, 8:2, w/w) when used together to mitigate metabolic-associated fatty liver disease in rats, and the effect is partially attributed to the modulation of the HIF-1 and PI3K/AKT/mTOR signaling pathways (60). Hosseini et al. discovered that resveratrol can mitigate NAFLD by inducing epigenetic modifications of the Nrf2-Keap1 signaling pathway, significantly reducing high glucose-induced triglyceride accumulation in HepG2 cells (61). According to these findings, it is plausible to hypothesize that resveratrol-regulated UPS could be a potential mechanism to improve NAFLD and alleviating metabolic disorders (Figure 5). However, the exact mechanism of resveratrol regulating UPS is still unknown. Further research to unveil how resveratrol regulate UPS to prevent metabolic disorders is needed.
2.4.2 Resveratrol regulates the UPS process to show anti-inflammatory and antioxidative effects
Moreover, resveratrol can also elicit anti-inflammatory and antioxidant effects through the UPS. Shi’s study revealed that resveratrol not only inhibits the overexpression of ubiquitin-specific protease 22 (USP22) and cytotoxicity induced by high D-glucose, but also suppresses the secretion of IL-1β, TNF-α, IL-6, and TGF-β1, providing protection against cell apoptosis and inflammation induced by high glucose (62). Moreover, resveratrol can downregulate the expression of the proteasome subunit PSMB8, thereby inhibiting inflammatory responses (58). The findings indicated that resveratrol exerts anti-inflammatory and antioxidant effects by modulating the UPS, demonstrating its potential value in protection of metabolic disorders (Figure 5).
2.5 Other polyphenols modulates UPS process to protect metabolic disorders
Theaflavins are a group of phenolic compounds commonly found in black tea. Studies have demonstrated that theaflavins possess a wide range of health-promoting effects, including antidiabetic, antioxidant, anticancer, and anti-inflammatory activities (63, 64). Theaflavins have been shown to have regulatory effect on UPS. Mizuno et al. (65) reported that TFDG promotes the downregulation of epidermal growth factor receptor (EGFR) through increasing EGFR ubiquitination. And theaflavins may suppress the fatty acid synthase gene through molecular mechanisms that could subsequently lead to the reduction of activity in the EGFR/PI3K/Akt/Sp-1 signal transduction pathways (66). Rutin is a natural phenolic compound present in various plants, including tea, buckwheat, and apples (67). Rutin has been proven to be an important UPS regulator. Research reported that rutin activate the UPS by positively influencing the Nrf2 transcription factor regulators, leading to increased translocation of Nrf2 to the nucleus (52, 68). Rutin additionally hinders carfilzomib-induced oxidative stress and inflammation by acting through the NOS-mediated NF-κB signaling pathway (69). Rutin displayed a multifaceted impact, encompassing the prevention or reversal of metabolic changes such as improved glucose tolerance (70) and reduced abdominal fat pads (71), countering alterations in cardiovascular and hepatic structure (72), as well as mitigating inflammation and oxidative stress in the heart and liver (73–75), while also normalizing liver markers (76). Therefore, utilizing rutin to target UPS could serve as an effective strategy for NAFLD and metabolic disorders treatment.
3 UPS modulates critical biological processes affecting metabolic disorders
Polyphenols ameliorate metabolic disorders through the UPS by regulating lipid metabolism, inflammation, oxidative stress, and insulin resistance during disease development. The UPS is responsible for protein degradation and homeostasis, and plays a significant role in the development of metabolic disorders. Consequently, we further review the latest findings by which the UPS affects metabolic disorders by modulating these key pathways, aiming to provide new mechanisms for understanding how polyphenols ameliorating metabolic disorders.
3.1 UPS and lipid metabolism
Disruptions of hepatic lipid metabolism are linked to metabolic disorders development (77). Previous researches indicated that UPS crucially regulates lipid metabolism, including cholesterol synthesis, uptake, and efflux (78, 79). The intricate cholesterol synthesis pathway involves over 20 enzymes, with emphasis on pivotal flux-controlling enzymes, such as squalene monooxygenase (SQLE) and 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR). Researchers have showed that both enzymes undergo ubiquitination and subsequent degradation (80, 81). HMGCR is a key substrate of UPS targeted by statins. And statins can degrade HMGCR and inhibit NASH through ubiquitin-proteasome pathway (82). The E3 ligase in sterol-dependent degradation is gp78 (79, 83), located in the ER membrane. Gp78 first interacts and then promotes the degradation of ER resident proteins, Insig-1 and Insig-2 (84, 85), which is a crucial event for sterol-dependent HMGCR degradation and pre-venting proteolytic activation of sterol regulatory element binding proteins (SREBP) (85). A previous study revealed that Gp78−/− mice exhibit typical characteristics of NAFLD, including lipid degeneration, hepatic inflammation, and fibrosis (86). Moreover, the E3 ligase HRD1 is implicated in the constitutive degradation of HMGCR (87, 88). SQLE converts squalene to squalene-2,3-epoxide, which is a crucial step in cholesterol biosynthesis. Recently, post-transcriptional SQLE regulation gained attention. Researchers found that the E3 ligase that mediating SM ubiquitylation was identified as MARCH6 (89, 90). MARCH6, an ER membrane protein, is part of the ERAD system (90), and loss of MARCH6 activity increases SQLE protein abundance in mammalian cells (89). Moreover, the pathological consequences of P450-ERAD disruption serve as a significant triggering factor for NAFLD (91).
The low density lipoprotein receptor (LDLR) pathway mediates cholesterol imports in mammalian cells (92). E3 ubiquitin ligase-inducible degrader of LDLR (Idol) induce endocytosis and lysosomal degradation of LDLR by polyubiquitinating (93). In mice lacking LDLR, high-fat diet induces degradation of hepatic mitochondrial proteins, leading to oxidative stress, lipid degeneration, and the development of metabolic disorders (94).
Abnormal cholesterol metabolism directly leads to metabolic disorders (95). Cellular cholesterol levels are finely tuned by the interplay of biosynthesis, uptake, and efflux, and are primarily regulated by two transcription factor families, which are liver X receptors α and β (LXRs) (96, 97) and SREBPs (97, 98). LXRs respond to elevated cholesterol levels by activating ABCA1, ABCG1, and the E3 ligase IDOL, subsequently reduce cholesterol burden. LXRs activation by endogenous ligands in response to increased cellular cholesterol induces IDOL, ABCA1, and ABCG1 expression, and reduce cholesterol burden (96, 99). Additionally, ubiquitin-dependent mechanisms control SREBP2 protein stability, processing, and transcriptional activity. In the ER, E3 ligases TRC8, MARCH6, and RNF145 negatively regulate SCAP and SREBP2 (79, 100–102). Furthermore, the mature nuclear SREBP2-N transcription factor, akin to its precursor, is unstable. Phosphorylated SREBP2-N undergoes ubiquitination by the Fbw7-Cullin-1 E3 ligase, leading to proteasomal degradation (103). A previous research indicated that loss of HSP90β promotes the degradation of mSREBPs via Akt-GSK3β-FBW7 pathway, which markedly reduced neutral lipid and cholesterol levels and improved NAFLD and metabolic disorders (104). In summary, ubiquitylation and degradation are crucial processes for maintaining cholesterol homeostasis, impact lipid synthesis (HMGCR, SQLE), uptake (LDLR), efflux (ABCA1, ABCG1), and transcriptional programming (SREBP, LXR).
The UPS also plays a crucial role in regulating fatty acid metabolism. Peroxisome proliferator-activated receptors (PPARγ) are vital transcription factors for regulating lipid and lipid-protein metabolism, undergo ubiquitin-mediated proteasomal degradation facilitated by CHIP (105). Additionally, the E3 ubiquitin ligase NEDD4 stabilizes PPARγ by inhibiting proteasomal degradation (106, 107). USP14, a key proteasome-associated DUB, is crucial for proteome lipid homeostasis (108, 109). Liu et al. (109) highlighted its essential role in hepatosteatosis by stabilizing fatty acid synthase (FASN). Additionally, Hu et al. showed that the E3 ligase TRIM28 mediates FASN ubiquitination and subsequent proteasomal degradation, thus disrupts lipid accumulation in hepatic cells and suppresses NAFLD (110).
Therefore, exploring the relationship between the UPS and lipid metabolism has the potential to unveil novel therapeutic approaches for preventing metabolic disorders through dietary polyphenols (Figure 6).
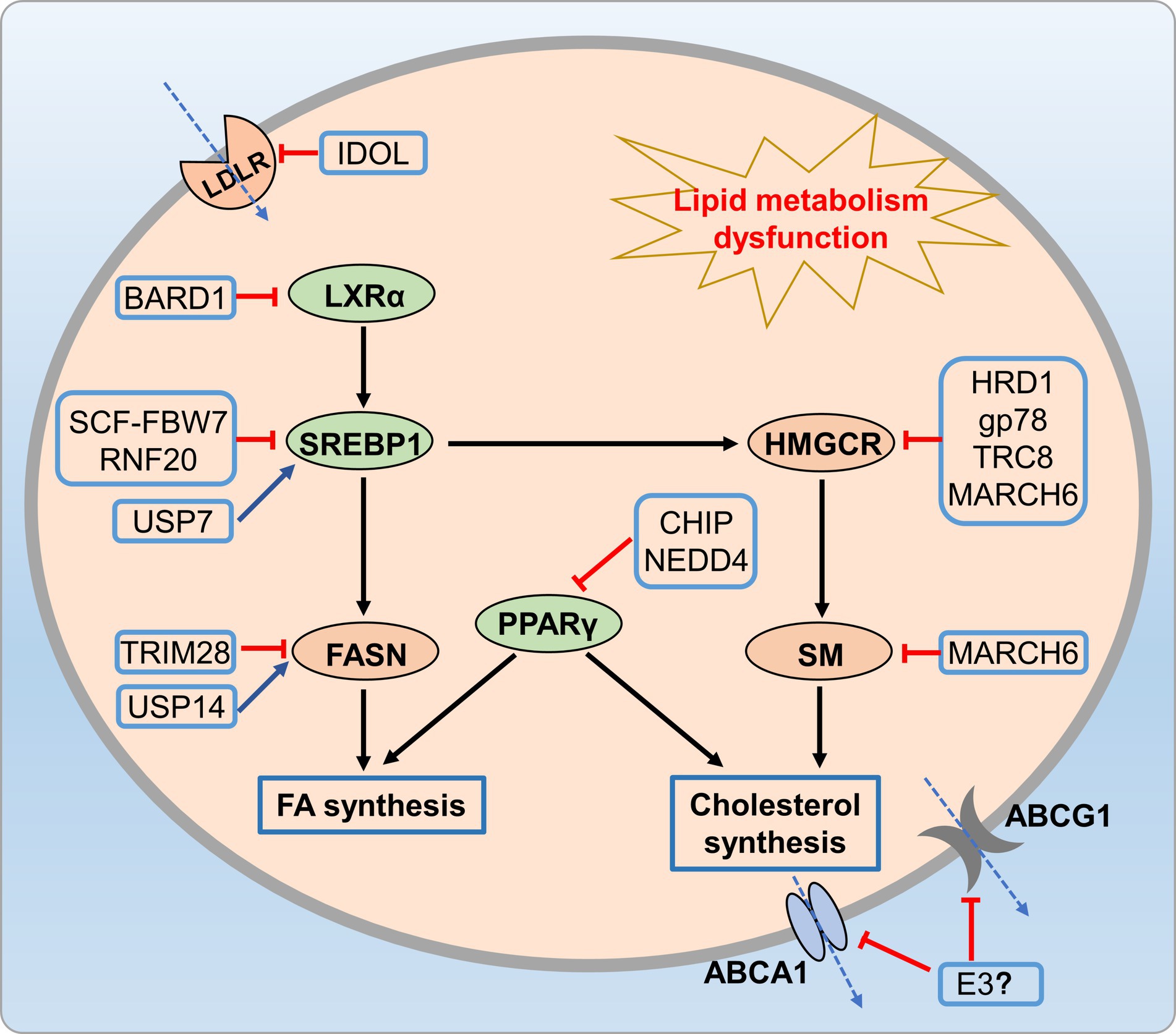
Figure 6. UPS-mediated lipid metabolic disorders. Many E3 ligases (HRD1, Gp78, and MARCH6) reduce cholesterol synthesis by promoting HMGCR ubiquitination and degradation, while CHIP and NEDD4 regulate cholesterol and fatty acid synthesis through ubiquitination of PPARγ. Additionally, E3 ligases like FBW7-Cullin-1, USP14, and TRIM28 reduce fatty acid synthesis by regulating SREBP-1 and FASN.
3.2 UPS and inflammation
Inflammation is a distinct characteristic of metabolic disorders. The disruption of UPS is associated with inflammation by degrading transcription factors in the inflammatory process (111, 112). Numerous studies emphasize the role of various E3 ubiquitin ligases in regulating inflammation. Cbl-b, Pellino3, and ITCH of E3 Ligases are closely associated with inflammation in metabolic disorders. Cbl-b, a RING E3 ligase, is implicated in protein tyrosine kinase activation, potentially influencing macrophage infiltration and activation (113). Cbl-b over-expression inhibited saturated fatty acid-induced toll-like receptor 4 (TLR4) signaling, as it mediates TLR4 ubiquitination and degradation in macrophages, leading to modulated TLR4 protein levels on the cell surface, thereby improving insulin resistance in the liver (113, 114). The pellino family regulated the TLR/IL-1R signaling pathway, subsequently impacted inflammatory cascades and immune response (115). Pellino3 deficiency in mice exacerbated inflammation, elevated IL-1β expression, and intensified insulin resistance induced by a high-fat diet (116). ITCH, a highly expressed HECT-type E3 ubiquitin ligase, increases anti-inflammatory M2 macrophages, subsequently prevents steatohepatitis when inactive in cells (117). Furthermore, E3 ligase CHIP regulates TLR2/4/7/9 signaling and inhibits NF-κB-mediated inflammation by promoting karyopherin α2 (KPNA2) degradation (118, 119). Conversely, ubiquitin ligases midline1 and siah2 enhance pro-inflammatory cytokine expression, positively regulating NF-κB and MAPK pathways (120, 121).
The NLPR3 inflammasome activation has been implicated in NAFLD progression, especially from hepatic steatosis to NASH (122). Thus, precise regulation of inflammasome is crucial to prevent NAFLD progression. The UPS has emerged as a key regulator of inflammasome activation, particularly in the modulating NLRP3 inflammasome complex (NLRP3, ASC, and procaspase-1). Upon activation, procaspase-1 cleaves and releases active caspase-1 that activates IL-1β and IL-18, aggravating hepatic inflammation and contributing to the innate immune response (123). E3 ubiquitin ligases also regulate NLRP3 inflammasome activation by targeting NLRP3, caspase-1, and ASC. TRIM31 binds to NLRP3, induces K48 poly-Ub and promotes NLRP3 proteasomal degradation (124). SCF-FBXL2 ubiquitinates NLRP3 in the resting state, which leading to proteasomal degradation (125, 126). Parkin, an E3 ligase, regulates NLRP3 through deubiquitinase A20, negatively controls priming and activation (127). The newly identified E3 ligase cullin interacts and ubiquitinates NLRP3 during priming, inhibits its activation (128). E3 ligases can also act as positive regulators, as shown by Juliana et al. (129), who demonstrated non-transcriptional priming for NLRP3 activation. Various E3 ligases, including pellino2, TRAF6, and TRIM33, are implicated in non-transcriptional priming of NLRP3. The NLRP3 inflammasome complex is regulated through ASC ubiquitination. NLRP3 and ASC undergo negative regulation through K63 polyubiquitination, resulting in degradation via autophagy, which is a mechanism that controls NLRP3 inflammasomes (130). Additionally, E3 ligases play a role in caspase-1 activation, as shown by Labbe et al. (131), who demonstrated that IAPs induce K63 polyubiquitination of caspase-1, a crucial step for inflammasome activation (Figure 7).
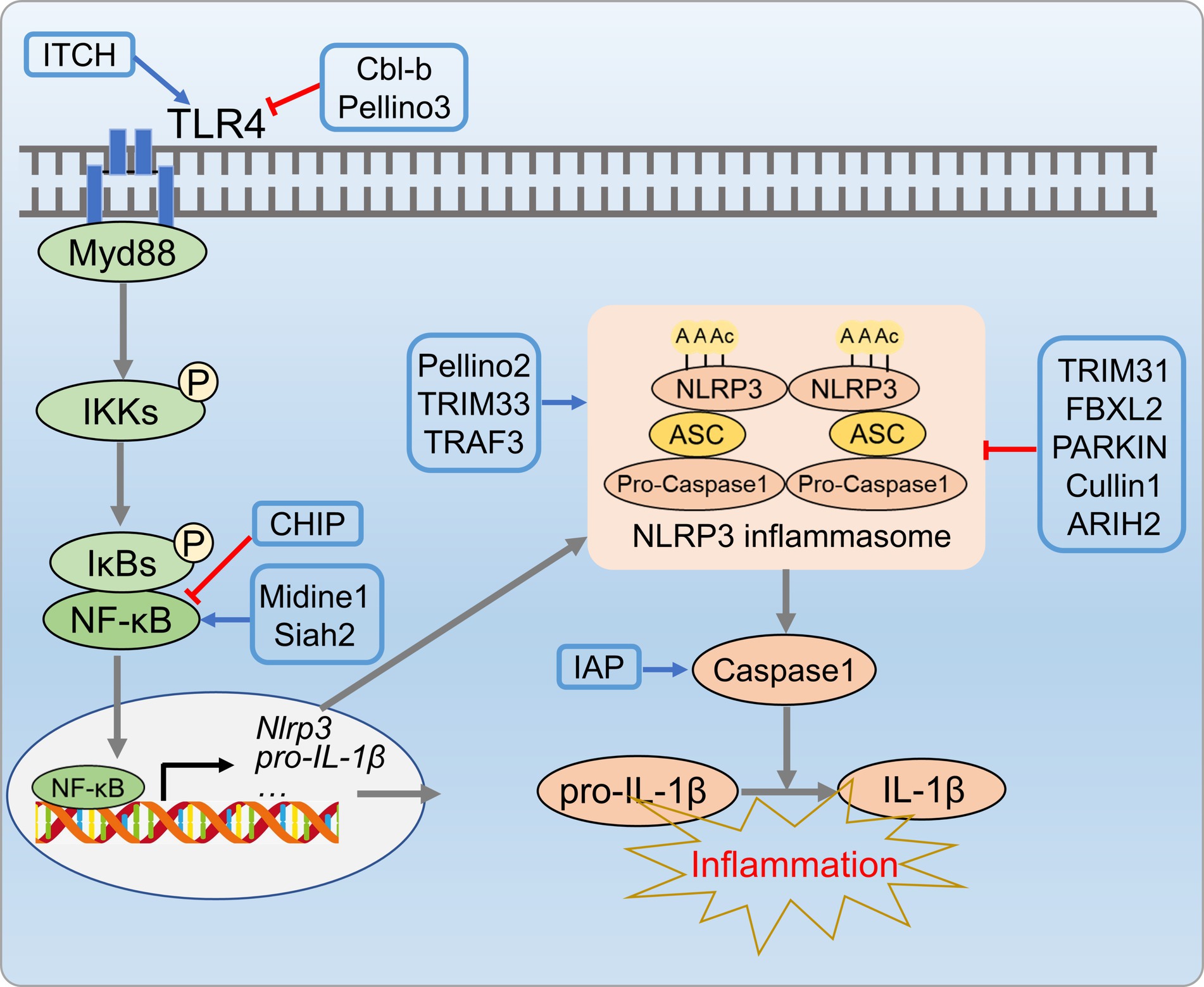
Figure 7. UPS-mediated inflammation processes in metabolic disorders. E3 ligases CL-b, Pellino3, and ITCH E3 inhibit inflammatory cascade reactions by regulating ubiquitination and degradation of TLR4. Additionally, ubiquitin ligases (TRIM31, Parkin, SCF-FBXL2, Cullin, etc.) suppress NLRP3 inflammasome activity by targeting NLRP3, caspase-1, and ASC.
Therefore, targeting the UPS to combat inflammation related metabolic disorders may provide new theoretical support for using dietary polyphenols to ameliorate these conditions.
3.3 UPS and oxidative stress
The UPS is crucial for maintaining redox homeostasis. Oxidative stress (OS) is a key contributor to development of metabolic disorders (132). In addition to the main antioxidant defense system, cells use proteolysis and transcription factors to counteract oxidative stress. Nuclear factor erythroid 2-related factor 2 (Nrf2) serves as a pivotal transcription factor activated in response to oxidative stress, directly influencing the onset and progression of NAFLD (133, 134). ROS increases hepatic oxidative stress, and Nrf2 regulates cellular resistance to oxidative damage by controlling antioxidant response element (ARE)-dependent genes (135). Normally, Nrf2 is cytoplasmic, bound to its inhibitor (Kelch-like ECH-associated protein 1Keap1) (136).TheKeap1/Nrf2 complex, involving the Cul3-RbX1 holoenzyme (an E3 ubiquitin ligase from the Cullin-RING box family), leads to Nrf2 ubiquitination and subsequent degradation by the 26S proteasome (136, 137). Oxidative stress causes Keap1 oxidation, which preventing its binding to Nrf2, lead Nrf2 release into the cytosol (137, 138). TRIM25 targets Keap1 directly through ubiquitination and degradation, resulting in Nrf2 activation, thereby enhancing cellular oxidative stress responses and causing damage to liver tissues (139).
Various kinases, including MAPKs, protein kinase C (PKC), or PI3K, phosphorylate Nrf2, and promote its nuclear translocation (140). In the nucleus, Nrf2 binds to the ARE in the gene promoters, enhancing transcription for the antioxidant response (138, 141). Specific E3 ubiquitin ligases like HRD1, MIB1, and TRIM22 facilitate NRF2 ubiquitination and degradation, inhibiting the antioxidant pathway (142–144). In cirrhotic livers, the activation of the XBP1-Hrd1 arm of ER stress transcriptionally upregulated Hrd1, leading to increased ubiquitination and degradation of Nrf2, thereby attenuating the Nrf2 signaling pathway (142). Furthermore, E3 ubiquitin ligase TRIM21 negatively regulates the p62-Keap1-Nrf2 antioxidant pathway by ubiquitinating p62. Conversely, TRIM16 and HACE1 enhance NRF2 protein stability by competitively binding, thereby promoting the antioxidant pathway (145, 146). Deubiquitinases DUB3 and USP11 in-crease NRF2 stability and transcriptional activity by reducing NRF2 ubiquitination (147, 148). Additionally, USP enzymes target Keap1 to influence oxidative stress pathways. TRIM25 and TRIM15 directly ubiquitinate and degrade Keap1, activating Nrf2 and enhancing the antioxidant defense system (139, 149). In metabolic disorders condition, impaired proteasome activity, compromised Nrf2 function, and inadequate response to oxidative stress expedite disease progression. Modulating the UPS to enhance antioxidant enzyme expression, to control Nrf2 activation, and to improve proteasome function can be benefit metabolic disorders by alleviating ROS-related effects (Figure 8).
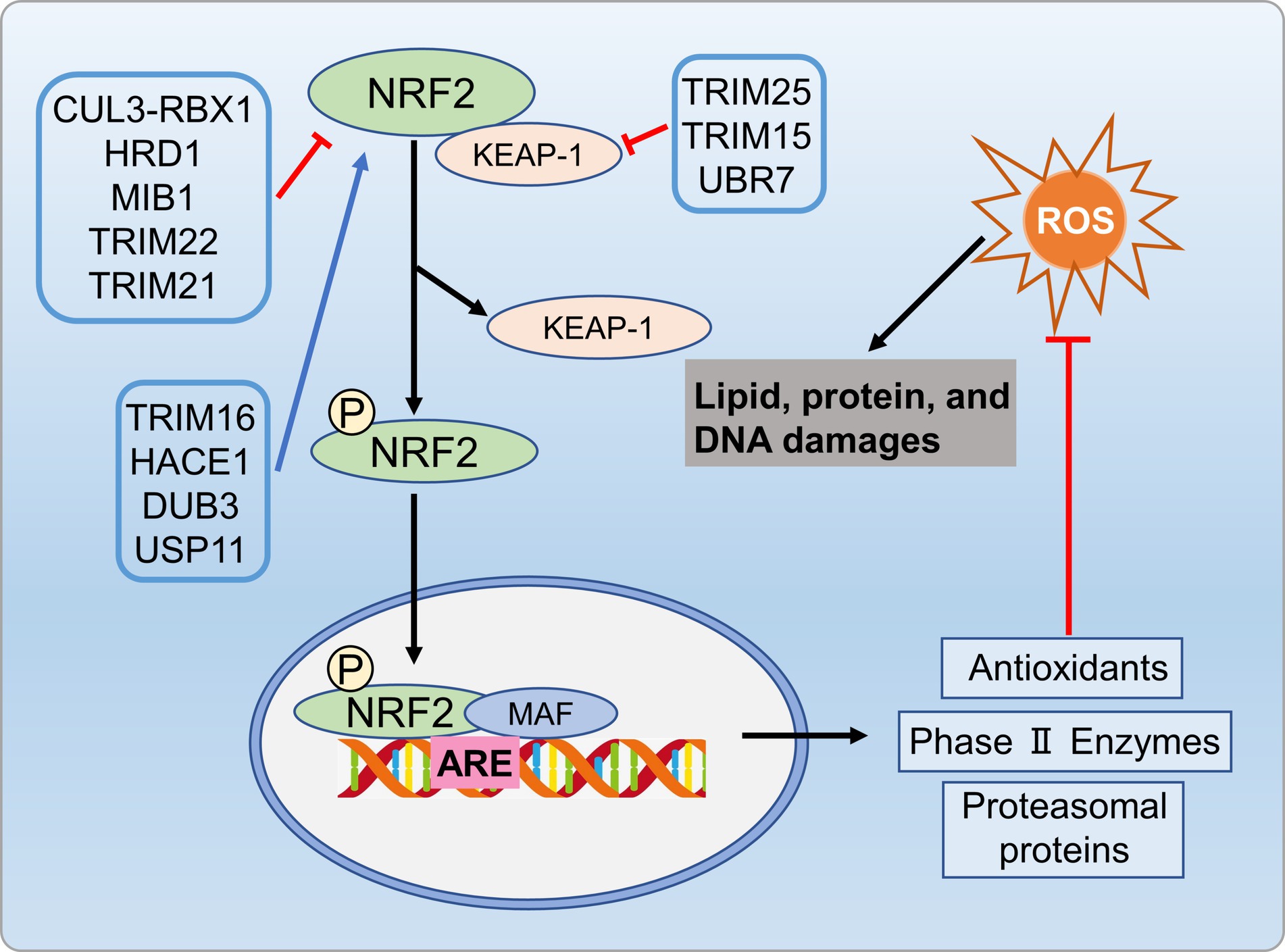
Figure 8. UPS regulates oxidative stress process involved in metabolic disorders. Specific E3 ubiquitin ligases like HRD1, MIB1, and TRIM22 promote NRF2 ubiquitination and degradation, inhibiting antioxidant pathways. Conversely, ubiquitinases TRIM16 and HACE1, as well as deubiquitinases DUB3 and USP11, enhance antioxidant pathways by regulating NRF2 protein stability. Additionally, TRIM25 and TRIM15 ubiquitinate and degrade Keap1, activating NRF2 and enhancing antioxidant defenses.
3.4 UPS and insulin resistance
Insulin resistance is closely associated with development of metabolic disorders (150). Research indicated that UPS is crucial in insulin resistance by regulating insulin signaling (151). Altered E3 ligase expression targeted insulin signaling molecules. Genetic deletion of these ligases can enhance or impair insulin action. Palmitate-induced insulin resistance involves upregulation of E3 ligases, which targeting AKT, IR, and IRS1, leading to ubiquitin-dependent degradation (152). Feeding Ube4A knockout mice with a high-fat diet (HFD) led to an augmented depot of white and brown adipose tissues, exacerbate insulin resistance and inflammation. Ube4A deficiency intensified hepatic steatosis, inflammation, and liver injury in mice. This effect was attributed to K63-linked ubiquitination (K63-Ub) of Akt and APPL1, further promoted insulin-induced AKT activation (153). E3 ubiquitin ligases contribute to insulin resistance via two main mechanisms, including direct targeting of insulin signaling molecules and indirect regulation of insulin signaling by targeting pro-inflammatory mediators. Firstly, insulin signaling molecules were directly targeted by E3 ubiquitin ligases, such as mitsugumin 53 (MG53) (154), CRL7 (155), Cbl proteins (156, 157), suppressors of cytokine signaling (SOCS) 1/3 (158), Fbxo40 (159), murine double minute 2 (MDM2) (160), NEDD4 (161, 162), and TRIM32 (163), which are known to be involved in the ubiquitin-dependent degradation of key insulin signaling molecules, including PI3K, AKT, IR, and IRS, contributed to insulin resistance. Drugs that can protect these molecules against UPS-mediated degradation might hold significant therapeutic promise for addressing insulin resistance syndromes and diabetes. Additionally, E3 ubiquitin ligases indirectly influence insulin signaling by targeting pro-inflammatory mediators linked to insulin resistance (Figure 9).
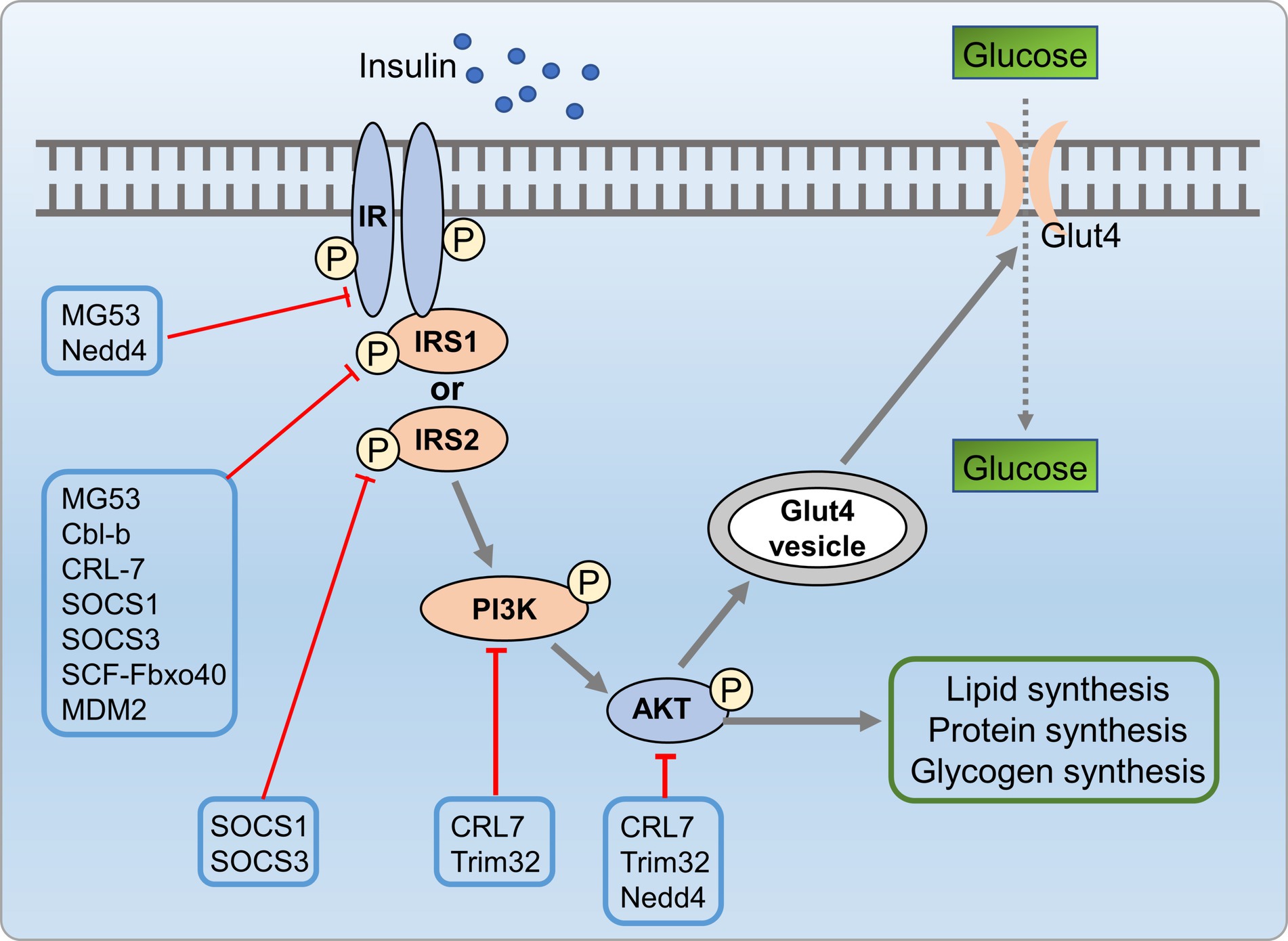
Figure 9. UPS mediated insulin resistance process involved in metabolic disorders. Insulin signaling molecules are directly targeted by E3 ubiquitin ligases such as MG53, CRL7, SOCS 1/3, MDM2, NEDD4, and TRIM32, which are involved in the ubiquitin-dependent degradation of key insulin signaling molecules, including PI3K, AKT, IR, and IRS, leading to insulin resistance.
4 Conclusion and perspectives
Metabolic disorders are increasing worldwide and negatively impact global economy and health. At present, the therapeutic treatment options for metabolic disorders are limited. The exclusive involvement of the UPS in metabolic disorders establishes it as a burgeoning subject, holding promise for drug discovery and emerging as a vital focus. Compelling evidence from both in vivo and in vitro settings demonstrates that natural polyphenols exhibit protective effects across various metabolic disorders, including inhibiting intrahepatic lipid accumulation, mitigating inflammatory factors, possessing antioxidative properties, and attenuating fibrotic progression (164, 165). So far, our understanding of the mechanisms of polyphenols preventing metabolic disorders remains to be further investigated in the future.
Despite notable progress, considerable gaps in comprehending UPS in metabolic disorders persist, necessitating urgent attention to address critical inquiries and effectively bridge knowledge disparities. First, it is crucial to thoroughly grasp the molecular mechanism of action of polyphenols and their potential interaction with the UPS. For example, in future research, we need to conduct more comprehensive and systematic studies to determine whether different polyphenols directly with UPS or indirectly promote the degradation of related proteins or act as proteasome regulators to directly control intracellular proteasome levels. In addition, by characterizing the upstream regulators or downstream targets of UPS, researchers can develop treatment approaches with improved efficacy and specificity. After gaining a deeper understanding of how polyphenols regulate upstream factors or downstream targets of the UPS, we can develop more effective agonists or inhibitors targeting these sites to achieve more precise regulation of metabolic disorders.
In the next testing phase, additional in vivo and in vitro experiments are crucial to assess molecule bioavailability, unravel their specific mechanism of action, and explore potential interactions with conventional proteasome inhibitors. Furthermore, clinical trials are needed to determine the efficacy and safety of UPS-targeted therapies for treating metabolic disorders in human patients. Future research should focus on developing new approaches to modulate the UPS pathway, including the use of natural compounds such as polyphenols, alongside the creation of small molecule inhibitors that specifically target enzymes within the UPS pathway. Additionally, further exploration is needed on the role of polyphenols in combination with other drugs to improve metabolic disorders. In summary, there is great potential for developing new therapies targeting metabolic disorders based on the regulation of the UPS pathway by polyphenols.
Author contributions
WG: Data curation, Formal analysis, Resources, Writing – original draft, Writing – review & editing. GW: Data curation, Methodology, Writing – original draft, Writing – review & editing. GC: Conceptualization, Formal analysis, Methodology, Writing – original draft, Writing – review & editing. XM: Data curation, Writing – original draft, Writing – review & editing. ZX: Conceptualization, Funding acquisition, Supervision, Writing – original draft, Writing – review & editing. SC: Conceptualization, Methodology, Project administration, Supervision, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This study was supported by a key joint grant for regional innovation from the National Natural Science Foundation of China (Grant No. U19A2034), China Agriculture Research System of MOF and MARA (CARS-19), a grant for University Synergy Innovation Program of Anhui Province (Grant No. GXXT-2019-49), a grant from “Research Funds of Joint Research Center for Food Nutrition and Health of IHM” (2023SJY02), and an open grant from State Key Laboratory of Tea Plant Biology and Utilization (Grant No. SKLTOF2022013).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Yu, Z, and Muehleman, V. Eating disorders and metabolic diseases. Int J Environ Res Public Health. (2023) 20:2446. doi: 10.3390/ijerph20032446
2. Powell, EE, Wong, VW, and Rinella, M. Non-alcoholic fatty liver disease. Lancet. (2021) 397:2212–24. doi: 10.1016/S0140-6736(20)32511-3
3. Tilg, H, Adolph, TE, and Moschen, AR. Multiple parallel hits hypothesis in nonalcoholic fatty liver disease: revisited after a decade. Hepatology. (2021) 73:833–42. doi: 10.1002/hep.31518
4. Shah, PA, Patil, R, and Harrison, SA. NAFLD-related hepatocellular carcinoma: the growing challenge. Hepatology. (2023) 77:323–38. doi: 10.1002/hep.32542
5. Arroyave-Ospina, JC, Wu, Z, Geng, Y, and Moshage, H. Role of oxidative stress in the pathogenesis of non-alcoholic fatty liver disease: implications for prevention and therapy. Antioxidants. (2021) 10:174. doi: 10.3390/antiox10020174
6. Kakiyama, G, Rodriguez-Agudo, D, and Pandak, WM. Mitochondrial cholesterol metabolites in a bile acid synthetic pathway drive non-alcoholic fatty liver disease: a revised "two-hit" hypothesis. Cells. (2023) 12:1434. doi: 10.3390/cells12101434
7. Sanyal, AJ . Past, present and future perspectives in nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol. (2019) 16:377–86. doi: 10.1038/s41575-019-0144-8
8. Shabalala, SC, Dludla, PV, Mabasa, L, Kappo, AP, Basson, AK, Pheiffer, C, et al. The effect of adiponectin in the pathogenesis of non-alcoholic fatty liver disease (NAFLD) and the potential role of polyphenols in the modulation of adiponectin signaling. Biomed Pharmacother. (2020) 131:110785. doi: 10.1016/j.biopha.2020.110785
9. Abenavoli, L, Larussa, T, Corea, A, Procopio, AC, Boccuto, L, Dallio, M, et al. Dietary polyphenols and non-alcoholic fatty liver disease. Nutrients. (2021) 13:494. doi: 10.3390/nu13020494
10. Rousseau, A, and Bertolotti, A. Regulation of proteasome assembly and activity in health and disease. Nat Rev Mol Cell Biol. (2018) 19:697–712. doi: 10.1038/s41580-018-0040-z
11. Li, Y, Li, S, and Wu, H. Ubiquitination-proteasome system (UPS) and autophagy two Main protein degradation machineries in response to cell stress. Cells. (2022) 11:851. doi: 10.3390/cells11050851
12. Das, D, Paul, A, Lahiri, A, Adak, M, Maity, SK, Sarkar, A, et al. Proteasome dysfunction under compromised redox metabolism dictates liver injury in NASH through ASK1/PPARγ binodal complementary modules. Redox Biol. (2021) 45:102043. doi: 10.1016/j.redox.2021.102043
13. Park, JS, Ma, H, and Roh, YS. Ubiquitin pathways regulate the pathogenesis of chronic liver disease. Biochem Pharmacol. (2021) 193:114764. doi: 10.1016/j.bcp.2021.114764
14. Li, K, Zhang, K, Wang, H, Wu, Y, Chen, N, Chen, J, et al. Hrd1-mediated ACLY ubiquitination alleviate NAFLD in db/db mice. Metabolism. (2021) 114:154349. doi: 10.1016/j.metabol.2020.154349
15. Wang, L, Zhang, X, Lin, ZB, Yang, PJ, Xu, H, Duan, JL, et al. Tripartite motif 16 ameliorates nonalcoholic steatohepatitis by promoting the degradation of phospho-TAK1. Cell Metab. (2021) 33:1372–1388.e7. doi: 10.1016/j.cmet.2021.05.019
16. Yang, Q, Chen, X, Zhang, Y, Hu, S, Hu, F, Huang, Y, et al. The E3 ubiquitin ligase ring finger protein 5 ameliorates NASH through ubiquitin-mediated degradation of 3-Hydroxy-3-Methylglutaryl CoA reductase degradation protein 1. Hepatology. (2021) 74:3018–36. doi: 10.1002/hep.32061
17. Zhang, L, Wu, M, Su, R, Zhang, D, and Yang, G. The efficacy and mechanism of proteasome inhibitors in solid tumor treatment. Recent Pat Anticancer Drug Discov. (2022) 17:268–83. doi: 10.2174/1574892816666211202154536
18. Ambrosio, FA, Costa, G, Gallo Cantafio, ME, Torcasio, R, Trapasso, F, Alcaro, S, et al. Natural agents as novel potential source of pro-teasome inhibitors with anti-tumor activity: focus on multiple myeloma. Molecules. (2023) 28:1438. doi: 10.3390/molecules28031438
19. Kwak, HJ, Choi, HE, Jang, J, Park, SK, Bae, YA, and Cheon, HG. Bortezomib attenuates palmitic acid-induced ER stress, inflammation and insulin resistance in myotubes via AMPK dependent mechanism. Cell Signal. (2016) 28:788–97. doi: 10.1016/j.cellsig.2016.03.015
20. Di Lorenzo, C, Colombo, F, Biella, S, Stockley, C, and Restani, P. Polyphenols and human health: the role of bioavailability. Nutrients. (2021) 13:273. doi: 10.3390/nu13010273
21. Wang, S, Du, Q, Meng, X, and Zhang, Y. Natural polyphenols: a potential prevention and treatment strategy for metabolic syndrome. Food Funct. (2022) 13:9734–53. doi: 10.1039/d2fo01552h
22. Chen, L, Liu, J, Mei, G, Chen, H, Peng, S, Zhao, Y, et al. Quercetin and non-alcoholic fatty liver disease: a review based on experimental data and bioinformatic analysis. Food Chem Toxicol. (2021) 154:112314. doi: 10.1016/j.fct.2021.112314
23. Machado, IF, Miranda, RG, Dorta, DJ, Rolo, AP, and Palmeira, CM. Targeting oxidative stress with polyphenols to fight liver diseases. Antioxidants. (2023) 12:1212. doi: 10.3390/antiox12061212
24. Sae-tan, S, Grove, KA, and Lambert, JD. Weight control and prevention of metabolic syndrome by green tea. Pharmacol Res. (2011) 64:146–54. doi: 10.1016/j.phrs.2010.12.013
25. Ntamo, Y, Jack, B, Ziqubu, K, Mazibuko-Mbeje, SE, Nkambule, BB, Nyambuya, TM, et al. Epigallocatechin gallate as a nutraceutical to potentially target the metabolic syndrome: novel insights into therapeutic effects beyond its antioxidant and anti-inflammatory proper-ties. Crit Rev Food Sci Nutr. (2022) 64:87–109. doi: 10.1080/10408398.2022.2104805
26. Payne, A, Nahashon, S, Taka, E, Adinew, GM, and Soliman, K. Epigallocatechin-3-Gallate (EGCG): new therapeutic perspectives for Neu-roprotection, aging, and Neuroinflammation for the modern age. Biomol Ther. (2022) 12:371. doi: 10.3390/biom12030371
27. Kuhn, DJ, Burns, AC, Kazi, A, and Dou, QP. Direct inhibition of the ubiquitin-proteasome pathway by ester bond-containing green tea poly-phenols is associated with increased expression of sterol regulatory element-binding protein 2 and LDL receptor. Biochim Biophys Acta. (2004) 1682:1–10. doi: 10.1016/j.bbalip.2003.12.006
28. Yee, WL, Wang, Q, Agdinaoay, T, Dang, K, Chang, H, Grandinetti, A, et al. Green tea catechins decrease apolipoprotein B-100 secretion from HepG2 cells. Mol Cell Biochem. (2002) 229:85–92. doi: 10.1023/a:1017920527201
29. Chang, YC, Liu, HW, Chan, YC, Hu, SH, Liu, MY, and Chang, SJ. The green tea polyphenol epigallocatechin-3-gallate attenuates age-associated muscle loss via regulation of miR-486-5p and myostatin. Arch Biochem Biophys. (2020) 692:108511. doi: 10.1016/j.abb.2020.108511
30. You, W, Liu, S, Ji, J, Ling, D, Tu, Y, Zhou, Y, et al. Growth arrest and DNA damage-inducible alpha regulates muscle repair and fat infil-tration through ATP synthase F1 subunit alpha. J Cachexia Sarcopenia Muscle. (2023) 14:326–41. doi: 10.1002/jcsm.13134
31. Zhang, J, Zhou, L, Huang, Z, Xu, Z, and Xiang, C. Epigallocatechin-3-gallate(EGCG) suppresses melanoma cell growth and metastasis by targeting TRAF6 activity. Oncotarget. (2016) 7:79557–71. doi: 10.18632/oncotarget.12836
32. Kumazoe, M, Nakamura, Y, Yamashita, M, Suzuki, T, Takamatsu, K, Huang, Y, et al. Green tea polyphenol Epigallocatechin-3-gallate suppresses toll-like receptor 4 expression via up-regulation of E3 ubiquitin-protein ligase RNF216. J Biol Chem. (2017) 292:4077–88. doi: 10.1074/jbc.M116.755959
33. Pettinari, A, Amici, M, Cuccioloni, M, Angeletti, M, Fioretti, E, and Eleuteri, AM. Effect of polyphenolic compounds on the proteolytic activi-ties of constitutive and immuno-proteasomes. Antioxid Redox Signal. (2006) 8:121–9. doi: 10.1089/ars.2006.8.121
34. Jabczyk, M, Nowak, J, Hudzik, B, and Zubelewicz-Szkodzińska, B. Curcumin in metabolic health and disease. Nutrients. (2021) 13:4440. doi: 10.3390/nu13124440
35. Chen, Y, Wu, R, Chen, W, Liu, Y, Liao, X, Zeng, B, et al. Curcumin prevents obesity by targeting TRAF4-induced ubiquitylation in m(6) A-dependent manner. EMBO Rep. (2021) 22:e52146. doi: 10.15252/embr.202052146
36. Zhang, D, Zhang, Y, Ye, M, Ding, Y, Tang, Z, Li, M, et al. Interference with Akt signaling pathway contributes curcumin-induced adipo-cyte insulin resistance. Mol Cell Endocrinol. (2016) 429:1–9. doi: 10.1016/j.mce.2016.04.013
37. Kong, D, Zhang, Z, Chen, L, Huang, W, Zhang, F, Wang, L, et al. Curcumin blunts epithelial-mesenchymal transition of hepatocytes to alleviate hepatic fibrosis through regulating oxidative stress and autophagy. Redox Biol. (2020) 36:101600. doi: 10.1016/j.redox.2020.101600
38. Zhao, JF, Ching, LC, Huang, YC, Chen, CY, Chiang, AN, Kou, YR, et al. Molecular mechanism of curcumin on the suppression of choles-terol accumulation in macrophage foam cells and atherosclerosis. Mol Nutr Food Res. (2012) 56:691–701. doi: 10.1002/mnfr.201100735
39. Ono, T, Takada, S, Kinugawa, S, and Tsutsui, H. Curcumin ameliorates skeletal muscle atrophy in type 1 diabetic mice by inhibiting protein ubiquitination. Exp Physiol. (2015) 100:1052–63. doi: 10.1113/EP085049
40. Cardaci, TD, Machek, SB, Wilburn, DT, Hwang, PS, and Willoughby, DS. Ubiquitin proteasome system activity is suppressed by curcumin following exercise-induced muscle damage in human skeletal muscle. J Am Coll Nutr. (2021) 40:401–11. doi: 10.1080/07315724.2020.1783721
41. Zhang, J, Zheng, J, Chen, H, Li, X, Ye, C, Zhang, F, et al. Curcumin targeting NF-κB/ubiquitin-proteasome-system axis ameliorates muscle atrophy in triple-negative breast cancer cachexia mice. Mediat Inflamm. (2022) 2022:2567150. doi: 10.1155/2022/2567150
42. Hasima, N, and Aggarwal, BB. Targeting proteasomal pathways by dietary curcumin for cancer prevention and treatment. Curr Med Chem. (2014) 21:1583–94. doi: 10.2174/09298673113206660135
43. Jana, NR, Dikshit, P, Goswami, A, and Nukina, N. Inhibition of proteasomal function by curcumin induces apoptosis through mitochondrial pathway. J Biol Chem. (2004) 279:11680–5. doi: 10.1074/jbc.M310369200
44. Hassan, FU, Rehman, MS, Khan, MS, Ali, MA, Javed, A, Nawaz, A, et al. Curcumin as an alternative epigenetic modulator: mechanism of action and potential effects. Front Genet. (2019) 10:514. doi: 10.3389/fgene.2019.00514
45. Liczbiński, P, Michałowicz, J, and Bukowska, B. Molecular mechanism of curcumin action in signaling pathways: review of the latest re-search. Phytother Res. (2020) 34:1992–2005. doi: 10.1002/ptr.6663
46. Andres, S, Pevny, S, Ziegenhagen, R, Bakhiya, N, Schäfer, B, Hirsch-Ernst, KI, et al. Safety aspects of the use of quercetin as a dietary supplement. Mol Nutr Food Res. (2018) 62:447. doi: 10.1002/mnfr.201700447
47. Gnoni, GV, Paglialonga, G, and Siculella, L. Quercetin inhibits fatty acid and triacylglycerol synthesis in rat-liver cells. Eur J Clin Investig. (2009) 39:761–8. doi: 10.1111/j.1365-2362.2009.02167.x
48. Chen, P, Chen, JB, Chen, WY, Zheng, QL, Wang, YQ, and Xu, XJ. Effects of quercetin on nuclear factor-κB p65 expression in renal ubiqui-tin-proteasome system of diabetic rats. Zhonghua Nei Ke Za Zhi. (2012) 51:460–5. doi: 10.3760/cma.j.issn.0578-1426.2012.06.015
49. Liu, P, Lin, H, Xu, Y, Zhou, F, Wang, J, Liu, J, et al. Frataxin-mediated PINK1-Parkin-dependent Mitophagy in hepatic steatosis: the protective effects of quercetin. Mol Nutr Food Res. (2018) 62:e1800164. doi: 10.1002/mnfr.201800164
50. Nakamura, M, Fukuma, Y, Notsu, K, and Kono, M. Quercetin and HSC70 coregulate the anti-inflammatory action of the ubiquitin-like protein MNSFβ. Mol Biol Rep. (2022) 49:1213–22. doi: 10.1007/s11033-021-06949-y
51. Dosenko, VE, Nagibin, VS, Tumanovskaia, LV, Viu, Z, and Moĭbenko, AA. The influence of quercetin on the activity of purified 20S, 26S proteasome and proteasomal activity in isolated cardiomyocytes. Biomed Khim. (2006) 52:138–45.
52. Martín-Aragón, S, Jiménez-Aliaga, KL, Benedí, J, and Bermejo-Bescós, P. Neurohormetic responses of quercetin and rutin in a cell line over-expressing the amyloid precursor protein (APPswe cells). Phytomed Int J Phytother Phytopharmacol. (2016) 23:1285–94. doi: 10.1016/j.phymed.2016.07.007
53. Peñalver, P, Belmonte-Reche, E, Adán, N, Caro, M, Mateos-Martín, ML, Delgado, M, et al. Alkylated resveratrol prodrugs and metabolites as potential therapeutics for neurodegenerative diseases. Eur J Med Chem. (2018) 146:123–38. doi: 10.1016/j.ejmech.2018.01.037
54. Suh, JH, Kim, KH, Conner, ME, Moore, DD, and Preidis, GA. Hepatic PPARα is destabilized by SIRT1 deacetylase in undernourished male mice. Front Nutr. (2022) 9:831879. doi: 10.3389/fnut.2022.831879
55. Suh, JH, Sieglaff, DH, Zhang, A, Xia, X, Cvoro, A, Winnier, GE, et al. SIRT1 is a direct coactivator of thyroid hormone receptor β1 with gene-specific actions. PLoS One. (2013) 8:e70097. doi: 10.1371/journal.pone.0070097
56. Floyd, ZE, Wang, ZQ, Kilroy, G, and Cefalu, WT. Modulation of peroxisome proliferator-activated receptor gamma stability and transcrip-tional activity in adipocytes by resveratrol. Metabolism. (2008) 57:S32–8. doi: 10.1016/j.metabol.2008.04.006
57. Qureshi, N, Desousa, J, Siddiqui, AZ, Morrison, DC, and Qureshi, AA. Reprograming of gene expression of key inflammatory signaling pathways in human peripheral blood mononuclear cells by soybean lectin and resveratrol. Int J Mol Sci. (2022) 23:12946. doi: 10.3390/ijms232112946
58. Silswal, N, Reddy, NS, Qureshi, AA, and Qureshi, N. Resveratrol downregulates biomarkers of Sepsis via inhibition of proteasome's proteases. Shock. (2018) 50:579–88. doi: 10.1097/SHK.0000000000001080
59. Seungkyu, LC, Young, CE, Chul, LS, Jun, KH, Haeng, LJ, and Hyung, CJ. Resveratrol inhibits hypoxia-induced vascular endothelial growth factor expression and pathological neovascularization. Yonsei Med J. (2015) 56:1678–85. doi: 10.3349/ymj.2015.56.6.1678
60. He, Y, Wang, H, Lin, S, Chen, T, Chang, D, Sun, Y, et al. Advanced effect of curcumin and resveratrol on mitigating hepatic steatosis in metabolic associated fatty liver disease via the PI3K/AKT/mTOR and HIF-1/VEGF cascade. Biomed Pharmacother. (2023) 165:115279. doi: 10.1016/j.biopha.2023.115279
61. Hosseini, H, Teimouri, M, Shabani, M, Koushki, M, Babaei Khorzoughi, R, Namvarjah, F, et al. Resveratrol alleviates non-alcoholic fatty liver disease through epigenetic modification of the Nrf2 signaling pathway. Int J Biochem Cell Biol. (2020) 119:105667. doi: 10.1016/j.biocel.2019.105667
62. Shi, JX, Wang, QJ, Li, H, and Huang, Q. Silencing of USP22 suppresses high glucose-induced apoptosis, ROS production and inflammation in podocytes. Mol BioSyst. (2016) 12:1445–56. doi: 10.1039/c5mb00722d
63. Takemoto, M, and Takemoto, H. Synthesis of Theaflavins and their functions. Molecules. (2018) 23:918. doi: 10.3390/molecules23040918
64. Ano, Y, Ohya, R, Kita, M, Taniguchi, Y, and Kondo, K. Theaflavins improve memory impairment and depression-like behavior by Regulat-ing microglial activation. Molecules. (2019) 24:24. doi: 10.3390/molecules24030467
65. Mizuno, H, Cho, YY, Zhu, F, Ma, WY, and Dong, Z. Theaflavin-3, 3′-digallate induces epidermal growth factor receptor downregulation. Mol Carcinog. (2010) 45:204–12. doi: 10.1002/mc.20174
66. Lin, JK, and Lin-Shiau, SY. Mechanisms of hypolipidemic and anti-obesity effects of tea and tea polyphenols. Mol Nutr Food Res. (2006) 50:211–7. doi: 10.1002/mnfr.200500138
67. Wu, CH, Lin, MC, Wang, HC, Yang, MY, Jou, MJ, and Wang, CJ. Rutin inhibits oleic acid induced lipid accumulation via reducing Lipo-genesis and oxidative stress in Hepatocarcinoma cells. J Food Sci. (2011) 76:T65–72. doi: 10.1111/j.1750-3841.2010.02033.x
68. Carrasco-Pozo, C, Gotteland, M, Castillo, RL, and Chen, C. 3,4-dihydroxyphenylacetic acid, a microbiota-derived metabolite of quercetin, protects against pancreatic β-cells dysfunction induced by high cholesterol. Exp Cell Res. (2015) 334:270–82. doi: 10.1016/j.yexcr.2015.03.021
69. Al-Harbi, NO, Imam, F, Al-Harbi, MM, Al-Shabanah, OA, Alotaibi, MR, As Sobeai, HM, et al. Rutin inhibits carfilzomib-induced oxida-tive stress and inflammation via the NOS-mediated NF-κB signaling pathway. Inflammopharmacology. (2019) 27:817–27. doi: 10.1007/s10787-018-0550-5
70. Tung, YT, Zeng, JL, Ho, ST, Xu, JW, Lin, IH, and Wu, JH. Djulis Hull improves insulin resistance and modulates the gut microbiota in high-fat diet (HFD)-induced Hyperglycaemia. Antioxidants. (2021) 11:45. doi: 10.3390/antiox11010045
71. Hashizume, Y, and Tandia, M. The reduction impact of monoglucosyl rutin on abdominal visceral fat: a randomized, placebo-controlled, double-blind, parallel-group. J Food Sci. (2020) 85:3577–89. doi: 10.1111/1750-3841.15429
72. Panchal, SK, Poudyal, H, Arumugam, TV, and Brown, L. Rutin attenuates metabolic changes, nonalcoholic steatohepatitis, and cardiovascular remodeling in high-carbohydrate, high-fat diet-fed rats. J Nutr. (2011) 141:1062–9. doi: 10.3945/jn.111.137877
73. Oluranti, OI, Alabi, BA, Michael, OS, Ojo, AO, and Fatokun, BP. Rutin prevents cardiac oxidative stress and inflammation induced by bi-sphenol a and dibutyl phthalate exposure via NRF-2/NF-κB pathway. Life Sci. (2021) 284:119878. doi: 10.1016/j.lfs.2021.119878
74. Ahmed, OM, Elkomy, MH, Fahim, HI, Ashour, MB, Naguib, IA, Alghamdi, BS, et al. Rutin and quercetin counter doxorubicin-induced liver toxicity in Wistar rats via their modulatory effects on inflammation, oxidative stress, apoptosis, and Nrf2. Oxidative Med Cell Longev. (2022) 2022:2710607–19. doi: 10.1155/2022/2710607
75. Kandemir, FM, Ileriturk, M, and Gur, C. Rutin protects rat liver and kidney from sodium valproate-induce damage by attenuating oxidative stress, ER stress, inflammation, apoptosis and autophagy. Mol Biol Rep. (2022) 49:6063–74. doi: 10.1007/s11033-022-07395-0
76. Küçükler, S, Kandemir, FM, Özdemir, S, Çomaklı, S, and Caglayan, C. Protective effects of rutin against deltamethrin-induced hepatotoxicity and nephrotoxicity in rats via regulation of oxidative stress, inflammation, and apoptosis. Environ Sci Pollut Res Int. (2021) 28:62975–90. doi: 10.1007/s11356-021-15190-w
77. Kitamura, H . Ubiquitin-specific proteases (USPs) and metabolic disorders. Int J Mol Sci. (2023) 24:3219. doi: 10.3390/ijms24043219
78. Zhao, Y, Wang, F, Gao, L, Xu, L, Tong, R, Lin, N, et al. Ubiquitin-specific protease 4 is an endogenous negative regulator of metabolic dysfunctions in nonalcoholic fatty liver disease in mice. Hepatology. (2018) 68:897–917. doi: 10.1002/hep.29889
79. van den Boomen, D, Volkmar, N, and Lehner, PJ. Ubiquitin-mediated regulation of sterol homeostasis. Curr Opin Cell Biol. (2020) 65:103–11. doi: 10.1016/j.ceb.2020.04.010
80. Gill, S, Stevenson, J, Kristiana, I, and Brown, AJ. Cholesterol-dependent degradation of squalene monooxygenase, a control point in Cho-lesterol synthesis beyond HMG-CoA reductase. Cell Metab. (2011) 13:260–73. doi: 10.1016/j.cmet.2011.01.015
81. Chua, NK, Coates, HW, and Brown, AJ. Squalene monooxygenase: a journey to the heart of cholesterol synthesis. Prog Lipid Res. (2020) 79:101033. doi: 10.1016/j.plipres.2020.101033
82. Li, XZ, Jiang, SY, Li, GQ, Jiang, QR, Li, JW, Li, CC, et al. Synthesis of heterocyclic ring-fused analogs of HMG499 as novel degraders of HMG-CoA reductase that lower cholesterol. Eur J Med Chem. (2022) 236:114323. doi: 10.1016/j.ejmech.2022.114323
83. Song, B, Sever, N, and DeBose-Boyd, RA. Gp78, a membrane-anchored ubiquitin ligase, associates with Insig-1 and couples Ster-ol-regulated ubiquitination to degradation of HMG CoA reductase. Mol Cell. (2005) 19:829–40. doi: 10.1016/j.molcel.2005.08.009
84. Tsai, YC, Leichner, GS, Pearce, MM, Wilson, GL, Wojcikiewicz, RJ, Roitelman, J, et al. Differential regulation of HMG-CoA reductase and Insig-1 by enzymes of the ubiquitin-proteasome system. Mol Biol Cell. (2012) 23:4484–94. doi: 10.1091/mbc.E12-08-0631
85. Zhou, ZS, Li, MX, Liu, J, Jiao, H, Xia, JM, Shi, XJ, et al. Competitive oxidation and ubiquitylation on the evolutionarily conserved cyste-ine confer tissue-specific stabilization of Insig-2. Nat Commun. (2020) 11:379. doi: 10.1038/s41467-019-14231-w
86. Zhang, T, Kho, DH, Wang, Y, Harazono, Y, Nakajima, K, Xie, Y, et al. Gp78, an E3 ubiquitin ligase acts as a gatekeeper suppressing non-alcoholic steatohepatitis (NASH) and liver cancer. PLoS One. (2015) 10:e0118448. doi: 10.1371/journal.pone.0118448
87. Menzies, SA, Volkmar, N, van den Boomen, DJ, Timms, RT, Dickson, AS, Nathan, JA, et al. The sterol-responsive RNF145 E3 ubiquitin ligase mediates the degradation of HMG-CoA reductase together with gp78 and Hrd1. eLife. (2018) 7:e40009. doi: 10.7554/eLife.40009
88. Wei, J, Chen, L, Li, F, Yuan, Y, Wang, Y, Xia, W, et al. HRD1-ERAD controls production of the hepatokine FGF21 through CREBH polyubiquitination. EMBO J. (2018) 37:e98942. doi: 10.15252/embj.201898942
89. Zelcer, N, Sharpe, LJ, Loregger, A, Kristiana, I, Cook, EC, Phan, L, et al. The E3 ubiquitin ligase MARCH6 degrades squalene monooxy-genase and affects 3-hydroxy-3-methyl-glutaryl coenzyme a reductase and the cholesterol synthesis pathway. Mol Cell Biol. (2014) 34:1262–70. doi: 10.1128/MCB.01140-13
90. Chua, NK, Hart-Smith, G, and Brown, AJ. Non-canonical ubiquitination of the cholesterol-regulated degron of squalene monooxygenase. J Biol Chem. (2019) 294:8134–47. doi: 10.1074/jbc.RA119.007798
91. Kwon, D, Kim, SM, and Correia, MA. Cytochrome P450 endoplasmic reticulum-associated degradation (ERAD): therapeutic and pathophys-iological implications. Acta Pharm Sin B. (2020) 10:42–60. doi: 10.1016/j.apsb.2019.11.002
92. Chang, XL, Liu, L, Wang, N, Chen, ZJ, and Zhang, C. The function of high-density lipoprotein and low-density lipoprotein in the maintenance of mouse ovarian steroid balance. Biol Reprod. (2017) 97:862–72. doi: 10.1093/biolre/iox134
93. Zelcer, N, Hong, C, Boyadjian, R, and Tontonoz, P. LXR regulates cholesterol uptake through Idol-dependent ubiquitination of the LDL re-ceptor. Science. (2009) 325:100–4. doi: 10.1126/science.1168974
94. Lee, K, Haddad, A, Osme, A, Kim, C, Borzou, A, Ilchenko, S, et al. Hepatic mitochondrial defects in a nonalcoholic fatty liver disease mouse model are associated with increased degradation of oxidative phosphorylation subunits. Mol Cell Proteomics. (2018) 17:2371–86. doi: 10.1074/mcp.RA118.000961
95. Li, H, Yu, XH, Ou, X, Ouyang, XP, and Tang, CK. Hepatic cholesterol transport and its role in non-alcoholic fatty liver disease and athero-sclerosis. Prog Lipid Res. (2021) 83:101109. doi: 10.1016/j.plipres.2021.101109
96. Calkin, AC, and Tontonoz, P. Transcriptional integration of metabolism by the nuclear sterol-activated receptors LXR and FXR. Nat Rev Mol Cell Biol. (2012) 13:213–24. doi: 10.1038/nrm3312
97. Bideyan, L, López Rodríguez, M, Priest, C, Kennelly, JP, Gao, Y, Ferrari, A, et al. Hepatic GATA4 regulates cholesterol and triglyceride homeostasis in collaboration with LXRs. Genes Dev. (2022) 36:1129–44. doi: 10.1101/gad.350145.122
98. Horton, JD, Goldstein, JL, and Brown, MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. (2002) 109:1125–31. doi: 10.1172/JCI15593
99. Sorrentino, V, Nelson, JK, Maspero, E, Marques, A, Scheer, L, Polo, S, et al. The LXR-IDOL axis defines a clathrin-, caveolae-, and dy-Namin-independent endocytic route for LDLR internalization and lysosomal degradation. J Lipid Res. (2013) 54:2174–84. doi: 10.1194/jlr.M037713
100. Lee Jason, P, Anne, B, Michael, R, Hooper Joan, E, Drabkin Harry, A, and Gemmill, RM. The TRC8 ubiquitin ligase is sterol regulated and interacts with lipid and protein biosynthetic pathways. Mol Cancer Res. (2010) 8:93–106. doi: 10.1158/1541-7786.MCR-08-0491
101. Loregger, A, Cook, E, Nelson, J, Moeton, M, Sharpe, L, Engberg, S, et al. A MARCH6 and IDOL E3 ubiquitin ligase circuit uncouples cholesterol synthesis from lipoprotein uptake in hepatocytes. Atherosclerosis. (2016) 252:e250. doi: 10.1016/j.atherosclerosis.2016.07.057
102. Zhang, L, Rajbhandari, P, Priest, C, Sandhu, J, Wu, X, Temel, R, et al. Inhibition of cholesterol biosynthesis through RNF145-dependent ubiquitination of SCAP. eLife. (2017) 6:e28766. doi: 10.7554/eLife.28766
103. Bengoechea-Alonso, MT, Aldaalis, A, and Ericsson, J. Loss of the Fbw7 tumor suppressor rewires cholesterol metabolism in cancer cells leading to activation of the PI3K-AKT signalling axis. Front Oncol. (2022) 12:990672. doi: 10.3389/fonc.2022.990672
104. Zheng, ZG, Zhang, X, Liu, XX, Jin, XX, Dai, L, Cheng, HM, et al. Inhibition of HSP90β improves lipid disorders by promoting mature SREBPs degradation via the ubiquitin-proteasome system. Theranostics. (2019) 9:5769–83. doi: 10.7150/thno.36505
105. Kim, JH, Shin, S, Seo, J, Lee, EW, Jeong, M, Lee, MS, et al. C-terminus of HSC70-interacting protein (CHIP) inhibits adipocyte Differen-tiation via ubiquitin- and proteasome-mediated degradation of PPARγ. Sci Rep. (2017) 7:40023. doi: 10.1038/srep40023
106. Li, JJ, Wang, R, Lama, R, Wang, X, Floyd, ZE, Park, EA, et al. Ubiquitin ligase NEDD4 regulates PPARγ stability and adipocyte Dif-ferentiation in 3T3-L1 cells. Sci Rep. (2016) 6:38550. doi: 10.1038/srep38550
107. Liu, J, Yao, Q, Xiao, L, Ma, W, Li, F, Lai, B, et al. PPARγ induces NEDD4 gene expression to promote autophagy and insulin action. FEBS J. (2020) 287:529–45. doi: 10.1111/febs.15042
108. Kamoshita, K, Ishii, KA, Tahira, Y, Kikuchi, A, Abuduwaili, H, Tajima-Shirasaki, N, et al. Insulin suppresses ubiquitination via the deubiquitinating enzyme ubiquitin-specific protease 14, independent of proteasome activity in H4IIEC3 hepatocytes. J Pharmacol Exp Ther. (2023) 385:5–16. doi: 10.1124/jpet.122.001088
109. Liu, B, Jiang, S, Li, M, Xiong, X, Zhu, M, Li, D, et al. Proteome-wide analysis of USP14 substrates revealed its role in hepatosteatosis via stabilization of FASN. Nat Commun. (2018) 9:4770. doi: 10.1038/s41467-018-07185-y
110. Hu, Y, He, W, Huang, Y, Xiang, H, Guo, J, Che, Y, et al. Fatty acid synthase-suppressor screening identifies sorting nexin 8 as a Thera-peutic target for NAFLD. Hepatology. (2021) 74:2508–25. doi: 10.1002/hep.32045
111. Lopez-Castejon, G . Control of the inflammasome by the ubiquitin system. FEBS J. (2020) 287:11–26. doi: 10.1111/febs.15118
112. Cockram, PE, Kist, M, Prakash, S, Chen, SH, Wertz, IE, and Vucic, D. Ubiquitination in the regulation of inflammatory cell death and cancer. Cell Death Differ. (2021) 28:591–605. doi: 10.1038/s41418-020-00708-5
113. Abe, T, Hirasaka, K, and Nikawa, T. Involvement of Cbl-b-mediated macrophage inactivation in insulin resistance. World J Diabetes. (2017) 8:97–103. doi: 10.4239/wjd.v8.i3.97
114. Abe, T, Hirasaka, K, Kohno, S, Ochi, A, Yamagishi, N, Ohno, A, et al. Ubiquitin ligase Cbl-b and obesity-induced insulin resistance. Endocr J. (2014) 61:529–38. doi: 10.1507/endocrj.ej14-0048
115. Kim, TW, Yu, M, Zhou, H, Cui, W, Wang, J, DiCorleto, P, et al. Pellino 2 is critical for toll-like receptor/interleukin-1 receptor (TLR/IL-1R)-mediated post-transcriptional control. J Biol Chem. (2012) 287:25686–95. doi: 10.1074/jbc.M112.352625
116. Yang, S, Wang, B, Humphries, F, Hogan, AE, Shea, D, Moynagh, PN, et al. The E3 ubiquitin ligase Pellino3 protects against obesity-induced inflammation and insulin resistance. Immunity. (2014) 41:973–87. doi: 10.1016/j.immuni.2014.11.013
117. Marino, A, Menghini, R, Fabrizi, M, Casagrande, V, Mavilio, M, Stoehr, R, et al. ITCH deficiency protects from diet-induced obesity. Diabetes. (2014) 63:550–61. doi: 10.2337/db13-0802
118. Yang, M, Wang, C, Zhu, X, Tang, S, Shi, L, Cao, X, et al. E3 ubiquitin ligase CHIP facilitates toll-like receptor signaling by recruiting and polyubiquitinating Src and atypical PKC{zeta}. J Exp Med. (2011) 208:2099–112. doi: 10.1084/jem.20102667
119. Liao, J, Su, X, Wang, M, Jiang, L, Chen, X, Liu, Z, et al. The E3 ubiquitin ligase CHIP protects against sepsis-induced myocardial dysfunc-tion by inhibiting NF-κB-mediated inflammation via promoting ubiquitination and degradation of karyopherin-α 2. Transl Res. (2022) 255:50–65. doi: 10.1016/j.trsl.2022.11.006
120. Collison, A, Hatchwell, L, Verrills, N, Wark, PA, de Siqueira, AP, Tooze, M, et al. The E3 ubiquitin ligase midline 1 promotes allergen and rhinovirus-induced asthma by inhibiting protein phosphatase 2A activity. Nat Med. (2013) 19:232–7. doi: 10.1038/nm.3049
121. Schmitz, ML, Dreute, J, Pfisterer, M, Günther, S, Kracht, M, and Chillappagari, S. SIAH ubiquitin E3 ligases as modulators of inflammatory gene expression. Heliyon. (2022) 8:e09029. doi: 10.1016/j.heliyon.2022.e09029
122. de Carvalho, RM, and Szabo, G. Role of the Inflammasome in liver disease. Annu Rev Pathol. (2022) 17:345–65. doi: 10.1146/annurev-pathmechdis-032521-102529
123. Gan, C, Cai, Q, Tang, C, and Gao, J. Inflammasomes and Pyroptosis of liver cells in liver fibrosis. Front Immunol. (2022) 13:896473. doi: 10.3389/fimmu.2022.896473
124. Song, H, Liu, B, Huai, W, Yu, Z, Wang, W, Zhao, J, et al. The E3 ubiquitin ligase TRIM31 attenuates NLRP3 inflammasome activation by promoting proteasomal degradation of NLRP3. Nat Commun. (2016) 7:13727. doi: 10.1038/ncomms13727
125. Han, S, Lear, TB, Jerome, JA, Rajbhandari, S, Snavely, CA, Gulick, DL, et al. Lipopolysaccharide primes the NALP3 Inflammasome by inhibiting its ubiquitination and degradation mediated by the SCFFBXL2 E3 ligase. J Biol Chem. (2015) 290:18124–33. doi: 10.1074/jbc.M115.645549
126. Jeon, S, Kang, J, and Lee, SB. BC-1215 inhibits ATP-induced IL-1β secretion via the FBXL2-mediated ubiquitination and degradation of not only NLRP3, but also pro-IL-1β in LPS-primed THP-1 cells. Biochem Biophys Res Commun. (2023) 657:128–35. doi: 10.1016/j.bbrc.2023.03.055
127. Mouton-Liger, F, Rosazza, T, Sepulveda-Diaz, J, Ieang, A, Hassoun, SM, Claire, E, et al. Parkin deficiency modulates NLRP3 inflam-masome activation by attenuating an A20-dependent negative feedback loop. Glia. (2018) 66:1736–51. doi: 10.1002/glia.23337
128. Wan, P, Zhang, Q, Liu, W, Jia, Y, Ai, S, Wang, T, et al. Cullin1 binds and promotes NLRP3 ubiquitination to repress systematic inflam-masome activation. FASEB J. (2019) 33:5793–807. doi: 10.1096/fj.201801681R
129. Juliana, C, Fernandes-Alnemri, T, Kang, S, Farias, A, Qin, F, and Alnemri, ES. Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J Biol Chem. (2012) 287:36617–22. doi: 10.1074/jbc.M112.407130
130. Shi, CS, Shenderov, K, Huang, NN, Kabat, J, Abu-Asab, M, Fitzgerald, KA, et al. Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol. (2012) 13:255–63. doi: 10.1038/ni.2215
131. Labbé, K, McIntire, CR, Doiron, K, Leblanc, PM, and Saleh, M. Cellular inhibitors of apoptosis proteins cIAP1 and cIAP2 are required for efficient caspase-1 activation by the inflammasome. Immunity. (2011) 35:897–907. doi: 10.1016/j.immuni.2011.10.016
132. Homma, T, and Fujii, J. Emerging connections between oxidative stress, defective proteolysis, and metabolic diseases. Free Radic Res. (2020) 54:931–46. doi: 10.1080/10715762.2020.1734588
133. Xu, D, Xu, M, Jeong, S, Qian, Y, Wu, H, Xia, Q, et al. The role of Nrf2 in liver disease: novel molecular mechanisms and therapeutic approaches. Front Pharmacol. (2018) 9:1428. doi: 10.3389/fphar.2018.01428
134. Ngo, V, and Duennwald, ML. Nrf2 and oxidative stress: a general overview of mechanisms and implications in human disease. Antioxidants. (2022) 11:2345. doi: 10.3390/antiox11122345
135. Gong, Y, and Yang, Y. Activation of Nrf2/AREs-mediated antioxidant signalling, and suppression of profibrotic TGF-β1/Smad3 pathway: a promising therapeutic strategy for hepatic fibrosis—a review. Life Sci. (2020) 256:117909. doi: 10.1016/j.lfs.2020.117909
136. Suzuki, T, Takahashi, J, and Yamamoto, M. Molecular basis of the KEAP1-NRF2 signaling pathway. Mol Cell. (2023) 46:133–41. doi: 10.14348/molcells.2023.0028
137. Korovila, I, Hugo, M, Castro, JP, Weber, D, Höhn, A, Grune, T, et al. Proteostasis, oxidative stress and aging. Redox Biol. (2017) 13:550–67. doi: 10.1016/j.redox.2017.07.008
138. Ma, Q . Role of nrf2 in oxidative stress and toxicity. Annu Rev PharmacolToxicol. (2013) 53:401–26. doi: 10.1146/annurev-pharmtox-011112-140320
139. Liu, Y, Tao, S, Liao, L, Li, Y, Li, H, Li, Z, et al. TRIM25 promotes the cell survival and growth of hepatocellular carcinoma through tar-geting Keap1-Nrf2 pathway. Nat Commun. (2020) 11:348. doi: 10.1038/s41467-019-14190-2
140. Zhang, J, Wang, X, Vikash, V, Ye, Q, Wu, D, Liu, Y, et al. ROS and ROS-mediated cellular signaling. Oxidative Med Cell Longev. (2016) 2016:4350965. doi: 10.1155/2016/4350965
141. Zgorzynska, E, Dziedzic, B, and Walczewska, A. An overview of the Nrf2/ARE pathway and its role in neurodegenerative diseases. Int J Mol Sci. (2021) 22:9592. doi: 10.3390/ijms22179592
142. Wu, T, Zhao, F, Gao, B, Tan, C, Yagishita, N, Nakajima, T, et al. Hrd1 suppresses Nrf2-mediated cellular protection during liver cirrhosis. Genes Dev. (2014) 28:708–22. doi: 10.1101/gad.238246.114
143. Liu, W, Zhao, Y, Wang, G, Feng, S, Ge, X, Ye, W, et al. TRIM22 inhibits osteosarcoma progression through destabilizing NRF2 and thus activation of ROS/AMPK/mTOR/autophagy signaling. Redox Biol. (2022) 53:102344. doi: 10.1016/j.redox.2022.102344
144. Wang, H, Huang, Q, Xia, J, Cheng, S, Pei, D, Zhang, X, et al. The E3 ligase MIB1 promotes proteasomal degradation of NRF2 and Sensi-tizes lung Cancer cells to Ferroptosis. Mol Cancer Res. (2022) 20:253–64. doi: 10.1158/1541-7786.MCR-21-0342
145. Jena, KK, Kolapalli, SP, Mehto, S, Nath, P, Das, B, Sahoo, PK, et al. TRIM16 controls assembly and degradation of protein aggregates by modulating the p62-NRF2 axis and autophagy. EMBO J. (2018) 37:e98358. doi: 10.15252/embj.201798358
146. Da, C, Pu, J, Liu, Z, Wei, J, Qu, Y, Wu, Y, et al. HACE1-mediated NRF2 activation causes enhanced malignant phenotypes and decreased radiosensitivity of glioma cells. Signal Transduct Target Ther. (2021) 6:399. doi: 10.1038/s41392-021-00793-z
147. Zhang, Q, Zhang, ZY, Du, H, Li, SZ, Tu, R, Jia, YF, et al. DUB3 deubiquitinates and stabilizes NRF2 in chemotherapy resistance of Colo-rectal cancer. Cell Death Differ. (2019) 26:2300–13. doi: 10.1038/s41418-019-0303-z
148. Meng, C, Zhan, J, Chen, D, Shao, G, Zhang, H, Gu, W, et al. The deubiquitinase USP11 regulates cell proliferation and ferroptotic cell death via stabilization of NRF2 USP11 deubiquitinates and stabilizes NRF2. Oncogene. (2021) 40:1706–20. doi: 10.1038/s41388-021-01660-5
149. Liang, M, Wang, L, Sun, Z, Chen, X, Wang, H, Qin, L, et al. E3 ligase TRIM15 facilitates non-small cell lung cancer progression through mediating Keap1-Nrf2 signaling pathway. Cell Commun Signal. (2022) 20:62. doi: 10.1186/s12964-022-00875-7
150. Sakurai, Y, Kubota, N, Yamauchi, T, and Kadowaki, T. Role of insulin resistance in MAFLD. Int J Mol Sci. (2021) 22:22. doi: 10.3390/ijms22084156
151. Yang, XD, Xiang, DX, and Yang, YY. Role of E3 ubiquitin ligases in insulin resistance. Diabet Obes Metab. (2016) 18:747–54. doi: 10.1111/dom.12677
152. Ishii, M, Maeda, A, Tani, S, and Akagawa, M. Palmitate induces insulin resistance in human HepG2 hepatocytes by enhancing ubiquitination and proteasomal degradation of key insulin signaling molecules. Arch Biochem Biophys. (2015) 566:26–35. doi: 10.1016/j.abb.2014.12.009
153. Mukherjee, S, Chakraborty, M, Msengi, EN, Haubner, J, Zhang, J, Jellinek, MJ, et al. Ube4A maintains metabolic homeostasis and facili-tates insulin signaling in vivo. Mol Metab. (2023) 75:101767. doi: 10.1016/j.molmet.2023.101767
154. Philouze, C, Turban, S, Cremers, B, Caliez, A, Lamarche, G, Bernard, C, et al. MG53 is not a critical regulator of insulin signaling pathway in skeletal muscle. PLoS One. (2021) 16:e0245179. doi: 10.1371/journal.pone.0245179
155. Scheufele, F, Wolf, B, Kruse, M, Hartmann, T, Lempart, J, Mühlich, S, et al. Evidence for a regulatory role of Cullin-RING E3 ubiquitin ligase 7 in insulin signaling. Cell Signal. (2014) 26:233–9. doi: 10.1016/j.cellsig.2013.11.005
156. Bonala, S, Lokireddy, S, McFarlane, C, Patnam, S, Sharma, M, and Kambadur, R. Myostatin induces insulin resistance via casitas B-lineage lymphoma b (Cblb)-mediated degradation of insulin receptor substrate 1 (IRS1) protein in response to high calorie diet intake. J Biol Chem. (2014) 289:7654–70. doi: 10.1074/jbc.M113.529925
157. Nikawa, T, and Ishidoh, K. Ubiquitin ligase Cbl-b and inhibitory Cblin peptides. Biochim Biophys Acta Proteins Proteom. (2020) 1868:140495. doi: 10.1016/j.bbapap.2020.140495
158. Dumpati, R, Ramatenki, V, Vadija, R, Vellanki, S, and Vuruputuri, U. Structural insights into suppressor of cytokine signaling 1 protein-iden-tification of new leads for type 2 diabetes mellitus. J Mol Recognit. (2018) 31:e2706. doi: 10.1002/jmr.2706
159. Zou, Y, Li, Z, Zou, Y, Hao, H, Li, N, and Li, Q. An FBXO40 knockout generated by CRISPR/Cas9 causes muscle hypertrophy in pigs without detectable pathological effects. Biochem Biophys Res Commun. (2018) 498:940–5. doi: 10.1016/j.bbrc.2018.03.085
160. Zhao, W, Xu, Q, Yang, J, Xie, X, Li, C, Zhang, W, et al. Murine double minute 2 aggravates adipose tissue dysfunction through ubiqui-tin-mediated six-transmembrane epithelial antigen of prostate 4 degradation. iScience. (2022) 25:104544. doi: 10.1016/j.isci.2022.104544
161. Jing, LJ, Ferry, RJ, Diao, S, Xue, B, Bahouth, SW, and Liao, FF. Nedd4 haploinsufficient mice display moderate insulin resistance, enhanced lipolysis, and protection against high-fat diet-induced obesity. Endocrinology. (2015) 156:1283–91. doi: 10.1210/en.2014-1909
162. Zhao, YY, Wu, DM, He, M, Zhang, F, Zhang, T, Liu, T, et al. Samotolisib attenuates acute liver injury through inhibiting Caspa-se-11-mediated Pyroptosis via regulating E3 ubiquitin ligase Nedd4. Front Pharmacol. (2021) 12:726198. doi: 10.3389/fphar.2021.726198
163. Cohen, S, Lee, D, Zhai, B, Gygi, SP, and Goldberg, AL. Trim32 reduces PI3K-Akt-FoxO signaling in muscle atrophy by promoting plakoglo-bin-PI3K dissociation. J Cell Biol. (2014) 204:747–58. doi: 10.1083/jcb.201304167
164. Bjørklund, G, and Chirumbolo, S. Role of oxidative stress and antioxidants in daily nutrition and human health. Nutrition. (2017) 33:311–21. doi: 10.1016/j.nut.2016.07.018
Keywords: polyphenols, metabolic disorders, ubiquitin-proteasome system, nonalcoholic fatty liver disease, lipid metabolism, inflammation
Citation: Gu W, Wu G, Chen G, Meng X, Xie Z and Cai S (2024) Polyphenols alleviate metabolic disorders: the role of ubiquitin-proteasome system. Front. Nutr. 11:1445080. doi: 10.3389/fnut.2024.1445080
Edited by:
Omar Guzmán Quevedo, Higher Technological Institute of Tacambaro, MexicoReviewed by:
Jagannath Misra, Purdue University Indianapolis, United StatesKaijun Wang, Hunan Agricultural University, China
Copyright © 2024 Gu, Wu, Chen, Meng, Xie and Cai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhongwen Xie, emhvbmd3ZW54aWVAYWhhdS5lZHUuY24=; Shanbao Cai, c2Jjc2JjQHVzdGMuZWR1LmNu
†These authors have contributed equally to this work