 Guido K. W. Frank
Guido K. W. Frank Barbara Scolnick
Barbara Scolnick- 1Department of Psychiatry, San Diego School of Medicine, University of California, San Diego, San Diego, CA, United States
- 2Boston University, Boston, MA, United States
Anorexia nervosa (AN) is a severe psychiatric disorder. However, we lack neurobiological models and interventions to explain and treat the core characteristics of food restriction, feeling fat, and body size overestimation. Research has made progress in understanding brain function involved in the pathophysiology of AN, but translating those results into biological therapies has been challenging. Studies have suggested that metabolic factors could contribute to developing and maintaining AN pathophysiology. Here, we describe a neurobiological model for why using a therapeutic ketogenic diet could address key alterations in brain function in AN and prevent the desire for weight loss and associated eating disorder-specific symptoms. This translational model is based on animal studies and human data and integrates behavioral traits, brain neural energy metabolism, and neurotransmitter function. Pilot data indicate that the intervention can dramatically reduce eating and body-related fears, although larger studies across illness stages still need to be conducted.
Introduction
Anorexia nervosa (AN) is a severe psychiatric illness characterized by food avoidance, severe emaciation, and a perception of being overweight despite a very low body weight (1). AN is a chronic disorder with frequent relapse, high disease burden, and treatment cost (2–6). Treatment effectiveness, however, is limited (7, 8). AN has a mortality rate twelve times higher than the death rate from all causes of death for females 15–24 years old (5, 6, 9). AN shows a complex interplay between neurobiological, psychological, and environmental factors, and little is known about the pathophysiology or biomarkers that characterize AN when underweight or weight recovered (10, 11). Notably, in individuals with AN after weight recovery (wrAN), fears of weight gain, body dissatisfaction, and body image distortion are often elevated similarly or even higher compared to the underweight state, can persist for many years, and pose a risk for renewed self-starvation and relapse (4–6, 12–15).
Brain research before and after weight restoration has indicated alterations in circuits that compute reward valence or motivational salience of stimuli (stimuli that propel an individual’s behavior toward or away from a particular stimulus), which, together with a conditioned fear of weight gain, may contribute to the vicious cycle of self-starvation (2, 16, 17). The neural basis of what drives self-starvation remains poorly understood (18). A better mechanistic understanding of what triggers and perpetuates core behaviors such as food restriction neurobiologically in AN and wrAN, besides an environmentally driven ideal of thinness, would help develop more effective treatments (11). However, important aspects of the AN pathophysiology are still largely not yet identified, and there is a lack of biological treatments available for AN when underweight, or relapse prevention after weight restoration, despite decades of brain research. In this hypothesis and theory article, we propose a neurobiological model based on animal studies and human pilot data and integrates behavioral traits, brain neural energy metabolism, and neurotransmitter function, supporting a therapeutic ketogenic diet in AN.
Neurobiology of AN
Brain research from various groups over the past decades has provided empirical data to better understand symptoms and behaviors in AN. Studies have found changes in the neurotransmitter systems for serotonin and dopamine in AN (8), altered brain structure, and more recently, tasks that engaged specific brain circuits that separated AN from healthy control groups (19–22). Those latter studies found evidence that AN is associated with altered brain function for processes involving the reward circuitry, cognition, emotion-regulating pathways, and regions that process executive function (21, 23, 24). Studies from our group, for instance, suggested that food avoidance and weight loss in AN alter circuits that process motivational salience (what stimuli to approach or avoid) and reward valence (whether expectations are violated) and involve cortical and subcortical regions that affect appetitive drive (16, 19, 25–33).
The primary brain regions that compute motivational salience and reward valence include the ventral striatum, insula, and orbitofrontal cortex (34). Another region, the amygdala, acts as an “early integrating” brain region for the salience of stimuli. It responds to expectation and triggers the dopamine response and associated behaviors in the nucleus accumbens and ventral striatum (35–37). Importantly, the frontal cortex processes whether an individual feels safe or whether a situation is associated with threat and fear, which activates those dopaminergic circuits to drive either approach or dread and avoidance (38, 39). Those mechanisms apply to AN, where fear of weight gain causes food and eating to become conditioned fear-inducing stimuli, leading to negative ruminations and ambiance (40, 41). The amygdala, in turn, responds to those fear and anxiety-inducing stimuli, modulates the dopaminergic motivational salience response, and triggers food avoidance (36, 42–44).
Individuals with AN overestimate their body size even when they are thin or underweight. Environmental factors, including thin-body messages from the media, may trigger those thoughts and condition a fear response (45–47). A “multisensory impairment of body perception” was proposed in some studies (48–50), but others suggested that affective factors drive body size overestimation in AN instead (50–53). Interestingly, some studies raised the possibility of a frontal cortical cognitive dysfunction in AN (54), and especially in restricting type AN, a psychotic-delusional component to the overvalued ideas of thinness could drive body size overestimation (55, 56). Those overvalued ideas may be driven by diminished sensory processing, which has also been associated with altered dopamine reward prediction error response (57). Brain dopamine activity is tied to glucose utilization, linking brain energy metabolism to a likely central aspect of AN neurobiology (57–59).
Anxious traits and stress may affect brain metabolism in AN
Negative affect and deficits in regulating emotions, together with elevated anxious traits, are considered important for the etiology of AN (60–62). Unpleasant feelings such as anxiety, sadness, fear, or anger contribute to the negative affect experience and may drive AN behaviors (63). Worry, for instance, was related to fasting and fear of gaining weight or becoming fat in a sample of individuals with AN or wrAN (64). Stress and resulting negative affect may thus drive negative body image and body size overestimation (65, 66), consistent with ecological momentary assessment research showing that negative affect is involved in maintaining restrictive eating across AN subtypes (67–69). In wrAN, negative affect assessed over 2 weeks was related to a self-perception of inefficiency (70). Fear predicted increased dietary restraint, whereas dietary restraint predicted increased guilt and hostility (71). The importance of negative affect has been recognized already in youth with AN, and negative affect may contribute to developing AN (41, 72). Furthermore, difficulties with negative affect and emotion regulation persist long into recovery and may pose significant long-term risk factors for relapse (73).
It has long been known that stress affects glucose metabolism, altering blood sugar levels (74). Recent research has refined those studies. For instance, animal studies found that 40% of mice had a stress-susceptible phenotype associated with elevated blood glucose but reduced brain glucose metabolism, suggesting a specific mechanism in susceptible individuals (75). It was subsequently hypothesized that stress-related disorders could be associated with altered glucose metabolism (76). Research in humans indicated, for instance, altered brain glucose metabolism after acute stress using the Trier Social Stress Test (77), or in individuals with a chronic stress condition, posttraumatic stress disorder, showing lower brain glucose metabolism after administration of the stress hormone hydrocortisone (78). A recent study in cancer patients showed that negative affect was negatively correlated with glucose metabolism across cortical and subcortical brain regions, indicating global effects (79).
Those studies may have direct implications for the pathophysiology of AN or wrAN as self-perceived stress and sensitivity to negative affect may create a condition of chronic stress affecting brain glucose metabolism. This hypothesis is supported by studies that showed elevated baseline levels of the stress hormone cortisol in AN, altered cortisol response to a stressor compared to controls, and altered stress axis response that persisted in wrAN (80). That research directly associated stress and “psychological burden” with the biological stress response in AN, including stress axis function.
It has previously been hypothesized that the pathophysiology of AN may include metabolic abnormalities, or AN may be a “metabolic disorder of psychological origin” (81), which has only recently again received attention (82, 83). We propose, based on the above-described literature, that elevated anxious traits and distress over negative affect in individuals prone to AN interfere with brain glucose utilization and, thus, normal brain metabolism and function (84, 85).
AN as a metabolic disorder
Several studies have found metabolic abnormalities in AN (81, 82) and associated elevated oxidative stress and inflammatory markers (86, 87). Recent genetic studies suggested that AN is associated with metabolic traits as a possible risk factor for developing the condition (88). A model that integrated biological and environmental factors hypothesized that anxiety and stress are critical factors in AN that drive altered glucose distribution between brain and body and induce changes in the lateral hypothalamus-pituitary–adrenal axis (89). That model is consistent with behavioral studies that indicated elevated intolerance of uncertainty and anxiety in AN, triggering the body’s biological stress response (90, 91). Stress leads to an increased allocation of glucose to the brain; however, while stress is associated with higher glucose needs, it is also associated with decreased glucose brain utilization, particularly in frontal cortical, thalamic, hippocampal, and temporal regions (85, 92, 93). In some individuals, this increased glucose requirement may deplete body energy resources; in others, reduced glucose utilization in the context of stress may lead to overeating (89, 90, 94). This model has been hypothesized to be relevant for psychiatric disorders, and computational models support the underlying premise (95, 96).
Research on the metabolic underpinnings of AN is still largely lacking. An animal study that evaluated glucose metabolism in nine female rats modeled AN brain response and randomized the animals to food restriction or regular food access (97). That study suggested increased glucose metabolism in the cerebellum but decreased metabolism in the hippocampus and striatum after food restriction. A few genetic studies suggested altered energy, including glucose homeostasis in AN (83, 98–101), and AN has been associated with altered glucose metabolism related to changes in insulin sensitivity (99). Others found mitochondrial dysfunction in AN related to oxidative stress and altered metabolic signaling (102), or metabolic dysfunction in the gut microbiome in AN that was opposite to that found in high-weight individuals (83). Those data, together with genetic markers, supported the possibility of critical metabolic targets that need to be identified for successful treatment development for AN (83). Only a few human brain imaging studies have investigated glucose metabolism in AN compared to healthy controls, and the samples were generally small and results inconsistent. One earlier study found glucose hypometabolism in AN (n = 10) that tended to normalize with weight gain (103), while another found glucose hypermetabolism in a small sample (n = 5) in cortical and subcortical regions (104). Another study in underweight AN (n = 14) found lower regional glucose metabolism in the anterior and posterior cingulate, dorsolateral prefrontal cortex, left middle temporal, and right superior temporal gyrus (105). After hormone replacement in six individuals with AN, regional glucose metabolism normalized in the anterior and posterior cingulate, premotor, parietal cortex, and caudate nucleus. A study that investigated brain glucose metabolism in AN before and after nucleus accumbens brain stimulation found increased regional glucose metabolism in AN (n = 6) versus healthy controls (n = 12) in the bilateral superior, medial and inferior frontal cortex, bilateral amygdala and hippocampus, left insula and bilateral putamen (106). The authors further reported that frontal cortical and hippocampal hypermetabolism decreased with nucleus accumbens stimulation.
Overall, the human neuroimaging results are mixed. The studies, in part, are decades old, and potential confounds such as nutritional status or comorbidity were not considered. Glucose metabolism has been linked to the neurotransmitters dopamine and serotonin, and altered neurotransmitter function found in AN as described above further supports the hypothesis of altered energy homeostasis, including glucose metabolism in AN (107, 108).
We propose that in individuals who have a predisposition for AN, there is reduced utilization of glucose in the brain despite high needs due to high anxious traits and sensitivity to stress and negative affect, which interfere with brain glucose metabolism (Figure 1) (91, 109–114).
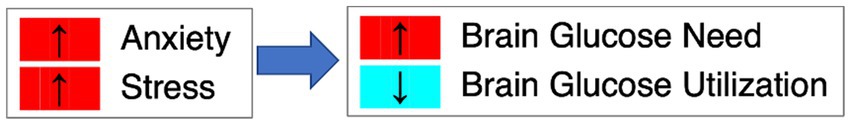
Figure 1. Anxiety, stress and brain glucose metabolism. We hypothesize that in AN, high anxiety and stress lead to elevated brain glucose need but reduced glucose utilization.
While there is evidence that the vicious cycle of weight loss followed by more food restriction is, at least in part, due to the interactions between conditioned fear of weight gain and altered dopamine circuit function, which may drive dread and avoidance (16), we propose that there are fundamental metabolic abnormalities in individuals who develop AN that drive the development of the illness, hinder recovery from AN and trigger relapse in wrAN. Individuals with AN often report that food restriction reduces anxiety and improves mood (115). This may be due to cognitive and emotional factors where weight loss is seen as a success toward a certain body shape or in part due to elevated cortisol release, although the results are mixed (116–119). Here, we postulate that individuals with AN may have a metabolic reason why it is so desirable for them to pursue the starvation state. Individuals who develop AN tend to score high on state and trait anxiety and have perfectionistic traits that drive fear of failure and anxiety (28, 42, 120–124).
We hypothesize that high state and trait anxiety levels create ongoing interference with brain glucose utilization in AN as a risk factor before, during, or after weight loss. If a person with that disposition loses weight and enters a ketosis state, the brain will use ketones as an alternative energy source that may be less affected by anxiety. Thus, the individual learns that starvation paradoxically provides a better subjective feeling of having sufficient energy, and food restriction becomes self-reinforcing. However, this state also depletes the body’s resources and eventually leads to death. We propose that providing a person with that disposition with ketone bodies while ensuring normal weight will remove the desire to self-starve and support weight maintenance.
The therapeutic ketogenic diet provides an alternative energy source to reduce anxiety and normalize inflammation
TKD is beneficial in neurological conditions such as seizure disorders, and there is emerging evidence that it might also be an effective intervention to treat psychiatric conditions (125–127).
TKD mimics some aspects of fasting (128). During fasting, the body faces an energy deficit in its cellular fuel supply as glucose and insulin levels decrease. The metabolism of white fat increases, and the resulting fatty acids are used to supplement the energy needs of most organs. The brain is unique, however, because fatty acids cannot easily cross the protective blood–brain barrier (129), and thus, the brain is highly sensitive to drops in glucose. For decades, it was thought the brain could only utilize glucose (130). This presented an enigma because human glucose stores are limited and can be sustained for only a few days. Yet, there have been many documented instances where individuals are capable of fasting (as long as they drink water) for several weeks before succumbing. This paradox was explained in an experiment performed on three obese patients who were starved for 6 weeks in a metabolic ward, and their internal carotid arteries and jugular veins were cannulated at the start and end of the experiment (131). The study demonstrated that beta-hydroxybutyrate and acetoacetate replaced glucose as the predominant fuel for brain metabolism.
The underlying principle of TKD is that most energy is supplied via fat in the diet, which is then broken down into fatty acids for energy consumption (132). However, sufficient calories are in the food to maintain normal weight. During fasting or TKD, fat metabolism increases, and fatty acids are transported to the liver. Fatty acids are composed of long chains of carbons. In the liver, fatty acids are ordinarily converted into acetyl-CoA, which enters the tricarboxylic acid (TCA) cycle. When fatty acid levels are elevated and exceed the metabolic capacity of the TCA cycle, acetyl-CoA is shunted to ketogenesis. Two acetyl-CoAs can combine through a thiolase enzyme to produce acetoacetyl-CoA, a precursor for acetoacetate synthesis (ACA) and β-hydroxybutyrate (BHB). Acetone, the other major ketone body, is produced primarily from spontaneous decarboxylation of ACA and can be eliminated as a volatile substrate through the lungs and kidneys. In the blood, ACA and BHB are transported from the vascular lumen to the brain interstitial space and both glia and neurons by monocarboxylic acid transporters (MCTs). MCT-1 is the principal carrier localized to the vascular endothelium. Within neurons, both ACA and BHB are transported directly into mitochondria and then converted to acetyl-CoA through several enzymatic steps. BHB is converted to ACA through D-β-hydroxybutyrate dehydrogenase, and ACA undergoes subsequent conversion to acetoacetyl-CoA through a succinyl-CoA transferase enzyme. Finally, acetoacetyl-CoA-thiolase converts acetoacetyl-CoA to two acetyl-CoA moieties, which then enter the TCA cycle (132).
TKD or exogenous ketones have been associated with marked changes in brain glucose metabolism. Specifically, elevated blood ketone levels resulted in lower brain glucose uptake in humans, which was studied using the radiotracer [18F]fluorodeoxyglucose ([18F]FDG) and positron emission tomography (PET). In one study, infusion of BHB ketone bodies reduced brain glucose uptake and enhanced blood flow, supporting the notion of TKD’s neuroprotective effects (133). A study that briefly applied a ketogenic diet found regionally specific effects of blood ketosis on lowering brain glucose uptake (134), including the precuneus, a brain region necessary for visuospatial function, episodic memory retrieval, and self-referential processing, affecting one’s perceptual image or mental concept of oneself (135), which could have implications for AN in the pathophysiology of body image distortion (136, 137). The ketogenic diet in that study was maintained only for 48 h, and results may differ after prolonged ketosis as in this study (134). A study testing the effects of ketogenic diet over 4 days led to global decreases in glucose metabolism across widespread cortical and subcortical regions, with the strongest decrease in the middle frontal gyrus (Brodmann area 8, 46) followed by the frontal pole (Brodmann area 10) and cuneus (Brodmann area 17) (138). Three weeks of a ketogenic diet in an animal model also led to widespread cortical reductions in glucose metabolism (139). Thus, short and longer-term ketogenic diets led to extensive regional glucose metabolism reductions, with longer duration associated with more extensive glucose metabolism decreases. The above-referenced study by Courchesne-Loyer et al. (138) suggests that the middle frontal cortex is most affected. However, there were large reductions across all frontal, temporal, parietal, occipital, cingulate, and subcortical regions tested. Those data indicate large global decreases without specific circuits delineated, although some areas were more affected than others (138).
The metabolic shift with TKD is associated with a variety of central nervous system and general effects on the body. Aside from the ketone bodies enhancing cell energy metabolism by replenishing the metabolic pathway, TKD has been associated with reducing oxidative stress and inflammatory processes and regulating neurotransmitter systems (140–142), which are all processes implicated in the pathophysiology of AN (21, 23, 86, 87). Furthermore, replacing glucose with ketone bodies via TKD to supply the brain with energy enhances γ-aminobutyric acid (GABA) in the brain via enhanced glutamate production converted to glutamine and GABA (143). GABA is a primary inhibitory neurotransmitter that reduces anxiety (144, 145). In the animal model, enhancing systemic ketone body levels reduced stress and anxiety (146, 147). In AN, altered GABA function has been reported in an animal model for AN, and enhancing GABA via ketosis might effectively reduce AN-specific and non-specific anxiety (148, 149). Other studies have found elevated inflammatory markers in AN, and elevating blood ketone levels has been shown to reduce inflammation (150).
Metabolism as neurobiological target in AN
It has been hypothesized that brain metabolic alterations, perhaps relating to cell mitochondria, have a critical role in psychiatric disorders (151, 152). Psychiatric disorders have been associated with inborn errors of metabolism, supporting the link between altered metabolism and psychiatric pathophysiology (153). Research has also increasingly recognized other abnormalities associated with psychiatric conditions, such as elevated inflammatory markers and markers for oxidative stress (154–156). It has been hypothesized that AN’s pathophysiology includes metabolic abnormalities (82), or that AN may be a “metabolic disorder of psychological origin” (81). Thus, there is growing evidence that nutrition and mental health are linked, and diet may be particularly appealing as a therapeutic intervention (157).
TKD has received increasing attention since it was found effective in pediatric epilepsy (158). Others have suggested that TKD could improve autism-related behaviors, symptoms associated with Alzheimer’s disease, or disorders related to mood or psychotic symptoms (159–161). These findings have led to the suggestion that TKD could be an effective metabolism-directed intervention for psychiatric conditions (162, 163).
A neurobiological model for TKD as a possible treatment intervention for AN
The TKD may be an effective treatment intervention for AN to normalize energy homeostasis and remove the need to self-starve for nutritional ketosis. Figure 2 provides a conceptual model of dysfunctional brain glucose metabolism and the therapeutic effects of TKD (85, 93, 132, 147, 164). (1) At baseline and under typical conditions, glucose is used by the brain mitochondria to generate energy and support brain function and associated behaviors. (2) In individuals with AN, high levels of anxiety and perfectionism lead to stress, which reduces glucose utilization despite high energy needs. (3) Body dissatisfaction and drive for thinness in susceptible individuals drive starvation. Transient imbalances between nutritional intake and energy requirements lead to the generation of ketones and the use of beta-hydroxybutyrate (BHB), acetone, and acetoacetate (ACA) as alternative energy compounds that enter the brain mitochondrial Krebs cycle and are better utilized than glucose and independent from the effects of stress. (4) Ketosis leads to elevated production of neuronal GABA via glutamine and glutamate, which may help with emotion regulation and reduce anxiety. (5) Ketosis leads to improved brain energy supply and elevated GABA production, which stabilizes neuronal function and causes positive feedback to promote further starvation-mediated ketosis. (6) A ketogenic diet that is energy-rich to accomplish weight maintenance in wrAN or weight gain in AN eliminates the need for ketosis via starvation, thus replacing “starvation ketosis” with “nutritional ketosis” (85, 93, 132, 147, 164).
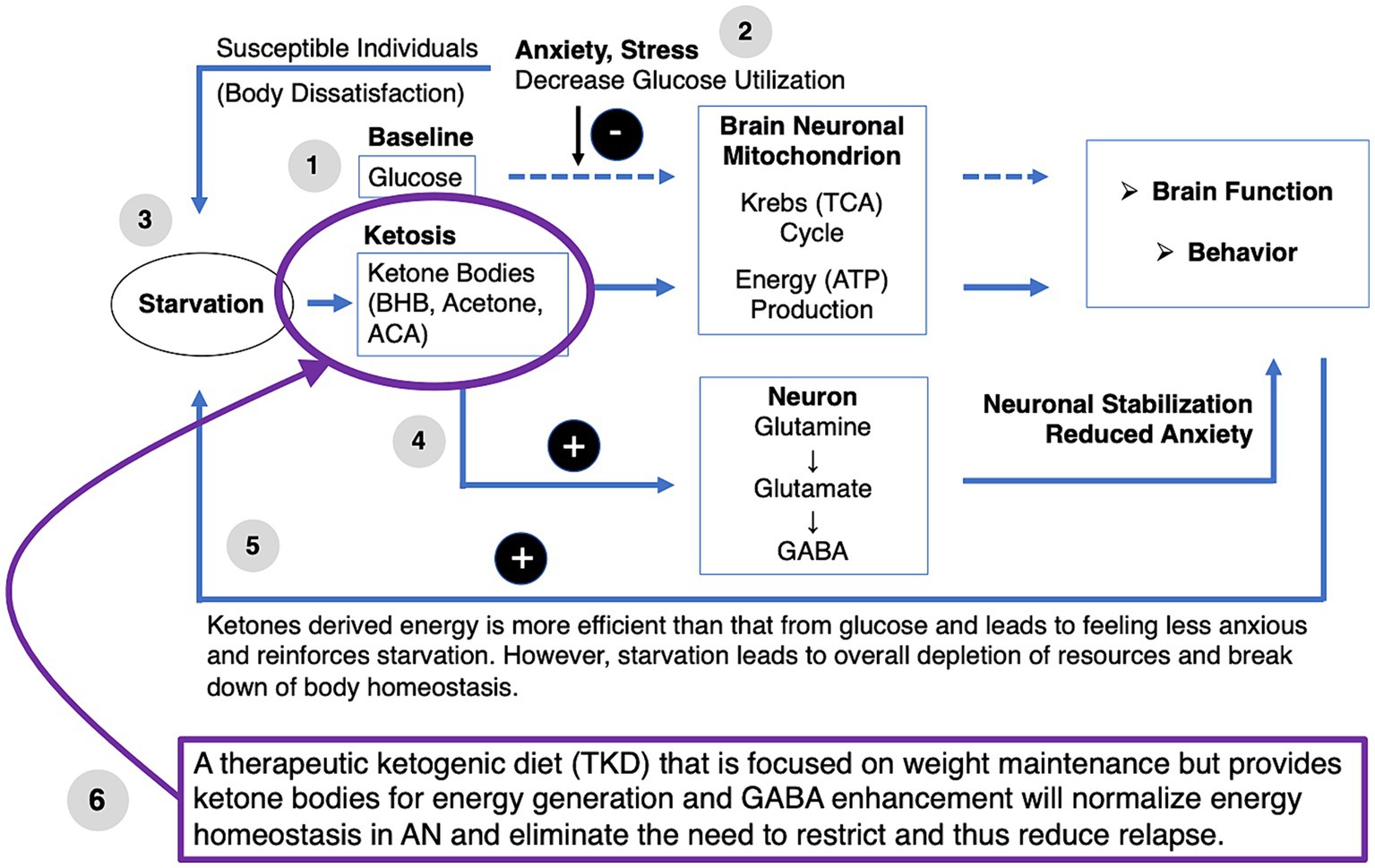
Figure 2. Therapeutic ketogenic diet in anorexie nervosa conceptual model. ACA, acetoacetate; BHB, beta-hydroxybutyrate; TCA, tricarboxylic acid cycle.
Supporting preliminary studies
A single case study suggested that TKD, followed by ketamine infusion, could help that patient recover (165). That individual remains recovered to this date (Dr. Scolnick, personal communications). That study laid the foundation for an IRB-approved protocol in five wrAN who were still highly affected by the illness (166). In that study, we conducted an open-label trial to test whether the case report response could be replicated. Those five wrAN adults with persistent eating disorder thoughts and behaviors adopted the TKD to maintain weight. In addition, participants received six ketamine infusions after 4 to 8 weeks of stable ketosis and were followed over six months. All participants completed the study protocol without significant adverse effects. The participants consumed the TKD for at least 8 weeks (4 to 8 weeks TKD alone, then with added ketamine for 4 weeks); two individuals continued TKD after the formal study intervention for a total of 4 months on TKD and two individuals for 6 months of TKD, suggesting good tolerability. The group showed significant improvements (repeated measures ANOVA) on the Clinical Impairment Assessment (p = 0.008), Eating Disorder Examination Questionnaire (EDEQ) Global score (p = 0.006), EDEQ-Eating Concerns (p = 0.005), EDEQ-Shape Concerns (p = 0.016), EDEQ-Weight Concerns (p = 0.032), Eating Disorders Recovery Questionnaire (EDRQ) Acceptance of Self and Body (0.027) and EDRQ-Social and Emotional Connection (p = 0.001). Weight remained stable during the trial. Figure 3 shows a change in composite scores and BMI over time. The baseline was at “0”; time point one indicates 4 weeks of TKD for all participants, timepoint 2 indicates 8 weeks of TKD for two subjects, and 4 weeks of TKD plus ketamine in 3 subjects; time points three and later are post study intervention assessments. EDEQ global score, Restraint, Eating Concern, Weight Concern, Acceptance of Self and Body, and Clinical Impairment showed steep improvements before adding ketamine, suggesting that TKD alone was highly effective.
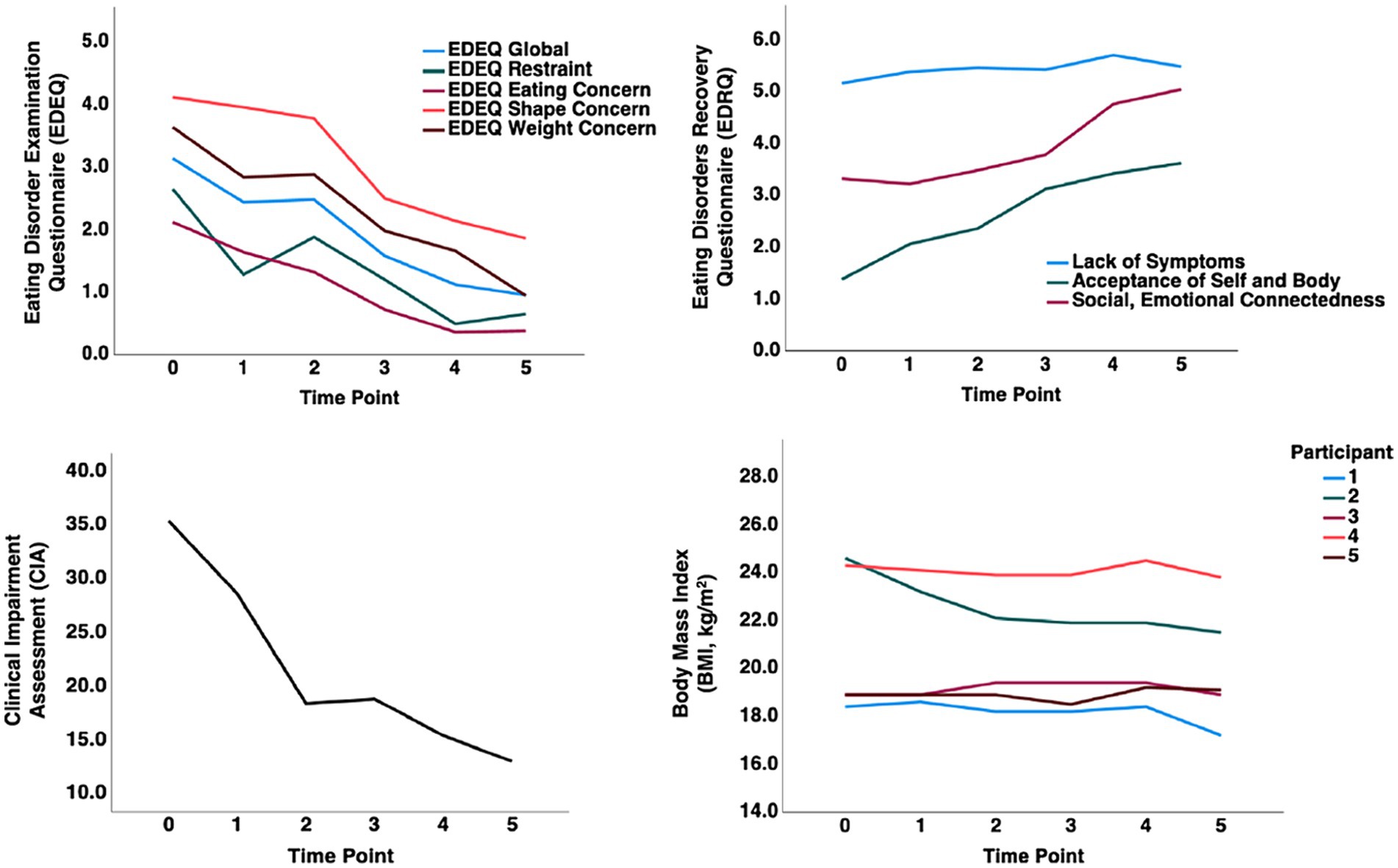
Figure 3. A pilot study showing therapeutic ketogenic diet and ketamine infusion effects in the treatment of chronic persistent eating disorder psychopathology in anorexia nervosa [Calabrese et al. (166), with permission]. Graphical representation of composite values for behavioral measures and body mass index (BMI) over time. Timepoint (TP) 0: Baseline, TP 1: 4 weeks TKD all subjects; TP 2: 8 weeks TKD (subjects 3, 4) or 4 weeks TKD + 4 weeks ketamine (subjects 1,2,5); TP 3: 1-month post-intervention; TP 4: 3 months post-intervention; TP 5: 6 months post-intervention.
Of note, in the case report and the case series (165, 166), ketamine infusions were added once the subjects had been on the TKD for at least 4 weeks. The choice was clinically driven and based on a small positive report of decreased obsessions/compulsions in a pilot study of twelve patients with AN (167). Ketamine is an N-methyl D-aspartate (NMDA) glutamate antagonist that has been in use since the 1960s as an anesthetic (168). Over the past two decades, ketamine has been in use as a rapid antidepressant agent (169). There have been accelerating efforts to discern the mechanism of action and focus on the effects of ketamine on energy metabolism, mitochondrial function, and glutamate/GABA function (170, 171). All these areas overlap with the effects of TKD, which corresponded to our clinical observation that the two modalities (TKD and ketamine) led to extended improvement. This warrants further study.
Four of the 5 study participants have remained recovered (low symptom scores, normal weight) for at least 12 months since the end of the study; one participant who stopped TKD after 8 weeks relapsed 4 months after treatment (unpublished data). The small study suggested that this novel treatment is safe and effective for wrAN adults with chronic AN-related psychopathology. The results from this open trial supported the idea that specific neurobiological underpinnings for AN can be modified with TKD.
Discussion
AN remains a severe psychiatric disorder without approved biological intervention. The above neurobiological model, with evidence from basic science and human genetic and preliminary clinical data, supports the possibility that brain metabolism may be a key target for intervention to treat this disorder and provide a treatment that targets the disorder’s pathophysiology mechanistically. The pilot data to date are from weight recovered individuals. We are currently conducting a follow-up study in a larger group of individuals in the wrAN group to test for reduction of thoughts, feelings, and behaviors that are specific to AN. Future studies will need to investigate individuals underweight with AN when we have further indications that this treatment is safe and effective. An important aspect to also consider is that while we can change neurobiology and provide effective treatment, individuals with AN have often learned to live with the disorder. Once those thoughts, feelings, and behaviors are diminishing, the individual has to re-organize their life, which can be anxiety-provoking in itself. In summary, there is much reason to believe that a TKD could support treatment outcomes in AN, and further study is needed to understand the underlying mechanisms in vivo and in relation to specific illness behaviors.
Author contributions
GF: Conceptualization, Writing – original draft, Writing – review & editing, Funding acquisition, Supervision. BS: Writing – original draft, Writing – review & editing, Conceptualization.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. Baszucki Family Foundation provided support for therapeutic ketogenic diet clinical research in anorexia nervosa; NIMH provided support for research on therapeutic ketogenic diet underlying brain metabolism.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. American Psychiatric Association. Desk reference to the diagnostic criteria from DSM-5-TR Washington, DC: American Psychiatric Association Publishing (2022).
2. Khalsa, SS, Portnoff, LC, McCurdy-McKinnon, D, and Feusner, JD. What happens after treatment? A systematic review of relapse, remission, and recovery in anorexia nervosa. J Eat Disord. (2017) 5:20. doi: 10.1186/s40337-017-0145-3
3. Hay, P, Mitchison, D, Collado, AEL, Gonzalez-Chica, DA, Stocks, N, and Touyz, S. Burden and health-related quality of life of eating disorders, including avoidant/restrictive food intake disorder (ARFID), in the Australian population. J Eat Disord. (2017) 5:21. doi: 10.1186/s40337-017-0149-z
4. Golden, NH. Eating disorders in adolescence and their sequelae. Best Pract Res Clin Obstet Gynaecol. (2003) 17:57–73. doi: 10.1053/ybeog.2003.0344
5. Arcelus, J, Mitchell, AJ, Wales, J, and Nielsen, S. Mortality rates in patients with anorexia nervosa and other eating disorders. A meta-analysis of 36 studies. Meta-analysis. Arch Gen Psychiatry. (2011) 68:724–31. doi: 10.1001/archgenpsychiatry.2011.74
6. Sullivan, PF. Mortality in anorexia nervosa. Am J Psychiatry. (1995) 152:1073–4. doi: 10.1176/ajp.152.7.1073
7. Powers, PS, and Bruty, H. Pharmacotherapy for eating disorders and obesity. Child Adolesc Psychiatr Clin N Am. (2009) 18:175–87. doi: 10.1016/j.chc.2008.07.009
8. Bulik, CM. Exploring the gene-environment nexus in eating disorders. J Psychiatry Neurosci. (2005) 30:335–9.
9. Rosling, AM, Sparen, P, Norring, C, and von Knorring, AL. Mortality of eating disorders: a follow-up study of treatment in a specialist unit 1974–2000. Int J Eat Disord. (2011) 44:304–10. doi: 10.1002/eat.20827
10. Bulik, CM, Hebebrand, J, Keski-Rahkonen, A, Klump, KL, Reichborn-Kjennerud, T, Mazzeo, SE, et al. Genetic epidemiology, endophenotypes, and eating disorder classification. Int J Eat Disord. (2007) 40:S52–60. doi: 10.1002/eat.20398
11. Strand, M, Zvrskovec, J, Hubel, C, Peat, CM, Bulik, CM, and Birgegard, A. Identifying research priorities for the study of atypical anorexia nervosa: a Delphi study. Int J Eat Disord. (2020) 53:1729–38. doi: 10.1002/eat.23358
12. Masheb, RM, Ramsey, CM, Marsh, AG, Snow, JL, Brandt, CA, and Haskell, SG. Atypical anorexia Nervosa, not so atypical after all: prevalence, correlates, and clinical severity among United States military veterans. Eat Behav. (2021) 41:101496. doi: 10.1016/j.eatbeh.2021.101496
13. Golden, NH, and Mehler, PS. Atypical anorexia nervosa can be just as bad. Cleve Clin J Med. (2020) 87:172–4. doi: 10.3949/ccjm.87a.19146
14. Keery, H, LeMay-Russell, S, Barnes, TL, Eckhardt, S, Peterson, CB, Lesser, J, et al. Attributes of children and adolescents with avoidant/restrictive food intake disorder. J Eat Disord. (2019) 7:31. doi: 10.1186/s40337-019-0261-3
15. Cornelissen, KK, Bester, A, Cairns, P, Tovee, MJ, and Cornelissen, PL. The influence of personal BMI on body size estimations and sensitivity to body size change in anorexia spectrum disorders. Body Image. (2015) 13:75–85. doi: 10.1016/j.bodyim.2015.01.001
16. Frank, GKW, Shott, ME, Stoddard, J, Swindle, S, and Pryor, TL. Association of Brain Reward Response with Body Mass Index and Ventral Striatal-Hypothalamic Circuitry among Young Women with Eating Disorders. JAMA Psychiatry. (2021) 78:1123–33. doi: 10.1001/jamapsychiatry.2021.1580
17. DeGuzman, M, Shott, ME, Yang, TT, Riederer, J, and Frank, GKW. Association of Elevated Reward Prediction Error Response with Weight Gain in adolescent anorexia Nervosa. Am J Psychiatry. (2017) 174:557–65. doi: 10.1176/appi.ajp.2016.16060671
18. Frank, GKW, Shott, ME, and DeGuzman, MC. Recent advances in understanding anorexia nervosa. F1000Res. (2019) 8:504. doi: 10.12688/f1000research.17789.1
19. Steward, T, Menchon, JM, Jimenez-Murcia, S, Soriano-Mas, C, and Fernandez-Aranda, F. Neural network alterations across eating disorders: a narrative review of fMRI studies. Curr Neuropharmacol. (2017) 16:1150–63. doi: 10.2174/1570159X15666171017111532
20. Monteleone, AM, Castellini, G, Volpe, U, Ricca, V, Lelli, L, Monteleone, P, et al. Neuroendocrinology and brain imaging of reward in eating disorders: a possible key to the treatment of anorexia nervosa and bulimia nervosa. Prog Neuro-Psychopharmacol Biol Psychiatry. (2018) 80:132–42. doi: 10.1016/j.pnpbp.2017.02.020
21. Frank, GKW. Neuroimaging and eating disorders. Curr Opin Psychiatry. (2019) 32:478–83. doi: 10.1097/YCO.0000000000000544
22. Bulik, CM, Coleman, JRI, Hardaway, JA, Breithaupt, L, Watson, HJ, Bryant, CD, et al. Genetics and neurobiology of eating disorders. Nat Neurosci. (2022) 25:543–54. doi: 10.1038/s41593-022-01071-z
23. Kaye, W. Neurobiology of anorexia and bulimia nervosa. Physiol Behav. (2008) 94:121–35. doi: 10.1016/j.physbeh.2007.11.037
24. Steinglass, JE, Berner, LA, and Attia, E. Cognitive neuroscience of eating disorders. Psychiatr Clin North Am. (2019) 42:75–91. doi: 10.1016/j.psc.2018.10.008
25. Berner, LA, and Marsh, R. Frontostriatal circuits and the development of bulimia nervosa. Front Behav Neurosci. (2014) 8:395. doi: 10.3389/fnbeh.2014.00395
26. Frank, GK. Advances from neuroimaging studies in eating disorders. CNS Spectr. (2015) 20:391–400. doi: 10.1017/S1092852915000012
27. Garcia-Garcia, I, Narberhaus, A, Marques-Iturria, I, et al. Neural responses to visual food cues: insights from functional magnetic resonance imaging. Eur Eat Disord Rev. (2013) 21:89–98. doi: 10.1002/erv.2216
28. Kaye, WH, Wierenga, CE, Bailer, UF, Simmons, AN, and Bischoff-Grethe, A. Nothing tastes as good as skinny feels: the neurobiology of anorexia nervosa. Trends Neurosci. (2013) 36:110–20. doi: 10.1016/j.tins.2013.01.003
29. Kaye, WH, Wierenga, CE, Bailer, UF, Simmons, AN, Wagner, A, and Bischoff-Grethe, A. Does a shared neurobiology for foods and drugs of abuse contribute to extremes of food ingestion in anorexia and bulimia nervosa? Biol Psychiatry. (2013) 73:836–42. doi: 10.1016/j.biopsych.2013.01.002
30. King, JA, Frank, GKW, Thompson, PM, and Ehrlich, S. Structural neuroimaging of anorexia Nervosa: future directions in the quest for mechanisms underlying dynamic alterations. Biol Psychiatry. (2017) 83:224–34. doi: 10.1016/j.biopsych.2017.08.011
31. Martin Monzon, B, Hay, P, Foroughi, N, and Touyz, S. White matter alterations in anorexia nervosa: a systematic review of diffusion tensor imaging studies. World J Psychiatry. (2016) 6:177–86. doi: 10.5498/wjp.v6.i1.177
32. Puglisi-Allegra, S, and Ventura, R. Prefrontal/accumbal catecholamine system processes emotionally driven attribution of motivational salience. Rev Neurosci. (2012) 23:509–26. doi: 10.1515/revneuro-2012-0076
33. Ventura, R, Morrone, C, and Puglisi-Allegra, S. Prefrontal/accumbal catecholamine system determines motivational salience attribution to both reward- and aversion-related stimuli. Proc Natl Acad Sci USA. (2007) 104:5181–6. doi: 10.1073/pnas.0610178104
34. Liu, X, Hairston, J, Schrier, M, and Fan, J. Common and distinct networks underlying reward valence and processing stages: a meta-analysis of functional neuroimaging studies. Neurosci Biobehav Rev. (2011) 35:1219–36. doi: 10.1016/j.neubiorev.2010.12.012
35. Kong, MS, and Zweifel, LS. Central amygdala circuits in valence and salience processing. Behav Brain Res. (2021) 410:113355. doi: 10.1016/j.bbr.2021.113355
36. O'Reilly, RC, Frank, MJ, Hazy, TE, and Watz, B. PVLV: the primary value and learned value Pavlovian learning algorithm. Behav Neurosci. (2007) 121:31–49. doi: 10.1037/0735-7044.121.1.31
37. Hazy, TE, Frank, MJ, and O'Reilly, RC. Neural mechanisms of acquired phasic dopamine responses in learning. Neurosci Biobehav Rev. (2010) 34:701–20. doi: 10.1016/j.neubiorev.2009.11.019
38. Castro, DC, Cole, SL, and Berridge, KC. Lateral hypothalamus, nucleus accumbens, and ventral pallidum roles in eating and hunger: interactions between homeostatic and reward circuitry. Front Syst Neurosci. (2015) 9:90. doi: 10.3389/fnsys.2015.00090
39. Baik, JH. Stress and the dopaminergic reward system. Exp Mol Med. (2020) 52:1879–90. doi: 10.1038/s12276-020-00532-4
40. Selby, EA, and Coniglio, KA. Positive emotion and motivational dynamics in anorexia nervosa: a positive emotion amplification model (PE-AMP). Psychol Rev. (2020) 127:853–90. doi: 10.1037/rev0000198
41. Seidel, M, Petermann, J, Diestel, S, Ritschel, F, Boehm, I, King, JA, et al. A naturalistic examination of negative affect and disorder-related rumination in anorexia nervosa. Eur Child Adolesc Psychiatry. (2016) 25:1207–16. doi: 10.1007/s00787-016-0844-3
42. Frank, GKW, Shott, ME, Pryor, T, Swindle, S, Nguyen, T, and Stoddard, J. Trait anxiety is associated with amygdala expectation and caloric taste receipt response across eating disorders. Neuropsychopharmacology. (2023) 48:380–90. doi: 10.1038/s41386-022-01440-z
43. Berridge, KC, Ho, CY, Richard, JM, and DiFeliceantonio, AG. The tempted brain eats: pleasure and desire circuits in obesity and eating disorders. Brain Res. (2010) 1350:43–64. doi: 10.1016/j.brainres.2010.04.003
44. Schultz, W. Getting formal with dopamine and reward. Neuron. (2002) 36:241–63. doi: 10.1016/S0896-6273(02)00967-4
45. Ben-Tovim, DI, and Walker, MK. A quantitative study of body-related attitudes in patients with anorexia and bulimia nervosa. Psychol Med. (1992) 22:961–9. doi: 10.1017/S0033291700038538
46. Hamilton, K, and Waller, G. Media influences on body size estimation in anorexia and bulimia. An experimental study. Br J Psychiatry. (1993) 162:837–40. doi: 10.1192/bjp.162.6.837
47. Legenbauer, T, Ruhl, I, and Vocks, S. Influence of appearance-related TV commercials on body image state. Behav Modif. (2008) 32:352–71. doi: 10.1177/0145445507309027
48. Gaudio, S, Brooks, SJ, and Riva, G. Nonvisual multisensory impairment of body perception in anorexia nervosa: a systematic review of neuropsychological studies. PLoS One. (2014) 9:e110087. doi: 10.1371/journal.pone.0110087
49. Zopf, R, Contini, E, Fowler, C, Mondraty, N, and Williams, MA. Body distortions in anorexia Nervosa: evidence for changed processing of multisensory bodily signals. Psychiatry Res. (2016) 245:473–81. doi: 10.1016/j.psychres.2016.09.003
50. Waldman, A, Loomes, R, Mountford, VA, and Tchanturia, K. Attitudinal and perceptual factors in body image distortion: an exploratory study in patients with anorexia nervosa. J Eat Disord. (2013) 1:17. doi: 10.1186/2050-2974-1-17
51. Goldzak-Kunik, G, Friedman, R, Spitz, M, Sandler, L, and Leshem, M. Intact sensory function in anorexia nervosa. Am J Clin Nutr. (2012) 95:272–82. doi: 10.3945/ajcn.111.020131
52. Gardner, RM, and Bokenkamp, ED. The role of sensory and nonsensory factors in body size estimations of eating disorder subjects. J Clin Psychol. (1996) 52:3–15. doi: 10.1002/(SICI)1097-4679(199601)52:1<3::AID-JCLP1>3.0.CO;2-X
53. Cash, T, and Deagle, E. The nature and extent of body-image disturbances in anorexia nervosa and bulimia nervosa: a meta-analysis. Int J Eat Disord. (1997) 22:107–26. doi: 10.1002/(SICI)1098-108X(199709)22:2<107::AID-EAT1>3.0.CO;2-J
54. Epstein, J, Wiseman, CV, Sunday, SR, Klapper, F, Alkalay, L, and Halmi, KA. Neurocognitive evidence favors "top down" over "bottom up" mechanisms in the pathogenesis of body size distortions in anorexia nervosa. Eat Weight Disord. (2001) 6:140–7. doi: 10.1007/BF03339763
55. Konstantakopoulos, G, Varsou, E, Dikeos, D, Ioannidi, N, Gonidakis, F, Papadimitriou, G, et al. Delusionality of body image beliefs in eating disorders. Psychiatry Res. (2012) 200:482–8. doi: 10.1016/j.psychres.2012.03.023
56. Steinglass, JE, Eisen, JL, Attia, E, Mayer, L, and Walsh, BT. Is anorexia nervosa a delusional disorder? An assessment of eating beliefs in anorexia nervosa. J Psychiatr Pract. (2007) 13:65–71. doi: 10.1097/01.pra.0000265762.79753.88
57. Heinz, A, Murray, GK, Schlagenhauf, F, Sterzer, P, Grace, AA, and Waltz, JA. Towards a unifying cognitive, neurophysiological, and computational neuroscience account of schizophrenia. Schizophr Bull. (2019) 45:1092–100. doi: 10.1093/schbul/sby154
58. Blum, K, Thanos, PK, and Gold, MS. Dopamine and glucose, obesity, and reward deficiency syndrome. Front Psychol. (2014) 5:919. doi: 10.3389/fpsyg.2014.00919
59. Kurachi, M, Yasui, S, Shibata, R, Murata, M, Hagino, H, Kurachi, T, et al. Comparative study of dopamine metabolism with local cerebral glucose utilization in rat brain following the administration of haloperidol decanoate. Biol Psychiatry. (1994) 36:110–7. doi: 10.1016/0006-3223(94)91191-6
60. Polivy, J, and Herman, CP. Causes of eating disorders. Annu Rev Psychol. (2002) 53:187–213. doi: 10.1146/annurev.psych.53.100901.135103
61. Oldershaw, A, Lavender, T, Sallis, H, Stahl, D, and Schmidt, U. Emotion generation and regulation in anorexia nervosa: a systematic review and meta-analysis of self-report data. Clin Psychol Rev. (2015) 39:83–95. doi: 10.1016/j.cpr.2015.04.005
62. Meule, A, Richard, A, Schnepper, R, Reichenberger, J, Georgii, C, Naab, S, et al. Emotion regulation and emotional eating in anorexia nervosa and bulimia nervosa. Eat Disord. (2019) 29:1–17. doi: 10.1080/10640266.2019.1642036
63. Watson, D, Clark, LA, and Tellegen, A. Development and validation of brief measures of positive and negative affect: the PANAS scales. J Pers Soc Psychol. (1988) 54:1063–70. doi: 10.1037/0022-3514.54.6.1063
64. Ralph-Nearman, C, Williams, BM, Ortiz, AML, Smith, AR, and Levinson, CA. Pinpointing core and pathway symptoms among sleep disturbance, anxiety, worry, and eating disorder symptoms in anorexia nervosa and atypical anorexia nervosa. J Affect Disord. (2021) 294:24–32. doi: 10.1016/j.jad.2021.06.061
65. Svaldi, J, Bender, C, Caffier, D, Ivanova, V, Mies, N, Fleischhaker, C, et al. Negative mood increases selective attention to negatively Valenced body parts in female adolescents with anorexia Nervosa. PLoS One. (2016) 11:e0154462. doi: 10.1371/journal.pone.0154462
66. Espeset, EM, Gulliksen, KS, Nordbo, RH, Skarderud, F, and Holte, A. Fluctuations of body images in anorexia nervosa: patients' perception of contextual triggers. Clin Psychol Psychother. (2012) 19:518–30. doi: 10.1002/cpp.760
67. Fitzsimmons-Craft, EE, Accurso, EC, Ciao, AC, Crosby, RD, Cao, L, Pisetsky, EM, et al. Restrictive eating in anorexia nervosa: examining maintenance and consequences in the natural environment. Int J Eat Disord. (2015) 48:923–31. doi: 10.1002/eat.22439
68. Engel, SG, Wonderlich, SA, Crosby, RD, Mitchell, JE, Crow, S, Peterson, CB, et al. The role of affect in the maintenance of anorexia nervosa: evidence from a naturalistic assessment of momentary behaviors and emotion. J Abnorm Psychol. (2013) 122:709–19. doi: 10.1037/a0034010
69. Haynos, AF, Berg, KC, Cao, L, Crosby, RD, Lavender, JM, Utzinger, LM, et al. Trajectories of higher- and lower-order dimensions of negative and positive affect relative to restrictive eating in anorexia nervosa. J Abnorm Psychol. (2017) 126:495–505. doi: 10.1037/abn0000202
70. Furtjes, S, Seidel, M, King, JA, et al. A naturalistic investigation of cognitive-affective dysfunction in anorexia nervosa: the role of inefficiency. Int J Eat Disord. (2020) 53:239–47. doi: 10.1002/eat.23189
71. Pila, E, Murray, SB, Le Grange, D, Sawyer, SM, and Hughes, EK. Reciprocal relations between dietary restraint and negative affect in adolescents receiving treatment for anorexia nervosa. J Abnorm Psychol. (2019) 128:129–39. doi: 10.1037/abn0000402
72. Stice, E, Gau, JM, Rohde, P, and Shaw, H. Risk factors that predict future onset of each DSM-5 eating disorder: predictive specificity in high-risk adolescent females. J Abnorm Psychol. (2017) 126:38–51. doi: 10.1037/abn0000219
73. Castro, TF, Miller, K, Araujo, MX, Brandao, I, and Torres, S. Emotional processing in recovered anorexia nervosa patients: a 15 year longitudinal study. Eur Eat Disord Rev. (2021) 29:955–68. doi: 10.1002/erv.2858
74. Sharma, K, Akre, S, Chakole, S, and Wanjari, MB. Stress-induced diabetes: a review. Cureus. (2022) 14:e29142. doi: 10.7759/cureus.29142
75. van der Kooij, MA, Jene, T, Treccani, G, Miederer, I, Hasch, A, Voelxen, N, et al. Chronic social stress-induced hyperglycemia in mice couples individual stress susceptibility to impaired spatial memory. Proc Natl Acad Sci USA. (2018) 115:E10187–96. doi: 10.1073/pnas.1804412115
76. van der Kooij, MA. The impact of chronic stress on energy metabolism. Mol Cell Neurosci. (2020) 107:103525. doi: 10.1016/j.mcn.2020.103525
77. Kern, S, Oakes, TR, Stone, CK, McAuliff, EM, Kirschbaum, C, and Davidson, RJ. Glucose metabolic changes in the prefrontal cortex are associated with HPA axis response to a psychosocial stressor. Psychoneuroendocrinology. (2008) 33:517–29. doi: 10.1016/j.psyneuen.2008.01.010
78. Yehuda, R, Harvey, PD, Golier, JA, et al. Changes in relative glucose metabolic rate following cortisol administration in aging veterans with posttraumatic stress disorder: an FDG-PET neuroimaging study. J Neuropsychiatry Clin Neurosci. (2009) 21:132–43. doi: 10.1176/jnp.2009.21.2.132
79. Reis, JC, Travado, L, Antoni, MH, Oliveira, FPM, Almeida, SD, Almeida, P, et al. Negative affect and stress-related brain metabolism in patients with metastatic breast cancer. Cancer. (2020) 126:3122–31. doi: 10.1002/cncr.32902
80. Schmalbach, I, Herhaus, B, Passler, S, et al. Cortisol reactivity in patients with anorexia nervosa after stress induction. Transl Psychiatry. (2020) 10:275. doi: 10.1038/s41398-020-00955-7
81. Farquharson, RF, and Hyland, HH. Anorexia nervosa. A metabolic disorder of psychologic origin. JAMA. (1938) 111:1085–92. doi: 10.1001/jama.1938.02790380027007
82. Duriez, P, Ramoz, N, Gorwood, P, Viltart, O, and Tolle, V. A metabolic perspective on reward abnormalities in anorexia Nervosa. Trends Endocrinol Metab. (2019) 30:915–28. doi: 10.1016/j.tem.2019.08.004
83. Bulik, CM, Flatt, R, Abbaspour, A, and Carroll, I. Reconceptualizing anorexia nervosa. Psychiatry Clin Neurosci. (2019) 73:518–25. doi: 10.1111/pcn.12857
84. Fehm, HL, Kern, W, and Peters, A. The selfish brain: competition for energy resources. Prog Brain Res. (2006) 153:129–40. doi: 10.1016/S0079-6123(06)53007-9
85. Carneiro-Nascimento, S, Opacka-Juffry, J, Costabile, A, Boyle, CN, Herde, AM, Ametamey, SM, et al. Chronic social stress in mice alters energy status including higher glucose need but lower brain utilization. Psychoneuroendocrinology. (2020) 119:104747. doi: 10.1016/j.psyneuen.2020.104747
86. Solmi, M, Veronese, N, Favaro, A, Santonastaso, P, Manzato, E, Sergi, G, et al. Inflammatory cytokines and anorexia nervosa: a meta-analysis of cross-sectional and longitudinal studies. Psychoneuroendocrinology. (2015) 51:237–52. doi: 10.1016/j.psyneuen.2014.09.031
87. Solmi, M, Veronese, N, Manzato, E, Sergi, G, Favaro, A, Santonastaso, P, et al. Oxidative stress and antioxidant levels in patients with anorexia nervosa: a systematic review and exploratory meta-analysis. Int J Eat Disord. (2015) 48:826–41. doi: 10.1002/eat.22443
88. Watson, HJ, Yilmaz, Z, Thornton, LM, et al. Genome-wide association study identifies eight risk loci and implicates metabo-psychiatric origins for anorexia nervosa. Nat Genet. (2019) 51:1207–14. doi: 10.1038/s41588-019-0439-2
89. Peters, A, Schweiger, U, Pellerin, L, Hubold, C, Oltmanns, KM, Conrad, M, et al. The selfish brain: competition for energy resources. Neurosci Biobehav Rev. (2004) 28:143–80. doi: 10.1016/j.neubiorev.2004.03.002
90. Peters, A, McEwen, BS, and Friston, K. Uncertainty and stress: why it causes diseases and how it is mastered by the brain. Prog Neurobiol. (2017) 156:164–88. doi: 10.1016/j.pneurobio.2017.05.004
91. Frank, GK, Roblek, T, Shott, ME, et al. Heightened fear of uncertainty in anorexia and bulimia nervosa. Int J Eat Disord. (2012) 45:227–32. doi: 10.1002/eat.20929
92. Thurston, JH, and Hauhart, RE. Effect of momentary stress on brain energy metabolism in weanling mice: apparent use of lactate as cerebral metabolic fuel concomitant with a decrease in brain glucose utilization. Metab Brain Dis. (1989) 4:177–86. doi: 10.1007/BF01000294
93. Warnock, GI, and Steckler, T. Stress-induced decreases in local cerebral glucose utilization in specific regions of the mouse brain. BMC Res Notes. (2011) 4:96. doi: 10.1186/1756-0500-4-96
94. Peters, A, Kubera, B, Hubold, C, and Langemann, D. The selfish brain: stress and eating behavior. Front Neurosci. (2011) 5:74. doi: 10.3389/fnins.2011.00074
95. Chung, M, and Gobel, B. Mathematical modeling of the human energy metabolism based on the selfish brain theory. Adv Exp Med Biol. (2012) 736:425–40. doi: 10.1007/978-1-4419-7210-1_25
96. Mansur, RB, and Brietzke, E. The "selfish brain" hypothesis for metabolic abnormalities in bipolar disorder and schizophrenia. Trends Psychiatry Psychother. (2012) 34:121–8. doi: 10.1590/s2237-60892012000300003
97. Barbarich-Marsteller, NC, Marsteller, DA, Alexoff, DL, Fowler, JS, and Dewey, SL. MicroPET imaging in an animal model of anorexia nervosa. Synapse. (2005) 57:85–90. doi: 10.1002/syn.20160
98. Yilmaz, Z, Halvorsen, M, Bryois, J, Yu, D, Thornton, LM, Zerwas, S, et al. Examination of the shared genetic basis of anorexia nervosa and obsessive-compulsive disorder. Mol Psychiatry. (2018) 25:2036–46. doi: 10.1038/s41380-018-0115-4
99. Zuniga-Guajardo, S, Garfinkel, PE, and Zinman, B. Changes in insulin sensitivity and clearance in anorexia nervosa. Metabolism. (1986) 35:1096–100. doi: 10.1016/0026-0495(86)90021-1
100. Lagou, V, Magi, R, Hottenga, JJ, et al. Sex-dimorphic genetic effects and novel loci for fasting glucose and insulin variability. Nat Commun. (2021) 12:24. doi: 10.1038/s41467-020-19366-9
101. Hubel, C, Gaspar, HA, Coleman, JRI, et al. Genomics of body fat percentage may contribute to sex bias in anorexia nervosa. Am J Med Genet B Neuropsychiatr Genet. (2019) 180:428–38. doi: 10.1002/ajmg.b.32709
102. Victor, VM, Rovira-Llopis, S, Saiz-Alarcon, V, Sangüesa, MC, Rojo-Bofill, L, Bañuls, C, et al. Altered mitochondrial function and oxidative stress in leukocytes of anorexia nervosa patients. PLoS One. (2014) 9:e106463. doi: 10.1371/journal.pone.0106463
103. Delvenne, V, Goldman, S, De Maertelaer, V, Simon, Y, Luxen, A, and Lotstra, F. Brain hypometabolism of glucose in anorexia nervosa: normalization after weight gain. Biol Psychiatry. (1996) 40:761–8. doi: 10.1016/0006-3223(95)00522-6
104. Herholz, K, Krieg, JC, Emrich, HM, et al. Regional cerebral glucose metabolism in anorexia nervosa measured by positron emission tomography. Biol Psychiatry. (1987) 22:43–51. doi: 10.1016/0006-3223(87)90128-4
105. Miller, KK, Deckersbach, T, Rauch, SL, Fischman, AJ, Grieco, KA, Herzog, DB, et al. Testosterone administration attenuates regional brain hypometabolism in women with anorexia nervosa. Psychiatry Res. (2004) 132:197–207. doi: 10.1016/j.pscychresns.2004.09.003
106. Zhang, HW, Li, DY, Zhao, J, Guan, YH, Sun, BM, and Zuo, CT. Metabolic imaging of deep brain stimulation in anorexia nervosa: a 18F-FDG PET/CT study. Clin Nucl Med. (2013) 38:943–8. doi: 10.1097/RLU.0000000000000261
107. Ter Horst, KW, Lammers, NM, Trinko, R, et al. Striatal dopamine regulates systemic glucose metabolism in humans and mice. Sci Transl Med. (2018) 10:eaar3752. doi: 10.1126/scitranslmed.aar3752
108. Yabut, JM, Crane, JD, Green, AE, Keating, DJ, Khan, WI, and Steinberg, GR. Emerging roles for serotonin in regulating metabolism: new implications for an ancient molecule. Endocr Rev. (2019) 40:1092–107. doi: 10.1210/er.2018-00283
109. Bijsterbosch, JM, Keizer, A, Boelen, PA, van den Brink, F, and Sternheim, LC. Understanding relations between intolerance of uncertainty and body checking and body avoiding in anorexia nervosa. J Eat Disord. (2022) 10:122. doi: 10.1186/s40337-022-00647-1
110. Konstantellou, A, Hale, L, Sternheim, L, Simic, M, and Eisler, I. The experience of intolerance of uncertainty for young people with a restrictive eating disorder: a pilot study. Eat Weight Disord. (2019) 24:533–40. doi: 10.1007/s40519-019-00652-5
111. Jacobs, MJ, Roesch, S, Wonderlich, SA, Crosby, R, Thornton, L, Wilfley, DE, et al. Anorexia nervosa trios: behavioral profiles of individuals with anorexia nervosa and their parents. Psychol Med. (2009) 39:451–61. doi: 10.1017/S0033291708003826
112. Lawson, EA, Holsen, LM, Santin, M, DeSanti, R, Meenaghan, E, Eddy, KT, et al. Postprandial oxytocin secretion is associated with severity of anxiety and depressive symptoms in anorexia nervosa. J Clin Psychiatry. (2013) 74:e451–7. doi: 10.4088/JCP.12m08154
113. Lilenfeld, LR, Kaye, WH, Greeno, CG, Merikangas, KR, Plotnicov, K, Pollice, C, et al. A controlled family study of anorexia nervosa and bulimia nervosa: psychiatric disorders in first-degree relatives and effects of proband comorbidity. Arch Gen Psychiatry. (1998) 55:603–10. doi: 10.1001/archpsyc.55.7.603
114. Steinglass, JE, Sysko, R, Mayer, L, Berner, LA, Schebendach, J, Wang, Y, et al. Pre-meal anxiety and food intake in anorexia nervosa. Appetite. (2010) 55:214–8. doi: 10.1016/j.appet.2010.05.090
115. Dignon, A, Beardsmore, A, Spain, S, and Kuan, A. “Why I won't eat”: patient testimony from 15 anorexics concerning the causes of their disorder. J Health Psychol. (2006) 11:942–56. doi: 10.1177/1359105306069097
116. Casper, RC, Chatterton, RT Jr, and Davis, JM. Alterations in serum cortisol and its binding characteristics in anorexia nervosa. J Clin Endocrinol Metab. (1979) 49:406–11. doi: 10.1210/jcem-49-3-406
117. Frank, GKW, DeGuzman, MC, Shott, ME, Laudenslager, ML, Rossi, B, and Pryor, T. Association of Brain Reward Learning Response with Harm Avoidance, weight gain, and hypothalamic effective connectivity in adolescent anorexia Nervosa. JAMA Psychiatry. (2018) 75:1071–80. doi: 10.1001/jamapsychiatry.2018.2151
118. Garfinkel, PE, Brown, GM, Stancer, HC, and Moldofsky, H. Hypothalamic-pituitary function in anorexia nervosa. Arch Gen Psychiatry. (1975) 32:739–44. doi: 10.1001/archpsyc.1975.01760240067005
119. Monteleone, AM, Ruzzi, V, Pellegrino, F, Patriciello, G, Cascino, G, del Giorno, C, et al. The vulnerability to interpersonal stress in eating disorders: the role of insecure attachment in the emotional and cortisol responses to the trier social stress test. Psychoneuroendocrinology. (2019) 101:278–85. doi: 10.1016/j.psyneuen.2018.12.232
120. Jappe, LM, Frank, GK, Shott, ME, et al. Heightened sensitivity to reward and punishment in anorexia nervosa. Int J Eat Disord. (2011) 44:317–24. doi: 10.1002/eat.20815
121. Khalsa, SS, Hassanpour, MS, Strober, M, Craske, MG, Arevian, AC, and Feusner, JD. Interoceptive anxiety and body representation in anorexia Nervosa. Front Psych. (2018) 9:444. doi: 10.3389/fpsyt.2018.00444
122. Lavender, JM, De Young, KP, Wonderlich, SA, et al. Daily patterns of anxiety in anorexia nervosa: associations with eating disorder behaviors in the natural environment. J Abnorm Psychol. (2013) 122:672–83. doi: 10.1037/a0031823
123. Klump, KL, Kaye, WH, and Strober, M. The evolving genetic foundations of eating disorders. Psychiatr Clin North Am. (2001) 24:215–25. doi: 10.1016/S0193-953X(05)70218-5
124. Schulze, UM, Calame, S, Keller, F, and Mehler-Wex, C. Trait anxiety in children and adolescents with anorexia nervosa. Eat Weight Disord. (2009) 14:e163–8. doi: 10.1007/BF03327817
125. Brietzke, E, Mansur, RB, Subramaniapillai, M, Balanzá-Martínez, V, Vinberg, M, González-Pinto, A, et al. Ketogenic diet as a metabolic therapy for mood disorders: evidence and developments. Neurosci Biobehav Rev. (2018) 94:11–6. doi: 10.1016/j.neubiorev.2018.07.020
126. Rho, JM, and Boison, D. The metabolic basis of epilepsy. Nat Rev Neurol. (2022) 18:333–47. doi: 10.1038/s41582-022-00651-8
127. Sethi, S, and Ford, JM. The role of ketogenic metabolic therapy on the brain in serious mental illness: a review. J Psychiatr Brain Sci. (2022) 7:e220009. doi: 10.20900/jpbs.20220009
128. Zhu, H, Bi, D, Zhang, Y, Kong, C, du, J, Wu, X, et al. Ketogenic diet for human diseases: the underlying mechanisms and potential for clinical implementations. Signal Transduct Target Ther. (2022) 7:11. doi: 10.1038/s41392-021-00831-w
129. Guest, J, Garg, M, Bilgin, A, and Grant, R. Relationship between central and peripheral fatty acids in humans. Lipids Health Dis. (2013) 12:79. doi: 10.1186/1476-511X-12-79
130. Mergenthaler, P, Lindauer, U, Dienel, GA, and Meisel, A. Sugar for the brain: the role of glucose in physiological and pathological brain function. Trends Neurosci. (2013) 36:587–97. doi: 10.1016/j.tins.2013.07.001
131. Owen, OE, Morgan, AP, Kemp, HG, Sullivan, JM, Herrera, MG, and Cahill, GF Jr. Brain metabolism during fasting. J Clin Invest. (1967) 46:1589–95. doi: 10.1172/JCI105650
132. Masino, SA, and Rho, JM. Metabolism and epilepsy: ketogenic diets as a homeostatic link. Brain Res. (1703) 1703:26–30. doi: 10.1016/j.brainres.2018.05.049
133. Svart, M, Gormsen, LC, Hansen, J, Zeidler, D, Gejl, M, Vang, K, et al. Regional cerebral effects of ketone body infusion with 3-hydroxybutyrate in humans: reduced glucose uptake, unchanged oxygen consumption and increased blood flow by positron emission tomography. A randomized, controlled trial. PLoS One. (2018) 13:e0190556. doi: 10.1371/journal.pone.0190556
134. Bennett, OA, Ramsay, SC, Malacova, E, Bourgeat, P, Goodman, SJ, Dunn, CJ, et al. Regional differences in the reduction in cerebral FDG uptake induced by the ketogenic diet. Eur J Hybrid Imaging. (2022) 6:29. doi: 10.1186/s41824-022-00150-5
135. Northoff, G. Self and brain: what is self-related processing? Trends Cogn Sci. (2011) 15:186–7. doi: 10.1016/j.tics.2011.03.001
136. Terhoeven, V, Nikendei, C, Faschingbauer, S, Huber, J, Young, KD, Bendszus, M, et al. Neurophysiological correlates of disorder-related autobiographical memory in anorexia nervosa. Psychol Med. (2021) 53:1–11. doi: 10.1017/S003329172100221X
137. Lee, S, Ran Kim, K, Ku, J, Lee, JH, Namkoong, K, and Jung, YC. Resting-state synchrony between anterior cingulate cortex and precuneus relates to body shape concern in anorexia nervosa and bulimia nervosa. Psychiatry Res. (2014) 221:43–8. doi: 10.1016/j.pscychresns.2013.11.004
138. Courchesne-Loyer, A, Croteau, E, Castellano, CA, St-Pierre, V, Hennebelle, M, and Cunnane, SC. Inverse relationship between brain glucose and ketone metabolism in adults during short-term moderate dietary ketosis: a dual tracer quantitative positron emission tomography study. J Cereb Blood Flow Metab. (2017) 37:2485–93. doi: 10.1177/0271678X16669366
139. Zhang, Y, Kuang, Y, Xu, K, Harris, D, Lee, Z, LaManna, J, et al. Ketosis proportionately spares glucose utilization in brain. J Cereb Blood Flow Metab. (2013) 33:1307–11. doi: 10.1038/jcbfm.2013.87
140. Newman, JC, and Verdin, E. Ketone bodies as signaling metabolites. Trends Endocrinol Metab. (2014) 25:42–52. doi: 10.1016/j.tem.2013.09.002
141. Bough, KJ, and Rho, JM. Anticonvulsant mechanisms of the ketogenic diet. Epilepsia. (2007) 48:43–58. doi: 10.1111/j.1528-1167.2007.00915.x
142. Rogawski, MA, Loscher, W, and Rho, JM. Mechanisms of action of Antiseizure drugs and the ketogenic diet. Cold Spring Harb Perspect Med. (2016) 6:a022780. doi: 10.1101/cshperspect.a022780
143. Hartman, AL, Gasior, M, Vining, EP, and Rogawski, MA. The neuropharmacology of the ketogenic diet. Pediatr Neurol. (2007) 36:281–92. doi: 10.1016/j.pediatrneurol.2007.02.008
144. Longo, LP. Anxiety: neurobiologic underpinnings. Psychiatr Ann. (1998) 28:130–8. doi: 10.3928/0048-5713-19980301-08
145. Judd, FK, Burrows, GD, and Norman, TR. The biological basis of anxiety. An overview. J Affect Disord. (1985) 9:271–84. doi: 10.1016/0165-0327(85)90058-8
146. Yamanashi, T, Iwata, M, Shibushita, M, Tsunetomi, K, Nagata, M, Kajitani, N, et al. Beta-hydroxybutyrate, an endogenous NLRP3 inflammasome inhibitor, attenuates anxiety-related behavior in a rodent posttraumatic stress disorder model. Sci Rep. (2020) 10:21629. doi: 10.1038/s41598-020-78410-2
147. Gumus, H, Ilgin, R, Koc, B, Yuksel, O, Kizildag, S, Guvendi, G, et al. A combination of ketogenic diet and voluntary exercise ameliorates anxiety and depression-like behaviors in Balb/c mice. Neurosci Lett. (2022) 770:136443. doi: 10.1016/j.neulet.2021.136443
148. Aoki, C, Chowdhury, TG, Wable, GS, and Chen, YW. Synaptic changes in the hippocampus of adolescent female rodents associated with resilience to anxiety and suppression of food restriction-evoked hyperactivity in an animal model for anorexia nervosa. Brain Res. (2017) 1654:102–15. doi: 10.1016/j.brainres.2016.01.019
149. Aoki, C, Sabaliauskas, N, Chowdhury, T, Min, JY, Colacino, AR, Laurino, K, et al. Adolescent female rats exhibiting activity-based anorexia express elevated levels of GABA(a) receptor alpha4 and delta subunits at the plasma membrane of hippocampal CA1 spines. Synapse. (2012) 66:391–407. doi: 10.1002/syn.21528
150. Yamanashi, T, Iwata, M, Kamiya, N, Tsunetomi, K, Kajitani, N, Wada, N, et al. Beta-hydroxybutyrate, an endogenic NLRP3 inflammasome inhibitor, attenuates stress-induced behavioral and inflammatory responses. Sci Rep. (2017) 7:7677. doi: 10.1038/s41598-017-08055-1
151. Rezin, GT, Amboni, G, Zugno, AI, Quevedo, J, and Streck, EL. Mitochondrial dysfunction and psychiatric disorders. Neurochem Res. (2009) 34:1021–9. doi: 10.1007/s11064-008-9865-8
152. Kim, Y, Vadodaria, KC, Lenkei, Z, Kato, T, Gage, FH, Marchetto, MC, et al. Mitochondria, metabolism, and redox mechanisms in psychiatric disorders. Antioxid Redox Signal. (2019) 31:275–317. doi: 10.1089/ars.2018.7606
153. van de Burgt, N, van Doesum, W, Grevink, M, van Niele, S, de Koning, T, Leibold, N, et al. Psychiatric manifestations of inborn errors of metabolism: a systematic review. Neurosci Biobehav Rev. (2023) 144:104970. doi: 10.1016/j.neubiorev.2022.104970
154. Pinto, JV, Moulin, TC, and Amaral, OB. On the transdiagnostic nature of peripheral biomarkers in major psychiatric disorders: a systematic review. Neurosci Biobehav Rev. (2017) 83:97–108. doi: 10.1016/j.neubiorev.2017.10.001
155. Fourrier, C, Singhal, G, and Baune, BT. Neuroinflammation and cognition across psychiatric conditions. CNS Spectr. (2019) 24:4–15. doi: 10.1017/S1092852918001499
156. Jorgensen, A, Baago, IB, Rygner, Z, Jorgensen, MB, Andersen, PK, Kessing, LV, et al. Association of Oxidative Stress-Induced Nucleic Acid Damage with Psychiatric Disorders in adults: a systematic review and Meta-analysis. JAMA Psychiatry. (2022) 79:920–31. doi: 10.1001/jamapsychiatry.2022.2066
157. Grajek, M, Krupa-Kotara, K, Bialek-Dratwa, A, et al. Nutrition and mental health: a review of current knowledge about the impact of diet on mental health. Front Nutr. (2022) 9:943998. doi: 10.3389/fnut.2022.943998
158. Martin-McGill, KJ, Jackson, CF, Bresnahan, R, Levy, RG, and Cooper, PN. Ketogenic diets for drug-resistant epilepsy. Cochrane Database Syst Rev. (2018) 11:CD001903. doi: 10.1002/14651858.CD001903.pub4
159. Ruskin, DN, Fortin, JA, Bisnauth, SN, and Masino, SA. Ketogenic diets improve behaviors associated with autism spectrum disorder in a sex-specific manner in the EL mouse. Physiol Behav. (2017) 168:138–45. doi: 10.1016/j.physbeh.2016.10.023
160. McDonald, TJW, and Cervenka, MC. Ketogenic diets for adult neurological disorders. Neurotherapeutics. (2018) 15:1018–31. doi: 10.1007/s13311-018-0666-8
161. Danan, A, Westman, EC, Saslow, LR, and Ede, G. The ketogenic diet for refractory mental illness: a retrospective analysis of 31 inpatients. Front Psych. (2022) 13:951376. doi: 10.3389/fpsyt.2022.951376
162. Norwitz, NG, Sethi, S, and Palmer, CM. Ketogenic diet as a metabolic treatment for mental illness. Curr Opin Endocrinol Diabetes Obes. (2020) 27:269–74. doi: 10.1097/MED.0000000000000564
163. Kraeuter, AK, Phillips, R, and Sarnyai, Z. Ketogenic therapy in neurodegenerative and psychiatric disorders: from mice to men. Prog Neuro-Psychopharmacol Biol Psychiatry. (2020) 101:109913. doi: 10.1016/j.pnpbp.2020.109913
164. Yudkoff, M, Daikhin, Y, Melo, TM, Nissim, I, Sonnewald, U, and Nissim, I. The ketogenic diet and brain metabolism of amino acids: relationship to the anticonvulsant effect. Annu Rev Nutr. (2007) 27:415–30. doi: 10.1146/annurev.nutr.27.061406.093722
165. Scolnick, B, Zupec-Kania, B, Calabrese, L, Aoki, C, and Hildebrandt, T. Remission from chronic anorexia Nervosa with ketogenic diet and ketamine: case report. Front Psych. (2020) 11:763. doi: 10.3389/fpsyt.2020.00763
166. Calabrese, L, Scolnick, B, Zupec-Kania, B, Beckwith, C, Costello, K, and Frank, GKW. Ketogenic diet and ketamine infusion treatment to target chronic persistent eating disorder psychopathology in anorexia nervosa: a pilot study. Eat Weight Disord. (2022) 27:3751–7. doi: 10.1007/s40519-022-01455-x
167. Mills, IH, Park, GR, Manara, AR, and Merriman, RJ. Treatment of compulsive behaviour in eating disorders with intermittent ketamine infusions. QJM. (1998) 91:493–503. doi: 10.1093/qjmed/91.7.493
168. Domino, EF. Taming the ketamine tiger. 1965. Anesthesiology. (2010) 113:678–84. doi: 10.1097/ALN.0b013e3181ed09a2
169. Zarate, CA Jr, Singh, JB, Carlson, PJ, et al. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry. (2006) 63:856–64. doi: 10.1001/archpsyc.63.8.856
170. Weckmann, K, Deery, MJ, Howard, JA, Feret, R, Asara, JM, Dethloff, F, et al. Ketamine's antidepressant effect is mediated by energy metabolism and antioxidant defense system. Sci Rep. (2017) 7:15788. doi: 10.1038/s41598-017-16183-x
Keywords: anorexia nervosa, metabolism, ketogenic, brain, behavior, treatment
Citation: Frank GKW and Scolnick B (2024) Therapeutic ketogenic diet as treatment for anorexia nervosa. Front. Nutr. 11:1392135. doi: 10.3389/fnut.2024.1392135
Edited by:
Beth Ann Zupec-Kania, Ketogenic Therapies, LLC, United StatesReviewed by:
Emilio Gutierrez, University of Santiago de Compostela, SpainNurul ‘Ain Azizan, University of Nottingham Malaysia Campus, Malaysia
Copyright © 2024 Frank and Scolnick. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guido K. W. Frank, Z2ZyYW5rQGhlYWx0aC51Y3NkLmVkdQ==; Barbara Scolnick, c2NvbG5pY2tiYXJiYXJhQGdtYWlsLmNvbQ==
†Present Address: Barbara Scolnick, Private Practice, Waban, MA, United States