 Silje Harvei
Silje Harvei Vemund Skogen1
Vemund Skogen1 Bjørg Egelandsdal
Bjørg Egelandsdal Harald Carlsen
Harald Carlsen- 1Faculty of Chemistry, Biotechnology and Food Science, Norwegian University of Life Sciences, As, Norway
- 2Faculty of Veterinary Medicine, Norwegian University of Life Sciences, As, Norway
Introduction: Lipopolysaccharides (LPS) present in the intestine are suggested to enter the bloodstream after consumption of high-fat diets and cause systemic inflammation and metabolic dysregulation through a process named “metabolic endotoxemia.” This study aimed to determine the role of orally administered LPS to mice in the early stage of chronic low-grade inflammation induced by diet.
Methods: We supplemented the drinking water with E. coli derived LPS to mice fed either high-fat Western-style diet (WSD) or standard chow (SC) for 7 weeks (n = 16–17). Body weight was recorded weekly. Systemic inflammatory status was assessed by in vivo imaging of NF-κB activity at different time points, and glucose dysregulation was assessed by insulin sensitivity test and glucose tolerance test near the end of the study. Systemic LPS exposure was estimated indirectly via quantification of LPS-binding protein (LBP) and antibodies against LPS in plasma, and directly using an LPS-sensitive cell reporter assay.
Results and discussion: Our results demonstrate that weight development and glucose regulation are not affected by LPS. We observed a transient LPS dependent upregulation of NF-κB activity in the liver region in both diet groups, a response that disappeared within the first week of LPS administration and remained low during the rest of the study. However, WSD fed mice had overall a higher NF-κB activity compared to SC fed mice at all time points independent of LPS administration. Our findings indicate that orally administered LPS has limited to no impact on systemic inflammation and metabolic dysregulation in mice fed a high-fat western diet and we question the capability of intestinally derived LPS to initiate systemic inflammation through a healthy and uncompromised intestine, even when exposed to a high-fat diet.
Introduction
Lipopolysaccharides (LPS) are the major components of the outer membrane of Gram-negative bacteria, naturally present in the human intestine as part of the microflora. LPS consists of a polysaccharide and a lipid component named lipid A. The host innate immune system recognizes LPS (lipid A) to mount a response against potentially pathogenic intruders. The recognition typically involves an initial binding to the LPS-binding protein (LBP), which delivers LPS to a complex consisting of CD14, MD-2 and Toll-like receptor 4 (TLR4; (1)). The LPS-TLR4 interaction initiates an inflammatory response characterized by increased levels of proinflammatory cytokines mediated by the transcriptional regulator nuclear factor-κB (NF-κB). This may lead to an acute local response or a systemic response depending on the LPS levels and where the LPS are found (2, 3). Minor increases of LPS in the circulatory system are proposed to induce low-grade inflammation, associated with metabolic dysregulation including insulin resistance and overweight (4–6).
LPS is seemingly detectable at very low concentrations in the circulation of healthy individuals, and it has been suggested that LPS can translocate through the intestinal wall and increase systemic LPS levels following ingestion of certain dietary patterns, leading to a phenomenon called “metabolic endotoxemia” (4). LPS translocation from the intestine into the blood may increase following ingestion of high fat, high caloric or high carbohydrate diets (6–8). Presently, dietary fat is the major dietary factor known to facilitate the translocation of LPS from the intestine (9–14) but seems to be augmented by diets high in sugar (15). Despite this being the common perception in the scientific community, the mechanisms connecting diet and elevated blood LPS with low-grade inflammation are still not well-understood. Of note, the original study demonstrating a role for LPS causing metabolic endotoxemia was based on systemic infusion of LPS using an implanted osmotic pump, thereby surpassing the physiological route for intestinal-derived LPS (4). In contrast, Dalby and coworkers demonstrated that diet-induced obesity is not dependent on TLR4 or the cluster of differentiation 14 (CD14) signaling (16), questioning the significance of LPS in causing or contributing to metabolic endotoxemia. Most of the studies supporting high-fat diet-induced metabolic endotoxemia are based on the experimental results of increased LPS in blood samples as determined by the Limulus amebocyte lysate (LAL) assay (4, 6, 17). The usefulness of the LAL assay is controversial as the test does not discriminate on the TLR4-stimulating properties of LPS. Detection of LPS from blood samples is regarded as difficult and susceptible to inaccuracies and is highly influenced by both inhibiting and enhancing factors (see reviews by Boutagy, McMillan (18) and Gnauck, Lentle and Kruger (19) for details). Furthermore, the same reviews note that the range of LPS blood concentrations in humans reported in the literature is highly variable, making it difficult to identify the quantifiable threshold between the normal and the inflammatory state.
The major source of circulating LPS is likely commensal bacteria residing in the gastrointestinal tract (GIT), which is a rich reservoir of Gram-negative bacteria (20). However, translocated LPS may also originate from the environment through the ingestion of common food items (21–23). Erridge and colleagues found that several common processed food items had significant amounts of TLR4-stimulants (21–23) suggesting that modern diets, characterized by processed- and pre-packaged foods, are associated with increased oral exposure to stimulants of TLR4, such as LPS. However, it has also been observed that different bacteria produce different types of LPS, some of which are less likely to elicit an inflammatory response through the classical LPS receptor, TLR4 (24, 25), corroborating with findings that concentrations of TLR-stimulants are low in healthy murine ileum (26).
To the best of our knowledge, few studies to date have explored systematically over several weeks whether LPS given through the oral route of mice can influence metabolic and inflammatory processes evoked by a high-fat diet. Given the higher tolerance of mice to LPS compared to humans, we administered a high yet physiologically relevant dose of LPS derived from E. coli through the drinking water to mice subjected to either a high-fat western-style diet (WSD) or a standard low-fat chow diet (SC). Our primary objective was to ensure that the mice faced a substantial LPS challenge and to examine whether LPS could induce systemic low-grade inflammation. Additionally, we hypothesized that the consumption of a high-fat western-style diet would accelerate the impact of LPS exposure on the development of low-grade inflammation by promoting the translocation of LPS into circulation.
Our findings indicate that intestinal LPS can indeed trigger a systemic inflammatory response; however, this effect is modest and transient. Over the long term, exposure to LPS did not influence metabolic dysregulation, as evidenced by normal body weight increase and glucose regulation. Consequently, we conclude that LPS is unlikely to play an active role in promoting metabolic dysfunction and chronic low-grade inflammation in a physiologically relevant setting, where LPS is administered in a manner that does not surpass the protective mechanisms associated with the intestinal barrier.
Methods
Animal experiments
Mice (C57BL/6JRj) were bred in-house by crossbreeding female wild-type (WT) with male heterozygous NF-κB-luciferase reporter mice (NF-κB-luc) (27). Female mice (C57BL/6JRj) for breeding were purchased from Janvier Labs (Le Genest-Saint-Isle, France). Both NF-κB-luc and WT mice were used in the experiments. All mice were housed in individually ventilated cages (Innocage; Innovive, San Diego, CA) supplied with hardwood chips as bedding in a temperature and humidity-controlled room (24°C ± 2, 50% RH ±5) on a 12 h light–dark cycle, with free access to food and water. Diets used in the experiments were standard chow (SC) (3.5 kcal/g; 7.5 E% fat [soy oil], 17.5 E% protein [whey powder and soy protein concentrate], 75 E% carbohydrates [soy and various grains], RM1, Special Diets Services, Essex, United Kingdom), or a Western-style diet (WSD) (Research Diets, New Brunswick, NJ) D12079B (4.5 kcal/g; 41 E% fat [milk fat and corn oil] 15 E% protein [casein], 44 E% carbohydrates [sucrose and corn starch/maltodextrin]). Detailed diet composition is provided in the Supplementary Tables S1, S2. Estimated daily water intake per animal was 4–6 mL. Escherichia coli O55:B5 LPS was from Sigma-Aldrich (St. Louis, MO; Cat# L2880).
Tolerance of oral LPS and detection of LPS in the small intestine
Nineteen female eight-month-old WT mice were divided into four groups, three experimental groups (n = 4/group) and one control group (n = 7). The three experimental groups were all supplemented with the same dose of LPS (in the drinking water 0.33 mg/mL; ~2 mg LPS/mouse/day), using three different exposure times (1, 4, or 8 days of LPS administration). The control was given water only. All mice were fed a standard chow diet until termination with subsequent sampling of intestinal content for LPS analysis. Body weights and general welfare (activity, body posture, self-grooming) were recorded during the LPS-administration.
Effect of long-term oral LPS and western-style diet on obesity and insulin resistance
Sixty-five male mice aged 22 weeks were first randomly divided into two dietary groups receiving either western-style diet (WSD) or low-fat standard chow (SC) diet for 2 weeks. After 2 weeks, half of the WSD fed mice received LPS supplemented in drinking water (0.33 mg/mL, WSD-LPS), and the rest were given water only (WSD-W). The same set up was chosen for the SC-fed mice; half of the SC-fed were given LPS in the drinking water (0.33 mg/mL, SC-LPS), whereas the rest were only given water (SC-W). These four groups (WSD-LPS, WSD-W, SC-LPS and SC-W; n = 16–17 in each group) where then followed for five additional weeks until termination at week 7. Assuming a water intake of 6 mL/day, each mouse in the LPS-groups was exposed to ~2 mg LPS/day (28). Each group consisted of eight WT mice and eight NF-κB-luc mice. Body weight was recorded once a week, and food consumption every 2–3 days. From our own experience, chow-pellets cause ~15% more food spillage (food crumbles collected from cage beddings) than western diet-pellets when measuring food intake from cage feeders. Thus, we subtracted 15% from calculated energy intake for the SC diet-groups. Intraperitoneal insulin tolerance test (IpITT) and oral glucose tolerance test (OGTT) were performed in non-anesthetized mice in weeks 5 and 6, respectively. Mice were euthanized in week 7 of the experiment, corresponding to 5 weeks after LPS initiation, and tissue and blood samples were collected from fed mice.
Detection of orally administered LPS in plasma
Fifteen female WT 6 month-old mice were divided into two experimental groups (LPS) with two different LPS doses: 0.1 (n = 5) or 1 mg/mL (n = 5) and a control group receiving regular tap water (n = 5) for 8 days. Mice were fed a standard chow diet, and they were not fasted before sampling or termination.
Ethics statement
All animal experiments were performed according to the national guidelines for animal welfare, and the protocol was approved by the Norwegian Food Safety Authority (FOTS ID 15600). Animal welfare was monitored by daily assessment of water and food intake, stool and fur quality, activity level and behavior.
Tissue and blood samples
Mice were anesthetized by intraperitoneal (i.p.) injection of a mixture (10 μL/g mouse) denoted ZRF cocktail (Zoletil Forte; Virbac, Carros, France, Rompun; Bayer, Oslo, Norway and Fentadon; Eurovet Animal Health, Bladel, The Netherlands) with the active substances Zolazepam (32 mg/kg), Tiletamin (32 mg/kg), Xylanine (4.5 mg/kg) and Fentanyl (26 μg/kg), followed by cardiac puncture and blood collection from the heart (0.5–1 mL). Mice were then euthanized by cervical dislocation before dissecting out the organs. Luminal content for LPS assessments in the small intestine were collected, beginning at the base of the stomach, from the proximal 2 cm of the small intestine, and the distal 2 cm of the small intestine, respectively for duodenum and ileum. The protocol for preparation of the luminal content samples is derived from (29). Briefly, samples were diluted 1:4 (weight:volume) in sterile phosphate-buffered saline (PBS) and centrifuged at 13,000 g for 20 min before decanting the supernatant and storage of samples at −80°C before continuing the protocol at day of analysis. Whole intestinal samples for the IAP enzyme assay were collected from the next proximal 3 cm of the duodenum and cut into 1 cm pieces. Samples were snap frozen and stored at −80°C until analysis. Plasma from cardiac blood collected in an EDTA-covered syringe with a 25G needle was prepared by centrifugation at 6,000 g, 4°C for 10 min and stored at −80°C for further analyses.
In vivo imaging of NF-κB activity
Imaging of transgenic mice expressing luciferase was performed with an IVIS Lumina Series III (PerkinElmer, Waltham, MA). Before imaging, the fur of the abdomen was shaved, and the mice anesthetized with 2.5% isoflurane (IsoFlo Vet 100% Isoflurane, Zoetis, Kalamazoo, MI). Mice were imaged in a fed state at 10 min post luciferin injection (15 mg/mL solution, 150 mg/kg mouse; D-Luciferin, Biosynth, Basel, Switzerland) with 5 min exposure time.
Intraperitoneal insulin tolerance test and oral glucose tolerance test
When testing for insulin resistance, mice were fasted for 4 h and i.p. injected insulin (0.1 U/mL solution, 0.75 U/kg mouse; human insulin, Sigma-Aldrich, Schnelldorf, Germany) was followed by glucose measurements. When testing for glucose tolerance, mice were fasted for 6 h and given an oral bolus of glucose (0.2 g/mL solution, 2 g/kg mouse; D-glucose, Sigma Aldrich). For both tests, blood glucose from tail vein blood samples were quantified (Accu Chek Aviva, Roche Diagnostics, Mannheim Germany) at 0, 15, 30, 60, 90, and 120 min post insulin or glucose administrations.
Measurement of plasma LBP, TNFα, and IL-6
Plasma LBP levels were measured by mouse LBP enzyme-linked immunosorbent assay (ELISA) kit (Biometec GmbH, Greifswald, Germany) and with mouse IL-6/TNFα Quantikine ELISA kit (R&D Systems, Minneapolis, MN). Plasma samples were diluted and assayed according to the manufacturer’s instructions.
Gene expression analysis by RT-qPCR
Total mRNA was extracted from RNAlater preserved ileal mucosa samples following the NucleoSpin RNA/Protein (Macherey-Nagel, Germany) protocol with some minor alterations. Samples were homogenized in RP1/BME mix with a 23 G needle; each centrifugation step were increased by 1–2 min at 11,000 or 13,000 × g; and RNA was eluted in 50 μL RNase-free H2O. For cDNA synthesis 200 ng of total mRNA was digested and reverse-transcribed with the iScript cDNA Synthesis kit (Bio-Rad Laboratories, CA, United States) according to the manufacturer’s instructions and used in Quantitative PCR on the LightCycler 480 system (Roche Diagnostics, Germany). The detection gene expression was performed using 5× HOT FIREPol EvaGreen qPCR Supermix (Solis BioDyne, Estonia). The following target genes were assessed: Tumor necrosis factor alpha (Tnfa) and interleukin 1 beta (Il1b). The housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (Gapdh) was used as a reference, and relative mRNA expression for both target genes in each sample were calculated using the formula where E denotes the primer efficiency. Primer sequences for Tnfa, Il1b, and Gapdh can be found in Supplementary Table S3. The mRNA extracts from all 16 mice per group were analyzed individually.
Measurement of duodenal IAP activity
IAP activity was measured with the SensoLyte p-nitrophenyl phosphatase (pNPP) Alkaline Phoshatase Kit (Anaspec, Fremont, CA) according to recommendations of the manufacturer. Briefly, duodenal tissue was homogenized in lysis buffer (400 μL/50 mg tissue), centrifuged at 10,000 g for 15 min at 4°C, before diluting the supernatant 1:800. Samples were incubated with pNPP reaction mixture for 20 min, and absorbance was read at 405 nm.
Measurement of plasma flagellin and LPS-specific immunoglobulins
Flagellin- and LPS-specific IgG levels were quantified by indirect ELISA based on previously published protocols (30, 31). Briefly, microtiter plates were coated overnight at 4°C with purified Escherichia coli (E. coli) flagellin (100 ng/well, orb107725, VWR, United Kingdom) or E. coli LPS (2 μg/well; serotypes O55:B5 [L4524] and O128:B12 [L2887], Sigma-Aldrich, St.Louis, MO, USA) in 0.05M carbonate-bicarbonate buffer pH 9.6. Plates were washed with wash buffer (PBS + 0.05% Tween®20 (Sigma-Aldrich, St.Louis, MO, USA)) and blocked for 1 h at 4°C with blocking buffer (1X ELISA Assay Buffer, Invitrogen™, Vienna, Austria (flagellin and LPS O128:B12 plates); 1X Reagent Diluent, R&D Systems, Minneapolis, MN, USA, (LPS O55:B5 plate)). Plasma samples were diluted 1:200 in corresponding blocking buffer and then applied to wells after another wash followed by incubation at 37°C for 1 hour. After incubation and another wash, the wells were incubated with anti-mouse IgG coupled to horseradish peroxidase (1:1000; Southern Biotechnology, Birmingham, AL, USA) at 37°C for 1 hour. Plates were washed before tetramethylbenzidine substrate (Thermo Scientific, Rockford, IL, USA (flagellin and LPS O128:B12 plates); R&D Systems, Minneapolis, MN, USA, (LPS O55:B5 plate)) was added to well for five minutes at room temperature and stopped using sulfuric acid (0.16M for flagellin and LPS O128:B12 plates; 1M for LPS O55:B5 plate (R&D Systems, Minneapolis, MN, USA)). Total immunoglobulins were quantified immediately using a microaplate reader (SpectraMax® M2, Molecular Devices, California, USA). Absorbance was measured at 450 nm and 540 nm, and IgG-levels are reported as optical density (OD) units corrected for background value blanks.
Quantification of TLR4-stimulants in plasma and small intestine
Biologically active LPS, i.e., LPS that induced TLR4 signaling, were determined in plasma, luminal duodenum, and ileum samples by employing a colorimetric cell-based reporter assay (HEK-Blue LPS Detection Kit 2, InvivoGen, San Diego, CA). Quantification was done according to the manufacturer’s instructions. In brief, diluted plasma and intestinal samples were added to human embryonic kidney (HEK-293) cells that express mTLR4 and an NF-κB-inducible secreted embryonic alkaline phosphatase reporter gene. After 18 h incubation, cell culture media was applied to QUANTI-Blue medium from said assay to measure alkaline phosphatase activity. Plates were read at a wavelength of 620 nm. The detection limit was 0.03 ng/mL according to the kit manufacturer.
Statistical analysis
Statistical analyses were performed using Prism version 9.4 (GraphPad Software, San Diego, CA). The results were expressed as mean ± standard error of the mean. The significance of difference between two groups was evaluated using the two-tailed Student’s t-test. Differences in continuous parameters among multiple groups were calculated using the one-way analysis of variance (ANOVA) with post hoc Tukey’s multiple comparisons test. Two-way ANOVA was used to assess effects of LPS exposure (LPS or water) in the two diet groups (WSD or SC). Significant interactions between LPS exposure and diet in two-way ANOVA were followed by the assessment of simple main effects (effect of LPS or water within each diet groups and effect of diet within LPS or water exposure groups) with Tukey’s correction for multiple comparisons. The results were considered as statistically significant at p < 0.05.
Results
Exogenous LPS reached the small intestine and mice tolerated continuous oral LPS administration
Our first step was to evaluate tolerance and acceptance to oral LPS. Purified E. coli LPS was administered in drinking water (0.33 mg/mL; ~2 mg LPS/mouse/day) for 1, 4, or 8 days. Daily visual inspection and objective measurements of water intake, weight development and general physical wellbeing established no concerns regarding potential toxic effects of LPS. To validate whether orally ingested LPS reached the intestine, we quantified small intestinal luminal LPS content using mTLR4 transfected reporter cells (HEKBlue™ TLR4 cells). LPS levels were significantly elevated in the small intestine of LPS administered mice compared to non-LPS exposed control mice, and most likely reached a steady state LPS concentration around 4 days following LPS exposure (p = 0.001 and p < 0.001 for duodenum and ileum, respectively) (Figure 1A).
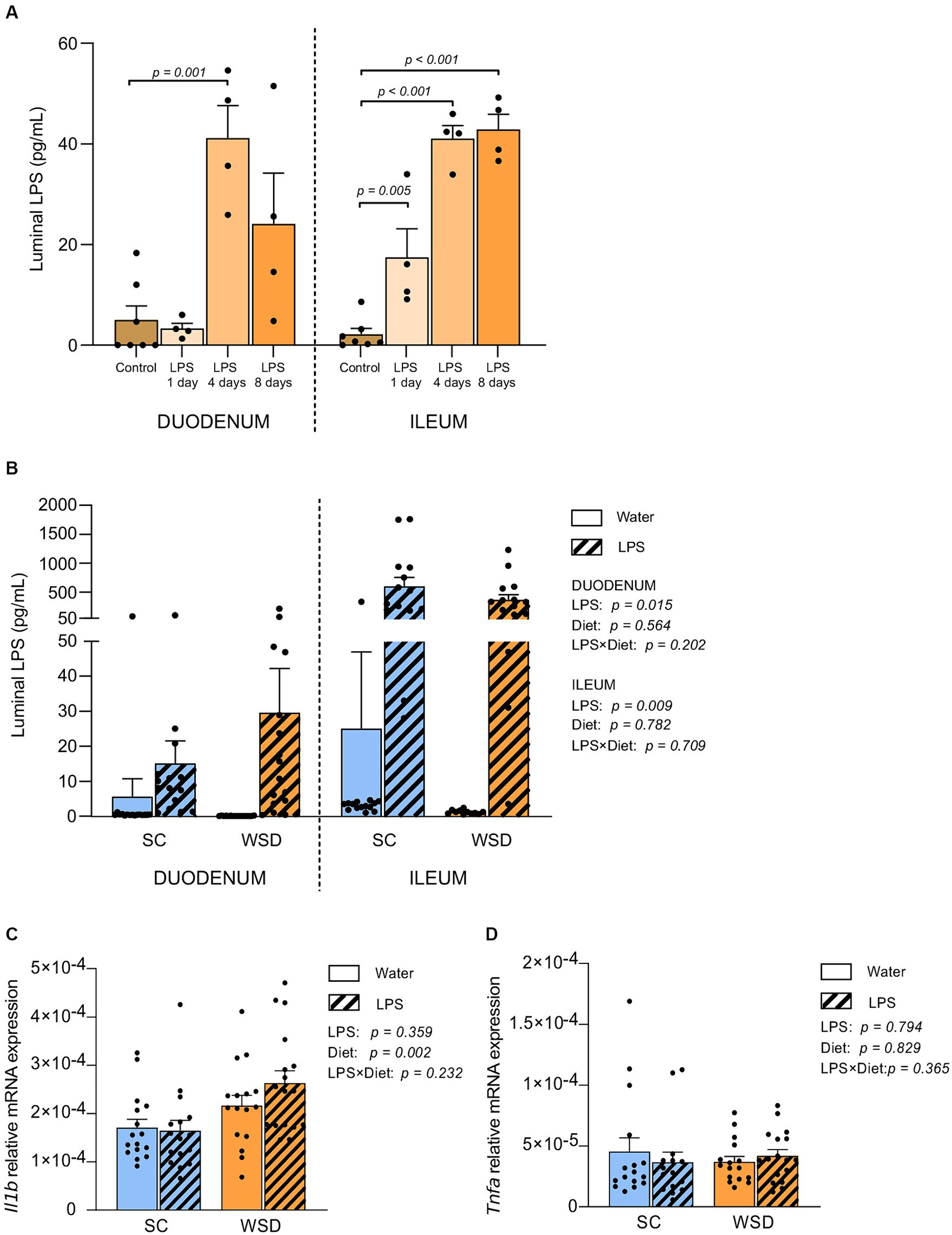
Figure 1. LPS content measured by the HEK-Blue™ TLR4 assay, and gene expression in the small intestine. (A) Duodenal and ileal LPS levels in luminal content from SC fed mice given LPS (0.33 mg/mL, estimated daily intake of 2 mg LPS) through drinking water for 1, 4 or 8 days. N = 4–7 per group. One-way ANOVA compared to the mean of control group (non-treated mice). (B) Duodenal and ileal LPS levels in mice fed WSD or SC diet for 7 weeks, and 35 days of oral LPS administration (0.33 mg/mL, estimated daily intake of 2 mg LPS), n = 11–17. Relative mRNA expression of (C) interleukin 1 beta (Il1b) and (D) Tumor necrosis factor alpha (Tnfa) in mucosa from the small intestine. n = 15–17. Two-way ANOVA with assessment of simple main effects (treatment, no-LPS versus LPS; diet, WSD versus SC) and interaction effect (treatment × diet) with Tukey’s correction for multiple comparisons (B–D). Data are presented as mean ± SEM. LPS, lipopolysaccharide; SC, standard chow; WSD, western-style diet.
Next, we assessed the levels of biologically active LPS (i.e., LPS recognized by TLR4 on HEK-Blue™-4 cells) in the intestine of animals from the long-term oral LPS and WSD experiment. Thirty-five days of oral E. coli LPS administration led to significantly higher levels of bioactive luminal LPS in both duodenum (p = 0.015) and ileum (p = 0.009) compared to non-LPS exposed mice for both diets (Figure 1B).
Western style diet (WSD) diet, but not oral LPS administration, led to a significant, but minor increase in IL-1β mRNA expression in small intestinal mucosa
Inflammatory cytokines are signaling proteins released from innate immune cells with key roles in regulating immune responses often elicited by microbial exposures. To address whether the elevated LPS levels in the ileum affected local inflammatory cytokine expression following a 5 week intervention with LPS administered in the drinking water fed western style diet (WSD) or a low-fat standard chow diet (SC). We measured the expression of TNFα and IL-1β in mucosal ileum tissue (32). Neither TNFα nor IL-1β were affected by LPS administration (Figures 1C,D). However, we detected a statistically significant increase in IL-1β gene expression in WSD-fed animals compared to SC-fed groups (p = 0.002; Figure 1C). TNFα was not expressed differently in animals fed WSD or SC (Figure 1D).
A WSD promoted weight gain, but oral LPS administration had no additional effect on body weight development or food intake
As expected, the WSD fed mice (WSD-W and WSD-LPS) gained significantly more weight than standard chow (SC) fed mice (SC-W and SC-LPS) during the 7 week dietary intervention (Figures 2A,B, p < 0.001). However, LPS administration did not affect weight gain (p = 0.413). Total body weight gain corresponded with total energy intake as the WSD fed mice had a higher energy intake than the SC mice (p = 0.002, Figure 2D), and administration of LPS had no significant effect on energy intake (p = 0.836, Figure 2D). Weekly energy intake of mice receiving WSD had the highest energy intake throughout the experiment regardless of LPS exposure, with a small exception at week 4 where no difference between groups was observed (Figure 2C).
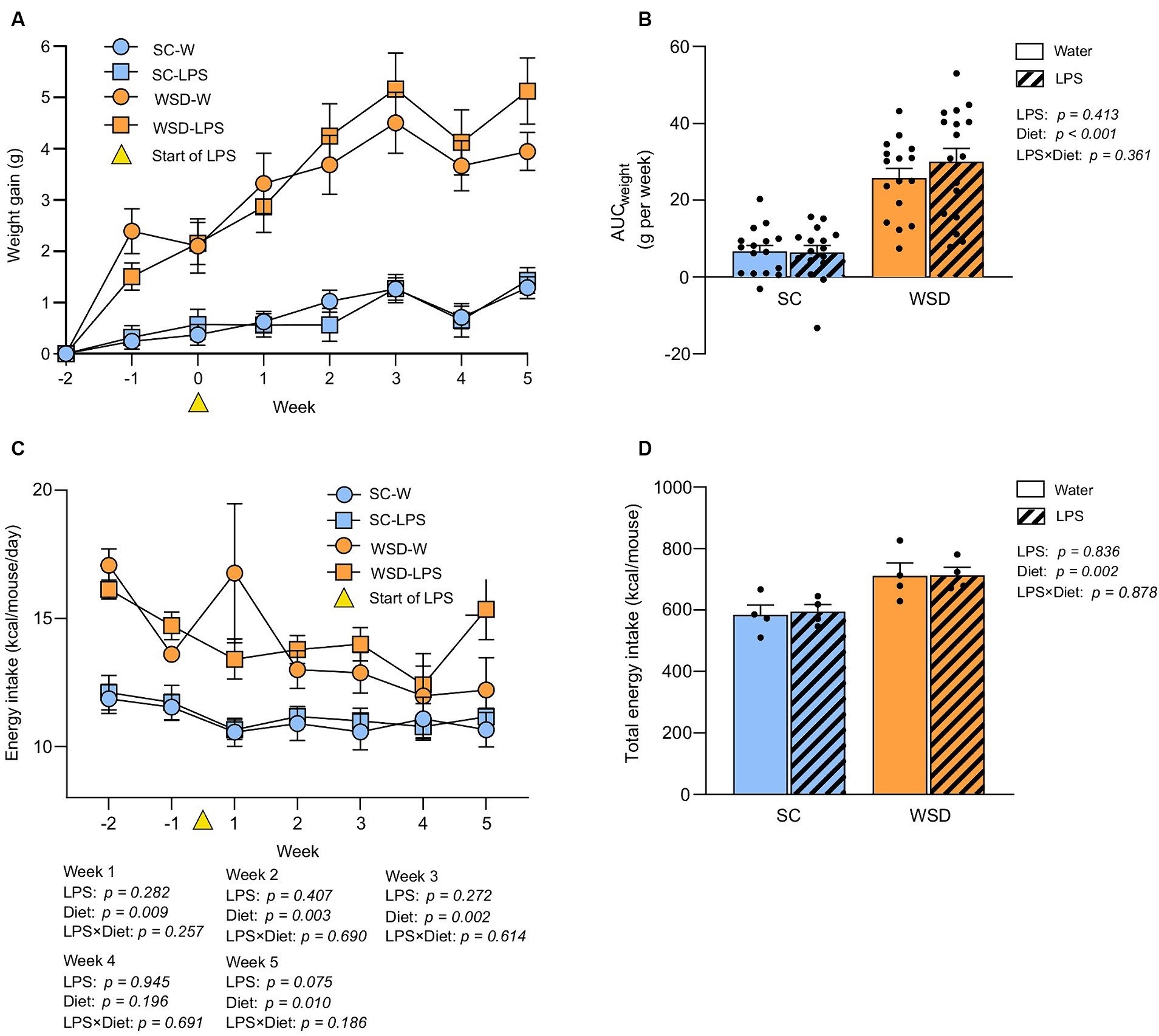
Figure 2. Weight and food intake assessed in mice fed WSD or SC diet for 7 weeks, and 35 days of oral LPS administration (0.33 mg/mL, estimated 2 mg LPS/mouse/day). (A) Body weight gain (G) of WSD or SC fed mice for 7 weeks. Week 0 (yellow triangle) indicates the start of LPS administration. (B) AUC body weight gain (g). (C) Weekly energy intake per diet group expressed as kcal/mouse/day. Week 0 (yellow triangle) indicates the start of LPS administration. (D) Total energy intake per diet group expressed as kcal/mouse. Two-way ANOVA with assessment of simple main effects (treatment, no-LPS versus LPS; diet, WSD versus SC) and interaction effect (treatment × diet) with Tukey’s correction for multiple comparisons (B–D). Data are presented as mean ± SEM. N = 16–17 per group. AUC, area under the curve; LPS, lipopolysaccharide; ns, non-significant; SD, standard chow; WSD, western-style diet.
Whole-body glucose metabolism and intestinal IAP activity were not affected by oral LPS administration or WSD feeding
We hypothesized that long-term exposure to orally administered LPS would elevate plasma LPS and induce insulin resistance as this would sufficiently challenge the tolerance of mice towards LPS. Accordingly, we evaluated the effect of chronic oral LPS administration on glycemic control and insulin resistance. We took advantage of the oral glucose tolerance test (OGTT), and the intraperitoneal insulin tolerance test (IpITT) to evaluate glucose regulation and whole-body insulin sensitivity, respectively, (Figures 3A–D). The OGGT showed no significant differences between different diets (p = 0.625) or treatment with LPS (p = 0.220). The effect of diet and LPS exposure collectively was also non-significant (p = 0.423; Figures 3A,B). In the IpITT the glucose curve (AUC) following insulin injection showed a clear effect of diet type (p = 0.003) where WSD mice displayed higher values than SC-fed mice, but with no effect of LPS exposure (p = 0.856) or any interacting effects between the two experimental factors (p = 0.712). The significant higher AUC values seen with WSD-feeding is likely attributed to the higher fasting glucose levels we observed in the WDS animals compared to SC-fed mice at the time point when doing the IpITT (p = 0.001, with one-way ANOVA).
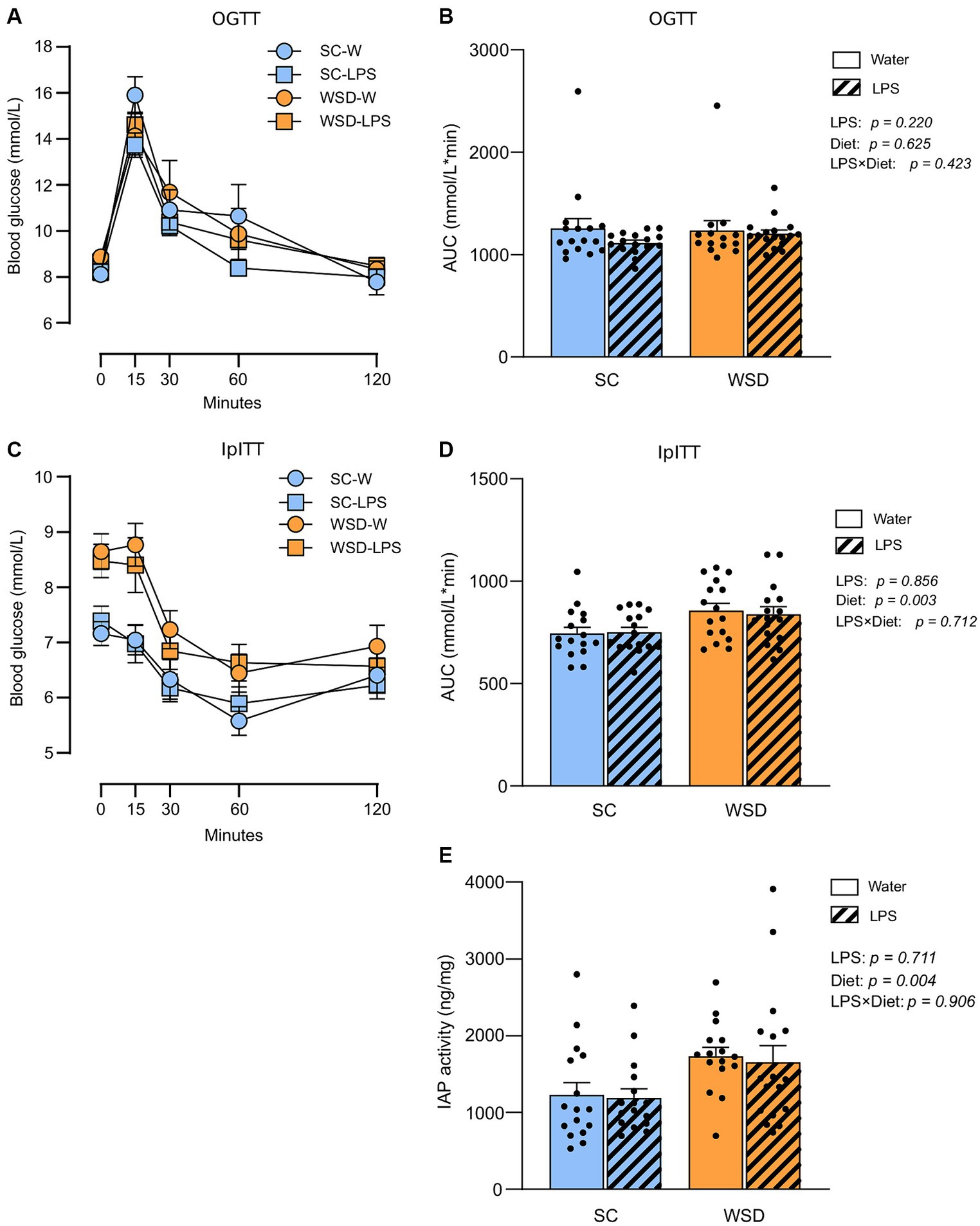
Figure 3. Oral glucose tolerance test (OGTT) and intraperitoneal insulin tolerance test (IpITT) in mice fed WSD or SC diet for 7 weeks, and 35 days of oral LPS administration (0.33 mg/mL, estimated 2 mg LPS/mouse/day). (A) OGTT. (B) Total glucose excursion after OGTT expressed as area under the curve. (C) IpITT. (D) AUC IpITT. (E) IAP activity from duodenal lumen content. Two-way ANOVA with assessment of simple main effects (treatment, no-LPS versus LPS; diet, WSD versus SC) and interaction effect (treatment × diet) with Tukey’s correction for multiple comparisons (B,D,E). Data are presented as mean ± SEM. AUC, area under the curve; IAP, intestinal alkaline phosphatase; LPS, lipopolysaccharide; ns, non-significant, WSD, western-style diet.
Intestinal alkaline phosphatase (IAP) detoxifies LPS by removing phosphate groups from the lipid A moiety, thereby inhibiting LPS-TLR4 interaction (33). Earlier studies have described increased IAP activity in high-fat fed rodents (34–36), or found increased IAP activity to be correlated with elevated LPS luminal concentrations in the murine small intestine (37). We hypothesized that IAP secretion would rise with increasing luminal LPS levels (37, 38) and fat feeding (39). We found no effect of LPS treatment (p = 0.711), or any interaction effect between diet and LPS (p = 0.906) (Figure 3E). However, the WSD groups displayed statistically significant higher IAP activity than the low-fat SC groups (p = 0.004).
NF-κB activity in liver- and abdominal region was transiently upregulated by oral LPS administration and WSD feeding
The transcription factor NF-κB is a primary regulator of inflammatory responses, and its activation is instrumental for the proinflammatory state associated with obesity and the detrimental effects on insulin resistance (2, 40). We hypothesized that mice fed the WSD would display increased global activation of the NF-κB signaling pathway, and that exposure to LPS would generate further increases in NF-κB activity. By exploiting a transgenic reporter model for NF-κB activity (NF-κB luc) (27), we assessed by in vivo imaging NF-κB activity in mice from all the experimental groups at a series of time points throughout the experiment.
We observed a significant and transient increase in NF-κB activity from the liver in LPS administered mice 24 h after start of oral LPS administration compared to mice receiving only water (p = 0.010) (Figures 4A,B). Additionally, we found that WSD significantly led to a higher NF-κB activity in the liver at this time point (p = 0.033), but the analyses revealed that no interaction effect between diet and LPS (p = 0.365) was evident, meaning that LPS was not dependent on WSD for the increased liver NF-κB activity. When comparing each experimental group, only SC-W and WSD-LPS were significantly different (p = 0.006), however, the results showed a trend between the two WSD groups at day 1 (p = 0.059). After 1 week no differences between groups were observed. However, we observed again a significant increase in NF-κB activity in the WSD groups relative to SC-fed control mice at day 35 (p = 0.004). The abdominal region includes the intestines which is connected to the mesentery. The mesenteric tissue is rich in lymph nodes and functions as the “first pass” organ for microbial substances entering the lymph from the intestinal tract. This immediate exposure to substances coming from the intestinal lumen, such as LPS, may therefore trigger NF-κB activation in lymphatic immune cells. Our results showed that NF-κB activity was elevated in the abdominal region throughout the experimental period in mice fed WSD (Figure 4B), with no additional increase caused by LPS. Intriguingly, we observed on the contrary that LPS treatment reduced the abdominal NF-κB signal at day 35 (p = 0.013), causing speculations of a compensating effect against LPS exposure with time.
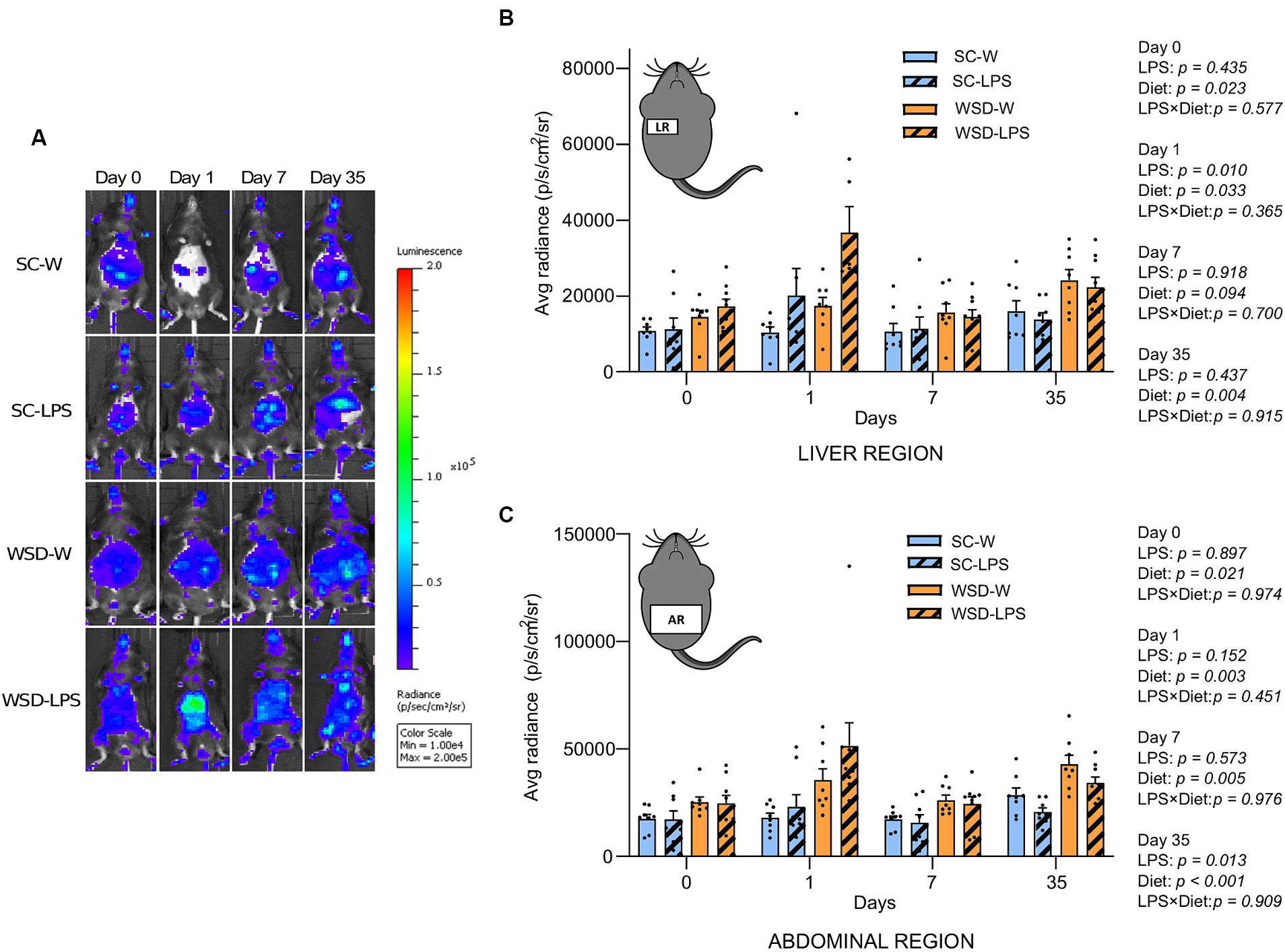
Figure 4. In vivo imaging of NF-κB activation during 35 days of LPS administration in drinking water (0.33 mg/mL, estimated 2 mg LPS/mouse/day). Day 0 indicates start of oral LPS administration Graphs display the photon emission in region of interest (ROIs) representing NF-κB activity in the liver region (A) and abdominal region (B) N = 8 per group. (C) Panel displays representative images of one mouse per group at different time points. Color bar indicates intensity of light emission from the mice. Two-way ANOVA with assessment of simple main effects (treatment, no-LPS versus LPS; diet, WSD versus SC) and interaction effect (treatment × diet) with Tukey’s correction for multiple comparisons (B,C). Data are presented as mean ± SEM. LPS, lipopolysaccharide; NF-κB, nuclear factor-kappaB; SC-W, standard chow + water; SC-LPS, standard chow + LPS; WSD, western-style diet; WSD-W, western-style diet + water; WSD-LPS, western-style diet + LPS.
As there are other inflammatory pathways independent of NF-κB activity, we included plasma IL-6 and TNFα measurements from day 35 to investigate if systemic inflammation was affected by the different treatments. Levels of plasma IL-6 were inside normal range for adult mice in all experimental groups (41, 42), and the results showed no significant differences between the groups (Figure 5B). Plasma TNFα levels were not possible to calculate as all samples ranged below detection limit.
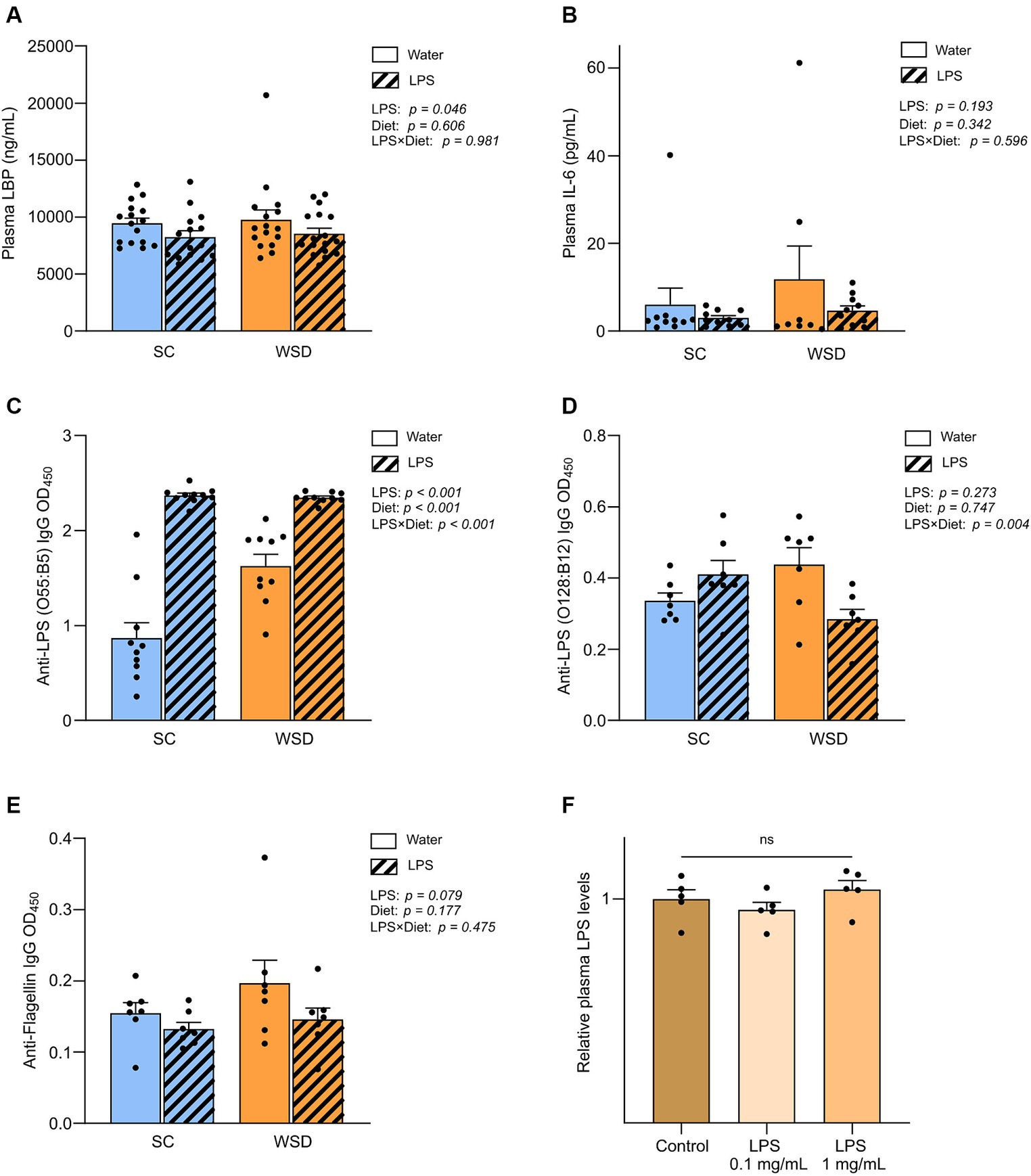
Figure 5. Plasma measurements of LBP, IL-6, immunoglobulins and LPS. (A) Measurement of plasma LBP, (B) IL-6, (C) Anti-LPS (O55:B5) IgG, (D) Anti-LPS IgG (O128:B12) and (E) Anti-flagellin IgG in mice fed WSD or chow diet for 7 weeks, and 35 days of oral LPS administration (0.33 mg/mL, estimated 2 mg LPS/mouse/day). Two-way ANOVA with assessment of simple main effects (treatment, no-LPS versus LPS; diet, WSD versus SC) and interaction effect (treatment × diet) with Tukey’s correction for multiple comparisons (A–E). (F) Relative plasma LPS levels with normalized values from mice administered with oral LPS for 8 days compared to control group (water). One-way ANOVA compared to the mean of control group (non-treated mice). Data are presented as mean ± SEM. N = 16–17 (LBP), 7 (IL-6, anti-flagellin IgG and anti-LPS IgG), and 5 (LPS) per group. IgG, immunoglobulin G; LBP, LPS-binding protein; LPS, lipopolysaccharide; ns, non-significant; WSD, western-style diet.
Oral LPS administration lowered lipopolysaccharide-binding protein levels in plasma, but LPS was not detected in circulation
Lipopolysaccharide-binding protein (LBP) is stable in blood and has a longer half-life in blood than LPS and is thus regarded as a useful biomarker of LPS exposure in circulation (43–45). Measurements of plasma LBP revealed firstly that the diet as such did not affect LBP levels (p = 0.606) (Figure 5A). However, in contrast to what we hypothesized exposure to LPS led to significantly lower levels of circulating LBP compared to non-LPS exposed mice (p = 0.046). To further explore the unexpected drop in LBP levels with LPS administration, we included a smaller follow-up experiment to measure LPS levels directly in plasma using a mTLR4-transfected cell-based colorimetric assay after 8 days of oral LPS administration (0.1 mg/mL and 1 mg/mL, estimated daily intake of 0.5 mg and 5 mg LPS, respectively). We were not able to quantify plasma LPS levels in any of the groups because values were below detection limit (0.001 ng/mL), indicating that LPS was either absent or bound up, making the LPS molecule unavailable for reacting with the TLR4-receptor. Relative comparison between group means showed that there were no significant differences in plasma LPS levels (p = 0.288) (Figure 5E).
Plasma antibody levels against LPS (E. coli O55:B5) increased in oral LPS administered animals, but bacterial flagellin antibodies were not altered by oral LPS administration or WSD feeding
As the bacterial protein flagellin, and LPS, are usually excluded from absorption by the gut epithelium, a period of gut-barrier dysfunction translocation of LPS and flagellin across the intestinal mucosa may activate the adaptive immune response resulting in the generation of anti-flagellin and anti-LPS antibodies. As circulating levels of flagellin- and LPS specific immunoglobulin G (IgG) may serve as markers of long-term systemic exposure to flagellin and LPS, we sought to measure their presence in the plasma samples as an indirect measure of long-term gut leakage. Since we administered LPS derived from E.coli, serotype O55:B5, we specifically looked for antibodies raised against this LPS. In addition we assessed antibodies against a related LPS (E.coli serotype O128:B12) and flagellin. The latter two were chosen as representatives of a general gut leakage. Antibody levels against E. coli O55:B5 LPS were significantly and robustly increased in the LPS administered mice compared with control mice, p < 0.001 (Figure 5C). Interestingly, levels of antibodies against LPS serotype O128:B12 or flagellin were not significantly altered when comparing diet groups and/or LPS exposure (Figure 5E). Intriguingly, we observed a trend that anti-flagellin levels were in fact lower in LPS administered mice compared to controls (p = 0.079), in line with our observations on LBP-levels.
Discussion
Consumption of high-fat, low-fiber, western-style diets (WSD) has been linked to obesity, low-grade inflammation, and poor glucose control, often attributed to the “leaky gut” hypothesis stating that intestinally derived bacteria or bacterial products can contribute to systemic inflammation (6, 46, 47). We expected that the higher content of dietary fat in the WSD would facilitate transportation of LPS over the intestinal wall and lead to systemic low-grade inflammation in mice administered LPS in the drinking water. We hypothesized that continuous oral LPS administration would further aggravate the proposed pro-inflammatory effect of the WSD, and even cause symptoms of metabolic disease in standard chow (SC)-fed mice. However, LPS supplementation did not assert any adverse consequences on glucose metabolism when testing for glucose intolerance and insulin resistance. Further, we did not detect strong indications of low-grade inflammation except for a transient upregulation of NF-κB activity in LPS administered mice during the first day of oral LPS exposure. These findings cohere with a comprehensive study in both mice and humans, which explored the impact of orally ingested LPS on intestinal barrier function and systemic inflammation (26). The authors found that chronic oral LPS intake promoted hepatic inflammation regardless of no substantial impact on energy intake, weight gain, intestinal inflammatory markers, intestinal barrier function, or glucose control assessed through HOMA-IR. Collectively, our results demonstrate that LPS situated in the intestine was not able to initiate systemic low-grade inflammation in animals receiving either WSD or SC.
There are a limited number of studies that address the direct link between small intestinal LPS concentrations and circulating LPS levels in models of high fat feeding. Exceptions are the forementioned study by Faraj et al. (26), and another study related to ours, where high fat fed mice were orally administered with E. coli strains that colonized the mouse gut and had differential expression of LPS (potent or weak). E. coli colonization led per se to increased inflammation in adipose tissue and dysregulated glucose control, but the latter effect was not dependent on LPS, which to a considerable extent corroborated with our findings (48). Yet, intestinal-derived LPS has been assigned a critical role in metabolic endotoxemia and low-grade inflammation (4, 6, 49, 50). Our measurements of LPS in luminal content from the small intestine showed that the number of TLR4-stimulants increased significantly in mice exposed to oral LPS obtained from E. coli both short term and long term, whereas non-exposed mice possessed only minor intestinal quantities of TLR4-stimulants. These findings may reflect that in our animals, most of the inherent LPS variants found in the intestinal lumen are not potent stimulators of TLR4. This observation is in line with other studies stating that the greater part of normal intestinal Gram-negative bacteria is of species from the bacteroidetes family that do not stimulate TLR4 signaling (24, 25). Hence, the ingested E. coli-derived LPS contributed to the elevated levels of TLR4-stimulatory environment of the small intestine in our experiment.
Although we observed large quantities of LPS in the small intestine, we could not through direct methods detect biologically active, TLR4-stimulating LPS in blood plasma. It should be noted that most studies supporting HFD-induced metabolic endotoxemia are based on LPS assessments by the limulus amebocyte lysate (LAL) assay (4, 6, 17). This method is dependent on the properties of the lipid A moiety and does not reflect the LPS’ ability to stimulate inflammation through the TLR4 signaling pathway, unlike the cell-based assays we have used. Consequently, the LAL test may overestimate the actual levels, and be misleading when correlating circulatory LPS levels and metabolic inflammation since the measured LPS is likely incapable of stimulating proinflammatory pathways in humans (51).
To further explore whether diet and LPS exposure would lead to other perturbations in the intestine we measured leakage of bacterial products into the circulation. In this context, we measured the plasma-levels of antibodies against LPS from serotype O55:B5, which was identical to the LPS administered in the water, and E.coli derived flagellin and LPS (O128:B12), the latter two representing general products from gut bacteria. Elevation in anti-flagellin and anti-LPS Ig concentrations have previously been observed in cases of chronic enteric conditions such as short-bowel syndrome (SBS) and inflammatory bowel diseases (IBD) (31, 52). Using flagellin- and LPS specific immunoglobulins as markers of long-term exposure to flagellin and LPS may therefore better capture long term gut leakage, than measuring either flagellin or LPS directly in blood as they can be more fluctuating (31). Antibodies against LPS O55:B5 was clearly higher in both mouse groups that had received LPS, suggesting that LPS given in the drinking water indeed has reached the circulation in sufficient quantities to raise an immune response against this particular LPS. However, the results also indicate that the presence of LPS in the circulation has later been neutralized and/or activated mechanisms of endotoxin tolerance. For flagellin and LPS (O128:B12) we see no increase in antibody-levels as a result of LPS administration. This indicates that LPS administration has not led to generally diminished intestinal barrier. On the contrary we observe a lower production of antibodies against flagellin in LPS administered mice. Collectively, our results imply that oral LPS exposure leads to only minor leakage into circulation of LPS and/or other bacterial products even with high-fat feeding, and that the animals quickly adapted to a higher LPS exposure, indicating acquired tolerance.
Supporting the notion that tolerance towards LPS was obtained is the unexpected results demonstrating reduced LBP levels in mice receiving oral LPS compared to controls not receiving LPS. Initially, this seemed contradictory as LPS promotes synthesis of LBP in liver and other tissues including the intestine (53–55). Therapeutic administration of high doses of LBP has also been shown to protect normal mice from LPS challenge (56). We can hypothesize that the low LBP levels are a consequence of acquired tolerance to chronic LPS administration, as previously demonstrated in a rat model of acute vs. chronic LPS exposure (57). Endotoxin tolerance is defined as the reduced capacity of a cell to respond to LPS after an initial exposure to this stimulus (58, 59). When the phenomenon endotoxin tolerance was introduced, the focus was largely on how treatments with low doses of LPS could prevent lethal outcomes of sepsis and fever. A broader understanding of endotoxin tolerance has been revealed through studies of possible beneficial mechanisms in areas of various chronic inflammatory diseases (60, 61) and other more specific areas such as cancer immunotherapy (58, 62). Endotoxin tolerance can be divided into early and late mechanisms of tolerance. Early tolerance is usually observed within hours of LPS administration and may last about a week. Early mechanisms involve altered cellular activation with suppression of cytokine production and fever independent of antibody production (63, 64). In contrast, late tolerance is induced many days after initiation of LPS treatment when early tolerance has generally ceased or declined. The molecular signature of endotoxin tolerance involves several mechanisms such as downregulation of TLR4 expression and decreased activation of intermediates in NF-κB signaling (58). Additionally, various negative regulatory molecules, for instance IL-1 receptor-associated kinase M (IRAK-M) and A20, are upregulated in cells tolerant to endotoxins and inhibit TLR signaling activation (58, 65, 66). Proposed mechanisms aside, exposure to immunostimulatory LPS has been shown to contribute to protection from immune-mediated diseases by modulating the immune system responsiveness (25). With the “hygiene hypothesis” in mind, early life exposure to potent immune activating LPS led through endotoxin tolerance to immune education and better protection against autoimmune disease than exposure to less inflammatory subtypes of LPS.
Had LBP concentrations been measured right after start of LPS exposure, we may assume that LBP levels would have been elevated in this initial phase as a protective mechanism after acute stress such as LPS challenge (53), before decreasing as a result of LPS tolerance. Moreover, our results demonstrated that plasma LBP levels were entirely independent on the assigned diet type and the final body weight of the mice and contrasts with many other studies demonstrating that high fat diets increase LBP levels in blood (16, 37). However, in line with our observations, a human intervention study showed that LBP levels were not elevated despite significant weight gain following a high-fat diet (11).
With regards to duodenal IAP, we initially hypothesized that this enzyme would have increased activity after oral LPS exposure to protect against toxic LPS activity (38, 67, 68). Surprisingly, IAP activity in duodenum was not affected by chronic LPS exposure despite elevated levels of LPS in the small intestine relative to non-LPS exposed animals. We instead observed a significant increase in small intestinal IAP activity in non-LPS exposed WSD-fed mice relative to SC-fed mice. Previous studies have shown that IAP regulates intestinal lipid absorption (34, 69, 70), and reduces weight gain in mice fed a high-fat diet (69). Moreover, IAP expression is increased in high fat diet fed mice, but it appears in some studies that this adaptive mechanism is not sufficient to counteract dietary fat stimulated LPS translocation from the intestine (33, 70).
NF-κB is a family of transcription factors instrumental in orchestrating the inflammatory response and is highly sensitive to LPS. We observed a temporary, inflammatory induction of NF-κB activation in the liver of mice receiving WSD-LPS diet after only one day of the feeding period. The transgenic NF-κB mice used in this study are sensitive to even minute changes in LPS quantity and can detect LPS levels in the range of 0.1–0.4 ng/mL using bioluminescent imaging tools (unpublished). This indicates that, if the orally derived LPS reached the bloodstream, the levels are below 0.1 ng/mL at the 5 week time point, which corresponds with our negative finding when measuring LPS directly in plasma with the HEK-Blue assay (detection limit 0.001 ng/mL). We can only speculate whether the upturn in NF-κB activation in both WSD-groups at the later stage of the experimental period might be indicative of an initiating, but not yet systemic, low-grade inflammation. This hypothesis is partly supported by another study in C57BL/6 mice which showed that the inflammatory response to a HF diet developed in two distinct phases; one immediate after a few days concurrent with an acute phase response, and a second increase after 12–16 weeks that correlated with WAT and muscle inflammation (71). The authors proposed that the initial response resulted from an acute and transient inflammation and activation of liver cells, which fits with our findings. As there are other inflammatory pathways independent of NF-κB activation, we analyzed plasma levels of two central proinflammatory cytokines: TNFα and IL-6. The results from these analyses supported our other findings that we could not find evidence of an ongoing low-grade systemic inflammation.
The results from our experiments point to that exposure to ingested LPS, high-fat diet or not, does not impair the intestinal barrier or trigger the development of chronic low-grade systemic inflammation. We wanted to lessen the possibility of missing whether our experimental factors could initiate a local intestinal inflammation that at a later stage could develop into a low-grade systemic inflammation. To do so we aimed to measure the state of intestinal inflammation through gene expression analyses of proinflammatory cytokines in the small intestinal mucosa. Others have found in murine models of intestinal inflammation, that TNFα and IL-1β appear to be protective during acute inflammation but exert pure proinflammatory functions in later stages of inflammation (32). In addition to studies researching the role of high fat diets and LPS in situations it is plausible that in subjects with weakened barriers (e.g., inflammatory bowel diseases such as Crohn’s disease or ulcerative colitis), leakage of LPS or other bacterial products from the intestine can have significant effects on systemic of low-grade systemic inflammation (see Introduction), both exposure to a western-style diet (72, 73) and intestinal LPS (74, 75) have previously been shown to increase intestinal inflammation through various possible mechanisms. When evaluating the local inflammatory status in small intestinal mucosa, we did not find any indication that WSD-feeding or oral exposure to LPS caused changes in proinflammatory factors of the intestinal immune defense.
Our results do not dismiss that LPS enter the circulation from the intestine, but the results point to the fact that the animals’ defense system managed the intruding LPS well and prevented, or slowed, the development of low-grade systemic inflammation. It is important to remember that these mice were metabolically healthy, and most likely had a robust intestinal barrier. The metabolic syndrome and associated diseases are characterized by a state of dyslipidemia. Recent studies have proposed that LPS is an relevant regulator of areas in lipid metabolism such as modifying plasma lipid levels (76) and activating cholesterol metabolism processes (77), further connecting LPS to dyslipidemia and systemic inflammation. Even though our results point to little or no metabolic dysregulation, a limitation of this study is that we did not explore LPS’ effect on lipid metabolism or markers of sustained inflammation such as gene expression in the liver or relevant biomarkers in plasma. It has been suggested that other factors in the modern lifestyle, such as exposure of processed food rich in certain food additives (e.g., emulsifiers), can lead to “leaky guts” and contribute to lifestyle diseases such as cardiovascular disease, cancer, and type 2 diabetes (30, 78). Hence, an uncontrolled increase in intestinal permeability to bacterial or other foreign molecules can play a significant role in disease development when consuming an unhealthy diet for an extended time. However, the present study creates new insights into mucosal tolerance levels to proinflammatory LPS in concert with dietary fat exposure, suggesting that LPS residing in the intestine might be less harmful than proclaimed. Further studies are now necessary to better understand the mechanisms behind restored immune responsiveness after LPS exposure.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
Ethics statement
The animal study was approved by Norwegian Food Safety Authority. The study was conducted in accordance with the Norwegian Regulation concerning the use of animals for scientific purposes and EU Directive 2010/63.
Author contributions
SH: Formal analysis, Methodology, Project administration, Writing – original draft, Writing – review & editing, Investigation. VS: Writing – review & editing, Methodology, Investigation. BE: Supervision, Writing – review & editing, Funding acquisition, Project administration. JP: Writing – review & editing. HC: Conceptualization, Formal analysis, Methodology, Project administration, Supervision, Writing – original draft, Writing – review & editing, Investigation. SB: Writing – review & editing, Investigation, Formal analysis, Methodology.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. The study was funded by internal grants (SEH PhD grant) from the Norwegian University of Life Sciences, Faculty of Chemistry, Biotechnology and Food Science. The grant for SEH was and internal PhD grant from the Faculty of Chemistry, Biotechnology and Food Science at NMBU.
Acknowledgments
We thank Lars Fredrik Moen, Sergio Rocha and Dimitrios Papoutsis for help and assistance in conducting the animal and cell experiments, Dimitrios Papoutsis for doing ELISA assays of IL-6 and Anne Mari Herfindal for help with figures. We also thank the Faculty of Chemistry, Biotechnology and Food Science, Norwegian University of Life Sciences for financial support of SEH. We used the assistance of the AI-based tool “ChatGPT” to identify and correct grammatical errors and to improve sentence structure in certain elements of “Introduction” and “Discussion” in this paper (79).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnut.2024.1376493/full#supplementary-material
References
1. Park, BS, Song, DH, Kim, HM, Choi, B-S, Lee, H, and Lee, J-O. The structural basis of lipopolysaccharide recognition by the TLR4–MD-2 complex. Nature. (2009) 458:1191–5. doi: 10.1038/nature07830
2. Baker, RG, Hayden, MS, and Ghosh, S. NF-κB, inflammation, and metabolic disease. Cell Metab. (2011) 13:11–22. doi: 10.1016/j.cmet.2010.12.008
3. Ghosh, S, and Hayden, MS. New regulators of NF-κB in inflammation. Nat Rev Immunol. (2008) 8:837–48. doi: 10.1038/nri2423
4. Cani, PD, Amar, J, Iglesias, MA, Poggi, M, Knauf, C, Bastelica, D, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. (2007) 56:1761–72. doi: 10.2337/db06-1491
5. Shi, H, Kokoeva, MV, Inouye, K, Tzameli, I, Yin, H, and Flier, JS. TLR4 links innate immunity and fatty acid–induced insulin resistance. J Clin Invest. (2006) 116:3015–25. doi: 10.1172/JCI28898
6. Cani, PD, Bibiloni, R, Knauf, C, Waget, A, Neyrinck, AM, Delzenne, NM, et al. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes. (2008) 57:1470–81. doi: 10.2337/db07-1403
7. Erridge, C, Attina, T, Spickett, CM, and Webb, DJ. A high-fat meal induces low-grade endotoxemia: evidence of a novel mechanism of postprandial inflammation. Am J Clin Nutr. (2007) 86:1286–92. doi: 10.1093/ajcn/86.5.1286
8. Ghanim, H, Abuaysheh, S, Sia, CL, Korzeniewski, K, Chaudhuri, A, Fernandez-Real, JM, et al. Increase in plasma endotoxin concentrations and the expression of toll-like receptors and suppressor of cytokine signaling-3 in mononuclear cells after a high-fat, high-carbohydrate meal: implications for insulin resistance. Diabetes Care. (2009) 32:2281–7. doi: 10.2337/dc09-0979
9. Amar, J, Burcelin, R, Ruidavets, JB, Cani, PD, Fauvel, J, Alessi, MC, et al. Energy intake is associated with endotoxemia in apparently healthy men. Am J Clin Nutr. (2008) 87:1219–23. doi: 10.1093/ajcn/87.5.1219
10. Laugerette, F, Furet, J-P, Debard, C, Daira, P, Loizon, E, Géloën, A, et al. Oil composition of high-fat diet affects metabolic inflammation differently in connection with endotoxin receptors in mice. Am J Physiol Endocrinol Metab. (2011) 302:E374–86. doi: 10.1152/ajpendo.00314.2011
11. Laugerette, F, Alligier, M, Bastard, JP, Drai, J, Chanséaume, E, Lambert-Porcheron, S, et al. Overfeeding increases postprandial endotoxemia in men: inflammatory outcome may depend on LPS transporters LBP and sCD14. Mol Nutr Food Res. (2014) 58:1513–8. doi: 10.1002/mnfr.201400044
12. Lyte, JM, Gabler, NK, and Hollis, JH. Postprandial serum endotoxin in healthy humans is modulated by dietary fat in a randomized, controlled, cross-over study. Lipids Health Dis. (2016) 15:186. doi: 10.1186/s12944-016-0357-6
13. Brown, BI. Nutritional management of metabolic endotoxemia: a clinical review. Altern Ther Health Med. (2017) 23:42–54.
14. Genser, L, Aguanno, D, Soula, HA, Dong, L, Trystram, L, Assmann, K, et al. Increased jejunal permeability in human obesity is revealed by a lipid challenge and is linked to inflammation and type 2 diabetes. J Pathol. (2018) 246:217–30. doi: 10.1002/path.5134
15. Sánchez-Tapia, M, Miller, AW, Granados-Portillo, O, Tovar, AR, and Torres, N. The development of metabolic endotoxemia is dependent on the type of sweetener and the presence of saturated fat in the diet. Gut Microbes. (2020) 12:1801301. doi: 10.1080/19490976.2020.1801301
16. Dalby, MJ, Aviello, G, Ross, AW, Walker, AW, Barrett, P, and Morgan, PJ. Diet induced obesity is independent of metabolic endotoxemia and TLR4 signalling, but markedly increases hypothalamic expression of the acute phase protein, SerpinA3N. Sci Rep. (2018) 8:15648. doi: 10.1038/s41598-018-33928-4
17. Vors, C, Pineau, G, Drai, J, Meugnier, E, Pesenti, S, Laville, M, et al. Postprandial endotoxemia linked with chylomicrons and lipopolysaccharides handling in obese versus lean men: a lipid dose-effect trial. J Clin Endocrinol Metabol. (2015) 100:3427–35. doi: 10.1210/jc.2015-2518
18. Boutagy, NE, McMillan, RP, Frisard, MI, and Hulver, MW. Metabolic endotoxemia with obesity: is it real and is it relevant? Biochimie. (2016) 124:11–20. doi: 10.1016/j.biochi.2015.06.020
19. Gnauck, A, Lentle, RG, and Kruger, MC. Chasing a ghost?—issues with the determination of circulating levels of endotoxin in human blood. Crit Rev Clin Lab Sci. (2016) 53:197–215. doi: 10.3109/10408363.2015.1123215
20. Ley, RE, Turnbaugh, PJ, Klein, S, and Gordon, JI. Human gut microbes associated with obesity. Nature. (2006) 444:1022–3. doi: 10.1038/4441022a
21. Erridge, C. Stimulants of toll-like receptor (TLR)-2 and TLR-4 are abundant in certain minimally-processed vegetables. Food Chem Toxicol. (2011) 49:1464–7. doi: 10.1016/j.fct.2011.02.018
22. Erridge, C. The capacity of foodstuffs to induce innate immune activation of human monocytes in vitro is dependent on food content of stimulants of toll-like receptors 2 and 4. Br J Nutr. (2011) 105:15–23. doi: 10.1017/S0007114510003004
23. Erridge, C. Accumulation of stimulants of toll-like receptor (TLR)-2 and TLR4 in meat products stored at 5 C. J Food Sci. (2011) 76:e2018. doi: 10.1111/j.1750-3841.2010.02018.x
24. d’Hennezel, E, Abubucker, S, Murphy, LO, and Cullen, TW. Total lipopolysaccharide from the human gut microbiome silences toll-like receptor signaling. MSystems. (2017) 2:e00046–17. doi: 10.1128/mSystems.00046-17
25. Vatanen, T, Kostic, AD, d’Hennezel, E, Siljander, H, Franzosa, EA, Yassour, M, et al. Variation in microbiome LPS immunogenicity contributes to autoimmunity in humans. Cell. (2016) 165:842–53. doi: 10.1016/j.cell.2016.04.007
26. Faraj, TA, Stover, CM, and Erridge, C. Dietary toll-like receptor stimulants promote hepatic inflammation and impair reverse cholesterol transport in mice via macrophage dependent Interleukin-1 production. Front Immunol. (2019) 10:1404. doi: 10.3389/fimmu.2019.01404
27. Carlsen, H, Moskaug, JØ, Fromm, SH, and Blomhoff, R. In vivo imaging of NF-κB activity. J Immunol. (2002) 168:1441–6. doi: 10.4049/jimmunol.168.3.1441
28. Bachmanov, AA, Reed, DR, Beauchamp, GK, and Tordoff, MG. Food intake, water intake, and drinking spout side preference of 28 mouse strains. Behav Genet. (2002) 32:435–43. doi: 10.1023/A:1020884312053
29. Erridge, C, Duncan, SH, Bereswill, S, and Heimesaat, MM. The induction of colitis and ileitis in mice is associated with marked increases in intestinal concentrations of stimulants of TLRs 2, 4, and 5. PLoS One. (2010) 5:e9125. doi: 10.1371/journal.pone.0009125
30. Chassaing, B, Koren, O, Goodrich, JK, Poole, AC, Srinivasan, S, Ley, RE, et al. Dietary emulsifiers impact the mouse gut microbiota promoting colitis and metabolic syndrome. Nature. (2015) 519:92–6. doi: 10.1038/nature14232
31. Ziegler, TR, Luo, M, Estívariz, CF, Moore, DA, Sitaraman, SV, Hao, L, et al. Detectable serum flagellin and lipopolysaccharide and upregulated anti-flagellin and lipopolysaccharide immunoglobulins in human short bowel syndrome. Am J Physiol Regul Integr Comp Physiol. (2008) 294:R402–10. doi: 10.1152/ajpregu.00650.2007
32. Bamias, G, Corridoni, D, Pizarro, TT, and Cominelli, F. New insights into the dichotomous role of innate cytokines in gut homeostasis and inflammation. Cytokine. (2012) 59:451–9. doi: 10.1016/j.cyto.2012.06.014
33. Lallès, J-P. Intestinal alkaline phosphatase: novel functions and protective effects. Nutr Rev. (2014) 72:82–94. doi: 10.1111/nure.12082
34. Lynes, M, Narisawa, S, Millán, JL, and Widmaier, EP. Interactions between CD36 and global intestinal alkaline phosphatase in mouse small intestine and effects of high-fat diet. Am J Phys Regul Integr Comp Phys. (2011) 301:R1738–47. doi: 10.1152/ajpregu.00235.2011
35. Shao, B, Lu, M, Katz, SC, Varley, AW, Hardwick, J, Rogers, TE, et al. A host lipase detoxifies bacterial lipopolysaccharides in the liver and spleen. J Biol Chem. (2007) 282:13726–35. doi: 10.1074/jbc.M609462200
36. Zhou, X, Han, D, Xu, R, Li, S, Wu, H, Qu, C, et al. A model of metabolic syndrome and related diseases with intestinal endotoxemia in rats fed a high fat and high sucrose diet. PLoS One. (2014) 9:e115148. doi: 10.1371/journal.pone.0115148
37. Guerville, M, Leroy, A, Sinquin, A, Laugerette, F, Michalski, M-C, and Boudry, G. Western-diet consumption induces alteration of barrier function mechanisms in the ileum that correlates with metabolic endotoxemia in rats. Am J Physiol Endocrinol Metab. (2017) 313:E107–20. doi: 10.1152/ajpendo.00372.2016
38. Koyama, I, Matsunaga, T, Harada, T, Hokari, S, and Komoda, T. Alkaline phosphatases reduce toxicity of lipopolysaccharides in vivo and in vitro through dephosphorylation. Clin Biochem. (2002) 35:455–61. doi: 10.1016/S0009-9120(02)00330-2
39. Alpers, D, Zhang, Y, and Ahnen, D. Synthesis and parallel secretion of rat intestinal alkaline phosphatase and a surfactant-like particle protein. Am J Physiol Endocrinol Metab. (1995) 268:E1205–14. doi: 10.1152/ajpendo.1995.268.6.E1205
40. Cai, D, Yuan, M, Frantz, DF, Melendez, PA, Hansen, L, Lee, J, et al. Local and systemic insulin resistance resulting from hepatic activation of IKK-β and NF-κB. Nat Med. (2005) 11:183–90. doi: 10.1038/nm1166
41. Jergović, M, Thompson, HL, Bradshaw, CM, Sonar, SA, Ashgar, A, Mohty, N, et al. IL-6 can singlehandedly drive many features of frailty in mice. GeroScience. (2021) 43:539–49. doi: 10.1007/s11357-021-00343-z
42. Qing, H, Desrouleaux, R, Israni-Winger, K, Mineur, YS, Fogelman, N, Zhang, C, et al. Origin and function of stress-induced IL-6 in murine models. Cell. (2020) 182:1660. doi: 10.1016/j.cell.2020.08.044
43. Gonzalez-Quintela, A, Alonso, M, Campos, J, Vizcaino, L, Loidi, L, and Gude, F. Determinants of serum concentrations of lipopolysaccharide-binding protein (LBP) in the adult population: the role of obesity. PLoS One. (2013) 8:e54600. doi: 10.1371/journal.pone.0054600
44. Sun, L, Yu, Z, Ye, X, Zou, S, Li, H, Yu, D, et al. A marker of endotoxemia is associated with obesity and related metabolic disorders in apparently healthy Chinese. Diabetes Care. (2010) 33:1925–32. doi: 10.2337/dc10-0340
45. Hamilton, MK, Boudry, G, Lemay, DG, and Raybould, HE. Changes in intestinal barrier function and gut microbiota in high-fat diet-fed rats are dynamic and region dependent. Am J Physiol Gastrointestinal Liver Physiol. (2015) 308:G840–51. doi: 10.1152/ajpgi.00029.2015
46. Rohr, MW, Narasimhulu, CA, Rudeski-Rohr, TA, and Parthasarathy, S. Negative effects of a high-fat diet on intestinal permeability: a review. Adv Nutr. (2020) 11:77–91. doi: 10.1093/advances/nmz061
47. Moreira, APB, Texeira, TFS, Ferreira, AB, Peluzio, MCG, and Alfenas, RCG. Influence of a high-fat diet on gut microbiota, intestinal permeability and metabolic endotoxaemia. Br J Nutr. (2012) 108:801–9. doi: 10.1017/S0007114512001213
48. Caesar, R, Reigstad, CS, Backhed, HK, Reinhardt, C, Ketonen, M, Lunden, GO, et al. Gut-derived lipopolysaccharide augments adipose macrophage accumulation but is not essential for impaired glucose or insulin tolerance in mice. Gut. (2012) 61:1701–7. doi: 10.1136/gutjnl-2011-301689
49. de La Serre, CB, Ellis, CL, Lee, J, Hartman, AL, Rutledge, JC, and Raybould, HE. Propensity to high-fat diet-induced obesity in rats is associated with changes in the gut microbiota and gut inflammation. American journal of physiology-gastrointestinal and liver. Physiology. (2010) 299:G440–8. doi: 10.1152/ajpgi.00098.2010
50. Kim, K-A, Gu, W, Lee, I-A, Joh, E-H, and Kim, D-H. High fat diet-induced gut microbiota exacerbates inflammation and obesity in mice via the TLR4 signaling pathway. PLoS One. (2012) 7:e47713. doi: 10.1371/journal.pone.0047713
51. Munford, RS. Endotoxemia—menace, marker, or mistake? J Leukoc Biol. (2016) 100:687–98. doi: 10.1189/jlb.3RU0316-151R
52. Dlugosz, A, Nowak, P, D’amato, M, Mohammadian Kermani, G, Nyström, J, Abdurahman, S, et al. Increased serum levels of lipopolysaccharide and antiflagellin antibodies in patients with diarrhea-predominant irritable bowel syndrome. Neurogastroenterol Motility. (2015) 27:1747–54. doi: 10.1111/nmo.12670
53. Schumann, RR. Lipopolysaccharide-binding protein. RS Jack, (Ed.) CD14 in the inflammatory response. 74. Basel, Karger: Chemical Immunology; (2000). 42–60
54. Li, X, Gong, J, Tu, B, Shi, Y, and Liu, C. In vivo expression of lipopolysaccharide binding protein and its gene induced by endotoxin. Chin J Traumatol. (2003) 6:280–3.
55. Vreugdenhil, AC, Dentener, MA, Snoek, AP, Greve, J-WM, and Buurman, WA. Lipopolysaccharide binding protein and serum amyloid a secretion by human intestinal epithelial cells during the acute phase response. J Immunol. (1999) 163:2792–8. doi: 10.4049/jimmunol.163.5.2792
56. Lamping, N, Dettmer, R, Schröder, N, Pfeil, D, Hallatschek, W, Burger, R, et al. LPS-binding protein protects mice from septic shock caused by LPS or gram-negative bacteria. J Clin Invest. (1998) 101:2065–71. doi: 10.1172/JCI2338
57. Järveläinen, HA, Fang, C, Ingelman-Sundberg, M, and Lindros, KO. Effect of chronic coadministration of endotoxin and ethanol on rat liver pathology and proinflammatory and anti-inflammatory cytokines. Hepatology. (1999) 29:1503–10. doi: 10.1002/hep.510290508
58. Biswas, SK, and Lopez-Collazo, E. Endotoxin tolerance: new mechanisms, molecules and clinical significance. Trends Immunol. (2009) 30:475–87. doi: 10.1016/j.it.2009.07.009
59. Pena, OM, Pistolic, J, Raj, D, Fjell, CD, and Hancock, RE. Endotoxin tolerance represents a distinctive state of alternative polarization (M2) in human mononuclear cells. J Immunol. (2011) 186:7243–54. doi: 10.4049/jimmunol.1001952
60. Larsson, L, Garaicoa-Pazmino, C, Asa'ad, F, and Castilho, RM. Understanding the role of endotoxin tolerance in chronic inflammatory conditions and periodontal disease. J Clin Periodontol. (2022) 49:270–9. doi: 10.1111/jcpe.13591
61. López-Collazo, E, and Del Fresno, C. Endotoxin tolerance and trained immunity: breaking down immunological memory barriers. Front Immunol. (2024) 15:1393283. doi: 10.3389/fimmu.2024.1393283
62. Roy, K, Kozłowski, HM, Jędrzejewski, T, Sobocińska, J, Maciejewski, B, Dzialuk, A, et al. Endotoxin tolerance creates favourable conditions for cancer development. Cancers. (2023) 15:5113. doi: 10.3390/cancers15205113
63. West, MA, and Heagy, W. Endotoxin tolerance: a review. Crit Care Med. (2002) 30:S64–73. doi: 10.1097/00003246-200201001-00009
65. Kobayashi, K, Hernandez, LD, Galán, JE, Janeway, CA, Medzhitov, R, and Flavell, RA. IRAK-M is a negative regulator of toll-like receptor signaling. Cell. (2002) 110:191–202. doi: 10.1016/S0092-8674(02)00827-9
66. Xiong, Y, and Medvedev, AE. Induction of endotoxin tolerance in vivo inhibits activation of IRAK4 and increases negative regulators IRAK-M, SHIP-1, and A20. J Leukoc Biol. (2011) 90:1141–8. doi: 10.1189/jlb.0611273
67. Bates, JM, Akerlund, J, Mittge, E, and Guillemin, K. Intestinal alkaline phosphatase detoxifies lipopolysaccharide and prevents inflammation in zebrafish in response to the gut microbiota. Cell Host Microbe. (2007) 2:371–82. doi: 10.1016/j.chom.2007.10.010
68. Guerville, M, and Boudry, G. Gastrointestinal and hepatic mechanisms limiting entry and dissemination of lipopolysaccharide into the systemic circulation. American journal of physiology-gastrointestinal and liver. Am J Physiol Gastrointestinal Liver Physiol. (2016) 311:G1–G15. doi: 10.1152/ajpgi.00098.2016
69. Narisawa, S, Huang, L, Iwasaki, A, Hasegawa, H, Alpers, DH, and Millán, JL. Accelerated fat absorption in intestinal alkaline phosphatase knockout mice. Mol Cell Biol. (2003) 23:7525–30. doi: 10.1128/MCB.23.21.7525-7530.2003
70. Lallès, J-P. Intestinal alkaline phosphatase: multiple biological roles in maintenance of intestinal homeostasis and modulation by diet. Nutr Rev. (2010) 68:323–32. doi: 10.1111/j.1753-4887.2010.00292.x
71. Williams, LM, Campbell, FM, Drew, JE, Koch, C, Hoggard, N, Rees, WD, et al. The development of diet-induced obesity and glucose intolerance in C57BL/6 mice on a high-fat diet consists of distinct phases. PLoS One. (2014) 9:e106159. doi: 10.1371/journal.pone.0106159
72. Liu, T-C, Kern, JT, Jain, U, Sonnek, NM, Xiong, S, Simpson, KF, et al. Western diet induces Paneth cell defects through microbiome alterations and farnesoid X receptor and type I interferon activation. Cell Host Microbe. (2021) 29:988–1001.e6. doi: 10.1016/j.chom.2021.04.004
73. Statovci, D, Aguilera, M, MacSharry, J, and Melgar, S. The impact of western diet and nutrients on the microbiota and immune response at mucosal interfaces. Front Immunol. (2017) 8:838. doi: 10.3389/fimmu.2017.00838
74. Candelli, M, Franza, L, Pignataro, G, Ojetti, V, Covino, M, Piccioni, A, et al. Interaction between lipopolysaccharide and gut microbiota in inflammatory bowel diseases. Int J Mol Sci. (2021) 22:6242. doi: 10.3390/ijms22126242
75. Bian, Y, Dong, Y, Sun, J, Sun, M, Hou, Q, Lai, Y, et al. Protective effect of kaempferol on LPS-induced inflammation and barrier dysfunction in a coculture model of intestinal epithelial cells and intestinal microvascular endothelial cells. J Agric Food Chem. (2019) 68:160–7. doi: 10.1021/acs.jafc.9b06294
76. Zheng, X, Li, Y, Shang, X, and Liu, R. Is lipopolysaccharide-induced lipid metabolism disorder in testis of rats a consequence of plasma lipid changes? J Inflamm Res. (2024) 17:765–76. doi: 10.2147/JIR.S441840
77. Hayakawa, S, Tamura, A, Nikiforov, N, Koike, H, Kudo, F, Cheng, Y, et al. Activated cholesterol metabolism is integral for innate macrophage responses by amplifying Myd88 signaling. JCI Insight. (2022) 7:138539. doi: 10.1172/jci.insight.138539
78. Cani, PD, and Everard, A. Keeping gut lining at bay: impact of emulsifiers. Trends Endocrinol Metab. (2015) 26:273–4. doi: 10.1016/j.tem.2015.03.009
Glossary
Keywords: obesity, LPS, intestinal permeability, low-grade inflammation, NF-κB activation
Citation: Harvei S, Skogen V, Egelandsdal B, Birkeland S, Paulsen JE and Carlsen H (2024) Chronic oral LPS administration does not increase inflammation or induce metabolic dysregulation in mice fed a western-style diet. Front. Nutr. 11:1376493. doi: 10.3389/fnut.2024.1376493
Edited by:
Michael Mathai, Victoria University, AustraliaReviewed by:
Sarah C. Pearce, United States Department of Agriculture, United StatesClett Erridge, Anglia Ruskin University, United Kingdom
Copyright © 2024 Harvei, Skogen, Egelandsdal, Birkeland, Paulsen and Carlsen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Harald Carlsen, aGFyYWxkLmNhcmxzZW5Abm1idS5ubw==