 Mengyao Wang
Mengyao Wang Ailin Zhao
Ailin Zhao Meng Li
Meng Li Ting Niu
Ting Niu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Nutr. , 23 March 2023
Sec. Clinical Nutrition
Volume 10 - 2023 | https://doi.org/10.3389/fnut.2023.1113228
This article is part of the Research Topic Nutritional Status in Disease: Development, Evaluation, Prognosis and Individualized Treatment in Clinical Practice View all 23 articles
In recent years, growing emphasis has been placed on amino acids and their role in hematologic malignancies. Cancer cell metabolism is altered during tumorigenesis and development to meet expanding energetic and biosynthetic demands. Amino acids not only act as energy-supplying substances, but also play a vital role via regulating key signaling pathways, modulating epigenetic factors and remodeling tumor microenvironment. Targeting amino acids may be an effective therapeutic approach to address the current therapeutic challenges. Here, we provide an updated overview of mechanisms by which amino acids facilitate tumor development and therapy resistance. We also summarize novel therapies targeting amino acids, focusing on recent advances in basic research and their potential clinical implications.
Uncontrollable cellular proliferation, invasion and metastasis are the characteristics of cancer cells (1, 2). To accommodate more rapid proliferation, tumor cells alter their metabolism to provide sufficient cellular structural substances (proteins, DNA, RNA, and lipids) and energy. Aberrant metabolism is also an important feature of cancer (2–4). The Warburg effect is the most well-known cancer-specific metabolic feature (2, 5). Cancer cells rewire their metabolic pathways, preferentially metabolizing glucose through aerobic glycolysis pathway to generate ATP efficiently, even under aerobic conditions (2). Besides glucose, cancer cells also develop increased reliance on amino acids (AAs) to meet enhanced demands for energy and cellular building blocks (6). Recently, increasing evidences show that AAs play vital roles in tumor development (6). AAs are not only involved in energy-generating and biosynthetic (4), but also exert cancer-promoting effects by regulating signaling pathways (7), epigenetic processes (8), cellular redox state (9) and immunosuppressive tumor microenvironment (TME) (10) (Figure 1).
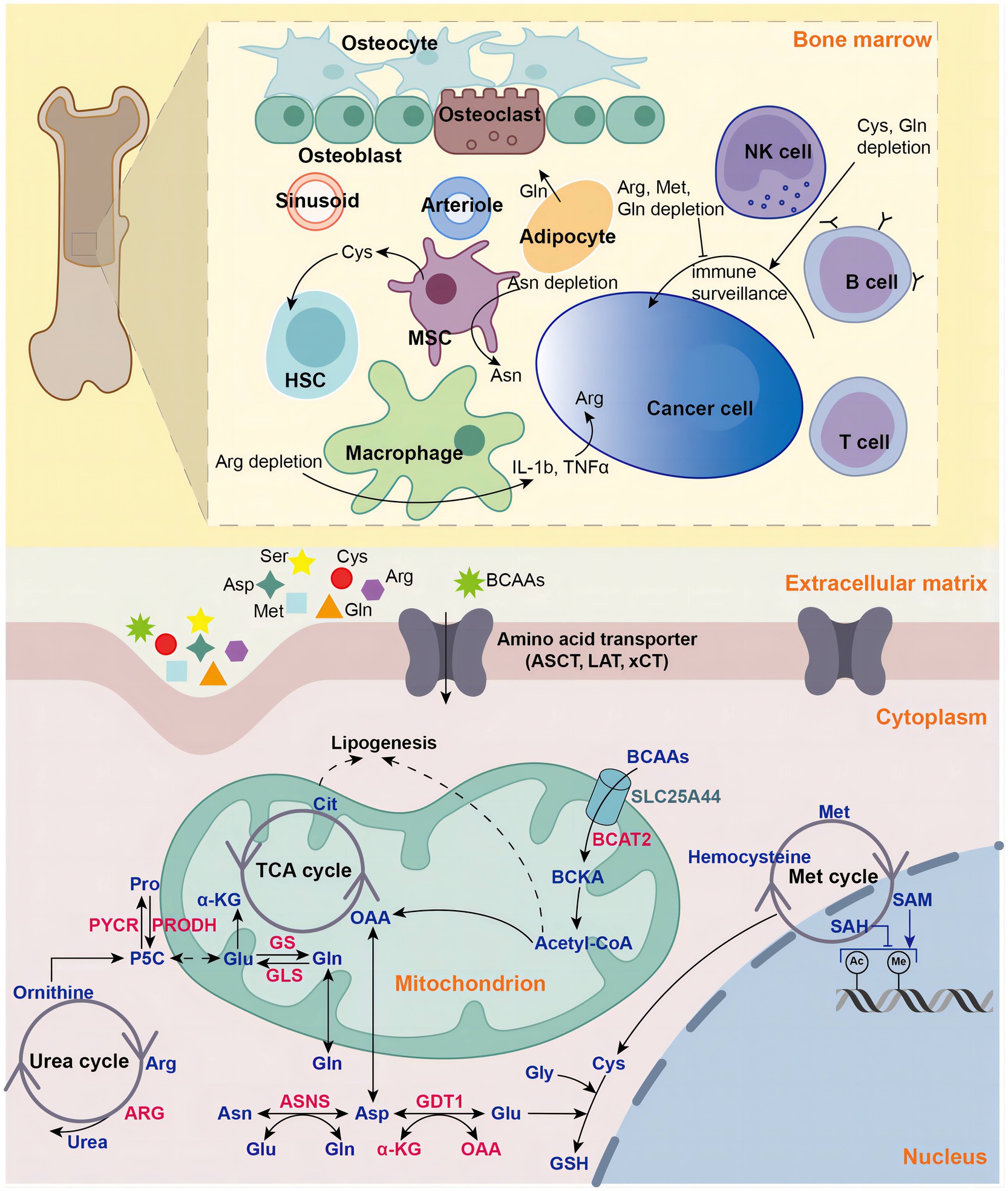
Figure 1. Amino acids in hematologic malignancies. Amino acids play vital roles in in tumorigenesis and development in hematologic malignancies, including energy supply, biosynthetic support, redox balance maintenance, epigenetic regulation and tumor microenvironment modulation. NK natural killer, MSC mesenchymal stromal cell, HSC hematopoietic stem cell, Gln glutamine, Glu glutamate, Gly glycine, Cys cysteine, Asp aspartate, Pro proline, Asp aspartate, Asn asparagine, Arg arginine, Met methionine, BCAA branched-chain amino acid, BCKA branched-chain ketoacid, aspartate transaminase (AST), GLS glutaminase, GS glutamine synthetase, ASNS asparagine synthetase, PRODH pyrroline-5-carboxylate dehydrogenase, PYCR pyrroline-5-carboxylate reductase, P5C pyrroline-5-carboxylate, ARG arginase, GSH glutathione, acetyl-coA acetyl-coenzyme A, α-KG alpha-ketoglutaric acid, OAA oxaloacetic acid, LAT large-neutral amino acid transporter, ASCT (alanine, serine, cysteine transporter), xCT cystine/glutamate antiporter, SLC25A44 solute carrier family 25 member 44, TCA tricarboxylic acid, Cit Citric Acid, SAM S-adenosylmethionine, SAH Sadenosyl homocysteine.
Requirements for AAs is different between normal and tumor cells, which can be exploited to develop anti-cancer therapies. Multiple strategies targeting AA metabolism have been developed, including dietary AA starvation, AA depletion, inhibition of AA transporters and synthases (4). In clinical practice, the utilization of therapies targeting AA is more common in hematologic malignancies and some have achieved remarkable therapeutic outcomes (11). L-asparaginase is the most successful example of AA deprivation therapy, which has changed the landscape of acute lymphoblastic leukemia (ALL) treatment (12). Promising new drugs are continually emerging. These novel agents not only demonstrate potential efficacy in monotherapy, but also show good results in reversing drug resistance and relieving immunosuppression (6). In this review, we discuss mechanisms of action of AAs in hematologic malignancies, focusing on novel roles beyond rewired metabolism. We also summarize deprivation strategies using heterologous agents and recent data from clinical trials.
Glutamine (Gln) is a nonessential AA. Intracellular glutamine mainly originates from the bloodstream (13). Gln is involved in a broad range of cellular activities, including energy supply, biosynthesis of other biomolecules and maintaining cellular redox homeostasis (14). Besides glucose, Gln is the most important energy supplyinig substance. As glucose is metabolized through anaerobic metabolism to produce lactate, Gln enters the tricarboxylic acid (TCA) cycle and provides TCA metabolites, supporting generation of lipids, proteins, and nucleic acids (13). Enhanced glutaminolysis is a prominent feature of various cancers (15).
Gln is the most abundant AA in serum and is avidly taken up by tumor cells (13). Gln is transported into cells and mitochondria through solute carrier (SLC) family members, mainly SLC1A5 (also known as alanine, serine, cysteine [Cys] transporter 2, ASCT2) (16, 17). In addition to transport, another important way for Gln entering cells is macropinocytosis. Macropinocytosis is a type of endocytosis in which extracellular fluid and nutrients are engulfed and taken up via large endocytic vesicles known as macropinosomes (18). L-type AA transporter 1 (LAT1), a heterodimer of SLC7A5 and SLC3A2, serves as an antiporter responsible for exporting Gln in exchange for leucine (19).
Gln dissociates into ammonium ions and glutamate under the catalysis of mitochondrial glutaminases (GLS) or cytoplasmic asparaginase (ASNase). Glutamate is then metabolized via two major pathways. Glutamate is converted into c-glutamylcysteine, which is utilized to generate glutathione (GSH) by GSH synthetase (13). By quenching reactive oxygen species and participating in the ascorbate-glutathione cycle, which eliminates peroxides, GSH protects both normal and malignant cells from oxidative injury. Leukemic cells have redox dysregulation, including aberrant GSH metabolism, thus making them sensitive to pro-oxidant therapies that further disrupt GSH pathways (20, 21).
When catalyzed by glutamate dehydrogenase (GDH), glutamate breaks into α-ketoglutarate (α-KG) and ammonia. When catalyzed by transaminases, glutamate transfers amino groups to generate other AAs (proline, aspartate, serine and alanine) and α-KG. α-KG produced by these two processes enters the TCA cycle, where it participates in mitochondrial oxidative phosphorylation (OXPHOS) and eventually generates ATP (15). Some hematologic tumor cells are addicted to Gln-fueled OXPHOS (9, 22). For example, in mantle cell lymphoma (MCL), lymphoma cells reprogram metabolically toward OXPHOS and glutaminolysis to gain advantage in generating energy and develop drug resistance to Bruton’s tyrosine kinase (BTK) inhibitors (9). Acute myeloid leukemia (AML) cells are sensitive to OXPHOS controlled by Gln (22). Interestingly, leukemia stem cells (LSCs) are characterized by a low rate of energy metabolism and a low cellular oxidative status. They are unable to utilize glycolysis and dependent on OXPHOS for energy generation (23). Furthermore, the isocitrate dehydrogenase (IDH) transforms isocitrate into α-KG. Mutated IDH, caused by IDH1/2 mutations, transforms α-KG into 2-hydroxyglutarate (2-HG), which represses demethylases of both DNA and histones, leading to aberrantly increased methylation levels of both DNA and histones and epigenetic dysregulation (24). IDH1 mutations and IDH2 mutations have been reported in 6%–9% and 8%–12% AML patients, respectively (25, 26). In low-risk or medium-risk AML patients with normal karyotype, IDH1/2 mutations were significantly related to worse prognosis (27, 28).
Similar to glucose, cancer cells also increase uptake and utilization of Gln to meet energy requirement and biosynthetic demands of rapid cell growth. Gln transporters and enzymes involved in glutaminolysis are highly expressed in many hematological malignancies (15). SLC overexpression was observed in most leukemia, lymphoma and myeloma and associated with poor prognosis (29). For example, Bolzoni et al. discovered that SLC1A5 was highly expressed in multiple myeloma (MM) cells and was needed for MM growth (30). Myc or Ras-driven tumors were particularly dependent on exogenous Gln (31). In Burkitt lymphoma, MYC increased the expression of both SLC1A5 and SLC7A5, promoted glutaminolysis and led to augmented tumor proliferation (32). In natural-killer T-cell lymphomas (NKTCLs), SLC1A1 acted as a central regulator of aberrant Gln metabolism (33). SLC1A1 overexpression in lymphoma not only enhanced tumor growth, but also promoted competition for Gln between lymphoma cells and CD8+ T cells. Impaired CD8+ T cell activity, together with PD-L1 downregulation, led to immunosuppression and tumor progression (33).
Considering the close connection between malignancies and Gln metabolism, disrupting Gln utilization in tumor can be an important means to impede tumor growth. Current therapeutical strategies are mainly focused on hindering cancer cells from obtaining and utilizing Gln, comprising the following aspects: (1) Blocking Gln transporters, including SLC1A5, SLC7A11 and cystine/glutamate antiporter (xCT) system. (2) Inhibition of enzymes involved in Gln metabolism. (3) Gln depletion: ASNase degrades Gln as well as Arg, which will be discussed in detail in the section of Arg-based therapeutic approaches (15, 34, 35). These therapeutic agents have shown excellent results in preclinical studies and some have already entered clinical trials. In the following discussion, we focus mainly on agents that have exhibited promising efficacy in hematological malignancies.
γ-Glutamyl-p-nitroanilide (GPNA) and V9302 are ASCT2 inhibitors. Results from preclinical studies have shown that blocking ASCT2 with GPNA inhibits proliferation and induces apoptosis in AML cells, thus prolong survival in AML mice (36). Blockade of ASCT2 with V-9302 also leads to effective tumor control (37, 38). It is noteworthy that, besides abrogating glutaminolysis, observed efficacy of V-9302 may be due to combinatorial blockade of multiple ASCT2 substrates. Elevated autophagy, increased oxidative stress and decreased mTOR activity were observed in V-9302-treated cancer, which implies a theoretical basis for rational combination therapy (37).
Multiple drugs act on enzymes involved in Gln metabolism. GLS inhibitors include CB-839 (Telaglenastat), bis-2-(5-phenylacetamido-1,3,4-thiadiazol-2-yl)ethyl sulfide (BPTES) and compound 968. [6-diazo-5-oxo-L-nor-leucine (DON)] and JHU083 have structural similarities with Gln, thereby inhibiting all related enzymes (35, 39).
CB-839 is the most well-studied GLS inhibitor. CB-839 exhibited potent anticancer activity against a variety of tumors in preclinical studies. Anticancer effects were more remarkable when combined with agents of other mechanisms of action (35). Increased mitochondrial respiration was found in proteasome inhibitors (PIs)-resistant MM cells (40). Gln was the primary fuel for mitochondrial respiration, cell proliferation and survival in both PI-sensitive and PI-resistant MM cells. CB-839 significantly blocked mitochondrial respiration and demonstrated dramatic synergy with PIs (40). AML cells were highly dependent on the Gln for their survival. Inhibiting Gln metabolism via CB-839 significantly impaired antioxidant GSH production in multiple types of AML, leading to accumulation of mitochondrial reactive oxygen species (ROS) and apoptosis. Together with the pro-oxidant drug, such as arsenic trioxide (ATO) and homoharringtonine (HHT), CB-839 induced more mitochondrial oxidative stress and led to more thorough leukemic cell elimination (41). In FLT3-internal tandem duplication (FLT3-ITD)-positive AML, a subtype of leukemia with notoriously dismal outcome, CB-839 also impaired GSH production, induced severe mitochondrial oxidative stress and cell apoptosis. More remarkably, CB-839 and FLT3 inhibitors, including AC220 and gilteritinib, exerted synergistic pro-apoptotic effects and displayed more potent anti-leukemia effect (42, 43). These redox-targeted combinations represented a novel therapeutic strategy with high efficiency, low toxicity and enormous potential for clinical translation (44). At present, CB-839 has entered clinical trials for treatment of leukemia, myelodysplastic syndromes (MDS), renal cell carcinoma, and non-small cell lung cancer. A phase II study (NCT0347993) was conducted to evaluate safety and efficacy of CB-839 (600 mg BID orally) in combination with standard Azacitidine (Aza) in advanced MDS patients (14). Primary efficacy outcome showed that 62.5% (10/16) patients achieved marrow complete response (CR) and 31.3% (5/16) had stable disease (SD). Adverse events analysis showed that the most common non-hematological adverse events (AEs) were gastrointestinal abnormalities (62.5%), including nausea, constipation, elevated ALT and anorexia. The most frequent hematological AEs were grade III-IV anemia (12.5%), neutropenia (37.5%) and thrombocytopenia (37.5%). 43.7% patients experienced grade III-IV infections. This interim analysis demonstrated that the combination of CB-839 and Aza was a promising treatment with acceptable safety profile and encouraging response rates.
DON is a glutamine antagonist with robust anticancer efficacy. But its clinical use was hampered due to its severe toxicity, especially gastrointestinal toxicity and mucositis (45). In recent years, various prodrugs for DON have been synthesized to enhance delivery of active compound to tumor lesions (39, 46). This delivery strategy enhanced the anti-tumor effects of DON while circumventing its toxicity, bringing it back on track. JHU083 is a dual promoeity prodrug modified from DON. JHU083 possessed several advantages over DON: enhanced oral bioavailability, tumor-targetability and central nervous system (CNS) penetration (47).
Arginine (Arg) is considered as a non-essential or semi-essential AA. The sources of Arg include dietary uptake, protein turnover and endogenous synthesis (48). Healthy individuals are capable of synthesizing Arg from Gln, glutamate, and proline. However, when the demand for Arg is increased, such as growth during infancy, pregnancy, and burn injuries, endogenous biosynthesis of Arg may be insufficient to support body needs, and dietary sources of Arg become essential (49). Arg is one of the most versatile AAs. Arg is involved in a variety of biochemical metabolic processes, such as the urea cycle and the TCA cycle (50). It also participates in regulating several important physiological activities, including cell proliferation, immunity and hormone secretion. Besides, Arg is the major precursor for synthesis of cancer-associated compounds such as polyamines and nitric oxide (NO) (50).
Normal cells synthesize Arg intracellularly from ornithine and citrulline under the catalysis of argininosuccinate synthase (ASS) 1, ornithine transcarbamylase (OTC), and argininosuccinate lyase (ASL). However, over 70% of cancers are deficient of these key enzymes, rendering them highly dependent on external Arg (51). The addiction of cancer cells for Arg is called Arg auxotrophism, Arg auxotrophism is a prominent feature of, hematologic malignancies which may be attributed to methylation of the ASS1 promoter (52, 53). For example, Mussai et al. identified that there was low or no expression of ASS and/or OTC in AML blasts. Bone marrow stroma may play a vital role in producing Arg to support AML expansion (51).
There are two major pathways of Arg catabolism involving four main enzymes: nitric oxide synthase (NOS), arginases (ARGs), arginine decarboxylase and arginine:glycine amidino transferase (OAT). One catabolic pathway is mediated by NOS, which catabolizes Arg into citrulline and NO (50). NO has two-way effects on tumor growth, depending on its concentration, time of exposure, cellular redox status and TME. In general, low concentration of NO promotes tumorigenesis and development, whereas high-level NO can cause DNA damage, induce apoptosis and activate immune defense (54). For example, in B-cell chronic lymphocytic leukemia (B-CLL), leukemic cells expressed inducible NOS and increased NO release. The anti-apoptotic effect of NO could be counteracted by NOS inhibitors and engagement of the APO-1/Fas pathway (54). However, exogenous supplementation of NO promoted cell death and potentiated the cytotoxic effect of fludarabine to B-CLL lymphocytes (55). ARGs compete with NOS to catabolize Arg decomposition. ARGs decompose Arg into urea and ornithine. Ornithine is an important intermediate. The vast majority of ornithine participates in the urea cycle: it is converted to citrulline by ornithine carbamoyltransferase (ODC) and subsequently involved in synthesis of Arg under the catalysis of ASS. Ornithine can also be converted into polyamines, which also promote cell proliferation and tumor growth (50).
Since most hematologic malignancies are auxotrophic for Arg, starving tumors by exhausting Arg is a potential therapeutic strategy (51). Two classes of Arg depleting agents, ARG and arginine deiminase (ADI), have achieved a certain level of success and moved into clinical trials (56, 57). PEG-ARG1 and BCT-100 are pegylated arginases. Rodriguez et al. found that in T-cell acute lymphoblastic leukemia (T-ALL), PEG-ARG1 triggered cell apoptosis and inhibited proliferation through phosphorylation of the eukaryotic-translation-initiation factor 2 alpha (eIF2a) (58). Combination of PEG-ARG1 with cytarabine (Ara-C) or phospho-eIF2a signaling significantly prolonged the survival of mice bearing T-ALL, representing a potential treatment therapy for this high-risk subtype of leukemia (59).
ADI, an enzyme derived from mycoplasma, rapidly degrades Arg intro citrulline (56, 57). To avoid adverse effects of foreign proteins, such as anaphylaxis and rapid clearance, ADI-PEG20, a pegylated form of ADI, has been developed. Results from preclinical studies showed that ADI-PEG 20 induced caspase-dependent cell apoptosis and autophagy in leukemia and lymphoma (50). ADI-PEG 20 depleted Arg and showed a significant killing effect on AML, which could be further enhanced when combined with cytarabine (60). In a phase II study (NCT01910012), single-agent ADI-PEG20 resulted in a moderate disease control rate (DCR) of 42.9% in relapsed/refractory/poor-risk AML patients (61). Its hematologic and non-hematologic toxicity was minimal when compared with chemotherapy.
Asparagine (Asn) is a multifunctional AA. It plays important roles in supporting cell proliferation and growth, especially in protein synthesis. Asn residues provide amide nitrogen that allows N-linked glycosylation, contributing to glycoprotein synthesis (62, 63). Intracellular Asn also acts as AA exchange factor. Asn is exported in exchange for extracellular AAs, especially serine, arginine and histidine, the process of which activates mTOR complex 1 (mTORC1) and promotes protein and nucleotide synthesis (64). Asn is also found to suppress apoptosis induced by Gln deprivation (65).
Asn is considered as a non-essential AA for normal human cells. Asn can be synthesized by Gln and aspartate under the catalysis of Asparagine synthetase (ASNS) in an ATP-dependent manner (65). ASNase hydrolyzes ASN into aspartate and ammonia. In some cancer cells, the expression of ASNS is low or even absent, rendering them unable to synthesize Asn by themselves. Once Asn from bloodstream is depleted, these cancer cells will suffer from Asn starvation and subsequently undergo apoptosis. Asn deprivation is an effective treatment strategy for cancers lack or deficient of ASNS expression, including hematological malignancies (12).
L-asparaginase (L-ASNase) is isolated from Escherichia coli and has been widely used in clinical practice for over 40 years. L-ASNase monotherapy is an effective therapeutic approach to treat ALL, especially pediatric ALL (12, 66, 67). With the illustration of mechanisms of action, L-ASNase has become the cornerstone of multi-agent chemotherapy regimen for various malignancies (12). For example, combination chemotherapies containing L-ASNase have became the standard treatment for NK/T-cell lymphoma (68–70). And L-ASNase was part of chemotherapy regimens for pediatric AML in come clinical trails (71, 72).
Despite favorable efficacy, there are still several issues that require further attention. First, the toxicity profiles require improvement, which includes severe immunological side effects as well as non-immune-related toxicities such as pancreatitis, liver toxicities, venous thromboembolism (VTE), and neurotoxicity (12, 67). Moreover, with the increase of drug exposure in clinical practice, secondary drug resistances are gradually emerging. The resistance mechanism is generally believed to include the following aspects (12). ASNS expression is upregulated up to 7-fold by tumor cells to develop resistance to ASNase in Asn-depleted environment (73). Bone marrow-derived mesenchymal stromal cells (MSCs) may secrete L-asparagine and rescue leukemic blasts from L-ASNase deprivation. As a foreign protein, production of autoantibodies accelerates clearance of L-ASNase (74). Besides, some previously unknown mechanisms of L-ASNase resistance have been gradually perceived and recognized. Huntingtin associated protein 1 (HAP1) has been identified as a biomarker for L-ASNase resistance in ALL (75). HAP1 loss suppresses formation of the ternary complex that mediates endoplasmic reticulum (ER) Ca2+ release, thus preventing cell apoptosis induced by L-ASNase (75). In extranodal NK/T-cell lymphoma, brain cytoplasmic RNA 1 (BCYRN1), a long non-coding RNA (lncRNA), induced the degradation of p53, promoted autophagy and counteracted the effects of Asn deprivation, resulting in L-ASNase resistance (76).
To address the above issues, multiple novel L-ASNase formulations have been developed. PEGylated L-ASNase was produced by covalently conjugating monomethoxypolyethyleneglycol (PEG) to E. coli L-ASNase. The PEGylated formulation reduced the immunogenicity and prolonged the half-life period (77). But the antibodies induced by native E. coli L-ASNase might cross-react with PEGylated L-ASNase. Erwinia chrysanthemi L-ASNase was a solution to E. coli L-ASNase. It might induce less complications and toxicities (like coagulation abnormalities, neurotoxicity, and pancreatitis) (12).
As a semiessential AA, Cys is indispensable to various reactions, including GSH generation, LSC maintenance, and redox homeostasis. Although Cys could be compensatively generated from methionine (Met), the overall demand could not be met. Furthermore, leukemic cells and lymphoma cells displayed translationally silenced Cys synthetic enzymes, making them more vulnerable to Cys starvation than normal cells (78).
The impact of Cys on hematologic malignancies could be divided into two aspects, Cys metabolism and Cys transport. Regarding Cys metabolism, the LSCs of AML were highly dependent on Cys metabolism in order to fuel OXPHOS. Although cancer cells have been regarded as highly dependent on glycolysis instead of OXPHOS to produce energy, as proposed by Warburg effect, various cancer stem cells have been reported to rely more on OXPHOS than glycolysis, including AML and melanoma (79). The de novo AML LSCs have been proved heavily dependent on Cys metabolism and other AA metabolism in order to conduct OXPHOS and to survive (80). Cys was transformed to GSH, which was significantly reduced upon Cys depletion. The lack of GSH inhibited electron transport chain complex (ETC) II, which in turn damaged OXPHOS and caused death of LSCs. The killing effect was demonstrated specific to LSCs, without affecting normal hematopoietic stem and progenitor cells (HSPCs) (81). Venetoclax combined with azacitidine could also impair AA uptake and reduce LSCs (80).
Besides the shortage of GSH, Cys depletion has been demonstrated to activate 5′ adenosine monophosphate-activated protein kinase (AMPK) pathway, which in turn triggered autophagy. Moreover, mTORC1, which negatively regulated autophagy, was downregulated upon Cys depletion (82).
Regarding Cys transport, the SLC7A11 gene encoded the transporter xCT, which imported Cys into cytoplasm while exported glutamate outside the cells. Since tumor cells exhibited higher ROS levels due to genetic or epigenetic change and aberrantly high metabolic activity, they required higher levels of reducing equivalents to repress the increased ROS, where the GSH made a great contribution to the reduction reaction. Since Cys was the component of GSH, tumor cells had a greater demand for Cys to generate sufficient GSH (11). Several studies have demonstrated increased xCT expression upon the surface of chemo-resistant tumor cells (83). Studies have found that SLC7A11 mutation was an independent risk factor of survival of AML patients (84). Thus, xCT could be a promising target for cancer therapy, especially when combined with other chemotherapy which increased ROS of tumor cells.
Based on the impact of Cys on hematologic malignancies, innovative treatment of hematologic malignancies targeting Cys has been investigated. In terms of Cys depletion, a PEG-engineered cyst(e)inase enzyme, which stably depleted extracellular L-Cys and L-cystine, significantly decreased GSH and increased ROS, leading to cell cycle arrest and apoptosis of various tumor cells (85). It doubled the survival of the aggressive CLL TCL1-Tg:p53−/− mice, and selectively eliminated AML LSCs without influence on survival or colony forming ability of normal HSPCs (80). This enzyme was reported as safe and irreversible, since concentrations of L-Cys and L-cystine returned to untreated level subsequent to two to 4 days, and no obvious side effects were found in mice models (85).
Regarding Cys transport inhibition, several agents have been demonstrated to repress xCT function. Sulfasalazine, though previously developed to treat inflammation, has been reported to selectively inhibit xCT, decrease GSH, increase ROS, decrease proliferation and stimulate apoptosis of primary effusion lymphoma (PEL) cells (86). Sulfasalazine also reduced progression in PEL mice models (87). Pardieu et al. demonstrated that sulfasalazine inhibited xCT and caused oxidative stress-dependent leukemic cell death in primary samples of AML patients. In AML patients with NPM1 mutations, Cys metabolism was upregulated with stronger cysteine dependency. Moreover, they found that sulfasalazine showed significant synergistic anti-leukemic effect with daunorubicin-cytarabine treatment in NPM1-mutated AML samples and patient-derived xenograft models (84). Erastin, though previously proved to trigger ferroptosis, has been recently found to inhibit xCT (88, 89). Erastin increased ROS and repressed viability of various solid tumor cells and AML cells (90–92). Sorafenib, a multi-receptor tyrosine kinase inhibitor, was also found to repress xCT function and stimulate ferroptosis (90). However, more investigation into the exact interaction between xCT function and ferroptosis in Cys starvation is needed to clarify the phenomenon.
Besides the Cys starvation to aggregate ROS and eliminate the existing tumor, Cys supplement has been reported to reduce ROS and delay tumor initiation. The N-acetyl-L-Cys (NAC) has been demonstrated to decrease ROS, repair DNA damage, and activate antioxidant enzymes in leukemic cells. Moreover, NAC could reduce leukemia initiation and organ damage, and prolonged survival in WEHI-3 leukemia mice models (93). Moreover, lymphoma incidence was relatively high in ataxia telangiectasia (AT) patients. Reliene et al. constructed an AT mice model and discovered that consistent dietary NAC supplement significantly decreased lymphoma incidence (94). However, the influence of NAC on tumor cells was controversial. Yedjou et al. aimed to investigate whether NAC supplement could reduce arsenic trioxide (ATO)-related toxicity in acute promyelocytic leukemia (APL) treatment. They discovered that the addition of NAC impeded ATO cytotoxicity, indicating the combination treatment of NAC with ATO was inappropriate to treat APL (95).
NAC has been widely used as antioxidants, and various studies have demonstrated that NAC could assist bone marrow reconstruction by promoting the function of bone marrow endothelial progenitor cells, especially for patients after allo-hematopoietic stem cell transplantation (HSCT) (96–98). A phase III, randomized, open-label trial was conducted in patients who received haploidentical HSCT. Patients underwent evaluation of endothelial cell proportion 2 weeks before conditioning treatment. Patients with endothelial cells less than 0.1% were recognized as high-risk, and were randomized into NAC prophylaxis group and non-prophylaxis group. Patients with endothelial cells more than 0.1% were considered as low-risk, and did not receive NAC prophylaxis. At 60 days after transplantation, the high-risk NAC prophylaxis group displayed significantly reduced poor graft function (PGF) and prolonged isolated thrombocytopenia (PIT) rate compared to high-risk non-prophylaxis group. Furthermore, the PGF and PIT rate was even lower in high-risk NAC prophylaxis group than low-risk group, indicating that NAC prophylaxis could overcome the disadvantage of poor endothelia cell function before transplantation (99).
Met is an essential AA, and serves as the building block of Cys as well as polyamine. The demand for Met relies heavily on dietary supply, and the only way to compensatively generate Met is the Met salvage pathway. In the salvage pathway of normal cells, 5,10-methylene-THF was transformed into 5-methyl-THF via methylenetetrahydrofolate reductase (MTHFR). And the 5-methyl-THF was later generated into Met via methionine synthetase (MS) (100). However, in the salvage pathway of tumor cells, function of these crucial enzymes is frequently inhibited, rendering tumor cells significantly dependent on exogeneous Met, making Met as a promising anti-cancer target and labeled Met as a potential tracer for PET/CT.
The innovative application of 11C-MET PET/CT to diagnosis and staging has been developed recently. Myeloma cells had a high demand for Met in order to generate immunoglobulin, and 11C-MET PET/CT showed higher sensitivity than 18F-FDG PET/CT. Various studies have demonstrated that 11C-MET PET/CT was more sensitive to detect focal lesions and extramedullary disease. The SUVmean of L2-L4 of 11C-MET PET/CT was significantly related to bone marrow plasma cell proportions, while that of 18F-FDG PET/CT showed no significant relationship between plasma cell proportion (101–103). Furthermore, studies have proved that higher total metabolic tumor volume and total lesion MET uptake were significant risk factors of PFS (104).
In lymphoma, the efficacy of 11C-MET PET/CT varies, depending on the site of lesions and the subtype of lymphoma. In CNS detection, 11C-MET PET/CT seemed superior to 18F-FDG PET/CT in differentiation diagnosis of CNS lymphoma from glioblastoma multiformes (GBMs). A study enrolled seven DLBCL and 15 GBM patients. 11C-MET PET/CT performed better than 18F-FDG PET/CT by completely differentiating DLBCL from GBM based on ΔSUVmax < 1.17, with no false negativity or false positivity (105). Furthermore, in primary CNS lymphoma patients, higher tumor-to-normal ratio of 11C-MET PET/CT was significantly related to worse PFS, while that of 18F-FDG PET/CT showed no significant relationship to prognosis (106). However, other studies claimed that 11C-MET PET/CT failed to differentiate primary CNS lymphoma or GBMs (107). In abdomen detection, 11C-MET PET/CT could not effectively differentiate the physiologic high Met uptake or malignant high uptake in pancreas and liver, limiting its application to abdomen detection (106).
Met is also indispensable to epigenetic regulation, because the S-adenosylmethionine (SAM) in the Met cycle serves as the only source to provide methyl residues to DNA and histone methylation. However, tumor cells displayed higher demand for Met and aberrantly stimulated Met adenosyltransferase (MAT), upstream of the SAM (108). The methyltransferase action was in turn overstimulated, which converted SAM mostly into S-adenosylhomoCys (SAH), leading to insufficient methyl residues from SAM and inability to methylate DNA or histone, interfering with the epigenetic regulation. For example, MLL-rearranged leukemia has been demonstrated to rely on the H3K79 methyltransferase DOTL1, and this disease entity was characterized as high demand for methyl residues to ensure DOTL1 function. Barve et al. explored whether inference with Met/SAM metabolism could inhibit MLL-rearranged leukemia. Not only Met depletion but also SAM inhibitor 3-deazaadenosine could decrease methylation level, inhibit proliferation and stimulate apoptosis of the MLL leukemic cells. Moreover, 3-deazaadenosine showed synergistic effect with 5 + 3 induction chemotherapy and extended survival of MLL-rearranged leukemia mice models (109). Dietary Met starvation has been demonstrated to impair H3K36me3, decrease total RNA concentration, stimulate apoptosis and cell cycle arrest of AML cells, delay AML progression in mice models without obvious adverse effects (110, 111). Thus, MAT inhibition might serve as a promising approach to treat hematologic malignancies with epigenetic alteration or IDH/TET mutations. SAM decarboxylase inhibitors have been explored in cancer treatment. SAM486A is an innovative second-generation SAM decarboxylase inhibitor, which has been tested in a phase II, multicenter clinical trial. The study enrolled 41 relapsed or refractory NHL patients, who received SAM486A monotherapy for eight cycles or until disease progression. The ORR was 18.9% with a tolerable safety profile (112). AG-270, a first-in-class MAT2A inhibitor which effectively decreased SAM and impeded tumor cell proliferation, is being investigated in lymphoma and solid tumor patients in an ongoing phase I trial (113).
Single nucleotide polymorphisms in various Met metabolic enzymes, including MS A2765G, MTRR A66G, MTHFR 677 or 1,298 gene polymorphism displayed different susceptibility to lymphoma, ALL, AML, and Chronic myeloid leukemia (CML), probably due to subtypes of malignancies, ethic factors, and gender (114–119).
Serine is a non-essential AA and serves as building blocks of glycine, phospholipids, and nucleotides. Tumor cells upregulate serine-related enzymes, including PHGDH, PSPH, and PSAT1, in order to meet the increased serine demand, indicating these enzymes as druggable target to treat hematologic malignancies. In FLT3-ITD-driven AML, FLT3-ITD upregulated serine synthesis via ATF4 (120). In T-ALL, PSPH transcription and translation were significantly increased in T-ALL cell lines and primary bone marrow cells of T-ALL patients. PSPH upregulation led to increase of serine synthesis, which not only directly promoted leukemic cell proliferation through increased purine and folate, but also indirectly promoted stromal cell proliferation through increased glycine (121). In MM, serine upregulation was associated with bortezomib resistance (122). These findings above indicated that serine metabolic enzymes could serve as promising target to treat hematologic malignancies.
WQ-2101 is a PHGDH inhibitor, and has been evaluated in FLT3-ITD-driven AML. Both WQ-2101 and PHGDH knockout could repress proliferation of leukemic cells in vitro and in vivo. Furthermore, WQ-2101 could increase the extent of DNA damage caused by cytarabine, increasing the sensitivity of leukemic cells to cytarabine (120). In T-ALL, inhibition of PSPH by shPSPH could repress proliferation and trigger apoptosis of leukemic cells in vitro. It could also alleviate splenomegaly and decrease leukemic cells in the bone marrow and spleen of T-ALL mice models (121).
Branched chain AAs (BCAAs) include valine, leucine, and isoleucine, and have been found related to aggressiveness of leukemia. BCAA transaminase 1 (BCAT1) and BCAA transaminase 2 (BCAT2) produce BCAAs via aminating branched chain keto acids. It has been reported that BCAT1 was overexpressed in CML patients and mouse models, and was related to worse prognosis. The oncogenic protein Musashi2 bound to BCAT1 and upregulate its expression, leading to progression of blast crisis CML. Inhibition of BCAT1 expression stimulated differentiation of blast cells and repressed blast crisis CML (123). Furthermore, BCAT1 and BCAT2 were upregulated by the overexpressed oncogenic m6A methyltransferase, METTL16, leading to BCAA reprogram in LSCs and leukemia-initiating cells. METTL16 knockout significantly repressed AML leukemogenesis and impeded LSC self-renewal (124). Studies have explored the BCAA-related metabolism in CD34+ cells of healthy controls, AML patients and ALL patients. The BCAA transporters, BCAT, BCAA concentration and α-KG were significantly higher in AML and ALL than those in healthy controls. Dietary BCAA starvation significantly repressed proliferation, development and self-renewal of LSCs in xenogeneic transplantation models (125). In a word, BCAA reprogram is relatively common in leukemia, which might maintain LSC stemness and promote leukemia transformation. BCAA starvation might be a promising strategy to postpone leukemogenesis.
Although AA starvation could impair viability of tumor cells, they could also possibly impair the function of immune cells, leading to immune escape. For example, since Met serves as the major source of methyl residue for DNA and histone methylation, starvation of Met inevitably caused epigenetic malfunction and impaired T cell function. To avoid the influence of Met starvation on normal cells, supplement of homocysteine, vitamin B12 and folate might assist Met salvage pathway in normal cells. Furthermore, cystine played an important role in T cell expansion as well as activation. Although Cys starvation has been reported effective to treat hematologic malignancies, cysteinase was also reported to promote anti-tumor immune response. The impact of AA starvation on immune cells has been controversial and requires further investigation to reveal their interaction.
Studies have found that chemoresistance is frequently related to the protection of tumor cells by the AA-providing bone marrow stromal cells. For example, Ede et al. explored the mechanism of chemoresistance to parthenolide in pediatric T-ALL patients. They discovered that the bone marrow mesenchymal stromal cells generated and provided Cys to leukemic cells, rendering them survival benefits (126). Moreover, in CLL, resistance to ASNase was attributable to the cystine provided to tumor cells by stromal cells (127). Thus, combination treatment of AA starvation with conventional chemotherapy might overcome chemoresistance and achieve a higher response rate.
Although AA starvation has shown some effect on treating hematologic malignancies, the half-life period of starvation was sometimes short in its current form, limiting its efficacy and feasibility in the future clinical practice. For example, the Cys returned to previous level only 2 days after injection of a cyst(e)inase into mice models, requiring improvement of its form to ensure more stable and sustainable effect in vivo[64].
AAs are indispensable to development and progression of hematologic malignancies, with tumor cells demanding significantly larger amount of various AAs for enhanced proliferation, abnormal signaling transduction, and epigenetic dysregulation. The innovative therapy targeting AAs mainly aims at AA metabolism (either starvation or supplement) and transportation. Numbers of AA starvation therapy has been approved for hematologic treatment, such as L-ASNase. Treatment aiming at AA supplement, such as NAC, or transportation, such as xCT inhibitors, is being investigated in clinical trials and showing encouraging results. The innovative AA therapy will provide a promising approach to further improve treatment of hematologic malignancies, especially when combined to traditional chemotherapy in a more stable form with longer half-life period.
MW: writing—original draft, writing—review and editing. AZ: writing—original draft, writing—review and editing. ML: writing—original draft. TN: validation, writing—review and editing. All authors contributed to the article and approved the submitted version.
This work was supported by Incubation Program for Clinical Trials (No. 19HXFH030), Achievement Transformation Project (No. CGZH21001), 1.3.5 Project for Disciplines of Excellence, West China Hospital, Sichuan University (No. ZYJC21007), Translational Research Grant of NCRCH (No. 2021WWB03), Chengdu Science and Technology Program (No. 2022-YF05-01444-SN), Key Research and Development Program of Sichuan Province (No. 2023YFS0031), and National Key Research and Development Program of China (No. 2022YFC2502603).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Hanahan, D, and Weinberg, RA. Hallmarks of cancer: the next generation. Cells. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
2. Schwartz, L, Supuran, CT, and Alfarouk, KO. The Warburg effect and the hallmarks of cancer. Anti Cancer Agents Med Chem. (2017) 17:164–70. doi: 10.2174/1871520616666161031143301
3. Park, JH, Pyun, WY, and Park, HW. Cancer metabolism: phenotype, signaling and therapeutic targets. Cells-Basel. (2020) 9:9. doi: 10.3390/cells9102308
4. Martinez-Outschoorn, UE, Peiris-Pages, M, Pestell, RG, Sotgia, F, and Lisanti, MP. Cancer metabolism: a therapeutic perspective. Nat Rev Clin Oncol. (2017) 14:11–31. doi: 10.1038/nrclinonc.2016.60
5. Warburg, O. On the origin of cancer cells. Science. (1956) 123:309–14. doi: 10.1126/science.123.3191.309
6. Lieu, EL, Nguyen, T, Rhyne, S, and Kim, J. Amino acids in cancer. Exp Mol Med. (2020) 52:15–30. doi: 10.1038/s12276-020-0375-3
7. Mossmann, D, Park, S, and Hall, MN. Mtor signalling and cellular metabolism are mutual determinants in cancer. Nat Rev Cancer. (2018) 18:744–57. doi: 10.1038/s41568-018-0074-8
8. Chen, CL, Hsu, SC, Chung, TY, Chu, CY, Wang, HJ, Hsiao, PW, et al. Arginine is an epigenetic regulator targeting tead4 to modulate oxphos in prostate cancer cells. Nat Commun. (2021) 12:2398. doi: 10.1038/s41467-021-22652-9
9. Zhang, L, Yao, Y, Zhang, S, Liu, Y, Guo, H, Ahmed, M, et al. Metabolic reprogramming toward oxidative phosphorylation identifies a therapeutic target for mantle cell lymphoma. Sci Transl Med. (2019) 11:11. doi: 10.1126/scitranslmed.aau1167
10. Lemos, H, Huang, L, Prendergast, GC, and Mellor, AL. Immune control by amino acid catabolism during tumorigenesis and therapy. Nat Rev Cancer. (2019) 19:162–75. doi: 10.1038/s41568-019-0106-z
11. Tabe, Y, Lorenzi, PL, and Konopleva, M. Amino acid metabolism in hematologic malignancies and the era of targeted therapy. Blood. (2019) 134:1014–23. doi: 10.1182/blood.2019001034
12. Van Trimpont, M, Peeters, E, de Visser, Y, Schalk, AM, Mondelaers, V, de Moerloose, B, et al. Novel insights on the use of l-asparaginase as an efficient and safe anti-cancer therapy. Cancers (Basel). (2022) 14:14. doi: 10.3390/cancers14040902
13. Cruzat, V, Macedo, RM, Noel, KK, Curi, R, and Newsholme, P. Glutamine: metabolism and immune function, supplementation and clinical translation. Nutrients. (2018):10. doi: 10.3390/nu10111564
14. Halama, A, and Suhre, K. Advancing cancer treatment by targeting glutamine metabolism-a roadmap. Cancers (Basel). (2022) 14:14. doi: 10.3390/cancers14030553
15. Altman, BJ, Stine, ZE, and Dang, CV. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer. (2016) 16:619–34. doi: 10.1038/nrc.2016.71
16. van Geldermalsen, M, Wang, Q, Nagarajah, R, Marshall, AD, Thoeng, A, Gao, D, et al. Asct2/slc1a5 controls glutamine uptake and tumour growth in triple-negative basal-like breast cancer. Oncogene. (2016) 35:3201–8. doi: 10.1038/onc.2015.381
17. Bhutia, YD, Babu, E, Ramachandran, S, and Ganapathy, V. Amino acid transporters in cancer and their relevance to “glutamine addiction”: novel targets for the design of a new class of anticancer drugs. Cancer Res. (2015) 75:1782–8. doi: 10.1158/0008-5472.CAN-14-3745
18. Recouvreux, MV, and Commisso, C. Macropinocytosis: a metabolic adaptation to nutrient stress in cancer. Front Endocrinol (Lausanne). (2017) 8:261. doi: 10.3389/fendo.2017.00261
19. Nicklin, P, Bergman, P, Zhang, B, Triantafellow, E, Wang, H, Nyfeler, B, et al. Bidirectional transport of amino acids regulates mtor and autophagy. Cells. (2009) 136:521–34. doi: 10.1016/j.cell.2008.11.044
20. Irwin, ME, Rivera-Del, VN, and Chandra, J. Redox control of leukemia: from molecular mechanisms to therapeutic opportunities. Antioxid Redox Signal. (2013) 18:1349–83. doi: 10.1089/ars.2011.4258
21. Chen, Y, Li, J, and Zhao, Z. Redox control in acute lymphoblastic leukemia: from physiology to pathology and therapeutic opportunities. Cells-Basel. (2021):10. doi: 10.3390/cells10051218
22. Jacque, N, Ronchetti, AM, Larrue, C, Meunier, G, Birsen, R, Willems, L, et al. Targeting glutaminolysis has antileukemic activity in acute myeloid leukemia and synergizes with bcl-2 inhibition. Blood. (2015) 126:1346–56. doi: 10.1182/blood-2015-01-621870
23. Lagadinou, ED, Sach, A, Callahan, K, Rossi, RM, Neering, SJ, Minhajuddin, M, et al. Bcl-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell. (2013) 12:329–41. doi: 10.1016/j.stem.2012.12.013
24. Xu, W, Yang, H, Liu, Y, Yang, Y, Wang, P, Kim, SH, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. (2011) 19:17–30. doi: 10.1016/j.ccr.2010.12.014
25. Patel, JP, Gönen, M, Figueroa, ME, Fernandez, H, Sun, Z, Racevskis, J, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. (2012) 366:1079–89. doi: 10.1056/NEJMoa1112304
26. Chou, WC, Lei, WC, Ko, BS, Hou, HA, Chen, CY, Tang, JL, et al. The prognostic impact and stability of isocitrate dehydrogenase 2 mutation in adult patients with acute myeloid leukemia. Leukemia. (2011) 25:246–53. doi: 10.1038/leu.2010.267
27. Marcucci, G, Maharry, K, Wu, YZ, Radmacher, MD, Mrózek, K, Margeson, D, et al. Idh1 and idh2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: a cancer and leukemia group b study. J Clin Oncol. (2010) 28:2348–55. doi: 10.1200/JCO.2009.27.3730
28. Abbas, S, Lugthart, S, Kavelaars, FG, Schelen, A, Koenders, JE, Zeilemaker, A, et al. Acquired mutations in the genes encoding idh1 and idh2 both are recurrent aberrations in acute myeloid leukemia: prevalence and prognostic value. Blood. (2010) 116:2122–6. doi: 10.1182/blood-2009-11-250878
29. Giuliani, N, Chiu, M, Bolzoni, M, Accardi, F, Bianchi, MG, Toscani, D, et al. The potential of inhibiting glutamine uptake as a therapeutic target for multiple myeloma. Expert Opin Ther Targets. (2017) 21:231–4. doi: 10.1080/14728222.2017.1279148
30. Bolzoni, M, Chiu, M, Accardi, F, Vescovini, R, Airoldi, I, Storti, P, et al. Dependence on glutamine uptake and glutamine addiction characterize myeloma cells: a new attractive target. Blood. (2016) 128:667–79. doi: 10.1182/blood-2016-01-690743
31. Yang, L, Venneti, S, and Nagrath, D. Glutaminolysis: a hallmark of cancer metabolism. Annu Rev Biomed Eng. (2017) 19:163–94. doi: 10.1146/annurev-bioeng-071516-044546
32. Jeong, SM, Lee, A, Lee, J, and Haigis, MC. Sirt4 protein suppresses tumor formation in genetic models of myc-induced b cell lymphoma. J Biol Chem. (2014) 289:4135–44. doi: 10.1074/jbc.M113.525949
33. Xiong, J, Wang, N, Zhong, HJ, Cui, BW, Cheng, S, Sun, R, et al. Slc1a1 mediated glutamine addiction and contributed to natural killer t-cell lymphoma progression with immunotherapeutic potential. EBioMedicine. (2021) 72:103614. doi: 10.1016/j.ebiom.2021.103614
34. Wang, D, Li, X, Gong, G, Lu, Y, Guo, Z, Chen, R, et al. An updated patent review of glutaminase inhibitors (2019-2022). Expert Opin Ther Pat. (2023) 33:17–28. doi: 10.1080/13543776.2023.2173573
35. Kim, GW, Lee, DH, Jeon, YH, Yoo, J, Kim, SY, Lee, SW, et al. Glutamine synthetase as a therapeutic target for cancer treatment. Int J Mol Sci. (2021) 22:22. doi: 10.3390/ijms22041701
36. Ni, F, Yu, WM, Li, Z, Graham, DK, Jin, L, Kang, S, et al. Critical role of asct2-mediated amino acid metabolism in promoting leukaemia development and progression. Nat Metab. (2019) 1:390–403. doi: 10.1038/s42255-019-0039-6
37. Schulte, ML, Fu, A, Zhao, P, Li, J, Geng, L, Smith, ST, et al. Pharmacological blockade of asct2-dependent glutamine transport leads to antitumor efficacy in preclinical models. Nat Med. (2018) 24:194–202. doi: 10.1038/nm.4464
38. Li, QQ, Pan, SY, Chen, QY, Zhou, W, and Wang, SQ. Effect of competitive antagonist of transmembrane glutamine flux v-9302 on apoptosis of acute myeloid leukemia cell lines hl-60 and kg-1. Zhongguo Shi Yan Xue Ye Xue Za Zhi. (2021) 29:685–9. doi: 10.19746/j.cnki.issn.1009-2137.2021.03.005
39. Tenora, L, Alt, J, Dash, RP, Gadiano, AJ, Novotná, K, Veeravalli, V, et al. Tumor-targeted delivery of 6-diazo-5-oxo-l-norleucine (don) using substituted acetylated lysine prodrugs. J Med Chem. (2019) 62:3524–38. doi: 10.1021/acs.jmedchem.8b02009
40. Thompson, RM, Dytfeld, D, Reyes, L, Robinson, RM, Smith, B, Manevich, Y, et al. Glutaminase inhibitor cb-839 synergizes with carfilzomib in resistant multiple myeloma cells. Oncotarget. (2017) 8:35863–76. doi: 10.18632/oncotarget.16262
41. Gregory, MA, Nemkov, T, Park, HJ, Zaberezhnyy, V, Gehrke, S, Adane, B, et al. Targeting glutamine metabolism and redox state for leukemia therapy. Clin Cancer Res. (2019) 25:4079–90. doi: 10.1158/1078-0432.CCR-18-3223
42. Zavorka Thomas, ME, Lu, X, Talebi, Z, Jeon, JY, Buelow, DR, Gibson, AA, et al. Gilteritinib inhibits glutamine uptake and utilization in flt3-itd-positive aml. Mol Cancer Ther. (2021) 20:2207–17. doi: 10.1158/1535-7163.MCT-21-0071
43. Gregory, MA, Nemkov, T, Reisz, JA, Zaberezhnyy, V, Hansen, KC, D’Alessandro, A, et al. Glutaminase inhibition improves flt3 inhibitor therapy for acute myeloid leukemia. Exp Hematol. (2018) 58:52–8. doi: 10.1016/j.exphem.2017.09.007
44. Butler, M, van der Meer, LT, and van Leeuwen, FN. Amino acid depletion therapies: starving cancer cells to death. Trends Endocrinol Metab. (2021) 32:367–81. doi: 10.1016/j.tem.2021.03.003
45. Sullivan, MP, Nelson, JA, Feldman, S, and Van Nguyen, B. Pharmacokinetic and phase i study of intravenous don (6-diazo-5-oxo-l-norleucine) in children. Cancer Chemother Pharmacol. (1988) 21:78–84. doi: 10.1007/BF00262746
46. Lemberg, KM, Vornov, JJ, Rais, R, and Slusher, BS. We’re not “don” yet: optimal dosing and prodrug delivery of 6-diazo-5-oxo-l-norleucine. Mol Cancer Ther. (2018) 17:1824–32. doi: 10.1158/1535-7163.MCT-17-1148
47. Hanaford, AR, Alt, J, Rais, R, Wang, SZ, Kaur, H, Thorek, D, et al. Orally bioavailable glutamine antagonist prodrug jhu-083 penetrates mouse brain and suppresses the growth of myc-driven medulloblastoma. Transl Oncol. (2019) 12:1314–22. doi: 10.1016/j.tranon.2019.05.013
48. Jahani, M, Noroznezhad, F, and Mansouri, K. Arginine: challenges and opportunities of this two-faced molecule in cancer therapy. Biomed Pharmacother. (2018) 102:594–601. doi: 10.1016/j.biopha.2018.02.109
49. Choi, BH, and Coloff, JL. The diverse functions of non-essential amino acids in cancer. Cancers (Basel). (2019):11. doi: 10.3390/cancers11050675
50. Chen, CL, Hsu, SC, Ann, DK, Yen, Y, and Kung, HJ. Arginine signaling and cancer metabolism. Cancers (Basel). (2021) 13:13. doi: 10.3390/cancers13143541
51. Szefel, J, Danielak, A, and Kruszewski, WJ. Metabolic pathways of l-arginine and therapeutic consequences in tumors. Adv Med Sci. (2019) 64:104–10. doi: 10.1016/j.advms.2018.08.018
52. de Santo, C, Booth, S, Vardon, A, Cousins, A, Tubb, V, Perry, T, et al. The arginine metabolome in acute lymphoblastic leukemia can be targeted by the pegylated-recombinant arginase i bct-100. Int J Cancer. (2018) 142:1490–502. doi: 10.1002/ijc.31170
53. Delage, B, Luong, P, Maharaj, L, O’Riain, C, Syed, N, Crook, T, et al. Promoter methylation of argininosuccinate synthetase-1 sensitises lymphomas to arginine deiminase treatment, autophagy and caspase-dependent apoptosis. Cell Death Dis. (2012) 3:e342. doi: 10.1038/cddis.2012.83
54. Atik, E, Ergin, M, Erdogan, S, and Tuncer, I. Inducible nitric oxide synthase and apoptosis in human b cell lymphomas. Mol Cell Biochem. (2006) 290:205–9. doi: 10.1007/s11010-005-9114-2
55. Adams, DJ, Levesque, MC, Weinberg, JB, Smith, KL, Flowers, JL, Moore, J, et al. Nitric oxide enhancement of fludarabine cytotoxicity for b-cll lymphocytes. Leukemia. (2001) 15:1852–9. doi: 10.1038/sj.leu.2402291
56. Kumari, N, and Bansal, S. Arginine depriving enzymes: applications as emerging therapeutics in cancer treatment. Cancer Chemother Pharmacol. (2021) 88:565–94. doi: 10.1007/s00280-021-04335-w
57. Phillips, MM, Sheaff, MT, and Szlosarek, PW. Targeting arginine-dependent cancers with arginine-degrading enzymes: opportunities and challenges. Cancer Res Treat. (2013) 45:251–62. doi: 10.4143/crt.2013.45.4.251
58. Morrow, K, Hernandez, CP, Raber, P, del Valle, L, Wilk, AM, Majumdar, S, et al. Anti-leukemic mechanisms of pegylated arginase i in acute lymphoblastic t-cell leukemia. Leukemia. (2013) 27:569–77. doi: 10.1038/leu.2012.247
59. Hernandez, CP, Morrow, K, Lopez-Barcons, LA, Zabaleta, J, Sierra, R, Velasco, C, et al. Pegylated arginase i: a potential therapeutic approach in t-all. Blood. (2010) 115:5214–21. doi: 10.1182/blood-2009-12-258822
60. Miraki-Moud, F, Ghazaly, E, Ariza-McNaughton, L, Hodby, KA, Clear, A, Anjos-Afonso, F, et al. Arginine deprivation using pegylated arginine deiminase has activity against primary acute myeloid leukemia cells in vivo. Blood. (2015) 125:4060–8. doi: 10.1182/blood-2014-10-608133
61. Tsai, HJ, Jiang, SS, Hung, WC, Borthakur, G, Lin, SF, Pemmaraju, N, et al. A phase ii study of arginine deiminase (adi-peg20) in relapsed/refractory or poor-risk acute myeloid leukemia patients. Sci Rep. (2017) 7:11253. doi: 10.1038/s41598-017-10542-4
62. Jiang, J, Batra, S, and Zhang, J. Asparagine: a metabolite to be targeted in cancers. Meta. (2021) 11:402. doi: 10.3390/metabo11060402
63. Lomelino, CL, Andring, JT, McKenna, R, and Kilberg, MS. Asparagine synthetase: function, structure, and role in disease. J Biol Chem. (2017) 292:19952–8. doi: 10.1074/jbc.R117.819060
64. Krall, AS, Xu, S, Graeber, TG, Braas, D, and Christofk, HR. Asparagine promotes cancer cell proliferation through use as an amino acid exchange factor. Nat Commun. (2016) 7:11457. doi: 10.1038/ncomms11457
65. Zhang, J, Fan, J, Venneti, S, Cross, JR, Takagi, T, Bhinder, B, et al. Asparagine plays a critical role in regulating cellular adaptation to glutamine depletion. Mol Cell. (2014) 56:205–18. doi: 10.1016/j.molcel.2014.08.018
66. Chiu, M, Taurino, G, Bianchi, MG, Kilberg, MS, and Bussolati, O. Asparagine synthetase in cancer: beyond acute lymphoblastic leukemia. Front Oncol. (2019) 9:1480. doi: 10.3389/fonc.2019.01480
67. Schmidt, MP, Ivanov, AV, Coriu, D, and Miron, IC. L-asparaginase toxicity in the treatment of children and adolescents with acute lymphoblastic leukemia. J Clin Med. (2021) 10:10. doi: 10.3390/jcm10194419
68. Jaccard, A, Gachard, N, Marin, B, Rogez, S, Audrain, M, Suarez, F, et al. Efficacy of l-asparaginase with methotrexate and dexamethasone (aspametdex regimen) in patients with refractory or relapsing extranodal nk/t-cell lymphoma, a phase 2 study. Blood. (2011) 117:1834–9. doi: 10.1182/blood-2010-09-307454
69. Zhou, Z, Li, X, Chen, C, Li, X, Zhang, L, Li, L, et al. Effectiveness of gemcitabine, pegaspargase, cisplatin, and dexamethasone (ddgp) combination chemotherapy in the treatment of relapsed/refractory extranodal nk/t cell lymphoma: a retrospective study of 17 patients. Ann Hematol. (2014) 93:1889–94. doi: 10.1007/s00277-014-2136-7
70. Lin, N, Song, Y, Zheng, W, Tu, M, Xie, Y, Wang, X, et al. A prospective phase ii study of l-asparaginase- chop plus radiation in newly diagnosed extranodal nk/t-cell lymphoma, nasal type. J Hematol Oncol. (2013) 6:44. doi: 10.1186/1756-8722-6-44
71. Alexander, TB, Wang, L, Inaba, H, Triplett, BM, Pounds, S, Ribeiro, RC, et al. Decreased relapsed rate and treatment-related mortality contribute to improved outcomes for pediatric acute myeloid leukemia in successive clinical trials. Cancer-Am Cancer Soc. (2017) 123:3791–8. doi: 10.1002/cncr.30791
72. Aplenc, R, Alonzo, TA, Gerbing, RB, Lange, BJ, Hurwitz, CA, Wells, RJ, et al. Safety and efficacy of gemtuzumab ozogamicin in combination with chemotherapy for pediatric acute myeloid leukemia: a report from the children’s oncology group. J Clin Oncol. (2008) 26:2390–5. doi: 10.1200/JCO.2007.13.0096
73. Haskell, CM, and Canellos, GP. L-asparaginase resistance in human leukemia--asparagine synthetase. Biochem Pharmacol. (1969) 18:2578–80. doi: 10.1016/0006-2952(69)90375-x
74. Iwamoto, S, Mihara, K, Downing, JR, Pui, CH, and Campana, D. Mesenchymal cells regulate the response of acute lymphoblastic leukemia cells to asparaginase. J Clin Invest. (2007) 117:1049–57. doi: 10.1172/JCI30235
75. Lee, JK, Kang, S, Wang, X, Rosales, JL, Gao, X, Byun, HG, et al. Hap1 loss confers l-asparaginase resistance in all by downregulating the calpain-1-bid-caspase-3/12 pathway. Blood. (2019) 133:2222–32. doi: 10.1182/blood-2018-12-890236
76. Wang, L, Yang, J, Wang, HN, Fu, RY, Liu, XD, Piao, YS, et al. Lncrna bcyrn1-induced autophagy enhances asparaginase resistance in extranodal nk/t-cell lymphoma. Theranostics. (2021) 11:925–40. doi: 10.7150/thno.46655
77. Vrooman, LM, Blonquist, TM, Stevenson, KE, Supko, JG, Hunt, SK, Cronholm, SM, et al. Efficacy and toxicity of pegaspargase and calaspargase pegol in childhood acute lymphoblastic leukemia: results of dfci 11-001. J Clin Oncol. (2021) 39:3496–505. doi: 10.1200/JCO.20.03692
78. Bonifacio, V, Pereira, SA, Serpa, J, and Vicente, JB. Cysteine metabolic circuitries: druggable targets in cancer. Br J Cancer. (2021) 124:862–79. doi: 10.1038/s41416-020-01156-1
79. Culp-Hill, R, D’Alessandro, A, and Pietras, EM. Extinguishing the embers: targeting aml metabolism. Trends Mol Med. (2021) 27:332–44. doi: 10.1016/j.molmed.2020.10.001
80. Jones, CL, Stevens, BM, D’Alessandro, A, Reisz, JA, Culp-Hill, R, Nemkov, T, et al. Inhibition of amino acid metabolism selectively targets human leukemia stem cells. Cancer Cell. (2018) 34:724–740.e4. doi: 10.1016/j.ccell.2018.10.005
81. Jones, CL, Stevens, BM, D’Alessandro, A, Culp-Hill, R, Reisz, JA, Pei, S, et al. Cysteine depletion targets leukemia stem cells through inhibition of electron transport complex ii. Blood. (2019) 134:389–94. doi: 10.1182/blood.2019898114
82. Ghislat, G, Patron, M, Rizzuto, R, and Knecht, E. Withdrawal of essential amino acids increases autophagy by a pathway involving ca2+/calmodulin-dependent kinase kinase-beta (camkk-beta). J Biol Chem. (2012) 287:38625–36. doi: 10.1074/jbc.M112.365767
83. Huang, Y, Dai, Z, Barbacioru, C, and Sadee, W. Cystine-glutamate transporter slc7a11 in cancer chemosensitivity and chemoresistance. Cancer Res. (2005) 65:7446–54. doi: 10.1158/0008-5472.CAN-04-4267
84. Pardieu, B, Pasanisi, J, Ling, F, Dal Bello, R, Penneroux, J, Su, A, et al. Cystine uptake inhibition potentiates front-line therapies in acute myeloid leukemia. Leukemia. (2022) 36:1585–95. doi: 10.1038/s41375-022-01573-6
85. Cramer, SL, Saha, A, Liu, J, Tadi, S, Tiziani, S, Yan, W, et al. Systemic depletion of l-cyst(e)ine with cyst(e)inase increases reactive oxygen species and suppresses tumor growth. Nat Med. (2017) 23:120–7. doi: 10.1038/nm.4232
86. Gout, PW, Buckley, AR, Simms, CR, and Bruchovsky, N. Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x(c)- cystine transporter: a new action for an old drug. Leukemia. (2001) 15:1633–40. doi: 10.1038/sj.leu.2402238
87. Dai, L, Cao, Y, Chen, Y, Parsons, C, and Qin, Z. Targeting xct, a cystine-glutamate transporter induces apoptosis and tumor regression for kshv/hiv-associated lymphoma. J Hematol Oncol. (2014) 7:30. doi: 10.1186/1756-8722-7-30
88. Yan, R, Xie, E, Li, Y, Li, J, Zhang, Y, Chi, X, et al. The structure of erastin-bound xct-4f2hc complex reveals molecular mechanisms underlying erastin-induced ferroptosis. Cell Res. (2022) 32:687–90. doi: 10.1038/s41422-022-00642-w
89. Chen, D, Fan, Z, Rauh, M, Buchfelder, M, Eyupoglu, IY, and Savaskan, N. Atf4 promotes angiogenesis and neuronal cell death and confers ferroptosis in a xct-dependent manner. Oncogene. (2017) 36:5593–608. doi: 10.1038/onc.2017.146
90. Dixon, SJ, Patel, DN, Welsch, M, Skouta, R, Lee, ED, Hayano, M, et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife. (2014) 3:e2523. doi: 10.7554/eLife.02523
91. Codenotti, S, Poli, M, Asperti, M, Zizioli, D, Marampon, F, and Fanzani, A. Cell growth potential drives ferroptosis susceptibility in rhabdomyosarcoma and myoblast cell lines. J Cancer Res Clin Oncol. (2018) 144:1717–30. doi: 10.1007/s00432-018-2699-0
92. Zheng, Z, Hong, X, Huang, X, Jiang, X, Jiang, H, Huang, Y, et al. Comprehensive analysis of ferroptosis-related gene signatures as a potential therapeutic target for acute myeloid leukemia: a bioinformatics analysis and experimental verification. Front Oncol. (2022) 12:930654. doi: 10.3389/fonc.2022.930654
93. Shih, WL, Chang, CD, Chen, HT, and Fan, KK. Antioxidant activity and leukemia initiation prevention in vitro and in vivo by n-acetyl-l-cysteine. Oncol Lett. (2018) 16:2046–52. doi: 10.3892/ol.2018.8864
94. Reliene, R, and Schiestl, RH. Antioxidant n-acetyl cysteine reduces incidence and multiplicity of lymphoma in atm deficient mice. DNA Repair (Amst). (2006) 5:852–9. doi: 10.1016/j.dnarep.2006.05.003
95. Yedjou, CG, Rogers, C, Brown, E, and Tchounwou, PB. Differential effect of ascorbic acid and n-acetyl-l-cysteine on arsenic trioxide-mediated oxidative stress in human leukemia (hl-60) cells. J Biochem Mol Toxicol. (2008) 22:85–92. doi: 10.1002/jbt.20223
96. Shi, MM, Kong, Y, Song, Y, Sun, YQ, Wang, Y, Zhang, XH, et al. Atorvastatin enhances endothelial cell function in posttransplant poor graft function. Blood. (2016) 128:2988–99. doi: 10.1182/blood-2016-03-702803
97. Zhao, HY, Lyu, ZS, Duan, CW, Song, Y, Han, TT, Mo, XD, et al. An unbalanced monocyte macrophage polarization in the bone marrow microenvironment of patients with poor graft function after allogeneic haematopoietic stem cell transplantation. Br J Haematol. (2018) 182:679–92. doi: 10.1111/bjh.15452
98. Kong, Y, Shi, MM, Zhang, YY, Cao, XN, Wang, Y, Zhang, XH, et al. N-acetyl-l-cysteine improves bone marrow endothelial progenitor cells in prolonged isolated thrombocytopenia patients post allogeneic hematopoietic stem cell transplantation. Am J Hematol. (2018) 93:931–42. doi: 10.1002/ajh.25056
99. Wang, Y, Kong, Y, Zhao, HY, Zhang, YY, Wang, YZ, Xu, LP, et al. Prophylactic nac promoted hematopoietic reconstitution by improving endothelial cells after haploidentical hsct: a phase 3, open-label randomized trial. BMC Med. (2022) 20:140. doi: 10.1186/s12916-022-02338-9
100. Fernandes, HS, Silva, TC, Fernandes, PA, Ramos, MJ, and Cerqueira, NM. Amino acid deprivation using enzymes as a targeted therapy for cancer and viral infections. Expert Opin Ther Pat. (2017) 27:283–97. doi: 10.1080/13543776.2017.1254194
101. Nakamoto, Y, Kurihara, K, Nishizawa, M, Yamashita, K, Nakatani, K, Kondo, T, et al. Clinical value of (1)(1)c-methionine pet/ct in patients with plasma cell malignancy: comparison with (1)(8)f-fdg pet/ct. Eur J Nucl Med Mol Imaging. (2013) 40:708–15. doi: 10.1007/s00259-012-2333-3
102. Lapa, C, Garcia-Velloso, MJ, Lückerath, K, Samnick, S, Schreder, M, Otero, PR, et al. (11)c-methionine-pet in multiple myeloma: a combined study from two different institutions. Theranostics. (2017) 7:2956–64. doi: 10.7150/thno.20491
103. Zhou, X, Dierks, A, Kertels, O, Kircher, M, Schirbel, A, Samnick, S, et al. 18f-fdg, 11c-methionine, and 68ga-pentixafor pet/ct in patients with smoldering multiple myeloma: imaging pattern and clinical features. Cancers (Basel). (2020) 12:12. doi: 10.3390/cancers12082333
104. Morales-Lozano, MI, Rodriguez-Otero, P, Sancho, L, Nuñez-Cordoba, JM, Prieto, E, Marcos-Jubilar, M, et al. (11)c-methionine pet/ct in assessment of multiple myeloma patients: comparison to (18)f-fdg pet/ct and prognostic value. Int J Mol Sci. (2022) 23:23. doi: 10.3390/ijms23179895
105. Okada, Y, Nihashi, T, Fujii, M, Kato, K, Okochi, Y, Ando, Y, et al. Differentiation of newly diagnosed glioblastoma multiforme and intracranial diffuse large b-cell lymphoma using (11)c-methionine and (18)f-fdg pet. Clin Nucl Med. (2012) 37:843–9. doi: 10.1097/RLU.0b013e318262af48
106. Ahn, SY, Kwon, SY, Jung, SH, Ahn, JS, Yoo, SW, Min, JJ, et al. Prognostic significance of interim 11c-methionine pet/ct in primary central nervous system lymphoma. Clin Nucl Med. (2018) 43:e259–64. doi: 10.1097/RLU.0000000000002154
107. Postnov, A, Toutain, J, Pronin, I, Valable, S, Gourand, F, Kalaeva, D, et al. First-in-man noninvasive initial diagnostic approach of primary cns lymphoma versus glioblastoma using pet with 18 f-fludarabine and l -[methyl- 11 c]methionine. Clin Nucl Med. (2022) 47:699–706. doi: 10.1097/RLU.0000000000004238
108. Wang, Z, Yip, LY, Lee, J, Wu, Z, Chew, HY, Chong, P, et al. Methionine is a metabolic dependency of tumor-initiating cells. Nat Med. (2019) 25:825–37. doi: 10.1038/s41591-019-0423-5
109. Barve, A, Vega, A, Shah, PP, Ghare, S, Casson, L, Wunderlich, M, et al. Perturbation of methionine/s-adenosylmethionine metabolism as a novel vulnerability in mll rearranged leukemia. Cells-Basel. (2019) 8:8. doi: 10.3390/cells8111322
110. Cunningham, A, Erdem, A, Alshamleh, I, Geugien, M, Pruis, M, Pereira-Martins, DA, et al. Dietary methionine starvation impairs acute myeloid leukemia progression. Blood. (2022) 140:2037–52. doi: 10.1182/blood.2022017575
111. Ulanovskaya, OA, Zuhl, AM, and Cravatt, BF. Nnmt promotes epigenetic remodeling in cancer by creating a metabolic methylation sink. Nat Chem Biol. (2013) 9:300–6. doi: 10.1038/nchembio.1204
112. Pless, M, Belhadj, K, Menssen, HD, Kern, W, Coiffier, B, Wolf, J, et al. Clinical efficacy, tolerability, and safety of sam486a, a novel polyamine biosynthesis inhibitor, in patients with relapsed or refractory non-hodgkin’s lymphoma: results from a phase ii multicenter study. Clin Cancer Res. (2004) 10:1299–305. doi: 10.1158/1078-0432.ccr-0977-03
113. Konteatis, Z, Travins, J, Gross, S, Marjon, K, Barnett, A, Mandley, E, et al. Discovery of ag-270, a first-in-class oral mat2a inhibitor for the treatment of tumors with homozygous mtap deletion. J Med Chem. (2021) 64:4430–49. doi: 10.1021/acs.jmedchem.0c01895
114. Elderdery, AY, Tebein, EM, Alenazy, FO, Elkhalifa, A, Shalabi, MG, Abbas, AM, et al. Impact of methionine synthase reductase polymorphisms in chronic myeloid leukemia patients. Genes (Basel). (2022) 13:13. doi: 10.3390/genes13101729
115. Lincz, LF, Scorgie, FE, Kerridge, I, Potts, R, Spencer, A, and Enno, A. Methionine synthase genetic polymorphism ms a2756g alters susceptibility to follicular but not diffuse large b-cell non-hodgkin’s lymphoma or multiple myeloma. Br J Haematol. (2003) 120:1051–4. doi: 10.1046/j.1365-2141.2003.04220.x
116. Matsuo, K, Suzuki, R, Hamajima, N, Ogura, M, Kagami, Y, Taji, H, et al. Association between polymorphisms of folate- and methionine-metabolizing enzymes and susceptibility to malignant lymphoma. Blood. (2001) 97:3205–9. doi: 10.1182/blood.v97.10.3205
117. Guo, SJ, Luo, SC, Liu, WY, Zuo, QN, and Li, XH. Methionine synthase a2756g polymorphism and lymphoma risk: a meta-analysis. Eur Rev Med Pharmacol Sci. (2017) 21:3075–82.
118. Li, M, Kong, XY, and Wang, SM. Analysis of the frequency distribution of five single-nucleotide polymorphisms of the mtrrgene in a chinese pediatric population with acute lymphoblastic leukemia. Pharmacotherapy. (2022) 42:442–52. doi: 10.1002/phar.2685
119. Ma, LM, Yang, HP, Yang, XW, and Ruan, LH. Methionine synthase a2756g polymorphism influences pediatric acute lymphoblastic leukemia risk: a meta-analysis. Biosci Rep. (2019) 39:39. doi: 10.1042/BSR20181770
120. Bjelosevic, S, Gruber, E, Newbold, A, Shembrey, C, Devlin, JR, Hogg, SJ, et al. Serine biosynthesis is a metabolic vulnerability in flt3-itd-driven acute myeloid leukemia. Cancer Discov. (2021) 11:1582–99. doi: 10.1158/2159-8290.CD-20-0738
121. Kampen, KR, Fancello, L, Girardi, T, Rinaldi, G, Planque, M, Sulima, SO, et al. Translatome analysis reveals altered serine and glycine metabolism in t-cell acute lymphoblastic leukemia cells. Nat Commun. (2019) 10:2542. doi: 10.1038/s41467-019-10508-2
122. Zaal, EA, Wu, W, Jansen, G, Zweegman, S, Cloos, J, and Berkers, CR. Bortezomib resistance in multiple myeloma is associated with increased serine synthesis. Cancer Metab. (2017) 5:7. doi: 10.1186/s40170-017-0169-9
123. Hattori, A, Tsunoda, M, Konuma, T, Kobayashi, M, Nagy, T, Glushka, J, et al. Cancer progression by reprogrammed bcaa metabolism in myeloid leukaemia. Nature. (2017) 545:500–4. doi: 10.1038/nature22314
124. Han, L, Dong, L, Leung, K, Zhao, Z, Li, Y, Gao, L, et al. Mettl16 drives leukemogenesis and leukemia stem cell self-renewal by reprogramming bcaa metabolism. Cell Stem Cell. (2023) 30:52–68.e13. doi: 10.1016/j.stem.2022.12.006
125. Kikushige, Y, Miyamoto, T, Kochi, Y, Semba, Y, Ohishi, M, Irifune, H, et al. Human acute leukemia utilizes branched-chain amino acid catabolism to maintain stemness through regulating prc2 function. Blood Adv. (2022). doi: 10.1182/bloodadvances.2022008242
126. Ede, BC, Asmaro, RR, Moppett, JP, Diamanti, P, and Blair, A. Investigating chemoresistance to improve sensitivity of childhood t-cell acute lymphoblastic leukemia to parthenolide. Haematologica. (2018) 103:1493–501. doi: 10.3324/haematol.2017.186700
Keywords: amino acid, hematologic malignancy, metabolism, therapy, glutaminolysis, glutathione
Citation: Wang M, Zhao A, Li M and Niu T (2023) Amino acids in hematologic malignancies: Current status and future perspective. Front. Nutr. 10:1113228. doi: 10.3389/fnut.2023.1113228
Edited by:
Rachel Culp-Hill, University of Colorado Denver, United StatesReviewed by:
Zhang Xiaohui, Peking University People’s Hospital, ChinaCopyright © 2023 Wang, Zhao, Li and Niu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ting Niu, bml1dGluZ0B3Y2hzY3UuY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.