 Yi-Shang Yan1,2
Yi-Shang Yan1,2 Chun Feng1,2
Chun Feng1,2 Dan-Qing Yu1,2
Dan-Qing Yu1,2 Shen Tian1,2Yin Zhou1,2Yi-Ting Huang1,2Yi-Ting Cai2Jian Chen2Miao-Miao Zhu3
Shen Tian1,2Yin Zhou1,2Yi-Ting Huang1,2Yi-Ting Cai2Jian Chen2Miao-Miao Zhu3 Min Jin1,2*
Min Jin1,2*- 1Department of Reproductive Medicine, The Second Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou, China
- 2Key Laboratory of Reproductive Genetics, Ministry of Education, School of Medicine, Zhejiang University, Hangzhou, China
- 3Department of Operating Theatre, The First Affiliated Hospital of Wenzhou Medical University, Wenzhou, China
Diabetes mellitus during pregnancy, which can be classified into pregestational diabetes and gestational diabetes, has become much more prevalent worldwide. Maternal diabetes fosters an intrauterine abnormal environment for fetus, which not only influences pregnancy outcomes, but also leads to fetal anomaly and development of diseases in later life, such as metabolic and cardiovascular diseases, neuropsychiatric outcomes, reproduction malformation, and immune dysfunction. The underlying mechanisms are comprehensive and ambiguous, which mainly focus on microbiota, inflammation, reactive oxygen species, cell viability, and epigenetics. This review concluded with the influence of intrauterine hyperglycemia on fetal structure development and organ function on later life and outlined potential mechanisms that underpin the development of diseases in adulthood. Maternal diabetes leaves an effect that continues generations after generations through gametes, thus more attention should be paid to the prevention and treatment of diabetes to rescue the pathological attacks of maternal diabetes from the offspring.
1. Introduction
Maternal diabetes mellitus, defined as glucose intolerance during pregnancy, can be classified into gestational diabetes mellitus (GDM) and pregestational diabetes mellitus (PGDM) (1, 2). The former is diagnosed when hyperglycemia is first detected during pregnancy and often returns to a normal glucose level after delivery (3). The latter condition happens when diabetic women, often with type 1 or type 2 diabetes mellitus (T1DM or T2DM), get pregnant (4). Intrauterine hyperglycemia may not only lead to adverse outcomes in pregnant patients and fetuses, such as preeclampsia (5, 6) and macrosomia (7, 8), but may also leave an impact on offspring in the long term (9). According to the theory of Developmental Origins of Health and Disease (10) and the thrifty phenotype hypothesis (11), a poor maternal condition and an intrauterine abnormal environment reprogram the fetus to a metabolic pattern that is adaptive to an insufficient nutrition (12). After birth, the thrifty phenotype lasts and encounters relatively abundant nutrients, and the body functions present a catch-up mode that leads to an eventually overnourished metabolic pathology (13, 14).
Fetal period is an important stage when poor environment in utero can decide the health destiny of offspring in the whole life (15). Apart from congenital anomalies that are associated with maternal diabetes mellitus and obesity (16, 17), numerous studies on clinical statistics have uncovered the phenomenon that women who experienced intrauterine hyperglycemia were more likely to acquire diabetes mellitus (18) and obesity (19). By comparing the siblings born before and after mothers acquired diabetes mellitus, the healthy condition of sibship differed significantly: after the mothers were diagnosed with diabetes mellitus, babies became vulnerable to acquire a higher BMI (20). In addition, cardiovascular diseases, malformation and dysfunction of organs and systems have aroused great concern among scientists. In this review, we will first discuss the adverse impact that intrauterine hyperglycemia leaves in an offspring (Table 1). Then, the potential mechanisms will be displayed, respectively (Table 2).
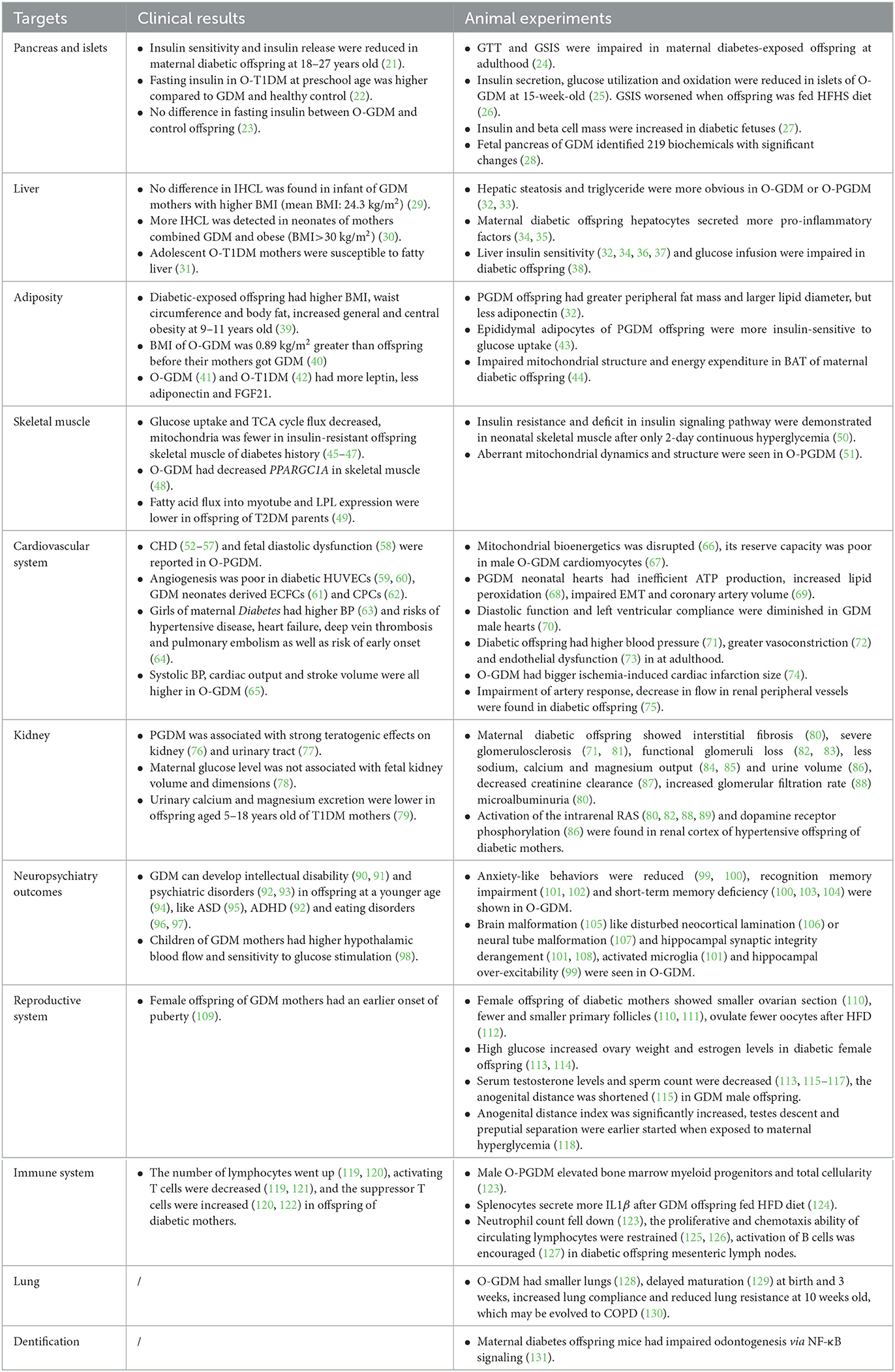
Table 1. The adverse effects of maternal diabetes on offspring.
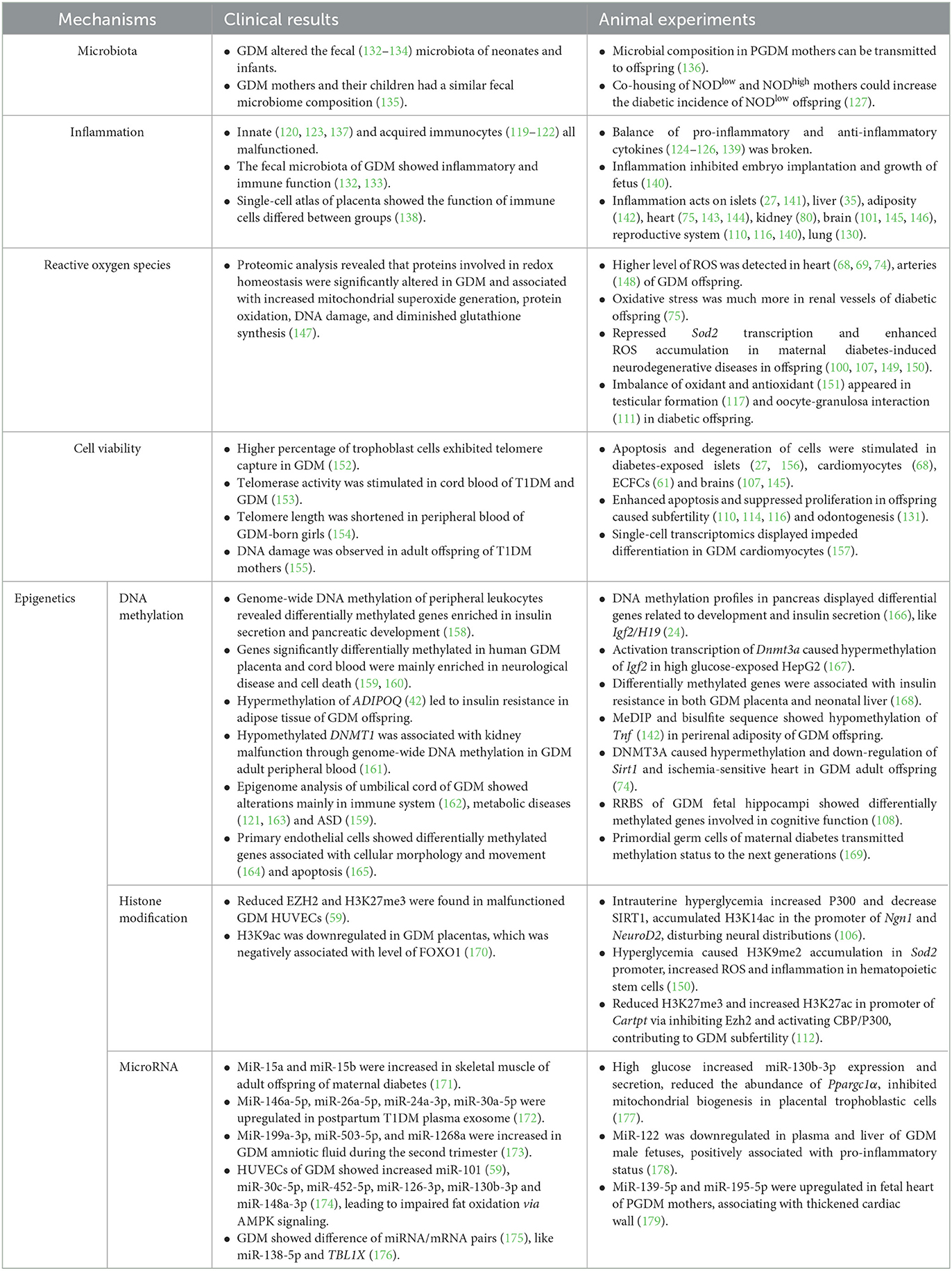
Table 2. The potential mechanisms of maternal diabetes act on offspring.
2. Methods
The National Center for Biotechnology Information (NCBI) search engine (PubMed) was used to extract relevant English-language articles, with keywords such as “Diabet*” or “intrauterine hyperglycemi*” or “hyperglycemi* in pregnancy” AND “Offspring” together with “islet or pancrea*”, “liver or hepatic”, “adipos* or fat or obes*”, “skeletal muscle”, “renal or kidney”, “heart or cardiovascular”, “immune or inflammation”, “testis or ovar* or reproducti*”, “neural”, and “microbiota” successively. We excluded articles that focused on the influence of paternal diabetes mellitus on offspring. The diet-induced animal model was classified into PGDM. After browsing texts, data extracted from each article included (1) type of research (clinical results or animal experiments), (2) methods for setting up an animal model, (3) subjects in clinical trials (maternal diabetes mellitus type, age, and sex of offspring), and (4) study outcomes/findings.
3. The adverse effects of maternal diabetes on offspring
3.1. Target on pancreas and islet
Diabetes mellitus is characterized by an insulin deficiency (180) and an insulin resistance (181). Islet consists of β cells, which are the only cell types that can secrete insulin to downregulate blood glucose. A follow-up cohort demonstrated that offspring of GDM and T1DM had reduced insulin sensitivity and insulin release at the age of 18–27 years (21). However, a clinical study reported that offspring of T1DM (O-T1DM) had a higher fasting serum insulin compared to offspring of GDM and heathy control at preschool age (22). No difference in fasting insulin profile was observed between the offspring of GDM (O-GDM) and control mothers in another clinical trial either (23). Due to anatomic position and ethical restrictions, scientists mimic intrauterine hyperglycemia by administration of streptozotocin (24, 115) or alloxan (111, 118) to specifically destroy the islet or feeding high fat high sucrose (HFHS) diet (112, 116) to induce PGDM or GDM in an animal model. Ding, et al. discovered that adult offspring of maternal diabetes presented impaired glucose tolerance and glucose-stimulated insulin secretion (GSIS) in mice (24). Research on rats came up with similar results: 15-week-old GDM offspring rats displayed an impairment in insulin secretion, glucose utilization, and oxidation in islets (25). Their GSIS capacity worsened when O-GDM was fed with a HFHS diet (26). Genes related to the islet development (24), metabolic enzymes (25), insulin secretion, and cell cycle (156) were demonstrated to be downregulated in the islet of Langerhans cells of O-GDM. By infusing glucose into maternal left uterine artery, hyperglycemic-exposed fetuses were observed to have higher levels of serum insulin and a larger β cell mass (27). The metabolomic analysis of fetal pancreas identified 219 biochemicals with significant changes. Among them, citrate acid and α-ketoglutarate were remarkably decreased in O-GDM, which suggested a metabolism-mediated mechanism (28).
3.2. Target on liver
Insulin secretion is regulated by pancreatic β cells according to blood glucose fluctuation, while target organs of insulin, including liver, adipose tissue, and skeletal muscle, bind with insulin through an insulin receptor, activate downstream signaling to uptake, and catabolize glucose (182).
As the main metabolic organ of glycogen synthesis, liver has been subjected to extensive research for years. A prospective longitudinal study showed no more intrahepatocellular lipid (IHCL) in infants of GDM mothers with a higher body mass index (BMI, mean BMI: 24.3 kg/m2; interquartile range: 21.7, 30.3) compared to control group (29). Another clinical study demonstrated that obese women (BMI>30 kg/m2) with GDM gave birth to neonates with more deposition of IHCL (30). In addition, adolescent O-T1DM may predispose to fatty liver (31). In animal models of mice (32) and rats (33), offspring livers of GDM or PGDM presented more detectable steatosis and hepatic triglyceride, accompanied by reduced lipid metabolic enzymes (32). Fat-laden hepatocytes could release hepatokines to induce pro-inflammatory signaling and hepatic insulin resistance (34). Overall, 20-week-old O-GDM mice were actually demonstrated to secrete more pro-inflammatory factors such as interleukin-1β (IL1β), IL6, and IL33 (35). Animal experiments suggested that, even though basal glucose utilization and insulin concentration were normal in O-GDM, glucose infusion into the liver was lower during a hyperinsulinemic clamp (38). This feature was consistent with a decreased level of insulin receptor and dephosphorylation of AKT in O-GDM livers in another study (36), which can be aggravated by postnatal HFHS diet (32). Forkhead Box O (FOXO) acts downstream of insulin signaling, which can be activated when insulin signaling is inhibited (182). In the offspring liver of maternal diabetes mellitus, phosphorylation of FOXO was repressed, its enzymatic activity was stimulated, and target genes were upregulated, which represented for a repressed insulin pathway (37).
3.3. Target on adipose tissue
A multinational cross-section study conducted in 12 countries showed a higher BMI, waist circumference, and body fat, increased general and central obesity in offspring at the age of 9–11 years once exposed to hyperglycemia in utero, but when adjusted for maternal normal weight, the said difference disappeared (39). A prospective cohort study of Swedish men displayed that the BMI of people who experienced intrauterine hyperglycemia was 0.89 kg/m2 (95% CI, 0.31 to 1.47) greater than the BMI of siblings born before their mother acquired GDM (40). In animal models, the offspring of PGDM (O-PGDM) had a greater peripheral fat mass and a larger lipid diameter, especially epididymal fat pads. Epididymal adipocytes from O-PGDM were also more responsive to an insulin-stimulated glucose uptake with increased levels of insulin receptor, Acetyl-CoA carboxylase, and glucose transporter 4 (43). The adipose tissue can secrete adipokines, such as leptin and adiponectin, which play a major role in glucose homeostasis and insulin sensitivity. The subcutaneous adipose tissue of O-PGDM was shown to secrete less adiponectin (32). In fact, a Danish follow-up study concluded that O-GDM had a higher leptin, a lower adiponectin, and FGF21 (41). Another clinical observation suggested that the plasma level of leptin increased, while the gene expression of adiponectin in subcutaneous adipose tissue became reduced in the offspring of both GDM and T1DM patients (42).
The brown adipose tissue (BAT) is involved in non-shivering thermogenesis and metabolic homeostasis. However, intrauterine hyperglycemia impaired the BAT mitochondrial structure and restrained its energy expenditure (44).
3.4. Target on skeletal muscle
The skeletal muscle accounts for almost half of the lean body mass in an adult and is responsible for the majority of issues related to postprandial and insulin-stimulated glucose disposal. The topic of how the skeletal muscle of an offspring reacts to intrauterine hyperglycemia has remained elusive yet. In insulin-resistant offspring of a T2DM parent or a grandparent, their insulin-stimulated rates of muscle glucose uptake and non-oxidative metabolism decreased as well as a deficit in muscle TCA cycle flux observed (45–47). As mitochondrion is the main organelle for catabolizing fatty acids, less mitochondria and impaired oxidative function can produce a lipotoxic deposit urging insulin insensitivity (183, 184). Insulin-resistant offspring with a family history of diabetes mellitus tend to generate a lower maximal oxygen (O2) consumption because of mitochondrial dysfunction (46). Another research has revealed that only O-GDM rather than O-T1DM presented a decreased expression of peroxisome proliferator-activated receptor γ coactivator-1α (PPARGC1A) in a skeletal muscle (48). Ppargc1α is known to be the key transcription cofactor of mitochondrial biogenesis (185). Another microarray in the offspring of T2DM parents showed that lipoprotein lipase (LPL) was lower, which resulted in a less fatty acid flux into the myotube (49). However, most of the muscle samples collected in these studies were biopsies taken from offspring with a family history of diabetes mellitus rather than from offspring with specific maternal diabetes mellitus, the result of which may be ambiguous. Animal models demonstrated that fetal and neonatal skeletal muscles were insulin resistant after only 2-day continuous intrauterine hyperglycemia (50). Aberrant mitochondrial dynamics and structure were seen in an O-PGDM mice skeletal muscle from F1 to F3 via abnormal oocytes (51).
3.5. Target on cardiovascular system
According to retrospective studies, PGDM was reported to be highly associated with congenital heart diseases (CHDs) (52–56) and fetal diastolic dysfunction (58). Even in non-diabetic pregnant women, an elevated glucose level in the first trimester was directly correlated with an increased risk of CHD in the offspring (57). In the animal model, isolated neonatal cardiomyocytes showed disrupted mitochondrial bioenergetics once exposed to late GDM (66), which caused mitochondria-mediated cell death under metabolic stress in aged male offspring (67). PGDM neonatal cardiomyocytes were reported to have cardiac dysfunction and hypertrophy with an inefficient ATP production and an increased lipid peroxidation (68). The fetal heart of PGDM mice exhibited increased reactive oxygen species (ROS) levels, impaired epicardial epithelial-to-mesenchymal transition (EMT), and decreased coronary artery volume (69). Actually, endothelial cells from human umbilical vein endothelial cells (HUVECs) also displayed damaged proliferation and migration and intensive apoptosis in hyperglycemic fetuses (59). A comparative analysis of gene expression showed poor endothelial tube formation and compromised angiogenic capabilities in isolated GDM HUVECs and mesenchymal stem cells from GDM neonates (60). The proteomic analysis of human fetal endothelial cells revealed an alteration in antioxidant signaling in GDM groups. When ROS attacked, nuclear factor erythroid-related factor 2 increased its expression, traveled into nucleus, and bound to an antioxidant response element sequence for restoration of vascular redox homeostasis (147). Endothelial colony-forming cells (ECFCs) play a key role in repairing damaged endothelium and forming new blood vessels. Researchers found enhanced senescence and worsened angiogenesis in GDM neonate-derived ECFCs (61). Another article found that a reduction in circulating progenitor cells (CPCs) in GDM neonates was correlated with loss of angiogenesis (62). Vascular dysfunction could lead to hypertension and heart insufficiency in an adult. It was reported that maternal hyperglycemia in pregnancy was associated with at least one higher blood pressure (BP) value in girls (63), but the difference in BP between groups became insignificant after adjusting for offspring BMI (186). A Danish follow-up study showed that maternal diabetes mellitus was more likely to develop a hypertensive disease, heart failure, deep vein thrombosis, and pulmonary embolism as well as an increased risk of early onset in offspring (64). Furthermore, during the Trier social stress test (TSST), there was an increase in systolic BP, cardiac output, and stroke volume in O-GDM, even after adjusting for various demographic factors such as sex, age, socioeconomic status, and BMI (65). Animal experiments bear witness to the clinical findings mentioned above. Echocardiography demonstrated a significant diastolic dysfunction in O-GDM male rats. In isolated hearts, the baseline cardiac function and left ventricular compliance were significantly diminished (70). Diabetic offspring was also shown to have a higher blood pressure (71), a greater vasoconstriction under electrical stimulation (72), an impaired vascular nitric oxid (NO)–ROS signaling (148), and endothelial dysfunction (73) in adulthood. HFD after birth induced cardiomyocyte hypertrophy, pro-inflammation status, and cardiac trauma in O-GDM (143). Ischemia-induced cardiac infarction size was bigger in O-GDM. Meanwhile, the ROS leveled up, and the heart function was significantly inhibited (74). The impairment of arterial response and a decrease in the flow in renal peripheral vessels were also shown in maternal diabetic offspring (75), which indicated that kidney was another target of intrauterine hyperglycemia.
3.6. Target on kidney
As the kidney plays an important role in the pathogenesis of hypertension and diabetic complications, the influence of maternal diabetes mellitus on kidney development in offspring has sprouted out. Epidemiologic studies suggested that O-PGDM was associated with strong teratogenic effects on the kidney (76) and the urinary tract (77). However, another study concluded that the maternal glucose level was not associated with volume and dimensions of fetal kidney (78). When children aged 5–18 years were affected, urinary calcium and magnesium excretion was lower in O-T1DM (79). In animal models, widespread interstitial fibrosis (80) and severe glomerulosclerosis (71, 81) with functional glomeruli loss (82, 83) were present at different periods in maternal diabetic offspring, leading to a decreased creatinine clearance (87), an increased glomerular filtration rate (88), and microalbuminuria (80). Urinary calcium and magnesium output was less (84, 85), similar to the excretion of basal sodium and urine volume (86). Apart from this fact, activation of the intrarenal renin–angiotensin system (RAS), together with intrarenal dopamine receptor phosphorylation (86) and increased transforming growth factor-β1 (Tgfβ1) (80), was evident in the cortex of hypertensive offspring of diabetic mothers (80, 82, 88, 89).
3.7. Target on neuropsychiatric outcomes
It also matters whether prenatal exposure to high glucose affects neuropsychiatric development. According to epidemiology, GDM led to adverse neurocognitive and behavioral outcomes in offspring, such as intellectual disability (90, 91) and psychiatric disorders (92, 93). A stratified analysis undertaken on diabetic mothers displayed a higher hazard ratio of autism spectrum disorder (ASD) in their offspring after adjustment (95). A retrospective study suggested that when maternal diabetes required medication, it was positively associated with the incidence of attention deficit/hyperactivity disorder in the offspring (92). Comparing GDM mothers treated with lifestyle or antidiabetic agents with their non-diabetic counterparts, the linear regression analysis showed that O-GDM mothers developed their neuropsychiatric conditions at a younger age (94). Exposure to diabetes mellitus in utero also led to eating disorders in the offspring at adolescence (96, 97). The hypothalamic blood flow in response to glucose in children of GDM mothers was obviously higher than control, combined with hypothalamic sensitivity to glucose stimulation (98). Based on experimental research, we arrived at conclusions as follows. First, anxiety-like behaviors were reduced in O-GDM adult mice as seen from the fact that they spent more time in center and open fields than control mice (99, 100). Male mice of GDM were reported to be more alarmed by the fear recall (105); second, impairment of recognition memory was demonstrated by the Novel Object Recognition (NOR) test as the diabetic offspring were not capable of distinguishing familiar from novel objects (101) or after they were fed HFD (102). Additionally, in the radical Y-maze test, O-GDM adult mice made more re-entry mistakes, which indicated a deficiency in short-term memory (100, 103, 104); third, the cell cycle was altered to a stage where apoptosis-related genes were promoted in GDM neonate-derived cells (107). Hippocampi derived from the offspring of diabetic mothers were characterized by cell death and susceptibility to LPS stimulation (145); fourth, hyperglycemia led to malformation of the brain (105), especially disturbed neocortical lamination (106), neural tube malformation (107), and hippocampal synaptic derangement (101, 108). Difference in the number of neurons in the ventromedial nucleus of the hypothalamus vanished after an islet transplantation in the diabetic group (187); fifth, fetal hippocampal formation was exposed to a neuroinflammatory environment because of hyperglycemia, so increased activated microglia and cytokines persisted into young adulthood (101), and O-GDM became more sensitive to an inflammatory response (145). However, another article suggested that microglia were not affected in GDM neonatal hippocampi (104); finally, both hippocampus and cortex were demonstrated to have a higher level of ROS and an enhanced oxidative metabolism together with inhibited superoxide dismutase (SOD) activity (100, 107). Importantly, overexpressing SOD2 in 6-week-old O-GDM can partly rescue its autism-like behavior in 8 weeks (149). There was also some disagreement among researchers on topics such as neural differentiation, where one researcher tested out that neuronal differentiation markers were less in PGDM fetal brains (107); while another stated that neural stem cells isolated from a 7-day-old fetus exited cell cycle in advance and entered premature differentiation after exposed to maternal hyperglycemia (106). In addition, hippocampal excitability was tested in cultured hippocampal neurons: action potentials were larger and the decay time was shorter in hyperglycemic-exposed offspring; and the resting potential was more hyperpolarized because outwards potassium channel density was larger (99).
3.8. Target on the reproductive system
As mentioned above, the influence of intrauterine adverse environment could inherit to the second generation (24, 103) through fetal primordial germ cells (169), thus it is reasonable for us to consider that maternal diabetes mellitus leaves negative effects on the reproductive system. Actually, a Danish national cohort study reported that female O-GDM had an earlier onset of puberty (109). In animal models, female offspring of diabetic mothers showed a smaller ovarian section (110). The number and diameter of primary follicles were also decreased (110, 111). O-GDM were more inclined to ovulate fewer oocytes after HFD, thus causing subfertility in adult life (112). Researchers attributed these phenomena to increases in apoptosis (110) and oxidative stress (111). Proteomic analysis showed that differentially expressed proteins identified in ovaries were involved in hydrogen peroxide catabolic process (151). A number of scientists have suggested that, because of lipidomic metabolites (113) and hyperactivity of insulin signaling (114), high glucose increased their ovarian weight and estrogen levels in diabetic female offspring. As for male, serum testosterone levels and sperm count were decreased in GDM male offspring (113, 115–117), possibly due to ROS (117) and apoptosis (116). Researchers once discovered that GDM inhibited glycogen synthase kinase-3β (GSK3β) signaling and delayed the differentiation of stem Leydig cells in fetal testis. Thus, the number of Leydig cells decreased and the anogenital distance was shortened (115). However, in another animal model, scientists suggested that the anogenital distance index was significantly increased, testes descent and preputial separation were started earlier when exposed to maternal hyperglycemia (118).
3.9. Target on hemopoietic and immune system
Diabetic mothers were believed to elevate the number of bone marrow myeloid progenitors and total cellularity in male offspring after a 10-month high fat diet (123). Hematopoiesis gives rise to circulating and tissue-resident immune cells (188). In fact, researchers also found that immune cells in offspring failed to function after exposed to maternal hyperglycemia at different periods. Splenocytes were induced to secrete more IL1β after a persistent HFD diet in O-PGDM (124). In the offspring of diabetic mothers, the neutrophil count dropped (123), while the lymphocyte count increased (119, 120). The percentage of activating T cells also decreased (119, 121), so did the capacity of proliferation after antigen stimulation (125, 126). T1DM mothers increased the suppressor T cells in offspring (120, 122). The proliferative ability of B cells was also found to be restrained in diabetic offspring (125). Non-obese diabetic (NOD) mouse colonies can be divided into two groups, NODlow indicates a lower incidence for T1DM development and NODhigh indicates a higher incidence for T1DM development (189). B cell activation was demonstrated to be encouraged in mesenteric lymph nodes of NODhigh and co-housed NODlow offspring (127). The disturbance of immune system led to higher pro-inflammatory cytokines, such as IL1β, IL6, and tumor necrosis factor-α (TNF-α), and lower anti-inflammatory cytokine such as IL2 (125, 126).
In addition to the organs and systems mentioned above, other organs that are affected by intrauterine hyperglycemia have been researched. Newborns and 3-week-old offspring of diabetic mothers had smaller lungs (128) and delayed maturation (129). Male O-GDM had increased lung compliance and reduced lung resistance at 10 weeks old, which may evolve into a chronic obstructive pulmonary disease (130). A high glucose environment was also demonstrated to activate the apoptosis of primary dental papilla cells and dental epithelial stem cells via nuclear factor κB (NF-κB) signaling pathway, resulting in impaired odontogenesis in maternal diabetic offspring mice (131).
4. Potential mechanisms
4.1. Microbiota
Recently, gut microbiota of mothers was thought to be associated with PGDM and GDM (190–192). Remarkably, the microbiota composition of neonates was associated with GDM pregnancy. One research study stated that GDM was related to increase in microbes that are involved in suppressing an early immune cell function in neonates (132); whereas the other research study suggested that a 7-day-old O-GDM showed a higher relative abundance of pro-inflammatory taxa (133). The fecal microbiota of infants aged 7 days and 9 months showed consistent difference between O-GDM and control, but some taxa did not (134). A cross-section study showed that genus Anaerotruncus was increased in 5-year-old children of GDM through gut microbiome. Mothers and their children had a more similar microbiome composition when compared with others (135). The mice model also demonstrated that gut microbiota could be transmitted via delivery and perinatal nursing. Multi-omics analysis supported that both glucometabolic deficits and microbial composition in PGDM mothers were subsequently transmitted to their offspring. By Cesarean delivery and cross-fostering, the microbiota vertical transmission and insulin resistance were blocked (136). There was yet another interesting phenomenon. Exposing NODlow mice to NODhigh after weaning cannot increase the incidence of T1D. However, if co-housing NODlow and NODhigh mothers in advance, the offspring of NODlow acquired increased the incidence of diabetes, which indicated that maternal microbiota and lactation environment were two important transmissible diabetes-promoting factors (127).
4.2. Inflammation
Another potential mechanism is inflammation. As we summarized above in the section “Target on hemopoietic and immune system”, immune cells were reprogrammed under intrauterine hyperglycemia. Not only in fetal or maternal cord blood, but also in children's peripheral blood, innate immunocytes such as leukocytes (120, 123, 137) and acquired immunocytes such as lymphocytes (119–122) were all dysfunctional, forming a status of imbalance of increased pro-inflammatory cytokines and decreased anti-inflammatory cytokines (124–126, 139). In periconception diabetic mice, the intrauterine pro-inflammatory factors, such as IL1α, TNFα, and interferon-γ (IFNγ), were increased. Thus, the blastocyst development was impaired, the embryo implantation was inhibited, and the growth and development of the fetus during middle and late gestational periods were delayed (140). Systematic pro-inflammatory cytokines or local immune cells acted on islets (27, 141), liver (35), peripheral adiposity (142), heart (75, 143, 144), kidney (80), brain (145, 146), reproductive tract (140), lung (130), and microbiota (133), resulting in damage to organ function.
4.3. Reactive oxygen species
Excess glucose in blood and enhanced intracellular glucose oxidation lead to a mitochondrial overproduction of superoxide (193). The ROS acts as an intracellular toxic substance (194) and amplifies mitochondrial damage of hyperglycemia and lipotoxicity. The role of ROS in the pathogenesis of metabolic diseases (195), including atherosclerosis (196) and neurodegenerative diseases (197), has been highlighted. Diabetes-exposed neonatal cardiomyocytes showed a decreased mitochondrial copy number and an impaired palmitate oxidation. Lipotoxicity (66) and ROS accumulation (69) in the heart resulted in cardiac dysfunction. Impaired NO–ROS signaling and increased superoxide production in GDM-exposed endothelial cells (147) and arteries led to hypertension (148). Furthermore, treatment with antioxidant or mitochondrial transfer in diabetic offspring could rescue deterioration in heart ischemic injury (74) and cardiomyocyte bioenergetics (68). Activation of the oxidative stress pathway was also demonstrated in renal vessels among diabetic offspring (75). SOD2, an enzyme that converts superoxide to less reactive hydrogen peroxide and diffuse freely, is strongly involved in the progression of neurodegenerative diseases (198). The transcription of Sod2 was found to be repressed through epigenetic mechanisms in maternal diabetes-induced autistic brain (100, 107), including neurons (149) and hematopoietic stem cells (150). In addition, the imbalance in oxidant and antioxidant was suggested to be an underlying factor resulting in testicular malformation (117) and altered oocyte–granulosa interaction (111) in diabetic offspring.
4.4. Cell viability
Telomere was thought to be related to cellular lifetime. Higher percentage of trophoblast cells exhibited telomere capture in GDM pregnancies (152). Telomerase activity was higher in cord blood from T1DM and GDM, but not T2DM pregnancies (153). Telomere length was observed to be shortened in GDM-born girls' peripheral blood at 9–16 years of age when compared to the control female. Telomere length showed significant association with maternal GDM status positively and insulin levels or HOMA-IR negatively (154). The absence of telomere shortening and oxidative DNA damage were observed in 16- to 23-year-old adult O-T1DM (155).
Cell cycle was another factor associated with cell viability. Cyclin-dependent kinase (Cdk), a cell cycle marker, was transcriptionally inhibited in diabetes-exposed islets (156). Inflammation (145) and ROS (107) in diabetes-exposed brains both could modulate cell cycle, contributing to apoptosis and neurodegeneration. In addition, cell cycle in reproduction plays an important role in gametogenesis. Intrauterine hyperglycemia impaired neonatal folliculogenesis (114) and initiation of meiosis (110) via promotion of apoptosis and inhibition of proliferation. In vivo and in vitro experiments also demonstrated that hyperglycemia suppressed proliferation and enhanced apoptosis in tooth germs via activation of NF-κB signaling pathway (131).
4.5. Epigenetics
Gestational complications are thought to influence the offspring by means of epigenetic changes accordingly. Epigenetics mainly consists of DNA methylation, histone modification, and microRNAs without alterations in DNA sequence.
4.5.1. DNA methylation
Genome-wide DNA methylation analysis of peripheral blood revealed the dysregulation of genes related to NOTCH and WNT signaling which were implicated in pancreatic development and insulin secretion (158). Another DNA methylation profile in pancreas of the mouse offspring showed that glucolipid metabolism-related pathways, such as pancreatic secretion and development, were influenced (166). Igf2 and H19, two imprinted genes, were methylated higher and expressed less in islets isolated from the mouse offspring of PGDM mothers (24). Exposed HepG2 cells to a high glucose resulted in many combinations of FOXO1 within DNA methyltransferase 3a (Dnmt3a) promoter regions. The activated transcription of Dnmt3a caused increased methylation of Igf2 (167). By overlapping differentially methylated genes in both placenta and neonatal liver, it was believed that GDM could significantly affect the biological function, on the top of which was endocrine disorder such as insulin resistance (168). An article suggested that the subcutaneous adipose tissue was regulated to be hypermethylated and thus downregulated adiponectin expression in GDM offspring, which led to insulin resistance in some sense (42). Tnf was hypomethylated and expressed higher in perirenal adiposity of the GDM offspring (142). Hypermethylation of sirtuin 1 (Sirt1) via DNMT3A contributed to a lower expression of Sirt1 and ischemia-sensitive heart in GDM offspring in later life (74). A lower methylated DNMT1 was also associated with alterations in the genome-wide DNA methylation profile in GDM adult peripheral blood cells, relating to an increased glomerular filtration rate and kidney dysfunction (161). Reduced representation bisulfite sequencing (RRBS) of GDM fetal hippocampi showed that altered methylated genes were involved in cognitive function. Metabolic profiling in this article also identified differential metabolites in fetal brain targeting epigenetic modifications (108).
By collecting clinical placenta and cord blood samples from newborns, researchers found that differentially methylated genes between groups in both tissues concentrated on immunological diseases, metabolic diseases, and endocrine disorders (163). Epigenome-wide and transcriptome-wide clinical analyses displayed that GDM was associated with alterations mainly in the immune system, such as the major histocompatibility complex (MHC) region (162). The umbilical blood of a GDM fetus showed hypomethylation of insulin CpG islands, which may contribute to hyperinsulinemia (121). Meta-analysis of cord blood from GDM newborns displayed lower methylation in olfactory receptor family 2 subfamily L member 13 (OR2L13) promoter region (159), which was also reported to be hypomethylated among whole blood cells from ASD patients (199). Another genome-wide DNA methylation profiling of GDM infants showed differentially methylated genes to be mainly enriched in the T1DM pathway, immune MHC-related pathway, neuron development-related pathway, and fetal growth (160). Epigenome-wide association study in cord blood revealed no shared and consistent epigenetic marks between GDM mothers and offspring. However, eight significant CpG sites in TFCP2, H3C6, LOC127841, UBE3C, and FAM13A genes were identified to be associated with GDM exposure (200). In addition, genome-wide methylation and transcriptome in primary fetoplacental arterial and vein endothelial cells between groups identified genes to be associated with cellular morphology and movement (164). Placenta-specific 8 (PLAC8) was hypomethylated in GDM ECFCs after exposed to intrauterine hyperglycemia, which partly resulted in apoptosis and senescence (165). Additionally, the status of methylation could be transmitted to F2 offspring via F1, whose sperm evolved from F1 primordial germ cells (PGCs) also experienced the same attack in the uterus (169).
4.5.2. Histone modification
Scientists also explained that intrauterine hyperglycemia may increase the expression of P300 and decrease the SIRT1 level in newborn neurons to increase histone H3 on lysine 14 (H3K14) acetylation. H3K14ac increased enrichment in neurogene 1 and neuronal differentiation 2 gene promoter region to activate their transcription to participate in neuronal differentiation, which disturbs the distribution of neocortical neurons (106).
Researchers also found that hyperglycemia led to histone H3 on lysine 9 di-methylation (H3K9me2) combination more in Sod2 promoter region in autistic mice of maternal diabetes. An increase in oxidative stress and inflammation caused by a lower transcription of Sod2 inhibited mitochondrial DNA copies and function in hematopoietic stem cells (150). Maternal diabetes also increased miR-101, targeting to capture histone methyltransferase enhancer of zester homolog-2 (Ezh2) mRNA and activate histone acetyltransferase. Reduced trimethylation of histone H3 on lysine 27 (H3K27me3) and increased acetylation of histone H3 on lysine 27 (H3K27ac) in CART prepropeptide promoter region induced Cartpt expression in subfertility female of diabetic mothers (112). Reduced EZH2 and H3K27me3 were also found in malfunctioning GDM HUVECs (59). Additionally, H3K9ac was downregulated in clinical GDM placentas, which was negatively associated with the level of FOXO1 (170).
4.5.3. MicroRNA
As a hotspot epigenetic modification in the past decade, microRNA was demonstrated to match with single complementary mRNA strand and induce retardation in its translation. From a follow-up study, miR-15a and miR-15b were increased in the skeletal muscle of offspring of a diabetic adult. The correlation analysis showed that the levels of miR-15a and miR-15b were positively associated with OGTT glucose, insulin, and C-peptide levels (171). The HUVECs of GDM and high glucose both displayed increased miR-101, but reduced EZH2b and H3K27me3, thereby associating with decrease in HUVEC functional capacities (59). In another research on HUVECs, GDM displayed higher levels of miR-30c-5p, miR-452-5p, miR-126-3p, miR-130b-3p, and miR-148a-3p, which inhibited the expression of AMP-activated protein kinase-α1 (AMPKα1) and decreased fatty-acid oxidation (174). In placental trophoblast cells, a high glucose increased miR-130b-3p expression and secretion, which reduced the abundance of PPARGC1A and mitochondrial biogenesis (177). miR-146a-5p, miR-26a-5p, miR-24a-3p, and miR-30a-5p were significantly upregulated in plasma exosome-enriched extracellular vesicles from T1DM mothers during the postpartum period (172), probably leading to an immune response at pregnancies. In addition, miR-199a-3p, miR-503-5p, and miR-1268a were increased in GDM amniotic fluid during the second trimester (173). The whole transcriptome profiles in GDM and healthy placenta showed a total of 2817 miRNAs with significant difference (175). GDM placenta showed that downregulated miR-138-5p and its corresponding mRNA, transducin β-like 1 X-linked, were upregulated. The miRNA–mRNA pair was required in proliferation (176). In animal models, the expression of miR-122 was downregulated in plasma and liver of GDM male fetuses and positively associated with pro-inflammatory status (178). The fetal heart of PGDM showed a dramatically thickened cardiac wall and upregulated miR-139-5p and miR-195-5p (179). From the results mentioned above, it seems that miRNA differs according to sex (173) and tissue (201).
5. Discussion and prospects
With progress in scientific technologies, transcriptome profiling of human placenta at the single-cell level demonstrated that immunocytes in the GDM placenta played an important role in pathophysiology (138). Single-cell transcriptomics also unveiled embryonic exposure to hyperglycemia perturbed cardiomyocyte differentiation, which might be associated with congenital heart disease (157).
In summary, maternal diabetes creates a hyperglycemic or a hyperinsulinemic environment for the development of the fetus. Glucose or insulin travels across the maternal–fetal barrier and acts on the fetal growth and organic function. Up to now, with clinical data and evidence of animal experiments, researchers have already demonstrated that intrauterine hyperglycemia or hyperinsulinemia caused systematic inflammation in the offspring, disrupted the balance of oxidant and antioxidant leading to an ROS accumulation and DNA damage, altered the expression of vital genes through DNA methylation, histone modification and miRNA, accelerated the apoptosis, and deferred renewal of cells. However, whether maternal glucose or insulin works directly on the offspring or whether there exist another pathogenic molecule, such as miRNA, or cytokines secreted by diabetic placenta, need to be explored. As we can see, different types of maternal diabetes mellitus showed different phenotypes in the offspring but specific mechanism has remained unclear. Regardless of inheritable virulence genes of T1DM or T2DM, the subject of how short-term intrauterine environment works on offspring for a long time is another research point. The affected organs in offspring by intrauterine hyperglycemia present disparate outcomes (Figure 1), meanwhile they influence each other: Dysfunction of islets in offspring exacerbates the glucose level and works on others' functioning. The poor reaction of peripheral insulin effectors lays stress on islets in return. Neural system is another victim of maternal diabetic environment. As a conductor, the dysregulated neural system could play a role in another development. The immune system also acts as both a victim and an initiator in the malformation and dysfunction of offspring. Furthermore, mechanisms we summarized could also work together. An adverse environment alters genetics or epigenetics in the placenta and offspring, bringing about pro-inflammatory gene expression and secretion and mitochondrial dysfunction. Furthermore, gut microbiota in offspring turns into acquiring pro-inflammation status. The ROS accumulation and inflammation result in a cellular inactivity and are short lived. There is no doubt that diabetic mothers provided an unhealthy environment for the fetus, so it is necessary for mothers to keep fit. Our previous study demonstrated that once the epigenetic imprint was left on the offspring by intrauterine hyperglycemia, even with insulin treatment to maintain normal maternal glucose, it is important for the offspring to keep a healthy diet in case of emergence of dysfunction (202). Thus, the question of how to intercept or mitigate the adverse effect of uncontrolled maternal diabetes mellitus is another topic worth emphasis in the future. Scientists suggested that the delivery mode and breast feeding by diabetic mothers can transmit a pro-inflammatory status to the offspring via microbiota. As microbiology is now a hotspot, whether there is need to cut intergenerational transmission through C-section and foster nursing remains to be verified. Although existing data can be controversial in some respects, conflicts can arise because of different criteria, classification standard, research method, or animal model. With the increasing occurrence and pronounced influence of gestational diabetes, more attention should be paid to and more effort should be bestowed on the precise mechanism and valid treatment, so that we can guarantee the health of both the mothers and the babies to the maximum extent.
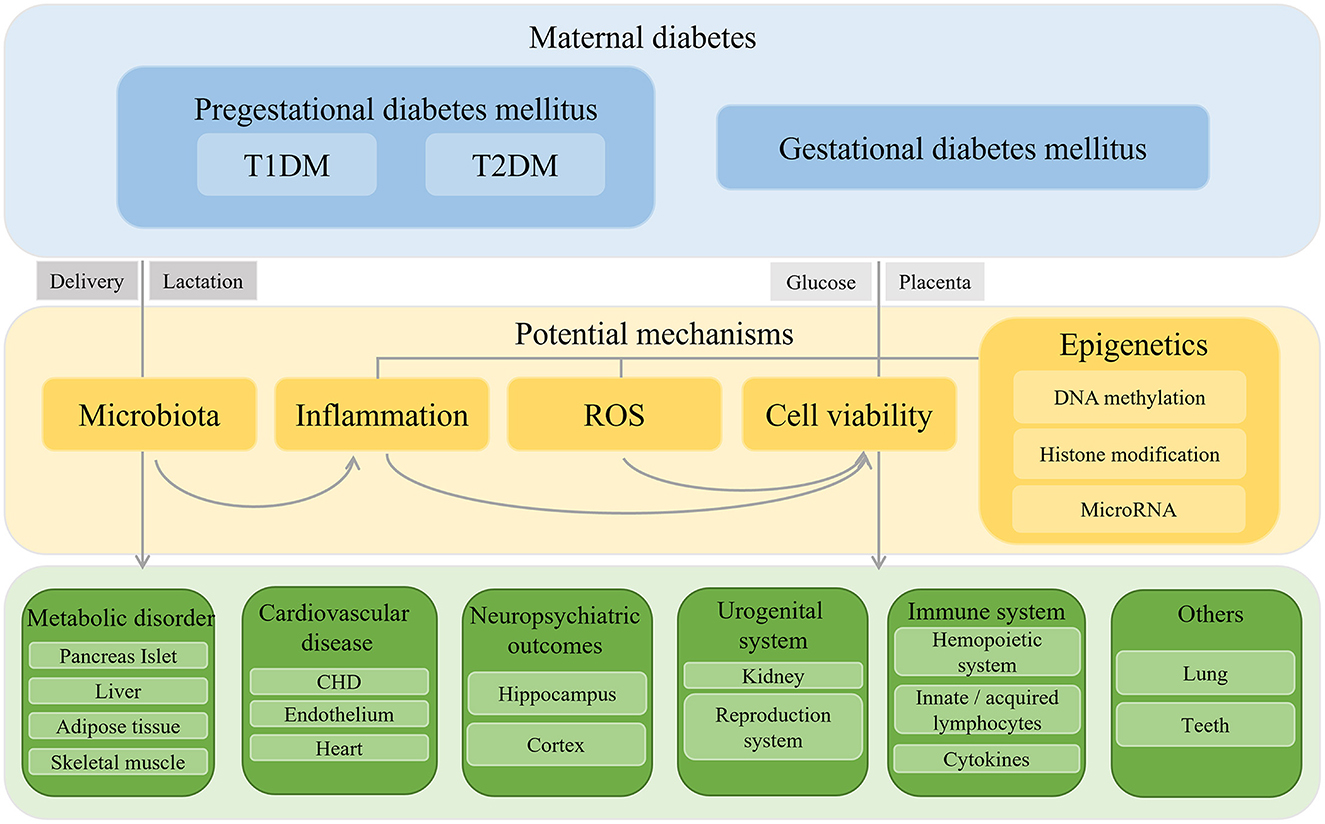
Figure 1. Schematic diaphragm of outcomes and mechanisms of offspring exposed to intrauterine hyperglycemia.
Author's note
Parts of phenotype have been received by a Chinese journal in Chinese.
Author contributions
Y-SY wrote the manuscript. CF and MJ wrote the review and editing. ST and YZ prepared the tables. Y-TH and D-QY designed the figure. M-MZ checked the tables. D-QY acquired the funding. Y-TC and JC supervised the manuscript. All the authors approved the final version of the manuscript.
Funding
This article was funded by National Natural Science Foundation of China Youth Fund (No. 81901498).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Weinert LS. International Association of Diabetes and Pregnancy Study Groups recommendations on the diagnosis and classification of hyperglycemia in pregnancy: comment to the international association of diabetes and pregnancy study groups consensus panel. Diabetes Care. (2010) 33:e97. doi: 10.2337/dc10-0544
2. Guariguata L, Linnenkamp U, Beagley J, Whiting DR, Cho NH. Global estimates of the prevalence of hyperglycaemia in pregnancy. Diabetes Res Clin Pract. (2014) 103:176–85. doi: 10.1016/j.diabres.2013.11.003
3. McIntyre HD, Catalano P, Zhang C, Desoye G, Mathiesen ER, Damm P. Gestational diabetes mellitus. Nat Rev Dis Primers. (2019) 5:47. doi: 10.1038/s41572-019-0098-8
4. Hart BN, Shubrook JH, Mason T. Pregestational diabetes and family planning. Clin Diabetes. (2021) 39:323–8. doi: 10.2337/cd20-0062
5. Oppermann M, Alessi J, Hirakata VN, Wiegand DM, Reichelt AJ. Preeclampsia in women with pregestational diabetes—a cohort study. Hypertens Pregnancy. (2020) 39:48–55. doi: 10.1080/10641955.2019.1704002
6. Ostlund I, Haglund B, Hanson U. Gestational diabetes and preeclampsia. Eur J Obstet Gynecol Reprod Biol. (2004) 113:12–6. doi: 10.1016/j.ejogrb.2003.07.001
7. Group HSCR, Metzger BE, Lowe LP, Dyer AR, Trimble ER, Chaovarindr U, et al. Hyperglycemia and adverse pregnancy outcomes. N Engl J Med. (2008) 358:1991–2002. doi: 10.1056/NEJMoa0707943
8. Song X, Shu J, Zhang S, Chen L, Diao J, Li J, et al. Pre-pregnancy body mass index and risk of macrosomia and large for gestational age births with gestational diabetes mellitus as a mediator: a prospective cohort study in central China. Nutrients. (2022) 14:1072. doi: 10.3390/nu14051072
9. Damm P, Houshmand-Oeregaard A, Kelstrup L, Lauenborg J, Mathiesen ER, Clausen TD. Gestational diabetes mellitus and long-term consequences for mother and offspring: a view from Denmark. Diabetologia. (2016) 59:1396–9. doi: 10.1007/s00125-016-3985-5
10. Barker DJ, Gluckman PD, Godfrey KM, Harding JE, Owens JA, Robinson JS. Fetal nutrition and cardiovascular disease in adult life. Lancet. (1993) 341:938–41. doi: 10.1016/0140-6736(93)91224-A
11. Hales CN, Barker DJ. The thrifty phenotype hypothesis. Br Med Bull. (2001) 60:5–20. doi: 10.1093/bmb/60.1.5
12. Satterfield MC, Edwards AK, Bazer FW, Dunlap KA, Steinhauser CB, Wu G. Placental adaptation to maternal malnutrition. Reproduction. (2021) 162:R73–83. doi: 10.1530/REP-21-0179
13. Gafni RI, Baron J. Catch-up growth: possible mechanisms. Pediatr Nephrol. (2000) 14:616–9. doi: 10.1007/s004670000338
14. Singhal A. Long-term adverse effects of early growth acceleration or catch-up growth. Ann Nutr Metab. (2017) 70:236–40. doi: 10.1159/000464302
15. Dabelea D, Crume T. Maternal environment and the transgenerational cycle of obesity and diabetes. Diabetes. (2011) 60:1849–55. doi: 10.2337/db11-0400
16. Araujo-Silva VC, Santos-Silva A, Lourenco AS, Barros-Barbosa CM, Moraes-Souza RQ, Soares TS, et al. Congenital anomalies programmed by maternal diabetes and obesity on offspring of rats. Front Physiol. (2021) 12:701767. doi: 10.3389/fphys.2021.701767
17. Zhang TN, Huang XM, Zhao XY, Wang W, Wen R, Gao SY. Risks of specific congenital anomalies in offspring of women with diabetes: a systematic review and meta-analysis of population-based studies including over 80 million births. PLoS Med. (2022) 19:e1003900. doi: 10.1371/journal.pmed.1003900
18. Clausen TD, Mathiesen ER, Hansen T, Pedersen O, Jensen DM, Lauenborg J, et al. High prevalence of type 2 diabetes and pre-diabetes in adult offspring of women with gestational diabetes mellitus or type 1 diabetes: the role of intrauterine hyperglycemia. Diabetes Care. (2008) 31:340–6. doi: 10.2337/dc07-1596
19. Hillier TA, Pedula KL, Schmidt MM, Mullen JA, Charles MA, Pettitt DJ. Childhood obesity and metabolic imprinting: the ongoing effects of maternal hyperglycemia. Diabetes Care. (2007) 30:2287–92. doi: 10.2337/dc06-2361
20. Dabelea D, Hanson RL, Lindsay RS, Pettitt DJ, Imperatore G, Gabir MM, et al. Intrauterine exposure to diabetes conveys risks for type 2 diabetes and obesity: a study of discordant sibships. Diabetes. (2000) 49:2208–11. doi: 10.2337/diabetes.49.12.2208
21. Kelstrup L, Damm P, Mathiesen ER, Hansen T, Vaag AA, Pedersen O, et al. Insulin resistance and impaired pancreatic beta-cell function in adult offspring of women with diabetes in pregnancy. J Clin Endocrinol Metab. (2013) 98:3793–801. doi: 10.1210/jc.2013-1536
22. Pirkola J, Vaarasmaki M, Leinonen E, Bloigu A, Veijola R, Tossavainen P, et al. Maternal type 1 and gestational diabetes: postnatal differences in insulin secretion in offspring at preschool age. Pediatr Diabetes. (2008) 9:583–9. doi: 10.1111/j.1399-5448.2008.00415.x
23. Kearney M, Perron J, Marc I, Weisnagel SJ, Tchernof A, Robitaille J. Association of prenatal exposure to gestational diabetes with offspring body composition and regional body fat distribution. Clin Obes. (2018) 8:81–7. doi: 10.1111/cob.12237
24. Ding GL, Wang FF, Shu J, Tian S, Jiang Y, Zhang D, et al. Transgenerational glucose intolerance with Igf2/H19 epigenetic alterations in mouse islet induced by intrauterine hyperglycemia. Diabetes. (2012) 61:1133–42. doi: 10.2337/db11-1314
25. Han J, Xu J, Long YS, Epstein PN, Liu YQ. Rat maternal diabetes impairs pancreatic beta-cell function in the offspring. Am J Physiol Endocrinol Metab. (2007) 293:E228–36. doi: 10.1152/ajpendo.00479.2006
26. Agarwal P, Brar N, Morriseau TS, Kereliuk SM, Fonseca MA, Cole LK, et al. Gestational diabetes adversely affects pancreatic islet architecture and function in the male rat offspring. Endocrinology. (2019) 160:1907–25. doi: 10.1210/en.2019-00232
27. Casasnovas J, Jo Y, Rao X, Xuei X, Brown ME, Kua KL. High glucose alters fetal rat islet transcriptome and induces progeny islet dysfunction. J Endocrinol. (2019) 240:309–23. doi: 10.1530/JOE-18-0493
28. Zhu H, Luo SS, Cheng Y, Yan YS, Zou KX, Ding GL, et al. Intrauterine hyperglycemia alters the metabolomic profile in fetal mouse pancreas in a gender-specific manner. Front Endocrinol. (2021) 12:710221. doi: 10.3389/fendo.2021.710221
29. Logan KM, Emsley RJ, Jeffries S, Andrzejewska I, Hyde MJ, Gale C, et al. Development of early adiposity in infants of mothers with gestational diabetes mellitus. Diabetes Care. (2016) 39:1045–51. doi: 10.2337/dc16-0030
30. Brumbaugh DE, Tearse P, Cree-Green M, Fenton LZ, Brown M, Scherzinger A, et al. Intrahepatic fat is increased in the neonatal offspring of obese women with gestational diabetes. J Pediatr. (2013) 162:930–6e1. doi: 10.1016/j.jpeds.2012.11.017
31. Knorr S, Bytoft B, Lohse Z, Boisen AB, Clausen TD, Jensen RB, et al. Fatty liver among adolescent offspring of women with type 1 diabetes (the EPICOM Study). Diabetes Care. (2019) 42:1560–8. doi: 10.2337/dc19-0571
32. Pereira TJ, Fonseca MA, Campbell KE, Moyce BL, Cole LK, Hatch GM, et al. Maternal obesity characterized by gestational diabetes increases the susceptibility of rat offspring to hepatic steatosis via a disrupted liver metabolome. J Physiol. (2015) 593:3181–97. doi: 10.1113/JP270429
33. Song Y, Li J, Zhao Y, Zhang Q, Liu Z, Li J, et al. Severe maternal hyperglycemia exacerbates the development of insulin resistance and fatty liver in the offspring on high fat diet. Exp Diabetes Res. (2012) 2012:254976. doi: 10.1155/2012/254976
34. Schuster S, Cabrera D, Arrese M, Feldstein AE. Triggering and resolution of inflammation in NASH. Nat Rev Gastroenterol Hepatol. (2018) 15:349–64. doi: 10.1038/s41575-018-0009-6
35. Dong X, Lin D, Sheng J, Xie Y. Intrauterine hyperglycemia induces liver inflammation in mouse male offspring. Int Immunopharmacol. (2021) 99:107974. doi: 10.1016/j.intimp.2021.107974
36. Yamashita H, Shao J, Qiao L, Pagliassotti M, Friedman JE. Effect of spontaneous gestational diabetes on fetal and postnatal hepatic insulin resistance in Lepr(db/+) mice. Pediatr Res. (2003) 53:411–8. doi: 10.1203/01.PDR.0000049667.58071.7D
37. Inoguchi Y, Ichiyanagi K, Ohishi H, Maeda Y, Sonoda N, Ogawa Y, et al. Poorly controlled diabetes during pregnancy and lactation activates the Foxo1 pathway and causes glucose intolerance in adult offspring. Sci Rep. (2019) 9:10181. doi: 10.1038/s41598-019-46638-2
38. Holemans K, Aerts L, Van Assche FA. Evidence for an insulin resistance in the adult offspring of pregnant streptozotocin-diabetic rats. Diabetologia. (1991) 34:81–5. doi: 10.1007/BF00500377
39. Zhao P, Liu E, Qiao Y, Katzmarzyk PT, Chaput JP, Fogelholm M, et al. Maternal gestational diabetes and childhood obesity at age 9–11: results of a multinational study. Diabetologia. (2016) 59:2339–48. doi: 10.1007/s00125-016-4062-9
40. Lawlor DA, Lichtenstein P, Langstrom N. Association of maternal diabetes mellitus in pregnancy with offspring adiposity into early adulthood: sibling study in a prospective cohort of 280,866 men from 248,293 families. Circulation. (2011) 123:258–65. doi: 10.1161/CIRCULATIONAHA.110.980169
41. Kampmann FB, Thuesen ACB, Hjort L, Bjerregaard AA, Chavarro JE, Frystyk J, et al. Increased leptin, decreased adiponectin and FGF21 concentrations in adolescent offspring of women with gestational diabetes. Eur J Endocrinol. (2019) 181:691–700. doi: 10.1530/EJE-19-0658
42. Houshmand-Oeregaard A, Hansen NS, Hjort L, Kelstrup L, Broholm C, Mathiesen ER, et al. Differential adipokine DNA methylation and gene expression in subcutaneous adipose tissue from adult offspring of women with diabetes in pregnancy. Clin Epigenetics. (2017) 9:37. doi: 10.1186/s13148-017-0338-2
43. Oliveira AC, Andreotti S, Chimin P, Sertie RA, Farias Tda S, Torres-Leal FL, et al. Neonatal streptozotocin-induced diabetes in mothers promotes metabolic programming of adipose tissue in male rat offspring. Life Sci. (2015) 136:151–6. doi: 10.1016/j.lfs.2015.06.024
44. Yu DQ, Lv PP, Yan YS, Xu GX, Sadhukhan A, Dong S, et al. Intrauterine exposure to hyperglycemia retards the development of brown adipose tissue. FASEB J. (2019) 33:5425–39. doi: 10.1096/fj.201801818R
45. Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med. (2004) 350:664–71. doi: 10.1056/NEJMoa031314
46. Befroy DE, Petersen KF, Dufour S, Mason GF, de Graaf RA, Rothman DL, et al. Impaired mitochondrial substrate oxidation in muscle of insulin-resistant offspring of type 2 diabetic patients. Diabetes. (2007) 56:1376–81. doi: 10.2337/db06-0783
47. Morino K, Petersen KF, Dufour S, Befroy D, Frattini J, Shatzkes N, et al. Reduced mitochondrial density and increased IRS-1 serine phosphorylation in muscle of insulin-resistant offspring of type 2 diabetic parents. J Clin Invest. (2005) 115:3587–93. doi: 10.1172/JCI25151
48. Kelstrup L, Hjort L, Houshmand-Oeregaard A, Clausen TD, Hansen NS, Broholm C, et al. Gene expression and DNA methylation of PPARGC1A in muscle and adipose tissue from adult offspring of women with diabetes in pregnancy. Diabetes. (2016) 65:2900–10. doi: 10.2337/db16-0227
49. Morino K, Petersen KF, Sono S, Choi CS, Samuel VT, Lin A, et al. Regulation of mitochondrial biogenesis by lipoprotein lipase in muscle of insulin-resistant offspring of parents with type 2 diabetes. Diabetes. (2012) 61:877–87. doi: 10.2337/db11-1391
50. Kua KL, Hu S, Wang C, Yao J, Dang D, Sawatzke AB, et al. Fetal hyperglycemia acutely induces persistent insulin resistance in skeletal muscle. J Endocrinol. (2019) 242:M1–M15. doi: 10.1530/JOE-18-0455
51. Saben JL, Boudoures AL, Asghar Z, Thompson A, Drury A, Zhang W, et al. Maternal metabolic syndrome programs mitochondrial dysfunction via germline changes across three generations. Cell Rep. (2016) 16:1–8. doi: 10.1016/j.celrep.2016.05.065
52. Ji H, Liang H, Yu Y, Wang Z, Yuan W, Qian X, et al. Association of maternal history of spontaneous abortion and stillbirth with risk of congenital heart disease in offspring of women with vs without type 2 diabetes. JAMA Netw Open. (2021) 4:e2133805. doi: 10.1001/jamanetworkopen.2021.33805
53. Chen L, Yang T, Chen L, Wang L, Wang T, Zhao L, et al. Risk of congenital heart defects in offspring exposed to maternal diabetes mellitus: an updated systematic review and meta-analysis. Arch Gynecol Obstet. (2019) 300:1491–506. doi: 10.1007/s00404-019-05376-6
54. Alyousif SMM, Aldokhel FT, Alkhanbashi OK, Alqahtani MHA, Aladawi AMM, Ashmawi AA, et al. The incidence of congenital heart defects in offspring among women with diabetes in Saudi Arabia. Cureus. (2021) 13:e14225. doi: 10.7759/cureus.14225
55. Oyen N, Diaz LJ, Leirgul E, Boyd HA, Priest J, Mathiesen ER, et al. Prepregnancy diabetes and offspring risk of congenital heart disease: a nationwide cohort study. Circulation. (2016) 133:2243–53. doi: 10.1161/CIRCULATIONAHA.115.017465
56. Luo L, Zhang S, Wang T, Diao J, Li J, Li Y, et al. Associations of maternal diabetes mellitus and adiponectin gene polymorphisms with congenital heart disease in offspring: A case-control study. Medicine (Baltimore). (2021) 100:e24672. doi: 10.1097/MD.0000000000024672
57. Helle EIT, Biegley P, Knowles JW, Leader JB, Pendergrass S, Yang W, et al. First trimester plasma glucose values in women without diabetes are associated with risk for congenital heart disease in offspring. J Pediatr. (2018) 195:275–8. doi: 10.1016/j.jpeds.2017.10.046
58. Senol G, Aslan Cetin B, Esin D, Tobas Selcuki NF, Tayyar A, Turhan U, et al. Evaluation of right side foetal myocardial performance index in pregestational and gestational diabetes mellitus. J Obstet Gynaecol. (2022) 42:91–6. doi: 10.1080/01443615.2021.1882971
59. Floris I, Descamps B, Vardeu A, Mitic T, Posadino AM, Shantikumar S, et al. Gestational diabetes mellitus impairs fetal endothelial cell functions through a mechanism involving microRNA-101 and histone methyltransferase enhancer of zester homolog-2. Arterioscler Thromb Vasc Biol. (2015) 35:664–74. doi: 10.1161/ATVBAHA.114.304730
60. Amrithraj AI, Kodali A, Nguyen L, Teo AKK, Chang CW, Karnani N, et al. Gestational diabetes alters functions in offspring's umbilical cord cells with implications for cardiovascular health. Endocrinology. (2017) 158:2102–12. doi: 10.1210/en.2016-1889
61. Ingram DA, Lien IZ, Mead LE, Estes M, Prater DN, Derr-Yellin E, et al. In vitro hyperglycemia or a diabetic intrauterine environment reduces neonatal endothelial colony-forming cell numbers and function. Diabetes. (2008) 57:724–31. doi: 10.2337/db07-1507
62. Acosta JC, Haas DM, Saha CK, Dimeglio LA, Ingram DA, Haneline LS. Gestational diabetes mellitus alters maternal and neonatal circulating endothelial progenitor cell subsets. Am J Obstet Gynecol. (2011) 204:254e8–e15. doi: 10.1016/j.ajog.2010.10.913
63. Tam WH, Ma RCW, Ozaki R, Li AM, Chan MHM, Yuen LY, et al. In utero exposure to maternal hyperglycemia increases childhood cardiometabolic risk in offspring. Diabetes Care. (2017) 40:679–86. doi: 10.2337/dc16-2397
64. Yu Y, Arah OA, Liew Z, Cnattingius S, Olsen J, Sorensen HT, et al. Maternal diabetes during pregnancy and early onset of cardiovascular disease in offspring: population based cohort study with 40 years of follow-up. BMJ. (2019) 367:l6398. doi: 10.1136/bmj.l6398
65. Krishnaveni GV, Veena SR, Jones A, Srinivasan K, Osmond C, Karat SC, et al. Exposure to maternal gestational diabetes is associated with higher cardiovascular responses to stress in adolescent indians. J Clin Endocrinol Metab. (2015) 100:986–93. doi: 10.1210/jc.2014-3239
66. Mdaki KS, Larsen TD, Wachal AL, Schimelpfenig MD, Weaver LJ, Dooyema SD, et al. Maternal high-fat diet impairs cardiac function in offspring of diabetic pregnancy through metabolic stress and mitochondrial dysfunction. Am J Physiol Heart Circ Physiol. (2016) 310:H681–92. doi: 10.1152/ajpheart.00795.2015
67. Louwagie EJ, Larsen TD, Wachal AL, Gandy TCT, Eclov JA, Rideout TC, et al. Age and sex influence mitochondria and cardiac health in offspring exposed to maternal glucolipotoxicity. iScience. (2020) 23:101746. doi: 10.1016/j.isci.2020.101746
68. Louwagie EJ, Larsen TD, Wachal AL, Gandy TCT, Baack ML. Mitochondrial transfer improves cardiomyocyte bioenergetics and viability in male rats exposed to pregestational diabetes. Int J Mol Sci. (2021) 22:2382. doi: 10.3390/ijms22052382
69. Moazzen H, Lu X, Liu M, Feng Q. Pregestational diabetes induces fetal coronary artery malformation via reactive oxygen species signaling. Diabetes. (2015) 64:1431–43. doi: 10.2337/db14-0190
70. Agoudemos M, Reinking BE, Koppenhafer SL, Segar JL, Scholz TD. Programming of adult cardiovascular disease following exposure to late-gestation hyperglycemia. Neonatology. (2011) 100:198–205. doi: 10.1159/000324863
71. Ramos-Alves FE, de Queiroz DB, Santos-Rocha J, Duarte GP, Xavier FE. Increased cyclooxygenase-2-derived prostanoids contributes to the hyperreactivity to noradrenaline in mesenteric resistance arteries from offspring of diabetic rats. PLoS ONE. (2012) 7:e50593. doi: 10.1371/journal.pone.0050593
72. de Queiroz DB, Sastre E, Caracuel L, Callejo M, Xavier FE, Blanco-Rivero J, et al. Alterations in perivascular innervation function in mesenteric arteries from offspring of diabetic rats. Br J Pharmacol. (2015) 172:4699–713. doi: 10.1111/bph.13244
73. Ramos-Alves FE, de Queiroz DB, Santos-Rocha J, Duarte GP, Xavier FE. Effect of age and COX-2-derived prostanoids on the progression of adult vascular dysfunction in the offspring of diabetic rats. Br J Pharmacol. (2012) 166:2198–208. doi: 10.1111/j.1476-5381.2012.01945.x
74. Chen Z, Gong L, Zhang P, Li Y, Liu B, Zhang L, et al. Epigenetic down-regulation of sirt 1 via dna methylation and oxidative stress signaling contributes to the gestational diabetes mellitus-induced fetal programming of heart ischemia-sensitive phenotype in late life. Int J Biol Sci. (2019) 15:1240–51. doi: 10.7150/ijbs.33044
75. Dib A, Payen C, Bourreau J, Munier M, Grimaud L, Fajloun Z, et al. In utero exposure to maternal diabetes is associated with early abnormal vascular structure in offspring. Front Physiol. (2018) 9:350. doi: 10.3389/fphys.2018.00350
76. Pylypjuk C, Sellers E, Wicklow B. Perinatal outcomes in a longitudinal birth cohort of first nations mothers with pregestational type 2 diabetes and their offspring: the next generation study. Can J Diabetes. (2021) 45:27–32. doi: 10.1016/j.jcjd.2020.05.001
77. Nielsen GL, Norgard B, Puho E, Rothman KJ, Sorensen HT, Czeizel AE. Risk of specific congenital abnormalities in offspring of women with diabetes. Diabet Med. (2005) 22:693–6. doi: 10.1111/j.1464-5491.2005.01477.x
78. Hokke S, de Zoysa N, Carr BL, Abruzzo V, Coombs PR, Allan CA, et al. Normal foetal kidney volume in offspring of women treated for gestational diabetes. Endocrinol Diabetes Metab. (2019) 2:e00091. doi: 10.1002/edm2.91
79. Mughal MZ, Eelloo JA, Roberts SA, Sibartie S, Maresh M, Sibley CP, et al. Intrauterine programming of urinary calcium and magnesium excretion in children born to mothers with insulin dependent diabetes mellitus. Arch Dis Child Fetal Neonatal Ed. (2005) 90:F332–6. doi: 10.1136/adc.2004.052795
80. Chen YW, Chenier I, Tran S, Scotcher M, Chang SY, Zhang SL. Maternal diabetes programs hypertension and kidney injury in offspring. Pediatr Nephrol. (2010) 25:1319–29. doi: 10.1007/s00467-010-1506-1
81. Nehiri T, Duong Van Huyen JP, Viltard M, Fassot C, Heudes D, Freund N, et al. Exposure to maternal diabetes induces salt-sensitive hypertension and impairs renal function in adult rat offspring. Diabetes. (2008) 57:2167–75. doi: 10.2337/db07-0780
82. Tran S, Chen YW, Chenier I, Chan JS, Quaggin S, Hebert MJ, et al. Maternal diabetes modulates renal morphogenesis in offspring. J Am Soc Nephrol. (2008) 19:943–52. doi: 10.1681/ASN.2007080864
83. Rocha SO, Gomes GN, Forti AL. do Carmo Pinho Franco M, Fortes ZB, de Fatima Cavanal M, et al. Long-term effects of maternal diabetes on vascular reactivity and renal function in rat male offspring. Pediatr Res. (2005) 58:1274–9. doi: 10.1203/01.pdr.0000188698.58021.ff
84. Bond H, Hamilton K, Balment RJ, Denton J, Freemont AJ, Garland HO, et al. Diabetes in rat pregnancy alters renal calcium and magnesium reabsorption and bone formation in adult offspring. Diabetologia. (2005) 48:1393–400. doi: 10.1007/s00125-005-1804-5
85. Bond H, Sibley CP, Balment RJ, Ashton N. Increased renal tubular reabsorption of calcium and magnesium by the offspring of diabetic rat pregnancy. Pediatr Res. (2005) 57:890–5. doi: 10.1203/01.PDR.0000157720.50808.97
86. Luo H, Chen C, Guo L, Xu Z, Peng X, Wang X, et al. Exposure to maternal diabetes mellitus causes renal dopamine d1 receptor dysfunction and hypertension in adult rat offspring. Hypertension. (2018) 72:962–70. doi: 10.1161/HYPERTENSIONAHA.118.10908
87. Martins Jde O, Panicio MI, Dantas MP, Gomes GN. Effect of maternal diabetes on female offspring. Einstein. (2014) 12:413–9. doi: 10.1590/S1679-45082014AO3200
88. Liao MC, Pang YC, Chang SY, Zhao XP, Chenier I, Ingelfinger JR, et al. AT2R deficiency in mice accelerates podocyte dysfunction in diabetic progeny in a sex-dependent manner. Diabetologia. (2021) 64:2108–21. doi: 10.1007/s00125-021-05483-7
89. Yan J, Li X, Su R, Zhang K, Yang H. Long-term effects of maternal diabetes on blood pressure and renal function in rat male offspring. PLoS ONE. (2014) 9:e88269. doi: 10.1371/journal.pone.0088269
90. Knorr S, Clausen TD, Vlachova Z, Bytoft B, Damm P, Beck-Nielsen H, et al. Academic achievement in primary school in offspring born to mothers with type 1 diabetes (the EPICOM Study): a register-based prospective cohort study. Diabetes Care. (2015) 38:1238–44. doi: 10.2337/dc15-0223
91. Clausen TD, Mortensen EL, Schmidt L, Mathiesen ER, Hansen T, Jensen DM, et al. Cognitive function in adult offspring of women with Type 1 diabetes. Diabet Med. (2011) 28:838–44. doi: 10.1111/j.1464-5491.2011.03300.x
92. Xiang AH, Wang X, Martinez MP, Getahun D, Page KA, Buchanan TA, et al. Maternal gestational diabetes mellitus, type 1 diabetes, and type 2 diabetes during pregnancy and risk of ADHD in offspring. Diabetes Care. (2018) 41:2502–8. doi: 10.2337/dc18-0733
93. Kong L, Norstedt G, Schalling M, Gissler M, Lavebratt C. The risk of offspring psychiatric disorders in the setting of maternal obesity and diabetes. Pediatrics. (2018) 142. doi: 10.1542/peds.2018-0776. [Epub ahead of print].
94. Nahum Sacks K, Friger M, Shoham-Vardi I, Abokaf H, Spiegel E, Sergienko R, et al. Prenatal exposure to gestational diabetes mellitus as an independent risk factor for long-term neuropsychiatric morbidity of the offspring. Am J Obstet Gynecol. (2016) 215:380e1–7. doi: 10.1016/j.ajog.2016.03.030
95. Xiang AH. Association of maternal diabetes with autism in offspring. JAMA. (2017) 317:537–8. doi: 10.1001/jama.2016.20122
96. Shapiro ALB, Sauder KA, Tregellas JR, Legget KT, Gravitz SL, Ringham BM, et al. Exposure to maternal diabetes in utero and offspring eating behavior: The EPOCH study. Appetite. (2017) 116:610–5. doi: 10.1016/j.appet.2017.05.005
97. Wang H, He H, Yu Y, Su X, Li F, Li J. Maternal diabetes and the risk of feeding and eating disorders in offspring: a national population-based cohort study. BMJ Open Diabetes Res Care. (2020) 8:e001738. doi: 10.1136/bmjdrc-2020-001738
98. Page KA, Luo S, Wang X, Chow T, Alves J, Buchanan TA, et al. Children exposed to aternal obesity or gestational diabetes mellitus during early fetal development have hypothalamic alterations that predict future weight gain. Diabetes Care. (2019) 42:1473–80. doi: 10.2337/dc18-2581
99. Chandna AR, Kuhlmann N, Bryce CA, Greba Q, Campanucci VA, Howland JG. Chronic maternal hyperglycemia induced during mid-pregnancy in rats increases RAGE expression, augments hippocampal excitability, and alters behavior of the offspring. Neuroscience. (2015) 303:241–60. doi: 10.1016/j.neuroscience.2015.06.063
100. Huerta-Cervantes M, Pena-Montes DJ, Montoya-Perez R, Trujillo X, Huerta M, Lopez-Vazquez MA, et al. Gestational diabetes triggers oxidative stress in hippocampus and cerebral cortex and cognitive behavior modifications in rat offspring: age- and sex-dependent effects. Nutrients. (2020) 12:376. doi: 10.3390/nu12020376
101. Vuong B, Odero G, Rozbacher S, Stevenson M, Kereliuk SM, Pereira TJ, et al. Exposure to gestational diabetes mellitus induces neuroinflammation, derangement of hippocampal neurons, and cognitive changes in rat offspring. J Neuroinflammation. (2017) 14:80. doi: 10.1186/s12974-017-0859-9
102. de Sousa RAL, de Lima EV, da Silva TP, de Souza RV, Figueiredo CP, Passos GF, et al. Late cognitive consequences of gestational diabetes to the offspring, in a new mouse model. Mol Neurobiol. (2019) 56:7754–64. doi: 10.1007/s12035-019-1624-0
103. Zou K, Ren J, Luo S, Zhang J, Zhou C, Tan C, et al. Intrauterine hyperglycemia impairs memory across two generations. Transl Psychiatry. (2021) 11:434. doi: 10.1038/s41398-021-01565-7
104. Sousa FJ, Correia RG, Cruz AF, Martins JM, Rodrigues MS, Gomes CA, et al. Sex differences in offspring neurodevelopment, cognitive performance and microglia morphology associated with maternal diabetes: Putative targets for insulin therapy. Brain Behav Immun Health. (2020) 5:100075. doi: 10.1016/j.bbih.2020.100075
105. Aviel-Shekler K, Hamshawi Y, Sirhan W, Getselter D, Srikanth KD, Malka A, et al. Gestational diabetes induces behavioral and brain gene transcription dysregulation in adult offspring. Transl Psychiatry. (2020) 10:412. doi: 10.1038/s41398-020-01096-7
106. Ji S, Zhou W, Li X, Liu S, Wang F, Li X, et al. Maternal hyperglycemia disturbs neocortical neurogenesis via epigenetic regulation in C57BL/6J mice. Cell Death Dis. (2019) 10:211. doi: 10.1038/s41419-019-1438-z
107. Jin Y, Wang G, Han SS, He MY, Cheng X, Ma ZL, et al. Effects of oxidative stress on hyperglycaemia-induced brain malformations in a diabetes mouse model. Exp Cell Res. (2016) 347:201–11. doi: 10.1016/j.yexcr.2016.08.002
108. Luo SS, Zou KX, Zhu H, Cheng Y, Yan YS, Sheng JZ, et al. Integrated multi-omics analysis reveals the effect of maternal gestational diabetes on fetal mouse hippocampi. Front Cell Dev Biol. (2022) 10:748862. doi: 10.3389/fcell.2022.748862
109. Grunnet LG, Hansen S, Hjort L, Madsen CM, Kampmann FB, Thuesen ACB, et al. Adiposity, dysmetabolic traits, and earlier onset of female puberty in adolescent offspring of women with gestational diabetes mellitus: a clinical study within the danish national birth cohort. Diabetes Care. (2017) 40:1746–55. doi: 10.2337/dc17-0514
110. Qiu XH Li ML, Li N, Sun Q, Zhou J, Ma RJ, et al. Maternal diabetes impairs the initiation of meiosis in murine female germ cells. Mol Med Rep. (2017) 16:5189–94. doi: 10.3892/mmr.2017.7245
111. Khaksar Z, Jelodar G, Hematian H, Poorahmadi M. Alterations of the ovarian histomorphometry at pre-puberty in rat offspring from diabetic mothers. Reprod Med Biol. (2013) 12:173–8. doi: 10.1007/s12522-013-0151-3
112. Sinha N, Biswas A, Nave O, Seger C, Sen A. Gestational diabetes epigenetically reprograms the cart promoter in fetal ovary, causing subfertility in adult life. Endocrinology. (2019) 160:1684–700. doi: 10.1210/en.2019-00319
113. Wang A, Luo B, Chen Z, Xia Y, Chen C, Qi H, et al. Rodents on a high-fat diet born to mothers with gestational diabetes exhibit sex-specific lipidomic changes in reproductive organs. Acta Biochim Biophys Sin. (2022) 54:1–12. doi: 10.3724/abbs.2022052
114. Xu J, Huang J, Pan Q, Du M, Li Z, Dong H. Gestational diabetes promotes germ cell cCyst breakdown and primordial follicle formation in newborn mice via the AKT signaling pathway. PLoS ONE. (2019) 14:e0215007. doi: 10.1371/journal.pone.0215007
115. Mo JY, Yan YS, Lin ZL, Liu R, Liu XQ, Wu HY, et al. Gestational diabetes mellitus suppresses fetal testis development in micedagger. Biol Reprod. (2022) 107:148–56. doi: 10.1093/biolre/ioac138
116. Mao J, Pennington KA, Talton OO, Schulz LC, Sutovsky M, Lin Y, et al. In utero and postnatal exposure to high fat, high sucrose diet suppressed testis apoptosis and reduced sperm count. Sci Rep. (2018) 8:7622. doi: 10.1038/s41598-018-25950-3
117. Turk G, Risvanli A, Ceribasi AO, Sonmez M, Yuce A, Guvenc M, et al. Effect of gestational diabetes mellitus on testis and pancreatic tissues of male offspring. Andrologia. (2018). doi: 10.1111/and.12976
118. Akindele OO, Kunle-Alabi OT, Udofia UA, Ahmed TT, Raji Y. Maternal hyperglycemia at different stages of gestation and its effects on male reproductive functions in rats. J Dev Orig Health Dis. (2015) 6:512–9. doi: 10.1017/S2040174415007217
119. Roll U, Scheeser J, Standl E, Ziegler AG. Alterations of lymphocyte subsets in children of diabetic mothers. Diabetologia. (1994) 37:1132–41. doi: 10.1007/BF00418377
120. Warncke K, Lickert R, Eitel S, Gloning KP, Bonifacio E, Sedlmeier EM, et al. Thymus growth and fetal immune responses in diabetic pregnancies. Horm Metab Res. (2017) 49:892–8. doi: 10.1055/s-0043-120671
121. Knoop J, Eugster A, Gavrisan A, Lickert R, Sedlmeier EM, Dietz S, et al. Maternal type 1 diabetes reduces autoantigen-responsive CD4(+) T cells in offspring. Diabetes. (2020) 69:661–9. doi: 10.2337/db19-0751
122. Luopajarvi K, Nieminen JK, Ilonen J, Akerblom HK, Knip M, Vaarala O. Expansion of CD4+CD25+FOXP3+ regulatory T cells in infants of mothers with type 1 diabetes. Pediatr Diabetes. (2012) 13:400–7. doi: 10.1111/j.1399-5448.2012.00852.x
123. Blue EK, Ballman K, Boyle F, Oh E, Kono T, Quinney SK, et al. Fetal hyperglycemia and a high-fat diet contribute to aberrant glucose tolerance and hematopoiesis in adult rats. Pediatr Res. (2015) 77:316–25. doi: 10.1038/pr.2014.185
124. Li Q, Pereira TJ, Moyce BL, Mahood TH, Doucette CA, Rempel J, et al. In utero exposure to gestational diabetes mellitus conditions TLR4 and TLR2 activated IL-1beta responses in spleen cells from rat offspring. Biochim Biophys Acta. (2016) 1862:2137–46. doi: 10.1016/j.bbadis.2016.08.004
125. Mahmoud MH, Badr G, El Shinnawy NA. Camel whey protein improves lymphocyte function and protects against diabetes in the offspring of diabetic mouse dams. Int J Immunopathol Pharmacol. (2016) 29:632–46. doi: 10.1177/0394632016671729
126. Badr G, Alwasel S, Ebaid H, Mohany M, Alhazza I. Perinatal supplementation with thymoquinone improves diabetic complications and T cell immune responses in rat offspring. Cell Immunol. (2011) 267:133–40. doi: 10.1016/j.cellimm.2011.01.002
127. De Riva A, Wallberg M, Ronchi F, Coulson R, Sage A, Thorne L, et al. Regulation of type 1 diabetes development and B-cell activation in nonobese diabetic mice by early life exposure to a diabetogenic environment. PLoS ONE. (2017) 12:e0181964. doi: 10.1371/journal.pone.0181964
128. Baack ML, Forred BJ, Larsen TD, Jensen DN, Wachal AL, Khan MA, et al. Consequences of a maternal high-fat diet and late gestation diabetes on the developing rat lung. PLoS ONE. (2016) 11:e0160818. doi: 10.1371/journal.pone.0160818
129. Trevino-Alanis M, Ventura-Juarez J, Hernandez-Pinero J, Nevarez-Garza A, Quintanar-Stephano A, Gonzalez-Pina A. Delayed lung maturation of foetus of diabetic mother rats develop with a diminish, but without changes in the proportion of type I and II pneumocytes, and decreased expression of protein D-associated surfactant factor. Anat Histol Embryol. (2009) 38:169–76. doi: 10.1111/j.1439-0264.2008.00902.x
130. Pascoe CD, Basu S, Schwartz J, Fonseca M, Kahnamoui S, Jha A, et al. Maternal diabetes promotes offspring lung dysfunction and inflammation in a sex-dependent manner. Am J Physiol Lung Cell Mol Physiol. (2022) 322:L373–L84. doi: 10.1152/ajplung.00425.2021
131. Chen G, Sun W, Liang Y, Chen T, Guo W, Tian W. Maternal diabetes modulates offspring cell proliferation and apoptosis during odontogenesis via the TLR4/NF-kappaB signalling pathway. Cell Prolif. (2017) 50:e12324. doi: 10.1111/cpr.12324
132. Soderborg TK, Carpenter CM, Janssen RC, Weir TL, Robertson CE, Ir D, et al. Gestational diabetes is uniquely associated with altered early seeding of the infant gut microbiota. Front Endocrinol. (2020) 11:603021. doi: 10.3389/fendo.2020.603021
133. Ponzo V, Ferrocino I, Zarovska A, Amenta MB, Leone F, Monzeglio C, et al. The microbiota composition of the offspring of patients with gestational diabetes mellitus (GDM). PLoS ONE. (2019) 14:e0226545. doi: 10.1371/journal.pone.0226545
134. Crusell MKW, Hansen TH, Nielsen T, Allin KH, Ruhlemann MC, Damm P, et al. Comparative studies of the gut microbiota in the offspring of mothers with and without gestational diabetes. Front Cell Infect Microbiol. (2020) 10:536282. doi: 10.3389/fcimb.2020.536282
135. Hasan S, Aho V, Pereira P, Paulin L, Koivusalo SB, Auvinen P, et al. Gut microbiome in gestational diabetes: a cross-sectional study of mothers and offspring 5 years postpartum. Acta Obstet Gynecol Scand. (2018) 97:38–46. doi: 10.1111/aogs.13252
136. Xue C, Xie Q, Zhang C, Hu Y, Song X, Jia Y, et al. Vertical transmission of the gut microbiota influences glucose metabolism in offspring of mice with hyperglycaemia in pregnancy. Microbiome. (2022) 10:122. doi: 10.1186/s40168-022-01318-8
137. Perna-Barrull D, Rodriguez-Fernandez S, Pujol-Autonell I, Gieras A, Ampudia-Carrasco RM, Villalba A, et al. Prenatal betamethasone interferes with immune system development and alters target cells in autoimmune diabetes. Sci Rep. (2019) 9:1235. doi: 10.1038/s41598-018-37878-9
138. Yang Y, Guo F, Peng Y, Chen R, Zhou W, Wang H, et al. Transcriptomic profiling of human placenta in gestational diabetes mellitus at the single-cell level. Front Endocrinol. (2021) 12:679582. doi: 10.3389/fendo.2021.679582
139. Nelson SM, Sattar N, Freeman DJ, Walker JD, Lindsay RS. Inflammation and endothelial activation is evident at birth in offspring of mothers with type 1 diabetes. Diabetes. (2007) 56:2697–704. doi: 10.2337/db07-0662
140. Brown HM, Green ES, Tan TCY, Gonzalez MB, Rumbold AR, Hull ML, et al. Periconception onset diabetes is associated with embryopathy and fetal growth retardation, reproductive tract hyperglycosylation and impaired immune adaptation to pregnancy. Sci Rep. (2018) 8:2114. doi: 10.1038/s41598-018-19263-8
141. Kagohashi Y, Udagawa J, Moriyama K, Otani H. Maternal environment affects endogenous virus induction in the offspring of type 1 diabetes model non-obese diabetic mice. Congenit Anom. (2005) 45:80–4. doi: 10.1111/j.1741-4520.2005.00071.x
142. Su R, Yan J, Yang H. Transgenerational glucose intolerance of tumor necrosis factor with epigenetic alteration in rat perirenal adipose tissue induced by intrauterine hyperglycemia. J Diabetes Res. (2016) 2016:4952801. doi: 10.1155/2016/4952801
143. Schutte T, Kedziora SM, Haase N, Herse F, Alenina N, Muller DN, et al. Diabetic pregnancy as a novel risk factor for cardiac dysfunction in the offspring-the heart as a target for fetal programming in rats. Diabetologia. (2021) 64:2829–42. doi: 10.1007/s00125-021-05566-5
144. Cerychova R, Bohuslavova R, Papousek F, Sedmera D, Abaffy P, Benes V, et al. Adverse effects of Hif1a mutation and maternal diabetes on the offspring heart. Cardiovasc Diabetol. (2018) 17:68. doi: 10.1186/s12933-018-0713-0
145. Glombik K, Trojan E, Kurek A, Budziszewska B, Basta-Kaim A. Inflammatory consequences of maternal diabetes on the offspring brain: a hippocampal organotypic culture study. Neurotox Res. (2019) 36:357–75. doi: 10.1007/s12640-019-00070-6
146. Money KM, Barke TL, Serezani A, Gannon M, Garbett KA, Aronoff DM, et al. Gestational diabetes exacerbates maternal immune activation effects in the developing brain. Mol Psychiatry. (2018) 23:1920–8. doi: 10.1038/mp.2017.191
147. Cheng X, Chapple SJ, Patel B, Puszyk W, Sugden D, Yin X, et al. Gestational diabetes mellitus impairs Nrf2-mediated adaptive antioxidant defenses and redox signaling in fetal endothelial cells in utero. Diabetes. (2013) 62:4088–97. doi: 10.2337/db13-0169
148. Katkhuda R, Peterson ES, Roghair RD, Norris AW, Scholz TD, Segar JL. Sex-specific programming of hypertension in offspring of late-gestation diabetic rats. Pediatr Res. (2012) 72:352–61. doi: 10.1038/pr.2012.93
149. Wang X, Lu J, Xie W, Lu X, Liang Y, Li M, et al. Maternal diabetes induces autism-like behavior by hyperglycemia-mediated persistent oxidative stress and suppression of superoxide dismutase 2. Proc Natl Acad Sci U S A. (2019) 116:23743–52. doi: 10.1073/pnas.1912625116
150. Lu J, Xiao M, Guo X, Liang Y, Wang M, Xu J, et al. Maternal diabetes induces immune dysfunction in autistic offspring through oxidative stress in hematopoietic stem cells. Front Psychiatry. (2020) 11:576367. doi: 10.3389/fpsyt.2020.576367
151. Clark KL, Talton OO, Ganesan S, Schulz LC, Keating AF. Developmental origins of ovarian disorder: impact of maternal lean gestational diabetes on the offspring ovarian proteome in micedagger. Biol Reprod. (2019) 101:771–81. doi: 10.1093/biolre/ioz116
152. Biron-Shental T, Liberman M, Elbaz M, Laish I, Sharony R, Amiel A. Telomere homeostasis in placentas from pregnancies with uncontrolled diabetes. Placenta. (2016) 44:13–8. doi: 10.1016/j.placenta.2016.05.009
153. Cross JA, Temple RC, Hughes JC, Dozio NC, Brennan C, Stanley K, et al. Cord blood telomere length, telomerase activity and inflammatory markers in pregnancies in women with diabetes or gestational diabetes. Diabet Med. (2010) 27:1264–70. doi: 10.1111/j.1464-5491.2010.03099.x
154. Hjort L, Vryer R, Grunnet LG, Burgner D, Olsen SF, Saffery R, et al. Telomere length is reduced in 9- to 16-year-old girls exposed to gestational diabetes in utero. Diabetologia. (2018) 61:870–80. doi: 10.1007/s00125-018-4549-7
155. Cross JA, Brennan C, Gray T, Temple RC, Dozio N, Hughes JC, et al. Absence of telomere shortening and oxidative DNA damage in the young adult offspring of women with pre-gestational type 1 diabetes. Diabetologia. (2009) 52:226–34. doi: 10.1007/s00125-008-1207-5
156. Nazari Z, Nabiuni M, Saeidi M, Golalipour MJ. Gestational diabetes leads to down-regulation of CDK4-pRB-E2F1 pathway genes in pancreatic islets of rat offspring. Iran J Basic Med Sci. (2017) 20:150–4. doi: 10.22038/ijbms.2017.8240
157. Manivannan S, Mansfield C, Zhang X, Kodigepalli KM, Majumdar U, Garg V, et al. Single-cell transcriptomic profiling unveils dysregulation of cardiac progenitor cells and cardiomyocytes in a mouse model of maternal hyperglycemia. Commun Biol. (2022) 5:820. doi: 10.1038/s42003-022-03779-x
158. del Rosario MC, Ossowski V, Knowler WC, Bogardus C, Baier LJ, Hanson RL. Potential epigenetic dysregulation of genes associated with MODY and type 2 diabetes in humans exposed to a diabetic intrauterine environment: an analysis of genome-wide DNA methylation. Metabolism. (2014) 63:654–60. doi: 10.1016/j.metabol.2014.01.007
159. Howe CG, Cox B, Fore R, Jungius J, Kvist T, Lent S, et al. Maternal gestational diabetes mellitus and newborn DNA methylation: findings from the pregnancy and childhood epigenetics consortium. Diabetes Care. (2020) 43:98–105. doi: 10.2337/dc19-0524
160. Weng X, Liu F, Zhang H, Kan M, Wang T, Dong M, et al. Genome-wide DNA methylation profiling in infants born to gestational diabetes mellitus. Diabetes Res Clin Pract. (2018) 142:10–8. doi: 10.1016/j.diabres.2018.03.016
161. Gautier JF, Porcher R, Abi Khalil C, Bellili-Munoz N, Fetita LS, Travert F, et al. Kidney dysfunction in adult offspring exposed in utero to type 1 diabetes is associated with alterations in genome-wide DNA methylation. PLoS ONE. (2015) 10:e0134654. doi: 10.1371/journal.pone.0134654
162. Binder AM, LaRocca J, Lesseur C, Marsit CJ, Michels KB. Epigenome-wide and transcriptome-wide analyses reveal gestational diabetes is associated with alterations in the human leukocyte antigen complex. Clin Epigenetics. (2015) 7:79. doi: 10.1186/s13148-015-0116-y
163. Ruchat SM, Houde AA, Voisin G, St-Pierre J, Perron P, Baillargeon JP, et al. Gestational diabetes mellitus epigenetically affects genes predominantly involved in metabolic diseases. Epigenetics. (2013) 8:935–43. doi: 10.4161/epi.25578
164. Cvitic S, Novakovic B, Gordon L, Ulz CM, Muhlberger M, Diaz-Perez FI, et al. Human fetoplacental arterial and venous endothelial cells are differentially programmed by gestational diabetes mellitus, resulting in cell-specific barrier function changes. Diabetologia. (2018) 61:2398–411. doi: 10.1007/s00125-018-4699-7
165. Blue EK, Sheehan BM, Nuss ZV, Boyle FA, Hocutt CM, Gohn CR, et al. Epigenetic regulation of placenta-specific 8 contributes to altered function of endothelial colony-forming cells exposed to intrauterine gestational diabetes mellitus. Diabetes. (2015) 64:2664–75. doi: 10.2337/db14-1709
166. Zhu Z, Chen X, Xiao Y, Wen J, Chen J, Wang K, et al. Gestational diabetes mellitus alters DNA methylation profiles in pancreas of the offspring mice. J Diabetes Complications. (2019) 33:15–22. doi: 10.1016/j.jdiacomp.2018.11.002
167. Jiang Y, Zhu H, Chen Z, Yu YC, Guo XH, Chen Y, et al. Hepatic IGF2/H19 Epigenetic alteration induced glucose intolerance in gestational diabetes mellitus offspring via FoxO1 mediation. Front Endocrinol. (2022) 13:844707. doi: 10.3389/fendo.2022.844707
168. Petropoulos S, Guillemin C, Ergaz Z, Dimov S, Suderman M, Weinstein-Fudim L, et al. Gestational diabetes alters offspring DNA methylation profiles in human and rat: identification of key pathways involved in endocrine system disorders, insulin signaling, diabetes signaling, and ILK signaling. Endocrinology. (2015) 156:2222–38. doi: 10.1210/en.2014-1643
169. Ren J, Cheng Y, Ming ZH, Dong XY, Zhou YZ, Ding GL, et al. Intrauterine hyperglycemia exposure results in intergenerational inheritance via DNA methylation reprogramming on F1 PGCs. Epigenetics Chromatin. (2018) 11:20. doi: 10.1186/s13072-018-0192-2
170. Hepp P, Hutter S, Knabl J, Hofmann S, Kuhn C, Mahner S, et al. Histone H3 lysine 9 acetylation is downregulated in GDM Placentas and Calcitriol supplementation enhanced this effect. Int J Mol Sci. (2018) 19:4061. doi: 10.3390/ijms19124061
171. Houshmand-Oeregaard A, Schrolkamp M, Kelstrup L, Hansen NS, Hjort L, Thuesen ACB, et al. Increased expression of microRNA-15a and microRNA-15b in skeletal muscle from adult offspring of women with diabetes in pregnancy. Hum Mol Genet. (2018) 27:1763–71. doi: 10.1093/hmg/ddy085
172. Frorup C, Mirza AH, Yarani R, Nielsen LB, Mathiesen ER, Damm P, et al. Plasma exosome-enriched extracellular vesicles from lactating mothers with type 1 diabetes contain aberrant levels of miRNAs during the postpartum period. Front Immunol. (2021) 12:744509. doi: 10.3389/fimmu.2021.744509
173. Joshi A, Azuma R, Akumuo R, Goetzl L, Pinney SE. Gestational diabetes and maternal obesity are associated with sex-specific changes in miRNA and target gene expression in the fetus. Int J Obes. (2020) 44:1497–507. doi: 10.1038/s41366-019-0485-y
174. Tryggestad JB, Vishwanath A, Jiang S, Mallappa A, Teague AM, Takahashi Y, et al. Influence of gestational diabetes mellitus on human umbilical vein endothelial cell miRNA. Clin Sci. (2016) 130:1955–67. doi: 10.1042/CS20160305
175. Tang L, Li P, Li L. Whole transcriptome expression profiles in placenta samples from women with gestational diabetes mellitus. J Diabetes Investig. (2020) 11:1307–17. doi: 10.1111/jdi.13250
176. Ding R, Guo F, Zhang Y, Liu XM, Xiang YQ, Zhang C, et al. Integrated transcriptome sequencing analysis reveals role of miR-138-5p/ TBL1X in placenta from gestational diabetes mellitus. Cell Physiol Biochem. (2018) 51:630–46. doi: 10.1159/000495319
177. Jiang S, Teague AM, Tryggestad JB, Chernausek SD. Role of microRNA-130b in placental PGC-1alpha/TFAM mitochondrial biogenesis pathway. Biochem Biophys Res Commun. (2017) 487:607–12. doi: 10.1016/j.bbrc.2017.04.099
178. Fornes D, Heinecke F, Roberti SL, White V, Capobianco E, Jawerbaum A. Proinflammation in maternal and fetal livers and circulating miR-122 dysregulation in a GDM rat model induced by intrauterine programming. Mol Cell Endocrinol. (2020) 510:110824. doi: 10.1016/j.mce.2020.110824
179. He L, Wang X, Jin Y, Xu W, Guan Y, Wu J, et al. Identification and validation of the miRNA-mRNA regulatory network in fetoplacental arterial endothelial cells of gestational diabetes mellitus. Bioengineered. (2021) 12:3503–15. doi: 10.1080/21655979.2021.1950279
180. DiMeglio LA, Evans-Molina C, Oram RA. Type 1 diabetes. Lancet. (2018) 391:2449–62. doi: 10.1016/S0140-6736(18)31320-5
181. Lee SH, Park SY, Choi CS. Insulin resistance: from mechanisms to therapeutic strategies. Diabetes Metab J. (2022) 46:15–37. doi: 10.4093/dmj.2021.0280
182. Batista TM, Haider N, Kahn CR. Defining the underlying defect in insulin action in type 2 diabetes. Diabetologia. (2021) 64:994–1006. doi: 10.1007/s00125-021-05415-5
183. Kitessa SM, Abeywardena MY. Lipid-induced insulin resistance in skeletal muscle: the chase for the culprit goes from total intramuscular fat to lipid intermediates, and finally to species of lipid intermediates. Nutrients. (2016) 8:466. doi: 10.3390/nu8080466
184. Roden M. Muscle triglycerides and mitochondrial function: possible mechanisms for the development of type 2 diabetes. Int J Obes. (2005) 29:S111–5. doi: 10.1038/sj.ijo.0803102
185. Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. (1999) 98:115–24. doi: 10.1016/S0092-8674(00)80611-X
186. Krishnaveni GV, Veena SR, Hill JC, Kehoe S, Karat SC, Fall CH. Intrauterine exposure to maternal diabetes is associated with higher adiposity and insulin resistance and clustering of cardiovascular risk markers in Indian children. Diabetes Care. (2010) 33:402–4. doi: 10.2337/dc09-1393
187. Harder T, Aerts L, Franke K, Van Bree R, Van Assche FA, Plagemann A. Pancreatic islet transplantation in diabetic pregnant rats prevents acquired malformation of the ventromedial hypothalamic nucleus in their offspring. Neurosci Lett. (2001) 299:85–8. doi: 10.1016/S0304-3940(01)01495-1
188. Park JE, Jardine L, Gottgens B, Teichmann SA, Haniffa M. Prenatal development of human immunity. Science. (2020) 368:600–3. doi: 10.1126/science.aaz9330
189. De Riva A, Varley MC, Bluck LJ, Cooke A, Deery MJ, Busch R. Accelerated turnover of MHC class II molecules in nonobese diabetic mice is developmentally and environmentally regulated in vivo and dispensable for autoimmunity. J Immunol. (2013) 190:5961–71. doi: 10.4049/jimmunol.1300551
190. Tsarna E, Christopoulos P. The role of gut microbiome in prevention, diagnosis and treatment of gestational diabetes mellitus. J Obstet Gynaecol. (2022) 42:719–25. doi: 10.1080/01443615.2021.1959534
191. Gurung M, Li Z, You H, Rodrigues R, Jump DB, Morgun A, et al. Role of gut microbiota in type 2 diabetes pathophysiology. EBioMedicine. (2020) 51:102590. doi: 10.1016/j.ebiom.2019.11.051
192. Dedrick S, Sundaresh B, Huang Q, Brady C, Yoo T, Cronin C, et al. The role of gut microbiota and environmental factors in type 1 diabetes pathogenesis. Front Endocrinol. (2020) 11:78. doi: 10.3389/fendo.2020.00078
193. Piconi L, Quagliaro L, Ceriello A. Oxidative stress in diabetes. Clin Chem Lab Med. (2003) 41:1144–9. doi: 10.1515/CCLM.2003.177
194. Sies H, Jones DP. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat Rev Mol Cell Biol. (2020) 21:363–83. doi: 10.1038/s41580-020-0230-3
195. Forrester SJ, Kikuchi DS, Hernandes MS, Xu Q, Griendling KK. Reactive oxygen species in metabolic and inflammatory signaling. Circ Res. (2018) 122:877–902. doi: 10.1161/CIRCRESAHA.117.311401
196. Kattoor AJ, Pothineni NVK, Palagiri D, Mehta JL. Oxidative stress in atherosclerosis. Curr Atheroscler Rep. (2017) 19:42. doi: 10.1007/s11883-017-0678-6
197. Nissanka N, Moraes CT. Mitochondrial DNA damage and reactive oxygen species in neurodegenerative disease. FEBS Lett. (2018) 592:728–42. doi: 10.1002/1873-3468.12956
198. Flynn JM, Melov S. SOD2 in mitochondrial dysfunction and neurodegeneration. Free Radic Biol Med. (2013) 62:4–12. doi: 10.1016/j.freeradbiomed.2013.05.027
199. Wong CC, Meaburn EL, Ronald A, Price TS, Jeffries AR, Schalkwyk LC, et al. Methylomic analysis of monozygotic twins discordant for autism spectrum disorder and related behavioural traits. Mol Psychiatry. (2014) 19:495–503. doi: 10.1038/mp.2013.41
200. Canouil M, Khamis A, Keikkala E, Hummel S, Lobbens S, Bonnefond A, et al. Epigenome-wide association study reveals methylation loci associated with offspring gestational diabetes mellitus exposure and maternal methylome. Diabetes Care. (2021) 44:1992–9. doi: 10.2337/dc20-2960
201. Shah KB, Chernausek SD, Teague AM, Bard DE, Tryggestad JB. Maternal diabetes alters microRNA expression in fetal exosomes, human umbilical vein endothelial cells and placenta. Pediatr Res. (2021) 89:1157–63. doi: 10.1038/s41390-020-1060-x
Keywords: maternal diabetes, offspring, fetal development, long-term outcomes, potential mechanisms
Citation: Yan Y-S, Feng C, Yu D-Q, Tian S, Zhou Y, Huang Y-T, Cai Y-T, Chen J, Zhu M-M and Jin M (2023) Long-term outcomes and potential mechanisms of offspring exposed to intrauterine hyperglycemia. Front. Nutr. 10:1067282. doi: 10.3389/fnut.2023.1067282
Received: 11 October 2022; Accepted: 06 April 2023;
Published: 15 May 2023.
Edited by:
Zheng Feei Ma, University of the West of England, United KingdomReviewed by:
Rufei Gao, Chongqing Medical University, ChinaGustavo Tadeu Volpato, Federal University of Mato Grosso, Brazil
Copyright © 2023 Yan, Feng, Yu, Tian, Zhou, Huang, Cai, Chen, Zhu and Jin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Min Jin, bWluX2ppbkB6anUuZWR1LmNu