 Julio Plaza-Díaz1,2,3
Julio Plaza-Díaz1,2,3 Manuel Manzano4
Manuel Manzano4 Francisco Javier Ruiz-Ojeda1,3,5,6
Francisco Javier Ruiz-Ojeda1,3,5,6 Maria D. Giron1
Maria D. Giron1 Rafael Salto1
Rafael Salto1 Jose M. López-Pedrosa4Angela Santos-Fandila4
Jose M. López-Pedrosa4Angela Santos-Fandila4 Maria Teresa Garcia-Corcoles1
Maria Teresa Garcia-Corcoles1 Ricardo Rueda4
Ricardo Rueda4 Ángel Gil1,3,5,7*
Ángel Gil1,3,5,7*- 1Department of Biochemistry and Molecular Biology II, School of Pharmacy, University of Granada, Granada, Spain
- 2Children’s Hospital of Eastern Ontario Research Institute, Ottawa, ON, Canada
- 3Instituto de Investigación Biosanitaria de Granada (ibs.Granada), Complejo Hospitalario Universitario de Granada, Granada, Spain
- 4Abbott Nutrition R&D, Granada, Spain
- 5Institute of Nutrition and Food Technology “José Mataix”, Biomedical Research Centre, University of Granada, Granada, Spain
- 6RG Adipocytes and Metabolism, Institute for Diabetes and Obesity, Helmholtz Diabetes Center at Helmholtz Center Munich, Munich, Germany
- 7CIBER Physiopathology of Obesity and Nutrition, Instituto de Salud Carlos III, Madrid, Spain
Introduction: The main cause of insulin resistance in childhood is obesity, which contributes to future comorbidities as in adults. Although high-calorie diets and lack of exercise contribute to metabolic disease development, food quality rather than the quantity of macronutrients is more important than food density. The purpose of the present study was to examine the effects of changing the quality of carbohydrates from rapidly to slowly digestible carbohydrates on the composition of the gut microbiota and the profiles of the functional pathways in growing rats with obesity due to a high-fat diet (HFD).
Methods: During the course of 4 weeks, rats growing on an HFD-containing carbohydrates with different digestive rates were fed either HFD-containing carbohydrates with a rapid digestion rate (OBE group) or HFD-containing carbohydrates with a slow digestion rate (OBE-ISR group). A non-obese group (NOB) was included as a reference, and rats were fed on a rodent standard diet (AIN93G). An analysis of gut microbiota was conducted using 16S rRNA-based metagenomics; a linear mixed-effects model (LMM) was used to determine changes in abundance between baseline and 4 weeks of treatment, and functional pathways were identified. Gut microbiota composition at bacterial diversity and relative abundance, at phylum and genus levels, and functional profiles were analyzed by integrating the Integrated Microbial Genomes (IMG) database.
Results: The groups showed comparable gut microbiota at baseline. At the end of the treatment, animals from the ISR group exhibited differences at the phylum levels by decreasing the diversity of Fisher’s index and Firmicutes (newly named as Bacillota), and increasing the Pielou’s evenness and Bacteroidetes (newly named as Bacteroidota); at the genus level by increasing Alistipes, Bifidobacterium, Bacteroides, Butyricimonas, Lachnoclostridium, Flavonifractor, Ruminiclostridium 5, and Faecalibaculum and decreasing Muribaculum, Blautia, and Ruminiclostridium 9. Remarkably, relative abundances of genera Tyzzerella and Angelakisella were higher in the OBE group compared to NOB and OBE-ISR groups. In addition, some microbiota carbohydrate metabolism pathways such as glycolysis, glucuronic acid degradation, pentose phosphate pathway, methanogenesis, and fatty acid biosynthesis exhibited increased activity in the OBE-ISR group after the treatment. Higher levels of acetate and propionate were found in the feces of the ISR group compared with the NOB and OBE groups.
Conclusion: The results of this study demonstrate that replacing rapidly digestible carbohydrates with slowly digestible carbohydrates within an HFD improve the composition of the gut microbiota. Consequently, metabolic disturbances associated with obesity may be prevented.
Introduction
Over the last century, non-communicable diseases (NCDs) have replaced communicable diseases as the primary cause of premature death worldwide, including heart disease, stroke, cancer, diabetes, and chronic lung disease. These diseases account for approximately 70% of all deaths worldwide (World Health Organization, WHO). NCDs and dysbiotic gut microbiota have been linked in several studies; therefore, establishing a causal association between dysbiotic microbiota and NCDs may represent a paradigm shift in the prevention and control of these diseases (1).
As a consequence of the obesity pandemic, insulin resistance, metabolic diseases, type 2 diabetes, non-alcoholic fatty liver disease (NAFLD), and certain cancers are at an increased risk (2). In this context, gut microbiota composition and its metabolites change in individuals with obesity, resulting in an increase of Firmicutes (newly named as Bacillota) to Bacteroidetes (newly named as Bacteroidota) ratio (3, 4), which is already established as an indicator of obesity and cardiometabolic disease in human and murine studies (5). Diet influences the human gut microbiota, and dietary interventions cause substantial changes in microbial diversity, both in the short and long term. These findings suggest that dietary interventions might improve the low microbial gene richness and the host metabolism of obese individuals (6).
A long-term high-fat diet (HFD) changes gut microbiota in experimental animals, such as rats and mice, leading to higher intestinal permeability and mucosal immune responses, contributing to obesity and chronic inflammation. As a result of increased permeability, lipopolysaccharide is translocated into the liver, kidney, and heart of obese rats. As a result, oxidative stress and lipid peroxidation are induced, and antioxidant enzyme activity is reduced. The structure of gut microbiota may be altered by HFD feeding, which may lead to the development of obesogenic bacteria and dysbiosis (7, 8). Furthermore, HFD increases the levels of phyla Bacillota and Proteobacteria (newly named as Pseudomonadota) and decreases the beneficial species of Bifidobacterium spp. and Lactobacillus gasseri (9). Indeed, HFD can induce gut microbial dysbiosis in more than 200 strains of mice with genetic variations (10), suggesting that dietary perturbations can lead to changes in gut microbiota despite variations in the host genome.
Generally, a metabolically healthy microbiota is mainly achieved by adhering to a diet that is low in animal fat, and animal protein, as well as fermentable dietary fiber. This serves as a growth substrate for bacteria and yeasts in the distal bowel. The opposite is true, and microbial dysbiosis is linked with a high-fat, high-protein diet, and sedentary lifestyle. Smoking, alcohol consumption, and relatively infrequent defecation are associated with a leaky mucosa, inflammation, and reduced production of short-chain fatty acids (SCFAs), including acetate and butyrate. In particular, dysbiotic microbiota is associated with a prolonged colonic transit time, which results in an increase in microbial proteolysis as a result of a shift in colonic metabolism. It has been reported that people who consume a high amount of protein and animal fat have a lower Bacteroidota to Bacillota ratio in their gut microbiota, whereas people who consume a high amount of fiber and carbohydrates (CHO) have a dominant genus of Prevotella (11).
Dietary fiber, in contrast to simple CHO, is not readily digested or absorbed by the host, but appears to affect the metabolic activities of intestinal bacteria or the interactions between bacteria and the host (12). In fact, slow-digesting CHO (ISR) influences endogenous substrate utilization during growth and development in pre-pubertal children, suggesting a beneficial effect on energy intake and CHO regulation/metabolism (13). Rats exhibit a higher abundance of Bifidobacterium spp. when inulin and fructooligosaccharide (FOS) are not hydrolyzed by mammalian enzymes but instead fermented by the bacterial community in the caecum and colon. A reduction in chronic intestinal inflammation was observed in both fructans, which reduced Clostridium cluster XI and C. difficile toxin gene expression (14). It has been shown that resistant maltodextrin is not digestible in small intestine and ferments in the colon. This stimulates the growth of probiotic microorganisms, particularly those belonging to the genera Lactobacillus and Bifidobacterium. As a result of this fermentation, the colon pH decreases, which in turn increases the amount of Bifidobacterium spp. found in the colon (15, 16).
According to the previous research, slow-digesting CHOs (ISR) provide a number of metabolic benefits during growth and development as well as a reduction in the predisposition to obesity later in life (17, 18). Recently, we have previously revealed that the replacement of rapid digestible CHO for slowly digestible CHO (ISR, containing isomaltulose, resistant maltodextrin, inulin, and FOS) in an HFD rat model prevented the diet-induced obesity-related adverse effects and improved lipid metabolism by different mechanisms in adipose tissue, muscle, and liver, which are related to modifications in key proteins, such as GLUT2 and GLUT4, FAS, SRBP1, AMPK, and Akt, among others (19). Currently, there are not enough basic studies analyzing microbial, metabolic, and inflammation data. Here, we aimed to evaluate the fecal microbiota and associated metabolic pathway changes after the consumption of those ISR in growing rats with HFD-induced obesity.
Materials and methods
Housing and experimental design
A total of thirty weanling male Wistar Han IGS (International Genetic Standard) rats (21–25 days of age) were provided by Charles Rivers (Orleans Cedex, France). Each animal was housed in a cage under a cycle of 12 h of light and 12 h of darkness. A temperature of 21°C was maintained in the room. The experimental procedures (approval code 29 October 2018/152) were carried out in accordance with the European Convention for the Protection of Vertebrate Animals used for Experimental and other Scientific Purposes (Directive 2010/63/EU) and to the Spanish National Research Council’s ethical guidelines for animal experimentation (RD 53/1 February 2013). Dietary obesity can be studied using an animal model induced by HFDs.
On the basis of the diet they received, rats (n = 10/group) were randomly assigned to three nutritional groups:
(i) The NOB group, a lean group of rats, was fed a standard rodent diet (AIN93G) (20).
(ii) The OBE group, an obese group of rats, was fed an HFD (21).
(iii) The rats in the OBE-ISR group were fed an HFD that included ISR.
The total fiber content of all diets was the same. In addition, both HFDs are isoenergetic. Table 1 contains all information regarding the diets. The nutritional intervention lasted for 4 weeks. Food and water were freely available to all rats, and their weight and food consumption were determined on a weekly basis. There has been previous publication on body composition, body fat mass, and lean body mass and metabolic parameters (19).
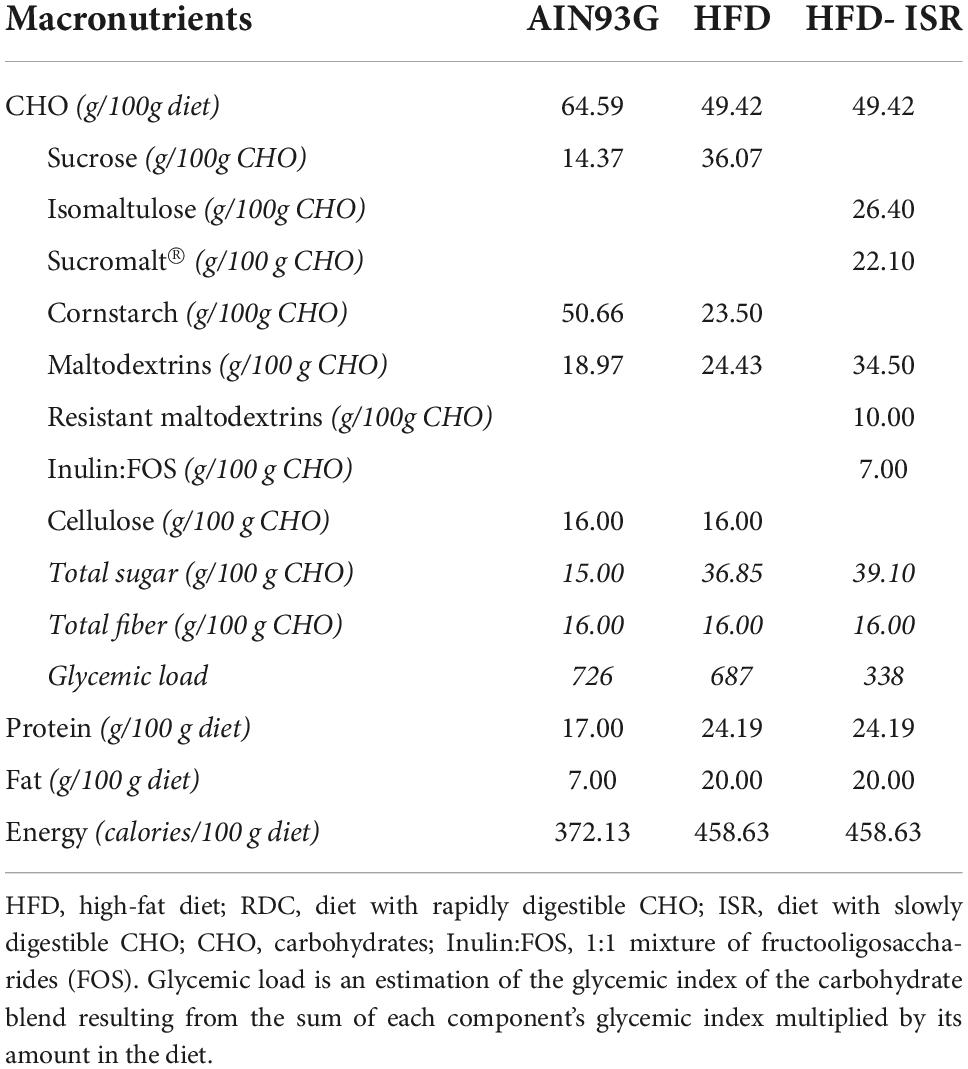
Table 1. Composition of diets.
Microbiota and related functional pathway profile analyses
A hermetically sealed, sterile container was used to collect fecal samples at the beginning and the end of the intervention. As soon as the samples were collected, they were immediately refrigerated after collection and kept at −80°C until they were used for analysis.
DNA extraction
A Stomacher-400 blender was used to homogenize the samples. As instructed by the manufacturer, DNA extraction was conducted using the QIAamp DNA Stool Mini Kit (QIAGEN, Barcelona, Spain), except for the incubation at 70°C. To ensure that gram-positive and gram-negative samples were lysed, the samples were mixed with the lysis buffer and incubated at 95°C. It was determined that the DNA yield can be calculated spectrophotometrically with a NanoDrop ND1000 spectrophotometer (Thermo Fisher Scientific, DE, United States) with absorbance ratios measured at A260/230 nm for salt and phenol contamination and A260/280 nm for protein contamination (22–24).
Sequencing
PCR was used to amplify the extracted DNA using the primers, 16S Amplicon PCR forward primer:
5′TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGC CTACGGGNGGCWGCAG, and 16S Amplicon PCR Reverse Primer: 5′GTCTCGTGGGCTCGGAGATGTGTATAAGAGAC AGGACTACHV GGGTATCTAATCC targeting the V3 and V4 hypervariable regions of the bacterial 16S rRNA gene. PCRs were conducted in 25 μL reaction volumes of which 12.5 μL were 2X KAPA HiFi HotStart ready mix (KAPA Biosystems, Woburn, MA, United States). In addition, 5 μL of forward and reverse primers (1 μM) were prepared, as well as 2.5 μL of extracted DNA (10 ng). PCR cycling conditions were also identical for each PCR: initial denaturation at 95°C for 3 min, followed by cycles of denaturation at 95°C for 30 s, annealing at 55°C for 30 s, and elongation at 72°C for 30 s, with a final extension at 72°C for 5 min. As part of the PCR-clean up, AMPure XP beads (Beckman Coulter, Indianapolis, IN, United States) were employed to remove free primers and primer-dimer species from the 16S V3 and V4 amplicons. Immediately after this, the index PCR was performed at 95°C for 3 min. This was followed by 8 cycles at 95°C for 30 s, 55°C for 30 s, 72°C for 30 s, and 72°C for 5 min and held at 4°C. The Nextera XT Index Kit (Illumina, San Diego, CA, United States) was used to attach dual indices and Illumina sequencing adapters. Before quantification, AMPure XP beads (Beckman Coulter, Indianapolis, IN, United States) were employed to purify the pooled PCR products. Using a paired-end (2 × 300 nt) Illumina MiSeq sequencing system (Illumina, San Diego, CA, United States), the resultant amplicons were sequenced at MiSeq (Illumina, San Diego, CA, United States) (22–24).
Bioinformatic analysis
Illumina bcl2fastq2 Conversion Software v2.20 was used to demultiplex raw sequences, and raw data were imported into QIIME 2 2020.8 open-source software (25) using the q2-tools-import script which uses the PairedEndFastqManifestPhred33 input format. Sequencing of 16S rRNA genes was performed on V3-V4. Denoising was performed using DADA2 (26), which uses a quality-aware model of Illumina amplicon errors to obtain a distribution of sequence variances, each differing by one nucleotide. To truncate the forward reads at position 288 and trim them at position 6, the q2-dada2-denoise script was executed following the retrieving quality scores. We trimmed reverse reads at position 7 after truncating them at position 220. To remove chimeras, we applied the “consensus” filter, which detects chimeras in samples individually and removes those found in a sufficient fraction of samples. In addition, forward and reverse reads are merged during this step. Phylogenies were constructed with FASTTREE2 (via q2- phylogeny) (27) using all amplicon sequence variants (ASVs) aligned with MAFFT (28) via q2- alignment. To classify ASVs, a naïve Bayes taxonomy classifier was used (via q2-feature-classifier) (29) against the SILVA 16S V3-V4 v132_99 (30) along with a similarity threshold of 99%. As part of the data filtering process, samples with fewer than 10,000 reads were excluded. Both weighted (quantitative) and unweighted (qualitative) variants of UniFrac were determined, where the former accounts for the abundance of observed organisms, whereas the latter only accounts for their presence or absence.
Functional profiles
PICRUSt2 was used to predict potential functional profiles for sequenced samples (31). To summarize, phylotypes were arranged into a reference tree containing 20,000 full 16S rRNA genes from prokaryotic genomes in the Integrated Microbial Genomes (IMG) database. Clusters of Orthologous Groups of proteins (COG) and the Enzyme Commission numbers (EC) databases were used to annotate the functional characteristics of these genomes. EC numbers were first regrouped into MetaCyc reactions to infer MetaCyc pathways. Each pathway abundance was calculated as the harmonic mean of the key reaction abundances in each sample. Phylotypes abundances were corrected by 16S rRNA gene copy numbers and then multiplied by functional predictions to estimate the abundance of each gene family per sample.
Quantitation of short-chain fatty acids
With some modifications, the method of Zeng and Cao (32) was used to analyze SCFAs in feces. In brief, a UPLC® Acquity system from Waters (Milford, MA, United States) was coupled to a triple quadrupole mass spectrometer detector (XEVO-TQS) with electrospray ionization (ESI) interface in positive ion mode. Tandem mass spectrometers were operated in multiple reaction monitoring (MRM) mode with unit mass resolution set for Q1 and Q3 quadrupoles. Continuous infusion of standard solutions (1 mg L–1) optimized the mass spectrometric conditions for each compound. An electrospray ionization voltage of 2000 volts was applied to the capillary. In the cone, nitrogen was used as an auxiliary gas at 150 L h–1 and as a desolvation gas at 1000 L h–1. The source temperature was 150°C, and the desolvation temperature was 500°C. As a collision gas, argon (99.999% purity) was used at an approximate rate of 0.15 mL min–1.
Waters UPLC BEH C18 column (2.1 mm × 100 mm i.d., 1.7 μm particle size) was used for chromatographic analysis. The flow rate was 300 μL min–1, column temperature was 35°C, sample temperature was 8°C, and the injection volume was 1 μL. There was a gradient mobile phase consisting of 0.1% (v/v) of formic acid (FA) in water with 10 mM ammonium formate as solvent A and 0.1% (v/v) of FA in methanol:isopropanol (MeOH:IPA, 9:1 v/v) as solvent B. Gradient conditions were as follows: 0.0–4.0 min, 32% to 60% B; 4.0–4.8 min, 60% to 65% B; 4.8–4.9 min, 65% to 98% B; 4.9–5.5 min, 98% B and 5.5–6 min, 98% to 32% B for re-equilibration. The total run time was 6 min. The weak solvent was a mixture of 30% of water and 70% of acetonitrile (MeCN), and the strong solvent was 100% MeOH.
For instrument control and data acquisition and analysis, MassLynx software version 4.1 was used. With only a few changes in dilutions, samples and standards were processed according to the protocol described in the method of Zheng et al. (32). The procedure involves derivatization with 0.1 M of O-Benzylhydroxylamine hydrochloride (BHA) in MeOH and 0.25 M of N-(3-Dimethylaminopropyl)-N’-ethylcarbodiimide hydrochloride (EDC) in MeOH at 25°C for 1 h. A 50% methanol solution in water was used to dilute the samples after incubation, and dichloromethane was used to extract them. A specific volume of 50% aqueous MeOH was added after the extract was evaporated to rehydrate the residual. to inject samples into the UPLC-MS/MS system, samples were collected in vials. In SCFA analysis, specific commercial standards are used to quantify each compound individually with its corresponding standard: acetic acid (AA) ref: 71251-5ML; propionic acid (PA) ref: 94425-5ML; iso-butyric acid (Iso-BA) ref: 46935-U-500MG; butyric acid (BA) ref: 19215-5ML; 2-methyl-butyric acid (2M-BA) ref: 49659-1ML; iso-valeric acid (Iso-VA) ref: 78651-5ML; valeric acid (VA) ref: 75054-5ML; 3-methyl-valeric acid (3M-VA) ref: 222453-5G; 4-methyl-valeric acid (4M-VA) ref: 277827-25G; hexanoic acid (HA) ref: 21529-5ML; all of them are from Sigma-Aldrich. In addition, internal standards have also been used to improve accuracy and precision: sodium acetate-d3 (d3-AA) ref: 176079-25G; propionic acid-d5 (d5-PA) ref: 596507-1G; butyric acid-d7 (d7-BA) ref: 488399-5G; hexanoic acid-d11 (d11-HA) ref: 448168-1G; they also are from Sigma-Aldrich. Due to the lack of internal standards in all the studied compounds, some of them were used when the compounds had similar structures. The calibration curve was done in a linear range of 0.0005–1.034 ug/ml for feces samples.
Determination of serum bile acid profiles
Metabolite profiles were determined as described previously (33, 34). In brief, OWL Metabolomics (Derio, Spain) assessed bile acids on an established rodent serum platform. A UHPLC-time-of-flight (TOF)–Mass spectroscopy (MS)-based platform (Agilent Technologies, Santa Clara, CA, United States) was used here to semi-quantify bile acid species in methanol serum extracts. The TargetLynx application manager for MassLynx 4.1 (Waters Corp., Milford, MA, United States) was used to handle the data obtained with the aforementioned platform. The data obtained during the process were processed with R software v3.2.0 (19).
Biochemical parameters
In this study, serum interleukin-(IL)-1β, monocyte attractant proteins 1 (MCP-1), leptin, and tumor necrosis factor-alpha (TNF-α) were measured using Bio-Plex 200 system (Bio-Rad, Hercules, CA, United States). Serum glucagon-like peptide-1 (GLP-1) was measured using an ELISA kit (Mercodia, Uppsala, Sweden) according to the manufacturer’s instructions (19).
Statistical analysis
Median and range are used to express data. To determine differences due to intervention time and treatment, we used a general linear model for repeated measures. For time and treatment × time, p-Values were calculated with LSD post hoc multiple comparisons with different letters indicating significant differences (p < 0.05).
Data regarding functional pathways profiles and SCFAs are presented as a mean and standard mean error (SEM). A statistically significant difference was considered to be p < 0.05. In the case of variables that were not normally distributed, they were log-transformed, and/or outliers were removed (without achieving a loss of value from samples of up to 15%). To ensure a clear understanding of the data, the values are presented untransformed. To assess differences at baseline in the relative abundance of bacteria (phylum and genus), as well as for the alpha indexes and beta diversity, the Mann–Whitney U test was applied. When testing differences in beta diversity, we used ADONIS-2 function from the vegan package using 10,000 permutations for calculating p-Values.
The Kruskal–Wallis test was used to analyze the differential relative abundance of general metabolic pathways. To correct for multiple comparisons, the Benjamini–Hochberg correction (FDR) was applied. GraphPad Prism 8 was used to create all metabolic pathway images. Unless otherwise specified in the figure legend, data are presented as mean ± SEM. As indicated in the respective figure legend, statistical significance was determined by a one-way ANOVA, followed by Tukey’s multiple comparison test. Differences reached statistical significance with p < 0.05. At the end of the intervention, Pearson’s correlations were used to examine relationships between diversity indices, microbiome variables, anthropometric, inflammation, metabolic parameters, serum bile acids, and fecal SCFA levels. Multiple testing was corrected by applying the FDR procedure (35). Using the corrplot function in R studio, associations were expressed. The graph shows only significant and corrected associations (36). Correlation values were shown within the graphs in red and blue colors, the negatively correlated in red (−1) and the positively correlated in blue (+1).
Results
In this study, we observed the replacement of rapidly digestible CHO for ISR within an HFD-promoted change in the gut microbiota composition. Table 2 represents the phylum-based abundances of Verrucomicrobia (newly named as Verrucomicrobiota), Pseudomonadota, Bacillota, Deferribacteres (newly named as Deferribacterota), Bacteroidota, and Actinobacteria (newly named as Actinobacteriota) in the NOB, OBE, and OBE-ISR at the baseline and the end of the treatment. Indeed, the OBE-ISR group showed significant changes in Bacteroidota, Actinobacteriota, Bacillota, and Pseudomonadota. At the phylum level, the relative abundance was similar to groups at the baseline. Based on the interaction time × treatment after 4 weeks, bacteria from the OBE-ISR group showed a decrease in Fisher’s index (p = 0.004) and Bacillota (p = 0.024) and an increase in Pielou’s evenness (p = 0.004) and Bacteroidota (p = 0.002) (Table 2). At the genus level, the groups had comparable gut microbiota at baseline. After 4 weeks of treatment, OBE and OBE-ISR groups had a higher proportion of sequences in the Alistipes genus than NOB group (p = 0.023). The OBE-ISR group exhibited an increase in the relative abundances of the genera Bifidobacterium (p = 0.045), Bacteroides (p = 0.001), Butyricimonas (p = 0.023), cc (p < 0.001), Flavonifractor (p = 0.003), Ruminiclostridium 5 (p = 0.039), and Faecalibaculum (p = 0.009). On the contrary, the OBE-ISR group revealed a decrease in the relative abundances of the genera Muribaculum (p = 0.01), Blautia (p = 0.024), and Ruminiclostridium 9 (p = 0.002). Interestingly, relative abundances of genera Tyzzerella (p = 0.006) and Angelakisella (p = 0.003) increased in the OBE group compared to NOB and OBE-ISR groups, and the OBE-ISR group exhibited similar levels to NOB group at the end of the intervention (Table 3). Remarkably, we observed that both Fisher’s alpha and species richness abundance in the OBE-ISR group were reduced compared to NOB and OBE groups at the end of intervention after 4 weeks (Figure 1 and Table 4). The visualization of weighted (ADONIS p = 0.001, r2 = 0.495) and unweighted (ADONIS p = 0.001, r2 = 0.429) UniFrac distance data demonstrated that fecal samples from the OBE-ISR group after 4 weeks of nutritional intervention were separated from those of NOB and OBE groups (Figure 2).
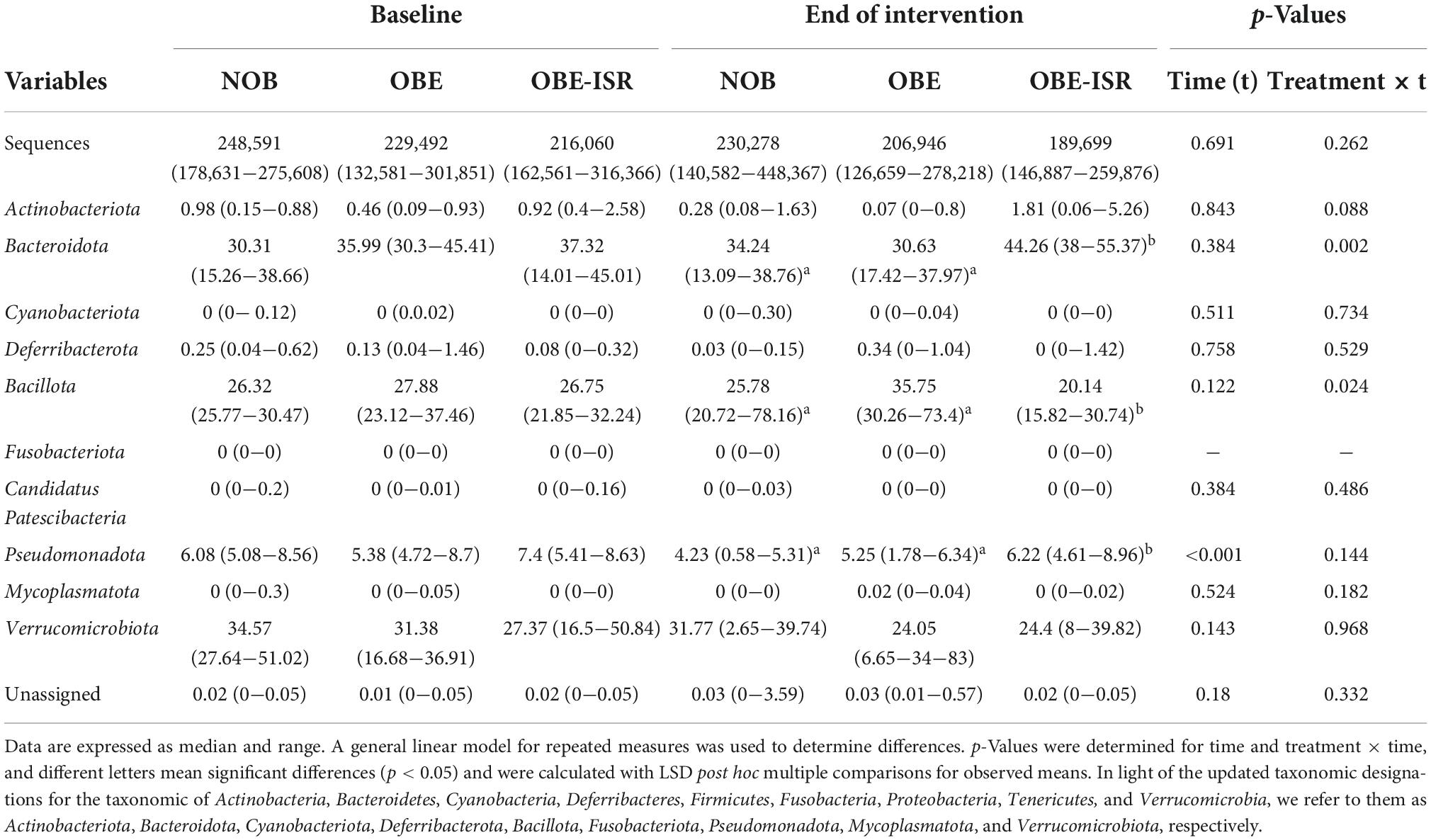
Table 2. Relative abundances at the phylum level.
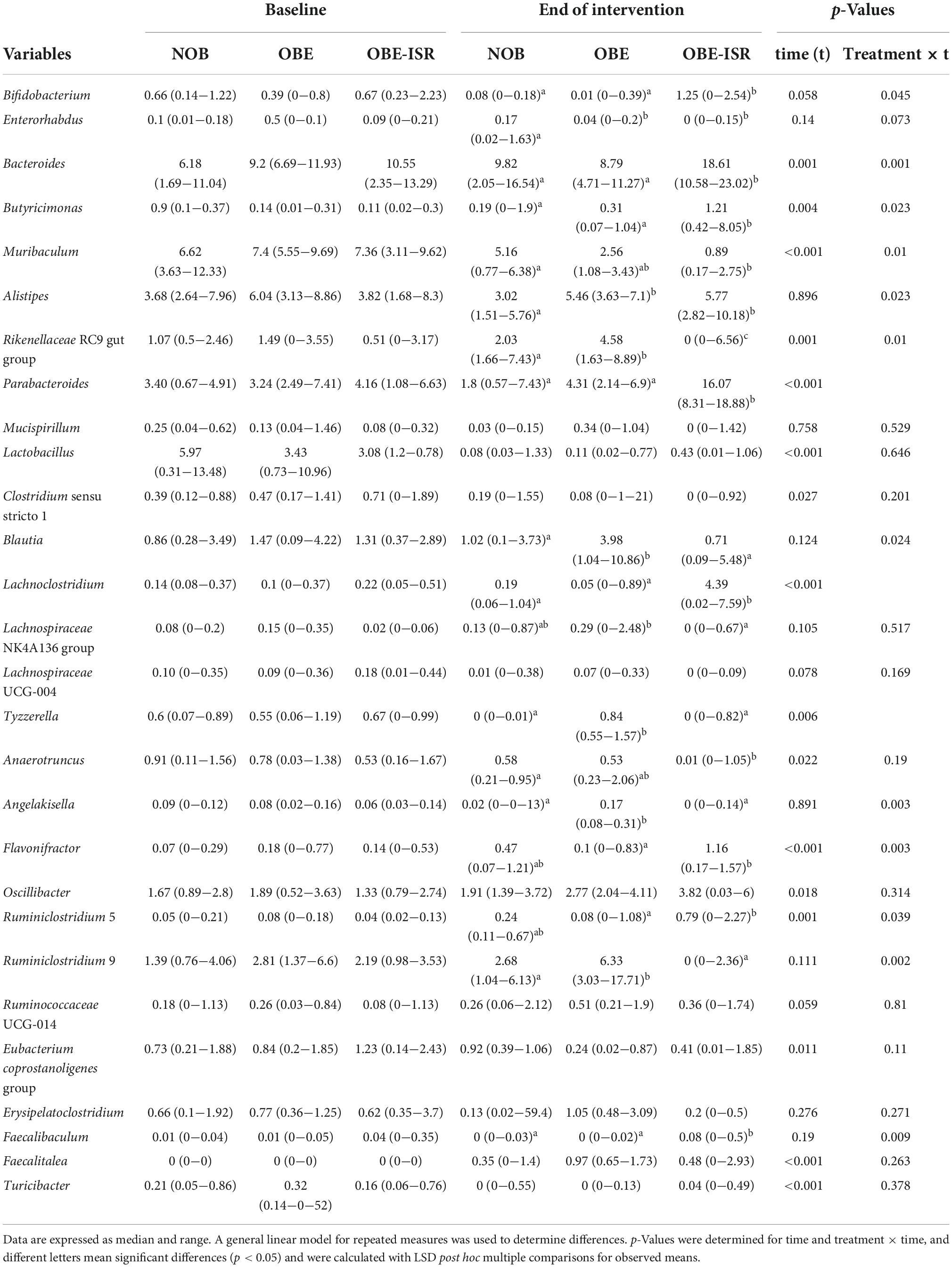
Table 3. Relative abundances at the genus level.
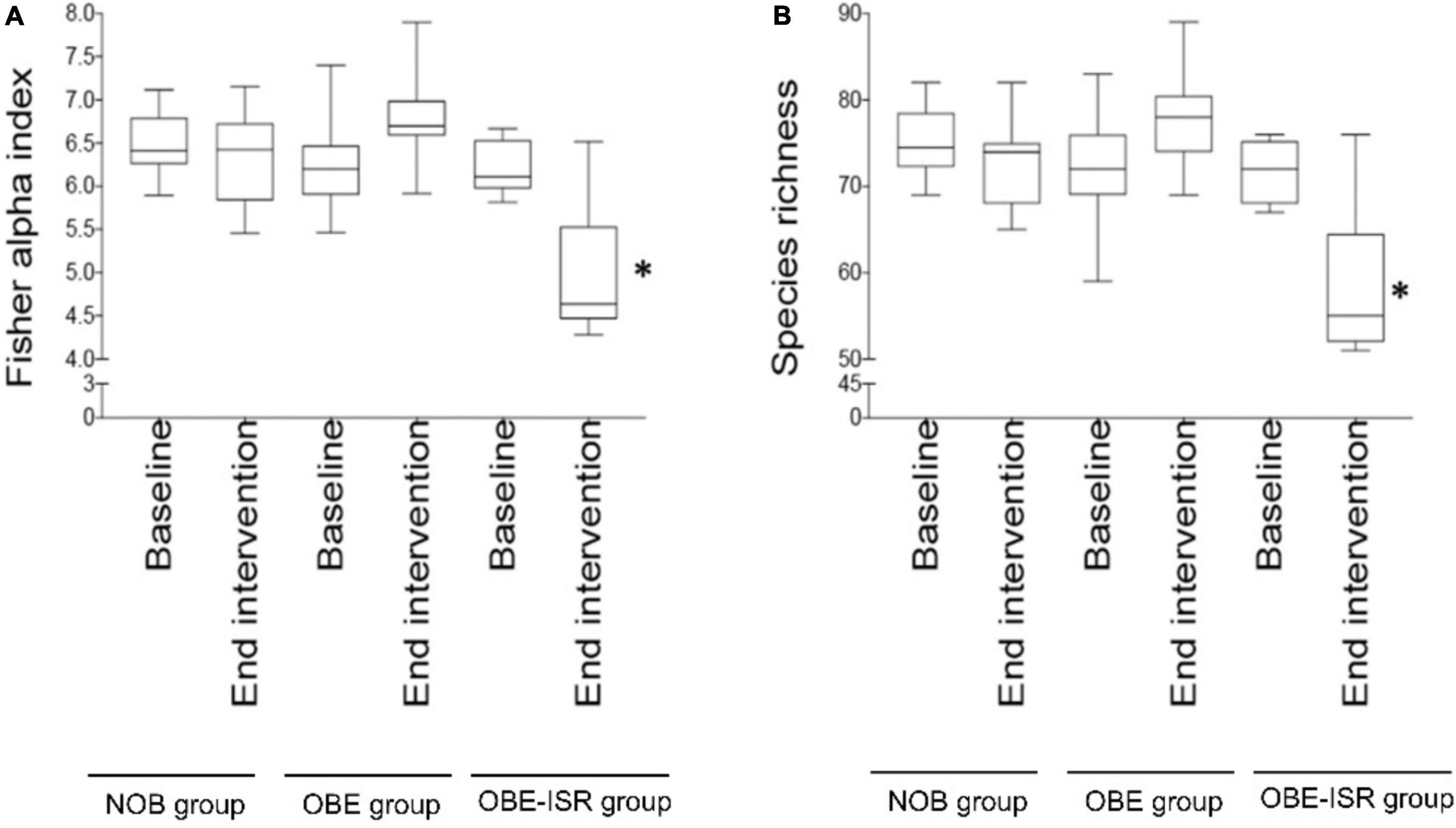
Figure 1. Diversity indices across the intervention. (A) Fisher’s alpha index abundance in the non-obese (NOB), obese (OBE), and obese with slowly digestible CHO (OBE-ISR) groups at the baseline and at the end of intervention after 4 weeks. (B) Species richness abundance in the non-obese (NOB), obese (OBE), and obese with slowly digestible CHO (OBE-ISR) groups at the baseline and at the end of intervention after 4 weeks. *p < 0.05.
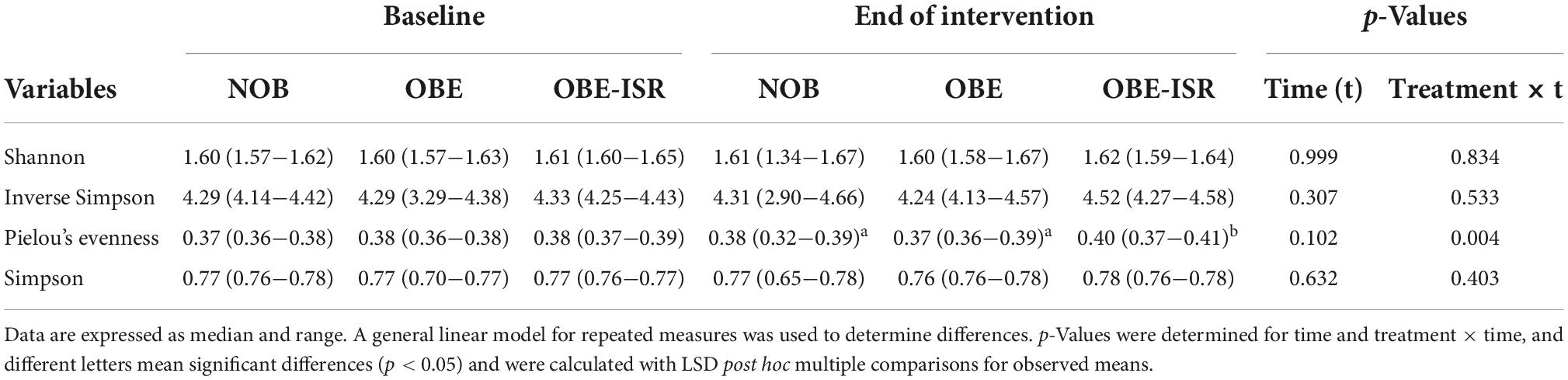
Table 4. Alpha indexes.
Figure 2. (A) Weighted UniFrac distances (ADONIS p = 0.001, r2 = 0.495). (B) Unweighted UniFrac distances (ADONIS p = 0.001, r2 = 0.429). Beta diversity is represented by principal coordinate analysis (PCoA) based on unweighted UniFrac distance.
At the end of the experiment, we observed significant increases in the activity of the different metabolic pathways in the OBE-ISR group, including glycolysis with the Entner–Doudoroff pathway, glucuronic acid degradation, D-galacturonate degradation, the super pathway of N-acetylglucosamine, N-acetylmannosamine and N-acetylneuraminate degradation, and the pentose phosphate pathway (PPP) (Figure 3). Moreover, key metabolic pathways such as methanogenesis, methanogenesis from acetate, fatty acid biosynthesis pathway, L-tyrosine degradation, D-glucarate, and tricarboxylic acid cycle (TCA) degradation were significantly enhanced in the more abundant bacteria isolated from the ISR-treated group after 4 weeks of the treatment (Figure 4).
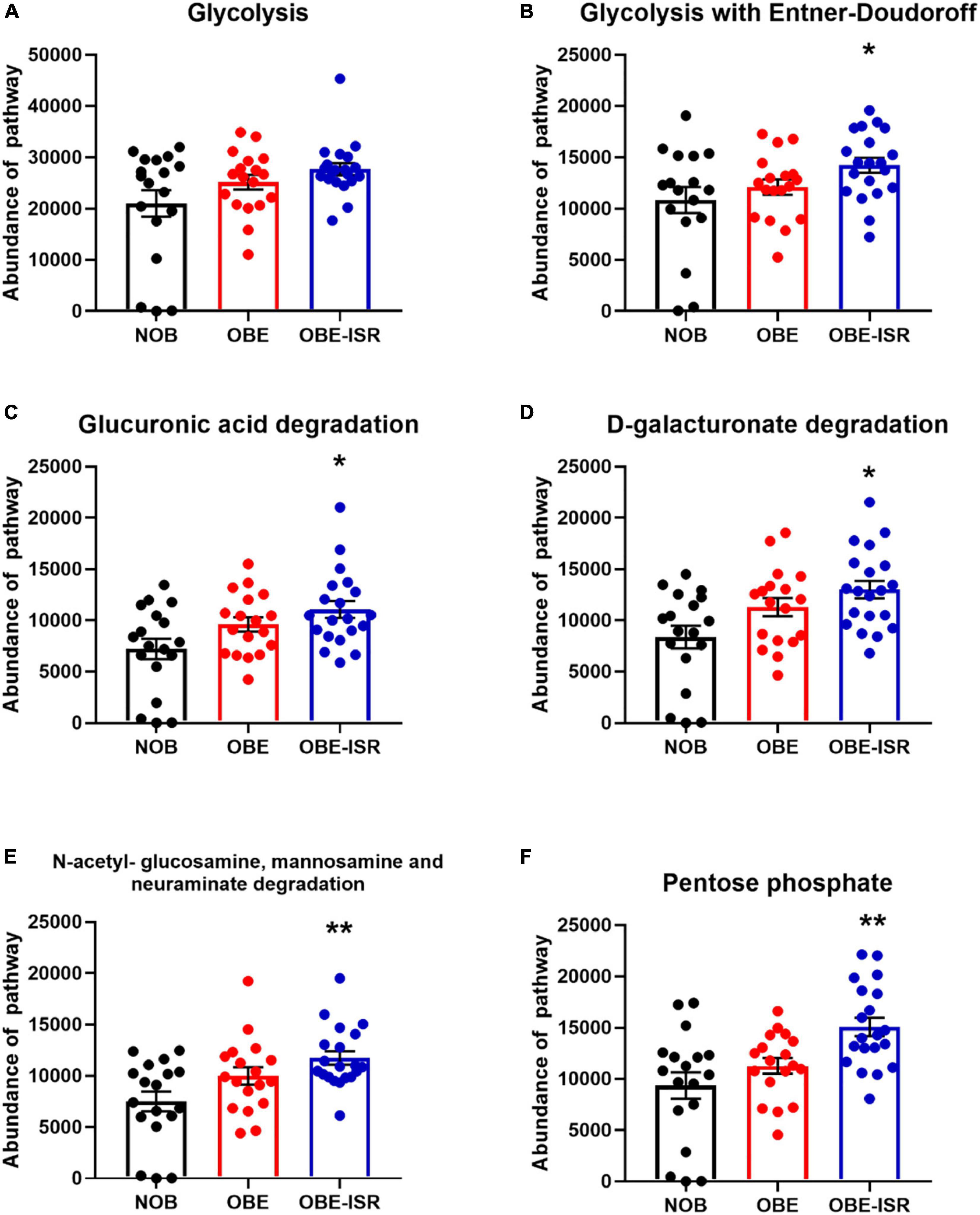
Figure 3. Major metabolic pathway categories relative to the abundance of each sample, including non-obese (NOB), obese (OBE), and experimental treatment slow-digesting carbohydrates OBE-ISR. (A) Glycolysis: Glycolysis I (from glucose-6-phosphate). (B) Glycolysis with Entner–Doudoroff: superpathway of glycolysis and the Entner–Doudoroff pathway. (C) Glucuronic acid degradation: Glucurocat pathway. (D) D-galacturonate degradation II: Galacturocat-pwy. (E) N-acetylglucosamine, mannosamine, and neuraminate degradation: Glcmannanaut-pwy, superpathway of N-acetylglucosamine, N-acetylmannosamine, and N-acetylneuraminate degradation. (F) Pentose phosphate pathway. *p < 0.05; **p < 0.01.
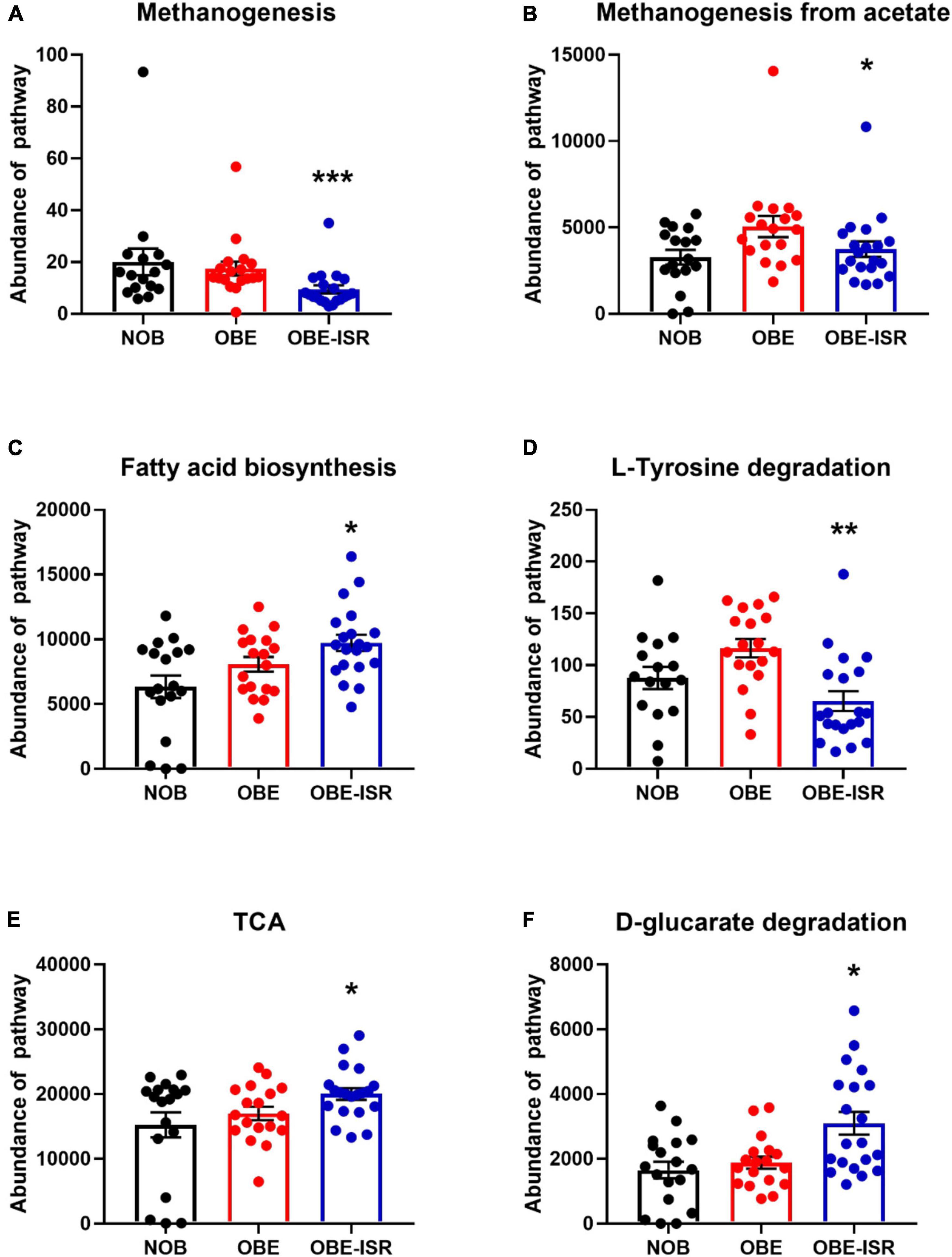
Figure 4. Major metabolic pathway categories relative to the abundance of each sample, including non-obese (NOB), obese (OBE), and experimental treatment of obese with slow-digesting carbohydrates OBE-ISR. (A) Methanogenesis-pwy, superpathway of methanogenesis. (B) Methanogenesis from acetate: Meth-acetate-pwy. (C) Fatty acid biosynthesis: superpathway of fatty acid biosynthesis initiation (Escherichia coli). (D) L-tyrosine degradation I: TYRFUMCAT-PWY. (E) TCA: Tricarboxylic acid cycle (prokaryotic). (F) D-glucarate degradation I. *p < 0.05; **p < 0.01.
In addition, we evaluated the fecal levels of SCFAs in these experimental groups. We observed that the OBE-ISR group had the highest levels of acetate and propionate, being statistically different from the other two groups NOB and OBE. However, no differences in butyrate levels were observed (Table 5). In addition, Pearson’s correlations between anthropometric, physiological, inflammatory and metabolic traits, and bacterial variables and serum levels of bile acids revealed some associations in NOB, OBE, and OBE-ISR groups (Supplementary Tables 1–3).
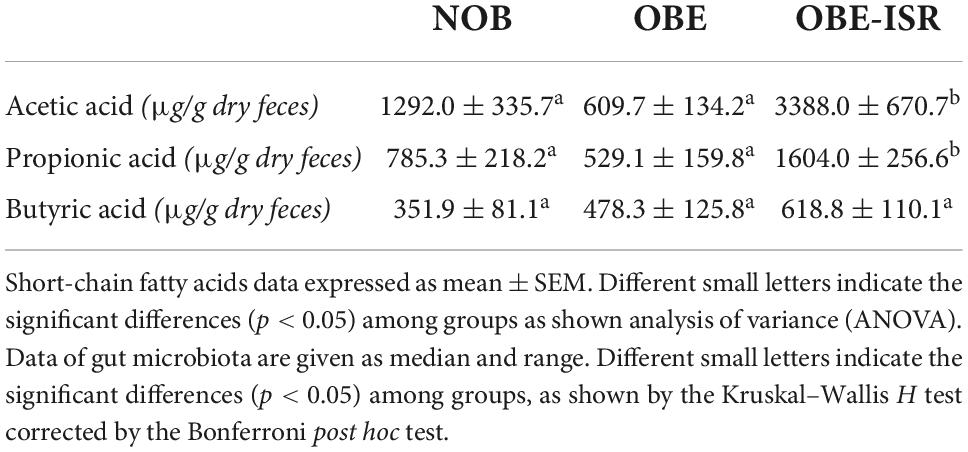
Table 5. Short-chain fatty acid (SCFA) production of experimental groups fed on the different experimental diets for 4 weeks to induce obesity.
Body weight was negatively correlated with Butyricimonas and Ruminiclostridium 5 in the NOB group. In the case of primary bile acids, which are taurocholic acid and glycocholic acid (derivatives of cholic acid) and taurochenodeoxycholic acid and glycochenodeoxycholic acid (derivatives of chenodeoxycholic acid) (37), and SCFA, in the NOB group, we observed that chenodeoxycholic acid exhibited a positive association with Muribaculum; glycocholic acid was positively correlated with Bifidobacterium; glycochenodeoxycholic acid was negatively correlated with Blautia; and Ruminiclostridium 5 was positively associated with taurocholic and taurodeoxycholic acids. Furthermore, TG (50:1, glycerolipids. Subclass: Triacylglycerols). Composition: TG (16:0 + 18:1 + 16:0) was negatively associated with Bacteroidota. PE (16:0/20:4, glycerophospholipids. Subclass 1-ether, 2-acylglycerophosphoethanolamine) was negatively correlated with the Rikenellaceae RC9 gut group, and acetic and propionic acid levels showed a negative association with methanogenesis and L-tyrosine degradation. Finally, acetic acid was positively associated with Butyricimonas (Supplementary Table 1). On the contrary, we found that GLP-1 and IL-1β were positively correlated with the Rikenellaceae RC9 gut group and Blautia in the NOB group, respectively. Bacteroides exhibited a negative correlation with glycolysis with the Entner–Doudoroff pathway, glucuronic acid degradation, D-galacturonate degradation, the super pathway of N-acetylglucosamine, N-acetylmannosamine and N-acetylneuraminate degradation, and PPP. In the same way, Flavonifractor was negatively associated with D-galacturonate degradation, methanogenesis from acetate, and the fatty acid biosynthesis pathway (Supplementary Table 1).
The OBE group revealed that taurodeoxycholic acid was negatively associated with Butyricimonas and positively associated with GLP-1 levels. Besides, Blautia showed a negative association with D-glucarate degradation (Supplementary Table 2).
In the case of the OBE-ISR group, we have observed that cholic acid exhibited a positive association with Faecalibaculum and glucuronic acid degradation, and chenodeoxycholic acid with glucuronic acid degradation, PPP, and Muribaculum. Ursodeoxycholic and hyodeoxycholic acids were highly correlated with glucuronic acid degradation, Bacteroidota, Muribaculum, and Faecalibaculum. Glycocholic acid was positively associated with methanogenesis, methanogenesis from acetate, and L-tyrosine degradation. Moreover, deoxycholic acid was positively associated with Butyricimonas, Ruminiclostridium 5, and Faecalibaculum. Glycochenodeoxycholic acid was positively correlated with methanogenesis. Bacteroidota showed a positive association with taurocholic and taurodeoxycholic acids, and the last one also with glucuronic acid degradation. PE (16:0/20:4) was correlated negatively with Bifidobacterium, Butyricimonas, and Faecalibaculum. Butyric acid was positively associated with Ruminiclostridium 5. On the contrary, acetic and propionic acids showed a negative association with TCA (Supplementary Table 3).
We also found that glucagon-like peptide-1 (GLP-1) was negatively associated with gut microbiota Bacteroides whereas TNF-α and IL-1β were positively correlated with Ruminiclostridium 5 in the ISR group. In addition, Bacillota and Ruminiclostridium 9 were positively related to the super pathway of N-acetylglucosamine, N-acetylmannosamine, and N-acetylneuraminate degradation, as well as Bacillota with galacturonate degradation and PPP. Finally, Flavonifractor was negatively correlated with L-tyrosine degradation in the ISR group (Supplementary Table 3).
Discussion
It has been proposed that gut microbiota is an integral component of human health. However, there is little information available regarding the composition of the gut microbiota during childhood. In spite of the suggestion that the microbiota reaches a relatively stable state in the first 3 years of life, other evidence indicates that it continues to develop during adolescence (38). In several studies, gut microbes and their hosts have been shown to interact metabolically, and dietary factors have a direct impact on the composition of the gut microbiota, which is thought to regulate systemic metabolism (39). For the development and growth of a child, CHO is one of the most significant sources of energy (40). Nonetheless, the quality of CHO is crucial for optimal glycemic control, insulin response, and weight management (13, 41, 42). In that context, we have previously reported that the OBE-ISR group improves insulin sensitivity and reduces dyslipidemia in growing rats, as well as GLP-1 augmented levels (19). Using a well-established HFD rodent model of childhood diet-induced obesity, we evaluated the effects of a specialized CHO diet on the gut microbiota composition by comparing its quality rather than its quantity (rapid digestible vs. slowly digestible CHO). As demonstrated here, the consumption of ISR within an HFD improved the composition of the gut microbiota as well as the functional pathway profile in obesity-growing rats, thereby potentially protecting against the development of metabolic disturbances.
Inflammation and decreased insulin sensitivity are linked with obesity and changes in fecal microbiota (43). Studies suggest that maintaining or restoring a healthy gut ecosystem may help to prevent the early onset of obesity, from a young age (44). There is evidence that insulin resistance in children is connected with a number of Pseudomonadota species, as well as a lower abundance of gram-negative bacteria and bacteria involved with butyrate production (45). It has been hypothesized that obese children have significantly higher Bacillota and lower Bifidobacterium than lean children (46). However, a meta-analysis of the gut microbiome revealed no correlation between these two phyla and obesity (47). As a result, there is a tendency for less bacterial diversity (4) and a reduced abundance of Prevotella (48). Both adults and children are affected by obesity, which results in changes in microbial diversity. In this matter, the causal relationship is still controversial; however, it is well-known that weight loss in people with obesity recovers gut microbiota composition toward a healthy status or similar to lean individuals (49).
Children who consume a Western-style diet have a more prevalent presence of Bacteroidaceae and Ruminococcaceae, as well as a lower presence of Prevotellaceae (50). The composition of the gut microbiota has been influenced by the consumption of certain types of fiber since early childhood. Human milk oligosaccharides and prebiotics added to infant formula are bifidogenic and inhibit pathogen growth. In addition, inulin increases Bifidobacterium abundance, while dietary fiber decreases fecal pH and increases alpha diversity and calcium absorption (50).
The use of rats after weaning may be able to mimic childhood conditions. After weaning, rats given an HFD developed many of the characteristics of the metabolic syndrome such as central obesity, systolic and diastolic hypertension, altered fasting glucose levels, hypertriglyceridemia, and decreased HDL cholesterol levels (51). During the present study, the nutritional intervention lasted for 4 weeks from weaning to infancy. Here, we observed that rats fed HFD supplemented with the ISR exhibited significant changes in Bacteroidota, Actinobacteriota, Bacillota, and Pseudomonadota, indicating an improvement of the gut microbiota profile disturbed by rapid digestible CHO/HFD-induced obesity. Indeed, obesity per se drives a lower relative abundance of Bacteroidota and reduced biodiversity in the gut microbiota (52). The interaction time × treatment showed that after 4 weeks of treatment, bacteria in the OBE-ISR group showed a decrease in Bacillota abundance, a decrease in Fisher’s index, and an increase in Pielou’s evenness and Bacteroidota, restoring the microbiota composition to a more “lean” or “healthy” state.
At the genus level, treatment of ISR after 4 weeks revealed a higher proportion of Alistipes compared to the NOB group, which might be a bacterial genus of particular interest in the field of obesity since Alistipes is a potential SCFA producer and it might play a role in reducing non-alcoholic steatohepatitis and/or liver fibrosis (53, 54).
Indeed, we observed a reduced hepatosteatosis in the OBE-ISR group with lower GLUT2, FAS, and SRBP1 (19). In addition, gut microbiota from the OBE-ISR group showed increased relative abundances of the genera Bifidobacterium, Bacteroides, Butyricimonas, Lachnoclostridium, Flavonifractor, Ruminiclostridium 5, and Faecalibaculum, indicating an improvement of these genera. Oligosaccharides such as FOS or inulin stimulate the growth of Bifidobacterium in the colon, reducing iron intake by enteric pathogens in children (55) and a rise in Bifidobacterium and Bacteroides in rats (14). It has been reported that the genus Butyricimonas is increased in overweight and obese individuals on a low-calorie weight loss diet with fiber content (56). Lachnoclostridium, a genus altered by calorie restriction (57), is more pronounced in response to non-digestible oligo- and polysaccharides in children with overweight (16). Regarding the genus Flavonifractor, which also exhibited higher relative abundance in the OBE-ISR group after 4 weeks of the treatment, it has been stated to be negatively correlated with body fat content and distribution in the pediatric population (58). Moreover, Flavonifractor genera may be associated with the lower fat deposition observed in the rats (19). Though the genus Ruminiclostridium 5 is known to produce SCFAs in the colon (59), it has been linked to obesity and cardiometabolic traits in children with normal weight and obesity, showing that the relative abundance of Ruminiclostridium 5 is associated with obesity and fasting plasma insulin (60). The relative abundance of genus Faecalibaculum is augmented in HFD feeding in mice (61) and in the galactooligosaccharides (GOS)-supplemented diet in rats (62), indicating that dietary prebiotics such as GOS may shift the gut microbiota composition to a more healthy status.
Regarding the effects on SCFAs, there was a significant increase in the levels of acetate and propionate in the ISR group, indicating a putative improvement of the intestinal barrier dysfunction and immune response (63, 64).
On the contrary, the OBE-ISR group revealed a decrease in the relative abundances of the genera Muribaculum, Blautia, and Ruminiclostridium 9. The results of a recent metatranscriptomic analysis of colonic microbiota are consistent with our findings; dietary fibers rich in inulin, among others, decreased Muribaculum genus in growing pigs (65), providing new insight into the effects of dietary fibers on animal health. There is, however, evident that the genus Blautia is more common in diets high in soluble fibers, which are beneficial to glucan fermentation, compared with rats fed dietary fiber derived from barley malts, brewer’s spent grain, or barley extracts (containing significant amounts of β-glucan, soluble arabinoxylan, and insoluble arabinoxylan) (66). In the case of Ruminiclostridium 9, a study revealed that consumption of foods containing type 2 resistant starch, in the context of HFD-fed rats, decreased the abundance of pathogen taxa associated with obesity, inflammation, and aging including Ruminiclostridium 9. In aged mice on HFD, modulating microbiota and metabolites improved systemic inflammation and intestinal permeability (67). Interestingly, relative abundances of genera Tyzzerella and Angelakisella augmented in the microbiota from the OBE group compared to NOB and OBE-ISR groups, establishing that the ISR group restores the abundance of these genera to the same levels as the NOB group (68).
Dietary fiber contributes to the diversity of the microbiome by providing a variety of substrates for fermentation reactions carried out by those microbes that possess the necessary enzymatic machinery to degrade these complex CHO (69). Despite the fact that dietary fibers usually drive greater species richness, it should be noted that the OBE-ISR group underwent an HFD diet, which has been linked to obesity, insulin resistance, and dyslipidemia, as well as reduced gut bacterial communities (43), including Bacteroidota groups (70). The Western diet or HFD has profound effects on the diversity and populations of gut microbiota, increasing the relative abundance of Bacillota and decreasing the abundance of Bacteroidota (71). Although the OBE-ISR group revealed a lower alpha diversity after the treatment, we should note that we observed an enrichment of beneficial bacteria such as Bifidobacterium and SCFA-producing bacteria. In line with this, it has been published that the richness of gut microbiota could be reduced in nutritional interventions based on ingredients that modulate microbiota (e.g., berberine) (72). Indeed, a lower alpha diversity does not necessarily mean better protection for the host. This is well-known in infants exclusively breastfed where a predominant bifidobacteria microbiota reduces its diversity decreasing the counts of potential bacterial pathogens. In addition, beta diversity represents how much the community changes between the groups. Weighted UniFrac takes into account the relative abundance of species/taxa shared between samples, while unweighted UniFrac only considers the presence or absence. Thus, the visualization of weighted and unweighted UniFrac distances data demonstrated that fecal samples from the OBE-ISR group at the end of 4 weeks of intervention were separated from those from NOB and OBE groups, indicating a significant shift in the relative abundance of bacteria in the OBE-ISR group after the intervention.
Beyond taxonomic composition, we conducted functional profiling of gut microbiota to identify metabolic pathways involved in the effects of ISR within an HFD feeding. This was done at the end of the treatment. In addition, we observed an increased number of bacteria associated with glycolysis, including the Entner–Doudoroff pathway, glucuronic acid degradation, D-galacturonate degradation, the super pathway of N-acetylglucosamine, N-acetylmannosamine and N-acetylneuraminate degradation, as well as PPP, indicating a higher activity of CHO metabolism pathways. As part of the metabolic pathway parallel to glycolysis known as the PPP (also referred to as phosphogluconate pathway and hexose monophosphate shunt), it maintains carbon homeostasis, provides precursors to nucleotide and amino acid biosynthesis, reduces molecules for anabolism, and combats oxidative stress (73). Likewise, the Entner–Doudoroff pathway is present in several bacteria in which it may be the main pathway of glucose catabolism under aerobic conditions and represents an offshoot of the oxidative branch of the PPP, generating NADPH from the oxidation of glucose-6-phosphate (74), and it is used in many biosynthetic reactions. We detected increased activity in the PPP, and fatty acid biosynthesis and TCA pathways in the ISR gut microbiota at the end of the treatment. This indicates that its microbiota metabolizes glucose and other sugars for energy production. The Entner–Doudoroff pathway is present in some gram-negative bacteria such as Pseudomonas, Rhizobium, and Agrobacterium, but not in gram-positive bacteria, with the exception of Enterococcus faecalis (73). In fact, we found that Bacteroides are negatively associated with glycolysis with the Entner–Doudoroff pathway, glucuronic acid degradation, D-galacturonate degradation, the super pathway of N-acetylglucosamine, N-acetylmannosamine and N-acetylneuraminate degradation, and PPP in the OBE group. Moreover, Flavonifractor was also negatively associated with D-galacturonate degradation, methanogenesis from acetate, and the fatty acid biosynthesis pathway in OBE group, and no associations were observed in either NOB or OBE-ISR group, confirming that the obesity group exhibited lower activity of these metabolic pathways.
Pellock and Redinbo (75) point out that gastrointestinal microbiota produces β-glucuronidase enzymes that remove glucuronic acid as a carbon source, as a result of which the effects of mammalian inactivation can be effectively reversed (75). Glucuronic acid enters the Entner–Doudoroff pathway and shunts the resulting pyruvate into the TCA cycle (76). The ISR exhibited increased glucuronic acid degradation pathway activity after 4 weeks, indicating an increased abundance of substrate for both the Entner–Doudoroff pathway and TCA, which exhibited higher activity in the group as was abovementioned. Similarly, D-galacturonate degradation activity is significantly higher in the OBE-ISR group. A key component of pectin D-galacturonate, which can be converted to pyruvate and glyceraldehyde-3-phosphate via 2-keto-3-deoxy-phosphogluconate, a characteristic intermediate in the Entner–Doudoroff pathway for sugar dissimilation (77).
N-acetylglucosamine, N-acetylmannosamine, and N-acetylneuraminate are convergent pathways for utilizing amino sugars. Both N-acetylglucosamine and N-acetylneuraminate are effective carbon sources for Escherichia coli, whereas N-acetylmannosamine is metabolized very slowly (78). Here, our treatment with ISR in rats fed HFD exhibits higher activity in these pathways compared to OBE and NOB groups, which may help to maintain the barrier function in the intestine (79).
On the contrary, key metabolic pathways such as methanogenesis, methanogenesis from acetate, and L-tyrosine degradation revealed significantly lower abundance in the ISR-treated group after 4 weeks of the treatment. D-glucarate degradation exhibited higher activity in the ISR-treated group after 4 weeks of the treatment, indicating a putative higher metabolic rate in the CHO metabolism. Of interest, D-glucarate is an effective antitumoral agent able to bind to environmental carcinogens like benzo[a]pyrene (80, 81). Methanogenesis pathway promotes the removal of gaseous fermentation by-products, carbon dioxide and hydrogen, from the distal gut ecosystem converting carbon dioxide plus hydrogen to methane by methanogenic Archaea (82, 83). Rather than host absorption, these substrates are utilized primarily by cross-feeding between gut microbiota members (84).
Bacteria degrade L-tyrosine mainly through the production of phenolic compounds. Here, ISR treatment decreased significantly the L-tyrosine degradation pathway in the context of HFD feeding in rats. A major microbial metabolite derived from tyrosine is phenol (phenol and p-cresol) (85). A high level of phenol is associated with chronic kidney disease, cardiovascular disease, and altered cellular immune responses (86). Recent research has identified 36 phenol-producing bacteria and 55 p-cresol-producing bacteria; strong phenol producers were found in the Coriobacteriaceae, Enterobacteriaceae, Fusobacteriaceae, and Clostridium clusters I and XIVa, and strong p-cresol producers were found in the Coriobacteriaceae and Clostridium clusters XI and XIVa (87). As a result, ISR could contribute to a reduction in the production of phenols by inhibiting the degradation of L-tyrosine and assist in identifying the relationship between microbiota and host disease.
In hepatocytes, primary bile acids, cholic and chenodeoxycholic acids, are synthesized from cholesterol. Those acids are normally coupled with glycine to produce glycocholic acid and glycochenodeoxycholic acid, and taurine to produce tauro-conjugates (37). The total pool of conjugated bile acids is dominated by glycine-conjugated bile acids (88). These acids are collected in the gallbladder and liberated into the intestine to facilitate absorption of dietary lipids and liposoluble vitamins, protection against bacterial overgrowth, and elimination of cholesterol from the body (88). In the distal ileum, approximately 95% of the bile acids are reabsorbed and return to the liver through the enterohepatic circulation (89). By the action of specific bacterial enzymes, the remaining 5% of primary bile acids are metabolized into the secondary bile acids, such as deoxycholic acid, lithocholic acid, and ursodeoxycholic acid (90).
Pearson’s correlations, adjusted using the FDR procedure, reported specific associations between bile acids and SCFA with microbial populations and metabolic pathways in NOB and OBE-ISR groups. However, no significant changes were observed in the OBE group. Here, we found associations of several bile acids with some phylum and some genera in the OBE-ISR group. The main genera implicated were Butyricimonas, Ruminiclostridium 5, Faecalibaculum, and Bacteroidota. In addition, butyric acid was positively associated with Ruminiclostridium 5. Similar results have been found previously with the same genus (91), supporting a new association between the changes in Ruminiclostridium 5 and the concomitant levels of butyric acid. A study in a healthy population has reported that Butyricimonas was correlated with a body mass index < 25 after the consumption of the Mediterranean diet. Likewise, adherence to the Mediterranean diet was associated with augmented levels of SCFAs (92). According to our study, the Butyricimonas genus is related to the bile acid levels, and according to recent reports, the genus is related to the body mass index in healthy volunteers. These associations in the OBE-ISR group between Butyricimonas and Ruminiclostridium 5 should be highlighted. This is because the NOB group showed a significant association between body weight and the same bacterial genera.
There are two dominant phyla in the gut microbiota, Bacillota and Bacteroidota (93). There is an opposite trend in the abundance of Bacteroidota with the elderly having an abundance 80% lower than children (94). The levels of Bacteroidota were significantly elevated in the ISR group, and they were positively associated with taurocholic and taurodeoxycholic acids.
In the case of GLP-1, we previously reported augmented levels for the OBE-ISR group (19), and in our study, we have found a negative association with Bacteroides, which are prevalent in the gut of individuals living with high-fat and protein diets (95, 96). It appears that Bacteroides spp. play a key role in the immunomodulation of the human immune system (96). In our study, Bacteroides levels are elevated at the end of the intervention in the OBE-ISR group, which may be related to GLP-1 secretion, which in turn regulates insulin secretion, appetite, and body weight (97–100).
The number of basic studies addressing the analysis of microbial, anthropometric, metabolic, and inflammation data is not enough. The need for general view analyses is currently an imperative issue in the nutrition field. Consequently, recent studies using a similar methodology have reported an increase in beneficial bacteria in the colonic microbiota, as well as increased activity of enzymes involved in liver lipid metabolism (101). Further studies with similar and more advanced methodologies are necessary.
Conclusion
In conclusion, we show that consumption of ISR within an HFD feeding in growing rats as a model of childhood obesity modifies the gut microbiota composition and provokes a shift in the functional pathway profile, which might have a protective effect against the development of metabolic disturbances in obesity (Figure 5). In addition, the increase in SCFAs resulting from ISR seems to be of relevance, since SCFAs have been shown to contribute to dietary energy harvest, modulate host adiposity, and alter the expression of host satiety hormones (102, 103). All together our results suggest that the ISR mix could preserve the gut microbiota from the dysbiosis resulting from obesity during childhood.

Figure 5. Simplified diagram of the main microbial metabolic pathways affected by HFD-ISR diet in the context of HFD. ISR containing isomaltulose, sucromalt, maltodextrin, resistant starch, inulin, and fructooligosaccharides rises glucose and fructose in the intestine, which it promotes the Entner–Doudoroff pathway, and pentose phosphate and glycolysis pathways by gut microbiota, increasing the pyruvate production and bacterial proliferation. Pyruvate increases the short-chain fatty acid (SCFA) production (butyrate, acetate, and propionate) and decreases the methanogenesis. Hence, the elevated SCFA promotes the carbohydrates fermentation and decreases the protein fermentation, lowering the L-tyrosine production, which may potentially reduce the p-cresol and phenol in the intestine. 2-keto-3-deoxy-phosphogluconate (KDPG); glyceraldehyde 3-phosphate (GAP); tricarboxylic acid (TCA) cycle. Black arrows represent the analyzed metabolic pathways. Gray arrows represent the putative products affected by the metabolic pathways according to the literature. SI, Small intestine.
Data availability statement
The data presented in this study are deposited in the National Center for Biotechnology Information (NCBI) repository, accession number Bioproject PRJNA866156.
Ethics statement
The animal study was reviewed and approved by the animal study was reviewed and approved by the Consejería de Agricultura, Ganaderia, Pesca y Desarrollo Sostenible, Junta de Andalucia, Spain.
Author contributions
MM, MTG-C, RR, and JML-P did the conceptualization and supervised the data. MM, RS, MDG, and ÁG carried out the data curation and investigated the data. JP-D, FR-O, AS-F, and MTG-C carried out the funding acquisition. RS, MDG, JML-P, and MM wrote the original draft. RS, MDG, RR, MM, JML-P, JP-D, FR-O, and ÁG wrote, reviewed, and edited the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by funds provided by the Abbott Laboratories S.A. JP-D was supported by a fellowship awarded to postdoctoral researchers at foreign universities and research centers from the “Fundación Ramón Areces”, Madrid, Spain. JP-D was part of the “UGR Plan Propio de Investigación 2016” and the “Excellence actions: Unit of Excellence on Exercise and Health (UCEES), University of Granada.” The funder had the following involvement with the study: design of the study and collection of the samples.
Conflict of interest
Authors MM, JML-P, AS-F, and RR are employees at the Abbott Laboratories S.A.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnut.2022.992682/full#supplementary-material
References
1. Huang R, Ju Z, Zhou PK. A gut dysbiotic microbiota-based hypothesis of human-to-human transmission of non-communicable diseases. Sci Total Environ. (2020) 745:141030. doi: 10.1016/j.scitotenv.2020.141030
2. Scully T, Ettela A, Leroith D, Gallagher EJ. Obesity, type 2 diabetes, and cancer risk. Front Oncol. (2020) 10:615375. doi: 10.3389/fonc.2020.615375
3. De Bandt JP, Waligora-Dupriet AJ, Butel MJ. Intestinal microbiota in inflammation and insulin resistance: relevance to humans. Curr Opin Clin Nutr Metab Care. (2011) 14:334–40. doi: 10.1097/MCO.0b013e328347924a
4. Magne F, Gotteland M, Gauthier L, Zazueta A, Pesoa S, Navarrete P, et al. The Firmicutes/Bacteroidetes ratio: a relevant marker of gut dysbiosis in obese patients? Nutrients. (2020) 12:1474. doi: 10.3390/nu12051474
5. Stojanov S, Berlec A, Štrukelj B. The influence of probiotics on the Firmicutes/Bacteroidetes ratio in the treatment of obesity and inflammatory Bowel disease. Microorganisms. (2020) 8:1715. doi: 10.3390/microorganisms8111715
6. Cotillard A, Kennedy SP, Kong LC, Prifti E, Pons N, Le Chatelier E, et al. Dietary intervention impact on gut microbial gene richness. Nature. (2013) 500:585–8. doi: 10.1038/nature12480
7. Luo Q, Cheng D, Huang C, Li Y, Lao C, Xia Y, et al. Improvement of colonic immune function with soy isoflavones in high-fat diet-induced obese rats. Molecules. (2019) 24:1474. doi: 10.3390/molecules24061139
8. Mao K, Gao J, Wang X, Li X, Geng S, Zhang T, et al. Bifidobacterium animalis subsp. lactis BB-12 has effect against obesity by regulating gut microbiota in two phases in human microbiota-associated rats. Front Nutr. (2021) 8:811619. doi: 10.3389/fnut.2021.811619
9. Wang J, Jia H. Metagenome-wide association studies: fine-mining the microbiome. Nat Rev Microbiol. (2016) 14:508–22. doi: 10.1038/nrmicro.2016.83
10. Carmody RN, Gerber GK, Luevano JM Jr., Gatti DM, Somes L, Svenson KL, et al. Diet dominates host genotype in shaping the murine gut microbiota. Cell Host Microbe. (2015) 17:72–84. doi: 10.1016/j.chom.2014.11.010
11. Fan Y, Pedersen O. Gut microbiota in human metabolic health and disease. Nat Rev Microbiol. (2021) 19:55–71. doi: 10.1038/s41579-020-0433-9
12. Hamaker BR, Tuncil YE. A perspective on the complexity of dietary fiber structures and their potential effect on the gut microbiota. J Mol Biol. (2014) 426:3838–50. doi: 10.1016/j.jmb.2014.07.028
13. Gillen ZM, Mustad VA, Shoemaker ME, Mckay BD, Leutzinger TJ, Lopez-Pedrosa JM, et al. Impact of slow versus rapid digesting carbohydrates on substrate oxidation in pre-pubertal children: a randomized crossover trial. Clin Nutr. (2021) 40:3718–28. doi: 10.1016/j.clnu.2021.05.004
14. Koleva PT, Valcheva RS, Sun X, Gänzle MG, Dieleman LA. Inulin and fructo-oligosaccharides have divergent effects on colitis and commensal microbiota in HLA-B27 transgenic rats. Br J Nutr. (2012) 108:1633–43. doi: 10.1017/S0007114511007203
15. Lee SM, Han HW, Yim SY. Beneficial effects of soy milk and fiber on high cholesterol diet-induced alteration of gut microbiota and inflammatory gene expression in rats. Food Funct. (2015) 6:492–500. doi: 10.1039/C4FO00731J
16. Adamberg K, Adamberg S, Ernits K, Larionova A, Voor T, Jaagura M, et al. Composition and metabolism of fecal microbiota from normal and overweight children are differentially affected by melibiose, raffinose and raffinose-derived fructans. Anaerobe. (2018) 52:100–10. doi: 10.1016/j.anaerobe.2018.06.009
17. Martin MJ, Manzano M, Bueno-Vargas P, Rueda R, Salto R, Giron M-D, et al. Feeding a slowly digestible carbohydrate diet during pregnancy of insulin-resistant rats prevents the excess of adipogenesis in their offspring. J Nutr Biochem. (2018) 61:183–96. doi: 10.1016/j.jnutbio.2018.05.018
18. Salto R, Girón MD, Ortiz-Moral C, Manzano M, Vílchez JD, Reche-Perez FJ, et al. Dietary complex and slow digestive carbohydrates prevent fat deposits during catch-up growth in rats. Nutrients. (2020) 12:2568. doi: 10.3390/nu12092568
19. Manzano M, Giron MD, Salto R, Vilchez JD, Reche-Perez FJ, Cabrera E, et al. Quality more than quantity: the use of carbohydrates in high-fat diets to tackle obesity in growing rats. Front Nutr. (2022) 9:809865. doi: 10.3389/fnut.2022.809865
20. Reeves PG, Nielsen FH, Fahey GC Jr. AIN-93 Purified Diets for Laboratory Rodents: Final Report of the American Institute of Nutrition ad hoc Writing Committee on the Reformulation of the AIN-76A Rodent Diet. Oxford: Oxford University Press (1993). doi: 10.1093/jn/123.11.1939
21. Buettner R, Schölmerich J, Bollheimer LC. High-fat diets: modeling the metabolic disorders of human obesity in rodents. Obesity. (2007) 15:798–808. doi: 10.1038/oby.2007.608
22. Plaza-Diaz J, Gomez-Fernandez A, Chueca N, Torre-Aguilar MJ, Gil A, Perez-Navero JL, et al. Autism spectrum disorder (ASD) with and without mental regression is associated with changes in the fecal microbiota. Nutrients. (2019) 11:337. doi: 10.3390/nu11020337
23. Pastor-Villaescusa B, Plaza-Díaz J, Egea-Zorrilla A, Leis R, Bueno G, Hoyos R, et al. Evaluation of the gut microbiota after metformin intervention in children with obesity: a metagenomic study of a randomized controlled trial. Biomed Pharmacother. (2021) 134:111117. doi: 10.1016/j.biopha.2020.111117
24. Plaza-Diaz J, Bernal MJ, Schutte S, Chenoll E, Genovés S, Codoñer FM, et al. Effects of whole-grain and sugar content in infant cereals on gut microbiota at weaning: a randomized trial. Nutrients. (2021) 13:1496. doi: 10.3390/nu13051496
25. Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol. (2019) 37:852–7.
26. Callahan BJ, Mcmurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods. (2016) 13:581–3. doi: 10.1038/nmeth.3869
27. Price MN, Dehal PS, Arkin AP. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS One. (2010) 5:e9490. doi: 10.1371/journal.pone.0009490
28. Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. (2013) 30:772–80. doi: 10.1093/molbev/mst010
29. Bokulich NA, Kaehler BD, Rideout JR, Dillon M, Bolyen E, Knight R, et al. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome. (2018) 6:1–17. doi: 10.1186/s40168-018-0470-z
30. Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. (2012) 41:D590–6. doi: 10.1093/nar/gks1219
31. Douglas GM, Maffei VJ, Zaneveld JR, Yurgel SN, Brown JR, Taylor CM, et al. PICRUSt2 for prediction of metagenome functions. Nat Biotechnol. (2020) 38:685–8. doi: 10.1038/s41587-020-0548-6
32. Zheng M, Lamb KE, Grimes C, Laws R, Bolton K, Ong KK, et al. Rapid weight gain during infancy and subsequent adiposity: a systematic review and meta-analysis of evidence. Obes Rev. (2018) 19:321–32. doi: 10.1111/obr.12632
33. Barr J, Caballería J, Martínez-Arranz I, Domínguez-Díez A, Alonso C, Muntané J, et al. Obesity-dependent metabolic signatures associated with nonalcoholic fatty liver disease progression. J Proteome Res. (2012) 11:2521–32. doi: 10.1021/pr201223p
34. Martínez-Uña M, Varela-Rey M, Cano A, Fernández-Ares L, Beraza N, Aurrekoetxea I, et al. Excess S-adenosylmethionine reroutes phosphatidylethanolamine towards phosphatidylcholine and triglyceride synthesis. Hepatology. (2013) 58:1296–305. doi: 10.1002/hep.26399
35. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B (Methodol). (1995) 57:289–300. doi: 10.1111/j.2517-6161.1995.tb02031.x
36. Protivinsky T, Wei T, Simko V, Levy M, Xie Y, Jin Y, et al. Package ‘Corrplot’. (2021). Available online at: https://cran.r-project.org/web/packages/corrplot/corrplot.pdf
37. Hofmann AF. The continuing importance of bile acids in liver and intestinal disease. Arch Intern Med. (1999) 159:2647–58. doi: 10.1001/archinte.159.22.2647
38. Rodríguez JM, Murphy K, Stanton C, Ross RP, Kober OI, Juge N, et al. The composition of the gut microbiota throughout life, with an emphasis on early life. Microb Ecol Health Dis. (2015) 26:26050. doi: 10.3402/mehd.v26.26050
39. Vernocchi P, Del Chierico F, Putignani L. Gut microbiota metabolism and interaction with food components. Int J Mol Sci. (2020) 21:3688. doi: 10.3390/ijms21103688
40. Malik VS, Pan A, Willett WC, Hu FB. Sugar-sweetened beverages and weight gain in children and adults: a systematic review and meta-analysis. Am J Clin Nutr. (2013) 98:1084–102. doi: 10.3945/ajcn.113.058362
41. Gellar L, Nansel TR. High and low glycemic index mixed meals and blood glucose in youth with type 2 diabetes or impaired glucose tolerance. J Pediatr. (2009) 154:455–8. doi: 10.1016/j.jpeds.2008.09.040
42. Stephen A, Alles M, De Graaf C, Fleith M, Hadjilucas E, Isaacs E, et al. The role and requirements of digestible dietary carbohydrates in infants and toddlers. Eur J Clin Nutr. (2012) 66:765–79. doi: 10.1038/ejcn.2012.27
43. Le Chatelier E, Nielsen T, Qin J, Prifti E, Hildebrand F, Falony G, et al. Richness of human gut microbiome correlates with metabolic markers. Nature. (2013) 500:541–6.
44. Álvarez-Mercado AI, Navarro-Oliveros M, Robles-Sánchez C, Plaza-Díaz J, Sáez-Lara MJ, Muñoz-Quezada S, et al. Microbial population changes and their relationship with human health and disease. Microorganisms. (2019) 7:68. doi: 10.3390/microorganisms7030068
45. Orsso CE, Peng Y, Deehan EC, Tan Q, Field CJ, Madsen KL, et al. Composition and functions of the gut microbiome in pediatric obesity: relationships with markers of insulin resistance. Microorganisms. (2021) 9:1490. doi: 10.3390/microorganisms9071490
46. Da Silva CC, Monteil MA, Davis EM. Overweight and obesity in children are associated with an abundance of Firmicutes and reduction of Bifidobacterium in their gastrointestinal microbiota. Child Obes. (2020) 16:204–10. doi: 10.1089/chi.2019.0280
47. Sze MA, Schloss PD. Looking for a signal in the noise: revisiting obesity and the microbiome. mBio. (2016) 7:e01018–16. doi: 10.1128/mBio.01018-16
48. Nakayama J, Yamamoto A, Palermo-Conde LA, Higashi K, Sonomoto K, Tan J, et al. Impact of westernized diet on gut microbiota in children on Leyte Island. Front Microbiol. (2017) 8:197. doi: 10.3389/fmicb.2017.00197
49. Liou AP, Paziuk M, Luevano JM Jr., Machineni S, Turnbaugh PJ, Kaplan LM. Conserved shifts in the gut microbiota due to gastric bypass reduce host weight and adiposity. Sci Transl Med. (2013) 5:178ra41. doi: 10.1126/scitranslmed.3005687
50. Davis EC, Dinsmoor AM, Wang M, Donovan SM. Microbiome composition in pediatric populations from birth to adolescence: impact of diet and prebiotic and probiotic interventions. Dig Dis Sci. (2020) 65:706–22. doi: 10.1007/s10620-020-06092-x
51. Cheng HS, Ton SH, Phang SCW, Tan JBL, Abdul Kadir K. Increased susceptibility of post-weaning rats on high-fat diet to metabolic syndrome. J Adv Res. (2017) 8:743–52. doi: 10.1016/j.jare.2017.10.002
52. Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, et al. A core gut microbiome in obese and lean twins. Nature. (2009) 457:480–4. doi: 10.1038/nature07540
53. Louis S, Tappu R-M, Damms-Machado A, Huson DH, Bischoff SC. Characterization of the gut microbial community of obese patients following a weight-loss intervention using whole metagenome shotgun sequencing. PLoS One. (2016) 11:e0149564. doi: 10.1371/journal.pone.0149564
54. Parker BJ, Wearsch PA, Veloo ACM, Rodriguez-Palacios A. The genus Alistipes: gut bacteria with emerging implications to inflammation, cancer, and mental health. Front Immunol. (2020) 11:906. doi: 10.3389/fimmu.2020.00906
55. Costa G, Vasconcelos Q, Abreu G, Albuquerque A, Vilarejo J, Aragão G. Changes in nutrient absorption in children and adolescents caused by fructans, especially fructooligosaccharides and inulin. Arch Pediatr. (2020) 27:166–9. doi: 10.1016/j.arcped.2020.01.004
56. Jaagura M, Viiard E, Karu-Lavits K, Adamberg K. Low-carbohydrate high-fat weight reduction diet induces changes in human gut microbiota. Microbiologyopen. (2021) 10:e1194. doi: 10.1002/mbo3.1194
57. Wang S, Huang M, You X, Zhao J, Chen L, Wang L, et al. Gut microbiota mediates the anti-obesity effect of calorie restriction in mice. Sci Rep. (2018) 8:13037–13037. doi: 10.1038/s41598-018-31353-1
58. Wei Y, Liang J, Su Y, Wang J, Amakye WK, Pan J, et al. The associations of the gut microbiome composition and short-chain fatty acid concentrations with body fat distribution in children. Clin Nutr. (2021) 40:3379–90. doi: 10.1016/j.clnu.2020.11.014
59. Liu D, Wen B, Zhu K, Luo Y, Li J, Li Y, et al. Antibiotics-induced perturbations in gut microbial diversity influence metabolic phenotypes in a murine model of high-fat diet-induced obesity. Appl Microbiol Biotechnol. (2019) 103:5269–83. doi: 10.1007/s00253-019-09764-5
60. Vazquez-Moreno M, Perez-Herrera A, Locia-Morales D, Dizzel S, Meyre D, Stearns JC, et al. Association of gut microbiome with fasting triglycerides, fasting insulin and obesity status in Mexican children. Pediatr Obes. (2021) 16:e12748. doi: 10.1111/ijpo.12748
61. Ye X, Liu Y, Hu J, Gao Y, Ma Y, Wen D. Chlorogenic acid-induced gut microbiota improves metabolic endotoxemia. Front Endocrinol (Lausanne). (2021) 12:762691. doi: 10.3389/fendo.2021.762691
62. Fuhren J, Schwalbe M, Boekhorst J, Rösch C, Schols HA, Kleerebezem M. Dietary calcium phosphate strongly impacts gut microbiome changes elicited by inulin and galacto-oligosaccharides consumption. Microbiome. (2021) 9:218. doi: 10.1186/s40168-021-01148-0
63. Parada Venegas D, De La Fuente MK, Landskron G, González MJ, Quera R, Dijkstra G, et al. Short chain fatty acids (SCFAs)-mediated gut epithelial and immune regulation and its relevance for inflammatory bowel diseases. Front Immunol. (2019) 10:277. doi: 10.3389/fimmu.2019.00277
64. Beisner J, Filipe Rosa L, Kaden-Volynets V, Stolzer I, Günther C, Bischoff SC. Prebiotic inulin and sodium butyrate attenuate obesity-induced intestinal barrier dysfunction by induction of antimicrobial peptides. Front Immunol. (2021) 12:678360. doi: 10.3389/fimmu.2021.678360
65. Xu J, Xu R, Jia M, Su Y, Zhu W. Metatranscriptomic analysis of colonic microbiota’s functional response to different dietary fibers in growing pigs. Anim Microbiome. (2021) 3:45. doi: 10.1186/s42523-021-00108-1
66. Teixeira C, Prykhodko O, Alminger M, Fåk Hållenius F, Nyman M. Barley products of different fiber composition selectively change microbiota composition in rats. Mol Nutr Food Res. (2018) 62:e1701023. doi: 10.1002/mnfr.201701023
67. Zhang Y, Chen L, Hu M, Kim JJ, Lin R, Xu J, et al. Dietary type 2 resistant starch improves systemic inflammation and intestinal permeability by modulating microbiota and metabolites in aged mice on high-fat diet. Aging (Albany NY). (2020) 12:9173–87. doi: 10.18632/aging.103187
68. Daniel N, Perazza LR, Varin TV, Trottier J, Marcotte B, St-Pierre P, et al. Dietary fat and low fiber in purified diets differently impact the gut-liver axis to promote obesity-linked metabolic impairments. Am J Physiol Gastrointest Liver Physiol. (2021) 320:G1014–33. doi: 10.1152/ajpgi.00028.2021
69. Walter J. Murine gut microbiota-diet trumps genes. Cell Host Microbe. (2015) 17:3–5. doi: 10.1016/j.chom.2014.12.004
70. Turnbaugh PJ, Bäckhed F, Fulton L, Gordon JI. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe. (2008) 3:213–23. doi: 10.1016/j.chom.2008.02.015
71. Hills RD Jr., Pontefract BA, Mishcon HR, Black CA, Sutton SC, Theberge CR. Gut microbiome: profound implications for diet and disease. Nutrients. (2019) 11:1613. doi: 10.3390/nu11071613
72. Zhang X, Zhao Y, Zhang M, Pang X, Xu J, Kang C, et al. Structural changes of gut microbiota during berberine-mediated prevention of obesity and insulin resistance in high-fat diet-fed rats. PLoS One. (2012) 7:e42529. doi: 10.1371/journal.pone.0042529
73. Stincone A, Prigione A, Cramer T, Wamelink MM, Campbell K, Cheung E, et al. The return of metabolism: biochemistry and physiology of the pentose phosphate pathway. Biol Rev Camb Philos Soc. (2015) 90:927–63. doi: 10.1111/brv.12140
74. Chavarría M, Nikel PI, Pérez-Pantoja D, De Lorenzo V. The Entner-Doudoroff pathway empowers Pseudomonas putida KT2440 with a high tolerance to oxidative stress. Environ Microbiol. (2013) 15:1772–85. doi: 10.1111/1462-2920.12069
75. Pellock SJ, Redinbo MR. Glucuronides in the gut: sugar-driven symbioses between microbe and host. J Biol Chem. (2017) 292:8569–76. doi: 10.1074/jbc.R116.767434
76. Peekhaus N, Conway T. What’s for dinner?: Entner-Doudoroff metabolism in Escherichia coli. J Bacteriol. (1998) 180:3495–502. doi: 10.1128/JB.180.14.3495-3502.1998
77. Valk LC, Luttik MAH, De Ram C, Pabst M, Van Den Broek M, Van Loosdrecht MCM, et al. A novel D-galacturonate fermentation pathway in Lactobacillus suebicus links initial reactions of the galacturonate-isomerase route with the phosphoketolase pathway. Front Microbiol. (2020) 10:3027. doi: 10.3389/fmicb.2019.03027
78. Plumbridge J, Vimr E. Convergent pathways for utilization of the amino sugars N-acetylglucosamine, N-acetylmannosamine, and N-acetylneuraminic acid by Escherichia coli. J Bacteriol. (1999) 181:47–54. doi: 10.1128/JB.181.1.47-54.1999
79. Coker JK, Moyne O, Rodionov DA, Zengler K. Carbohydrates great and small, from dietary fiber to sialic acids: how glycans influence the gut microbiome and affect human health. Gut Microbes. (2021) 13:1–18. doi: 10.1080/19490976.2020.1869502
80. Gupta KP, Singh J. Modulation of carcinogen metabolism and DNA interaction by calcium glucarate in mouse skin. Toxicol Sci. (2004) 79:47–55. doi: 10.1093/toxsci/kfh098
81. Yang W, Zhou G, Zou S, Yang W, Liu A, Sun S, et al. Metabonomics of d-glucaro-1,4-lactone in preventing diethylnitrosamine-induced liver cancer in rats. Pharm Biol. (2018) 56:643–8. doi: 10.1080/13880209.2018.1525414
82. Turroni F, Ribbera A, Foroni E, Van Sinderen D, Ventura M. Human gut microbiota and bifidobacteria: from composition to functionality. Antonie Van Leeuwenhoek. (2008) 94:35–50. doi: 10.1007/s10482-008-9232-4
83. Horz H-P. Archaeal lineages within the human microbiome: absent, rare or elusive? Life. (2015) 5:1333–45. doi: 10.3390/life5021333
84. Oliphant K, Allen-Vercoe E. Macronutrient metabolism by the human gut microbiome: major fermentation by-products and their impact on host health. Microbiome. (2019) 7:91. doi: 10.1186/s40168-019-0704-8
85. Windey K, De Preter V, Verbeke K. Relevance of protein fermentation to gut health. Mol Nutr Food Res. (2012) 56:184–96. doi: 10.1002/mnfr.201100542
86. Verbeke KA, Boobis AR, Chiodini A, Edwards CA, Franck A, Kleerebezem M, et al. Towards microbial fermentation metabolites as markers for health benefits of prebiotics. Nutr Res Rev. (2015) 28:42–66. doi: 10.1017/S0954422415000037
87. Saito Y, Sato T, Nomoto K, Tsuji H. Identification of phenol–and p-cresol-producing intestinal bacteria by using media supplemented with tyrosine and its metabolites. FEMS Microbiol Ecol. (2018) 94:fiy125. doi: 10.1093/femsec/fiy125
88. Wahlström A, Sayin SI, Marschall HU, Bäckhed F. intestinal crosstalk between bile acids and microbiota and its impact on host metabolism. Cell Metab. (2016) 24:41–50. doi: 10.1016/j.cmet.2016.05.005
89. Zwicker BL, Agellon LB. Transport and biological activities of bile acids. Int J Biochem Cell Biol. (2013) 45:1389–98. doi: 10.1016/j.biocel.2013.04.012
90. Ridlon JM, Kang DJ, Hylemon PB. Bile salt biotransformations by human intestinal bacteria. J Lipid Res. (2006) 47:241–59. doi: 10.1194/jlr.R500013-JLR200
91. Fang S, Chen X, Ye X, Zhou L, Xue S, Gan Q. Effects of gut microbiome and short-chain fatty Acids (SCFAs) on finishing weight of meat rabbits. Front Microbiol. (2020) 11:1835. doi: 10.3389/fmicb.2020.01835
92. Garcia-Mantrana I, Selma-Royo M, Alcantara C, Collado MC. Shifts on gut microbiota associated to mediterranean diet adherence and specific dietary intakes on general adult population. Front Microbiol. (2018) 9:890. doi: 10.3389/fmicb.2018.00890
93. The Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature. (2012) 486:207–14. doi: 10.1038/nature11234
94. Vaiserman A, Romanenko M, Piven L, Moseiko V, Lushchak O, Kryzhanovska N, et al. Differences in the gut Firmicutes to Bacteroidetes ratio across age groups in healthy Ukrainian population. BMC Microbiol. (2020) 20:221. doi: 10.1186/s12866-020-01903-7
95. Conlon MA, Bird AR. The impact of diet and lifestyle on gut microbiota and human health. Nutrients. (2014) 7:17–44. doi: 10.3390/nu7010017
96. Zafar H, Saier MH Jr. Gut Bacteroides species in health and disease. Gut Microbes. (2021) 13:1–20. doi: 10.1080/19490976.2020.1848158
97. Shah M, Vella A. Effects of GLP-1 on appetite and weight. Rev Endocr Metab Disord. (2014) 15:181–7. doi: 10.1007/s11154-014-9289-5
98. Drucker DJ. Mechanisms of action and therapeutic application of glucagon-like peptide-1. Cell Metab. (2018) 27:740–56. doi: 10.1016/j.cmet.2018.03.001
99. Salehi M, Purnell JQ. The role of glucagon-like peptide-1 in energy homeostasis. Metab Syndr Relat Disord. (2019) 17:183–91. doi: 10.1089/met.2018.0088
100. Nauck MA, Quast DR, Wefers J, Pfeiffer AFH. The evolving story of incretins (GIP and GLP-1) in metabolic and cardiovascular disease: a pathophysiological update. Diabetes Obes Metab. (2021) 23(Suppl. 3):5–29. doi: 10.1111/dom.14496
101. Huo R, Zhang M, Zhang Y, Bai X, Zhang Y, Guo X. Effects of oat complex high-fiber formula powder on the composition of intestinal microbiota and enzyme activities in mice induced by a high-fat diet. Front Nutr. (2022) 9:871556. doi: 10.3389/fnut.2022.871556
102. Bliss ES, Whiteside E. The gut-brain axis, the human gut microbiota and their integration in the development of obesity. Front Physiol. (2018) 9:900. doi: 10.3389/fphys.2018.00900
Keywords: obesity, dietary carbohydrates, gastrointestinal microbiome, metabolism, microbiota, pediatric obesity, rats
Citation: Plaza-Díaz J, Manzano M, Ruiz-Ojeda FJ, Giron MD, Salto R, López-Pedrosa JM, Santos-Fandila A, Garcia-Corcoles MT, Rueda R and Gil Á (2022) Intake of slow-digesting carbohydrates is related to changes in the microbiome and its functional pathways in growing rats with obesity induced by diet. Front. Nutr. 9:992682. doi: 10.3389/fnut.2022.992682
Received: 12 July 2022; Accepted: 02 November 2022;
Published: 30 November 2022.
Edited by:
Clara G. De Los Reyes-Gavilan, Spanish National Research Council (CSIC), SpainReviewed by:
Thais Manzano Parisotto, Sao Francisco University, BrazilLorella Paparo, University of Naples Federico II, Italy
Copyright © 2022 Plaza-Díaz, Manzano, Ruiz-Ojeda, Giron, Salto, López-Pedrosa, Santos-Fandila, Garcia-Corcoles, Rueda and Gil. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ángel Gil, YWdpbEB1Z3IuZXM=