 Chen Zhaojun1,2
Chen Zhaojun1,2 Feng Xin
Feng Xin Singab Abdel-nasser
Singab Abdel-nasser Liu Xiong
Liu Xiong- 1College of Food Science, Southwest University, Chongqing, China
- 2Guizhou Provincial Academy of Agricultural Sciences, Guiyang, China
- 3Department of Pharmacognosy, Ain Shams University, Cairo, Egypt
Sanshools, long-chain polyunsaturated amides in Zanthoxylum bungeanum (prickly ash), have important bioactivity. The objective was to assess inhibitory effects and molecular mechanisms of sanshools isolated from supercritical fluid (SCF) extract on human colon adenocarcinoma cells (HCT-116) cultured in vitro. Cells were exposed to various concentrations (0, 50, 90, or 130 μM) of sanshools for 24 or 48 h, with assessment of apoptosis and cell cycle arrest as well as regulatory gene and protein expression associated with apoptosis and the cell cycle. Sanshools profoundly inhibited growth of HCT-116 cells, with hydroxy-γ-sanshool (HRS) being the optimal active component (IC50 = 88.01 μM) inhibiting cell proliferation and having no cytotoxic effect to normal cells (IC50 = 481.52 μM) by CCK-8 assay. In HCT-116 cells, HRS inhibited cell growth, induced morphological distortion, and arrested the cell cycle at G1 phase (50.31 ± 4.13% vs. 72.16 ± 8.14% in Control and 130 μM HRS, respectively), and also caused programmed cell death in a dose-dependent manner. The percentage of apoptotic cells were remarkably increased after treated with HRS (6.2, 11.9, 19.8, and 30.7% for 0, 50, 90, and 130 μM, respectively). Moreover, in HCT-116 cells, HRS significantly inhibited mRNA and protein levels of Cyclin D1, CDK4, PCNA, and increased mRNA and protein levels of P21, P53, Fas, and Caspase 8. Furthermore, inhibitors of P53 and Caspase 8 proteins significantly mitigated the HRS-induced cell cycle arrest and apoptosis. In conclusion, our study provides evidence that HRS induced human colorectal cancer cell apoptosis by up-regulating P53 and Caspase 8.
Introduction
Colorectal cancer (CRC), a malignant tumor on the wall of the rectum or colon, ranks as the second leading cause of cancer incidence and the third most common cancer worldwide (1). Currently, the dominant and most effective treatment strategy is pharmacotherapy with 5-fluorouracil, leucovorin, irinotecan, and oxaliplatin (2). However, these compounds have many side effects and disadvantages, including bone marrow suppression, gastrointestinal tract lesions, hair loss, nausea, high cost, and development of clinical resistance (3). Therefore, there is an impetus to search for new compounds with low toxicity that could safely inhibit CRC development.
Prevention and treatment of CRC can be done with several natural bioactive compounds derived from plants and microorganisms, including quercetin, kaempferol, phenolic acids, ellagic acid, and protocatechuic acid (3, 4). Underlying mechanisms may be activation-induced apoptosis and cell cycle arrest (5), through cell signaling pathways (NF-κB, Wnt/β-catenin, ERK/MAPK, and PI3K/Akt/mTOR) (6). Apoptosis is mediated through three crucial pathways: intrinsic mitochondrial pathway, extrinsic death receptor pathway, and intrinsic endoplasmic reticulum pathway. Caspase 9 is the intrinsic pathway whereas Caspase 8 is the extrinsic pathway; both pathways converge to Caspase 3, causing breakdown of DNA and proteins, resulting in phagocytosis by macrophages.
The cell cycle is a complex process in growth and proliferation of cancer cells, involving numerous regulatory proteins, including cdks and cyclin (6). Sequential activation of cyclin-dependent kinases and phosphorylation of critical substrates facilitates orderly progression of the cell cycle (7, 8). Furthermore, complexes formed by CDK4 and D-type cyclins have been strongly implicated in the control of cell proliferation during the G1 phase (9, 10).
Prickly ash, called “Huajiao” in China, is mainly distributed in Asia, America, Oceania, and Africa. This plant belongs to the family Rutaceae and genus Zanthoxylum (11, 12), and is used as medicine, spice, or woody oil. Reported bioactivities of Zanthoxylum-based extracts include antioxidant, antitumor, antibacterial, anti-inflammatory, and hypoglycemic effects (11). Sanshools, the dominant sector of polyunsaturated amides (13) characterized by three conjugated double bonds, have abundant pharmacological effects, including unique tingling and numbing. Yields, species, and biological activities of sanshools are affected by extraction (13, 14). Sanshools regulated blood glucose (15) and serum lipid (15) concentrations and induced apoptosis in hepatoma cells (16) and other human cancer cell lines including DLD-1, HepG2, and Caco-2 (12). However, sanshool-induced apoptosis has not been well characterized. In addition, there are >10 kinds of sanshools (11) with various structural properties having distinct physiological activities. Therefore, there is a compelling need to explore the structure-activity relationship of various sanshools.
Objectives were to compare induced apoptosis effects of three sanshools derived from one species of prickly ash and explore mechanisms promoting CRC apoptosis.
Materials and methods
Chemicals
Supercritical fluid (SCF) extract of Zanthoxylum bungeanum was purchased from Zhengzhou Xuemailong Food Spice Co., Ltd. in Henan Province, China. Both HCT-116 (human colon adenocarcinoma cell line), and HEK293T cells were obtained from the National Collection of Authenticated Cell Cultures in Shanghai, China. McCoy's 5A media, DMEM media, fetal bovine serum (FBS), and 0.25% trypsin solution were obtained from Invitrogen (Carlsbad, CA, USA). The Caspase 8 inhibitor Z-IETD-FMK and P53 inhibitor PFTα hydrobromide were obtained from MedChemExpress (Monmouth Junction, NJ, USA). Primary antibodies (anti-CDK4, anti-cyclin D1, anti-PCNA, anti-BAX, anti-Caspase 3, anti-Cleaved Caspase 3, anti-P53, anti-Bcl-2, anti-Caspase 9, anti-Cleaved Caspase 9, anti-Fas, anti-P21, anti-Caspase 8, anti-Cleaved Caspase 8, and anti-β-actin), anti-mouse, and anti-rabbit secondary were obtained from the Beyotime Institute of Biotechnology (Shanghai, China).
Sample preparation
Sanshools were extracted as described (15), with a minor modification. The SCF extracts (60 g) were mixed with water-free silica (120 g), and thrice defatted with 400 mL anhydrous ether. The defatted sample was homogenized with 200 mL methanol at 55°C for 6 h; the crude extract was refluxed with 60 mL petroleum ether overnight after filtering and vacuum evaporation. The supernatant was collected and crystallized at −20°C. The suspension was dried using dry N2 gas at 4°C.
Further purification was done by semi-preparative HPLC (Waters Technologies, Milford, MA, USA). The mobile phase consisted of (A) water and (B) acetonitrile. A Waters preparative C18 column (9.4 ×250 mm, 5 μm) was used with the following gradient elution program: 0 min, 35% B; 30 min, 75% B; 40 min, 100% B; 45 min, 100% B; and 50 min, 35% B. Purified extract was subjected to mass spectrometry using an API 4000 QTrap mass spectrometer (AB/Sciex, Framingham, MA, USA) fitted with an electrospray ionization (ESI) source, using the following parameters: turbo ion spray probe, 400°C, ion spray voltage, 4500 V, entrance potential, 6 V, and collision energy of 10 V. In addition, nuclear magnetic resonance (NMR) including 13C NMR and 1H NMR was done as described 24 with a VNMRS600 NMR spectrometer (Agilent, Palo Alto, USA) to identify the purified extract.
Cell proliferation assay
Inhibition of growth of HCT-116 cells was assessed using the Cell Counting Kit8 (APExBIO, Houston, TX, USA). Briefly, 96-well plates were plated with 5 × 103 cells per well and incubated for 12 h. Then, cells were treated with HRS (0, 30, 50, 70, 90, 110, 130, 150, or 170 μM) for 24 h. Thereafter, 10 μL of CCK8 reagent was added and incubated at 37°C for 2 h. Absorbance was measured by a microplate reader at a wavelength of 450 nm. Inhibition effects of HRS on HCT-116 were calculated by comparison to a control group.
Morphological assessment
Morphological changes in HCT-116 cells and HEK293T cells treated with HRS (0, 50, 90, or 130 μM) for 12 or 24 h were observed using an inverted optical microscope at 200 × magnification (Nikon 80i, Tokyo, Japan).
Cell cycle analysis
The HCT-116 cells and HEK293T cells with a density of 3 × 105 cells/well were exposed to HRS (0, 50, 90, or 130 μM) for 24 h at 37°C. Then, cells were treated with trypsin, gathered, and fixed with 5 mL 80% ethanol at 4°C for 18 h, followed by washing with PBS. Propidium iodide (PI) was added and cells incubated for 30 min in the dark. Cell cycle distribution was assessed by flow cytometry (FACS Canto II plus, BD BioSciences, USA) with a cell cycle kit (BD, NJ, USA).
Hoechst 33342 staining assay
Apoptosis staining was detected by Hoechst 33342 (Solarbio, Beijing, China). In brief, after treatment with HRS (0, 50, 90, or 130 μM) for 24 h, cells were stained with Hoechst 33342 staining solution for 20 min at 4°C. Then, cells were washed with PBS and observed with fluorescence microscopy at 200 × magnification (Nikon 80i, Tokyo, Japan).
Apoptosis analysis
After treatment with a series of HRS solutions (0, 50, 90, or 130 μM) for 24 h, the HCT-116 cells were harvested. Washed cells were resuspended with 400 μL of 1 × Annexin V binding buffer at a final concentration of 3 × 105 cells/mL. Then, 5 μL Annexin V-FITC (BD, New Jersey, USA) was added and incubated in the dark for 15 min at 2–8°C. Thereafter, 5 μL PI (BD, New Jersey, USA) was added to the mixture and incubated for 10 min. Cell apoptosis was examined using flow cytometry (FACS Canto II plus, BD BioSciences, USA).
The mRNA extraction and quantitative real-time PCR
Cells (density, 3 × 105 cells/well) were treated with HRS (0, 50, 90, or 130 μM) and cultured in a 6-well plate for 24 h. Total RNA was isolated by TRIzol (Takara, Dalian, China). Synthesis of cDNA was conducted by PrimeScript™ RT Reagent Kit (Thermo Fisher Scientific Inc., Waltham, MA, USA) and RT-PCR was done with an Applied Biosystems® QuantStudio™ 5 Flex Real-Time PCR System (Thermo Fisher Scientific Inc.). The reaction conditions were: 2 min at 95°C, followed by 40 cycles of 15 s at 95°C, and 1 min at 60°C. Relative expression of the selected gene was calculated with the 2−ΔΔCt method, with β-actin used as a control. Primer sequences are shown (Table 1).

Table 1. Primers used for real-time PCR.
Protein extraction and western blot
Western blot analysis was done as described (17). Treated cells were washed thrice with PBS, followed by scraping with ice-cold RIPA buffer containing 1 mM protease phosphatase inhibitor and protease inhibitor. Protein concentrations were determined with a BCA protein quantification kit (Solarbio, Beijing, China). Furthermore, after boiling for 10 min, supernatant proteins were separated electrophoretically via SDS-PAGE and placed onto polyvinylide fluoride (PVDF) membranes. These membranes were blocked with 5% dried skim milk containing 0.1% Tween-20 (PBST) for 2 h at 37°C. The membrane was then incubated overnight at 4°C with primary antibodies. Finally, membranes were treated with a secondary antibody for 1 h at 37°C. Visualization was done with a High-sensitivity Chemiluminescence imaging system ChemiDocXRS+ (Bio-Rad, California, USA).
Inhibition of caspase 8 and p53
Inhibitors were used as described (18, 19), with a minor modification. After exposure to 90 μM HRS for 22 h, HCT-116 cells were treated with P53 inhibitor (Pifithrin-α hydrobromide) (30 μM) or with Caspase 8 inhibitor (40 μM) (Z-IETD-FMK) for 2 h. Thereafter, analysis was done with Western blotting using p53 and Caspase 8 antibodies.
Statistical analyses
All dates are expressed as mean ± standard deviation (SD) (n ≥ 3). One-way ANOVA and Student's t-test were used to detect the differences between various treatments. All analyses were done with SPSS17 (SPSS Inc., Chicago, IL, USA) and p < 0.05 was considered significant.
Results
HRS extraction from supercritical fluid extract
The purity of extracted sanshools was reported as 93.42%. Peaks 1, 2, and 3 were situated at 19.825, 20.353, and 21.069, respectively (Figure 1A), and isolated by semi-preparative HPLC into three compounds to perform the cell inhibition assay. The purity of HAS, HBS, and HRS were determined as 95.32, 96.15, and 96.36%. Inhibitory effects of the three compounds on HCT-116 cell proliferation were concentration-dependent (Figures 2A–C). Compound 3 had the lowest IC50 value (88.01 μM) for HCT-116 cell lines compared to the other two compounds, whereas the IC50 for HEK293T was 481.52 μM (Figure 2D), indicating it was non-toxic to normal cell lines. Compound 3 was then identified by HPLC (Figure 1B) and MS, revealing an [M+H] + ion at m/z 290.97 (Figure 1C), with further identification by NMR in Figures 3A,B. 1H NMR (500 MHz, Methanol-d4) δ: 7.15 (dd, J = 15.1, 10.8 Hz, 1H), 6.44–6.35 (m, 1H), 6.26 (dd, J = 15.2, 10.7 Hz, 1H), 6.23–6.08 (m, 3H), 6.03 (dd, J = 12.9, 9.7 Hz, 2H), 5.73 (dq, J = 13.9, 6.9 Hz, 1H), 5.39 (dt, J = 10.9, 7.5 Hz, 1H), 3.32–3.26 (m, 2H), 2.39–2.26 (m, 4H), 1.78 (dd, J = 6.6, 1.3 Hz, 3H), 1.20 (s, 6H).
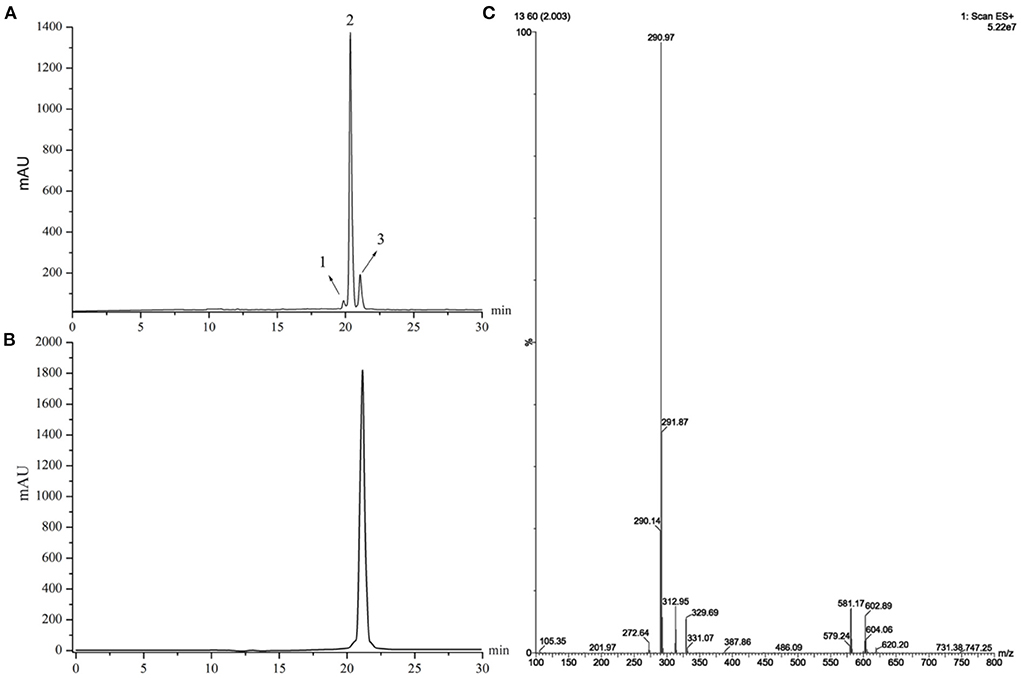
Figure 1. (A) HPLC chromatograms of a supercritical fluid extracted from Zanthoxylum bungeanum. (B) HPLC chromatograms of Compound 3. (C) MS spectra of Compound 3.
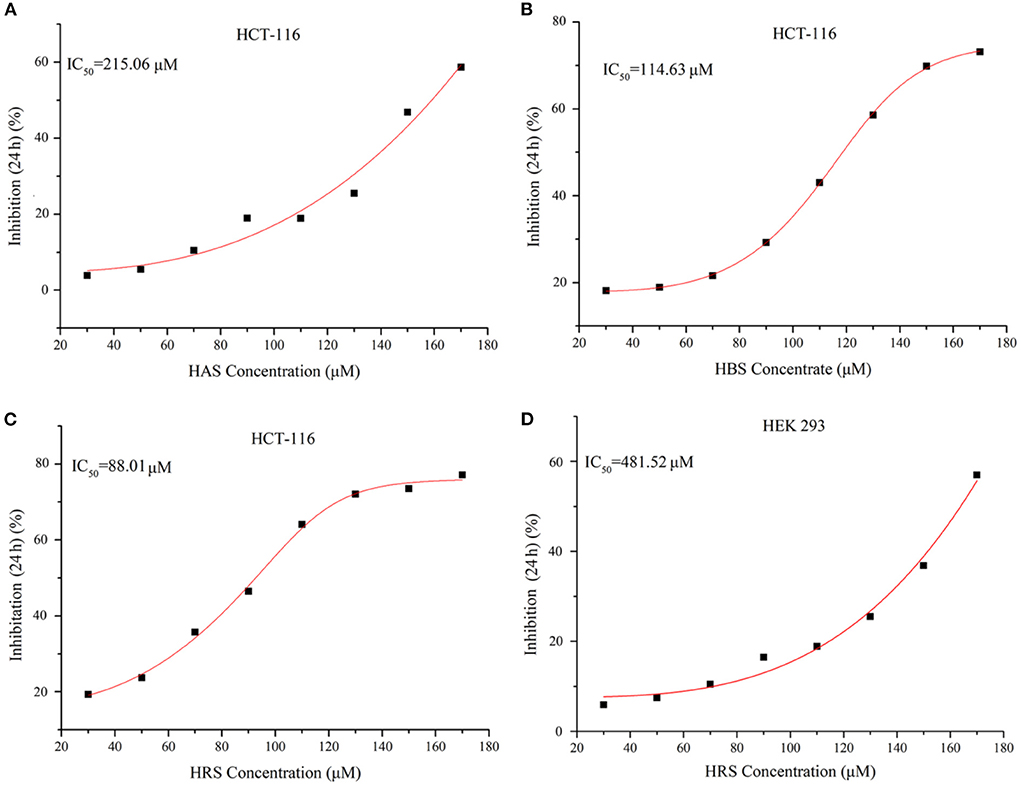
Figure 2. (A) Cell growth inhibition curves of Compound 1 (HAS) on HCT-116 cells. (B) Cell growth inhibition curves of Compound 2 (HBS) on HCT-116 cells. (C) Cell growth inhibition curves of Compound 3 (HRS) on HCT-116 cells. (D) Cell growth Inhibition curves of Compound 3 (HRS) on HEK293 cells (normal cells).
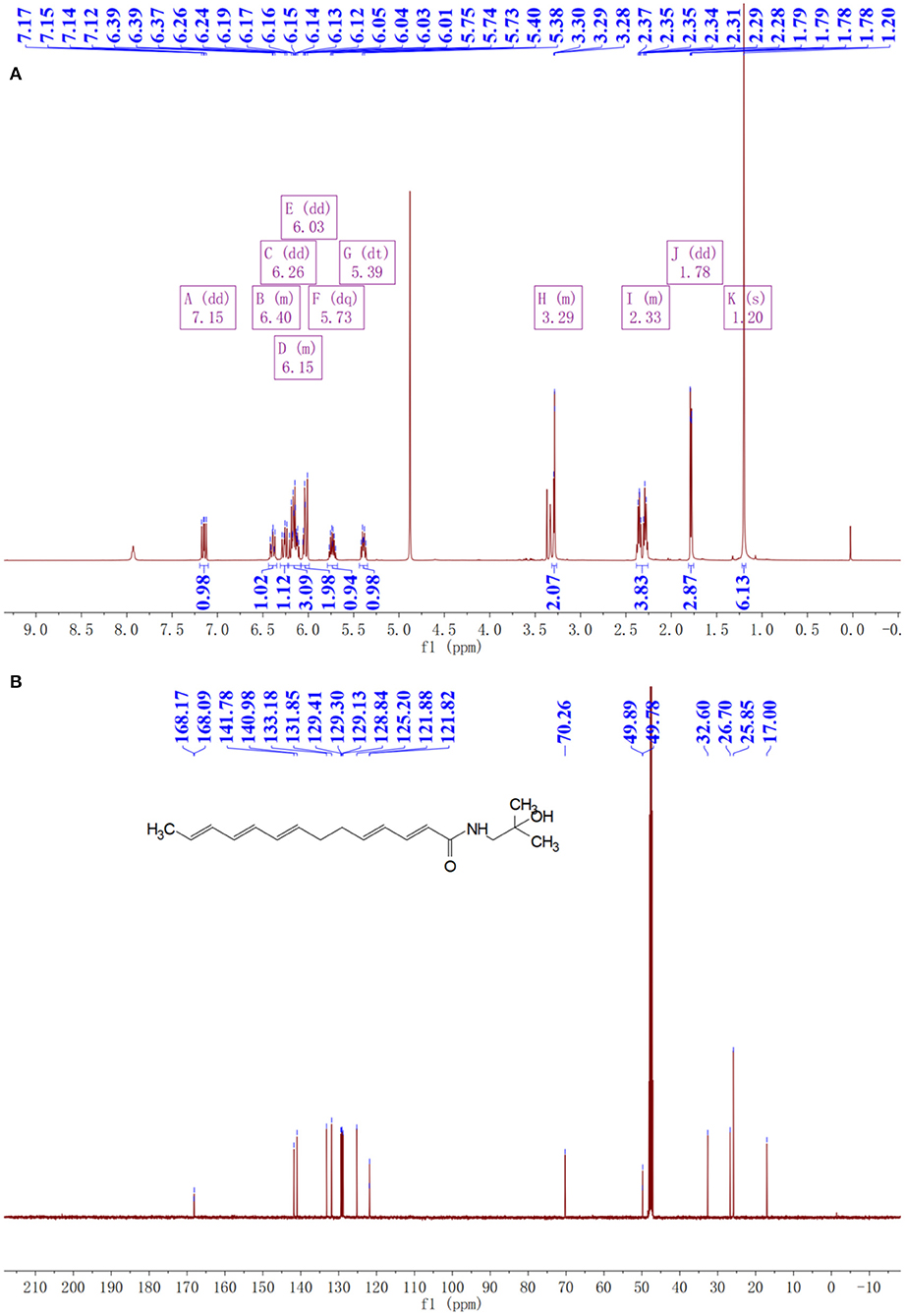
Figure 3. NMR spectra of prepared hydroxy-γ-sanshool (HRS). (A) 1H NMR spectra. (B) 1C NMR spectra.
13C NMR (126 MHz, MeOD) δ: 168.17, 168.09, 141.78, 140.98, 133.18, 131.85, 129.41, 129.30, 129.13, 128.84, 125.20, 121.88, 121.82, 70.26, 49.89, 49.78, 32.60, 26.70, 25.85, 17.00. Compound 3 was identified as hydroxy-γ-sanshool based on the analysis of MS and NMR as well as a previous report (20).
Effects of HRS on morphology of HCT-116 cells
Effect of HRS on the morphology of HCT-116 cells is shown (Figure 4). Whereas, numerous cells in the control group had an irregular polygon attached to the wall and an integrated cytoskeleton, the morphology of cells exposed to HRS (50 μM) changed slightly after treatment for 12 h, with obvious reductions in cell number after 24 h. Shrinkage and detachment of cells were evident when the concentration was increased to 90 μM. A significant decrease in cell numbers and greater subversive morphology changes with 130 μM; specifically, chromatin condensation, loss of nuclear construction and formation of apoptosis bodies appeared. The morphology and number of HEK293T cells were not effected after treatment with 0, 50, 90, or 130 μM HRS (Supplementary Figure S1).
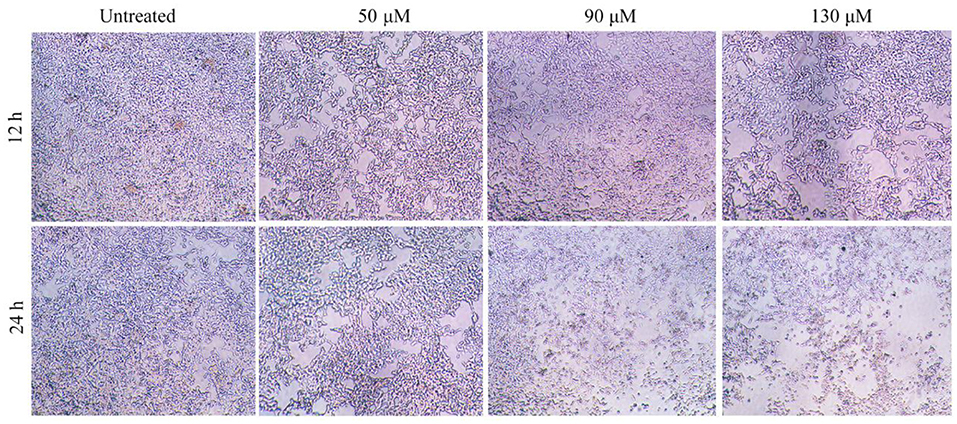
Figure 4. Effects of hydroxy-γ-sanshool (HRS) on morphological changes in HCT-116 cells. The original magnification was × 200.
Effects of HRS on the HCT-116 cell cycle
Treatment with HRS induced cell cycle arrest at the G1 phase; percentage of cells in various growth phases was determined (Figures 5A,B). The number of cells in the G1 phase were significantly increased (17.71, 34.86, and 44.50% for 50, 90, and 130 μM, respectively) compared to the untreated group on the HCT-116 cells. However, the proportion of cells in G0/G1, S, and G2/M phases were not observably changed for HEK293T cells after treatment with 0, 50, 90, or 130 μM HRS, indicating it could not stop or slow normal cell growth (Supplementary Figure S2).
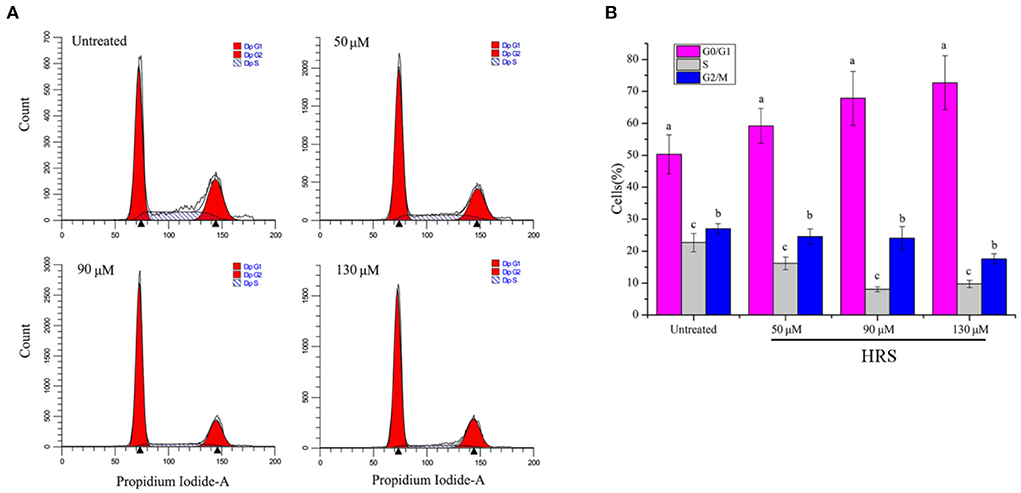
Figure 5. Hydroxy-γ-sanshool (HRS) induced cell cycle arrest at G1 phase in HCT-116 cell lines. (A) Cells in various cell cycle stages after HRS-treatment. (B) Percentages of cells in various cell cycle stages after treatment with HRS for 24 h. a−cWithin a concentration, columns without a common superscript differed (p < 0.05).
We further investigated the molecular regulation mechanism with real-time PCR and western blot analysis in HCT-116 cells treated with HRS. Relative mRNA expression of p21, TGFβ, p15, p16, GSK3β, GADD45A, SMAD4, and p53 were dose-dependently up-regulated, whereas PCNA, CyclinD1, CDC25A, CDK2, and CDK4 were significantly down-regulated (Figure 6A).
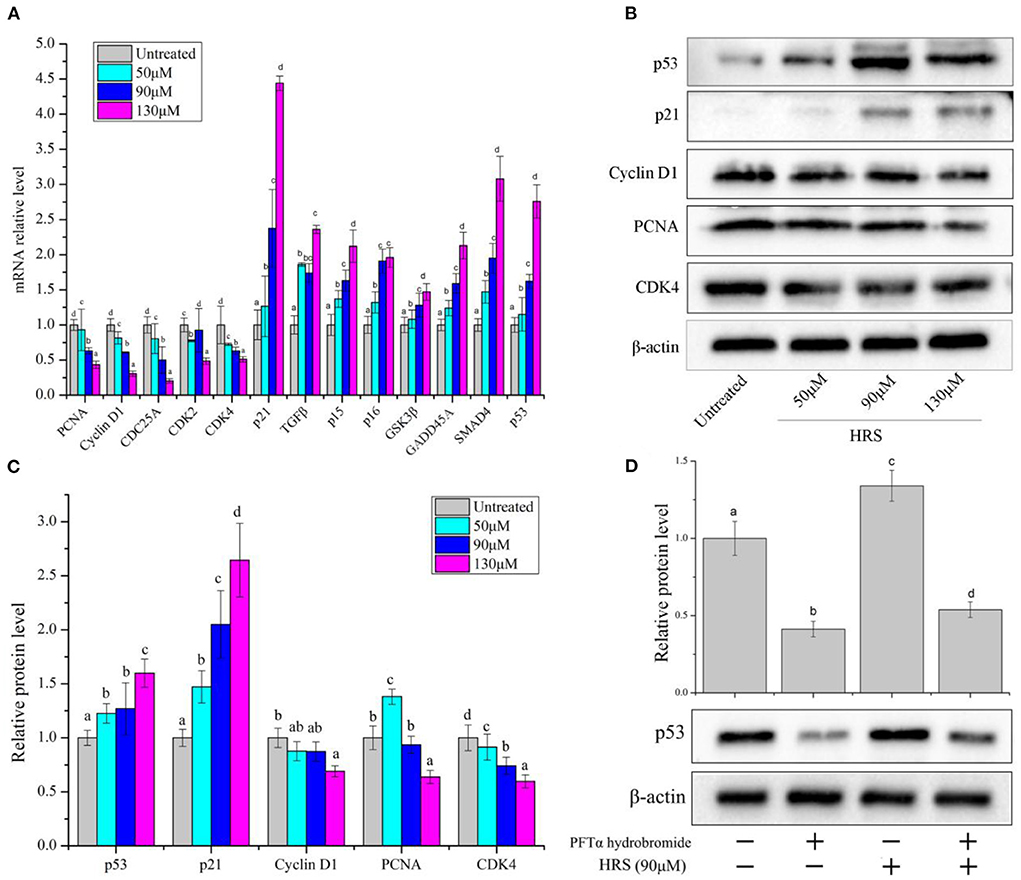
Figure 6. Expression of cell-cycle related protein and genes in HCT-116 cells after treatment with hydroxy-γ-sanshool (HRS). (A) Relative mRNA level of related genes in HCT-116 cells. (B) Western blot of effects on HRS on cell-cycle related proteins in HCT-116 cells. (C) Levels of cell-cycle related proteins. (D) Protein level of p53 in HCT-116 cells, with or without a p53 inhibitor (PFTα hydrobromide). HCT-116 cells were treated with HRS at designated concentrations and compared to untreated cells. All data were expressed as means ± SD for three replicated experiments. a−dWithin a cluster, columns without a common superscript differed (p < 0.05).
Based on the above analyses, protein expressions of selected genes (p53, p21, PCNA, CDK4, and cyclinD1) were evaluated (Figures 6B,C). Protein levels of CDK4, cyclin D1, and PCNA in treated cells were decreased; however, p53 and p21 were increased with exposure to HRS for 24 h. The p53/p21 pathway is essential in cell cycle arrest at the G1 phase. P53 can activate p21, which in return inhibits the activity of CDK4 and cyclin D1 (21). Subsequently, the PFTα hydrobromide (p53 inhibitor) remarkably decreased the HRS-induced increment of p53 (Figure 6D). Based on these results, it was suggested that the growth of HCT-116 cells was suppressed mainly through arresting cell cycle in the G1 phase, increasing the protein levels of p53 and p21, and down-regulating CDK4, cyclinD1, and PCNA.
Effects of HRS on HCT-116 cell apoptosis
To confirm whether the growth inhibitory effect of HRS was due to programmed cell death, Hoechst 33342 staining and flow cytometry were used. There were 6.2, 11.9, 19.8, and 30.7% apoptotic cells after treatment with 0, 50, 90, and 130 μM HRS, respectively (Figure 7A). In addition, the apoptotic cells were detected by Hoechst 33342 staining (Figure 7B). The apoptotic cells containing apoptotic features such as nuclear shrinkage and chromatin condensation were observed, the apoptotic cell number was increased compared to the control group, and the nuclei were generally fragmented and stained more intensely following exposure to 50, 90, or 130 μM. Moreover, the edges of the nucleus were less clearly defined after treatment with 130 μM HRS. These findings suggested that HRS could induce apoptosis and necrosis of HCT-116 cells. Moreover, the nuclei of HEK293T cells appeared round and evenly stained after treatment with 0, 50, 90, and 130 μM HRS, which indicated that the apoptosis of mornal cell could not be affected by HRS (Supplementary Figure S3).
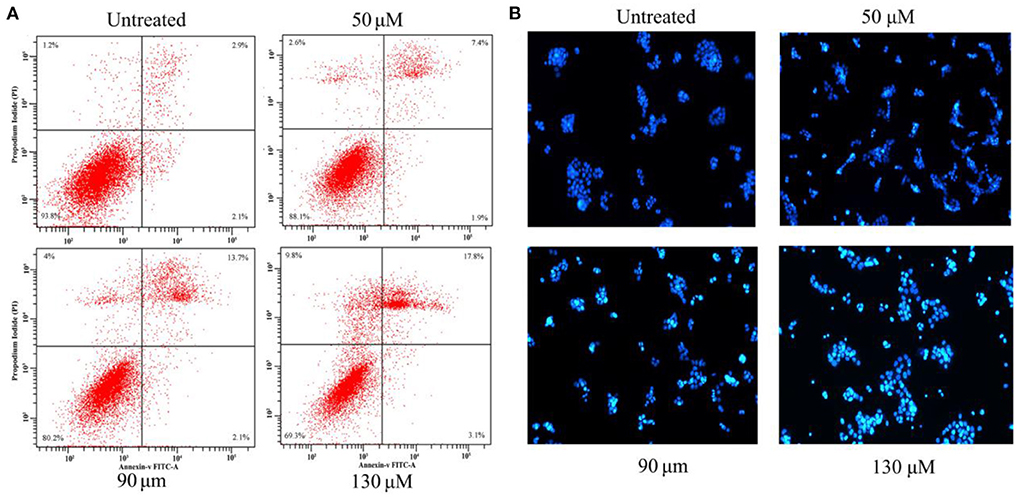
Figure 7. (A) Apoptosis in HCT-116 cells treated with various concentrations of hydroxy-γ-sanshool (HRS) for 24 h. The effect of HRS on apoptosis was assessed by flow cytometry after staining with Annexin V-PI. (B) Morphologic examination of treated HCT-116 cells stained with Hoechst 33342.
Effects of HRS on the mitochondrial pathway
There are two common initiation pathways (mitochondrial and death receptor) that lead to the execution of apoptosis (22). In terms of the mitochondrial pathway, expression levels of Bax, Bcl-2, Caspase 3, Caspase 9, Smac, Bid, Cyt c, DFF45, and PUMA were assessed by RT-PCR. The mRNA levels of Bax, Caspase 3, Caspase 9, Smac, Bid, Cyt c, and PUMA were increased by HRS except for Bcl-2 and DFF45 (Figure 8A). Similarly, expression of pivotal proteins in the mitochondrial pathway was further investigated. Up-regulation of protein levels of Bax, Caspase 3, Cleaved Caspase 3, Caspase 9, and Cleaved Caspase 9 and down-regulation of Bcl-2 confirmed results of RT-PCR (Figures 8B,C). These findings suggested that the mitochondrial pathway has an essential role in HRS-induced apoptosis and led us to further focus on the receptor-mediated pathway.
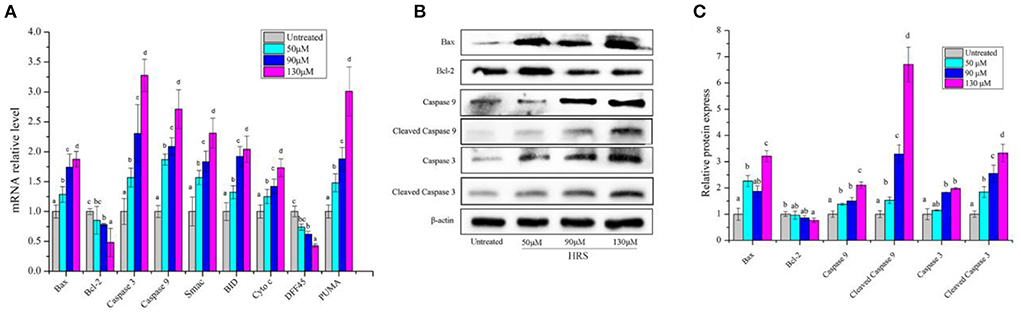
Figure 8. The mRNA and protein expressions of mitochondrial pathway in HCT-116 cells treated with hydroxy-γ-sanshool (HRS). (A) The mRNA level of mitochondrial pathway related genes. (B) Effects of HRS on related proteins in HCT-116 cells. (C) Levels of mitochondrial pathway related proteins. HCT-116 cells were treated with HRS at designated concentrations and compared to untreated cells. All data were expressed as means ± SD from three replicated experiments. a−dWithin a cluster, columns without a common superscript differed (p < 0.05).
Effects of HRS on death receptor pathway
In the death receptor pathway, mRNA levels of several genes (TRAIL, TRAIL-R, Fas-L, Fas, FADD, and Caspase 8) (Figure 9A) and protein levels of Fas, Caspase 8, and Cleaved Caspase 8 (Figures 9B,C) were increased by HRS after treatment for 24 h. Furthermore, a Caspase 8 inhibitor (Z-IETD-FMK) was used to confirm the role of Caspase 8 and Cleaved Caspase 8 in the death receptor pathway in HRS-induced apoptosis (Figure 9D). Pretreatment with Z-IETD-FMK significantly decreased Caspase 8 and Cleaved Caspase 8 protein expression, which was reduced to some extent with addition of HRS, implicating involvement of the death receptor pathway.
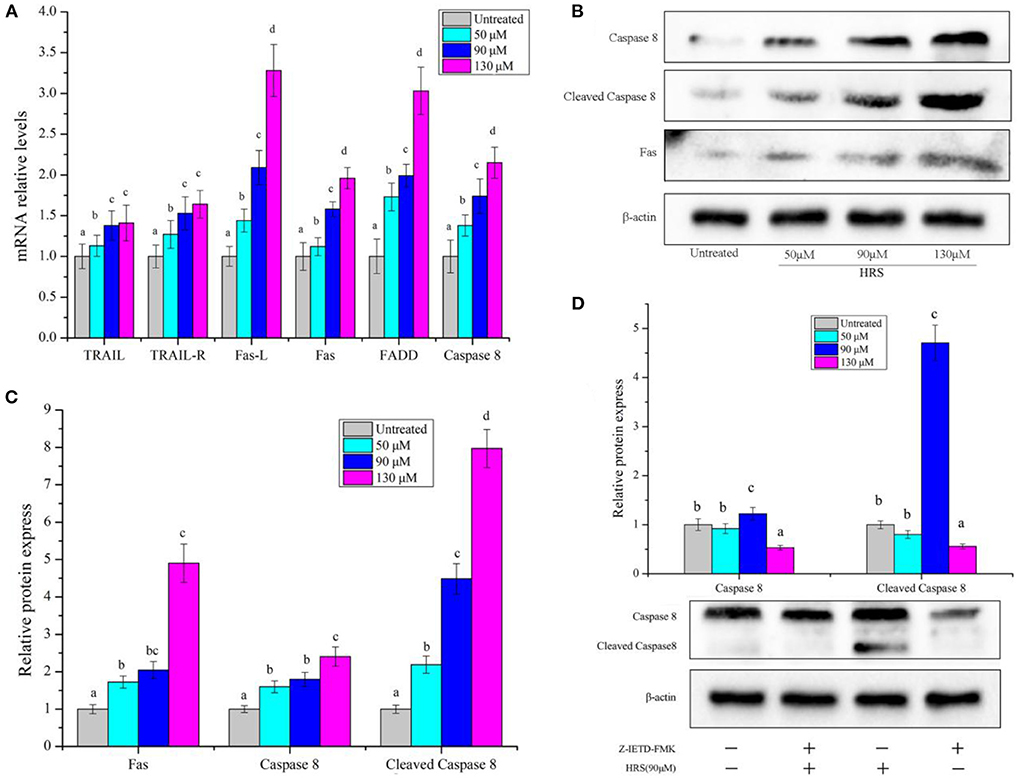
Figure 9. The mRNA level of death receptor pathway genes and protein in HCT-116 cells treated with hydroxy-γ-sanshool (HRS). (A) The mRNA level of genes in HCT-116 cells. (B) Western blot analyses of related genes. (C) Relative protein expression of related genes. (D) Protein level of Caspase 8 in HCT-116 cells, with or without an inhibitor (Z-IETD-FMK). HCT-116 cells were treated with HRS at designated concentrations and compared to untreated cells. All data were expressed as means ± SD from three replicated experiments. a−dWithin a cluster, columns without a common superscript differed (p < 0.05).
Discussion
Consumption of various natural products may prevent cancer as they contain various phytochemicals. Prickly ash belongs to the Rutaceae family and is widely distributed in the tropical and subtropical areas of Asia and America (23). It is listed in the Pharmacopeia, with >30 prescriptions for the treatment of dyspepsia, vomiting, toothache, ascariasis, eczema, abdominal pain, etc. (24). Sanshools are the primary compounds of polyunsaturated amides, regarded as the critical constituents in the majority of the biological activities of prickly ash. In several studies, sanshool extracts had noticeable anticancer effects in human cancer cell lines DLD-1, HepG2, and Caco-2 (12). Furthermore, according to the modular variation, >10 types of sanshools with various structures have been identified. Sanshools structure is commonly allocated into three separate parts: vanillyl group, amide bond, and fatty chain. However, comparison of anticancer activity sanshools as a function of their structural distinctions is not well characterized. Therefore, the current study is an attempt to compare inhibitory effects of sanshools (HAS, HBS, and HRS) on a colon cancer cell line (HCT-116) and investigate the possible mode of apoptosis induction.
In our studies, the three sanshools had distinctly different inhibition potency (IC50 were 215.06, 114.63, and 88.01 μM, respectively), despite all being isolated from the same species of prickly ash. The inhibition effect of HBS was superior to HAS, attributed to the difference between cis- and trans-isomer (25). In contrast, the cis-isomer HAS was superior to its trans-isomer HBS on inhibiting a neurofibromatosis type 1 (NF1)-defective tumor cell (26). Perhaps these opposing results were due to the differences in study cell lines and their pathways (27). The IC50 of HRS was lowest compared to HAS and HBS, attributed to the quantity and location of carbon-carbon double bonds in the sanshool molecule (28). Compounds with more carbon-carbon double bonds are readily peroxidized, yielding more and different peroxides that induce cell apoptosis (28). Furthermore, variations in locations of carbon-carbon double bonds affect functional groups and alter various biological activities (29). Regardless, sanshool had low toxicity and no significant inhibitory effect on normal cells, indicating high selectivity toward cancer cells.
The cell cycle is a collection of ordered processes that coordinate DNA replication and cell division. However, a human cancer cell has deregulation of the cell cycle and unscheduled proliferation. The present study produced convincing evidence that HRS induced cell cycle arrest in the G1 phase and inhibited proliferation of HCT-116 cells, consistent with previous results (16). Although effects of HRS-induced cell cycle arrest were indicated by these results, underlying mechanisms remain to be elaborated. In present study, HRS treatment of HCT-116 cells induced dose-responsive cell-cycle arrest by decreasing mRNA and protein expression levels of CDK4 and CyclinD1. Expression of Cyclin D1 and CDK4 (16) were investigated because they are the key genes of the G1 phase and form active cyclinD1/CDK4 complexes (30), ultimately initiating G1-S transition (31). In addition, HRS increased protein and mRNA levels of p53 and p21, while decreasing expression of PCNA in HCT-116 cells, indicating that p53 may be the key factor affecting the cell cycle. P53 controls CDk4 activity and PCNA-dependent DNA replication to alter cell cycle progression via regulation of protein p21.
Next, we further determined the role of p53 in the cell cycle arrest process. The tumor suppressor p53 functions is a gene-specific transcription factor (32). Two physiological outcomes of expression of p53 in tumor cells are G1 arrest and apoptosis, whereas, the trigger of G1 arrest by p53 is much better than its ability to induce apoptosis. P53 induces the expression of p21, an inhibitor of cyclin-dependent kinases 2, 3, 4, and 6 (33). Thus, a G1 arrest can result from the p53-induced expression of p21 and we choose p53 as the key protein to research its effect on cell cycle regulation. Pifithrin-alpha (PFT) a useful p53 inhibitor (34), decreased protein expression of p53 and suppressed the HRS-induced cell cycle arrest in HCT-116 cells. Therefore, HRS-induced arrest of the cell cycle in the G1 phase was attributed to increased expression of p53 in HCT-116 cells. Similarly, transforming acidic coiled-coil (TACC) depletion-induced G1 arrest and cell death was reduced in cells, indicating that induction of G1 arrest and cell death require p53 (35). In addition, inhibiting expression of p53 protein in SGC7901 cells blocked cell arrest and cell apoptosis (34).
The extrinsic (receptor-mediated) and intrinsic (mitochondria-mediated) pathways of apoptosis are well described (22); in the present study, HRS initiated apoptosis of HCT-116 cells through both pathways. The mitochondrial pathway is initiated by internal stimuli and regulated by Bcl-2 family proteins, including anti-apoptotic proteins (Bcl-2, Bcl-XL, and Bcl-W) and pro-apoptotic proteins (Bax, Bid, etc.) (36). In this study, HRS improved the mRNA expression of Bax, Caspase 3, Caspase 9, BID, Cyt c, and PUMA except for Bcl-2 (Figure 8). These anti- and pro-apoptotic proteins may regulate mitochondrial release of Cyt-c that in turn activates Caspase 9 and 3 (37), the molecular “scissors” of apoptosis. Protein expression of Bax, Caspase 9, Cleaved Caspase 9, Bcl-2, Caspase 3, and Cleaved Caspase 3 was consistent with changes in mRNA, indicating that the Bcl-2 family of proteins was involved in HRS-induced apoptosis. Furthermore, in our study, HRS increased relative mRNA expression of Smac, and decreased the level of DFF45. Smac improves caspase activation by combining inhibitors of apoptosis proteins. Ultimately, Caspase 3-mediated cleavage of DFF45 releases DFF40, leading to chromatin condensation and triggering cell death (38). Based on these findings, we inferred that the mitochondria-dependent apoptotic pathway was activated by HRS in HCT-116 cells. Similarly, sanshools extracted from the leaf of Zanthoxylum also induced apoptosis of Jurkat cells through a mitochondria-dependent pathway (39).
The death receptor pathway, the key executor of apoptosis, is another important signaling mechanism in anticancer therapy. Genes such as TRAIL, TRAIL-R1, Fas-L, Fas, FADD, and Caspase 8, and protein levels of Fas, Caspase 8, and Cleaved Caspase 8 were all up regulated by HRS (Figure 9), implicating initiation of death-receptor mediated pathway of apoptosis. Members of the death receptor family, Fas and TRAILR1 containing the death domain (DD) integrated with FasL and TRAIL, respectively, and once bound, activated Caspase 8 which initiates the pivotal pro-apoptotic protein caspase 3 (40), followed by programmed cell death. This is apparently the first report that Caspase 8 and 3 were activated and Fas was significantly increased in HRS-treated HCT-116 cells. Caspase 8 is regarded as the primary mediator of extrinsic apoptosis (41). It belongs to the caspase family of proteases and plays a key role in the regulation of programmed cell death during normal development (41). Extrinsic apoptosis relies on the formation of a death-inducing signaling complex (DISC), which always includes Caspase 8 (42). In addition, Caspase-8 can cleave the protein BID and cleaved BID (cBID) then activates the effector proteins BAX and BAK, which mediate mitochondrial outer membrane permeabilization, which promote the formation of the socalled “apoptosome”, and the activation of caspases that orchestrate the death of the cell (43). Therefore, we explored whether HRS-induced extrinsic apoptosis through activation of Caspase 8. Pretreatment of HCT-116 with Z-IETD-FMK (Caspase 8 inhibitor) reduced Caspase 8 protein concentration and abolished HRS-induced death receptor apoptosis. Therefore, we concluded that HRS-induced death receptor apoptosis was mediated via activation of Caspase 8, consistent with previous studies (44). Following treatment with Z-IETD-FMK, the number of apoptotic hepatocellular carcinoma cells decreased, and release of cytochrome c into the cytoplasm was prevented (45). Furthermore, Cadmium (Cd)-induced apoptotic morphological changes and increase of domain death agonist (tBID/BID) were mitigated by a Caspase 8 inhibitor (46).
Conclusions
In this study, HRS, an important active component of sanshools, was isolated, identified and used to explore an inhibition effect on HCT-116 cells proliferation. Clearly, HRS induced cell cycle arrest at the G1 phase and promoted apoptosis in dose- and time-dependent manners. Activation of p53 appeared to be involved in arresting the cell cycle in the G1 phase, and both the death receptor and mitochondrial pathway were involved in HRS-induced apoptosis. Therefore, we concluded that HRS has potential as a functional food. Based on the current outcomes, a flow diagram accounting for the anti-cancer effects of HRS, including induction of cell cycle arrest and apoptosis, has been prepared (Figure 10). Nevertheless, the inhibition effect needs further study to better understand underlying mechanisms.
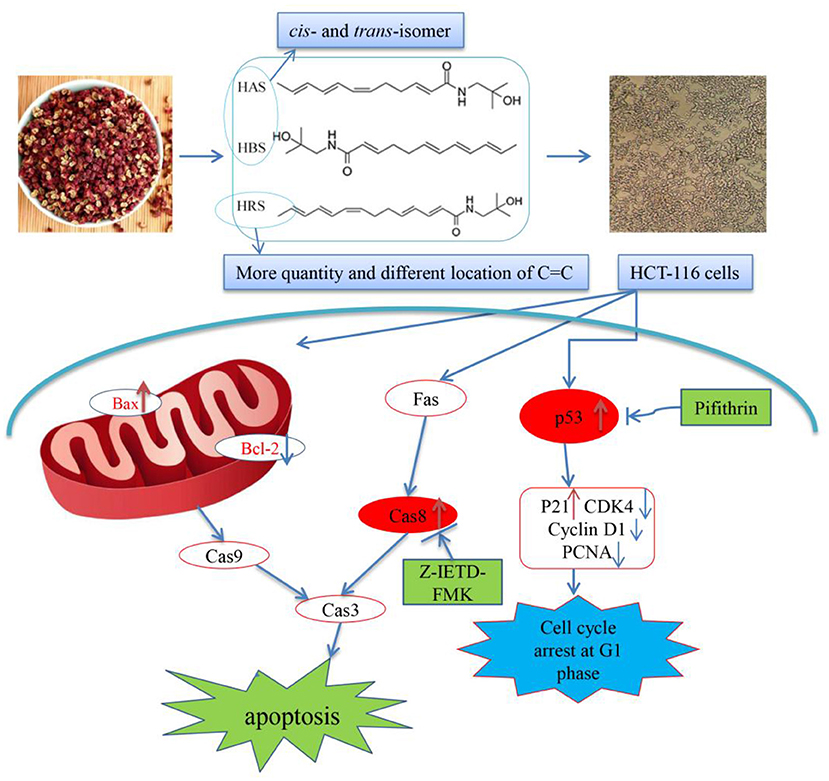
Figure 10. Proposed mechanism of the induced apoptotic effects of hydroxy-γ-sanshool (HRS) on HCT-116 cells.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author contributions
CZ: conceptualization and writing-original draft preparation. TL: methodology and software. FX: visualization. FX and LZ: investigation. SA-n: data curation. LX: supervision. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Science and Technology Department of Guizhou Province [2020]1Y039 and Guizhou Science and Technology Cooperation Platform Talent Project ([2019] No. 5201).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnut.2022.914638/full#supplementary-material
References
1. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2021. CA Cancer J Clin. (2021) 71:7–33. doi: 10.3322/caac.21654
2. Gingras D, Béliveau R. Colorectal cancer prevention through dietary and lifestyle modifications. Cancer Microenviron. (2011) 4:133–9. doi: 10.1007/s12307-010-0060-5
4. Tao J, Li Y, Li S, Li H. Plant foods for the prevention and management of colon cancer. J Funct Foods. (2018) 42:95–110. doi: 10.1016/j.jff.2017.12.064
5. Pietenpol JA, Stewart AZ. Cell cycle checkpoint signaling: cell cycle arrest versus apoptosis. Toxicology. (2002) 181:475–81. doi: 10.1016/S0300-483X(02)00460-2
6. Afrin S, Giampieri F, Gasparrini M, Forbes-Hernandez TY, Varela-López A, Quiles JL, et al. Chemopreventive and therapeutic effects of edible berries: a focus on colon cancer prevention and treatment. Molecules. (2016) 21:169. doi: 10.3390/molecules21020169
7. Schafer KA. The cell cycle: a review. Vet Pathol. (1998) 35:461−78. doi: 10.1177/030098589803500601
8. Vermeulen K, Van Bockstaele DR, Berneman NZ. The cell cycle: a review of regulation, deregulation and therapeutic targets in cancer. Cell Proliferat. (2003) 36:131–49. doi: 10.1046/j.1365-2184.2003.00266.x
9. Motokura T, Arnold A. Cyclins and oncogenesis. Biochimica et Biophysica Acta (BBA)-Rev Cancer. (1993) 1155:63–78. doi: 10.1016/0304-419X(93)90022-5
11. Chruma JJ, Cullen DJ, Bowman L, Toy HP. Polyunsaturated fatty acid amides from the Zanthoxylum genus–from culinary curiosities to probes for chemical biology. Nat Prod Rep. (2018) 35:54–74. doi: 10.1039/C7NP00044H
12. Nozaki R, Kono T, Bochimoto H, Watanabe T, Oketani K, Sakamaki Y, et al. Zanthoxylum fruit extract from Japanese pepper promotes autophagic cell death in cancer cells. Oncotarget. (2016) 7:70437. doi: 10.18632/oncotarget.11926
13. Sun X, Zhang D, Zhao L, Shi B, Xiao J, Shi J, et al. Development of differential pulse voltammetric method for rapid quantification of total hydroxyl-sanshools in Sichuan Pepper. LWT. (2020) 130:109640. doi: 10.1016/j.lwt.2020.109640
14. Ma Y, Li X, Hou L, Wei A. Extraction solvent affects the antioxidant, antimicrobial, cholinesterase and HepG2 human hepatocellular carcinoma cell inhibitory activities of Zanthoxylum bungeanum pericarps and the major chemical components. Ind Crop Prod. (2019) 142:111872. doi: 10.1016/j.indcrop.2019.111872
15. You Y, Ren T, Zhang S, Shirima GG, Cheng Y, Liu X. Hypoglycemic effects of Zanthoxylum alkylamides by enhancing glucose metabolism and ameliorating pancreatic dysfunction in streptozotocin-induced diabetic rats. Food Funct. (2015) 6:3144–54. doi: 10.1039/C5FO00432B
16. You Y, Zhou M, Lu H, Shirima GG, Cheng Y, Liu X. Sanshool from Zanthoxylum L induces apoptosis in human hepatocarcinoma HepG2 cells. Food Sci Biotechnol. (2015) 24:2169–75. doi: 10.1007/s10068-015-0289-3
17. Liu X, Xing Y, Li M, Zhang Z, Wang J, Ri M, et al. Licochalcone A inhibits proliferation and promotes apoptosis of colon cancer cell by targeting programmed cell death-ligand 1 via the NF-κB and Ras/Raf/MEK pathways. J Ethnopharmacol. (2021) 273:113989. doi: 10.1016/j.jep.2021.113989
18. Gary RK, Jensen AD. The p53 inhibitor pifithrin-α forms a sparingly soluble derivative via intramolecular cyclization under physiological conditions. Mol Pharmaceut. (2005) 2:462–74. doi: 10.1021/mp050055d
19. Bajt ML, Lawson JA, Vonderfecht SL, Gujral JS, Jaeschke H. Protection against Fas receptor–mediated apoptosis in hepatocytes and nonparenchymal cells by a caspase-8 inhibitor in vivo: evidence for a postmitochondrial processing of caspase-8. Toxicol Sci. (2000) 58:109–17. doi: 10.1093/toxsci/58.1.109
20. Mizutani K, Fukunaga Y, Tanaka O, Takasugi N, Saruwatari Y, Fuwa T, et al. Amides from huajiao, pericarps of Zanthoxylum bungeanum MAXI. Chem Pharm Bull. (1988) 36:2362–5. doi: 10.1248/cpb.36.2362
21. He G, Siddik ZH, Huang Z, Wang R, Koomen J, Kobayashi R, et al. Induction of p21 by p53 following DNA damage inhibits both Cdk4 and Cdk2 activities. Oncogene. (2005) 24:2929–43. doi: 10.1038/sj.onc.1208474
22. Wong RS. Apoptosis in cancer: from pathogenesis to treatment. J Exp Clin Canc Res. (2011) 30:87. doi: 10.1186/1756-9966-30-87
23. Kono R, Nomura S, Okuno Y, Kagiya T, Nakamura M, Utsunomiya H, et al. Two Japanese pepper (Zanthoxylum piperitum) fruit-derived compounds attenuate IgE-mediated allergic response in vitro and in vivo via inhibition of mast cell degranulation. Eur J Pharmacol. (2020) 885:173435. doi: 10.1016/j.ejphar.2020.173435
24. Chinese Pharmacopoeia Commission. Chinese Pharmacopoeia. Beijing: China Medical Science Press (2015), 191–193.
25. Birringer M, EyTina JH, Salvatore BA, Neuzil J. Vitamin E analogues as inducers of apoptosis: structure–function relation. Brit Cancer J. (2003) 88:1948–55. doi: 10.1038/sj.bjc.6600981
26. Devkota KP, Wilson J, Henrich CJ, McMahon JB, Reilly KM, Beutler AJ. Isobutylhydroxyamides from the pericarp of nepalese zanthoxylum armatum inhibit NF1-defective tumor cell line growth. J Nat Prod. (2013) 76:59–63. doi: 10.1021/np300696g
27. Maximov PY, Abderrahman B, Fanning SW, Sengupta S, Fan P, Curpan RF, et al. Endoxifen, 4-hydroxytamoxifen and an estrogenic derivative modulate estrogen receptor complex mediated apoptosis in breast cancer. Mol Pharmacol. (2018) 94:812–22. doi: 10.1124/mol.117.111385
28. Hawkins RA, Sangster K, Arends JM. Apoptotic death of pancreatic cancer cells induced by polyunsaturated fatty acids varies with double bond number and involves an oxidative mechanism. J Pathol. (1998) 185:61–70.
29. Lin Q, Li P, Fang M, Zhang D, Xia Y. Deep profiling of aminophospholipids reveals a dysregulated desaturation pattern in breast cancer cell lines. Anal Chem. (2021) 17:478–87. doi: 10.1021/acs.analchem.1c03494
30. Day PJ, Cleasby A, Tickle IJ, O'Reilly M, Coyle JE, Holding FP, et al. Crystal structure of human CDK4 in complex with a D-type cyclin. Proc Natl Acad Sci. (2009) 106:4166−70. doi: 10.1073/pnas.0809645106
31. Liu L, Shi R, Chen Z. Paeonol exerts anti-tumor activity against colorectal cancer cells by inducing G0/G1 phase arrest and cell apoptosis via inhibiting the Wnt/β-catenin signaling pathway. Int J Mol Med. (2020) 46:675–84. doi: 10.3892/ijmm.2020.4629
32. Diller L, Kassel J, Nelson CE, Gryka MA, Litwak G, Gebhardt M, et al. p53 functions as a cell cycle control protein in osteosarcomas. Mol Cell Biol. (1990) 10:5772–81. doi: 10.1128/mcb.10.11.5772-5781.1990
33. Shaw PH. The role of p53 in cell cycle regulation. Pathol Res Pract. (1996) 192:669–75. doi: 10.1016/S0344-0338(96)80088-4
34. Zhu B, Xing C, Lin F, Fan X, Zhao K, Qin Z. Blocking NF-κB nuclear translocation leads to p53-related autophagy activation and cell apoptosis. World J Gastroenterol. (2011) 17:478. doi: 10.3748/wjg.v17.i4.478
35. Suhail TV, Singh P, Manna KT. Suppression of centrosome protein TACC3 induces G1 arrest and cell death through activation of p38–p53–p21 stress signaling pathway. Eur J Cell Biol. (2015) 94:90–100. doi: 10.1016/j.ejcb.2014.12.001
36. Lowe SW, Lin WA. Apoptosis in cancer. Carcinogenesis. (2000) 21:485–95. doi: 10.1093/carcin/21.3.485
37. Woo M, Hakem R, Soengas MS, Duncan GS, Shahinian A, Kägi D, et al. Essential contribution of caspase 3/CPP32 to apoptosis and its associated nuclear changes. Gene Dev. (1998) 12:806–19. doi: 10.1101/gad.12.6.806
38. Kulbay M, Johnson B, Fiola S, Diaz RJ, Bernier J. DFF40 deficiency in cancerous T cells is implicated in chemotherapy drug sensitivity and resistance through the regulation of the apoptotic pathway. Biochem Pharmacol. (2021) 194:114801. doi: 10.1016/j.bcp.2021.114801
39. Mallya R, Bhitre JM. Cytotoxic activity and initiation of apoptosis via intrinsic pathway in jurkat cells by leaf extract of Zanthoxylum rhetsa DC. Nutr Cancer. (2021) 73:1768–79. doi: 10.1080/01635581.2020.1808231
40. Gao J, Zhao Y, Wang C, Ji H, Yu J, Liu C, Liu A. A novel synthetic chitosan selenate (CS) induces apoptosis in A549 lung cancer cells via the Fas/FasL pathway. Int J Biol Macromol. (2020) 158:689–97. doi: 10.1016/j.ijbiomac.2020.05.016
41. Mandal R, Barrón JC, Kostova I, Becker S, Strebhardt K. Caspase-8: The double-edged sword. Biochim Biophys Acta (BBA) Rev Cancer. (2020) 1873:188357. doi: 10.1016/j.bbcan.2020.188357
42. Salvesen GS, Walsh CM. Functions of caspase 8: The identified and the mysterious. Semin Immunol. (2014) 26:246-52 doi: 10.1016/j.smim.2014.03.005
43. Tummers B, Green RD. Caspase-8: regulating life and death. Immunol Rev. (2017) 277:76–89. doi: 10.1111/imr.12541
44. Slee EA, Adrain C, Martin SJ. Executioner caspase-3,−6, and−7 perform distinct, non-redundant roles during the demolition phase of apoptosis. J Biol Chem. (2001) 276:7320–6. doi: 10.1074/jbc.M008363200
45. Zhang H, Wu J, Wen M, Su L, Luo H. Galangin induces apoptosis in hepatocellular carcinoma cells through the caspase 8/t-Bid mitochondrial pathway. J Asian Nat Prod Res. (2012) 14:626–33. doi: 10.1080/10286020.2012.682152
Keywords: hydroxy-γ-sanshool, Zanthoxylum bungeanum, apoptosis, cell arrest, colorectal cancer cells
Citation: Zhaojun C, Lulin T, Xin F, Abdel-nasser S, Zunguo L and Xiong L (2022) Hydroxy-γ-sanshool from Zanthoxylum bungeanum (prickly ash) induces apoptosis of human colorectal cancer cell by activating P53 and Caspase 8. Front. Nutr. 9:914638. doi: 10.3389/fnut.2022.914638
Received: 07 April 2022; Accepted: 06 July 2022;
Published: 01 August 2022.
Edited by:
Wuyang Huang, Jiangsu Academy of Agricultural Sciences (JAAS), ChinaReviewed by:
Mohammad Reza Sam, Urmia University, IranFanxing Xu, Shenyang Pharmaceutical University, China
Copyright © 2022 Zhaojun, Lulin, Xin, Abdel-nasser, Zunguo and Xiong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liu Xiong, bGl1eGlvbmc4NDhAaG90bWFpbC5jb20=