 Arun J. Thirunavukarasu
Arun J. Thirunavukarasu A. Catharine Ross
A. Catharine Ross Rose M. Gilbert
Rose M. Gilbert- 1Corpus Christi College, University of Cambridge, Cambridge, United Kingdom
- 2University of Cambridge School of Clinical Medicine, Cambridge, United Kingdom
- 3Department of Nutritional Sciences, The Pennsylvania State University, University Park, PA, United States
- 4Cambridge University Hospitals NHS Foundation Trust, Cambridge, United Kingdom
- 5NIHR Biomedical Research Centre at Moorfields Eye Hospital NHS Foundation Trust and UCL Institute of Ophthalmology, London, United Kingdom
The first discovered vitamin, vitamin A, exists in a range of forms, primarily retinoids and provitamin carotenoids. The bioactive forms of vitamin A, retinol and retinoic acid, have many critical functions in body systems including the eye and immune system. Vitamin A deficiency is associated with dysfunctional immunity, and presents clinically as a characteristic ocular syndrome, xerophthalmia. The immune functions of vitamin A extend to the gut, where microbiome interactions and nutritional retinoids and carotenoids contribute to the balance of T cell differentiation, thereby determining immune status and contributing to inflammatory disease around the whole body. In the eye, degenerative conditions affecting the retina and uvea are influenced by vitamin A. Stargardt’s disease (STGD1; MIM 248200) is characterised by bisretinoid deposits such as lipofuscin, produced by retinal photoreceptors as they use and recycle a vitamin A-derived chromophore. Age-related macular degeneration features comparable retinal deposits, such as drusen featuring lipofuscin accumulation; and is characterised by parainflammatory processes. We hypothesise that local parainflammatory processes secondary to lipofuscin deposition in the retina are mediated by T cells interacting with dietary vitamin A derivatives and the gut microbiome, and outline the current evidence for this. No cures exist for Stargardt’s or age-related macular degeneration, but many vitamin A-based therapeutic approaches have been or are being trialled. The relationship between vitamin A’s functions in systemic immunology and the eye could be further exploited, and further research may seek to leverage the interactions of the gut-eye immunological axis.
Introduction
The first discovered vitamin, known initially as “fat-soluble A” (1, 2), and later as vitamin A, is comprised of fat-soluble organic compounds, or “vitamers,” chemically related to vitamin A1, retinol (3, 4). Although much was learned about the nutritional activity of this essential component of the diet in the first two decades following its discovery, it was not until the 1930’s that the chemical structure was elucidated by Karrer, who was awarded a Novel Prize for this and other discoveries (5). The chemical name (IUPAC nomenclature) of retinol is (2E,4E,6E,8E)-3,7-dimethyl-9-(2,6,6-trimethylcyclohexen-1-yl)nona-2,4,6,8-tetrae n-1-ol. The structure (Figure 1) contains an unsaturated ring more commonly referred to as a β-ionone ring, a methyl substituted unsaturated side chain, and a terminal alcohol moiety. This hydroxyl group is the site of enzyme-catalyzed oxidation reactions that yield retinal and, subsequently, retinoic acid (RA), considered the two major forms of vitamin A in terms of physiological activity. Vitamin A deficiency manifests as a characteristic clinical syndrome, and an intake that is either too high or low can have a wide range of pathological effects.
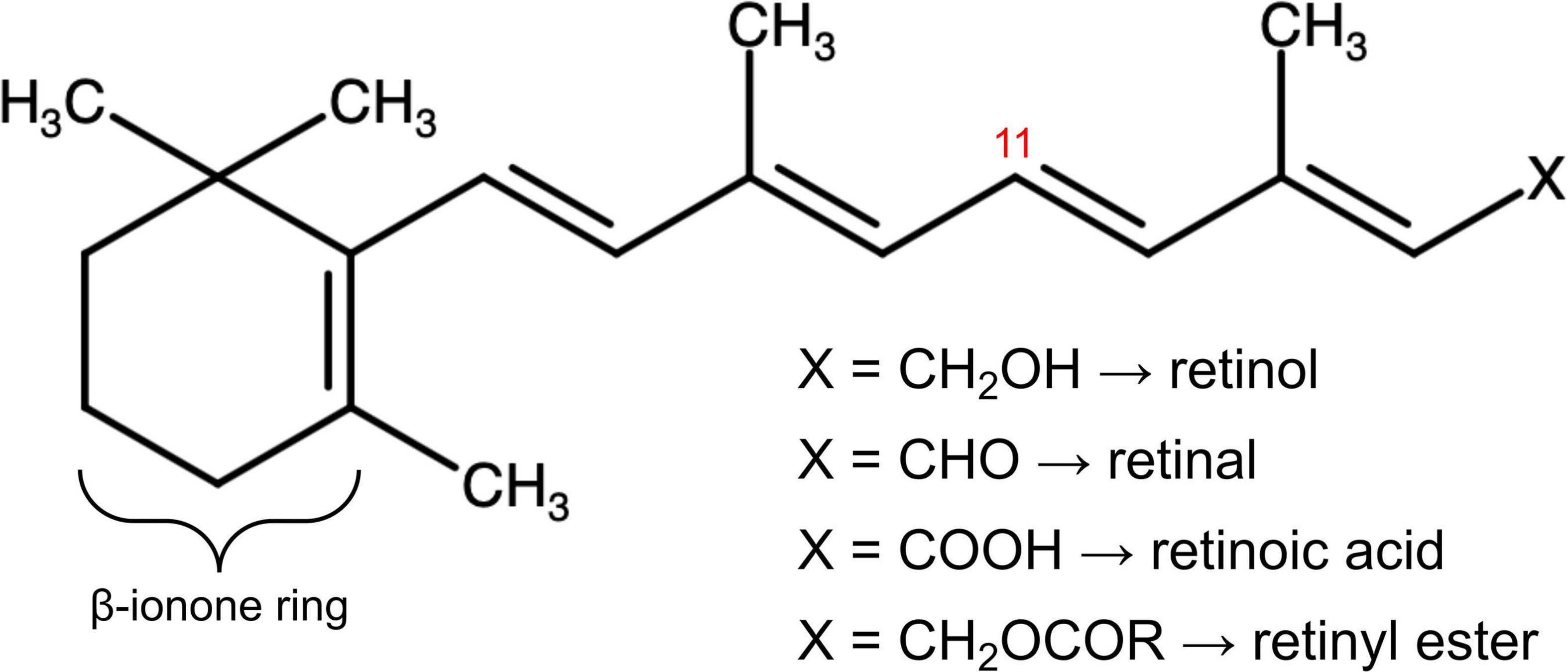
Figure 1. The molecular structure of vitamin A in its multiple forms. The chemical group denoted X defines retinol, retinal, retinoic acid, and retinyl esters depending on its composition. Stereoisomeric 11-cis and 11-trans retinoids depend on the three-dimensional arrangement of the vitamin A molecule around the double bond at the eleventh carbon residue, labelled in red. The ß-ionone ring, modified in synthetic forms of vitamin A, is also labelled.
Retinoic acid functions as a ligand for nuclear RA receptors (RARs), which are among the most studied of nuclear hormone receptors, mediating both the organismal and cellular effects of intracellular retinoic acids and their synthetic analogues (6). The RAR family has three main isoforms: α, β, and γ, capable of forming heterodimers with retinoid X receptors (RXRs). RAR-RXR heterodimers interact with retinoic acid response elements (RAREs) within the promoters of RA-responsive genes (6, 7). Retinoids regulate many essential biological processes, acting as potent gene expression modulators during embryogenesis, organogenesis, cell growth, differentiation and apoptosis, including the embryonic development and normal metabolism of the eye (6, 8).
The neurosensory retina is a layered tissue which lines the back of the eye, communicating with the brain via the optic nerve. Blood is supplied to the neurosensory retina by retinal blood vessels originating from the central retinal artery. Transport across retinal blood vessels is limited by endothelial tight junctions, which constitute the inner blood–retinal barrier. The optic nerve is comprised of ganglion cell axons, the “output neurons” of the retina. The retina is built up of three layers of cell bodies and two plexiform layers of synapses, clearly defined on histological sections. The innermost layer is the ganglion cell layer. The inner nuclear layer, featuring the cell bodies of bipolar, amacrine, and horizontal cells, is sandwiched by an internal plexiform layer and ganglion cell layer on one side, and outer plexiform layer and outer nuclear layer on the other. The outer nuclear layer features the cell bodies of most of the photoreceptors: the rods and cones. A third class of photoreceptor, intrinsically photoreceptive ganglion cells, is situated in the ganglion cell layer (9, 10). All photoreceptors depend on a vitamin A-derived chromophore to detect light (11). The chromophore associated with light transduction proteins, opsins, and undergoes photoisomerisation during transduction. Expended chromophore then enters the visual cycle, a sequence of reactions which facilitates regeneration and re-association with opsins for further transduction (Figure 2). While in rods, the visual cycle involves the rod itself and retinal pigment epithelium (RPE) overlying the neurosensory retina, cones operate with Müller cells instead, a sort of glial support cell in the retina (12). Accumulation of retinoid products of these photoisomerisation and visual cycle processes underlies the pathophysiology of Stargardt’s disease (STGD), and similar products are associated with age-related macular degeneration (AMD).
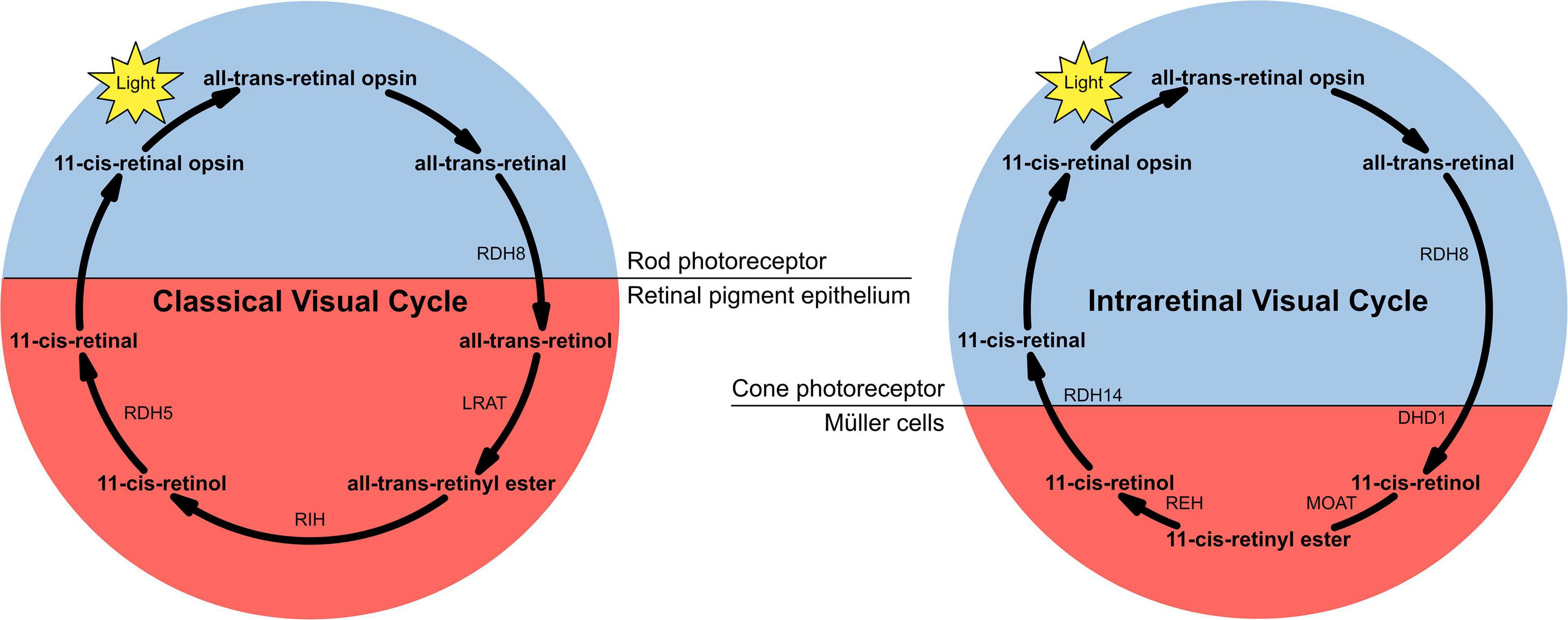
Figure 2. The two versions of the visual cycle, classical and intraretinal, a series of chemical reactions, mostly enzyme-catalysed, underlying chromophore regeneration in rods and cones, respectively. Intra- and extra-photoreceptor processes are shaded blue and red, respectively. Rods work with the retinal pigmented epithelium in the classical visual cycle, whereas cones work with Müller cells in the intraretinal visual cycle.
T-lymphocytes (T-cells) are a main cell type of the adaptive immune system and circulate in the peripheral blood, constituting approximately 80% of the total lymphocyte population (13). They express a T-cell receptor (TCR), which interacts with peptides complexed with Major Histocompatibility Complex (MHC) antigens (14) and has the capacity to “recognise” diverse antigens from the host body, pathogens, tumours, and the environment. In addition to facilitating adaptive immune responses, T cells are implicated in many inflammatory and autoimmune diseases (15, 16). Various subsets of T-cell are defined by the cell surface markers, cytokines, and transcription factors that they express. The T-helper 17 (Th17) cell subset is characterised by two master regulator orphan nuclear receptors: retinoid orphan receptor (ROR) γt and RORα, named for their homology to the nuclear retinoid receptor proteins; and by the expression of the RORγt transcription factor and its signature cytokine, IL-17 (17–20). The Th17 lineage, in addition to the Th1 subset (characterised by expression of the T-bet transcription factor), is implicated in chronic inflammatory and autoimmune disease. There is evidence that both of these Th subsets may act as effector cells in the pathogenesis of ocular autoimmunity (21, 22). T-regulatory (Treg) cells function as a natural mechanism to suppress autoreactive T-cells and are characterised by expression of CD4 and CD25 surface receptors, a forkhead box P3 (FoxP3) transcription regulator and production of cytokines such as IL-10 and transforming growth factor β (TGF-β) (21, 23, 24). The bioactive metabolite of vitamin A, RA, has been shown to oppose Th17 cell commitment (19) and, as discussed further below, is capable of inducing Treg that are critical for maintaining immune homeostasis and for opposing the induction of autoimmune T cells (20).
Fully differentiated T cells have the capacity to modify their gene expression to adapt to new conditions. Differentiation of naïve CD4+ cells into T-cell subsets such as effector Th1 and Th17, or induced Treg (iTreg) cells may depend on epigenetic regulation in response to changing environmental conditions, in addition to determination by cell fate programmes (25, 26). Therefore, the ability of the local environment to provide an adequate supply of RA to promote iTreg maintenance, or to be conducive to the conversion of Th17 cells into iTreg cells, is important (20). This is especially significant within the mucosal environment of the intestine, where dietary nutrients such as vitamin A and the bacteria of the microbiome act to co-regulate the balance of pro-inflammatory and anti-inflammatory T cells (27, 28). There are likely a myriad of interactions between the host and microbiome that shape the immune profile of an individual at any one time, and the gut microbiome is specifically implicated in ocular inflammatory diseases, including AMD (29, 30).
In this review, vitamin A physiology is described, including intake, absorption, and delivery to dependent tissues; ocular usage of vitamin A, with an emphasis on retinal phototransduction and the visual cycle; as well as the role of vitamin A in T-cell mediated inflammation. The gut-eye axis is discussed, and its reliance on normal vitamin A metabolism and T cell function is explored. The pathophysiology of STGD (STGD1; MIM 248200) and AMD are then outlined, as diseases associated with chronic inflammatory and local parainflammatory processes, driven by aberrant generation of vitamin A derivatives, and/or retinoid-related disturbances in T-cell homeostasis in the microbiome, influenced by dietary vitamin A. Disruption in the interrelationships between nutrition, the microbiome, and host metabolism may underlie degenerative retinal diseases like STGD and AMD, presenting interesting therapeutic targets. Potential retinoid-based treatments for STGD and AMD are discussed, some of which have been and are currently being trialled in human patients. Potential future directions for research are posited, based on this evidenced vitamin A-related pathophysiology and therapy. In particular, a nutritional approach to improving outcomes is considered, with an overview of the complexity associated with dealing with patients exhibiting different phenotypes, and at different disease stages.
Nutritional forms of vitamin A and their sources
Forms of vitamin A present in the diet include preformed retinol, contained in foods of animals origin and mainly in the form of retinyl esters, and the provitamin A carotenoids such as β-carotene. De novo synthesis of carotenoids is restricted to plants and microorganisms, meaning that animals like humans must consume either preformed vitamin A, obtained in foods of animal origin, or provitamin compounds (5). Important dietary sources of preformed vitamin A, are found in the highest concentrations in avian and mammalian liver, instant powdered breakfast drinks, ready-to-eat cereals, and margarine (31). The provitamin carotenoids that can give rise to retinol through metabolism include β-carotene, α-carotene, γ-carotene, and β-cryptoxanthin (31, 32), which are found in highest concentrations in green leafy and yellow vegetables (33). Several other dietary carotenoids, such as the xanthophylls zeathanthin, lutein, and lycopene, are absorbed into plasma and exert protective antioxidant activities; however, these xanthophylls are not precursors of retinol or its metabolites. As described further below, retinol is metabolized to two bioactive compounds, retinal, the form of the vitamin that is essential for vision, and RA, which is essential as a signalling ligand for nuclear retinoic acid receptors, RARs.
Nutritional supplements
Vitamin A present in nutritional supplements is generally of manufactured origin, produced in the form of retinyl palmitate or retinyl acetate for greater stability. When vitamin A is given as a supplement to young children 6 months–5 years of age, as recommended by WHO in low- and middle-income countries in which deficiency is a public health problem, the form is most often retinyl palmitate, provided as a high-dose capsule once every 4–6 months (34).
Non-nutritional retinoids
In addition to the naturally occurring forms above, many synthetic vitamin A derivatives, referred to as retinoids (35), have been synthesized and tested for their potential medicinal value. These can be classified in generations, with the first generation being natural compounds listed above including the acidic acid form RA; second generation compounds derived from the first generation, such as acitretin (36), which generally have a modified ring structure in place of the β-ionone ring; a third generation derived from the second, such as bexarotene (37); and recently a single fourth generation compound, trifarotene (38). Several of these retinoids are used in clinical contexts, including in the treatment of certain skin diseases (39) and cancers (40).
Metabolism of dietary vitamin A
Vitamin A metabolism comprises a complex series of reactions which include the cleavage of carotenoids, the esterification of retinol and hydrolysis of retinyl esters, a series of redox reactions that interconvert retinol and retinal in a reversible manner, and a terminal oxidation reaction which coverts retinal into retinoic acid. Additional enzymatic conjugation reactions serve to produce water-miscible forms of vitamin A for excretion. The enzymes principally responsible for these interconversions are indicated in Figure 3. Notably, retinol and retinal can cycle between these forms, while esterification of retinol provides a storage form of the vitamin, often the most abundant form in most tissues, which can then be drawn upon through retinyl ester hydrolysis to produce retinol, as required. Carotenoids, retinyl esters, and retinol itself can be considered as precursors for the two principal bioactive forms, retinal and RA. Both forms are critical for the health of the eye, therefore dietary vitamin A can be traced from uptake to its delivery to the eye, in addition to the reactions that take place in the cells of the retina and cornea.
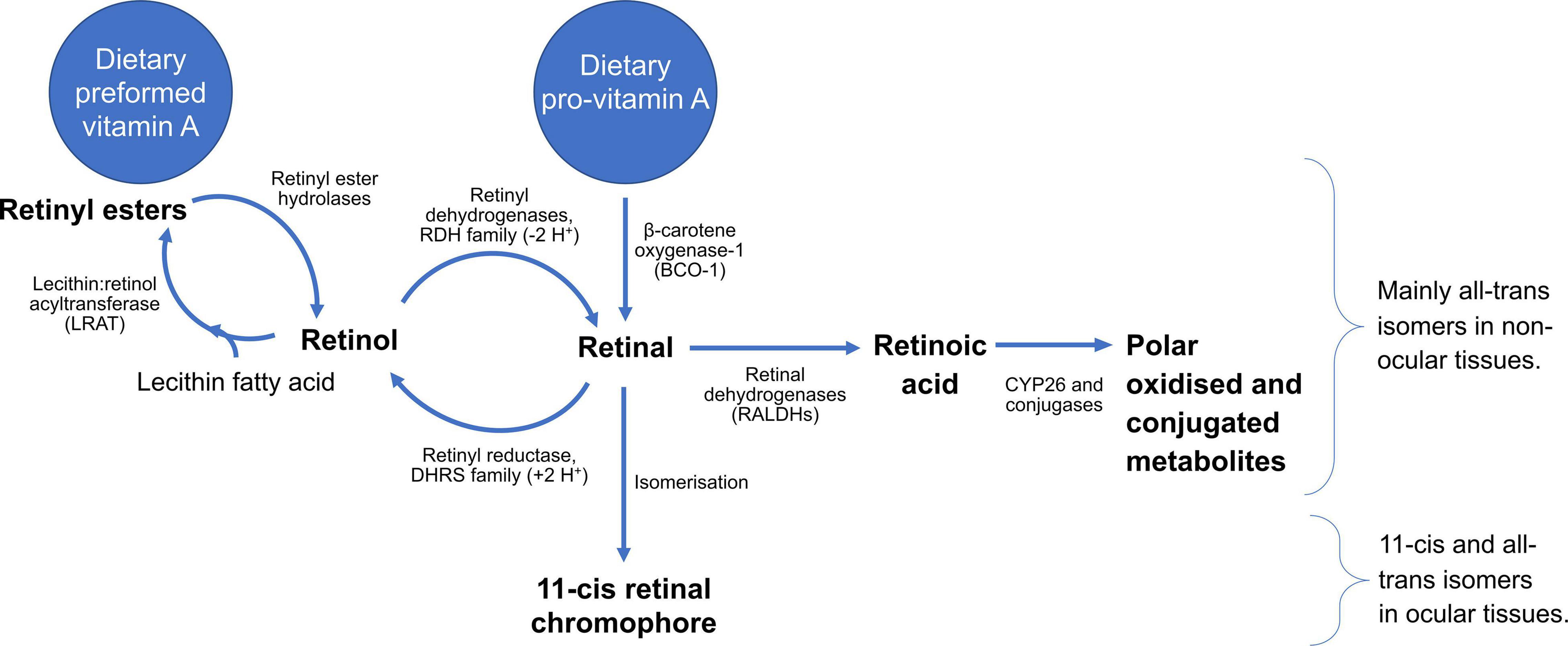
Figure 3. Summary of the metabolism of vitamin A, running from different dietary sources through generation of bioactive metabolites, as well as reactions required for clearance. All-trans stereoisomers predominate away from the eye, whereas the eye, and particularly the retina, feature 11-cis and all-trans isomers, crucial for vision.
A vast array of proteins are involved in the processes underlying absorption, such as for carotenoid cleavage (5, 41, 42), retinyl ester hydrolysis (43–45), facilitated diffusion into cells (46–48), intracellular trafficking of retinol in the enterocytes (31), re-esterification of retinoids (49, 50), and sequestering of free retinol (51). Mutations in the genes encoding these proteins may affect vitamin A absorption (52–54), and potentially lead to vitamin A deficiency syndromes, as discussed below (55).
Intestinal processing
In the lumen, dietary vitamin A, mostly as retinol esters, must be solubilised with bile salts and products of lipid digestion to form mixed micelles, then, once the retinyl esters are in micellar form, they can be hydrolysed by pancreatic lipases to liberate retinol, which is then absorbed from the micelle across the enterocyte’s brush border and into the cytosol. β-carotene also requires micellisation for absorption, but the carotene molecule enters the enterocyte intact through the brush border transmembrane transporter ABCA1 (31). Inside the cell, most of the newly absorbed β-carotene is cleaved at the central 15,15′ double bond by the soluble enzyme β-carotene monooxygenase-I (BCO-1); this cleavage immediately produces two molecules of retinal that can then be reduced by retinal reductase to form retinol. β-Carotene may also undergo eccentric cleavage, in which case at most one molecule of retinal is formed, however, this route is considered secondary (56). Moreover, small amounts of retinoic acid are generated during the absorption of vitamin A and this metabolite may play a regulatory role in the intestine or be transported from the intestine via the portal vein (57).
As retinol is insoluble in the aqueous cytosol, its intracellular trafficking and further metabolism require that it be bound to a cellular retinol-binding protein (CRBP). In enterocytes the predominant form of CRBP is CRBP-II, which binds retinol that is derived either from metabolism of preformed vitamin A or carotenoids, and chaperones it to the esterifying enzyme lecithin:retinol acyltransferase (LRAT), which produces newly esterified retinol (58). Although the absorptive pathways for retinol and β-carotene converge at the CRBP-II–LRAT step, there are some differences in overall efficiency. For preformed vitamin A, the efficiency of absorption is typically around 70%, whereas for carotenoids, it is typically 20–30%, being lower as the ingested dose increases (5, 59). A feedback mechanism has recently been described whereby retinoic acid acting as a ligand for RAR induces an inhibitory transcription factor, ISX, that blocks the transcription of the ABCA1 and BCO-1 required for carotene uptake and cleavage, and subsequent retinol esterification (60).
Moreover, in humans and a few other mammals, a small fraction of β-carotene escapes cleavage, becoming incorporated directly into the lipid core of nascent chylomicrons. In this manner, the chylomicrons, which are present in plasma only in the postprandial state, transport not only triglycerides and other newly absorbed dietary lipids, but small quantities of fat-soluble vitamins including retinyl esters and carotene, the quantity of which depends on the amounts in the recently ingested meal (5). β-Carotene can be transformed sequentially to provide the photoreceptor pigments, as can preformed vitamin A (61).
After the newly formed chylomicrons enter the circulation, their triglycerides are rapidly hydrolysed by lipoprotein lipase to provide fatty acids to tissues, leaving in the circulation the so-called chylomicron remnants that are taken up by numerous tissues, with most entering the hepatocyte through lipoprotein receptors. Once retinol has entered the liver, after further metabolic processing (5), retinol can either be stored in hepatic stellate cells as retinyl esters, mediated by CRBP (type 1) and LRAT, or it can be secreted into plasma bound to its transport protein, retinol-binding protein (62). Esterification predominates under conditions of adequate vitamin A intake, and significant vitamin A stores may build up in the hepatic stellate cells over time. When, however, tissues are depleted of vitamin A, the newly absorbed retinyl esters in the chylomicron remnants are hydrolysed and the retinol is shunted towards the holo-RBP secretory pathway and into plasma (5).
Plasma transport
When vitamin A is required by tissues, the hepatic retinyl ester pool in the hepatic stellate cells is drawn upon by hydrolysis reactions, and the retinol is transferred back to the hepatocytes where newly synthesised retinol-binding protein (RBP) binds the retinol and the complex, known as holo-RBP, is secreted into plasma. Holo-RBP consists of one molecule of all-trans-retinol per molecule of RBP, which then associates in plasma with a co-transport protein, transthyretin (63). In the fasting state and, in fact, during most of the day except in the postprandial period after consumption of vitamin A-containing meals, the majority of plasma vitamin A is in the form of holo-RBP (5). In healthy humans, holo-RBP concentration is quite steady over time, between 1 and 3 μmol/L, and is regulated in this relatively narrow range over a much wider range of liver vitamin A concentrations (64). Because RBP synthesis is essential for the secretion of holo-RBP, it has been of interest to understand conditions that may compromise the synthesis and secretion of holo-RBP and thereby result in low serum retinol and RBP concentrations. Conditions that limit holo-RBP production include inadequate protein or energy consumption, and zinc deficiency, and renal diseases leading to hyperfiltration or reduced reuptake in the proximal tubules. Additionally, both experimental and clinical data have shown that circulating levels of holo-RBP, retinoids, and carotenoids are low during infection or inflammation, independently of vitamin A nutritional status (65), and may affect the response to vitamin A supplementation (66). This is due in part to RBP’s nature as a negative acute phase protein; its concentration falls rapidly in response to inflammatory stimuli (67). Thus, low holo-RBP levels in patients with inflammatory conditions should be interpreted with caution, as the underlying cause could be nutritional or it could be due to inflammation itself (63).
Vitamin A and the eye
Retinol in the retina
The retina must acquire retinol from holo-RBP circulating through the capillaries in the choroid. A receptor for RBP, Stra6, was described, cloned and shown to be most abundantly present in the basolateral plasma membrane of the retinal pigmented epithelial (RPE) cells (68). Stra6 binds RBP and allows for the uptake of retinol, which very likely is immediately picked up by CRBP-1 on the cytosolic side and, then, similar to other tissues, esterified by LRAT (69). Recent studies have identified that Stra6 transports retinol both into and out of cells, with calmodulin as an interacting regulatory protein (70).
The retina is unique in having an abundance of 11-cis-retinoids, the essential chromophore for all photoreceptor cells; vitamin A is thus an absolutely required nutrient for vision (71). The RPE cells contain retinyl esters in both the all-trans- and the 11-cis configuration, suggesting that LRAT also esterifies 11-cis-retinol produced by isomerisation in the retina. The concentration of vitamin A is the highest in the eye of any tissue, reaching as high as 3 millimolar in the retina (72, 73), mostly in the form of 11-cis-retinal bound in a Schiff base with rhodopsin in rods or photopsins in cones. Opsins are G-protein coupled receptors (GPCRs), a class of protein which underlies vision in all animals, including in man (11). In addition to rhodopsin in rods for vision in scotopic-mesopic light intensities, and three photopsins in cones for colour-tuned vision in mesopic-photopic light intensities, humans express a fifth opsin, melanopsin, which facilitates non-image forming vision (10, 74–77). The molecular interaction between the specific GPCR and chromophore tunes light sensitivity to different wavelengths (78, 79). Each of the five human opsins has a different and characteristic absorption spectrum due to their slightly different interactions with the 11-cis-retinal chromophore; dependent on a relatively small number of key amino acid residues (80, 81). Long, medium, and short-wavelength sensitive photopsins underlie colour vision by facilitating visual contrast owing to preferential responses to red, green, and blue light, respectively (77, 79). At lower light intensities, rods facilitate vision with rhodopsin-dependent transduction, due to both specific photoreceptor characteristics and downstream signalling pathways adapted to optimise sensitivity (82, 83). The human retina is thus described as “duplex,” as it depends on two photoreceptor classes at different ambient light levels, developed in discrete evolutionary steps (84).
At the molecular level, a photon of light provides the energy to switch the chromophore’s structure, from 11-cis to all-trans retinol. This increases the physical strain on the molecule, likened to a spring, with relief on tension conferred by altering the conformation of the opsin, which thus activates a signalling cascade (85). As the photoisomerisation transduction process results, at least transiently, in the formation of “free” retinoids, and as aldehydes in particular are highly reactive molecules, especially in the presence of amines, researchers have looked for and identified a number of adduct products of retinal and cellular amines. Those identified include A2E, a bisretinoid condensation product of retinal and phosphatidyl ethanolamine (PE), its isomer iso-A2E, and other products such as lipofuscin (86). These by-products of the photoreceptor processes are difficult for cells to clear and, though reversible, may accumulate. A2E, lipofuscin, and other bisretinoids are discussed below as the metabolites responsible for the pathogenesis of Stargardt’s disease.
Corneal vitamin A dependence
The cornea is essentially an avascular tissue, yet it also requires retinol for the normal structure and function of the conjunctival and corneal epithelial cells. The synthesis of RBP by the lacrimal gland and the presence of RBP in tear fluid was noted some time ago (87, 88). While relatively little is understood of the mechanisms by which RBP enters the lacrimal glands and is taken up by corneal cells from tear fluid, it seems likely that tears may play an important role in providing retinol to this epithelium. The lacrimal gland, cornea and its cells contain retinoid receptors and respond to all-trans-retinoic acid (89–91). Conjunctival impression cytology, in which cells lifted from the outer edge of the conjunctiva are stained for mucins, has been used as a clinical tool for assessing vitamin A deficiency (92); effective as vitamin A deficiency presents clinically as corneal xerosis, colloquially called “dry eye.” The broader ocular syndrome associated with vitamin A deficiency, xerophthalmia, which involves the cornea and retina, is described in greater depth below.
Vitamin A’s influence on T cell function
Shortly after vitamin A was initially identified in the early 1900s, it was termed “the anti-infective vitamin” (93). It has important roles in innate immunity with regards to barrier (i.e., skin), neutrophil, macrophage, and NK cell function (93). Furthermore, it is thought be a key player in immune regulation through its effects on adaptive immunity, particularly via T-helper (Th) cells (7, 93). Vitamin A mediates these effects directly in the bioactive form of RA, and, also, via vitamin D and the vitamin D receptor (VDR). To mediate an anti-inflammatory effect, active vitamin D, calcitriol binds to the VDR to form a VDR-D complex which then binds to the RXR receptor, activated by its ligand RA. It is this heterodimer complex, RXR–RA/VDR-D, dependent on vitamin D and vitamin A, that controls the expression of several genes involved in inflammatory and autoimmune processes in chronic diseases by inhibiting the pro-inflammatory transcription factor NF-κB and down-regulating the synthesis of inflammatory molecules (7, 94, 95). It is also worth noting that, given their common RXR nuclear binding partners, calcitriol and retinoic acid may antagonize each other’s effects (7, 96).
Retinoic acid is a critical signalling factor which drives differentiation of iTreg cells (20, 97–100). iTreg differentiation is favoured, for example, in the presence of TGF-β and adequate amounts of RA, but low IL-6 (20). RA indirectly promotes TGF-β-mediated Treg cell conversion by suppressing pro-inflammatory cytokine production, thereby stabilising FoxP3 expression. Generation of iTreg cells also occurs through direct binding of FoxP3 to RORα and RORγt, inhibiting their transcriptional activity (101, 102). RA produced by specific dendritic cell (DC) subsets, directly and indirectly modulates FoxP3 expression and increases the activation of extracellular-related kinase (ERK) signalling to promote FoxP3 expression (100, 103, 104). RA increases demethylation and histone acetylation of the FoxP3 gene locus and promotor, respectively (100). RA attenuates the deceleration of FoxP3 expression during Treg cell expansion and in inflammatory conditions, with superior efficacy compared with rapamycin, an mTORC1 inhibitor known to stabilise FoxP3 expression (100). RA also suppresses Th17 cell development by increasing TGF-β signalling and reducing the level of expression of the IL-6 receptor (105, 106). It is thought that when conditions favour immune tolerance, there is a balance between effector T cells and Treg cells, whereas imbalances occur in autoimmunity, which could be due to inadequate numbers of Treg cells, defects in Treg function or phenotype or reduced responsiveness of effector T cells to Treg-mediated suppression (107). Therefore relative increase in iTreg and the inhibition of Th17 development mediated by RA, can shift the balance of these cells toward more effective immune homeostasis (20).
The T cell-mediated gut-eye axis
It is well recognised that the human immune system has a highly co-evolved relationship with the microbes that inhabit the human intestine, or microbiome, resulting in the co-maintenance of homeostasis between the host and resident microbes (108). This relationship is shaped throughout development and into adulthood, and contributes to the function of the gastrointestinal immune system, as well as playing an important role in health and disease throughout life (108). Dysbiosis is defined as disruption to the microbiome’s homeostasis, caused by imbalanced microbiota structure or function, and is an emerging feature of many non-communicable inflammatory diseases (109). The gut microbiome composition varies according to macro/micronutrient dietary habits (110–115). Different bacterial taxa modulate immune functionality, driving pro or anti-inflammatory activity (28, 116, 117). Thus, the composition of the microbiota community determines, in part, the level of resistance to infection and susceptibility to inflammatory diseases.
RA seems to play a predominant role in the homeostasis and homing of lymphoid populations of the gut-associated lymphoid tissue (GALT). It is synthesised in abundance by gut DCs (118, 119), induces the specific gut-homing molecules CCR9 and α4β7 integrin expressed by T cells, and also promotes GALT-related activity in B cells (7, 118). The balance between Th17 and iTreg cells and their ability to cross-regulate one another have emerged as crucial factors for normal mucosal immunity (19, 98, 101). The local microenvironment, which may include the cytokine milieu and the microbiome, has been shown to regulate the Treg/Th17 balance and influence cell plasticity; disruption of this balance may lead to the development of inflammatory disease (120–123). Paradoxical expression of effector CD4 T-cell transcription factors, such as T-bet and RORγt, by Treg has been suggested to enhance Treg suppressive capacity in murine models of inflammation (123–126). These murine models demonstrate that intestinal “double positive” RORγt+FoxP3+ Treg cells induced in vivo by the local microbiota display a stable suppressive phenotype and exist in dynamic balance with pathogenic Th17 (122, 125).
Induction of Treg cells in the small intestine appears to depend more on dietary antigens than microbes, according to experimental results in murine models. RA acts in concert with TGF-β for Treg cell generation (27, 119, 127), where abundant vitamin A from the diet is converted to RA by local epithelial cells and DCs (5, 128, 129). Moreover, RA induces the expression of CCR9 on Treg cells, which is required for their migration to small intestine (27, 127). RA-induced Treg cell differentiation underlies oral tolerance, as mice treated with a vitamin A-deficient diet display defects in the induction of oral tolerance (130). Thus, dietary antigens and RA-dependent Treg cells in the environment of the small intestine may modulate inflammation at distant sites immune-privileged sites, such as the retina. This is one potential mechanism of many by which the gut microbiome affects retinal health, potentially contributing to a wide range of chronic eye diseases, including AMD (29, 30).
Ocular immune privilege is a complex phenomenon that involves multiple components, starting with sequestration behind an efficient blood–retina barrier, through active local inhibition by soluble and surface-bound molecules that actively inhibit activation and function of adaptive and innate immune cells, and culminating in systemic regulation via induction of Treg (131). Failure of ocular immunological tolerance may lead to classical autoimmune-type inflammation, which may manifest in the eye as uveitis, an inflammatory disease of the choroid, which is often associated with inflammation of the overlying retina. It is hypothesised that RPE cells produce RA, thereby enabling bystander T cells to be converted into Tregs through TGFβ promotion, which can then participate in the establishment of immune tolerance in the eye (132). There is evidence that RORγt-expressing Th17 cells act as the effector cells in ocular inflammation (133), including B27+ anterior uveitis (17, 98, 101, 134). In a large clinical study of patients with non-infectious uveitis, it was shown that systemic Th17 cells were not associated with active ocular inflammation and that double positive FoxP3 + RORγt + Treg cells were not significantly associated with clinical remission of ocular inflammation (135). However, systemic Th17 cells from patients with active uveitic disease were observed to express FoxP3 during the disease resolution phase, in response to immunosuppressive treatment (135). There has been interest in glucocorticoid-resistant Th17 cells refractory to Treg suppression (136, 137), and these have been previously investigated in sight-threatening uveitis (138). Furthermore, a subset of “non-classic” Th17-derived Th1 have been described, which are hypomethylated at RORC, the gene encoding RORγ, and are thought to play a pivotal role in the establishment and persistence of inflammatory disease (139). These non-classic Th1 express CD161, a marker of steroid-resistant human Th17, induced by RORγ signalling (140). CpG methylation at RORC in systemic Treg appears to indicate clinical remission or clinical response to systemic corticosteroids in non-infectious sight-threatening uveitis (135), highlighting the potential role of RA in T-cell mediated ocular inflammation.
Vitamin A deficiency and excess
Although the body stores vitamin A as retinyl ester in liver and to a lesser extent in adipose, lung, and other tissues, these stores may become depleted over time if the diet is consistently deficient in vitamin A, or stores may never develop in children who are not adequately breastfed and/or do not receive an adequate diet after weaning (141). Food deprivation, unbalanced diet, and malabsorption syndromes are the most common causes of deficiency (142–144). Worldwide, dietary deficiency is by far the most common cause, epidemic in developing countries and particularly affecting young children (145, 146). Dietary deficiency may also be observed in individuals with low general food intake, such as in anorexia or bulimia nervosa (142). Fat malabsorption syndromes can result in hypovitaminosis A, such as due to chronic pancreatitis, inflammatory bowel disease, and liver disease (142, 147).
Dietary advice regarding optimal vitamin A intake varies between jurisdictions, and is summarised in Table 1 (33, 148, 149). As discussed above, the many forms of vitamin A distribute differently across a wide range of foods. To deal with this variability, the vitamin A activity of biological substances and of the daily nutritional requirement may be expressed in terms of “retinol equivalents” (RE). 1 μg RE may be considered equivalent to 1 μg of retinol, 6 μg of β-carotene, or 12 μg of other provitamin A carotenoids (149). Daily intake for vitamin A can be met with any combination of preformed vitamin A or carotenoids that produces the correct quantity in RE terms.

Table 1. Summary of dietary advice regarding daily vitamin A intake from the United States of America (National Institutes of Health Office of Dietary Supplements, NIH ODS), United Kingdom (Scientific Advisory Committee on Nutrition, SACN), and European Union (European Food Safety Authority Panel on Dietetic Products, Nutrition, and Allergies, EFSA NDA).
Vitamin A deficiency affects many physiological processes. The body’s epithelial tissues, including the cornea and retina, as well as the immune system are affected, resulting in xerophthalmia and a dysregulated immune response (95, 150). In addition, aberrant vitamin A levels in pregnant patients leads to developmental defects in the embryo (146).
Xerophthalmia
Although xerophthalmia literally means “dry eye,” it is generally used as a term to describe the collective ocular syndromes observed in hypovitaminosis A (151), with clinical features classified by the World Health Organisation (Table 2) (152). The bulbar conjunctiva lining the globe of the eye, and not the palpebral conjunctiva lining the eyelid, is affected by xerosis, or dryness (X1A), sometimes difficult to detect (152). The superficial conjunctiva may accumulate keratin, forming foamy patches known as Bitot’s spots (X1B) (153, 154). Acute vitamin A deficiency presents with corneal pathology. Corneal xerosis (X2) is most common, as in the conjunctiva. Dryness of the corneal surface predisposes to ulceration, an ophthalmic emergency (155), and keratomalacia, characterised by diffuse keratinisation of the corneal surface, permanently impairing vision; these pathologies are categorised based on the proportion of the cornea affected (X3A/X3B). Ulcers and keratomalacia lead to permanent corneal scarring (XS). The retina is affected due to the requirement of vitamin A to produce the chromophore discussed above, essential for phototransduction. Deficiency leads to nyctalopia, or night blindness (XN) as rods are more sensitive to deficiency, and these photoreceptors underlie vision in scotopic light levels. More rarely, structural damage to rods is observed, known as xerophthalmic fundus (XF).

Table 2. World Health Organisation classification of xerophthalmia, based on clinical ophthalmological features. Shaded cells correspond to retinal pathology.
Xerophthalmia is treated with large dose vitamin A supplementation, up to 200,000 IU (150, 154). Oral and intramuscular supplementation is equally effective (150). While vitamin A toxicity following supplementation is unlikely in patients with symptomatic deficiency before treatment, acutely high intake has theoretical risks associated with vitamin A’s roles in inflammation, as well as the contribution of retinoid products to Stargardt’s disease, discussed below.
Immune dysregulation
It has been observed that in populations where vitamin A availability from food is low, infectious diseases can precipitate vitamin A deficiency by decreasing intake, decreasing absorption, and increasing excretion (93). Vitamin A deficiency has profound effects on innate immunity, including impeding normal regeneration of mucosal barriers damaged by infection, impairing mucosal inflammatory responses contributing to the synthesis of antibodies, especially IgA and by diminishing the function of neutrophils, macrophages, and natural killer cells (93). Vitamin A deficiency in viral infections, such as measles, has been shown to increase disease severity and appropriately timed supplementation during recovery has been found to reduce mortality and hasten recovery (156). The role of Vitamin A in T-cell-mediated immunity is discussed above, with reference to Treg and Th17. In addition to these effects, vitamin A deficiency has been shown to diminish Th2-mediated antibody responses and may also impair Th1 responses, whereas high dietary vitamin A may enhance Th2-mediated responses (7, 93, 157). This is thought to contribute to the increased mortality observed in vitamin A-deficient children and pregnant women.
Hypervitaminosis A
Vitamin A deficiency is treated with supplementation, and vitamin A supplements are widely used with no adverse effects. However, acute toxicity is possible. Excessive vitamin A intake is generally associated with excessive use of vitamin A-containing supplements (158, 159). Persistently high intakes can result in overt vitamin A toxicity, described as hypervitaminosis A syndrome (160), and may exacerbate Stargardt’s disease (161).
Women of child-bearing age are cautioned not to consume high amounts of vitamin A-rich foods such as organ meats. The use of prescription retinoids is known to be associated with birth defects and thus their use by persons of reproductive age is highly regulated, and generally not recommended outside of settings where vitamin A deficiency is a public health problem (162). On the other hand, β-carotene has not been shown to be teratogenic or to produce other symptoms of toxicity and therefore it may be considered a “safe” form of vitamin A (163). Consistently high intakes of carotenoids may result in hypercarotenaemia and deposition of carotenoids in skin, causing yellowing (164), but the condition is benign (165).
Parainflammatory degenerative retinal disease
Stargardt’s disease
Stargardt disease (STGD) is the most common single-gene heritable disease of the retina, with three genetic bases: STGD1 caused by autosomal recessive ATP-binding cassette, sub-family A, member 4 (ABCA4) mutations (166); STGD3 caused by autosomal dominant mutations in “elongation of very long chain fatty acid protein 4” (ELOVL4) (167); STGD4 caused by autosomal dominant mutations in prominin 1 (PROM1) (168). STGD2 was attributed erroneously to a distinct genetic locus, but is in fact attributable to ELOVL4 mutations, and is now diagnosed as STGD3. STGD1 is the most common form of Stargardt’s (169), driven by a lack of functional ABCA4. Clinically, patients present with bilateral central visual loss and dyschromatopsia, due to macular atrophy (170, 171). On examination, the fundus exhibits characteristic yellow-white lesions, centred on the retina (172). Onset is usually early in life, with peaks in childhood and early adulthood; later onset is associated with better prognosis (173, 174). The disease tends to progress gradually, outward from the central macula, often over years (171, 175–177). Wide variability in disease trajectory, between patients with particular mutations as well as within individuals with similar genetics indicates dependence on both genetic and environmental factors (178, 179).
ABCA4 encodes an active transporter highly expressed in the retina (180), specifically localising in the outer segments of rod and cone photoreceptors according to immunofluorescence studies (181, 182). The protein functions as a retinoid “flippase,” facilitating excretion of all-trans-retinal following light responses (183), as well as excess 11-cis-retinal which otherwise accumulates in dark conditions (184). Unlike most ATP-binding cassette proteins, ABCA4 acts as an importer, specifically of N-retinylidene-phosphatidylethanolamine (NRPE) (185), a Schiff-base conjugate of PE and retinal (186), in either an N-cis (NcRPE) or N-trans (NtRPE) conformation, associated with 11-cis and 11-trans-retinal, respectively (187). The protein functions as a retinoid “flippase” of NRPE from the luminal to the cytoplasmic side of the phospholipid bilayer that comprises the outer segment disc membrane (187), facilitating excretion of all-trans-retinal following light responses (183), as well as excess 11-cis-retinal which otherwise accumulates in dark conditions (184). All-trans retinal produced by photoisomerisation binds to PE to form NtRPE in the outer segment disc membrane, which ABCA4 “flips” towards the cytoplasm for entry into the visual cycle, to regenerate chromophore (187). 11-cis-retinal chromophore can be produced to excess in the darkness; it also binds with PE to form NcRPE in the outer segment membrane, which is similarly “flipped” towards the cytoplasm by ABCA4, where it is isomerised into NtRPE before dissociating into PE and all-trans-retinal which re-entering the visual cycle (187). This “flipping” function of ABCA4 is illustrated in Figure 4.
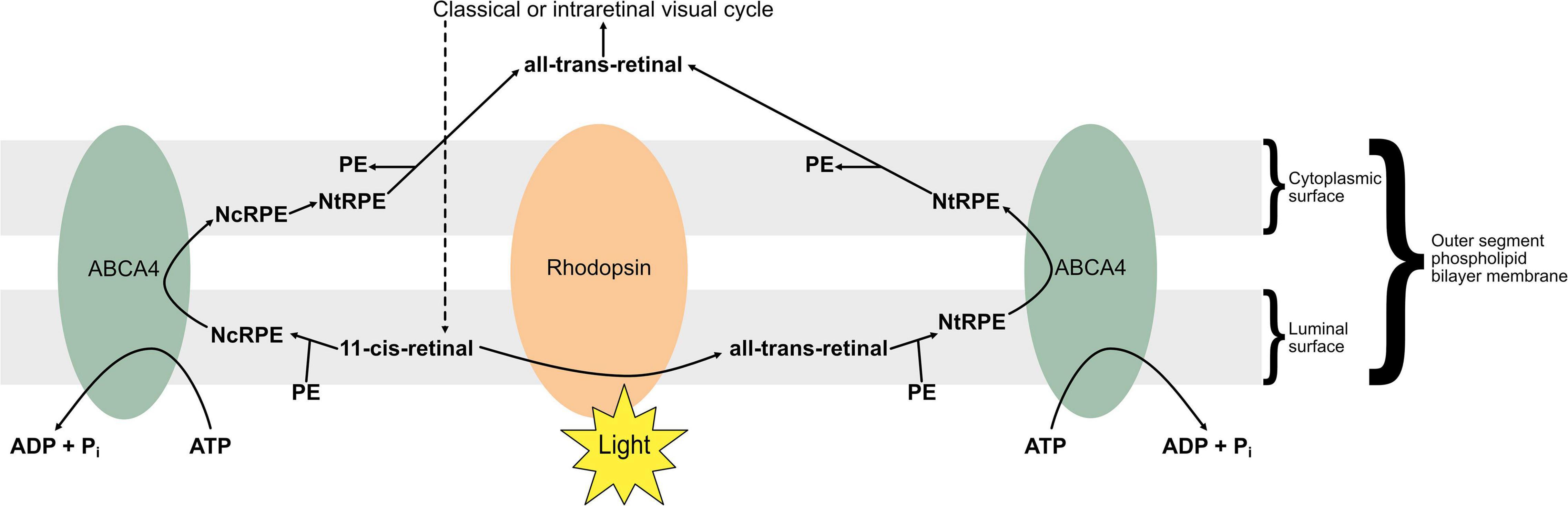
Figure 4. The “flipping” function of ABCA4. 11-cis retinal accumulates in darker conditions as opsins are saturated, whereas all-trans retinal is produced by photoisomerisation in lighter conditions. Retinal associates with membranal phosphatidylethanolamine (PE) to form N-cis-retinylidene-PE (NcRPE) and N-trans-retinylidene-PE (NtRPE), which ABCA4 flips across the membrane at the cost of ATP hydrolysis. NcRPE and NtRPE dissociate, and 11-cis retinal undergoes chemical isomerisation, allowing all-trans retinal to reenter the visual cycle.
A lack of functional ABCA4 at the rim region of outer segment disc membranes underlies the majority of STGD1 cases (187): this can be due to loss of ATPase activity, NRPE “flipping” activity, mislocalisation of protein, or protein misfolding (182, 186, 188, 189). As a result, NRPE, therefore retinal, is not removed from disc membranes as efficiently, leading to retinoid accumulation. These retinoids may give rise to pyridinium bisretinoids including A2PE (190). A2PE undergoes hydrolysis by phospholipase D to form A2E, a fluorophore and major component of lipofuscin (187, 190, 191), which characteristically accumulates in STGD1 (183, 192). Lipofuscin and other bisretinoids like A2E have a toxic effect on the photoreceptors and are thought to underlie the STGD1 disease process (184, 189, 192–194). It is lipofuscin accumulation underlying the characteristic yellow-white lesions on fundoscopy in STGD1, giving the retina a beaten-bronze appearance (172, 195).
Age-related macular degeneration
Age-related macular degeneration is a common cause of vision loss in the ageing population, growing in prevalence (196, 197). Patients initially experience subtle disturbances to their vision, which progress over time to permanent loss of central vision, as disease locates primarily to the central retina, or macula, as in STGD (198). AMD occurs in two distinct forms, “dry” and “wet,” with wet AMD contributing to the vast majority of severe central visual loss (199). In dry AMD, progressive loss of vision occurs due to degeneration of the choriocapillaries, outer retina, and photoreceptors (200). Outer retinal atrophy is generally observed earliest, with eventual progression to “geographic atrophy,” where the outer retina and RPE are completely atrophic (201). Wet AMD is characterised by neovascularisation, driven by immune cells recruited to the damaged macula. Cytokines underlie immune cell recruitment and neovascularisation, with vascular endothelial growth factor (VEGF) being especially involved in wet AMD (198). The earliest sign of all forms of AMD is drusen: subretinal deposits of lipids, proteins, and carbohydrates which appear as white or yellow deposits on fundoscopy, comparable to lipofuscin (198, 202).
The aetiology of AMD is complicated, with a wide range of risk factors, both modifiable (e.g., smoking) and non-modifiable (e.g., family history) (199, 203). Particular genotypes are associated with AMD, and as in STGD, ABCA4 has been specifically implicated (204, 205). Accumulation of bisretinoids such as A2E is associated with AMD as well as STGD, and administration of exogenous A2E induces ocular changes similar to AMD and STGD in animal models (206–208). However, defining a causal link between ABCA4 mutations and AMD has proven more challenging, as some researchers find no association (209, 210), perhaps due to genetic heterogeneity in the AMD phenotype, or incomplete penetrance of disease-causing alleles (211). An added complication lies in the comparable presentations of Stargardt’s and AMD, albeit usually in younger and older patients, respectively, with consensus favouring the role of ABCA4 as limited to Stargardt’s and related retinal dystrophies and mimicking, rather than causing AMD (211, 212). ABCA4 is nevertheless included in the EYE-RISK genotype assay for AMD, as retinal dystrophies are an important differential diagnosis in patients presenting with macular pathology (212).
Retinoic acid and parainflammation in the retina
Parainflammation, defined as an adaptive tissue response to noxious stress, between basal and inflammatory states, has been proposed as a mechanism for the pathogenesis of degenerative retinal disease (213, 214). Furthermore, immunosenescence (altered immune functions during ageing) is thought to play an important role in AMD, which could be considered a systemic immunologic disease, with local expression in the retina, due to the decline of the ocular down-regulatory immune environment (30). Oxidative stress in the retina increases with age, with reactive oxygen species (ROS) affecting metabolic processes (215, 216). ROS are produced physiologically, such as due to lipofuscin accumulation or due to environmental influences, including ultraviolet (UV) radiation from sun exposure, or other modifiable AMD risk factors such as smoking (217). Inflammatory processes are also thought to contribute, particularly from tissue-resident immune cells and supporting cells, such as Müller glial cells (216). Macrophages appear to be particularly involved in AMD, with preferential differentiation of the pro-inflammatory M1 phenotype, as opposed to the M2 phenotype associated with regular ageing (218, 219). As the retina is exposed to physiological stress, immune activation may exacerbate cellular damage, leading to loss of function (214). In Stargardt disease, the A2E vitamin A cycle byproducts form abnormally in the young retina, whereas in AMD, the byproducts form, to a similar degree, in the aged retina and it is hypothesised that the difference in the two diseases is explained by the age of the eye in which the vitamin A cycle byproducts (208). Determining the effects of dietary vitamin A on parainflammation in STGD1 and AMD is complex as it requires consideration of the pleotrophic effects of RA-signalling and its targets (in the eye, gut or other immunological sites), the age of the eye, the extent of local retinal disease, as well systemic immunological factors. Whereas RA is known to act directly on macrophages at mucosal and other immunological sites to modulate inflammation (93, 220), the effect of RA on local immune antigen cells residing in damaged RPE may well be pro-inflammatory, as a result of aberrant Vitamin A processing. On the other hand, the parainflammatory response to deposition of toxic metabolic byproducts could potentially be modulated by RA-dependent iTreg generation in the microbiome environment. Dietary vitamin A is likely to influence local retinal parainflammatory processes through the effects of RA signalling on the microbiome environment, systemic T cell homeostasis and resident retinal immune cells but whether the overall effect of RA is beneficial or detrimental to retinal disease is yet to be determined.
Clinical applications and directions for research
No curative treatments exist for STGD or AMD. Current research may lead to improved options relating to pharmacology, stem cells, laser treatment, and implantable intraocular lens telescopes (221). The quality of evidence for potential treatments is variable (221). Nutritional and retinoid- and carotenoid-related approaches are explored below, with human evidence summarised in Tables 3, 4.

Table 3. Human-based experimental studies relating to vitamin A treatment for STGD, with PICO characteristics summarised.
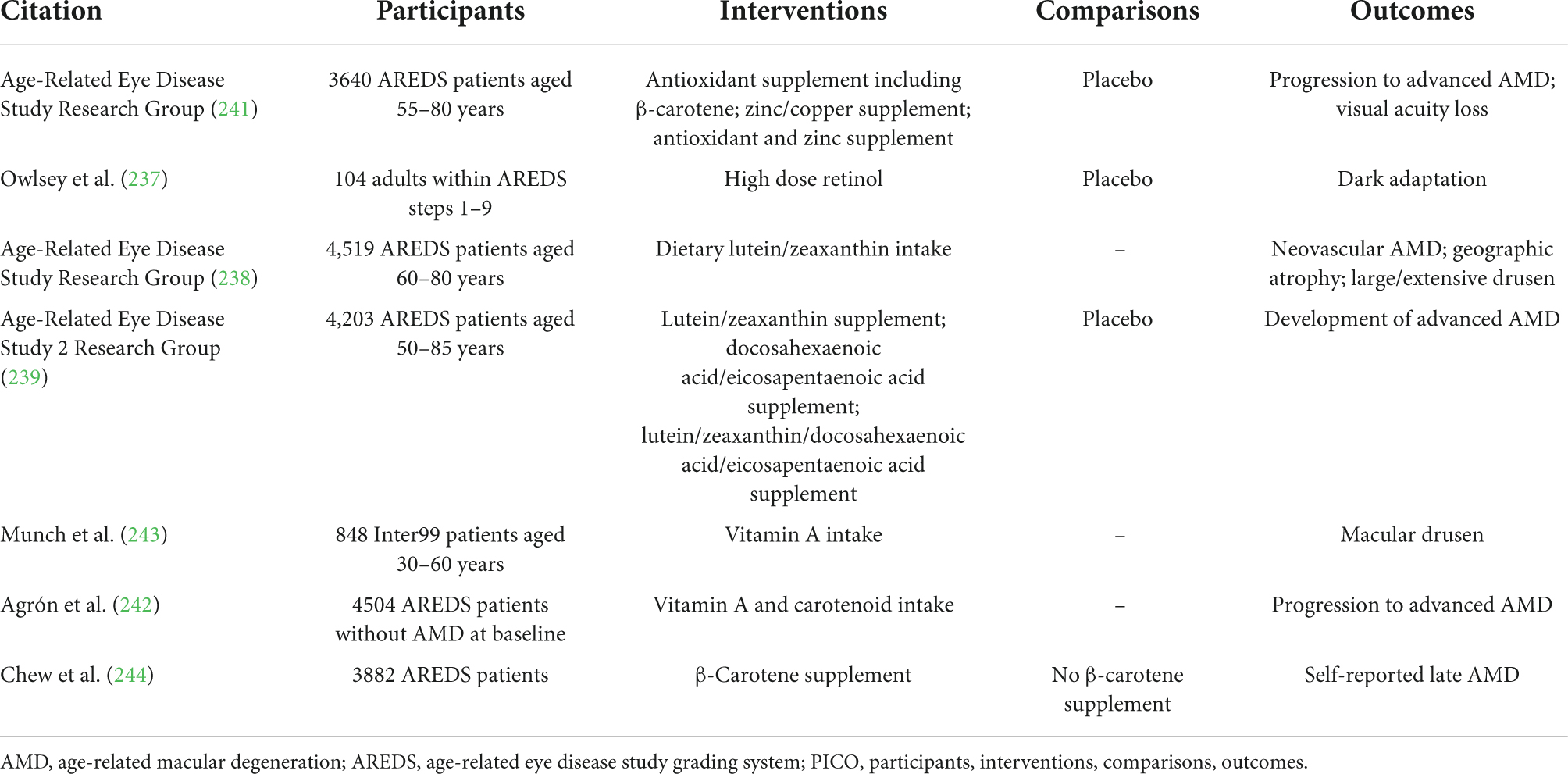
Table 4. Human-based studies relating to vitamin A in AMD, with PICO characteristics summarised.
Stargardt’s disease
Given that the pathophysiology of STGD depends on accumulation of retinoids in photoreceptor outer segments, various research groups have hypothesised that vitamin A levels ought to correlate negatively with outcome in patients lacking functional ABCA4. A mouse-model for STGD1 has been developed, by functionally knocking out the ABCA4 gene (222). Although genetically and phenotypically similar to human STGD1 patients, the validity of this model has been questioned as ABCA4-knockout mice are healthy unless exposed to very bright light (223). The relevance of ABCA4-knockout models to other STGD genotypes, with pathogenic mutations outside ABCA4, is also questionable (169). In ABCA4-knockout mice, no association is apparent between vitamin A supplementation and retinal morphology and structure (161, 224), although greater lipofuscin accumulation following vitamin A supplementation was noted in one study (161). Studies in humans are not much more conclusive than results in animal models. A small cross-sectional study of 24 patients assessing vitamin A intake with diet questionnaires found low vitamin A intake associated with better visual acuity (225). However, a larger prospective cohort study (n = 259) found vitamin A supplementation had no effect on visual acuity at baseline, or over 12 months (226).
Another potential strategy is based on deuterium-enriched vitamin A (C20-D3-vitamin A), which reduces vitamin A dimerisation through competitive inhibition, thereby slowing A2E biosynthesis (227). C20-D3-vitamin A has the added benefit of reacting normally throughout the visual cycle, therefore not impinging upon chromophore generation which is required for visual transduction (228). In ABCA4-knockout mice, C20-D3-vitamin A has exhibited conflicting results: one study finding greater electroretinogram responses with C20-D3-vitamin A supplement relative to non-deuterated vitamin A supplement control (229); one study finding no differences between similar groups, though vitamin A dimerisation was inhibited (230); another study finding that C20-D3-vitamin A inhibited prodromal pathological features (231). Following a successful phase 1 clinical trial assessing the safety and pharmacokinetics of C20-D3-vitamin A (232), a larger phase 2 clinical trial assessing deuterated vitamin A’s long-term safety and tolerability is currently recruiting (233).
Carotenoid supplementation, which provides a source of retinoids, has been suggested as a potential nutritional approach to improving STGD outcomes by protecting the macula (221), due to observed lower carotenoid serum levels in STGD patients compared to controls (234). While carotenoid supplementation has a positive effect on visual performance in ABCA4-knockabout mice (235), no nutritional trials have been conducted as of yet (219). Although there is conflicting evidence regarding dietary vitamin A and STGD outcomes (225, 226), laboratory evidence of vitamin A supplementation driving greater lipofuscin accumulation (161), albeit without significant effects on retinal function (57, 224) has led to recommendations for STGD patients to avoid retinoid supplements (221). Whether or not potential differential effects of dietary carotenoids and retinoids are related to differences in the gut microbiome of STGD patients is not clear; if demonstrated, this would represent another avenue for experimental enquiry.
Age-related macular degeneration
Investigating the effects of nutritional supplements in AMD is complicated, as although serum levels of retinoids or carotenoids may increase quickly, changes in the macular may occur over months, and effects on visual function could take even longer, perhaps years, to manifest (236). Nevertheless, retinoid and carotenoid deficiency are implicated in the progression of AMD (237, 238), and evidence from large randomised controlled trials of nutritional supplementation in ophthalmology patients, AREDS1 and AREDS2 (239, 240), suggests that sufficient levels of vitamin A or β-carotene was associated with decreased risk of late AMD, neovascular AMD, geographic atrophy, and large drusen accumulations (239, 241, 242). This agrees with the results of a meta-analysis of six longitudinal cohort studies examining the impact of carotenoids on AMD prevalence (229). However, one study found a greater risk of drusen in patients with high vitamin A intake, perhaps suggesting that supplementation increases the risk in patients with very early stage disease (243). In terms of the specific nutrient to supplement, zeaxanthin is preferred to β-carotene, as the latter tends to compete with other supplemented nutrients for absorption (221). Lutein and zeaxanthin supplementation appears to associate with better outcomes in late AMD than β-carotene (244).
It remains an open question as to how nutrition can be optimised to improve outcomes in STGD1 and AMD. While retinoids are critical for vision, as explained above, it may be the case that ingesting too much accelerates the pathogenesis of STGD1 (161, 225). Retinoids and carotenoids are often deficient in AMD, carotenoid supplementation appears to be beneficial (229, 244), and vitamin A supplementation inhibits IL-17 and ROR expression in atherosclerosis, which features comparable macrophage-driven pathophysiology to AMD (245). However, certain data suggest that there may be a risk of worsening outcomes in patients with early-stage disease (243). The role of the gut microbiome in affecting treatment options remains unclear, and no attempts have been made to leverage observed differences in STGD and AMD patients in terms of clinical trials. Further research is required to (a) determine the optimal amount of dietary and retinal vitamin A to maintain normal physiology while slowing, or at least without accelerating disease processes; (b) explore whether different sources of vitamin A have different effects on Stargardt’s, for instance comparing vitamin A retinoids and provitamin carotenoids; (c) differentiate between different disease phenotypes and disease phases, and associate differential effects of vitamin A and associated therapies on their pathophysiology accordingly; and (d) investigate the influence of the gut microbiome on STGD and AMD, and develop effective therapeutics based on observed differences.
Conclusion
The group of fat-soluble compounds referred to as vitamin A is essential to humans, and has particularly significant roles in vision and T-cell mediated inflammation, as well as in a wide range of other physiological systems. Its importance is illustrated by the deficiency syndrome xerophthalmia, particularly affecting the eyes. In addition to maintaining the cornea, vitamin A is critical for the function of the retina, as it provides the raw material to produce the chromophore, 11-cis-retinal. Photoreceptor cells require the chromophore to transduce light energy for vision, and regenerate chromophore in collaboration with the RPE and Müller cells through a series of reactions known as the visual cycle. Bisretinoid byproducts of photoisomerisation and the visual cycle include A2E and lipofuscin, which can accumulate with associated toxicity to rods and cones. Vitamin A also has a critical signalling role in the immune system, with particular effects on T cell differentiation across the body, including in the intestine, in close association with the gut microbiome. Vitamin A deficiency is associated with susceptibility to infections, causing significant morbidity and mortality worldwide, as well as predisposition to autoimmunity and hypersensitivity.
In STGD1, a lack of a membrane exchange protein leads to accumulation of bisretinoids, ultimately derived from vitamin A, which culminates in blindness progressing from the central visual field as the macula is affected first. AMD similarly affects the macula, and features drusen comparable to the lipofuscin deposits of STGD, but instead primarily containing lipids and other waste products. Various retinoid and carotenoid-based approaches to treating STGD and AMD have been posited. In particular, zeaxanthin supplementation is well-evidenced as beneficial for AMD patients, while vitamin A supplementation may worsen STGD1 outcomes. However, deuterium-enriched vitamin A could target STGD1 with high specificity, and is currently in the clinical trial stage. No accepted or trialled therapies have leveraged the altered gut microbiome in STGD or AMD, and this may represent an interesting area for further research. As no curative treatments exist for STGD or dry AMD, novel approaches are necessary to improve outcomes for patients, and optimising nutrition represents an important facet of management.
Author contributions
AJT and RMG undertook editing. AJT and ACR produced figures. AJT organised references. All authors contributed to research and writing and approved the final draft for submission.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. McCollum EV, Davis M. The necessity of certain lipins in the diet during growth. Nutr Rev. (1913) 15:167–75. doi: 10.1016/S0021-9258(18)88553-2
2. McCollum EV, Simmonds N, Pitz W. THE RELATION OF THE UNIDENTIFIED DIETARY FACTORS, THE FAT-SOLUBLE A, AND WATER SOLUBLE B, OF THE DIET TO THE GROWTH PROMOTING PROPERTIES OF MILK. J Biol Chem. (1916) 27:33–43. doi: 10.1016/S0021-9258(18)86888-0
3. Carazo A, Macáková K, Matoušová K, Krčmová LK, Protti M, Mladěnka P. Vitamin A update: forms, sources, kinetics, detection, function, deficiency, therapeutic use and toxicity. Nutrients. (2021) 13:1703. doi: 10.3390/nu13051703
4. Jakobsen J, Melse-Boonstra A, Rychlik M. Challenges to quantify total vitamin activity: how to combine the contribution of diverse vitamers? Curr Dev Nutr. (2019) 3:nzz086. doi: 10.1093/cdn/nzz086
5. Ross AC. Vitamin A. In: Modern Nutrition in Health and Disease. 11th ed. Baltimore, MD: Lippincott Williams & Wilkins (2014). p. 260–77.
6. Huang P, Chandra V, Rastinejad F. Retinoic acid actions through mammalian nuclear receptors. Chem Rev. (2014) 114:233–54. doi: 10.1021/cr400161b
7. Mora JR, Iwata M, von Andrian UH. Vitamin effects on the immune system: vitamins A and D take centre stage. Nat Rev Immunol. (2008) 8:685–98. doi: 10.1038/nri2378
8. Ghyselinck NB, Duester G. Retinoic acid signaling pathways. Development. (2019) 146:dev167502. doi: 10.1242/dev.167502
9. Berson DM, Dunn FA, Takao M. Phototransduction by retinal ganglion cells that set the circadian clock. Science. (2002) 295:1070–3. doi: 10.1126/science.1067262
10. Hattar S, Liao HW, Takao M, Berson DM, Yau KW. Melanopsin-containing retinal ganglion cells: architecture, projections, and intrinsic photosensitivity. Science. (2002) 295:1065–70. doi: 10.1126/science.1069609
11. Spudich JL, Yang CS, Jung KH, Spudich EN. Retinylidene proteins: structures and functions from archaea to humans. Annu Rev Cell Dev Biol. (2000) 16:365–92. doi: 10.1146/annurev.cellbio.16.1.365
12. Tsin A, Betts-Obregon B, Grigsby J. Visual cycle proteins: Structure, function, and roles in human retinal disease. J Biol Chem. (2018) 293:13016–21. doi: 10.1074/jbc.AW118.003228
13. Lewis DE. Organisation of the immune system. In: RR Rich editor. Clinical Immunology: Principles and Practice. (Philadelphia, PA: Elseviser Saunders) (2013).
14. Swainson L. T-cell development. In: R Rich editor. Clinical Immunology: Principles and Practice. Philadelphia, PA: Elsevier Saunders (2013).
15. Rosenblum MD, Remedios KA, Abbas AK. Mechanisms of human autoimmunity. J Clin Invest. (2015) 125:2228–33. doi: 10.1172/JCI78088
16. Kumar BV, Connors T, Farber DL. Human T cell development, localization, and function throughout life. Immunity. (2018) 48:202–13. doi: 10.1016/j.immuni.2018.01.007
17. Ziegler SF, Buckner JH. FOXP3 and the regulation of Treg/Th17 differentiation. Microbes Infect. (2009) 11:594–8. doi: 10.1016/j.micinf.2009.04.002
18. Kimura A, Kishimoto T. Th17 cells in inflammation. Int Immunopharmacol. (2011) 11:319–22. doi: 10.1016/j.intimp.2010.10.004
19. Kimura A, Kishimoto T. IL-6: regulator of Treg/Th17 balance. Eur J Immunol. (2010) 40:1830–5. doi: 10.1002/eji.201040391
20. Ross AC. Vitamin A and retinoic acid in T cell–related immunity. Am J Clin Nutr. (2012) 96:1166S–72S. doi: 10.3945/ajcn.112.034637
21. Caspi R. Autoimmunity in the immune privileged eye: pathogenic and regulatory T cells. Immunol Res. (2008) 42:41–50. doi: 10.1007/s12026-008-8031-3
22. Damsker JM, Hansen AM, Caspi RR. Th1 and Th17 cells: adversaries and collaborators. Ann N Y Acad Sci. (2010) 1183:211–21. doi: 10.1111/j.1749-6632.2009.05133.x
23. Agarwal RK, Caspi RR. Rodent models of experimental autoimmune uveitis. Methods Mol Med. (2004) 102:395–419. doi: 10.1385/1-59259-805-6:395
24. Vignali DAA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol. (2008) 8:523–32. doi: 10.1038/nri2343
25. Wilson CB, Rowell E, Sekimata M. Epigenetic control of T-helper-cell differentiation. Nat Rev Immunol. (2009) 9:91–105. doi: 10.1038/nri2487
26. Murphy KM, Stockinger B. Effector T cell plasticity: flexibility in the face of changing circumstances. Nat Immunol. (2010) 11:674–80. doi: 10.1038/ni.1899
27. Mucida D, Park Y, Kim G, Turovskaya O, Scott I, Kronenberg M, et al. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science. (2007) 317:256–60. doi: 10.1126/science.1145697
28. Belkaid Y, Hand TW. Role of the microbiota in immunity and inflammation. Cell. (2014) 157:121–41. doi: 10.1016/j.cell.2014.03.011
29. Zisimopoulos A, Klavdianou O, Theodossiadis P, Chatziralli I. The role of the microbiome in age-related macular degeneration: a review of the literature. OPH. (2021) 244:173–8. doi: 10.1159/000515026
30. Scuderi G, Troiani E, Minnella AM. Gut microbiome in retina health: the crucial role of the gut-retina axis. Front Microbiol. (2022) 12:726792. doi: 10.3389/fmicb.2021.726792
31. Harrison EH. Mechanisms involved in the intestinal absorption of dietary vitamin A and provitamin A carotenoids. Biochim Biophys Acta. (2012) 1821:70–7. doi: 10.1016/j.bbalip.2011.06.002
32. D’Ambrosio DN, Clugston RD, Blaner WS. Vitamin A metabolism: an update. Nutrients. (2011) 3:63–103. doi: 10.3390/nu3010063
33. National Institutes of Health.Office of Dietary Supplements – Vitamin A and Carotenoids [Internet]. Bethesda, MD: National Institutes of Health (2022).
34. McLean E, Klemm R, Subramaniam H, Greig A. Refocusing vitamin A supplementation programmes to reach the most vulnerable. BMJ Glob Health. (2020) 5:e001997. doi: 10.1136/bmjgh-2019-001997
36. Chiricozzi A, Panduri S, Dini V, Tonini A, Gualtieri B, Romanelli M. Optimizing acitretin use in patients with plaque psoriasis. Dermatol Ther. (2017) 30:e12453. doi: 10.1111/dth.12453
37. Dragnev KH, Petty WJ, Shah SJ, Lewis LD, Black CC, Memoli V, et al. A proof-of-principle clinical trial of bexarotene in patients with non-small cell lung cancer. Clin Cancer Res. (2007) 13:1794–800. doi: 10.1158/1078-0432.CCR-06-1836
39. Szymański Ł, Skopek R, Palusińska M, Schenk T, Stengel S, Lewicki S, et al. Retinoic acid and its derivatives in skin. Cells. (2020) 9:E2660. doi: 10.3390/cells9122660
40. Hunsu VO, Facey COB, Fields JZ, Boman BM. Retinoids as chemo-preventive and molecular-targeted anti-cancer therapies. Int J Mol Sci. (2021) 22:7731. doi: 10.3390/ijms22147731
41. Yan W, Jang GF, Haeseleer F, Esumi N, Chang J, Kerrigan M, et al. Cloning and characterization of a human beta, beta-carotene-15,15’-dioxygenase that is highly expressed in the retinal pigment epithelium. Genomics. (2001) 72:193–202. doi: 10.1006/geno.2000.6476
42. Kiefer C, Hessel S, Lampert JM, Vogt K, Lederer MO, Breithaupt DE, et al. Identification and characterization of a mammalian enzyme catalyzing the asymmetric oxidative cleavage of provitamin A. J Biol Chem. (2001) 276:14110–6. doi: 10.1074/jbc.M011510200
43. Weng W, Li L, van Bennekum AM, Potter SH, Harrison EH, Blaner WS, et al. Intestinal absorption of dietary cholesteryl ester is decreased but retinyl ester absorption is normal in carboxyl ester lipase knockout mice. Biochemistry. (1999) 38:4143–9. doi: 10.1021/bi981679a
44. van Bennekum AM, Fisher EA, Blaner WS, Harrison EH. Hydrolysis of retinyl esters by pancreatic triglyceride lipase. Biochemistry. (2000) 39:4900–6. doi: 10.1021/bi9927235
45. Reboul E, Berton A, Moussa M, Kreuzer C, Crenon I, Borel P. Pancreatic lipase and pancreatic lipase-related protein 2, but not pancreatic lipase-related protein 1, hydrolyze retinyl palmitate in physiological conditions. Biochim Biophys Acta. (2006) 1761:4–10. doi: 10.1016/j.bbalip.2005.12.013
46. Hollander D, Muralidhara KS. Vitamin A1 intestinal absorption in vivo: influence of luminal factors on transport. Am J Physiol. (1977) 232:E471–7. doi: 10.1152/ajpendo.1977.232.5.E471
47. During A, Harrison EH. Mechanisms of provitamin A (carotenoid) and vitamin A (retinol) transport into and out of intestinal Caco-2 cells. J Lipid Res. (2007) 48:2283–94. doi: 10.1194/jlr.M700263-JLR200
48. During A, Hussain MM, Morel DW, Harrison EH. Carotenoid uptake and secretion by CaCo-2 cells: beta-carotene isomer selectivity and carotenoid interactions. J Lipid Res. (2002) 43:1086–95. doi: 10.1194/jlr.M200068-JLR200
49. O’Byrne SM, Wongsiriroj N, Libien J, Vogel S, Goldberg IJ, Baehr W, et al. Retinoid absorption and storage is impaired in mice lacking lecithin:retinol acyltransferase (LRAT). J Biol Chem. (2005) 280:35647–57. doi: 10.1074/jbc.M507924200
50. Wongsiriroj N, Piantedosi R, Palczewski K, Goldberg IJ, Johnston TP, Li E, et al. The molecular basis of retinoid absorption: a genetic dissection. J Biol Chem. (2008) 283:13510–9. doi: 10.1074/jbc.M800777200
51. Lissoos TW, Davis AE, Levin MS. Vitamin A trafficking in Caco-2 cells stably transfected with cellular retinol binding proteins. Am J Physiol. (1995) 268:G224–31. doi: 10.1152/ajpgi.1995.268.2.G224
52. Borel P, Moussa M, Reboul E, Lyan B, Defoort C, Vincent-Baudry S, et al. Human plasma levels of vitamin E and carotenoids are associated with genetic polymorphisms in genes involved in lipid metabolism. J Nutr. (2007) 137:2653–9. doi: 10.1093/jn/137.12.2653
53. Ferrucci L, Perry JRB, Matteini A, Perola M, Tanaka T, Silander K, et al. Common variation in the beta-carotene 15,15’-monooxygenase 1 gene affects circulating levels of carotenoids: a genome-wide association study. Am J Hum Genet. (2009) 84:123–33. doi: 10.1016/j.ajhg.2008.12.019
54. Leung WC, Hessel S, Méplan C, Flint J, Oberhauser V, Tourniaire F, et al. Two common single nucleotide polymorphisms in the gene encoding beta-carotene 15,15’-monoxygenase alter beta-carotene metabolism in female volunteers. FASEB J. (2009) 23:1041–53. doi: 10.1096/fj.08-121962
55. Borel P, Desmarchelier C. Genetic variations associated with vitamin a status and vitamin A bioavailability. Nutrients. (2017) 9:E246. doi: 10.3390/nu9030246
56. Wu L, Guo X, Wang W, Medeiros DM, Clarke SL, Lucas EA, et al. Molecular aspects of β, β-carotene-9’, 10’-oxygenase 2 in carotenoid metabolism and diseases. Exp Biol Med (Maywood). (2016) 241:1879–87. doi: 10.1177/1535370216657900
57. Wang XD. Review: absorption and metabolism of beta-carotene. J Am Coll Nutr. (1994) 13:314–25. doi: 10.1080/07315724.1994.10718416
58. Ruiz A, Winston A, Lim Y-H, Gilbert BA, Rando RR, Bok D. Molecular and biochemical characterization of lecithin retinol acyltransferase. J Biol Chem. (1999) 274:3834–41. doi: 10.1074/jbc.274.6.3834
59. Ross AC, Moran NE. Our current dietary reference intakes for vitamin A–now 20 years old. Cur Dev Nutr. (2020) 4:nzaa096. doi: 10.1093/cdn/nzaa096
60. Ramkumar S, Moon J, Golczak M, von Lintig J. LRAT coordinates the negative-feedback regulation of intestinal retinoid biosynthesis from β-carotene. J Lipid Res. (2021) 62:100055. doi: 10.1016/j.jlr.2021.100055
61. Biesalski HK, Chichili GR, Frank J, von Lintig J, Nohr D. Conversion of beta-carotene to retinal pigment. Vitam Horm. (2007) 75:117–30. doi: 10.1016/S0083-6729(06)75005-1
62. Steinhoff JS, Lass A, Schupp M. Retinoid homeostasis and beyond: how retinol binding protein 4 contributes to health and disease. Nutrients. (2022) 14:1236. doi: 10.3390/nu14061236
63. Tanumihardjo SA, Russell RM, Stephensen CB, Gannon BM, Craft NE, Haskell MJ, et al. Biomarkers of nutrition for development (BOND)-vitamin A review. J Nutr. (2016) 146:1816S–48S. doi: 10.3945/jn.115.229708
64. Olsen T, Blomhoff R. Retinol, retinoic acid, and retinol-binding protein 4 are differentially associated with cardiovascular disease, type 2 diabetes, and obesity: an overview of human studies. Adv Nutr. (2020) 11:644–66. doi: 10.1093/advances/nmz131
65. Rubin LP, Ross AC, Stephensen CB, Bohn T, Tanumihardjo SA. Metabolic effects of inflammation on vitamin a and carotenoids in humans and animal models. Adv Nutr. (2017) 8:197–212. doi: 10.3945/an.116.014167
66. Rosales FJ, Ross AC. Acute inflammation induces hyporetinemia and modifies the plasma and tissue response to vitamin A supplementation in marginally vitamin A-deficient rats. J Nutr. (1998) 128:960–6. doi: 10.1093/jn/128.6.960
67. Thurnham DI. Micronutrients and immune function: some recent developments. J Clin Pathol. (1997) 50:887–91. doi: 10.1136/jcp.50.11.887
68. Chen Y, Clarke OB, Kim J, Stowe S, Kim Y-K, Assur Z, et al. Structure of the STRA6 receptor for retinol uptake. Science. (2016) 353:aad8266. doi: 10.1126/science.aad8266
69. Kawaguchi R, Zhong M, Kassai M, Ter-Stepanian M, Sun H. STRA6-catalyzed vitamin A influx, efflux, and exchange. J Membr Biol. (2012) 245:731–45. doi: 10.1007/s00232-012-9463-1
70. Zhong M, Kawaguchi R, Costabile B, Tang Y, Hu J, Cheng G, et al. Regulatory mechanism for the transmembrane receptor that mediates bidirectional vitamin A transport. Proc Natl Acad Sci USA. (2020) 117:9857–64. doi: 10.1073/pnas.1918540117
72. Saari JC. Vitamin A metabolism in rod and cone visual cycles. Annu Rev Nutr. (2012) 32:125–45. doi: 10.1146/annurev-nutr-071811-150748
73. Saari JC. Vitamin A and vision. Subcell Biochem. (2016) 81:231–59. doi: 10.1007/978-94-024-0945-1_9
74. Güler AD, Ecker JL, Lall GS, Haq S, Altimus CM, Liao H-W, et al. Melanopsin cells are the principal conduits for rod-cone input to non-image-forming vision. Nature. (2008) 453:102–5. doi: 10.1038/nature06829
75. Merbs SL, Nathans J. Absorption spectra of human cone pigments. Nature. (1992) 356:433–5. doi: 10.1038/356433a0
76. Hubbell WL, Altenbach C, Hubbell CM, Khorana HG. Rhodopsin structure, dynamics, and activation: a perspective from crystallography, site-directed spin labeling, sulfhydryl reactivity, and disulfide cross-linking. Adv Protein Chem. (2003) 63:243–90. doi: 10.1016/S0065-3233(03)63010-X
77. Nathans J, Thomas D, Hogness DS. Molecular genetics of human color vision: the genes encoding blue, green, and red pigments. Science. (1986) 232:193–202. doi: 10.1126/science.2937147
78. Kochendoerfer GG, Lin SW, Sakmar TP, Mathies RA. How color visual pigments are tuned. Trends Biochem Sci. (1999) 24:300–5. doi: 10.1016/S0968-0004(99)01432-2
79. Nathans J. The evolution and physiology of human color vision: insights from molecular genetic studies of visual pigments. Neuron. (1999) 24:299–312. doi: 10.1016/S0896-6273(00)80845-4
80. Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, et al. Crystal structure of rhodopsin: a g protein-coupled receptor. Science. (2000) 289:739–45. doi: 10.1126/science.289.5480.739
81. Katayama K, Gulati S, Ortega JT, Alexander NS, Sun W, Shenouda MM, et al. Specificity of the chromophore-binding site in human cone opsins. J Biol Chem. (2019) 294:6082–93. doi: 10.1074/jbc.RA119.007587
82. Enroth-Cugell C, Hertz BG, Lennie P. Convergence of rod and cone signals in the cat’s retina. J Physiol. (1977) 269:297–318. doi: 10.1113/jphysiol.1977.sp011903
83. Ingram NT, Sampath AP, Fain GL. Why are rods more sensitive than cones? J Physiol. (2016) 594:5415–26. doi: 10.1113/JP272556
84. Valen R, Eilertsen M, Edvardsen RB, Furmanek T, Rønnestad I, van der Meeren T, et al. The two-step development of a duplex retina involves distinct events of cone and rod neurogenesis and differentiation. Dev Biol. (2016) 416:389–401. doi: 10.1016/j.ydbio.2016.06.041
85. Röhrig UF, Guidoni L, Laio A, Frank I, Rothlisberger UA. Molecular spring for vision. J Am Chem Soc. (2004) 126:15328–9. doi: 10.1021/ja048265r
86. Crouch RK, Koutalos Y, Kono M, Schey K, Ablonczy Z. A2E and lipofuscin. Prog Mol Biol Transl Sci. (2015) 134:449–63. doi: 10.1016/bs.pmbts.2015.06.005
87. Lee SY, Ubels JL, Soprano DR. The lacrimal gland synthesizes retinol-binding protein. Exp Eye Res. (1992) 55:163–71. doi: 10.1016/0014-4835(92)90104-Z
88. Ubels JL, MacRae SM. Vitamin A is present as retinol in the tears of humans and rabbits. Curr Eye Res. (1984) 3:815–22. doi: 10.3109/02713688409000793
89. Ubels JL, Dennis MH, Rigatti BW, Vergnes JP, Beatty R, Kinchington PR. Nuclear retinoic acid receptors in the lacrimal gland. Curr Eye Res. (1995) 14:1055–62. doi: 10.3109/02713689508998530
90. Bossenbroek NM, Sulahian TH, Ubels JL. Expression of nuclear retinoic acid receptor and retinoid X receptor mRNA in the cornea and conjunctiva. Curr Eye Res. (1998) 17:462–9. doi: 10.1076/ceyr.17.5.462.5189
91. Kim SW, Seo KY, Rhim T, Kim EK. Effect of retinoic acid on epithelial differentiation and mucin expression in primary human corneal limbal epithelial cells. Curr Eye Res. (2012) 37:33–42. doi: 10.3109/02713683.2011.620728
92. Fuchs GJ, Ausayakhun S, Ruckphaopunt S, Tansuhaj A, Suskind RM. Relationship between vitamin A deficiency, malnutrition, and conjunctival impression cytology. Am J Clin Nutr. (1994) 60:293–8. doi: 10.1093/ajcn/60.2.293
93. Stephensen CB. Vitamin A, infection, and immune function. Annu Rev Nutr. (2001) 21:167–92. doi: 10.1146/annurev.nutr.21.1.167
94. Riccio P, Rossano R. Diet, gut microbiota, and vitamins D + A in multiple sclerosis. Neurotherapeutics. (2018) 15:75–91. doi: 10.1007/s13311-017-0581-4
95. Cantorna MT, Snyder L, Arora J. Vitamin A and vitamin D regulate the microbial complexity, barrier function, and the mucosal immune responses to ensure intestinal homeostasis. Crit Rev Biochem Mol Biol. (2019) 54:184–92. doi: 10.1080/10409238.2019.1611734
96. Bastie JN, Balitrand N, Guidez F, Guillemot I, Larghero J, Calabresse C, et al. 1 alpha,25-dihydroxyvitamin D3 transrepresses retinoic acid transcriptional activity via vitamin D receptor in myeloid cells. Mol Endocrinol. (2004) 18:2685–99. doi: 10.1210/me.2003-0412
97. Benson MJ, Pino-Lagos K, Rosemblatt M, Noelle RJ. All-trans retinoic acid mediates enhanced T reg cell growth, differentiation, and gut homing in the face of high levels of co-stimulation. J Exp Med. (2007) 204:1765–74. doi: 10.1084/jem.20070719
98. Mucida D, Park Y, Cheroutre H. From the diet to the nucleus: vitamin A and TGF-beta join efforts at the mucosal interface of the intestine. Semin Immunol. (2009) 21:14–21. doi: 10.1016/j.smim.2008.08.001
99. Shi H, Chi H. Metabolic control of treg cell stability, plasticity, and tissue-specific heterogeneity. Front Immunol. (2019) 10:2716. doi: 10.3389/fimmu.2019.02716
100. Lu L, Lan Q, Li Z, Zhou X, Gu J, Li Q, et al. Critical role of all-trans retinoic acid in stabilizing human natural regulatory T cells under inflammatory conditions. Proc Natl Acad Sci USA. (2014) 111:E3432–40.
101. Weaver CT, Harrington LE, Mangan PR, Gavrieli M, Murphy KM. Th17: an effector CD4 T cell lineage with regulatory T cell ties. Immunity. (2006) 24:677–88. doi: 10.1016/j.immuni.2006.06.002
102. Weaver CT, Hatton RD. Interplay between the TH17 and TReg cell lineages: a (co-)evolutionary perspective. Nat Rev Immunol. (2009) 9:883–9. doi: 10.1038/nri2660
103. Lu L, Ma J, Wang X, Wang J, Zhang F, Yu J, et al. Synergistic effect of TGF-beta superfamily members on the induction of Foxp3+ Treg. Eur J Immunol. (2010) 40:142–52. doi: 10.1002/eji.200939618
104. Beijer MR, Kraal G, den Haan JMM. Vitamin A and dendritic cell differentiation. Immunology. (2014) 142:39–45. doi: 10.1111/imm.12228
105. Xiao S, Jin H, Korn T, Liu SM, Oukka M, Lim B, et al. Retinoic acid increases Foxp3+ regulatory T cells and inhibits development of Th17 cells by enhancing TGF-beta-driven Smad3 signaling and inhibiting IL-6 and IL-23 receptor expression. J Immunol. (2008) 181:2277–84. doi: 10.4049/jimmunol.181.4.2277
106. Hill JA, Hall JA, Sun C-M, Cai Q, Ghyselinck N, Chambon P, et al. Retinoic acid enhances foxp3 induction indirectly by relieving inhibition from CD4+CD44hi cells. Immunity. (2008) 29:758–70. doi: 10.1016/j.immuni.2008.09.018
107. Chavele KM, Ehrenstein MR. Regulatory T-cells in systemic lupus erythematosus and rheumatoid arthritis. FEBS Lett. (2011) 585:3603–10. doi: 10.1016/j.febslet.2011.07.043
108. Silva MJ, Carneiro MB, dos Anjos Pultz B, Pereira Silva D, Lopes ME, dos Santos LM. The multifaceted role of commensal microbiota in homeostasis and gastrointestinal diseases. J Immunol Res. (2015) 2015:321241. doi: 10.1155/2015/321241
109. West CE, Renz H, Jenmalm MC, Kozyrskyj AL, Allen KJ, Vuillermin P, et al. The gut microbiota and inflammatory noncommunicable diseases: associations and potentials for gut microbiota therapies. J Allergy Clin Immunol. (2015) 135:3–13;quiz14. doi: 10.1016/j.jaci.2014.11.012
110. Hildebrandt MA, Hoffmann C, Sherrill-Mix SA, Keilbaugh SA, Hamady M, Chen YY, et al. High-fat diet determines the composition of the murine gut microbiome independently of obesity. Gastroenterology. (2009) 137:e1–2. doi: 10.1053/j.gastro.2009.08.042
111. Clarke SF, Murphy EF, Nilaweera K, Ross PR, Shanahan F, O’Toole PW, et al. The gut microbiota and its relationship to diet and obesity: new insights. Gut Microbes. (2012) 3:186–202. doi: 10.4161/gmic.20168
112. Molan AL, Liu Z, Plimmer G. Evaluation of the effect of blackcurrant products on gut microbiota and on markers of risk for colon cancer in humans. Phytother Res. (2014) 28:416–22. doi: 10.1002/ptr.5009
113. Andriessen EM, Wilson AM, Mawambo G, Dejda A, Miloudi K, Sennlaub F, et al. Gut microbiota influences pathological angiogenesis in obesity-driven choroidal neovascularization. EMBO Mol Med. (2016) 8:1366–79. doi: 10.15252/emmm.201606531
114. Li L, Krause L, Somerset S. Associations between micronutrient intakes and gut microbiota in a group of adults with cystic fibrosis. Clin Nutr. (2017) 36:1097–104. doi: 10.1016/j.clnu.2016.06.029
115. Rowan S, Jiang S, Korem T, Szymanski J, Chang ML, Szelog J, et al. Involvement of a gut-retina axis in protection against dietary glycemia-induced age-related macular degeneration. Proc Natl Acad Sci USA. (2017) 114:E4472–81. doi: 10.1073/pnas.1702302114
116. Slingerland AE, Schwabkey Z, Wiesnoski DH, Jenq RR. Clinical evidence for the microbiome in inflammatory diseases. Front Immunol. (2017) 8:400. doi: 10.3389/fimmu.2017.00400
117. Clemente JC, Manasson J, Scher JU. The role of the gut microbiome in systemic inflammatory disease. BMJ. (2018) 360:j5145. doi: 10.1136/bmj.j5145
118. Iwata M, Hirakiyama A, Eshima Y, Kagechika H, Kato C, Song S-Y. Retinoic acid imprints gut-homing specificity on T cells. Immunity. (2004) 21:527–38. doi: 10.1016/j.immuni.2004.08.011
119. Coombes JL, Siddiqui KRR, Arancibia-Cárcamo CV, Hall J, Sun C-M, Belkaid Y, et al. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J Exp Med. (2007) 204:1757–64. doi: 10.1084/jem.20070590
120. Lochner M, Peduto L, Cherrier M, Sawa S, Langa F, Varona R, et al. In vivo equilibrium of proinflammatory IL-17+ and regulatory IL-10+ Foxp3+ RORgamma t+ T cells. J Exp Med. (2008) 205:1381–93. doi: 10.1084/jem.20080034
121. Leung S, Liu X, Fang L, Chen X, Guo T, Zhang J. The cytokine milieu in the interplay of pathogenic Th1/Th17 cells and regulatory T cells in autoimmune disease. Cell Mol Immunol. (2010) 7:182–9. doi: 10.1038/cmi.2010.22
122. Omenetti S, Pizarro TT. The Treg/Th17 axis: a dynamic balance regulated by the gut microbiome. Front Immunol. (2015) 6:639. doi: 10.3389/fimmu.2015.00639
123. Sefik E, Geva-Zatorsky N, Oh S, Konnikova L, Zemmour D, McGuire AM, et al. Individual intestinal symbionts induce a distinct population of RORγ+ regulatory T cells. Science. (2015) 349:993–7. doi: 10.1126/science.aaa9420
124. Koch MA, Tucker-Heard G, Perdue NR, Killebrew JR, Urdahl KB, Campbell DJ. The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat Immunol. (2009) 10:595–602. doi: 10.1038/ni.1731
125. Ohnmacht C, Park J-H, Cording S, Wing JB, Atarashi K, Obata Y, et al. The microbiota regulates type 2 immunity through RORγt+ T cells. Science. (2015) 349:989–93. doi: 10.1126/science.aac4263
126. Levine AG, Medoza A, Hemmers S, Moltedo B, Niec RE, Schizas M, et al. Stability and function of regulatory T cells expressing the transcription factor T-bet. Nature. (2017) 546:421–5. doi: 10.1038/nature22360
127. Kang SG, Lim HW, Andrisani OM, Broxmeyer HE, Kim CH. Vitamin A metabolites induce gut-homing FoxP3+ regulatory T cells. J Immunol. (2007) 179:3724–33. doi: 10.4049/jimmunol.179.6.3724
128. Jaensson-Gyllenbäck E, Kotarsky K, Zapata F, Persson EK, Gundersen TE, Blomhoff R, et al. Bile retinoids imprint intestinal CD103+ dendritic cells with the ability to generate gut-tropic T cells. Mucosal Immunol. (2011) 4:438–47. doi: 10.1038/mi.2010.91
129. McDonald KG, Leach MR, Brooke KWM, Wang C, Wheeler LW, Hanly EK, et al. Epithelial expression of the cytosolic retinoid chaperone cellular retinol binding protein II is essential for in vivo imprinting of local gut dendritic cells by lumenal retinoids. Am J Pathol. (2012) 180:984–97. doi: 10.1016/j.ajpath.2011.11.009
130. Cassani B, Villablanca EJ, Quintana FJ, Love PE, Lacy-Hulbert A, Blaner WS, et al. Gut-tropic T cells that express integrin α4β7 and CCR9 are required for induction of oral immune tolerance in mice. Gastroenterology. (2011) 141:2109–18. doi: 10.1053/j.gastro.2011.09.015
131. Stein-Streilein J, Streilein JW. Anterior chamber associated immune deviation (ACAID): regulation, biological relevance, and implications for therapy. Int Rev Immunol. (2002) 21:123–52. doi: 10.1080/08830180212066
132. Kawazoe Y, Sugita S, Keino H, Yamada Y, Imai A, Horie S, et al. Retinoic acid from retinal pigment epithelium induces T regulatory cells. Exp Eye Res. (2012) 94:32–40. doi: 10.1016/j.exer.2011.11.002
133. Amadi-Obi A, Yu CR, Liu X, Mahdi RM, Clarke GL, Nussenblatt RB, et al. TH17 cells contribute to uveitis and scleritis and are expanded by IL-2 and inhibited by IL-27/STAT1. Nat Med. (2007) 13:711–8. doi: 10.1038/nm1585
134. Zhuang Z, Wang Y, Zhu G, Gu Y, Mao L, Hong M, et al. Imbalance of Th17/Treg cells in pathogenesis of patients with human leukocyte antigen B27 associated acute anterior uveitis. Sci Rep. (2017) 7:40414. doi: 10.1038/srep40414
135. Gilbert RM, Zhang X, Sampson RD, Ehrenstein MR, Nguyen DX, Chaudhry M, et al. Clinical remission of sight-threatening non-infectious uveitis is characterized by an upregulation of peripheral T-regulatory cell polarized towards T-bet and TIGIT. Front Immunol. (2018) 9:907. doi: 10.3389/fimmu.2018.00907
136. Ramesh R, Kozhaya L, McKevitt K, Djuretic IM, Carlson TJ, Quintero MA, et al. Pro-inflammatory human Th17 cells selectively express P-glycoprotein and are refractory to glucocorticoids. J Exp Med. (2014) 211:89–104. doi: 10.1084/jem.20130301
137. Basdeo SA, Moran B, Cluxton D, Canavan M, McCormick J, Connolly M, et al. Polyfunctional, pathogenic CD161+ Th17 lineage cells are resistant to regulatory T cell-mediated suppression in the context of autoimmunity. J Immunol. (2015) 195:528–40. doi: 10.4049/jimmunol.1402990
138. Lee RW, Schewitz LP, Nicholson LB, Dayan CM, Dick AD. Steroid refractory CD4+ T cells in patients with sight-threatening uveitis. Invest Ophthalmol Vis Sci. (2009) 50:4273–8. doi: 10.1167/iovs.08-3152
139. Mazzoni A, Santarlasci V, Maggi L, Capone M, Rossi MC, Querci V, et al. Demethylation of the RORC2 and IL17A in human CD4+ T lymphocytes defines Th17 origin of nonclassic Th1 cells. J Immunol. (2015) 194:3116–26. doi: 10.4049/jimmunol.1401303
140. Maggi L, Santarlasci V, Capone M, Peired A, Frosali F, Crome SQ, et al. CD161 is a marker of all human IL-17-producing T-cell subsets and is induced by RORC. Eur J Immunol. (2010) 40:2174–81. doi: 10.1002/eji.200940257
141. Wiseman EM, Bar-El Dadon S, Reifen R. The vicious cycle of vitamin a deficiency: a review. Crit Rev Food Sci Nutr. (2017) 57:3703–14. doi: 10.1080/10408398.2016.1160362
142. Faustino JF, Ribeiro-Silva A, Dalto RF, de Souza MM, Furtado JMF, de Rocha GM, et al. Vitamin A and the eye: an old tale for modern times. Arq Bras Oftalmol. (2016) 79:56–61. doi: 10.5935/0004-2749.20160018
143. Cella W, Urbano AP, Vinhadelli WS, Donatti M, Rocha EM. Xerophthalmia secondary to short bowel syndrome. J Pediatr Ophthalmol Strabismus. (2002) 39:125–7. doi: 10.3928/0191-3913-20020301-17
144. Whitcher JP, Srinivasan M, Upadhyay MP. Corneal blindness: a global perspective. Bull World Health Organ. (2001) 79:214–21.
145. Spannaus-Martin DJ, Cook LR, Tanumihardjo SA, Duitsman PK, Olson JA. Vitamin A and vitamin E statuses of preschool children of socioeconomically disadvantaged families living in the midwestern United States. Eur J Clin Nutr. (1997) 51:864–9. doi: 10.1038/sj.ejcn.1600503
146. Akhtar S, Ahmed A, Randhawa MA, Atukorala S, Arlappa N, Ismail T, et al. Prevalence of vitamin A deficiency in south asia: causes, outcomes, and possible remedies. J Health Popul Nutr. (2013) 31:413–23. doi: 10.3329/jhpn.v31i4.19975
147. Suan EP, Bedrossian EH, Eagle RC, Laibson PR. Corneal perforation in patients with vitamin A deficiency in the United States. Arch Ophthalmol. (1990) 108:350–3. doi: 10.1001/archopht.1990.01070050048028
148. Scientific Advisory Committee on Nutrition.Review of Dietary Advice on Vitamin A [Internet]. (2005) Scientific Advisory Committee on Nutrition. Available online at: https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/338853/SACN_Review_of_Dietary_Advice_on_Vitamin_A.pdf (accessed June 9, 2022).
149. EFSA Panel on Dietetic Products, Nutrition, and Allergies [NDA]. Scientific opinion on dietary reference values for vitamin A. EFSA J. (2015) 13:4028. doi: 10.2903/j.efsa.2015.4028
150. Sommer A, Muhilal, Tarwotjo I, Djunaedi E, Glover J. Oral versus intramuscular vitamin A in the treatment of xerophthalmia. Lancet. (1980) 1:557–9. doi: 10.1016/S0140-6736(80)91053-3
151. McLaren DS, Kraemer K, Druck R, Ag BG. Xerophthalmia. Manual Vitamin A Deficiency Disord. (2012) 103:65–75. doi: 10.1159/000185254
154. Chakraborty U, Chandra A. Bitot’s spots, dry eyes, and night blindness indicate vitamin A deficiency. Lancet. (2021) 397:e2. doi: 10.1016/S0140-6736(21)00041-6
155. Ahmed F, House RJ, Feldman BH. Corneal abrasions and corneal foreign bodies. Prim Care. (2015) 42:363–75. doi: 10.1016/j.pop.2015.05.004
156. Stephensen CB, Lietz G. Vitamin A in resistance to and recovery from infection: relevance to SARS-CoV2. Br J Nutr. (2021) 126:1663–72. doi: 10.1017/S0007114521000246
157. Huang Z, Liu Y, Qi G, Brand D, Zheng SG. Role of vitamin A in the immune system. J Clin Med. (2018) 7:258. doi: 10.3390/jcm7090258
158. Penniston KL, Tanumihardjo SA. The acute and chronic toxic effects of vitamin A. Am J Clin Nutr. (2006) 83:191–201. doi: 10.1093/ajcn/83.2.191
159. Priyadarshani AMB. Insights of hypercarotenaemia: a brief review. Clin Nutr ESPEN. (2018) 23:19–24. doi: 10.1016/j.clnesp.2017.12.002
160. Silverman AK, Ellis CN, Voorhees JJ. Hypervitaminosis A syndrome: a paradigm of retinoid side effects. J Am Acad Dermatol. (1987) 16:1027–39. doi: 10.1016/S0190-9622(87)70133-9
161. Radu RA, Yuan Q, Hu J, Peng JH, Lloyd M, Nusinowitz S, et al. Accelerated accumulation of lipofuscin pigments in the RPE of a mouse model for ABCA4-mediated retinal dystrophies following Vitamin A supplementation. Invest Ophthalmol Vis Sci. (2008) 49:3821–9. doi: 10.1167/iovs.07-1470
162. Bastos Maia S, Rolland Souza AS, de Costa Caminha MF, da Lins Silva S, de Callou Cruz RSBL, dos Carvalho Santos C, et al. Vitamin A and pregnancy: a narrative review. Nutrients. (2019) 11:681. doi: 10.3390/nu11030681
163. Heywood R, Palmer AK, Gregson RL, Hummler H. The toxicity of beta-carotene. Toxicology. (1985) 36:91–100. doi: 10.1016/0300-483X(85)90043-5
164. Mazzone A, Dal Canton A. Hypercarotenemia. N Engl J Med. (2002) 346:821–821. doi: 10.1056/NEJMicm950425
165. Bendich A. The safety of β−carotene. Nutr Cancer. (1988) 11:207–14. doi: 10.1080/01635588809513989
166. Lewis RA, Shroyer NF, Singh N, Allikmets R, Hutchinson A, Li Y, et al. Genotype/phenotype analysis of a photoreceptor-specific ATP-binding cassette transporter gene, ABCR, in stargardt disease. Am J Hum Genet. (1999) 64:422–34. doi: 10.1086/302251
167. Logan S, Anderson RE. Dominant stargardt macular dystrophy (STGD3) and ELOVL4. Adv Exp Med Biol. (2014) 801:447–53. doi: 10.1007/978-1-4614-3209-8_57
168. Imani S, Cheng J, Shasaltaneh MD, Wei C, Yang L, Fu S, et al. Genetic identification and molecular modeling characterization reveal a novel PROM1 mutation in Stargardt4-like macular dystrophy. Oncotarget. (2018) 9:122–41. doi: 10.18632/oncotarget.22343
169. Zaneveld J, Siddiqui S, Li H, Wang X, Wang H, Wang K, et al. Comprehensive analysis of stargardt macular dystrophy patients reveals new genotype-phenotype correlations and unexpected diagnostic revisions. Genet Med. (2015) 17:262–70. doi: 10.1038/gim.2014.174
170. Fishman GA, Stone EM, Grover S, Derlacki DJ, Haines HL, Hockey RR. Variation of clinical expression in patients with Stargardt dystrophy and sequence variations in the ABCR gene. Arch Ophthalmol. (1999) 117:504–10. doi: 10.1001/archopht.117.4.504
171. Fujinami K, Sergouniotis PI, Davidson AE, Mackay DS, Tsunoda K, Tsubota K, et al. The clinical effect of homozygous ABCA4 alleles in 18 patients. Ophthalmology. (2013) 120:2324–31. doi: 10.1016/j.ophtha.2013.04.016
172. Tanna P, Strauss RW, Fujinami K, Michaelides M. Stargardt disease: clinical features, molecular genetics, animal models and therapeutic options. Br Ophthalmol. (2017) 101:25–30. doi: 10.1136/bjophthalmol-2016-308823
173. Rotenstreich Y, Fishman GA, Anderson RJ. Visual acuity loss and clinical observations in a large series of patients with stargardt disease. Ophthalmology. (2003) 110:1151–8. doi: 10.1016/S0161-6420(03)00333-6
174. Westeneng-van Haaften SC, Boon CJF, Cremers FPM, Hoefsloot LH, den Hollander AI, Hoyng CB. Clinical and genetic characteristics of late-onset stargardt’s disease. Ophthalmology. (2012) 119:1199–210. doi: 10.1016/j.ophtha.2012.01.005
175. Cideciyan AV, Swider M, Aleman TS, Tsybovsky Y, Schwartz SB, Windsor EAM, et al. ABCA4 disease progression and a proposed strategy for gene therapy. Hum Mol Genet. (2009) 18:931–41. doi: 10.1093/hmg/ddn421
176. Cukras CA, Wong WT, Caruso R, Cunningham D, Zein W, Sieving PA. Centrifugal expansion of fundus autofluorescence patterns in stargardt disease over time. Arch Ophthalmol. (2012) 130:171–9. doi: 10.1001/archophthalmol.2011.332
177. Strauss RW, Ho A, Muñoz B, Cideciyan AV, Sahel J-A, Sunness JS, et al. The natural history of the progression of atrophy secondary to stargardt disease (ProgStar) studies: design and baseline characteristics: progstar report no. 1. Ophthalmology. (2016) 123:817–28. doi: 10.1016/j.ophtha.2015.12.009
178. Singh R, Fujinami K, Chen LL, Michaelides M, Moore AT. Longitudinal follow-up of siblings with a discordant Stargardt disease phenotype. Acta Ophthalmol. (2014) 92:e331–2. doi: 10.1111/aos.12280
179. Fakin A, Robson AG, Fujinami K, Moore AT, Michaelides M, Chiang J, et al. Phenotype and progression of retinal degeneration associated with nullizigosity of ABCA4. Invest Ophthalmol Vis Sci. (2016) 57:4668–78. doi: 10.1167/iovs.16-19829
180. Allikmets R, Singh N, Sun H, Shroyer NF, Hutchinson A, Chidambaram A, et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet. (1997) 15:236–46. doi: 10.1038/ng0397-236
181. Molday LL, Rabin AR, Molday RS. ABCR expression in foveal cone photoreceptors and its role in Stargardt macular dystrophy. Nat Genet. (2000) 25:257–8. doi: 10.1038/77004
182. Sun H, Nathans J. ABCR: rod photoreceptor-specific ABC transporter responsible for Stargardt disease. Methods Enzymol. (2000) 315:879–97. doi: 10.1016/S0076-6879(00)15888-4
183. Weng J, Mata NL, Azarian SM, Tzekov RT, Birch DG, Travis GH. Insights into the function of Rim protein in photoreceptors and etiology of Stargardt’s disease from the phenotype in abcr knockout mice. Cell. (1999) 98:13–23. doi: 10.1016/S0092-8674(00)80602-9
184. Boyer NP, Higbee D, Currin MB, Blakeley LR, Chen C, Ablonczy Z, et al. Lipofuscin and N-retinylidene-N-retinylethanolamine (A2E) accumulate in retinal pigment epithelium in absence of light exposure: THEIR ORIGIN IS 11-cis-RETINAL. J Biol Chem. (2012) 287:22276–86. doi: 10.1074/jbc.M111.329235
185. Beharry S, Zhong M, Molday RS. N-retinylidene-phosphatidylethanolamine is the preferred retinoid substrate for the photoreceptor-specific ABC transporter ABCA4 (ABCR). J Biol Chem. (2004) 279:53972–9. doi: 10.1074/jbc.M405216200
186. Quazi F, Lenevich S, Molday RS. ABCA4 is an N-retinylidene-phosphatidylethanolamine and phosphatidylethanolamine importer. Nat Commun. (2012) 3:925. doi: 10.1038/ncomms1927
187. Molday RS. Insights into the molecular properties of ABCA4 and Its role in the visual cycle and stargardt disease. Prog Mol Biol Transl Sci. (2015) 134:415–31. doi: 10.1016/bs.pmbts.2015.06.008
188. Zhong M, Molday LL, Molday RS. Role of the C terminus of the photoreceptor ABCA4 transporter in protein folding, function, and retinal degenerative diseases. J Biol Chem. (2009) 284:3640–9. doi: 10.1074/jbc.M806580200
189. Quazi F, Molday RS. Differential phospholipid substrates and directional transport by ATP-binding cassette proteins ABCA1, ABCA7, and ABCA4 and disease-causing mutants. J Biol Chem. (2013) 288:34414–26. doi: 10.1074/jbc.M113.508812
190. Ben-Shabat S, Parish CA, Vollmer HR, Itagaki Y, Fishkin N, Nakanishi K, et al. Biosynthetic studies of A2E, a major fluorophore of retinal pigment epithelial lipofuscin. J Biol Chem. (2002) 277:7183–90. doi: 10.1074/jbc.M108981200
191. Parish CA, Hashimoto M, Nakanishi K, Dillon J, Sparrow J. Isolation and one-step preparation of A2E and iso-A2E, fluorophores from human retinal pigment epithelium. Proc Natl Acad Sci USA. (1998) 95:14609–13. doi: 10.1073/pnas.95.25.14609
192. Radu RA, Mata NL, Bagla A, Travis GH. Light exposure stimulates formation of A2E oxiranes in a mouse model of stargardt’s macular degeneration. Proc Natl Acad Sci USA. (2004) 101:5928–33. doi: 10.1073/pnas.0308302101
193. Schütt F, Davies S, Kopitz J, Holz FG, Boulton ME. Photodamage to human RPE cells by A2-E, a retinoid component of lipofuscin. Invest Ophthalmol Vis Sci. (2000) 41:2303–8.
194. Sparrow JR, Nakanishi K, Parish CA. The lipofuscin fluorophore A2E mediates blue light-induced damage to retinal pigmented epithelial cells. Invest Ophthalmol Vis Sci. (2000) 41:1981–9.
195. Federspiel CA, Bertelsen M, Kessel L. Vitamin A in stargardt disease-an evidence-based update. Ophthalmic Genet. (2018) 39:555–9. doi: 10.1080/13816810.2018.1488174
196. Colijn JM, Buitendijk GHS, Prokofyeva E, Alves D, Cachulo ML, Khawaja AP, et al. Prevalence of age-related macular degeneration in Europe: the past and the future. Ophthalmology. (2017) 124:1753–63.
197. Wong WL, Su X, Li X, Cheung CMG, Klein R, Cheng C-Y, et al. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: a systematic review and meta-analysis. Lancet Glob Health. (2014) 2:e106–16. doi: 10.1016/S2214-109X(13)70145-1
198. Flores-Bellver M, Mighty J, Aparicio-Domingo S, Li KV, Shi C, Zhou J, et al. Extracellular vesicles released by human retinal pigment epithelium mediate increased polarised secretion of drusen proteins in response to AMD stressors. J Extracell Vesicles. (2021) 10:e12165. doi: 10.1002/jev2.12165
199. Flaxel CJ, Adelman RA, Bailey ST, Fawzi A, Lim JI, Vemulakonda GA, et al. Age-related macular degeneration preferred practice pattern®. Ophthalmology. (2020) 127:1–65. doi: 10.1016/j.ophtha.2019.09.024
200. Al-Zamil WM, Yassin SA. Recent developments in age-related macular degeneration: a review. CIA. (2017) 12:1313–30. doi: 10.2147/CIA.S143508
201. Sadda SR, Guymer R, Holz FG, Schmitz-Valckenberg S, Curcio CA, Bird AC, et al. Consensus definition for atrophy associated with age-related macular degeneration on OCT: classification of atrophy report 3. Ophthalmology. (2018) 125:537–48. doi: 10.1016/j.ophtha.2017.09.028
202. Booij JC, Baas DC, Beisekeeva J, Gorgels TGMF, Bergen AAB. The dynamic nature of Bruch’s membrane. Prog Retin Eye Res. (2010) 29:1–18. doi: 10.1016/j.preteyeres.2009.08.003
203. Heesterbeek TJ, Lorés-Motta L, Hoyng CB, Lechanteur YTE, den Hollander AI. Risk factors for progression of age-related macular degeneration. Ophthalmic Physiol Opt. (2020) 40:140–70. doi: 10.1111/opo.12675
204. Rivera A, White K, Stöhr H, Steiner K, Hemmrich N, Grimm T, et al. A comprehensive survey of sequence variation in the ABCA4 (ABCR) gene in Stargardt disease and age-related macular degeneration. Am J Hum Genet. (2000) 67:800–13. doi: 10.1086/303090
205. Allikmets R, Shroyer NF, Singh N, Seddon JM, Lewis RA, Bernstein PS, et al. Mutation of the stargardt disease gene (ABCR) in age-related macular degeneration. Science. (1997) 277:1805–7. doi: 10.1126/science.277.5333.1805
206. Mihai DM, Washington I. Vitamin A dimers trigger the protracted death of retinal pigment epithelium cells. Cell Death Dis. (2014) 5:e1348–1348. doi: 10.1038/cddis.2014.314
207. Penn J, Mihai DM, Washington I. Morphological and physiological retinal degeneration induced by intravenous delivery of vitamin A dimers in rabbits. Dis Model Mech. (2015) 8:131–8. doi: 10.1242/dmm.017194
208. Zhang D, Mihai DM, Washington I. Vitamin A cycle byproducts explain retinal damage and molecular changes thought to initiate retinal degeneration. Biol Open. (2021) 10:bio058600. doi: 10.1242/bio.058600
209. Stone EM, Webster AR, Vandenburgh K, Streb LM, Hockey RR, Lotery AJ, et al. Allelic variation in ABCR associated with Stargardt disease but not age-related macular degeneration. Nat Genet. (1998) 20:328–9. doi: 10.1038/3798
210. Guymer RH, Héon E, Lotery AJ, Munier FL, Schorderet DF, Baird PN, et al. Variation of codons 1961 and 2177 of the stargardt disease gene is not associated with age-related macular degeneration. Arch Ophthalmol. (2001) 119:745–51. doi: 10.1001/archopht.119.5.745
211. Gorin MB. The ABCA4 gene and age-related macular degeneration: innocence or guilt by association. Arch Ophthalmol. (2001) 119:752–3. doi: 10.1001/archopht.119.5.752
212. de Breuk A, Acar IE, Kersten E, Schijvenaars MMVAP, Colijn JM, Haer-Wigman L, et al. Development of a genotype assay for age-related macular degeneration. Ophthalmology. (2021) 128:1604–17. doi: 10.1016/j.ophtha.2020.07.037
213. Medzhitov R. Origin and physiological roles of inflammation. Nature. (2008) 454:428. doi: 10.1038/nature07201
214. Chen M, Xu H. Parainflammation, chronic inflammation, and age-related macular degeneration. J Leukocyte Biol. (2015) 98:713–25. doi: 10.1189/jlb.3RI0615-239R
215. Nussenblatt RB, Ferris F. Age-related macular degeneration and the immune response: implications for therapy. Am J Ophthalmol. (2007) 144:618–26. doi: 10.1016/j.ajo.2007.06.025
216. Nussenblatt RB, Liu B, Li Z. Age-related macular degeneration: an immunologically driven disease. Curr Opin Invest Drugs. (2009) 10:434–42.
217. Datta S, Cano M, Ebrahimi K, Wang L, Handa JT. The impact of oxidative stress and inflammation on RPE degeneration in non-neovascular AMD. Prog Retin Eye Res. (2017) 60:201–18. doi: 10.1016/j.preteyeres.2017.03.002
218. Cao X, Shen D, Patel MM, Tuo J, Johnson TM, Olsen TW, et al. Macrophage polarization in the maculae of age-related macular degeneration: a pilot study. Pathol Int. (2011) 61:528–35. doi: 10.1111/j.1440-1827.2011.02695.x
219. Zhuang Y, Lyga J. Inflammaging in skin and other tissues – the roles of complement system and macrophage. Inflamm Allergy Drug Targets. (2014) 13:153–61. doi: 10.2174/1871528113666140522112003
220. de Oliveira LM, Teixeira FME, Sato MN. Impact of retinoic acid on immune cells and inflammatory diseases. Mediat Inflamm. (2018) 2018:3067126–3067126. doi: 10.1155/2018/3067126
221. Waugh N, Loveman E, Colquitt J, Royle P, Yeong JL, Hoad G, et al. Treatments for dry age-related macular degeneration and stargardt disease: a systematic review. Health Technol Assess. (2018) 22:1–168. doi: 10.3310/hta22270
222. Charbel Issa P, Barnard AR, Singh MS, Carter E, Jiang Z, Radu RA, et al. Fundus autofluorescence in the Abca4-/- Mouse model of stargardt disease—correlation with accumulation of A2E, retinal function, and histology. Invest Ophthalmol Vis Sci. (2013) 54:5602–12. doi: 10.1167/iovs.13-11688
223. Tsybovsky Y, Molday RS, Palczewski K. The ATP-binding cassette transporter ABCA4: structural and functional properties and role in retinal disease. Adv Exp Med Biol. (2010) 703:105–25. doi: 10.1007/978-1-4419-5635-4_8
224. Radu RA, Han Y, Bui TV, Nusinowitz S, Bok D, Lichter J, et al. Reductions in serum vitamin A arrest accumulation of toxic retinal fluorophores: a potential therapy for treatment of lipofuscin-based retinal diseases. Invest Ophthalmol Vis Sci. (2005) 46:4393–401. doi: 10.1167/iovs.05-0820
225. Sofi F, Sodi A, Franco F, Murro V, Biagini D, Miele A, et al. Dietary profile of patients with Stargardt’s disease and retinitis pigmentosa: is there a role for a nutritional approach? BMC Ophthalmol. (2016) 16:13. doi: 10.1186/s12886-016-0187-3
226. Kong X, Strauss RW, Cideciyan AV, Michaelides M, Sahel J-A, Munoz B, et al. Visual acuity change over 12 months in the prospective progression of atrophy secondary to stargardt disease (ProgStar) study: progstar report number 6. Ophthalmology. (2017) 124:1640–51. doi: 10.1016/j.ophtha.2017.04.026
227. Kaufman Y, Ma L, Washington I. Deuterium enrichment of vitamin A at the C20 position slows the formation of detrimental vitamin A dimers in wild-type rodents. J Biol Chem. (2011) 286:7958–65. doi: 10.1074/jbc.M110.178640
228. Saad L, Washington I. Can vitamin A be improved to prevent blindness due to age-related macular degeneration, stargardt disease and other retinal dystrophies? In: C Bowes Rickman, MM LaVail, RE Anderson, C Grimm, J Hollyfield, J Ash editors. Retinal Degenerative Diseases. (Cham: Springer International Publishing) (2016). p. 355–61. doi: 10.1007/978-3-319-17121-0_47
229. Ma L, Kaufman Y, Zhang J, Washington I. C20-D3-vitamin A slows lipofuscin accumulation and electrophysiological retinal degeneration in a mouse model of stargardt disease. J Biol Chem. (2011) 286:7966–74. doi: 10.1074/jbc.M110.178657
230. Charbel Issa P, Barnard AR, Herrmann P, Washington I, MacLaren RE. Rescue of the Stargardt phenotype in Abca4 knockout mice through inhibition of vitamin A dimerization. Proc Natl Acad Sci USA. (2015) 112:8415–20. doi: 10.1073/pnas.1506960112
231. Zhang D, Robinson K, Washington I. C20D3-vitamin A prevents retinal pigment epithelium atrophic changes in a mouse model. Transl Vis Sci Technol. (2021) 10:8. doi: 10.1167/tvst.10.14.8
232. National Institutes of Health.A Phase 1, Open Label, Repeat Dose Study to Investigate the Safety and Pharmacokinetics of 4-week Daily Dosing of ALK-001 in Healthy Volunteers. Bethesda, MD: National Institutes of Health (2015).
233. National Institutes of Health.A Phase 2 Multicenter, Double-Masked, Randomized, Placebo-Controlled Study to Investigate the Long Term Safety, Tolerability, Pharmacokinetics and Effects of ALK-001 on the Progression of Stargardt Disease [Internet]. Bethesda, MD: National Institutes of Health (2021).
234. Aleman TS, Cideciyan AV, Windsor EAM, Schwartz SB, Swider M, Chico JD, et al. Macular pigment and lutein supplementation in ABCA4-associated retinal degenerations. Invest Ophthalmol Vis Sci. (2007) 48:1319–29. doi: 10.1167/iovs.06-0764
235. Arunkumar R, Gorusupudi A, Li B, Blount JD, Nwagbo U, Kim HJ, et al. Lutein and zeaxanthin reduce A2E and iso-A2E levels and improve visual performance in Abca4-/-/Bco2-/- double knockout mice. Exp Eye Res. (2021) 209:108680. doi: 10.1016/j.exer.2021.108680
236. Scripsema NK, Hu D-N, Rosen RB. Lutein, zeaxanthin, and meso-zeaxanthin in the clinical management of eye disease. J Ophthalmol. (2015) 2015:e865179. doi: 10.1155/2015/865179
237. Owsley C, McGwin G, Jackson GR, Heimburger DC, Piyathilake CJ, Klein R, et al. Effect of short-term, high-dose retinol on dark adaptation in aging and early age-related maculopathy. Invest Ophthalmol Vis Sci. (2006) 47:1310–8. doi: 10.1167/iovs.05-1292
238. Age-Related Eye Disease Study Research Group. The relationship of dietary carotenoid and vitamin A, E, and C intake With age-related macular degeneration in a case-control study: AREDS report No. 22. Arch Ophthalmol. (2007) 125:1225–32. doi: 10.1001/archopht.125.9.1225
239. Age-Related Eye Disease Study 2 Research Group. Lutein + zeaxanthin and omega-3 fatty acids for age-related macular degeneration: the age-related eye disease study 2 (AREDS2) randomized clinical trial. JAMA. (2013) 309:2005–15. doi: 10.1001/jama.2013.4997
240. The Age-Related Eye Disease Study Research Group. The age-related eye disease study (AREDS): design implications AREDS report no. 1. Control Clin Trials. (1999) 20:573–600. doi: 10.1016/S0197-2456(99)00031-8
241. Age-Related Eye Disease Study Research Group. A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E, beta carotene, and zinc for age-related macular degeneration and vision loss: AREDS report no. 8. Arch Ophthalmol. (2001) 119:1417–36. doi: 10.1001/archopht.119.10.1417
242. Agrón E, Mares J, Clemons TE, Swaroop A, Chew EY, Keenan TDL. Dietary nutrient intake and progression to late age-related macular degeneration in the age-related eye disease studies 1 and 2. Ophthalmology. (2021) 128:425–42. doi: 10.1016/j.ophtha.2020.08.018
243. Munch IC, Toft U, Linneberg A, Larsen M. Precursors of age-related macular degeneration: associations with vitamin A and interaction with CFHY402H in the Inter99 eye study. Acta Ophthalmol. (2016) 94:657–62. doi: 10.1111/aos.13198
244. Chew EY, Clemons TE, Agrón E, Domalpally A, Keenan TDL, Vitale S, et al. Long-term outcomes of adding lutein/zeaxanthin and ω-3 fatty acids to the AREDS supplements on age-related macular degeneration progression: AREDS2 Report 28. JAMA Ophthalmol. (2022):e221640.[Epub ahead of print].
Keywords: vitamin A, retina, gut microbiome, T cell, Stargardt’s disease, age - related macular degeneration, carotenoids, retinoids
Citation: Thirunavukarasu AJ, Ross AC and Gilbert RM (2022) Vitamin A, systemic T-cells, and the eye: Focus on degenerative retinal disease. Front. Nutr. 9:914457. doi: 10.3389/fnut.2022.914457
Received: 06 April 2022; Accepted: 29 June 2022;
Published: 18 July 2022.
Edited by:
Yannan (Jessica) Jin, De Montfort University, United KingdomReviewed by:
Xu Zhang, The Scripps Research Institute, United StatesRohan Kumar Raman, ICAR-Research Complex for Eastern Region, India
Copyright © 2022 Thirunavukarasu, Ross and Gilbert. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rose M. Gilbert, cm9zZS5naWxiZXJ0QG5ocy5uZXQ=