 Masako Suzuki*
Masako Suzuki* Meika Tomita
Meika Tomita- Department of Genetics, Albert Einstein College of Medicine, Bronx, NY, United States
Vitamin A, an essential fat-soluble micronutrient, plays a critical role in the body, by regulating vision, immune responses, and normal development, for instance. Vitamin A deficiency (VAD) is a major cause of xerophthalmia and increases the risk of death from infectious diseases. It is also emerging that prenatal exposure to VAD is associated with disease risks later in life. The overall prevalence of VAD has significantly declined over recent decades; however, the rate of VAD is still high in many low- and mid-income countries and even in high-income countries among specific ethnic/race groups. While VAD occurs when dietary intake is insufficient to meet demands, establishing a strong association between food insecurity and VAD, and vitamin A supplementation is the primary solution to treat VAD, genetic contributions have also been reported to effect serum vitamin A levels. In this review, we discuss genetic variations associated with vitamin A status and vitamin A bioactivity-associated genes, specifically those linked to uptake of the vitamin in the small intestine and its storage in the liver, as well as their potential contribution to vitamin A deficiency risks among different ethnic groups.
Introduction
Malnutrition refers to deficiencies, excess, or imbalance of energy or nutrition intake. In 2020, about one in three people in the world (2.37 billion) did not have access to adequate food (1). Iron, iodine, folate, vitamin A, and zinc deficiencies are the most significant micronutrient-related malnutrition conditions in the world, specifically in low-income and middle-income countries (2–4). Vitamin A, an essential fat-soluble micronutrient, plays a critical role in the body, effecting vision, immune responses, and normal development. Vitamin A deficiency (VAD) is a major cause of xerophthalmia and increases the risk of death from infectious diseases. In addition, prenatal exposure to VAD is associated with preterm birth, lung and kidney functions of the fetus, and mortality of offspring born to mothers with human immunodeficiency virus infections (5–7). While VAD prevalence rates have reduced in the last two decades, about 30% of children world-wide under the age of 5 years are still vitamin A deficient, and 1.7% of all deaths are attributable to VAD in this age group (3). VAD occurs when dietary intake is insufficient to meet demands; thus, food insecurity is strongly associated with its prevalence. The amount of vitamin A we need in the body depends on age and sex. The Recommended Daily Allowances (RDAs) recommended by FDA is 900 μg of Retinol Activity Equivalents (RAE) for adult men, 700 μg for adult women, 770 μg for pregnant women, 1300 μg for breastfeeding women, and 400 μg for infants aged 6 months or less (8). An RDA is the average daily dietary intake level to meet sufficient nutrition requirements for nearly all (97–98%) healthy individuals in a group. RAE for each vitamin A form can convert as: 1 μg RAE = 1 μg retinol, 2 μg β-carotene from a supplement, 12 μg β-carotene from food, 24 μg α-carotene from food, or 24 μg β-cryptoxanthin from food (8, 9). There are two major forms of vitamin A in the human diet, preformed vitamin A, mainly as retinol, retinyl esters and small amount of retinoic acid, and provitamin A carotenoids, mainly as β-carotene. The proportion of preformed vitamin A and provitamin A carotenoid intake is dependent on our diet. Preformed vitamin A is derived from animal products and provitamin A carotenoids from plant-derived food [reviewed in (10)]. Since our body can downregulate bioconversion of provitamin A carotenoids (11, 12), high intakes of fruit and vegetables will usually not significantly contribute to hypervitaminosis A. However, it has been reported that high-dose supplementation of β-carotene increases cancer incidence in the lung and stomach (13–16), suggesting that excess supplementation of provitamin A carotenoid could be harmful to humans. In contrast to provitamin A, if the dietary intake of preformed vitamin A becomes high through foods, such as animal liver, fortified foods, or supplements, the body will store the excess in the liver, reaching levels defining hypervitaminosis A (defined as ≥1 μg/g liver) (17, 18). Ingested preformed vitamin A and provitamin A carotenoid are processed for normal physiological functions or storage (bioavailability). While the metabolic pathways by which each form of vitamin A is metabolized are partly different until they are converted to retinal, both preformed vitamin A and provitamin A carotenoids are metabolized to the active vitamin A molecules, retinoic acid that directly regulates gene activity and function, in the body [reviewed in (19)].
Although vitamin A deficiency in high-income countries is believed to be a rare condition, it has been reported that certain ethnic groups have high rates of vitamin A deficiency even in developed countries (20–22). Several genetic variations to play a role in serum vitamin A levels have been reported in humans (23, 24). As we reported previously, the allele frequencies of some genetic variations associated with vitamin A bioavailability vary among ethnic groups (22). Moreover, Obrochta et al. reported that strain and tissue-specific variations of retinol and all trans-retinoic acid in serum and tissues of five inbred mouse strains feeding two different vitamin A concentration diets to the mothers (25). Since all mouse strains fed equal amounts of vitamin A in the study, their findings suggest the existence of genetic variation-dependent vitamin A variations. This review focuses on genetic variations associated with vitamin A status and vitamin A bioactivity-associated genes, specifically vitamin A uptake in the small intestine and storage in the liver, and their potential contribution to vitamin A deficiency risks among different ethnic groups.
Intestinal Absorption of β-Carotene
In the human diet, retinyl palmitate and β-carotene are the dominant forms of vitamin A (26). While retinyl palmitate is enzymatically converted to retinol in the intestinal lumen before absorption by enterocytes, β-carotene is partially converted to retinol in the enterocytes. Interestingly, high inter-individual variability in carotenoid absorption from nutritional intake has been observed in human studies (27–30). Moreover, reported β-carotene absorption rates differ between individuals as well as between studies, for instance, 3.4% (n = 12 individuals) (30) to 90.0% (n = 5 individuals) (28) following the oral administration of a pharmacologic dose of β-carotene. These interindividual efficiency ranges were much higher than that of the preformed vitamin A (retinol) absorption efficiency (70 to 90%) (8, 31). Intriguingly, O'Neill and Thurnham further reported that while the interindividual variations of absorption rates of β-carotene are high, the response to β-carotene was constant within an individual and reproducible over time (30). De Pee & West reviewed and noted that a number of factors influence the bioavailability of carotenoids, which they grouped in the mnemonic SLAMENGHI: Species of carotenoids, molecular Linkage, Amount of carotenoids consumed in a meal, Matrix in which the carotenoid is incorporated, Effectors of absorption and bioconversion, the Nutrient status of the host, Genetic factors, Host-related factors, and mathematical Interactions (32). The genetic contribution to interindividual variability of β-carotene status has also been proposed [reviewed in (33)]. While a part of β-carotene incorporated in mixed micelles reaches the apical membrane as free molecules, the scavenger receptor class B type I (SR-BI) protein, encoded by SCARB1, and cluster determinant 36 (CD36), also known as FAT (fatty acid transporter), are involved in cellular uptake of β-carotene in the small intestine (29, 34). Associations between SNPs and haplotypes in SCARB1 and CD36 and plasma concentrations of provitamin A carotenoids have been identified (29, 35). Study participants bearing the C allele of the SNP rs61932577 at the SCARB1 intron 5 had lower β-carotene concentrations than men homozygous for the T allele, while participants bearing rs5888C/rs4238001C/rs61932577T haplotype had lower plasma β-cryptoxanthin than the participants did bearing the rs5888T/rs4238001C/rs61932577C haplotype (35). The same research group also reported that the CD36 haplotype affects plasma provitamin A carotenoid (29).
In the enterocyte, the β-carotene taken up is converted into retinaldehyde by the β-Carotene 15,15′ oxygenase 1 enzyme [BCO1, previously named β-Carotene 15,15'-monooxygenase 1 (BCMO1)] and the retinaldehyde is then reduced to retinol by a retinal reductase (36, 37). We summarize the known genetic polymorphisms of the BCO1 gene, the associations of retinoid status, and the linkage disequilibrium of the known genetic polymorphisms in Figure 1A. It has been suggested that the reduced enzymatic activity of BCO1 might cause the large interindividual variability of β-carotene absorption as a consequence of genetic polymorphisms in the gene (38). Importantly, the study reported that these polymorphisms, rs12934922 A/T (R267S) and rs7501331 C/T (A379V), are common variants (i.e., variants with a frequency of >1% in the population). They reported that 379V and 267S + 379V reduced the ability to convert β-carotene (38). Using the 1,000 Genomes Project dataset, we found that the low BCO1 activity genotype allele frequency is higher in European Ancestry, especially the Finnish group who have the highest percent (267S + 379V 9.09, and 379V 37.9%) out of all 25 ethnic/racial groups (Figure 1B). The aforementioned study which found that β-carotene supplementation increases lung cancer incidence was conducted on Finnish cohorts suggesting a subset of participants carry low BCO1 activity genotype (15, 16). A Bco1 knock-out mouse model study showed that while β-carotene supplementation to mice lacking Bco1 function reduced the inflammatory response, it increased concentration of β-carotene in lung and serum and retinyl ester concentration in the lung compared to wild type mice (39). This result suggests that a person who carries the genotype that reduces the activity of the Bco1 enzyme may more readily accumulate β-carotene and retinyl ester in the lung following β-carotene supplementation. Therefore, assessing BCO1 genotypes in participants who developed lung cancer in the β-carotene supplementation study is needed to test whether the BCO1 genotype also modulates lung cancer incidence. The associations between other common variants of the gene and genes adjacent to the BCO1 gene and serum β-carotene status also have been extensively reported (40–45). The rs6564851 common variant located about 8 kb upstream of the BCO1 gene is reported to be associated with circulating levels of β-carotene in different ethnic groups (40, 43, 44). The rs6564851 variant is located within a haplotype that includes other genetic polymorphisms (Figure 1A). Hendrickson et al. carefully tested the associations between BCO1 common variants and plasma carotenoid concentrations and developed gene score systems that predict plasma carotenoid concentrations in women of European descent (42). In their model, the rs12934922 T allele and rs4889286 T allele predict higher plasma β-carotene (42). Interestingly, the proportions of these allele combinations vary between ethnic/race groups, with the allele frequencies of the “lower” plasma β-carotene combination highest among African ancestry groups (Figure 1C). These findings show that testing the associations of genotypes with plasma β-carotene in a multi-ethnic population is needed. Besides the common variants, one missense pathogenic variant of BCO1, rs119478057, has been identified in a patient with hypercarotenemia and hypovitaminosis A (46, 47). The rs119478057 T allele is a rare variant with a frequency less than 0.01 in the European population.
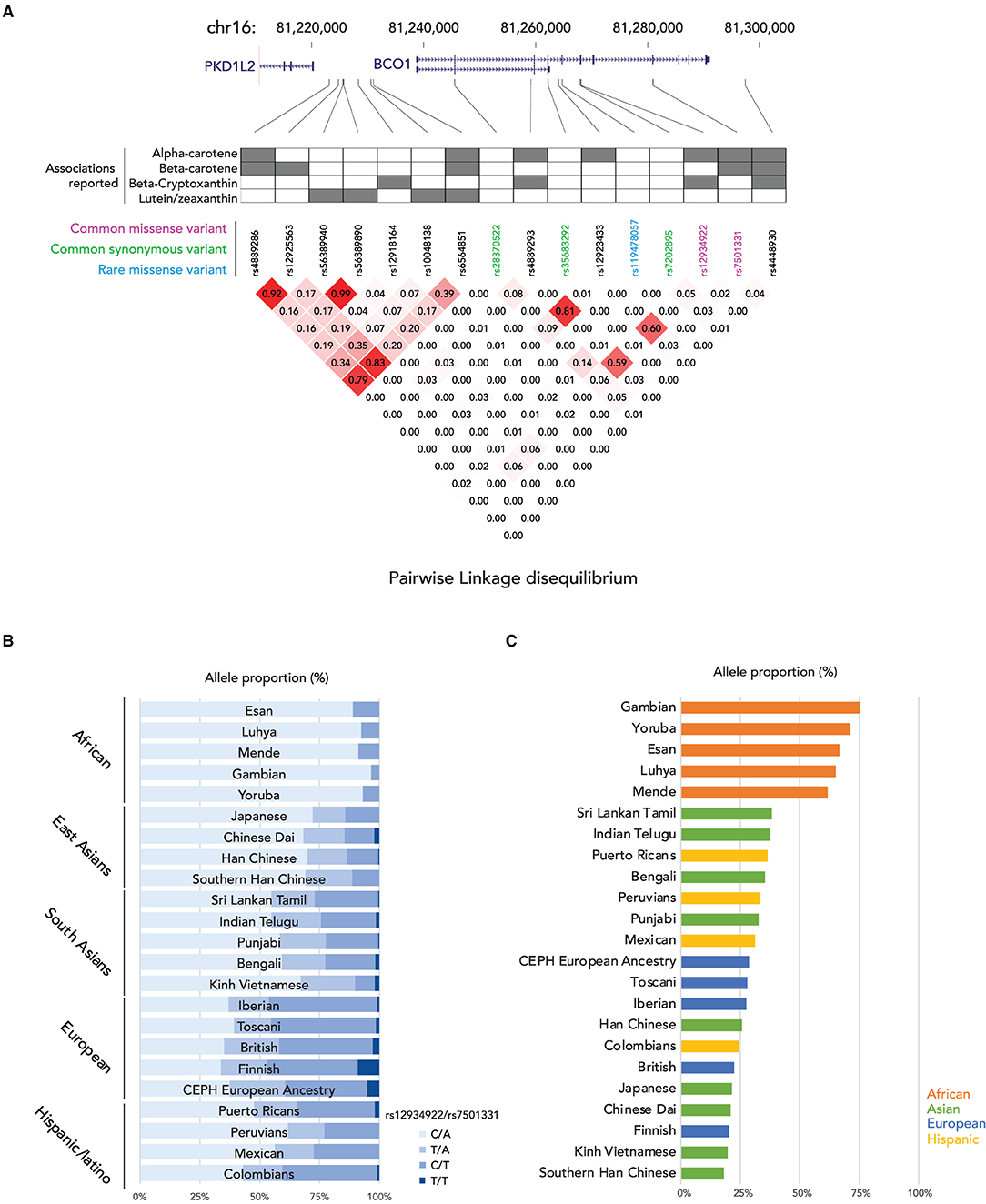
Figure 1. Summary of genetic variations of BCO1 gene. (A) (top) locations of genetic variants in BCO1 locus; (middle) the association to the provitamin A status of each genetic variation; (bottom) Linkage Disequilibrium (LD) plot, the numbers in the boxes indicate the LD status and darker red shading illustrate higher LD; (B) Proportions of BCO1 gene activity genotypes by ethnic groups. (C) Proportion of low BCO1 gene activity haplotype defined by Hendrickson's estimation by ethnic groups.
β-carotene-9',10'-oxygenase 2 (BCO2) is another carotenoid converting enzyme located in the mitochondria; it cleaves β-carotene asymmetrically in position 9',10' generating long-chain apocarotenoids. BCO1 and BCO2 also differ for their substrate specificity. Lutein, lycopene, β-cryptoxanthin, and zeaxanthin are all specific substrates for BCO2, which BCO1 cannot cleave (48). While livestock studies suggested that BCO2 mutations are associated with carotenoid metabolism alterations, genetic variation associations in humans' carotenoid statuses are not elucidated yet. Table 1 lists the reported genetic variants and the reported associations on provitamin A intestinal absorption.
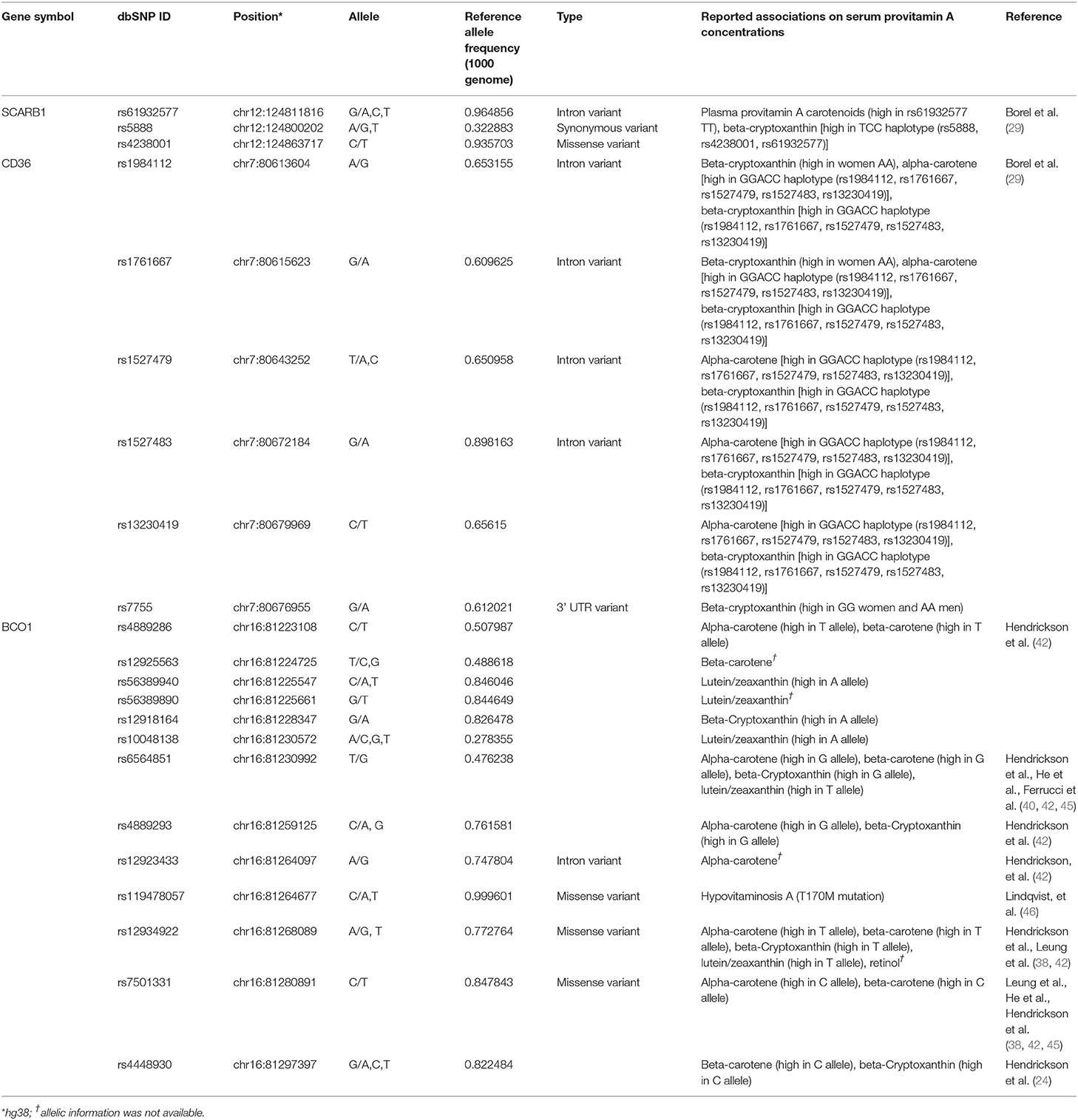
Table 1. A list of sequence polymorphisms and the reported associations on provitamin A intestinal absorption.
Intestinal Absorption of Retinol
Dietary retinyl esters are enzymatically converted to retinol in the intestinal lumen before absorption by enterocytes. Pancreatic lipase, encoded by PNLIP, and pancreatic lipase-related protein 2, encoded by PNLIPRP2, hydrolyze retinyl palmitate to retinol (49). PNLIP, on chromosome 10q26.1, encodes a 465-amino acid protein and is predominantly expressed in the pancreas (50). One missense variant of PNLIP (rs746000327 C/T) associated with pancreatic lipase deficiency has been identified in two brothers from a consanguineous family of Arab ancestry (51). The frequency of rs746000327 T allele is less than 0.0001 in NHLBI Trans-Omics for Precision Medicine (TOPMed, PRJNA400167) Whole Genome Sequencing (WGS) Project and the Genome Aggregation Database (gnomAD, PRJNA398795). Currently, no sequence variations with clinical significance on PNLIPRP2 have been reported. In enterocytes, retinol that has been taken up binds to retinol-binding protein 2 [RBP2; initially called cellular retinol-binding protein, type II (CRBPII) (52)]. This retinol-RBP2 complex serves as a substrate for microsomal lecithin:retinol acyltransferase (LRAT). RBP2 is exclusively expressed in the small intestinal absorptive cells with specific localization (highly expressed in duodenum and jejunum and less expressed in the ileum). It is one of the most abundant soluble proteins in the small intestine (53, 54). It has been suggested that the expression of RBP2 is regulated predominantly by dietary fatty acids but little by dietary retinoids, as reviewed by Takase et al. (55). Currently, no sequence variations with clinical significance have been identified in RBP2. Retinol bound to RBP2 converted from retinyl palmitate is re-esterified by the enzyme lecithin:retinol acyltransferase (LRAT) and acyl coA:retinol acyltransferase (ARAT). While retinol complexed with CRBP2 is the preferred substrate of LRAT, uncomplexed retinol may be esterified by ARAT. LRAT may mainly esterify in the intestine when the retinol is present in normal amounts. In contrast, when retinol is present at high levels, and CRBP becomes saturated, ARAT may function to esterify the excess (26). Three rare pathogenic variants of LRAT have been identified in European populations; a single nucleotide mutation, rs104893848, and two 2-bp deletions, rs761717462 and rs1560870755. Two variants, rs104893848 and rs761717462, were identified as early-onset severe retinal dystrophy-associated mutations (56). The rs104893848 is a missense variant causing the Ser175Arg (S175R) amino acid change. The rs761717462 variant is a 2-bp deletion (396delAA) which causes a frameshift after codon 133 to encode 11 amino acids unrelated to the wildtype sequence followed by a premature stop codon. The S175R missense variant shows no acyltransferase activity in an in vitro study (56). The third variant, rs1560870755, a Leber congenital amaurosis-associated mutation, is a mutation causing a frameshift at codon 73 and a premature termination codon at residue 120. The mutation results in the loss of essential amino acids for accepting the acyl group from the lecithin (57). In humans, two transferases can also have acyl-CoA:retinol acyltransferase activities have been identified, diacylglycerol O-acyltransferase 1 (DGAT1) (58–60) and acyl-CoA wax alcohol acyltransferase 2 (AWAT2) (61). While both genes have the acyl-CoA:retinol acyltransferase activities, DGAT1 plays an important intestinal ARAT in vivo (60). One pathogenic variant of DGAT1, rs148665132, has been identified in a study of a family of Ashkenazi Jewish descent with congenital diarrheal disorders; however, the effect of the variant on acyl-CoA:retinol acyltransferase activity is not yet elucidated (62). Table 2 lists these pathogenic sequence polymorphisms of intestinal retinol absorption-related genes.
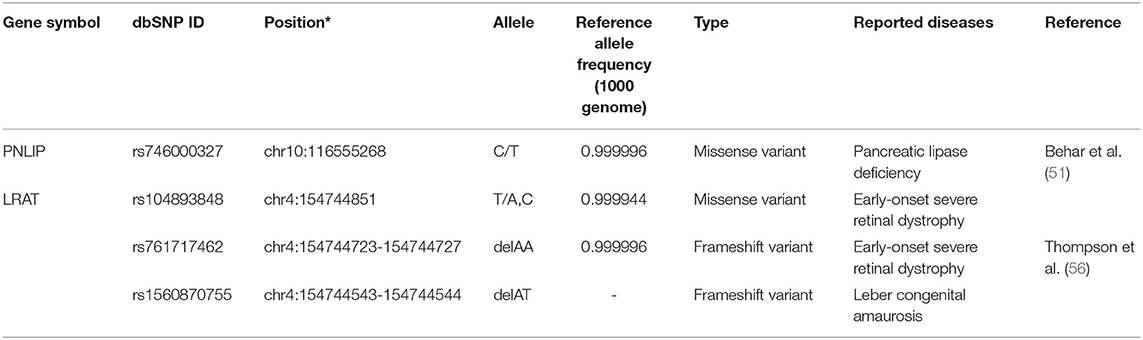
Table 2. A list of pathogenic sequence polymorphisms of intestinal retinol absorption related genes.
Transport, Absorption, and Storage In The Liver
As mentioned earlier, newly absorbed vitamin A in the small intestine is packaged into chylomicrons and secreted to the lymphatic system by enterocytes. Once the chylomicrons reach the circulation, a part of the triglycerides are hydrolyzed, and the lipoprotein particles shrink to form chylomicron remnants. About 70% of retinoids packaged into chylomicron remnants in the bloodstream are taken up by the liver, the main organ involved in storing retinoids and regulating serum vitamin A levels (63, 64). Besides the liver, chylomicron remnants are delivered to the lung, heart, kidney, adipose tissue, muscle, spleen, and bone marrow (65, 66). In the liver, hepatocytes play the leading role in the uptake of chylomicrons, where retinyl ester is hydrolyzed again to form retinol by several enzymes, such as carboxyl ester lipases and carboxylesterases, and hepatic lipases [reviewed in (67)]. In the hepatocyte, formed retinol is bound to cellular retinol-binding protein 1 (CRBP1 or RBP1) and then is transferred to retinol-binding protein 4 (RBP4 or RBP) to secrete into e the circulation or transferred to hepatic stellate cells where retinol is re-esterified by LRAT. From the hepatocyte, retinol bound to the RBP4 is secreted into the circulation and delivered to the target cells and organs. RBP4 is the sole specific retinol-binding protein in the blood, and RBP-bound retinol accounts for approximately 99% of all retinoids present in the blood in the fasting state (68). Genetic variations of RBP4 and serum retinol levels have been identified in European populations and estimated that 2.3% of the variance in serum retinol levels in the cohort are accounted for the genetic variations of RBP4 and transthyretin (TTR)(24), and low serum retinol level allele frequency varies between race/ethnic groups (22). Delivery of retinol to the target cells is mediated by the binding of retinol-bound- (holo-) RBP and an RBP receptor called STRA6 (Stimulated by Retinoic Acid 6) (69). STRA6 is ubiquitously expressed, and its expression status is regulated by the retinoid metabolisms (69). While there are five genetic variants listed on the NHGRI-EBI Catalog of Published Genome-Wide Association Studies, rs351242 (70), rs351237 (71), rs11635868 (72), rs10910 (73), and rs11638831 (74), no associations between these genetic variants and STRA6 function have been described. Interestingly, a missense double-nucleotide polymorphism (g.1157G>A and g.1156G>A; p.Gly304Lys, rs151341424) in STRA6 was found in the Irish Traveler family with isolated microphthalmia and coloboma (MCOPCB8) (75), a rare autosomal recessive disorder (76). Casey reported that the G304K mutant STRA6 protein is mislocalized and has severely reduced vitamin A uptake activity (75). Besides rs151341424, four common missense variants, rs736118, rs351236, rs971756, and rs76336272, have been identified. Of those, rs971756 and rs736118 are reported to be linked to human diseases (77–84), but associations of the variants to STRA6 function were not described.
Hepatic stellate cells play a major role in storing vitamin A in the liver. It has been estimated that approximately 70% of vitamin A in the body of healthy and well-nourished individuals is stored in the liver, specifically a small subset of cells, hepatic stellate cells (85). Hepatic LRAT expression is regulated by vitamin A status, a positive feedback loop to increase retinyl ester synthesis when cellular retinoic acid concentration is high (86–88). Two lipases mainly perform the mobilization of retinyl ester stores: the patatin-like phospholipase domain-containing 2 (PNPLA2)/adipose triglyceride lipase (ATGL) and patatin-like phospholipase domain-containing 3 (PNPLA3) proteins (89, 90). Currently, 11 genetic variants in ATGL/PNPLA2 are reported to associate with neutral lipid storage disease with myopathy (91–94); but no associations of retinyl ester mobilization and these genetic variations have been reported. A missense genetic variation of the PNPLA3 (I148M, rs738409) gene, one of the most prominent genetic risk factors associated with non-alcoholic liver disease (NAFLD), elevates retinyl ester levels in the liver with reduced serum retinol levels (90, 95, 96). While 32 missense variations have been identified in the PNPLA3 gene, rs738409 is the only pathogenic genetic variant of the PNPLA3 gene. Table 3 lists the reported genetic variants associated with the transport, absorption, and storage of vitamin A in the liver.
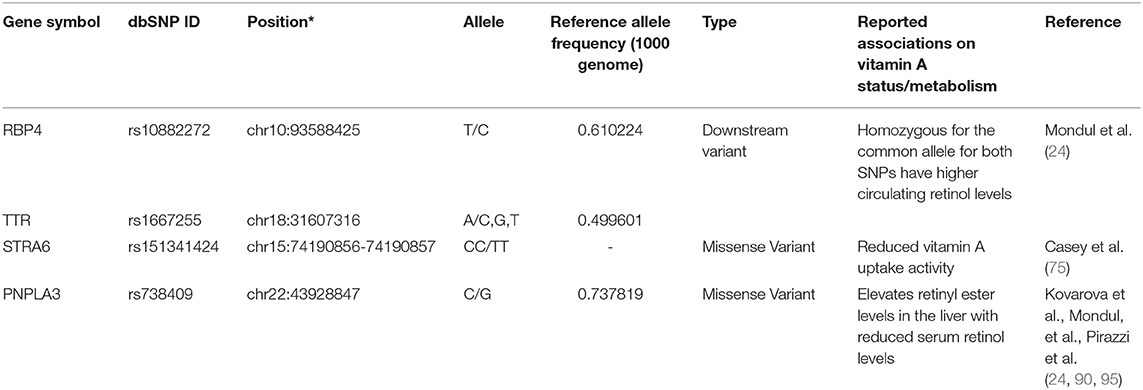
Table 3. A list of sequence polymorphisms and the reported associations on transport, absorption, and storage of vitamin A in liver.
Genetic Variants and Expression Status of The Vitamin a Intake-Related Genes
The associations between genetic variants and gene expression status have been well-characterized. In the previous sections, we discussed the associations of genetic variants on the vitamin A metabolism related to gene functions and human disease. This section focuses on the genetic variants that alter gene expression status. Genetic variants associated with gene expression status, expression quantitative trait loci (eQTLs), have been identified in the human genome (97–100). We searched for eQTLs of the vitamin A metabolism-related genes in the Genotype-Tissue Expression (GTEx) project database (97). We identified eQTLs clusters for RBP4 (liver) and PNLIPRP2 (pancreas) (Figure 2, Supplementary Table 1). The RBP4 gene has two eQTLs, rs11187538 and rs11187547, located in a haplotype block including the previously reported genetic variant associated with lower RBP4 levels, rs10882272 (24). Allele frequency analysis showed that Eastern Asian ancestry has a higher frequency of the low-expression haplotype (rs11187538C/rs11187547A) (Figure 2A). While no genetic variants with clinical significance on PNLIPRP2 have been reported, there are 173 eQTLs in its most highly expressed tissue, the pancreas. Haplotype analysis indicates that the PNLIPRP2 eQTL cluster contains two large blocks (Figure 2B). The block located upstream of the gene (Block 1) is positively associated with the expression with the reference allele, and the block downstream of the gene (Block 2) is negatively associated with the reference allele. Based on the 1,000 Genomes allele frequency database, the haplotype allele frequencies of the blocks vary by race/ethnicity. Europeans and South Asians have higher frequencies with low expression haplotypes in Block 1, and East Asians have higher frequencies with low expression haplotypes in Block 2. These findings suggest that the genetic variations might contribute to the vitamin A metabolism through modifications of the expression of these vitamin A intake-related genes.
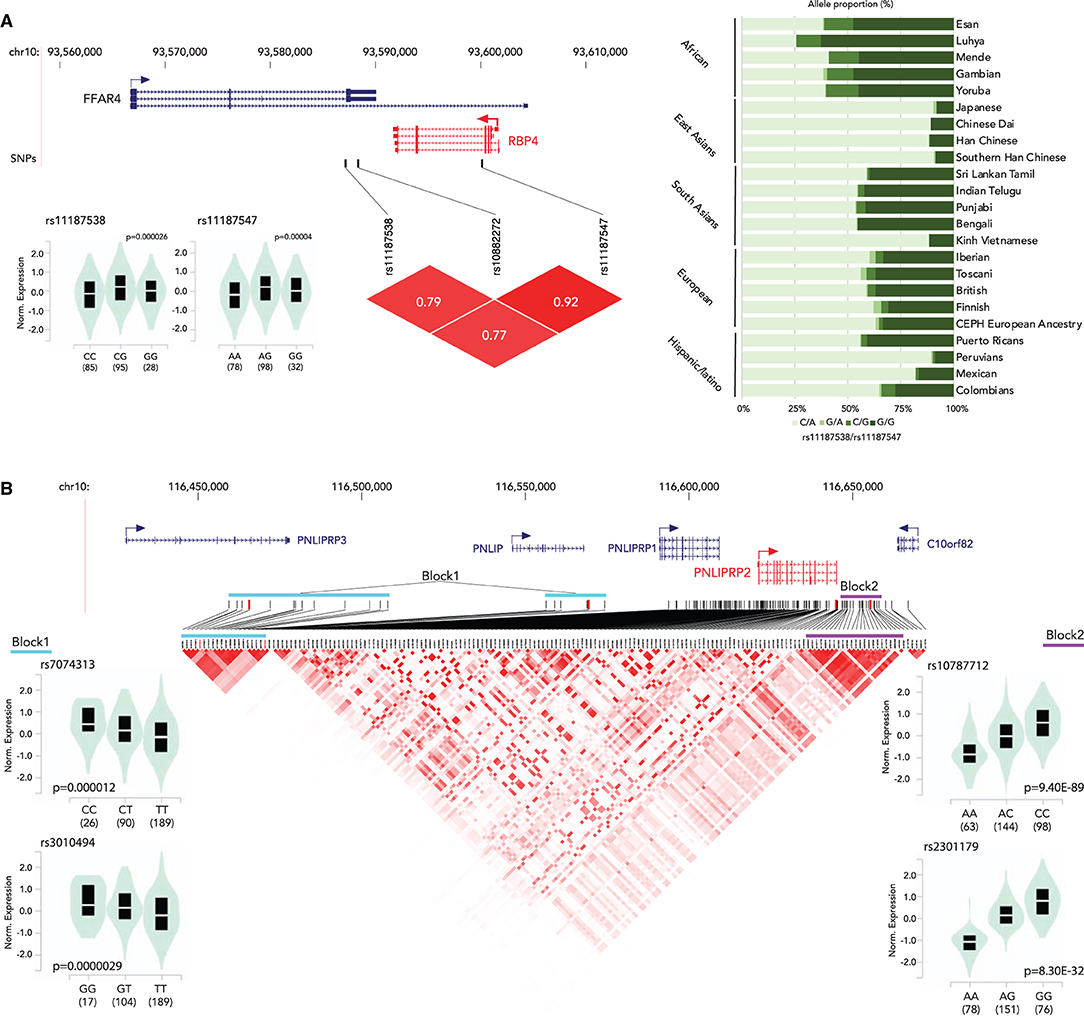
Figure 2. Summary of eQTLs of RBP4 (A) and PNLIPRP2 (B). (A) (top) Locations of eQTLs in RBP4; (bottom left) normalized expression status of RBP4 by genotype; (bottom right) an LD plot; (right) haplotype proportion by race/ethnicity group. (B) (top) Locations of eQTLs in PNLIPRP2; (bottom left) normalized expression status of PNLIPRP2 by genotype (block 1); (bottom center) a Linkage disequilibrium (LD) plot of eQTLs; (bottom right) normalized expression status of PNLIPRP2 by genotype (block 2).
Discussion
While the prevalence of VAD has significantly declined over recent decades (3), it remains a concern from a population health perspective that the rate of VAD remains high in many low- and mid-income countries (3, 101, 102) and in specific ethnic/race groups within high-income countries (22). West reported that approximately 127 million pre-school-aged children and 7 million pregnant women were VAD in 2003 (103). The WHO estimated in 2009 that 5.2 million preschool children and 9.8 million pregnant women were affected with the VAD-associated phenotype of night blindness (104). Many interventions have been implemented to reduce VAD in lower-income countries (101); but these interventions did not consider the individual susceptibilities associated with genetic variation and race/ethnicities. As described in this review, genetic variation near retinoid metabolism-related genes significantly contributes to the function and expression levels of these genes. Excess amounts of vitamin A can be toxic to human health and have adverse effects on normal development (reviewed in [105]). Moreover, the genome-wide association study that identified genetic variants contributing to serum retinol levels was performed in European ancestry, in which the overall rate of VAD is low (24). This raises the possibility that this kind of study may not fully detect all of the genetic variants that contribute to VAD. Although vitamin A supplementation is a primary treatment for VAD and a solution to reduce the prevalence rate of VAD in populations bearing a high prevalence of VAD, knowing the person's vitamin A metabolism-related gene variants helps to perform the treatment and intervention effectively and safely. In addition, vitamin A metabolism is not fully elucidated yet. Genetic variations associated with the process that has not been described fully could contribute to the VAD. We propose a major need to perform genome-wide association studies in multiethnic and admixed populations to fill this knowledge gap.
Author Contributions
MT: data curation, visualization, and writing—review and editing. MS: conceptualization, visualization, writing—original draft preparation, writing—review and editing, and funding acquisition. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Institutes of Health under award number R01HL145302. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors thank Drs. John M. Greally and Loredana Quadro for their advice.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnut.2022.861619/full#supplementary-material
References
1. Food Food and Agriculture Organization of the United Nations International Fund for Agricultural Development United Nations International Children's Emergency Fund Programme WF Organization WH. The state of food security and nutrition in the world 2021. Food Agri Org. (2021), p. 240. doi: 10.4060/cb4474en
2. Freeland-Graves JH, Sachdev PK, Binderberger AZ, Sosanya ME. Global diversity of dietary intakes and standards for zinc, iron, and copper. J Trace Elem Med Biol. (2020) 61:126515. doi: 10.1016/j.jtemb.2020.126515
3. Stevens GA, Bennett JE, Hennocq Q, Lu Y, De-Regil LM, Rogers L, et al. Trends and mortality effects of vitamin A deficiency in children in 138 low-income and middle-income countries between 1991 and 2013: a pooled analysis of population-based surveys. Lancet Glob Health. (2015) 3:e5283:15 doi: 10.1016/S2214-109X(15)00039-X
4. Black RE, Victora CG, Walker SP, Bhutta ZA, Christian P, de Onis M, et al. Maternal and child undernutrition and overweight in low-income and middle-income countries. Lancet. (2013) 382:427–51. doi: 10.1016/S0140-6736(13)60937-X
5. Semba RD, Miotti PG, Chiphangwi JD, Liomba G, Yang LP, Saah AJ, et al. Infant mortality and maternal vitamin A deficiency during human immunodeficiency virus infection. Clin Infect Dis. (1995) 21:966Dis Ddoi: 10.1093/clinids/21.4.966
6. Verma RP, McCulloch KM, Worrell L, Vidyasagar D. Vitamin A deficiency and severe bronchopulmonary dysplasia in very low birthweight infants. Am J Perinatol. (1996) 13:966–72.
7. Radhika MS, Bhaskaram P, Balakrishna N, Ramalakshmi BA, Devi S, Kumar BS. Effects of vitamin A deficiency during pregnancy on maternal and child health. BJOG. (2002) 109:689–93. doi: 10.1111/j.1471-0528.2002.01010.x
8. Institute of Medicine (US) Panel on Micronutrients. Dietary Reference Intakes for Vitamin A, Vitamin K, Arsenic, Boron, Chromium, Copper, Iodine, Iron, Manganese, Molybdenum, Nickel, Silicon, Vanadium, and Zinc. Washington, DC: National Academies Press (US) (2001). doi: 10.17226/10026
9. West CE, Eilander A, van Lieshout M. Consequences of revised estimates of carotenoid bioefficacy for dietary control of vitamin A deficiency in developing countries. J Nutr. (2002) 132:2920S−6S. doi: 10.1093/jn/132.9.2920S
10. Harrison EH. Mechanisms involved in the intestinal absorption of dietary vitamin A and provitamin A carotenoids. Biochim Biophys Acta. (2012) 1821:70–7. doi: 10.1016/j.bbalip.2011.06.002
11. Lobo GP, Hessel S, Eichinger A, Noy N, Moise AR, Wyss A, et al. is a retinoic acid-sensitive gatekeeper that controls intestinal beta, beta-carotene absorption and vitamin A production. FASEB J. (2010) 24:1656–656:doi: 10.1096/fj.09-150995
12. Novotny JA, Harrison DJ, Pawlosky R, Flanagan VP, Harrison EH, Kurilich AC. Beta-carotene conversion to vitamin A decreases as the dietary dose increases in humans. J Nutr. (2010) 140:915–8. doi: 10.3945/jn.109.116947
13. Tanvetyanon T, Bepler G. Beta-carotene in multivitamins and the possible risk of lung cancer among smokers vs. former smokers: a meta-analysis and evaluation of national brands. Cancer. (2008) 113:150–7. doi: 10.1002/cncr.23527
14. Middha P, Weinstein SJ MP, Wein S, Albanes D, Mondul AM. βAM. l AM. supplementation and lung cancer incidence in the alpha-tocopherol, beta-carotene cancer prevention study: the role of tar and nicotine. Nicotine Tob Res. (2019) 21:1045–045:doi: 10.1093/ntr/nty115
15. Alpha-Tocopherol, Beta Carotene Cancer Prevention Study Group. The effect of vitamin E and beta carotene on the incidence of lung cancer and other cancers in male smokers. N Engl J Med. (1994) 330:1029–35. doi: 10.1056/NEJM199404143301501
16. Albanes D, Heinonen OP, Taylor PR, Virtamo J, Edwards BK, Rautalahti M, et al. Alpha-Tocopherol and beta-carotene supplements and lung cancer incidence in the alpha-tocopherol, beta-carotene cancer prevention study: effects of base-line characteristics and study compliance. J Natl Cancer Inst. (1996) 88:1560–560:doi: 10.1093/jnci/88.21.1560
17. Tanumihardjo SA, Kaliwile C, Boy E, Dhansay MA, van Stuijvenberg ME. Overlapping vitamin A interventions in the United States, Guatemala, Zambia, and South Africa: case studies. Ann N Y Acad Sci. (2019) 1446:102–16. doi: 10.1111/nyas.13965
18. van Stuijvenberg ME, Dhansay MA, Nel J, Suri D, Grahn M, Davis CR, et al. South African preschool children habitually consuming sheep liver and exposed to vitamin A supplementation and fortification have hypervitaminotic A liver stores: a cohort study. Am J Clin Nutr. (2019) 110:91–101. doi: 10.1093/ajcn/nqy382
19. D2Ambrosio DN, Clugston RD, Blaner WS. Vitamin A metabolism: an update. Nutrients. (2011) 3:63–103. doi: 10.3390/nu3010063
20. Hanson C, Lyden E, Abresch C, Anderson-Berry A. Serum retinol concentrations, race, and socioeconomic status in of women of childbearing age in the united states. Nutrients. (2016) 8:508. doi: 10.3390/nu8080508
21. Spannaus-Martin DJ, Cook LR, Tanumihardjo SA, Duitsman PK, Olson JA. Vitamin A and vitamin E statuses of preschool children of socioeconomically disadvantaged families living in the midwestern United States. Eur J Clin Nutr. (1997) 51:864–9. doi: 10.1038/sj.ejcn.1600503
22. Suzuki M, Wang T, Garretto D, Isasi CR, Cardoso WV, Greally JM, Quadro L. Disproportionate Vitamin A deficiency in women of specific ethnicities linked to differences in allele frequencies of Vitamin A-related polymorphisms. Nutrients. (2021) 13:1743. doi: 10.3390/nu13061743
23. Gueguen S, Leroy P, Gueguen R, Siest G, Visvikis S, Herbeth B. Genetic and environmental contributions to serum retinol and alpha-tocopherol concentrations: the Stanislas Family Study. Am J Clin Nutr. (2005) 81:1034–034:doi: 10.1093/ajcn/81.5.1034
24. Mondul AM Yu K, Wheeler W, Zhang H, Weinstein SJ, Major JM, Cornelis MC, et al. Genome-wide association study of circulating retinol levels. Hum Mol Genet. (2011) 20:4724–724:doi: 10.1093/hmg/ddr387
25. Obrochta KM, Kane MA, Napoli JL. Effects of diet and strain on mouse serum and tissue retinoid concentrations. PLoS ONE. (2014) 9:e99435. doi: 10.1371/journal.pone.0099435
26. Blomhoff R, Green M, Berg T, Norum K. Transport and storage of vitamin A. Science. (1990) 250:399–404. doi: 10.1126/science.2218545
27. Novotny JA, Dueker SR, Zech LA, Clifford AJ. Compartmental analysis of the dynamics of beta-carotene metabolism in an adult volunteer. J Lipid Res. (1995) 36:1825–38.
28. Faulks RM, Hart DJ, Wilson PD, Scott KJ, Southon S. Absorption of all-trans and 9-cis beta-carotene in human ileostomy volunteers. Clin Sci. (1997) 93:585–91. doi: 10.1042/cs0930585
29. Borel P, Lietz G, Goncalves A, Szabo de, Edelenyi F, Lecompte S, Curtis P, et al. CD36 and SR-BI are involved in cellular uptake of provitamin A carotenoids by Caco-2 and HEK cells, and some of their genetic variants are associated with plasma concentrations of these micronutrients in humans. J Nutr. (2013) 143:448–56. doi: 10.3945/jn.112.172734
30. O'Neill ME, Thurnham DI. Intestinal absorption of beta-carotene, lycopene and lutein in men and women following a standard meal: response curves in the triacylglycerol-rich lipoprotein fraction. Br J Nutr. (1998) 79:149–59. doi: 10.1079/bjn19980026
31. Ross AC. Vitamin A: physiology, dietary sources, and requirements, in Encyclopedia of Human Nutrition. Elsevier. 333–9. doi: 10.1016/B978-0-12-375083-9.00273-7
32. de Pee S, West CE. Dietary carotenoids and their role in combating vitamin A deficiency: a review of the literature. Eur J Clin Nutr. (1996) 50(Suppl 3) :S38–53.
33. Borel P. Genetic variations involved in interindividual variability in carotenoid status. Mol Nutr Food Res. (2012) 56:228–40. doi: 10.1002/mnfr.201100322
34. van Bennekum A, Werder M, Thuahnai ST, Han C-H, Duong P, Williams DL, et al. Class B scavenger receptor-mediated intestinal absorption of dietary beta-carotene and cholesterol. Biochemistry. (2005) 44:4517–517:doi: 10.1021/bi0484320
35. Borel P, Moussa M, Reboul E, Lyan B, Defoort C, Vincent-Baudry S, et al. Human plasma levels of vitamin E and carotenoids are associated with genetic polymorphisms in genes involved in lipid metabolism. J Nutr. (2007) 137:2653–9. doi: 10.1093/jn/137.12.2653
36. dela Seña C, Riedl KM, Narayanasamy S, Curley RW, Schwartz SJ, Harrison EH. The human enzyme that converts dietary provitamin A carotenoids to vitamin A is a dioxygenase. J Biol Chem. (2014) 289:13661–6. doi: 10.1074/jbc.M114.557710
37. Paik J, During A, Harrison EH, Mendelsohn CL, Lai K, Blaner WS. Expression and characterization of a murine enzyme able to cleave beta-carotene. The formation of retinoids. J Biol Chem. (2001) 276:32160–8. doi: 10.1074/jbc.M010086200
38. Leung WC, Hessel S, Meung C, Flint J, Oberhauser V, Tourniaire F, et al. Two common single nucleotide polymorphisms in the gene encoding beta-carotene 15,15m-monoxygenase alter beta-carotene metabolism in female volunteers. FASEB J. (2009) 23:1041–53. doi: 10.1096/fj.08-121962
39. van Helden YGJ, Heil SG, van Schooten FJ, Kramer E, Hessel S, Amengual J, et al. Knockout of the Bcmo1 gene results in an inflammatory response in female lung, which is suppressed by dietary beta-carotene. Cell Mol Life Sci. (2010) 67:2039–56. doi: 10.1007/s00018-010-0341-7
40. Ferrucci L, Perry JRB, Matteini A, Perola M, Tanaka T, Silander K, et al. Common variation in the beta-carotene 15,15o-monooxygenase 1 gene affects circulating levels of carotenoids: a genome-wide association study. Am J Hum Genet. (2009) 84:123–33. doi: 10.1016/j.ajhg.2008.12.019
41. Lietz G, Oxley A, Leung W, Hesketh J. Single nucleotide polymorphisms upstream from the β-carotene 15,15'-monoxygenase gene influence provitamin A conversion efficiency in female volunteers. J Nutr. (2012) 142:161S−5S. doi: 10.3945/jn.111.140756
42. Hendrickson SJ, Hazra A, Chen C, Eliassen AH, Kraft P, Rosner BA, et al. β-Carotene 15,15'-monooxygenase 1 single nucleotide polymorphisms in relation to plasma carotenoid and retinol concentrations in women of European descent. Am J Clin Nutr. (2012) 96:1379–89. doi: 10.3945/ajcn.112.034934
43. Graßmann S, Pivovarova-Ramich O, Henze A, Raila J, Ampem Amoako Y, King Nyamekye R, et al. SNP rs6564851 in the BCO1 gene is associated with varying provitamin a plasma concentrations but not with retinol concentrations among adolescents from rural Ghana. Nutrients. (2020) 12:1786. doi: 10.3390/nu12061786
44. Yabuta S, Urata M, Wai Kun RY, Masaki M, Shidoji Y. Common SNP rs6564851 in the BCO1 gene affects the circulating levels of β-carotene and the daily intake of carotenoids in healthy Japanese women. PLoS ONE. (2016) 11:e0168857. doi: 10.1371/journal.pone.0168857
45. He F, Xiao R-D, Lin T, Xiong W-M, Xu Q-P, Li X, et al. Dietary patterns, BCMO1 polymorphisms, and primary lung cancer risk in a Han Chinese population: a case-control study in Southeast China. BMC Cancer. (2018) 18:445. doi: 10.1186/s12885-018-4361-2
46. Lindqvist A, Sharvill J, Sharvill DE, Andersson S. Loss-of-function mutation in carotenoid 15,15'-monooxygenase identified in a patient with hypercarotenemia and hypovitaminosis A. J Nutr. (2007) 137:2346–50. doi: 10.1093/jn/137.11.2346
48. Wu L, Guo X, Wang W, Medeiros DM, Clarke SL, Lucas EA, et al. Molecular aspects of β, β-carotene-9', 10'-oxygenase 2 in carotenoid metabolism and diseases. Exp Biol Med (Maywood). (2016) 241:1879–87. doi: 10.1177/1535370216657900
49. Reboul E, Berton A, Moussa M, Kreuzer C, Crenon I, Borel P. Pancreatic lipase and pancreatic lipase-related protein 2, but not pancreatic lipase-related protein 1, hydrolyze retinyl palmitate in physiological conditions. Biochim Biophys Acta. (2006) 1761:4–10. doi: 10.1016/j.bbalip.2005.12.013
50. Lowe ME, Rosenblum JL, Strauss AW. Cloning and characterization of human pancreatic lipase cDNA. J Biol Chem. (1989) 264:20042–8
51. Behar DM, Basel-Vanagaite L, Glaser F, Kaplan M, Tzur S, Magal N, et al. Identification of a novel mutation in the PNLIP gene in two brothers with congenital pancreatic lipase deficiency. J Lipid Res. (2014) 55:307–12. doi: 10.1194/jlr.P041103
52. Blaner WS, Brun P-J, Calderon RM, Golczak M. Retinol-binding protein 2 (RBP2): biology and pathobiology. Crit Rev Biochem Mol Biol. (2020) 55:197–218. doi: 10.1080/10409238.2020.1768207
53. Crow JA, Ong DE. Cell-specific immunohistochemical localization of a cellular retinol-binding protein (type two) in the small intestine of rat. Proc Natl Acad Sci USA. (1985) 82:4707–11. doi: 10.1073/pnas.82.14.4707
54. Reboul E. Absorption of vitamin A and carotenoids by the enterocyte: focus on transport proteins. Nutrients. (2013) 5:3563–81. doi: 10.3390/nu5093563
55. Takase S, Suruga K, Goda T. Regulation of vitamin A metabolism-related gene expression. Br J Nutr. (2000) 84(Suppl 2):S217–21. doi: 10.1079/096582197388572
56. Thompson DA Li Y, McHenry CL, Carlson TJ, Ding X, Sieving PA, Apfelstedt-Sylla E, et al. Mutations in the gene encoding lecithin retinol acyltransferase are associated with early-onset severe retinal dystrophy. Nat Genet. (2001) 28:123–4. doi: 10.1038/88828
57. SA, Humbert G, Surget M-O, Bazalgette C, Bazalgette C, Arnaud B, et al. Screening genes of the retinoid metabolism: novel LRAT mutation in leber congenital amaurosis. Am J Ophthalmol. (2006) 142:702–4. doi: 10.1016/j.ajo.2006.04.057
58. Orland MD, Anwar K, Cromley D, Chu C-H, Chen L, Billheimer JT, et al. Acyl coenzyme A dependent retinol esterification by acyl coenzyme A: diacylglycerol acyltransferase 1. Biochim Biophys Acta. (2005) 1737:76–82. doi: 10.1016/j.bbalip.2005.09.00
59. Yen CLE, Monetti M, Burri BJ, Farese RV. The triacylglycerol synthesis enzyme DGAT1 also catalyzes the synthesis of diacylglycerols, waxes, and retinyl esters. J Lipid Res. (2005) 46:1502–11. doi: 10.1194/jlr.M500036-JLR200
60. Wongsiriroj N, Piantedosi R, Palczewski K, Goldberg IJ, Johnston TP Li E, Blaner WS. The molecular basis of retinoid absorption: a genetic dissection. J Biol Chem. (2008) 283:13510–9. doi: 10.1074/jbc.M800777200
61. Arne JM, Widjaja-Adhi MAK, Hughes T, Huynh KW, Silvaroli JA, Chelstowska S, et al. Allosteric modulation of the substrate specificity of acyl-CoA wax alcohol acyltransferase 2. J Lipid Res. (2017) 58:719–30. doi: 10.1194/jlr.M073692
62. Haas J, Frese KS, Park YJ, Keller A, Vogel B, Lindroth AM, et al. Alterations in cardiac DNA methylation in human dilated cardiomyopathy. EMBO Mol Med. (2013) 5:413–29. doi: 10.1002/emmm.20120155
63. Harrison EH, Gad MZ, Ross AC. Hepatic uptake and metabolism of chylomicron retinyl esters: probable role of plasma membrane/endosomal retinyl ester hydrolases. J Lipid Res. (1995) 36:1498–506.
64. Blomhoff R, Helgerud P, Rasmussen M, Berg T, Norum KR. In vivo uptake of chylomicron [3H]retinyl ester by rat liver: evidence for retinol transfer from parenchymal to non-parenchymal cells. Proc Natl Acad Sci USA. (1982) 79:7326–30. doi: 10.1073/pnas.79.23.7326
65. Goodman DW, Huang HS, Shiratori T. Tissue distribution and metabolism of newly absorbed vitamin a in the rat. J Lipid Res. (1965) 6:390–6.
66. Hussain MM, Mahley RW, Boyles JK, Fainaru M, Brecht WJ, Lindquist PA. Chylomicron-chylomicron remnant clearance by liver and bone marrow in rabbits. Factors that modify tissue-specific uptake. J Biol Chem. (1989) 264:9571–82.
67. Merrill AH, Bowman BB, Preusch PC. Mechanistic aspects of vitamin and coenzyme utilization and function: a symposium in recognition of the distinguished career of Donald B. McCormick. J Nutr. (2000) 130:321S−2S. doi: 10.1093/jn/130.2.321S
68. Quadro L, Hamberger L, Colantuoni V, Gottesman ME, Blaner WS. Understanding the physiological role of retinol-binding protein in vitamin A metabolism using transgenic and knockout mouse models. Mol Aspects Med. (2003) 24:421–30. doi: 10.1016/s0098-2997(03)00038-4
69. Kawaguchi R, Yu J, Honda J, Hu J, Whitelegge J, Ping P, et al. membrane receptor for retinol binding protein mediates cellular uptake of vitamin A. Science. (2007) 315:820–5. doi: 10.1126/science.1136244
70. Kennedy OJ, Pirastu N, Poole R, Fallowfield JA, Hayes PC, Grzeszkowiak EJ, et al. Coffee consumption and kidney function: a mendelian randomization study. Am J Kidney Dis. (2020) 75:753y Disdoi: 10.1053/j.ajkd.2019.08.025
71. Wuttke M, Li Y, Li M, Sieber KB, Feitosa MF, Gorski M, et al. A catalog of genetic loci associated with kidney function from analyses of a million individuals. Nat Genet. (2019) 51:957tog of genet1038/s41588-019-0407-x
72. Weafer J, Gray JC, Hernandez K, Palmer AA, MacKillop J, de Wit H. Hierarchical investigation of genetic influences on response inhibition in healthy young adults. Exp Clin Psychopharmacol. (2017) 25:512armacdoi: 10.1037/pha0000156
73. Han X, Qassim A, An J, Marshall H, Zhou T, Ong J-S, et al. Genome-wide association analysis of 95 of genetic influences on response inhibition in healthy young adults. adults. inhHum Mol Genet. (2019) 28:3680–680:doi: 10.1093/hmg/ddz193
74. Kichaev G, Bhatia G, Loh P-R, Gazal S, Burch K, Freund MK, et al. Leveraging polygenic functional enrichment to improve GWAS power. Am J Hum Genet. (2019) 104:65 Genedoi: 10.1016/j.ajhg.2018.11.008
75. Casey J, Kawaguchi R, Morrissey M, Sun H, McGettigan P, Nielsen JE, et al. First implication of STRA6 mutations in isolated anophthalmia, microphthalmia, and coloboma: a new dimension to the STRA6 phenotype. Hum Mutat. (2011) 32:1417–417:doi: 10.1002/humu.21590
76. Richardson R, Sowden J, Gerth-Kahlert C, Moore AT, Moosajee M. Clinical utility gene card for: non-syndromic microphthalmia including next-generation sequencing-based approaches. Eur J Hum Genet. (2017) 25:201. doi: 10.1038/ejhg.2016.201
77. Golzio C, Martinovic-Bouriel J, Thomas S, Mougou-Zrelli S, Grattagliano-Bessieres B, Bonniere M, et al. Matthew-Wood syndrome is caused by truncating mutations in the retinol-binding protein receptor gene STRA6. Am J Hum Genet. (2007) 80:1179–179:doi: 10.1086/518177
78. White T, Lu T, Metlapally R, Katowitz J, Kherani F, Wang T-Y, et al. Identification of STRA6 and SKI sequence variants in patients with anophthalmia/microphthalmia. Mol Vis. (2008) 14:2458–458:Srinivasan BS, Doostzadeh J, Absalan F, Mohandessi S, Jalili R, Bigdeli S, et al. Whole genome survey of coding SNPs reveals a reproducible pathway determinant of Parkinson disease. Hum Mutat. (2009) 30:228tenomdoi: 10.1002/humu.20840
79. Nair AK, Sugunan D, Kumar H, Anilkumar G. Case-control analysis of SNPs in GLUT4, RBP4 and STRA6: association of SNPs in STRA6 with type 2 diabetes in a South Indian population. PLoS ONE. (2010) 5:e11444. doi: 10.1371/journal.pone.0011444
80. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. (2015) 17:405df medoi: 10.1038/gim.2015.30
81. Huang H-W, Liang B-Y, Li Y-X. Association of Polymorphisms in STRA6 and RARRES2 genes with type 2 diabetes in Southern Han Chinese. Biomed Res Int. (2016) 2016:6589793. doi: 10.1155/2016/6589793
82. Hu S, Yan J, You Y, Yang G, Zhou H, Li X, et al. Association of polymorphisms in STRA6 gene with gestational diabetes mellitus in a Chinese Han population. Medicine (Baltimore). (2019) 98:e14885. doi: 10.1097/MD.0000000000014885
83. Mu-Herneal dFernn JB DJB -Ch, DMondrag-Fonseca O, Mayns-Lobo Y, Ortega A, et al. STRA6 polymorphisms are associated with EGFR mutations in locally-advanced and metastatic non-small cell lung cancer patients. Front Oncol. (2020) 10:579561. doi: 10.3389/fonc.2020.579561
84. Blaner WS, O WS, Olung Wongsiriroj N, Kluwe J, DJ D J D DM, Jiang H, et al. Hepatic stellate cell lipid droplets: a specialized lipid droplet for retinoid storage. Biochim Biophys Acta. (2009) 1791:46709s doi: 10.1016/j.bbalip.2008.11.001
85. Randolph RK, Ross AC. Vitamin A status regulates hepatic lecithin: retinol acyltransferase activity in rats. J Biol Chem. (1991) 266:16453–645Dawson HD, Yamamoto Y, Zolfaghari R, Rosales FJ, Dietz J, Shimada T, et al. Regulation of hepatic vitamin A storage in a rat model of controlled vitamin A status during aging. J Nutr. (2000) 130:1280–28 doi: 10.1093/jn/130.5.1280
86. Zolfaghari R, Ross AC. Lecithin:retinol acyltransferase from mouse and rat liver. CDNA cloning and liver-specific regulation by dietary vitamin a and retinoic acid. J Lipid Res. (2000) 41:2024–024:doi: 10.1016/S0022-2275(20)32364-6
87. Taschler U, Schreiber R, Chitraju C, Grabner GF, Romauch M, Wolinski H, et al. Adipose triglyceride lipase is involved in the mobilization of triglyceride and retinoid stores of hepatic stellate cells. Biochim Biophys Acta. (2015) 1851:93715s Adoi: 10.1016/j.bbalip.2015.02.017
88. Pirazzi C, Valenti L, Motta BM, Pingitore P, Hedfalk K, Mancina RM, et al. PNPLA3 has retinyl-palmitate lipase activity in human hepatic stellate cells. Hum Mol Genet. (2014) 23:4077–077:doi: 10.1093/hmg/ddu121
89. Fischer J, Lefcher C, Morava E, Mussini J-M, Lafor L P, Negre-Salvayre A, et al. The gene encoding adipose triglyceride lipase (PNPLA2) is mutated in neutral lipid storage disease with myopathy. Nat Genet. (2007) 39:28etteddoi: 10.1038/ng1951
90. Akiyama M, Sakai K, Ogawa M, McMillan JR, Sawamura D, Shimizu H. Novel duplication mutation in the patatin domain of adipose triglyceride lipase (PNPLA2) in neutral lipid storage disease with severe myopathy. Muscle Nerve. (2007) 36:856ervedoi: 10.1002/mus.20869
91. Lin P, Li W, Wen B, Zhao Y, Fenster DS, Wang Y, et al. Novel PNPLA2 gene mutations in Chinese Han patients causing neutral lipid storage disease with myopathy. J Hum Genet. (2012) 57:679netiodoi: 10.1038/jhg.2012.84
92. Reilich P, Horvath R, Krause S, Schramm N, Turnbull DM, Trenell M, et al. The phenotypic spectrum of neutral lipid storage myopathy due to mutations in the PNPLA2 gene. J Neurol. (2011) 258:1987–9878. doi: 10.1007/s00415-011-6055-4
93. Kovarova M, KM Kova MI, KI Kova M A, Machicao F, HF Hc H-U, Schleicher E, et al. The genetic variant I148M in PNPLA3 is associated with increased hepatic retinyl-palmitate storage in humans. J Clin Endocrinol Metab. (2015) 100:E156800:indoi: 10.1210/jc.2015-2978
94. Mondul A, Mancina RM, Merlo A, Dongiovanni P, Rametta R, Montalcini T, et al. PNPLA3 I148M Variant influences circulating retinol in adults with non-alcoholic fatty liver disease or obesity. J Nutr. (2015) 145:1687–6875. doi: 10.3945/jn.115.210633
95. GTEx Consortium. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. (2015) 348:648–60. doi: 10.1126/science.1262110
96. GTEx Consortium. The genotype-tissue expression (GTEx) project. Nat Genet. (2013) 45:580–5. doi: 10.1038/ng.2653
97. Zhu Z, Zhang F, Hu H, Bakshi A, Robinson MR, Powell JE, et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat Genet. (2016) 48:481ttiodoi: 10.1038/ng.3538
98. Jansen RC, Nap JP. Genetical genomics: the added value from segregation. Trends Genet. (2001) 17:388enethdoi: 10.1016/s0168-9525(01)02310-1
99. Galicia L, Grajeda R, de Romade DL. Nutrition situation in Latin America and the Caribbean: current scenario, past trends, and data gaps. Rev Panam Salud Publica. (2016) 40:104blicaWirth JP, Petry N, Tanumihardjo SA, Rogers LM, McLean E, Greig A, Garrett GS, Klemm RDW, Rohner F. Vitamin A supplementation programs and country-level evidence of vitamin A deficiency. Nutrients (2017) 9:190. doi: 10.3390/nu9030190
100. West KP. Vitamin A deficiency disorders in children and women. Food Nutr Bull. (2003) 24:S78724:3ull Bul1177/15648265030244S204
101. World Health Organization. WHO Global Prevalence of Vitamin A Deficiency in Populations at Risk 1995–2005. WHO Global Database on Vitamin A Deficiency. Washington, DC: World Health Organization (2009).
Keywords: retinoid, vitamin A deficiency, genetic variations, micronutrient, ethnicity
Citation: Suzuki M and Tomita M (2022) Genetic Variations of Vitamin A-Absorption and Storage-Related Genes, and Their Potential Contribution to Vitamin A Deficiency Risks Among Different Ethnic Groups. Front. Nutr. 9:861619. doi: 10.3389/fnut.2022.861619
Received: 24 January 2022; Accepted: 23 March 2022;
Published: 28 April 2022.
Edited by:
Christelle Guillet, University of Auvergne, FranceReviewed by:
Thomas Olsen, University of Oslo, NorwayMoncef Feki, Tunis El Manar University, Tunisia
Copyright © 2022 Suzuki and Tomita. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Masako Suzuki, bWFzYWtvLnN1enVraUBlaW5zdGVpbm1lZC5lZHU=