
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Nutr. , 28 April 2022
Sec. Food Chemistry
Volume 9 - 2022 | https://doi.org/10.3389/fnut.2022.845150
This article is part of the Research Topic Analytical Methods, Formation Mechanisms and Control Strategies for Endogenous Hazardous Substances Produced during the Thermal Processing of Foods View all 14 articles
 Bingyao Du1,2,3Lu Meng1,3Haoming Wu1,3Huaigu Yang4Huimin Liu1,3
Bingyao Du1,2,3Lu Meng1,3Haoming Wu1,3Huaigu Yang4Huimin Liu1,3 Nan Zheng1,3Yangdong Zhang1,3
Nan Zheng1,3Yangdong Zhang1,3 Shengguo Zhao1,3
Shengguo Zhao1,3 Jiaqi Wang1,3*
Jiaqi Wang1,3*Milk is rich in fat, protein, minerals, vitamins, peptides, immunologically active substances, and other nutrients, and it plays an important role in satisfying human nutrition and health. However, dairy product safety incidents caused by microbial contamination have occurred. We found that the total bacterial numbers in the pasteurized product were low and far below the limit requirements of the food safety standards of the European Union, the United States, and China. At the genus level, the primary microbial groups found in milk samples were Acinetobacter, Macrococcus, Pseudomonas, and Lactococcus, while in the equipment rinse water and air samples there was contamination by Stenotrophomonas and Acinetobacter. The Source Tracker model analysis indicated that the microorganisms in the final milk products were significantly related to the contamination in product tanks and raw milk. Therefore, it is the hope that this work can provide guidance to pinpoint contamination problems using the proper quality control sampling at specific stages in the pasteurization process.
Milk, which is a nutrient source for protein, minerals, vitamins, and immunologically active substances, is considered a perfect food and plays an important role in meeting human nutritional needs (1–3). However, it is a good growth media for microorganisms because of the rich nutrients.
Contaminated milk may rapidly deteriorate and pose a potential health threat (4–8). According to reports, the detection rate of pathogens in pasteurized milk in Brazil is 4.7%, and pasteurized milk in China is often reported to be contaminated with pathogens (9–11). These bacteria are not naturally present in the mammary glands of healthy cows, and therefore, it is necessary to track the source of the microorganisms in milk, as they may find their way into milk as a result of poor milking and farming practices (12).
Spanu et al. used polymerase chain reaction (PCR) and pulsed-field gel electrophoresis technology to analyze the contamination sources of Listeria monocytogenes in cheese, and found that the processing environment of the cheese factory was the main source of microbial contamination (13). Maya analyzed the source of contamination of methicillin-resistant Staphylococcus aureus in pasteurized milk through multilocus sequence typing and pulsed-field gel electrophoresis technology, and found that cross-contamination after processing was one of the main sources of contamination (14). Wang et al. used 16S rRNA-PacBio Single Molecule Real-Time sequencing method to trace the microbes in milk powder and found that raw milk or processing equipment was one of the main sources of contamination (15). These studies are based on the typing and identification of strains based on the culture method or single sequencing technology, which has certain limitations.
In recent years, the Source Tracker modeling based on 16S rDNA sequencing and analysis of microbial contamination sources for raw milk have been widely used. Du et al. used Source Tracker modeling based on 16S rDNA sequencing to analyze the sources of microbial contamination in raw milk and found that the teat liner and teat dip cup were the main sources of contamination (16). Wu et al. used Source Tracker modeling based on 16S rDNA sequencing to analyze the source of microbial contamination in farm milk using automatic milking systems and found that airborne dust was the most important source of contamination (17).
Traceability analysis or determination of the presence of a particular substance or microorganism along a sample processing chain has been widely applied in microbiological studies. However, there have been few studies on the traceability of microorganisms in pasteurized milk and finding the particular processing step in which the contamination most likely occurs. The objective of the current study was to evaluate the traceability of microorganisms in pasteurized milk and analyze the individual processing steps, utilizing the Source Tracker model based on 16S rDNA sequencing, and find the potential sources of microbial contamination, which will help us formulate reasonable disinfection and cleaning procedures to improve the quality of dairy products.
Milk samples were collected at 8 critical control points along the milk processing pathway at the dairy processors in Zhengzhou City, Henan province in May 2019. The ambient temperature was 25°C. The control points included milk tanker (30 tons, raw milk, M1), storage tank (30 tons, raw milk, M2), storage tank for processing (30 tons, raw milk, M3), milk in balance tank (72°C/15 s, M4), milk in balance tank (75°C/15 s, M5), milk in balance tank (80°C/15 s, M6), milk in product tank (72°C/15 s, M7), milk in product tank (75°C/15 s, M8), milk in product tank (80°C/15 s, M9), pasteurized milk (72°C/15 s, M10), pasteurized milk (75°C/15 s, M11), pasteurized milk (80°C/15 s, M12), equipment water rinses (W), and processing factory air (A). The cleaning frequency of the tank was once a day. The milk was stored in the tank for 2 h before processing. A quantity of 200 ml of clean in place (CIP) water from the dairy factory was collected every day, and stored in sterile sampling bottles. For the collection method of air samples, referring to Du et al. (16), milk samples (50 ml) were collected in triplicate each day at each sampling point for 4 consecutive days and immediately stored at −20°C until analysis.
The counts of total viable bacteria(TBC) was determined following the National Standards of the Republic of China (GB 4789.2-2016) (18), alkaline phosphatase was detected following the International Dairy Federation (IDF) procedure (IDF, method 209, 2007) (19), and lactoperoxidase was determined following the Tianjin Dairy Science and Technology Innovation Association Group Standard (T/TDSTIA, method 001, 2021) (20).
Milk samples (10 ml each) were centrifuged at 14,000 × g for 5 min, and the precipitate was used for DNA extraction using the HiPure Stool DNA Kits (Magen, Guangzhou, China) according to the manufacturer's instructions. Extracted DNA was amplified using the following PCR cycling program: 94°C for 2 min, followed by 30 cycles at 98°C for 10 s, 62°C for 30 s, and 68°C for 30 s, and a final extension at 68°C for 5 min. The PCR primers were specific for the 16S rDNA V3-V4 region: (5′-3′) 341F: CCTACGGGNGGCWGCAG, 806R: GGACTACHVGGGTATCTAAT (21). PCR reactions utilized KOD Polymerase (Millipore Sigma, Rockville, MD, USA). Reagents were provided with the commercial enzyme kit. DNA recovery of amplicons was obtained following agarose gel electrophoresis using an AxyPrep DNA gel extraction kit (Axygen Biosciences, Union City, AZ, USA). FASTP was used to filter the original data, and reads containing >10% of unknown nucleotides (nt) were deleted as were strings of 20 nt with a mass <50% of the Q value (22, 23). Pairs of clean reads were combined into the original tag with a minimum overlap of 10 nt and a mismatch error of 2%. The original tag sequences were then filtered through QIIME (version 1.9.1) (24) using the reference database v.r20110519 (http://drive5.com/uchime/uchime_download.html) (25) to obtain high quality clean labels. Noise reduction, chimera detection, and aggregation into operational taxonomic units (OTU) containing 97% identities were performed using UPARSE (v. 9.2.64) (26).
The SILVA database (v. 132) (27) was used to classify representative sequences as biological using the naive Bayes model using confidence threshold ranges from 0.8 to 1. Krona (v.2.6) was used to visualize the abundance of each taxonomic group (28). Stacked bar charts were visualized using R project ggplot2 (v.2.2.1) (17, 29) and Omicsmart (http://www.omicsmart.com). Pheatmap software (v. 1.0.12) from Omicsmart was used to indicate species richness.
The principal coordinate (CAP) graphs were visualized using ggplot2 (30, 31). Pearson correlation coefficient >0.7 was considered significant. Similarly, hierarchical clustering and heat map construction were completed. The Source Tracker modeling based on 16S rDNA sequencing can be used for the analysis of microbial contamination sources for raw milk and pasteurized milk, and can calculate the similarities between different bacterial communities (32). The Source Tracker model was based on the Bayesian algorithm (https://github.com/danknights/sourcetracker, accessed as of July 2021) to predict the composition ratio of the target samples from each source sample, according to the microbial community structure distribution of the target samples and the source samples. The colored sector area indicated the proportion of each source in the samples, and explored the analysis of the source of microbial contamination in the target sample (32–34).
We tested the total number of colony counts in raw milk and pasteurized product for 4 consecutive days. The average total number of colonies in raw milk was 2.1 × 103 CFU/ml, and the average total number of colonies in the pasteurized products was 2.1 × 102 CFU/ml (72°C for 15 s), 18 CFU/ml (75°C for 15 s), and 2 CFU/ml (80°C for 15 s). The alkaline phosphatase in all pasteurized products was negative (<350 mU/L), and the average lactoperoxidase of the pasteurized products were 6,278.7 U/L (72°C for 15 s), 3,644.2 U/L (75°C for 15 s), and 0 U/L (80°C for 15 s). An analysis of the microbiota of all samples indicated the presence of 10 phyla that were considered as abundant. These included Chloroflexi, Gemmatimonadetes, Actinobacteria, Cyanobacteria,Planctomycetes, Verrucomicrobia, Acidobacteria, Bacteroidetes, Firmicutes, and Proteobacteria. All milk samples had the two major phyla groups of Proteobacteria and Firmicutes. Air and water samples had the largest number of OTUs present, comprising primarily of the three major phyla groups of Proteobacteria, Firmicutes, and Bacteroidetes. A comparison with the raw milk samples indicated that Firmicutes abundance in pasteurized samples gradually decreased while Bacteroidetes almost disappeared, even though Firmicutes and Proteobacteria remained the primary phyla. In addition, the relative abundance of Bacteroidetes in the water and air samples was higher than in the milk samples (Figure 1A). We also found 30 genera that were considered as abundant, of which, Acinetobacter, Macrococcus, Pseudomonas, and Lactococcus were the most abundant genera found in samples from the milk tanker, storage tanks, storage tank for processing, and milk in balance tank. Lysinibacillus was a relatively abundant genus in milk samples from the product tank. Acinetobacter was the most abundant genera in pasteurized samples. Stenotrophomonas and Acinetobacter were the most abundant genera in water and air samples, respectively (Figure 1B). Among the water samples, the relative abundance of Stenotrophomonas was higher than in other samples, but the relative abundance of Acinetobacter was lower than in other samples.
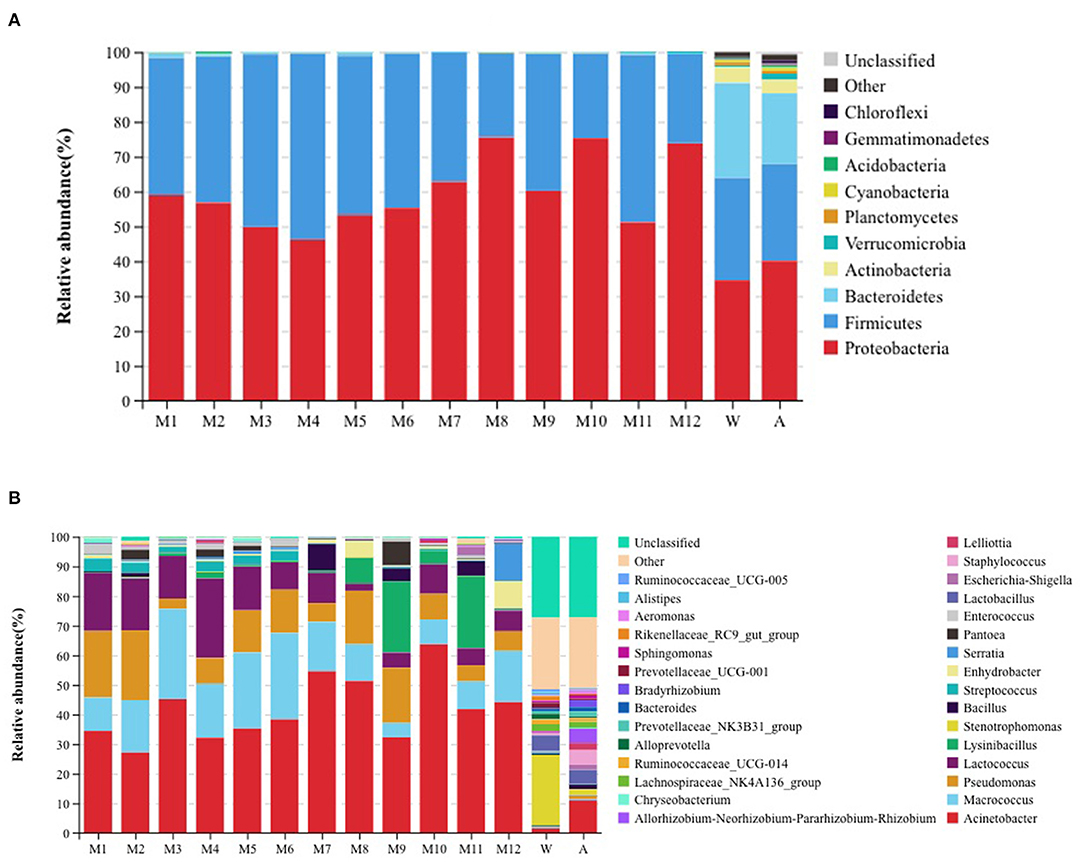
Figure 1. Relative abundance of operational taxonomic units at the (A) phylum and (B) genus level for the different samples. W: equipment water rinses, A: processing factory air. M1: milk tanker (raw milk); M2: storage tank (raw milk); M3: storage tank for processing (raw milk); M4: milk in balance tank (72°C/15 s); M5: milk in balance tank (75°C/15 s); M6: milk in balance tank (80°C/15 s); M7: milk in product tank (72°C/15 s); M8: milk in product tank (75°C/15 s); M9: milk in product tank (80°C/15 s); M10: pasteurized milk (72°C/15 s); M11: pasteurized milk (75°C/15 s); and M12: pasteurized milk (80°C/15 s).
The diversity within samples and between samples was evaluated using alpha diversity analyses. We found differences in microbiota richness between raw milk in the farm milk tanks and the air samples, when compared with all of the other samples, using the Chao1 index (Figure 2). Compared with raw milk, the Chao1 indices for the samples following pasteurization decreased, indicating a reduction in bacterial abundance.
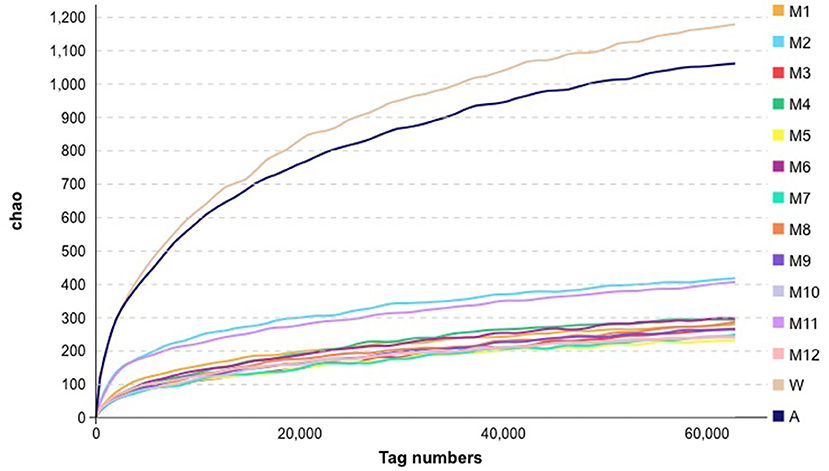
Figure 2. Alpha analysis metrics for bacteria in the samples collected for this study. See Figure 1 for abbreviations.
We used clustering heat map analysis to further explore the differences in the community structure (the top 20 relative abundances) of microorganisms in different samples at the genus level (Figure 3), and found that milk tanker sample (raw milk), storage tank sample (raw milk), storage tank for processing sample (raw milk), milk in balance tank sample (72°C/15 s), milk in balance tank sample (75°C/15 s), and milk in balance tank sample (80°C/15 s) were apparently clustered together due to Pseudomonas, Lactococcus, Streptococcus, and Enterococcus. Equipment water rinses sample and processing factory air sample were apparently clustered together due to Ruminococcaceae_UCG-014, Lachnospiraceae_NK4A136_group, and Lactobacillus.
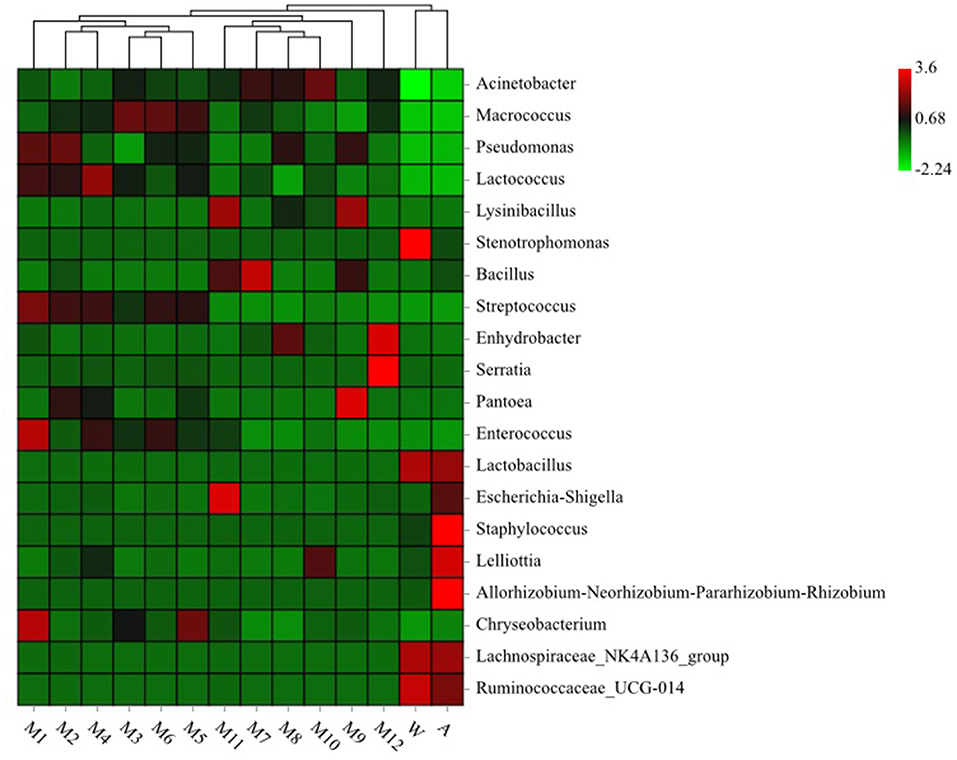
Figure 3. Heatmap analysis for bacteria genera in different samples. See Figure 1 for abbreviations.
Canonical analysis of principal coordinates (CAP) indicated the top 10 most abundant genera (Acinetobacter, Macrococcus, Pseudomonas, Lactococcus, Lysinibacillus, Stenotrophomonas, Bacillus, Streptococcus, and Enhydrobacter) associated with milk, which were clustered in contrast to the rinse water and the processing factory air represented by Stenotrophomonas, which was considered as an independent group with a 70% similarity (Figure 4). Acinetobacter, Macrococcus, Pseudomonas, Lactococcus, and Streptococcus, which are regarded as typical bacterial taxa of other samples, were not abundant in the processing factory air.
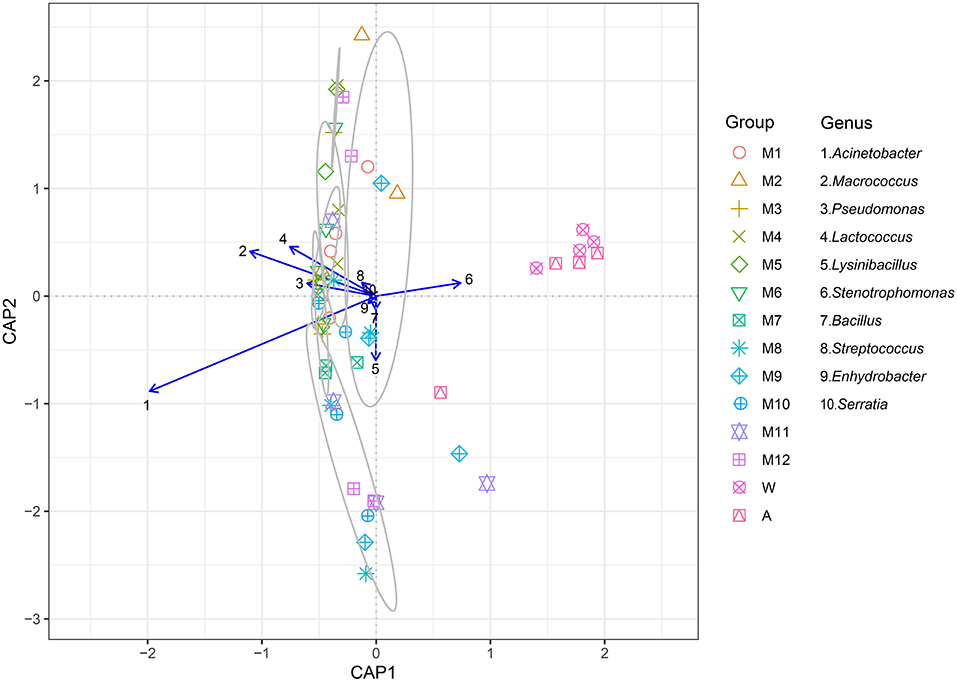
Figure 4. The canonical analysis of principal coordinates (CAP) explained the correlation of the level of the 10 primary bacterial genera (Acinetobacter, Macrococcus, Pseudomonas, Lactococcus, Lysinibacillus, Stenotrophomonas, Bacillus, Streptococcus, Enhydrobacter, and Serratia) in different samples. The samples in the gray circle are regarded as a group because the similarity was >70%. See Figure 1 for abbreviations.
Sources of environmental contamination in milk were assessed using the Source Tracker model. The sources included all factory environmental samples, raw milk sample, and processed samples, while the sinks were represented by including pasteurized milk samples. The Source Tracker model considered each individual community as a mixed community deposited from other known or unknown environmental sources. The Source Tracker model analysis indicated that the microorganisms in the products were significantly correlated to those in the product tank, followed by milk tanker, which was the second most important source of contaminants (Figure 5).
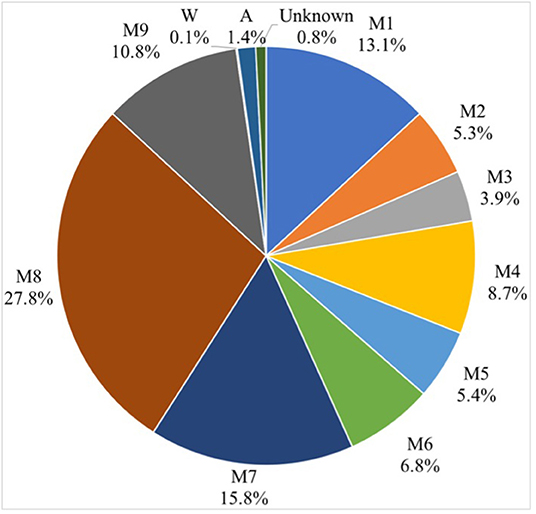
Figure 5. Source Tracker results show the contribution of inferred sources of microbial contamination in dairy products. See Figure 1 for abbreviations.
This study utilized high-throughput sequencing technology to analyze the microbial communities of milk from the delivery tanks to the final stage of pasteurization at a milk processing facility. The primary phyla found in raw milk were Proteobacteria and Firmicutes, while Proteobacteria, Firmicutes, and Bacteroidetes predominated the air and equipment rinse water. The Proteobacteria was the most dominant phylum in the pasteurized milk samples, followed by Firmicutes. After pasteurization of 15 s at 72, 75, and 80°C, the Proteobacteria and Firmicutes population remained constant. Previous studies have shown that the primary bacterial phyla were Proteobacteria and Firmicutes (35), while Pseudomonas, Acinetobacter, Lactococcus, Corynebacterium, and Streptococcus were the most abundant genera in raw milk (36). At the same time, the researchers compared the community structure of microbes in the raw milk collected from two dairy farms and found that Moraxellaceae, Streptococcaceae, Pseudomonadaceae, Staphylococcaceae, and Enterobacteriaceae were the main microorganisms, but the relative abundance was different. The most obvious were Enterobacteriaceae and Moraxellaceae. The relative abundances of Enterobacteriaceae in the raw milk from the two dairy farms were 82.8 and 0.6%, respectively, and the relative abundances of Moraxellaceae in the raw milk of the two dairy farms were 35.7 and 5.4%, respectively (16). The relative abundance of microorganisms in raw milk was different, which may be related to the cleanliness of farm production management, milk tankers, and transport vehicles. Enterobacteriaceae and Pseudomonadaceae are mainly derived from the environment. Comparing the microbial community structure of the same pasture in summer and winter, the microorganisms with relatively high abundance in summer milk were Staphylococcaceae, Ruminococcaceae, Aerococcaceae, Lachnospiraceae, and Corynebacteriaceae, while in winter the microorganisms were Staphylococcaceae, Ruminococcaceae, Aerococcaceae, and Lactonospirillaceae. The microorganisms in raw milk were relatively stable (30). In addition, researchers investigated the microbial community structure from the dairy farm to the final cheese products. Phylum Firmicutes was found as a main phylum, and the ratio between dairy farms and finished cheeses were 31% and 92%, respectively. The relative proportions of Lactococcus, Lactobacillus, Streptococcus, and Leuconostoc in cheese products accounts for 69–98% (37). Our results were consistent with another study, in which it was also reported that Gram-negative bacteria were more abundant than Gram-positive bacteria in all samples. Microorganisms in raw milk are mainly Proteobacteria, Firmicutes, Cyanobacteria, Actinobacteria, Bacteroidetes, and Verrucomicrobia, while Proteobacteria and Firmicutes are still the dominant phyla in pasteurized milk samples (38). Another study found that the primary bacterial communities in raw milk were Firmicutes, Actinobacteria, Bacteroidetes, Proteobacteria, and Tenericutes (39). Our study found that similar bacteria dominated the microbial communities of raw milk and pasteurized milk. Therefore, it is necessary to formulate good farm management practices, good production practices, and food safety systems to ensure the safety of dairy products.
Studies of the microorganisms of dairy products have also been reported. For example, Kamimura et al. used 16S rDNA sequencing to analyze the importance of microbial diversity in the processing environment, raw materials and final cheese to the characteristics and quality of cheese. They found that the microbial community structure of raw milk, whey, and environmental samples were significantly different. Both cheese and mature cheese showed a more stable and similar microbial community structure. Streptococcus and Lactococcus had high relative abundance throughout the cheese processing process (40). Zheng et al. used 16S rDNA sequencing to analyze the diversity of bacterial and fungal communities related to the quality and flavor during cheese maturation, and they found that Lactobacillus, Streptococcus, and Kluyveromyces were the main microorganisms in cheese (41). Schoen et al. used 16S rDNA sequencing to analyze the microbial community structure of the floor drain and biofilm samples of the Austrian cheese processing plant, and it was found that the microbial community was mainly composed of related bacteria in cheese such as Lactobacillus and Streptococcus thermophilus (42). Porcellato et al. used 16S rDNA sequencing to analyze the changes in the microbial community structure in raw milk and pasteurized milk, and demonstrated that Bacillus cereus was the dominant one in pasteurized milk, compared with pasteurized milk stored at 4°C. In comparison, the diversity of microorganisms was higher in pasteurized milk stored at 8°C (43). In this study, the predominant genera found in the milk sample were Acinetobacter, Macrococcus, Pseudomonas, and Lactococcus, while Stenotrophomonas and Acinetobacter were the most abundant genera in air and rinse water. Macrococcus are Gram-positive cocci belonging to the Staphylococcaceae family and are closely related to Staphylococcus but are not considered human pathogens. Pseudomonas, Lactococcus, and Acinetobacter are known psychrophilic bacteria and are relatively abundant in raw milk (44, 45). In the latter, there are also many species of Pseudomonas that produce heat-stable proteolytic and lipolytic enzymes and are responsible for milk quality defects including increased viscosity, sedimentation, aging gelation, fat separation, and increased bitterness. These are the most common reasons for milk spoilage and shortened shelf-life of milk products (46). In addition, Pseudomonas are dominant in the microflora of raw milk (38). In our study, Acinetobacter, Macrococcus, Pseudomonas, and Lactococcus were the most abundant in the microflora of raw milk and products. However, compared with raw milk, the relative abundance of Pseudomonas and Lactococcus were significantly reduced.
Viazis et al. analyzed the inactivation of specific microorganisms (Escherichia coli, Listeria monocytogenes, Staphylococcus aureus, and Streptococcus agalactiae) in milk for human consumption through traditional pasteurization, and found that pasteurization of the microorganisms inoculated milk (62.5°C for 30 min) can kill all microorganisms (47). Gabriel et al. analyzed the inactivation of specific microorganisms by pasteurization. The inactivation rate of Salmonella enterica, Escherichia coli O157:H7, Listeria monocytogenes, Pseudomonas aeruginosa, and Staphylococcus aureus after heating of heavily contaminated milk at 62.5°C for 30 min was 90.8–99.9% (48). Stabel and Lambertz through different pasteurization intensities (i.e., 62.7°C for 30 min, 65.5°C for 16 s, 71.7°C for 15 s, 71.7°C for 20 s, and 74.4°C for 15 s, respectively). Analysis of the inactivation of specific microorganisms (e.g., Mycobacterium avium subsp. paratuberculosis) found that pasteurization can significantly reduce the survival rate of the organisms, but the inactivation is not complete (49). Heat treatment of milk samples heavily contaminated by pathogenic bacteria shows that traditional pasteurization processes cannot completely eliminate biological hazards. The total number of bacteria in the dairy products in our study was very low and far below the limit requirements of the food safety standards established by the European Union, the United States, and China. We did not detect the presence of any pathogenic bacteria, such as Salmonella, Staphylococcus aureus, and Cronobacter. This may be related to the improved management methods developed by the dairy farm and processing factory management personnel. For example, the pasture uses sandy bedding, which does not allow bacteria to breed easily. In addition, the cleaning and disinfection procedures during milking also effectively control the raw milk.
The Source Tracker modeling indicated that the microorganisms in the products were significantly correlated to that in the product tank. The product tank becomes a source of microbial contamination due to defects in the management program, e.g., the cleaning-in-place procedures were not followed, the time was too short, rendering these practices ineffective, or the product tank had a secondary contamination source. Our results were consistent with a previous study of bacterial source tracking analysis for milk powder based on 16S rDNA-based single-molecule real-time sequencing technology, indicating that the primary microbes in milk powder originated in the raw milk (15). The Source Tracker model based on 16S rDNA sequencing technology was previously used to analyze potential contamination sources in pasteurized raw milk, and the contributions of cow pasture environmental samples to microbial contamination in raw milk were traced. The teat liners and the teat dip cups were found to be the most important sources of contamination, and this is most likely related to farm cleaning procedures or management practices (16). Source Tracker was also used to analyze the potential sources of microbial contamination in raw milk using an automatic milking system, and the effects of rumen fluid, drinking water, feed, bedding, air, and feces were analyzed. Air was found to be the most important source of contamination. If the cleaning measures of the automatic milking system were complete, the microbial impact on raw milk was relatively small (17).
In this study, we evaluated the traceability of microorganisms in pasteurized milk, and traced the influence of different steps in the processing lines on the microorganisms in pasteurized milk, using the Source Tracker model based on 16S rDNA sequencing technology. We observed significant differences in richness of microbiota in the samples between the raw milk and the processing factory air samples, even though the uniformity and coverage of bacteria in raw milk and the final products were similar. The Source Tracker model analysis indicated that the microorganisms in the products were significantly related to the product tank. Our analysis can assist in localizing potential sources of microbial contamination and act as guidance for quality control sampling to avoid food quality problems.
The original contributions presented in the study are publicly available. This data can be found here: Genome Sequence Archive in BIG Data Center, CRA005796.
BD: conceptualization, methodology, investigation, and writing—original draft. LM: methodology. HL: data curation. HW and HY: supervision. NZ: conceptualization. YZ and SZ: writing—review, supervision, and funding acquisition. JW: conceptualization and funding acquisition. All authors contributed to the article and approved the submitted version.
This study was funded by the Scientific Research Project for Major Achievements of Agricultural Science and Technology Innovation Program (CAAS-ZDXT2019004), the Agricultural Science and Technology Innovation Program (ASTIP-IAS12), and the Modern Agro-Industry Technology Research System of the PR China (CARS-36).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Loss G, Depner M, Ulfman LH, van Neerven RJJ, Hose AJ, Genuneit J, et al. Consumption of unprocessed cow's milk protects infants from common respiratory infections. J Allergy Clin Immunol. (2015) 135:56–62. doi: 10.1016/j.jaci.2014.08.044
2. Waser M, Michels KB, Bieli C, Floistrup H, Pershagen G, von Mutius E, et al. Inverse association of farm milk consumption with asthma and allergy in rural and suburban populations across Europe. Clin Exp Allergy. (2007) 37:661–70. doi: 10.1111/j.1365-2222.2006.02640.x
3. Zhan J-w, Shen Y-y, Li X, Zhang H, Niu H, Fang L-y, et al. Microbiome and metabolic changes of milk in response to dietary supplementation with bamboo leaf extract in dairy cows. Front Nutr. (2021) 8:e723446. doi: 10.3389/fnut.2021.723446
4. UN News. Deadly E. coli Outbreak Caused by Rare Strain of Bacterium, UN Confirms. (2011). Available online at: https://news.un.org/en/story/2011/06/377302 (accessed June 3, 2011).
5. UN News. Children Under Five Account for One Third of Deaths from Foodborne Diseases. (2015). Available online at: https://news.un.org/en/story/2015/12/517172 (accessed December 3, 2015).
6. He Y, Ren Y, Guo B, Yang Y, Ji Y, Zhang D, et al. Development of a specific nanobody and its application in rapid and selective determination of Salmonella enteritidis in milk. Food Chem. (2020) 310:125942. doi: 10.1016/j.foodchem.2019.125942
7. Kuehn B. Drug-resistant infections from raw milk. J Am Med Assoc. (2018) 319:1191. doi: 10.1001/jama.2018.2363
8. Costard S, Espejo L, Groenendaal H, Zagmutt FJ. Outbreak-related disease burden associated with consumption of unpasteurized cow's milk and cheese, United States, 2009-2014. Emerg Infect Dis. (2017) 23:957–64. doi: 10.3201/eid2306.151603
9. Montanhini MTM, Bersot LdS. Evaluation of psychrotrophic behavior and lipolytic and proteolytic activity of Bacillus cereus isolated from refrigerated dairy products. Acta Scientiarum-Technol. (2013) 35:163–7. doi: 10.4025/actascitechnol.v35i1.13752
10. Gao T, Ding Y, Wu Q, Wang J, Zhang J, Yu S, et al. Prevalence, virulence genes, antimicrobial susceptibility, and genetic diversity of bacillus cereus isolated from pasteurized milk in China. Front Microbiol. (2018) 9:e00533. doi: 10.3389/fmicb.2018.00533
11. Zhou G, Liu H, He J, Yuan Y, Yuan Z. The occurrence of Bacillus cereus, B. thuringiensis and B mycoides in Chinese pasteurized full fat milk. Int J Food Microbiol. (2008) 121:195–200. doi: 10.1016/j.ijfoodmicro.2007.11.028
12. Quigley L, O'Sullivan O, Stanton C, Beresford TP, Ross RP, Fitzgerald GF, et al. The complex microbiota of raw milk. FEMS Microbiol Rev. (2013) 37:664–98. doi: 10.1111/1574-6976.12030
13. Spanu C, Scarano C, Ibba M, Spanu V, De Santis EPL. Occurrence and traceability of Listeria monocytogenes strains isolated from sheep's milk cheese-making plants environment. Food Control. (2015) 47:318–25. doi: 10.1016/j.foodcont.2014.07.027
14. El-Mokdad MM. Implementing a Traceability System in a Small to Medium Dairy Plant in Lebanon with Isolation and Characterization of Staphylococcus aureus. Beirut: American University of Beirut (2014).
15. Wang J, Zheng Y, Xi X, Hou Q, Xu H, Zhao J, et al. Application of PacBio Single Molecule Real-Time (SMRT) sequencing in bacterial source tracking analysis during milk powder production. Food Control. (2018) 93:226–34. doi: 10.1016/j.foodcont.2018.05.044
16. Du B, Meng L, Liu H, Zheng N, Zhang Y, Guo X, et al. Impacts of milking and housing environment on milk microbiota. Animals. (2020) 10:2339. doi: 10.3390/ani10122339
17. Wu H, Nguyen QD, Tran TTM, Tang MT, Tsuruta T, Nishino N. Rumen fluid, feces, milk, water, feed, airborne dust, and bedding microbiota in dairy farms managed by automatic milking systems. Animal Sci J. (2019) 90:445–52. doi: 10.1111/asj.13175
18. National Standards of the Republic of China. GB 4789.2:Determination of Microbiological Properties (Total Bacterial Count). Beijing: National Standards of the Republic of China (2016).
19. International Dairy Federation IDF. 209:2007, Milk and Milk-based Drinks-Determination of Alkaline Phosphatase Activity-Enzymatic Photo-activated System (EPAS) Method. Brussels: International Dairy Federation. (2007).
20. Tianjin Dairy Science and Technology Innovation Association T/TDSTIA. 001: Determination of Lactoperoxidase in Milk and Dairy Products. Tianjin: Tianjin Dairy Science and Technology Innovation Association (2021).
21. Guo M, Wu F, Hao G, Qi Q, Li R, Li N, et al. Bacillus subtilis improves immunity and disease resistance in rabbits. Front Immunol. (2017) 8:e00354. doi: 10.3389/fimmu.2017.00354
22. Chen S, Zhou Y, Chen Y, Gu J. Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. (2018) 34:884–90. doi: 10.1093/bioinformatics/bty560
23. Magoc T, Salzberg SL. FLASH fast length adjustment of short reads to improve genome assemblies. Bioinformatics. (2011) 27:2957–63. doi: 10.1093/bioinformatics/btr507
24. Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. (2010) 7:335–6. doi: 10.1038/nmeth.f.303
25. Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R, UCHIME. improves sensitivity and speed of chimera detection. Bioinformatics. (2011) 27:2194–200. doi: 10.1093/bioinformatics/btr381
26. Edgar RC. UPARSE highly accurate OTU sequences from microbial amplicon reads. Nat Methods. (2013) 10:996–1000. doi: 10.1038/nmeth.2604
27. Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, et al. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. (2007) 35:7188–96. doi: 10.1093/nar/gkm864
28. Ondov BD, Bergman NH, Phillippy AM. Interactive metagenomic visualization in a Web browser. BMC Bioinformatics. (2011) 12:85. doi: 10.1186/1471-2105-12-385
29. ggplot2 (2013). Available online at: http://ggplot2.org/ (accessed 2013).
30. Nguyen TT, Wu H, Nishino N. An investigation of seasonal variations in the microbiota of milk, feces, bedding, and airborne dust. Asian-Australas J Anim Sci. (2019) 33:1858–65. doi: 10.5713/ajas.19.0506
32. Knights D, Kuczynski J, Charlson ES, Zaneveld J, Mozer MC, Collman RG, et al. Bayesian community-wide culture-independent microbial source tracking. Nature Methods. (2011) 8:761–5. doi: 10.1038/nmeth.1650
33. Dominguez-Bello MG, De Jesus-Laboy KM, Shen N, Cox LM, Amir A, Gonzalez A, et al. Partial restoration of the microbiota of cesarean-born infants via vaginal microbial transfer. Nat Med. (2016) 22:250–3. doi: 10.1038/nm.4039
34. Metcalf JL, Xu ZZ, Weiss S, Lax S, Van Treuren W, Hyde ER, et al. Microbial community assembly and metabolic function during mammalian corpse decomposition. Science. (2016) 351:158–62. doi: 10.1126/science.aad2646
35. Li N, Wang Y, You C, Ren J, Chen W, Zheng H, et al. Variation in raw milk microbiota throughout 12 months and the impact of weather conditions. Sci Rep. (2018) 8:1–10. doi: 10.1038/s41598-018-20862-8
36. McHugh AJ, Feehily C, Fenelon MA, Gleeson D, Hill C, Cotter PD. Tracking the dairy microbiota from farm bulk tank to skimmed milk powder. mSystems. (2020) 5:1–16. doi: 10.1128/mSystems.00226-20
37. Falardeau J, Keeney K, Trmcic A, Kitts D, Wang S. Farm-to-fork profiling of bacterial communities associated with an artisan cheese production facility. Food Microbiol. (2019) 83:48–58. doi: 10.1016/j.fm.2019.04.002
38. Ding R, Liu Y, Yang S, Liu Y, Shi H, Yue X, et al. High-throughput sequencing provides new insights into the roles and implications of core microbiota present in pasteurized milk. Food Res Int. (2020) 137:109586. doi: 10.1016/j.foodres.2020.109586
39. Kable ME, Srisengfa Y, Laird M, Zaragoza J, McLeod J, Heidenreich J, et al. The core and seasonal microbiota of raw bovine milk in tanker trucks and the impact of transfer to a milk processing facility. MBio. (2016) 7:e00836–16. doi: 10.1128/mBio.00836-16
40. Kamimura BA, Cabral L, Noronha MF, Baptista RC, Nascimento HM, Sant'Ana AS. Amplicon sequencing reveals the bacterial diversity in milk, dairy premises and Serra da Canastra artisanal cheeses produced by three different farms. Food Microbiol. (2020) 89:103453. doi: 10.1016/j.fm.2020.103453
41. Zheng X, Liu F, Shi X, Wang B, Li K, Li B, et al. Dynamic correlations between microbiota succession and flavor development involved in the ripening of Kazak artisanal cheese. Food Res Int. (2018) 105:733–42. doi: 10.1016/j.foodres.2017.12.007
42. Schoen K, Schornsteiner E, Dzieciol M, Wagner M, Mueller M, Schmitz-Esser S. Microbial communities in dairy processing environment floor-drains are dominated by product-associated bacteria and yeasts. Food Control. (2016) 70:210–5. doi: 10.1016/j.foodcont.2016.05.057
43. Porcellato D, Aspholm M, Skeie SB, Mellegard H. Application of a novel amplicon-based sequencing approach reveals the diversity of the Bacillus cereus group in stored raw and pasteurized milk. Food Microbiol. (2019) 81:32–9. doi: 10.1016/j.fm.2018.01.014
44. Meng Y, Chen X, Sun Z, Li Y, Chen D, Fang S, et al. Exploring core microbiota responsible for the production of volatile flavor compounds during the traditional fermentation of Koumiss. LWT-Food Sci Technol. (2021) 135:49. doi: 10.1016/j.lwt.2020.110049
45. Zheng X, Ge Z, Lin K, Zhang D, Chen Y, Xiao J, et al. Dynamic changes in bacterial microbiota succession and flavour development during milk fermentation of Kazak artisanal cheese. Int Dairy J. (2021) 113:878. doi: 10.1016/j.idairyj.2020.104878
46. Zhang C, Bijl E, Svensson B, Hettinga K. The extracellular protease AprX from pseudomonas and its spoilage potential for UHT milk: a review. Compreh Rev Food Sci Food Safety. (2019) 18:834–52. doi: 10.1111/1541-4337.12452
47. Viazis S, Farkas BE, Jaykus LA. Inactivation of bacterial pathogens in human milk by high-pressure processing. J Food Prot. (2008) 71:109–18. doi: 10.4315/0362-028X-71.1.109
48. Gabriel AA, Bayaga CLT, Magallanes EA, Aba RPM, Tanguilig KMN. Fates of pathogenic bacteria in time-temperature-abused and Holder-pasteurized human donor, infant formula and full cream cow's milk. Food Microbiol. (2020) 89:103450. doi: 10.1016/j.fm.2020.103450
Keywords: milk, microbial, contamination, 16s rDNA sequencing, Source Tracker
Citation: Du B, Meng L, Wu H, Yang H, Liu H, Zheng N, Zhang Y, Zhao S and Wang J (2022) Source Tracker Modeling Based on 16S rDNA Sequencing and Analysis of Microbial Contamination Sources for Pasteurized Milk. Front. Nutr. 9:845150. doi: 10.3389/fnut.2022.845150
Received: 29 December 2021; Accepted: 31 March 2022;
Published: 28 April 2022.
Edited by:
Hao Dong, Zhongkai University of Agriculture and Engineering, ChinaReviewed by:
Xin Yi Lan, Hunan Agricultural University, ChinaCopyright © 2022 Du, Meng, Wu, Yang, Liu, Zheng, Zhang, Zhao and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiaqi Wang, amlhcWl3YW5nQHZpcC4xNjMuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.