 Carlos A. Labarrere
Carlos A. Labarrere Ghassan S. Kassab
Ghassan S. Kassab- California Medical Innovations Institute, San Diego, CA, United States
Many local and systemic diseases especially diseases that are leading causes of death globally like chronic obstructive pulmonary disease, atherosclerosis with ischemic heart disease and stroke, cancer and severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) causing coronavirus disease 19 (COVID-19), involve both, (1) oxidative stress with excessive production of reactive oxygen species (ROS) that lower glutathione (GSH) levels, and (2) inflammation. The GSH tripeptide (γ- L-glutamyl-L-cysteinyl-glycine), the most abundant water-soluble non-protein thiol in the cell (1–10 mM) is fundamental for life by (a) sustaining the adequate redox cell signaling needed to maintain physiologic levels of oxidative stress fundamental to control life processes, and (b) limiting excessive oxidative stress that causes cell and tissue damage. GSH activity is facilitated by activation of the Kelch-like ECH-associated protein 1 (Keap1)-Nuclear factor erythroid 2-related factor 2 (Nrf2)-antioxidant response element (ARE) redox regulator pathway, releasing Nrf2 that regulates expression of genes controlling antioxidant, inflammatory and immune system responses. GSH exists in the thiol-reduced (>98% of total GSH) and disulfide-oxidized (GSSG) forms, and the concentrations of GSH and GSSG and their molar ratio are indicators of the functionality of the cell. GSH depletion may play a central role in inflammatory diseases and COVID-19 pathophysiology, host immune response and disease severity and mortality. Therapies enhancing GSH could become a cornerstone to reduce severity and fatal outcomes of inflammatory diseases and COVID-19 and increasing GSH levels may prevent and subdue these diseases. The life value of GSH makes for a paramount research field in biology and medicine and may be key against systemic inflammation and SARS-CoV-2 infection and COVID-19 disease. In this review, we emphasize on (1) GSH depletion as a fundamental risk factor for diseases like chronic obstructive pulmonary disease and atherosclerosis (ischemic heart disease and stroke), (2) importance of oxidative stress and antioxidants in SARS-CoV-2 infection and COVID-19 disease, (3) significance of GSH to counteract persistent damaging inflammation, inflammaging and early (premature) inflammaging associated with cell and tissue damage caused by excessive oxidative stress and lack of adequate antioxidant defenses in younger individuals, and (4) new therapies that include antioxidant defenses restoration.
Introduction
Glutathione (GSH) is a unique molecule essential for life that participates in key aspects of cellular homeostasis, having a paramount role in defense against the oxidative damage that occurs during all different diseases including coronavirus disease 19 (COVID-19) disease. GSH has a central participation in trans-hydrogenation reactions needed to maintain a reduced state of sulfhydryl groups of other molecules, proteins and enzymes, as well as formation of deoxyribonucleotides and vitamin reduction (1–5). GSH has the function of “master antioxidant” in all tissues and is involved in antioxidant defense, detoxication of xenobiotics, intracellular redox homeostasis, cysteine carrier/storage, cell signaling, protein folding and function, gene expression, cell differentiation/proliferation, immune response and antiviral defense, that make it a “Samsonian (mighty) little molecule” (1, 2). The high (millimolar) concentration of the reduced form highlights its central role in the control of those processes (5–8). The central role of GSH in oxidative stress and inflammation, in the pathophysiology of inflammatory diseases and COVID-19, and in host immune response and disease severity and mortality, makes GSH a little but powerful player in maintaining health and avoiding disease. In this review we will focus on (a) GSH depletion as a fundamental risk factor for diseases like chronic obstructive pulmonary disease and atherosclerosis (ischemic heart disease and stroke), (b) importance of oxidative stress and antioxidants in severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection and COVID-19 disease, (c) significance of GSH to counteract persistent damaging inflammation, inflammaging and early (premature) inflammaging associated with cell and tissue damage caused by excessive oxidative stress and lack of adequate antioxidant defenses in younger individuals, and (d) new therapies that include antioxidant defenses restoration.
Glutathione brief history
Glutathione was discovered in 1888 by de Rey-Pailhade and initially named “philothion” (from the Greek words meaning “love” and “sulfur”) because of its reactivity with sulfur to form hydrogen sulfide (4, 9). Subsequently, Hopkins reported this substance as a dipeptide containing glutamate and cysteine, and he named it “glutathione” (10), which is a tripeptide consisting of glutamate, cysteine, and glycine (11, 12). Harington and Mead finally described the correct chemical structure of the tripeptide in 1935 (13). GSH was virtually forgotten for 40 years until in 1969, Kosower and Kosower (14) emphasized the scant GSH research in those days. GSH research had a great momentum especially in the 1980s, with studies carried out by Meister and his collaborators who contributed to understanding the physiological functions and the metabolism (4).
Glutathione composition and synthesis
The GSH (γ- L-glutamyl-L-cysteinyl-glycine) is a water-soluble tripeptide formed by the amino-acids glutamic acid, cysteine and glycine (Figure 1) present in the cytoplasm of all cells. GSH is found in all mammalian tissues as the most abundant non-protein thiol that defends against oxidative stress and possess a distinctive stability provided by a γ-carboxyl bond within the molecule (Figure 1A). GSH exists in the thiol-reduced and disulfide-oxidized (GSSG) forms (1, 2); and it’s free and bound to proteins (Figure 1). The reduced form GSH is the active form of the molecule, it is the most abundant and it is found inside the cells in millimolar concentrations in the range of 1–10 mM (highest concentration in liver) (5–8), while extracellularly they are found in micromolar (GSH in plasma: 10–30 μM) levels (5, 15, 16). The active group of the molecule is represented by the thiol group (-SH) of the cysteine residue (Figure 1) which provides its reductive power. The millimolar GSH intracellular concentrations, the low plasma micromolar concentrations and the low GSH redox potential (E′0 = −240 mV) make GSH an ideal and perfect cellular redox buffer (5, 16–18). GSH accounts for >98% of total GSH (3, 19–23). Eukaryotic cells have three major reservoirs of GSH. Most (80–85%) of the cellular GSH are in the cytosol, 10–15% is in the mitochondria and a small percentage is in the endoplasmic reticulum (Figure 2) (3, 24–28). GSSG is found mainly extracellularly. The redox state of the GSH/GSSG couple can serve as an important indicator of redox environment (29–31), and changes in this couple correlate with multiple cellular processes, including cell differentiation (32–37), cell proliferation (32–37), and cell apoptosis (38–42). GSH deficiency as evidenced by a decrease in the GSH/GSSG ratio manifests itself largely through an increased susceptibility to oxidative stress, and the resulting damage is thought to be involved in SARS-CoV-2 infection leading to COVID-19 disease (6, 7, 39, 43–53). In addition, imbalances in GSH levels affect immune system function, and are thought to play a role in the aging process and the diseases of aging, one of the principal risk factors for the development and progression of COVID-19 disease.
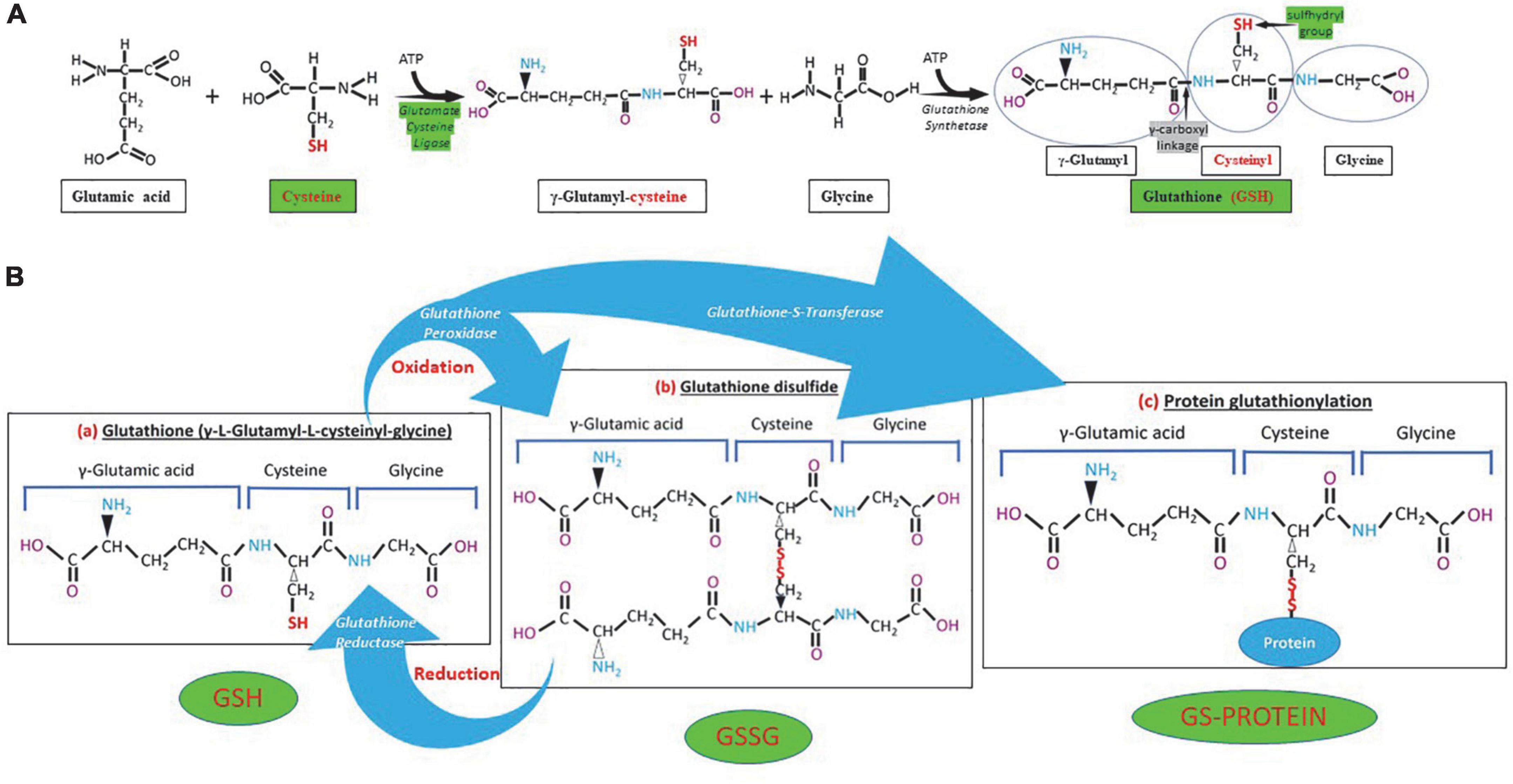
Figure 1. Glutathione (GSH) synthesis, chemical structure and different forms of GSH. (A) GSH is synthesized in the cytosol in two steps. The first step is the formation of γ-glutamylcysteine from glutamate and cysteine by the enzyme γ-glutamylcysteine synthetase (glutamate cysteine ligase). The second step in GSH synthesis is regulated by glutathione synthetase. Glutathione cysteine ligase and cysteine (green) are the limiting factors in GSH synthesis. The γ-carboxyl linkage (gray) and the sulfhydryl group (green) provide stability and reductive power to the molecule, respectively. (B) Chemical structure of reduced (GSH), oxidized (GSSG) glutathione and GS-protein generated by protein glutathionylation. Glutathione peroxidase oxidizes GSH and glutathione reductase reduces GSSG, while glutathione-S-transferase participates in protein glutathionylation.
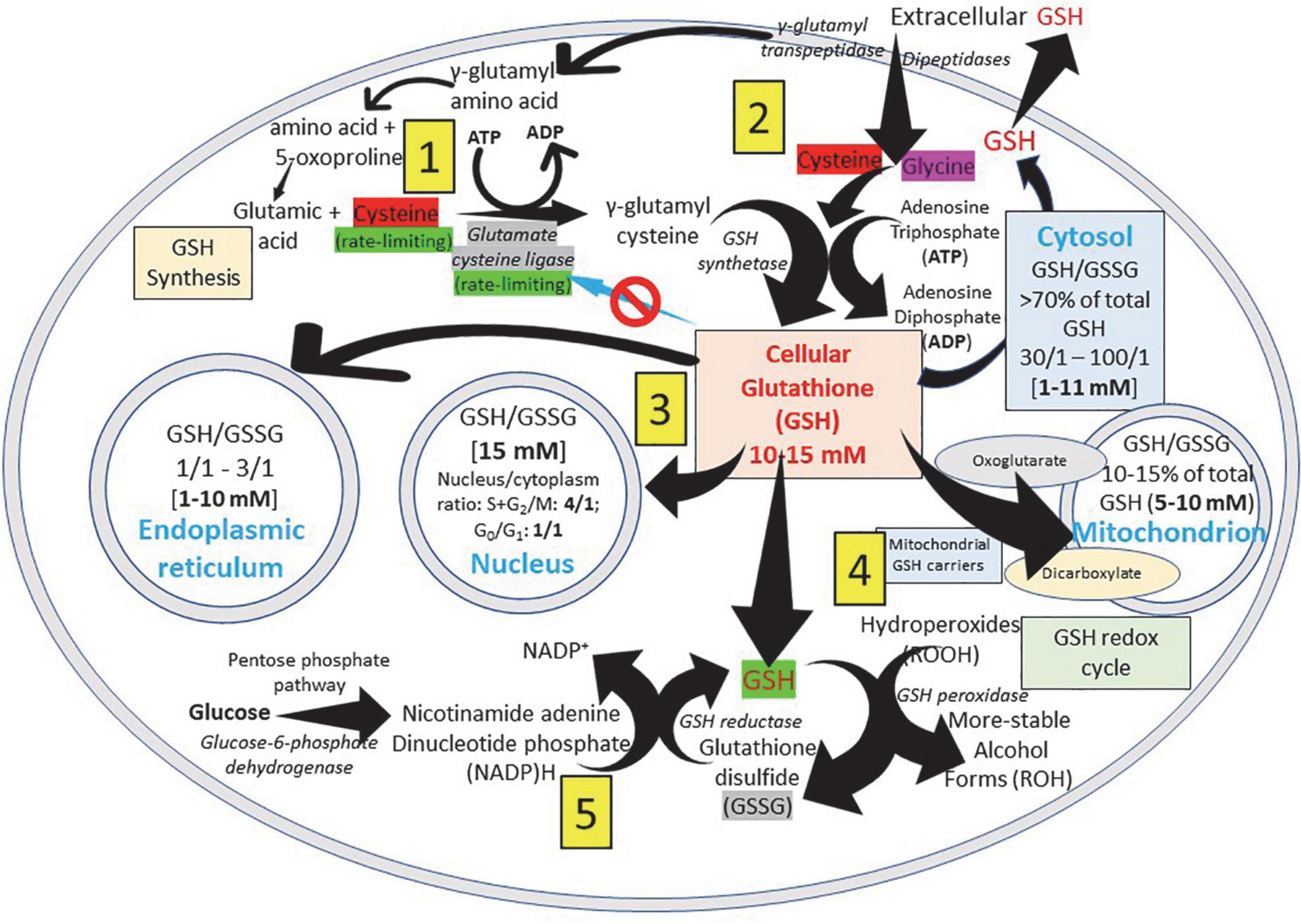
Figure 2. Glutathione distribution in subcellular compartments. GSH (γ- L-glutamyl-L-cysteinyl-glycine), a water-soluble tripeptide formed by the amino-acids glutamic acid, cysteine and glycine, is considered the major non-protein low molecular weight modulator of redox processes and the most important thiol reducing agent of the cell. (1) ATP-dependent GSH biosynthesis occurs in the cytosol of the cell and cysteine (red) and glutamate cysteine ligase (gray) are rate-limiting factors for its production. (2) Extracellular GSH is enzymatically degraded on the surface of the cells by γ-glutamyl transpeptidase generating the γ-glutamyl fraction (taken into the cell as γ-glutamyl-amino acid that can be metabolized to release the amino acid and 5-oxoproline, which can then be converted into glutamate to be used in the synthesis of GSH) and the cysteinyl-glycine fraction; and by dipeptidases splitting cysteinyl-glycine generating cysteine and glycine that are taken into the cell. (3) To allow normal cell function, it is essential to maintain an optimal GSH: GSSG ratio throughout all cell compartments. (4) The inner mitochondrial membrane system transport, that involves dicarboxylate and 2-oxoglutarate anion transporters, allows the passage of negatively charged GSH from the cytosol to the mitochondria. (5) GSH is present in both reduced (GSH) and oxidized (GSSG) states, and reduced GSH is maintained by GSH reductase, a cytosolic NADPH-dependent enzyme. GSSG returns to the reduced state by the NADPH-dependent activity of glutathione reductase. NADPH is rapidly regenerated from NADP + using electrons derived from catabolism of substrate molecules, such as glucose or isocitric and malic acid (pentose phosphate pathway). Reduced GSH neutralizes cellular hydroperoxides through GSH peroxidase activity.
Glutathione is synthesized in the cytosol of all cells from their precursor amino acids: glutamic acid, cysteine and glycine by consecutive action of two enzymes: γ-glutamyl-cysteine (γ-GluCys) synthetase (also known as glutamate cysteine ligase, GCL) that in a first step uses glutamate and cysteine as a substrate to form the dipeptide γ-glutamyl-cysteine; and glutathione synthetase that in a second step combines γ-glutamyl-cysteine with glycine for forming GSH (54, 55) (Figure 3). ATP (adenosine triphosphate) acts as a co-substrate for both enzymes (Figures 2, 3). Under normal physiological conditions, the rate of synthesis of GSH is determined to a large extent by two factors: (a) the activity of GCL and (b) the availability of the cysteine substrate. Therefore, the intracellular levels of GSH are regulated by the negative feedback of GSH itself on the GCL enzyme (1, 4, 55–57) and by the availability of the amino acid L-cysteine (1, 4, 58). The GCL enzyme is a heterodimer formed by two subunits: the heavy subunit or glutamate cysteine ligase catalytic subunit (GCLC, 73 kDa) and the light subunit or glutamate cysteine ligase modulating subunit (GCLM, 30 kDa). The heavy subunit has the active site responsible for the union between the amino group of the cysteine and the γ-carboxyl group of glutamate. The GCLM subunit has no enzymatic activity but has an important regulatory function increasing the efficiency of the GCLC subunit. This subunit is required for optimal activity and feedback inhibition by GSH (59). GSH inhibits GCL by competing with glutamate in the active site of GCLC (1, 57–60). The enzyme glutathione synthetase (GS) is formed by two identical subunits (52 kDa/subunit) and is not regulated by intracellular levels of GSH. The active site of the enzyme that binds glycine to the dipeptide γ-L-glutamyl-L-cysteine is highly specific (57). GCL is considered the speed limiting enzyme of synthesis since overexpression of GS does not increase GSH levels while overexpression of GCL increases the synthesis of GSH (61) (Figure 3). ATP is the energy donor for both enzymes. As mentioned above, GSH cellular concentrations are regulated by GSH-mediated GCL inhibition (Figures 2, 3). Thus, the biological control of intracellular GSH homeostasis through consumption and supply is an intricately balanced process that prevents oxidative stress. Cellular GSH (cytosol, mitochondria, endoplasmic reticulum, nucleus; Figures 2, 4) availability is maintained by de novo synthesis from precursor amino acids, (glutamate, cysteine, and glycine), reduction of GSSG by glutathione reductase (GR), and uptake from exogenous GSH sources across plasma membranes (Figure 4) (62, 63). The three amino acids are adsorbed by transporters. Additionally, intestinal epithelial cells can import intact GSH from the lumen via specific plasma membrane transporters (7).
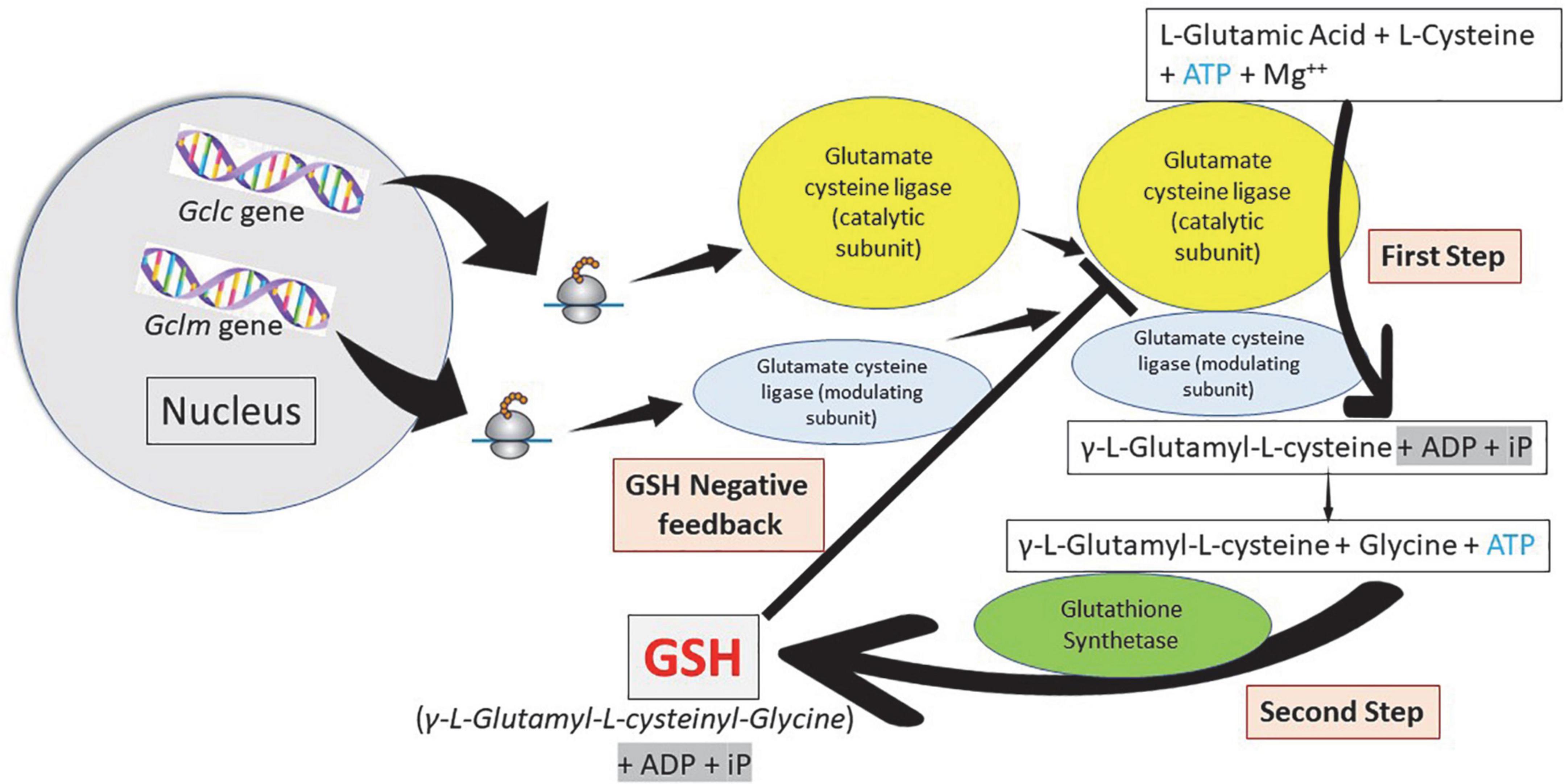
Figure 3. Glutathione synthesis: A two-step pathway. Homeostasis of cellular glutathione. Synthesis and regulation of the cell concentrations. Glutamate cysteine ligase (γ-glutamyl cysteine synthetase) constitute the first step in the synthesis of glutathione (GSH) forming γ-L-glutamyl-L-cysteine using adenosine triphosphate (ATP). Glutathione synthetase constitute the second step forming GSH, also using ATP. Cellular GSH concentration regulates the function of glutamate cysteine ligase.
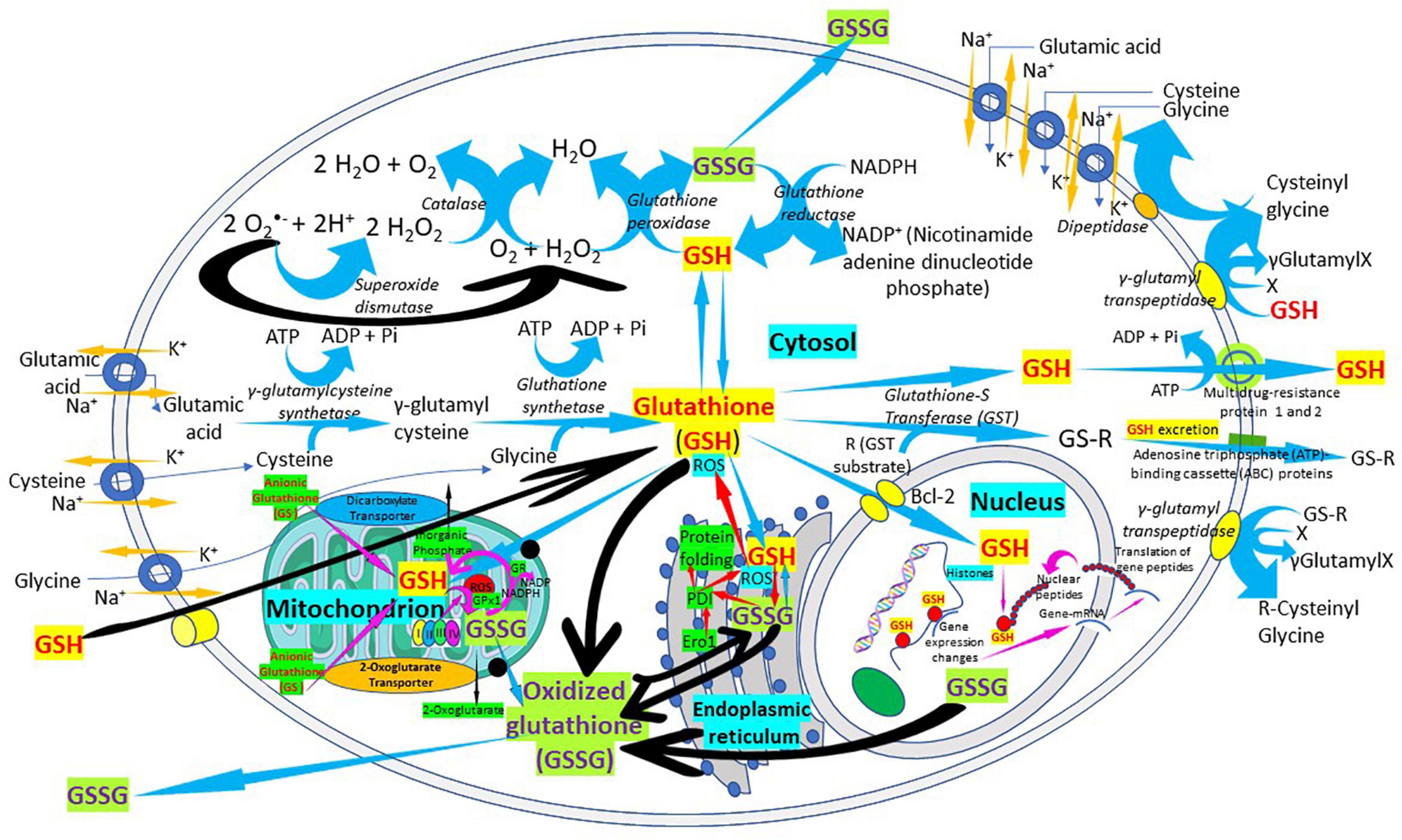
Figure 4. The “Glutathione Pathway.” Glutathione synthesis, γ-glutamyl pathway, cellular distribution, antioxidant properties, catabolism of xenobiotics, and glutathione recycling in the cell. The figure shows a schematic representation of the “glutathione pathway.” Glutathione (GSH) is synthesized from glutamate, cysteine, and glycine by γ-glutamyl-cysteine synthetase (glutamate cysteine ligase) and glutathione synthetase. Glutathione redox state is regulated, in part, by glutathione peroxidases, forming oxidized glutathione (GSSG), and by a reaction catalyzed by glutathione reductase. Glutathione is conjugated to substrates both through the action of the glutathione S-transferases and through non-enzymatic reactions. Glutathione conjugates can be excreted from the cells by members of the ATP-binding cassette (ABC) transporter family.
Glutathione cellular distribution
Glutathione is found in almost all cellular compartments, including the nucleus (5, 54, 64–68) (Figures 2, 4). The GSH transport between the various cell compartments is vital to buffer reactive oxygen species (ROS) and facilitate redox signaling in order to control cell growth, development and defense, as well as regulate cell proliferation. GSH is predominantly in its thiol-reduced form inside the cells, except in the lumen of the endoplasmic reticulum where it exists only in its GSSG form (Figures 2, 4). The GSH content existing in millimolar concentrations varies among different organs; liver being among organs with the highest content (56). GSH content also varies among different areas of the same tissues; periportal hepatocytes may contain nearly twice the centrilobular concentration, enterocytes at the villus tip have a higher content than the crypts, and renal proximal tubular cells have more GSH than other parts of the nephron (56). Mitochondria contain 10–15% of the intracellular GSH reaching a concentration of 10–12 mM (54) while in the cytosol the concentration is 7 mM (54, 56). This difference in concentration is associated with the absence of catalase inside the mitochondria, what leaves GSH in charge of all inactivation of the hydrogen peroxide generated during the oxidative processes that occur in the mitochondrial matrix (57).
The concentration of GSH in the mitochondrial compartment is more important for cell survival than the GSH found in the cytosol. Since mitochondria do not have the enzymes involved in the synthesis of GSH, all the GSH found in the mitochondrial compartment comes from the cytosol. A system transport present in the inner mitochondrial membrane, that involves dicarboxylate and 2-oxoglutarate anion transporters, allows the passage of negatively charged GSH from the cytosol to the mitochondria. The first incorporates GSH into the mitochondria by inorganic phosphate exchange and the second by exchange of 2-oxoglutarate (27, 28, 64) (Figures 2, 4). While the greater amount of cellular reduced GSH is found in the cytosol and mitochondria, the endoplasmic reticulum becomes a reservoir of small concentrations of the oxidized form of GSH (GSSG). The ratio of reduced GSH to the disulfide form (GSH/GSSG) within the endoplasmic reticulum ranges from 1:1 to 4:1, whereas the overall cellular GSH/GSSG ratio ranges from 30:1 to 100:1 (26) (Figure 2). There is a preferential transport of GSSG from the cytosol to the endoplasmic reticulum to maintain an adequate environment for protein disulfide bond formation and protein folding (69–71). There is little data about the concentrations of GSH in the nucleus and endoplasmic reticulum largely because of a lack of adequate techniques to accurately determine the GSH pool at those locations (15, 69, 72, 73). There are great variations in nuclear GSH concentration and its regulation mechanisms during the cell cycle since cells starting the proliferation phase have high levels of nuclear GSH, while resting cells have similar or lower GSH levels in the nucleus than in the cytoplasm (68, 72, 73). High nuclear GSH concentrations are vital since increase in total GSH is necessary for the cells to progress from the G1- (with low GSH levels) to the S-phase; addition of GSSG causes the cell cycle to arrest at G1; and excessive and prolonged oxidation arrest cell cycle triggering cell death (68, 72, 73). GSH behaves as “redox sensor” at the DNA synthesis onset by maintaining nuclear architecture providing the appropriate redox environment for DNA replication and safeguarding DNA integrity (72) and is a key regulator of epigenetic events critical in cell proliferation regulation and cellular resistance to apoptosis (73).
Glutathione and the γ-glutamyl cycle
The synthesis, transport and catabolism of GSH occur in a series of enzymatic steps and transports of membrane that are collectively called γ-glutamyl cycle (Figure 5) (1, 74, 75). The γ-glutamyl cycle was postulated by Meister (76) and it accounts for the GSH biosynthesis and degradation. The GSH biosynthesis has been described previously. After its synthesis, GSH is transported to the intracellular compartments, mitochondria, endoplasmic reticulum and nucleus, but most of it is released through transporters toward the extracellular space. In contrast to the synthesis, that occurs only intracellularly, the degradation or catabolic part of the GSH cycle, takes place partially extracellularly and partially inside cells. The extracellular degradation of GSH occurs on the surface of the cells that express the enzyme γ-glutamyl transpeptidase and the dipeptidases found in the external plasma membrane (1) (Figure 5). After the plasma membrane carrier-mediated GSH release from the cell, GSH becomes accessible to the active site of γ-glutamyl transpeptidase, which catalyzes the breakdown of the GSH γ-glutamyl bond forming two fractions: The γ-glutamyl fraction and the cysteinyl-glycine by transferring the γ-glutamyl fraction to an amino acid acceptor, forming γ-glutamyl-amino acid. Once inside the cell, the γ-glutamyl-amino acid can be metabolized to release the amino acid and 5-oxoproline, which can then be converted into glutamate to be used in the synthesis of GSH. On the other hand, also in the extracellular space, the cysteinyl-glycine fraction is split by the enzyme dipeptidase generating cysteine and glycine. The cells incorporate cysteine and most of the intracellular cysteine is incorporated into the synthesis of GSH. Depending on the metabolic needs of the cell, the cysteine can be used for protein synthesis and part can be degraded to sulfate and taurine. The cycle γ-glutamyl allows GSH to be used as a continuous source of cysteine. The γ-glutamyl amino acid is taken up by cells through a specific transport mechanism. Cysteinyl glycine is also taken up by cells. Inside the cell, the γ-glutamyl amino acid is hydrolyzed by γ-glutamyl cyclo-transferase and converted into oxoproline and a free amino acid. Oxoproline is a cyclic form of glutamate and is converted into glutamate via oxoprolinase (Figure 5). The γ-glutamyl cycle was initially postulated by Meister as a mechanism for amino acid transport (76). However, this presents major problems. The most important is the energetic one. The γ-glutamyl cycle requires the use of three ATP molecules per turn of the cycle. Thus, the uptake of an amino acid would require the use of three high-energy phosphate bonds. In favor of the cycle was the fact that addition of γ-glutamyl transpeptidase inhibitors in vivo caused a decrease in amino acid transfer into cells. The gamma-glutamyl cycle should be considered not as a mechanism for amino acid transport but rather a generator of extracellular signals, gamma-glutamyl amino acids, that are converted intracellularly to 5-oxoproline, which activates uptake and/or metabolism of amino acids (1, 74, 75). The γ-glutamyl amino acids or oxoproline could be signaling molecules to activate the transport of amino acids through membranes. Oxoproline catalytically activates the uptake of amino acids through the placental barrier, and the transfer of amino acids through the blood–brain barrier is activated by oxoproline (2, 75). Thus, the γ-glutamyl cycle, apart from explaining the synthesis and degradation of glutathione, may serve as a generator of signals to activate amino acid transport into cells (2, 75). GSH turnover may be considered as a multi-organ process. In fact, in liver, an organ in which glutathione synthesis is most active, the degradation is very slow due to the very low activity of γ-glutamyl transpeptidase. In the kidney, however, γ-glutamyl transpeptidase is very high. Thus, the γ-glutamyl cycle may be considered as a multi-organ cycle in which the synthetic part occurs in liver and the catabolic part occurs in kidney amongst other tissues.
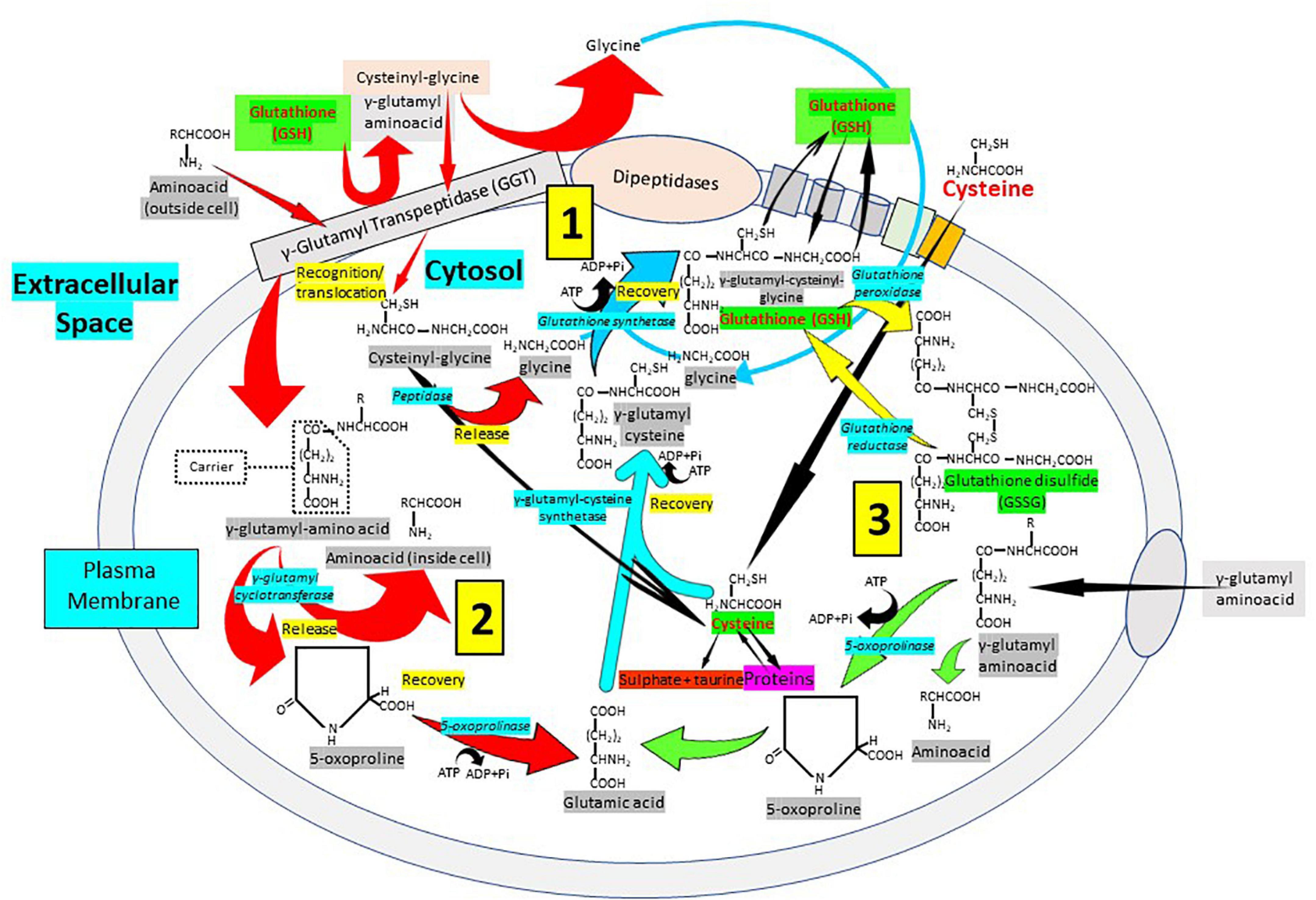
Figure 5. Cellular glutathione synthesis and recycling: The importance of the γ-glutamyl pathway. The degradation or catabolic part of the GSH cycle, takes place partially extracellularly and partially inside cells. (1) The extracellular degradation of GSH occurs on the surface of the cells that express the enzyme γ-glutamyl transpeptidase (GGT) and the dipeptidases found in the external plasma membrane. Following plasma membrane carrier-mediated GSH release from the cell, GSH becomes accessible to the active site of γ-glutamyl transpeptidase, which catalyzes GSH breakdown into γ-glutamyl fraction and cysteinyl-glycine by transferring the γ-glutamyl fraction to an amino acid acceptor, forming γ-glutamyl-amino acid. The cysteinyl-glycine fraction is split by the enzyme dipeptidase generating cysteine and glycine. (2) The γ-glutamyl-amino acid can be metabolized to release the amino acid and 5-oxoproline, which can then be converted into glutamate to be used in the synthesis of GSH. (3) The cells incorporate cysteine and most of the intracellular cysteine is used for the synthesis of GSH. Cysteine can be used for protein synthesis and part can be degraded to sulfate and taurine. The cycle γ-glutamyl allows GSH to be used as a continuous source of cysteine. The γ-glutamyl amino acid is taken up by cells through a specific transport mechanism. Cysteinyl glycine is also taken up by cells. Inside the cell, the γ-glutamyl amino acid is hydrolyzed by γ-glutamyl cyclo-transferase and converted into oxoproline, a cyclic form of glutamate converted into glutamate via oxoprolinase, and a free amino acid.
Glutathione and damaging inflammation in lower respiratory diseases
Glutathione Samsonian (mighty) power is centered in the thiol (sulfhydryl) group of the cysteine amino acid. GSH participates in numerous key processes where the thiol reducing potential is utilized. Several lung disorders are believed to be characterized by an increase in alveolar oxidant burden, potentially depleting alveolar and lung GSH. Low GSH has been linked to abnormalities in the lung surfactant system and the interaction between GSH and antiproteases in the epithelial lining fluid of patients. Normal levels of intracellular GSH may exert a critical negative control on the elaboration of proinflammatory cytokines. The increase of intracellular ROS is believed to correlate with the activation of nuclear factor (NF)-kappa B, a transcription activator linked to the elaboration of several cytokines (Figure 6). There is now sufficient data to strongly implicate free radical injury in the genesis and maintenance of several lung disorders in humans. This information is substantial and will help the development of clinical studies examining a variety of inflammatory lung disorders.
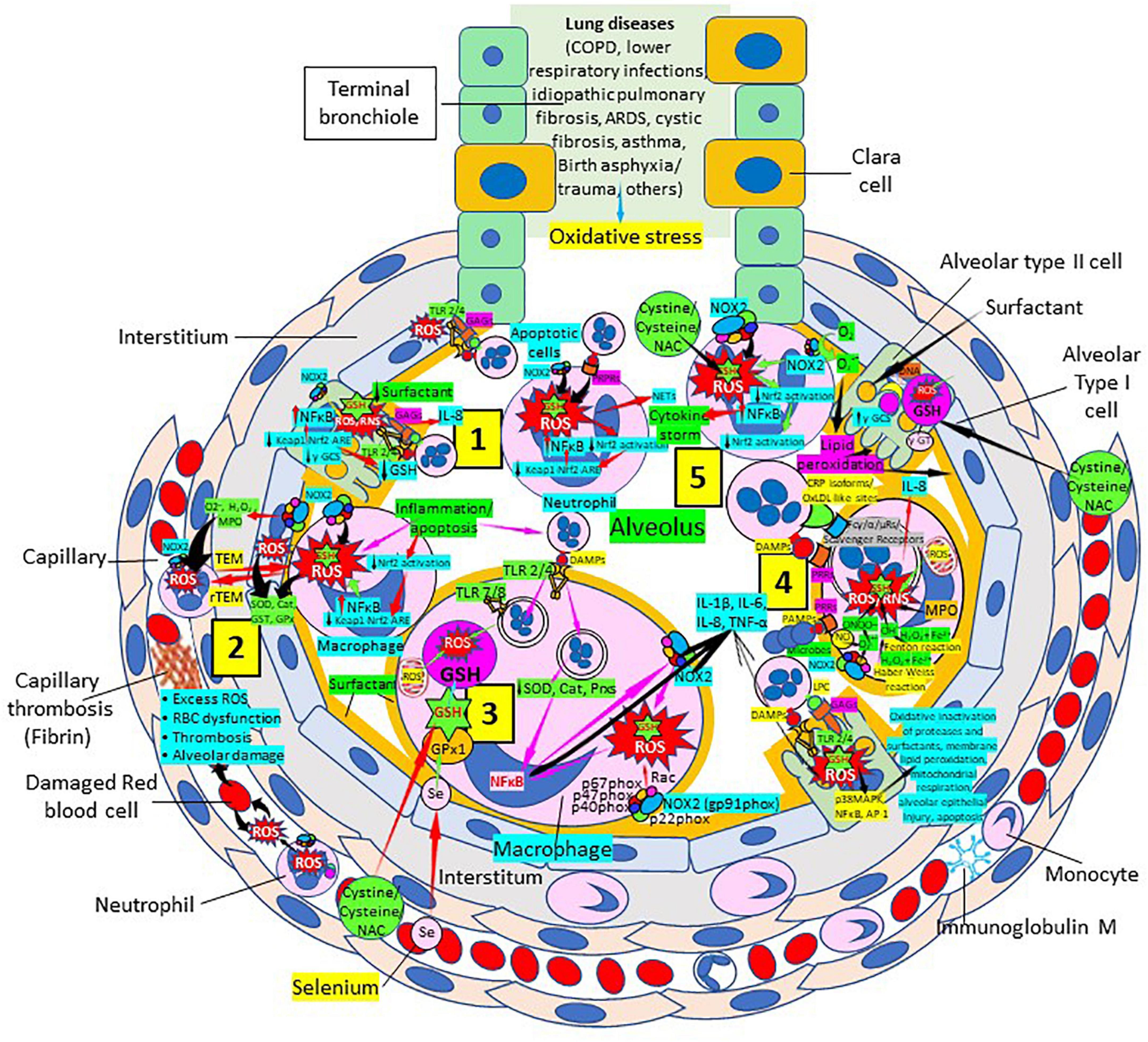
Figure 6. Oxidative stress, reduced glutathione (GSH) and lung diseases. (1) Lung diseases affect alveolar cells increasing reactive oxygen species (ROS) production, reduce Kelch-like ECH-associated protein 1 (Keap1)-Nuclear factor erythroid 2-related factor 2 (Nrf2)-antioxidant response element (ARE) redox regulator pathway and become defective for surfactant production. Damaged/apoptotic cells cause alveolar cell activation of nuclear factor (NF)-κB and release cytokines like interleukin (IL)-8. Alveolar type I cells augment ROS production via toll-like receptors (TLRs) 1 and 2. Inflammation enhances neutrophil extracellular trap (NET) release and increases ROS production. (2) Inflammation associated to lung diseases augments macrophage’s ROS production, inhibiting Nrf2 activation and enhancing NF-κB upregulation. ROS are counterbalanced by enzymes like superoxide dismutase (SOD), catalase (Cat), glutathione S-transferase (GST), and glutathione peroxidase (GPx) to protect cells from oxidative damage caused by nicotinamide adenine-dinucleotide phosphate (NADPH) oxidase 2 (NOX2), superoxide (O2–), hydrogen peroxide (H2O2), and myeloperoxidase (MPO). Capillary neutrophils migrate to and from alveoli by trans-endothelial (TEM) and reverse transmigration (rTEM), respectively. Inflammation can cause excessive ROS production in capillaries, red blood cell (RBC) dysfunction, thrombosis and alveolar damage. (3) Activated alveolar macrophages release increased levels of IL-1β, IL-6, IL-8, and tumor necrosis factor (TNF)-α. Inflammation-associated activated macrophages (via TLRs) reduce enzymes like SOD and Cat, among others, and activate NF-κB. NOX2 activation increases ROS production that enhance NF-κB activation. Glutathione (GSH) precursors (Cystine, cysteine, N-acetyl cysteine, NAC), and selenium (Se) restore GSH and GPx, respectively, to counteract the effects of ROS. (4) Alveolar macrophages engulf microbes and apoptotic cells via Fc (γ/α/μ) and scavenger receptors and/or pattern recognition protein receptors (PRPRs) leading to increased ROS production and cytokine release. MPO, nitric oxide (NO), O2–, and H2O2 through the Fenton and Haber-Weiss reactions that generate hydroxyl radicals, participate in ROS and RNS generation. Lung disease-associated inflammation and apoptosis [via TLRs and glycosaminoglycans (GAGs)] enhance alveolar cell ROS production that via p38MAPK, NF- κB, and AP-1 activation, contribute to epithelial injury and further inflammation. (5) Neutrophils contribute to O2– production, lipid peroxidation and increased oxidative stress to promote a cytokine storm (249). Administration of GSH precursors [cystine, cysteine, NAC; see (3), (4), and (5)] facilitate GSH formation to reduce oxidative stress. Abbreviations: PRRs, pattern recognition receptors; ɣ-GCS, ɣ-glutamyl cysteine synthetase; DAMPs, damage associated molecular patterns; Prxs, peroxiredoxins; NAC, N-acetyl cysteine; ɣ-GT, ɣ-glutamyl transpeptidase; PAMPs, pathogen associated molecular patterns; LPC, lysophosphatidylcholine.
Oxidative stress and inflammation are considered fundamental mediators of chronic obstructive pulmonary disease (COPD) pathophysiology (77–91). The lungs are directly exposed to tobacco smoke and air pollutants that are main sources of ROS. ROS directly cause lung damage as a result of DNA, lipid, carbohydrate, and protein alterations, and activate local inflammatory responses that contribute to COPD development and progression (79–83). ROS can further activate epithelial cells and macrophages facilitating neutrophil, monocyte, and lymphocyte recruitment, and the recruited activated inflammatory cells subsequently enhance additional ROS generation, increasing the pro-oxidant burden (80–85). ROS and RNS production are facilitated by pattern recognition receptors [C-reactive protein (CRP), toll-like receptors] capable of recognizing pathogen (bacterial/viral)-associated molecular patterns and/or damage-associated molecular patterns in apoptotic or damaged cells (92–100) (Figure 6). The phosphocholine head group in phospholipids of normal healthy cell membranes is not accessible but, when cells are damaged and die, enhanced availability of lysophosphatidylcholine and disruption of the lipid bilayer expose phosphocholine residues to which CRP avidly binds (99). These events lead to a state of persistent inflammation and chronic oxidative stress (82–85), characterized by increased ROS production, reduced GSH peroxidase activity, selenium deficiency and reduced GSH levels (80–91). Asymptomatic smokers and stable COPD patients showed increased GSH levels in bronchoalveolar lavage, while patients with severe/very severe exacerbation periods had significantly decreased levels (101). Patients with decreased GSH and increased oxidative stress also showed increased neutrophil influx and IL-8 levels (101). Alveolar macrophages derived from circulating monocytes recruited into the lungs by monocyte chemotactic factors produced by lung cells are increased 20-fold in COPD patients and release ROS as superoxide anions and hydrogen peroxide (102). Antioxidant therapies should be effective in preventing COPD disease progression and exacerbations. Although prolonged treatment with oral N-acetylcysteine (NAC) prevents acute exacerbations of chronic bronchitis, it remains controversial for the treatment of COPD (91, 103–105). A combination of antioxidants including thiol-based antioxidants, mitochondria-targeted antioxidants and Nrf2 activators should be more effective in the treatment of COPD patients (91).
In acute respiratory distress syndrome (ARDS), there is extensive overproduction of free radicals and reduced extracellular and intracellular GSH leading to oxidative cell damage (106). ROS such as hydrogen peroxide and hypochlorous acid may play a key role in the pathogenesis of the acute lung injury (107). It has been shown that alveolar epithelial lining fluid of patients with ARDS is deficient in total GSH compared to normal subjects (107), and neutrophil-mediated oxidants release leads to GSH deficiency and lung cell injury (107). The global antioxidant capacity of the epithelial lining fluid, despite an increase in single antioxidant compounds, seems unable to fully counterbalance the increased oxidative burden (108). NAC benefited ARDS patients as evidenced by intracellular (inside red blood cells) and extracellular (plasma) antioxidant defense biomarkers and outcome. NAC treatment increased extracellular total antioxidant power and total thiol molecules and enhanced intracellular GSH and patients’ outcome (106). NAC treatment improved oxygenation and decreased mortality in ARDS patients; and patients with glutathione-S-transferase M1 (proinflammatory-cytokine producer macrophages) null and M1 and T1 (Type 1 helper CD4 + lymphocytes) double null polymorphisms had increased mortality suggesting that antioxidant therapy becomes fundamental for those patients (109). A depressed antioxidant defense and dysfunctional iron regulation in ARDS might cause greater inflammation and anemia (110).
Glutathione is an important antioxidant in the lungs, but its concentration is low in the airways of patients with cystic fibrosis, since GSH is transported into the airways by the cystic fibrosis transmembrane conductance regulator, which is mutated in cystic fibrosis patients (111). The concentration of GSH that is normally about 400 μM in the epithelial lining fluid, over a 100-fold higher than in plasma, is low in the airways of patients with cystic fibrosis from an early age (112–114). Extracellular glutathione S-transferase omega-1, a cytosolic enzyme that modulates the S-thiolation status of intracellular factors involved in the inflammatory response, and its polymorphisms have been associated with an increased risk to develop COPD and could have a biological and clinical significance in cystic fibrosis (115). Low GSH, neutrophil infiltration, myeloperoxidase activity and inflammation increase oxidative stress overwhelming the antioxidant defense, and hypochlorous acid mediated GSH oxidation and its attachment to proteins contribute to further GSH deficiency (114). The lack of efficacy of inhaled GSH in patients with cystic fibrosis could be explained by the high concentrations of the GSH-degrading enzyme γ-glutamyltransferase present in lung fluids of those patients (116–123), and then, the use of precursors of GSH synthesis like NAC and cystine could be more effective in the synthesis of GSH (124). Lack of oral GSH supplementation effects upon growth or changes in serum or fecal inflammatory markers in children with cystic fibrosis with pancreatic insufficiency (125) could be probably explained by the inability of the cells to uptake extracellular GSH to be used inside the cells. Decreased GSH content in the apical fluid in cystic fibrosis could be the result of abnormal GSH transport associated with a defective cystic fibrosis transmembrane conductance regulator as mentioned previously (126).
An oxidant/antioxidant imbalance characterized by oxidative stress and low GSH levels is involved in the pathogenesis of idiopathic pulmonary fibrosis, since data show marked GSH deficiency in the lower respiratory tract of those patients (127). Glutathione-S-transferase π (GSTP) that participates in the conjugation of GSH to reactive cysteines (S-glutathionylation) seems to play an important role in idiopathic pulmonary fibrosis lung fibrogenesis, since GSTP immunoreactivity is increased in the lungs of idiopathic pulmonary fibrosis patients, notably within type II epithelial cells (128, 129). GSTP inhibition via the airways may be a novel therapeutic strategy for the treatment of idiopathic pulmonary fibrosis (128, 129). The use of GSH precursors like N-acetyl cysteine, enhancers of nuclear factor erythroid 2-related factor 2 (Nrf2) like sulforaphane, melatonin, and many more molecules involved in antioxidant defense were proposed as supplementation of other idiopathic pulmonary fibrosis therapies (130). Inhaled (nebulized or aerosolized) reduced GSH to augment the deficient GSH levels of the lower respiratory tract has been used effectively in numerous pulmonary diseases and respiratory conditions like HIV seropositive individuals, cystic fibrosis and idiopathic pulmonary fibrosis, among others (131–133). GSH has clearly a regulatory role in inflammation and immunity (134). GSH acts as an inhibitor of extended inflammation directing components of innate immunity like polymorphonuclear neutrophils specifically to the site of infection/damage allowing a proper response to infection. GSH then directs the migration of inflammatory polymorphonuclear neutrophils away from the lung, where they cause ARDS, and toward the site of infection, where they kill microorganisms. As a result, it develops more immunity and less inflammation, with the concomitant increased survival; in addition, GSH becomes not just an inhibitor of inflammation but a regulator of innate immunity in a direction that benefits the host (134).
Glutathione and atherosclerosis
Cardiovascular diseases are the leading causes of death in the US compared to any other cause (135). Cardiovascular complications are thought to result from increased free radical levels that impair redox homeostasis, that represents the interaction between oxidative stress and reductive stress. A prolonged oxidative or reductive stress will alter the homeostatic redox mechanism to cause cardiovascular complications. GSH, the most abundant antioxidant in the heart, plays a fundamental role in normalizing a redox homeostatic mechanism that was shifted toward oxidative or reductive stress. This may lead to impairment of cellular signaling mechanisms and accumulation of misfolded proteins causing proteotoxicity associated with cardiac dysfunction (136–143). Oxidative stress is crucial in atherogenesis (144–151), suggesting that a specific antioxidant/prooxidant imbalance, characterized by a weak GSH-related enzymatic antioxidant shield present in human atherosclerotic lesions, may be involved in atherogenic processes in humans (152). A higher level of oxidative stress as evidenced by elevated plasma malondialdehyde levels and low levels of GSH, α-tocotrienol and GSH peroxidase activity in patients under 45 years old may play a role in the development of premature coronary artery disease and be potential biomarkers for premature coronary artery disease (153). Similarly, coronary artery disease patients with single, double, or triple-vessel stenosis and patients with acute coronary syndrome had a significant increase in malondialdehyde levels and the percentage of malondialdehyde release, associated with a marked decrease in GSH concentration, total antioxidant capacity and erythrocyte GSH peroxidase activity compared with controls (154). Interestingly, differences in prooxidative parameters were more profound in acute coronary syndrome patients compared with coronary artery disease patients indicating that the acute form of coronary artery disease is more susceptible to oxidative damage, suggesting that use of antioxidant therapy may be warranted to reduce oxidative stress in this disorder (154).
Glutathione might inhibit the effects of cerebral infarction and enhance antiapoptotic signaling after ischemic stroke, suggesting that GSH may be a potent therapeutic antioxidant that can attenuate severe pathologies after ischemic stroke, and stimulating GSH synthesis through administration of GSH precursors and micronutrients like selenium can optimize GSH and GSH peroxidase for optimal antioxidant defense in cerebral ischemia (155, 156). Low total GSH and high homocysteine levels are considered as novel risk markers for acute stroke severity, and low total and reduced GSH levels may be potential risk markers for stroke severity and insufficient functional independence in large-artery atherosclerosis (157, 158). Since GSH is the final product of the homocysteine metabolism in the transsulfuration pathway by transferring sulfur from homocysteine to cysteine, a deficiency in transsulfuration pathway leads to excessive homocysteine production (hyperhomocysteinemia) and reduced GSH synthesis (159, 160). Homocysteine is a sulfur-containing amino acid tightly involved in methionine metabolism. Indeed, if there is a methionine deficit, homocysteine can be re-methylated to form methionine, and if there is an adequate amount of methionine, homocysteine is used to produce cysteine (161). Hyperhomocysteinemia decreases GSH peroxidase activity leading to the prevalence of GSSG on GSH with the GSH/GSSG impaired ratio causing some common cardiovascular and neurodegenerative disorders (159). N-acetyl-cysteine administration supplies the cysteine necessary for GSH synthesis and concomitantly reduces hyperhomocysteinemia, improving GSH peroxidase activity and reducing oxidative stress (159). Furthermore, the well documented efficacy of combined folic acid, B6, and B12-vitamin supplementation to reduce hyperhomocysteinemia could enhance GSH activity and reduce oxidative stress (161). Recently, it was shown that cysteine uptake via excitatory amino acid carrier 1 suppresses ischemia-induced neuronal death through promotion of hippocampal GSH synthesis in ischemic animal models (162). Alterations in the normal function of excitatory amino acid carrier 1 affect cysteine transport and GSH synthesis impairing zinc homeostasis (the thiol group of GSH can function as a principal Zn2+ chelator for the maintenance of Zn2+ homeostasis in neurons) and oxidative stress, enhancing susceptibility to ischemia-induced neuronal cell death in the hippocampus (162–164). Increased GSH synthesis neutralizes reactive oxygen and nitrogen species and regulates zinc homeostasis promoting neuroprotection after ischemia/reperfusion (162).
Atherosclerosis represents a state of intense oxidative stress characterized by vascular wall lipid and protein oxidation that contributes to chronic inflammation within the arterial wall, in which CRP is a major player (Figure 7). The balance of the different CRP isoforms, monomeric (mCRP) or native pentameric (nCRP) within the plaque determines the preponderance of a proinflammatory or anti-inflammatory effect, respectively (165). CRP is synthesized in smooth muscle cells of atherosclerotic lesions with active disease, foam cells, macrophages, lymphocytes, monocytes, and endothelial cells within the atherosclerotic plaque (166–170). CRP binds and aggregates oxidized low-density lipoprotein (ox-LDL) and enhances macrophage oxLDL uptake, promoting mitogen-activated protein kinase activation (171) required for foam cell formation (172). OxLDL enhances toll like receptor 4 expression further facilitating foam cell formation and development and progression of atherosclerosis (173, 174). CRP binding to oxLDL and apoptotic cells occurs through phosphorylcholine, and binding to this ligand starts phagocytosis (100, 170, 175–181). The different CRP isoforms, nCRP, non-native pentameric CRP (nnCRP) and mCRP (175–178, 181–184), may explain their protective and destructive effects, with nCRP being primarily antiinflammatory inducing Th2/M2 responses, while mCRP being typically proinflammatory inducing Th1/M1 responses (170, 185–189). Pentameric nCRP and CRP peptides 77–82, 174–185, and 201–206 can control the inflammatory response resolving inflammation by reducing inflammatory cell endothelial adhesion and tissue migration, and the described CRP-mediated enhanced monocyte chemotaxis could be explained by local generation of mCRP (190, 191). Pro-inflammatory and proatherogenic mCRP, but not nCRP, induces ROS monocyte/macrophage production and facilitates macrophage uptake of necrotic cells (170, 192) contributing to foam cell formation, atherosclerotic plaque formation and plaque rupture or destabilization (190, 191). Foam cell formation during atherogenesis could be also explained in part by uptake of CRP-opsonized native LDL (193). The dissociation/relaxation of nCRP into nnCRP occurs on necrotic, apoptotic, and ischemic cells, membranes of activated platelets, monocytes, and endothelial cells, and on the surface of microparticles, via phosphorylcholine binding and seems to be, as mCRP, proinflammatory (194–196). Pentameric nCRP does not possess intrinsic proinflammatory properties, while nnCRP and mCRP do (170, 196). The mCRP isoform, unlike nCRP, has a stimulatory effect on platelets, facilitates thrombus growth through platelet stimulation, and is the more potent reagent, both increasing monocyte activation and ROS production, generated through myeloperoxidase-mediated respiratory burst and raft-associated reduced nicotinamide adenine dinucleotide phosphate (NADPH)-oxidase during oxLDL-mediated foam cell formation (100, 170, 197–204). ROS activity in the vessel wall contributes to the formation of oxidized LDL, a major contributor to the pathogenesis of atherosclerosis (205, 206). Thrombus formation and the subsequent activation of the coagulation cascade with final generation of fibrin is facilitated by the mCRP-mediated enhancement of tissue factor on the endothelial cell surface, platelet aggregation and thrombus growth (207, 208) (Figure 7). OxLDL components and their interaction with toll-like receptors (TLRs) 2 and 4, CD36 and other cellular receptors further mediate thromboinflammation enhancing tissue and organ damage culminating in organ failure, i.e., myocardial infarction, stroke, and pulmonary artery embolism (174, 209–212).
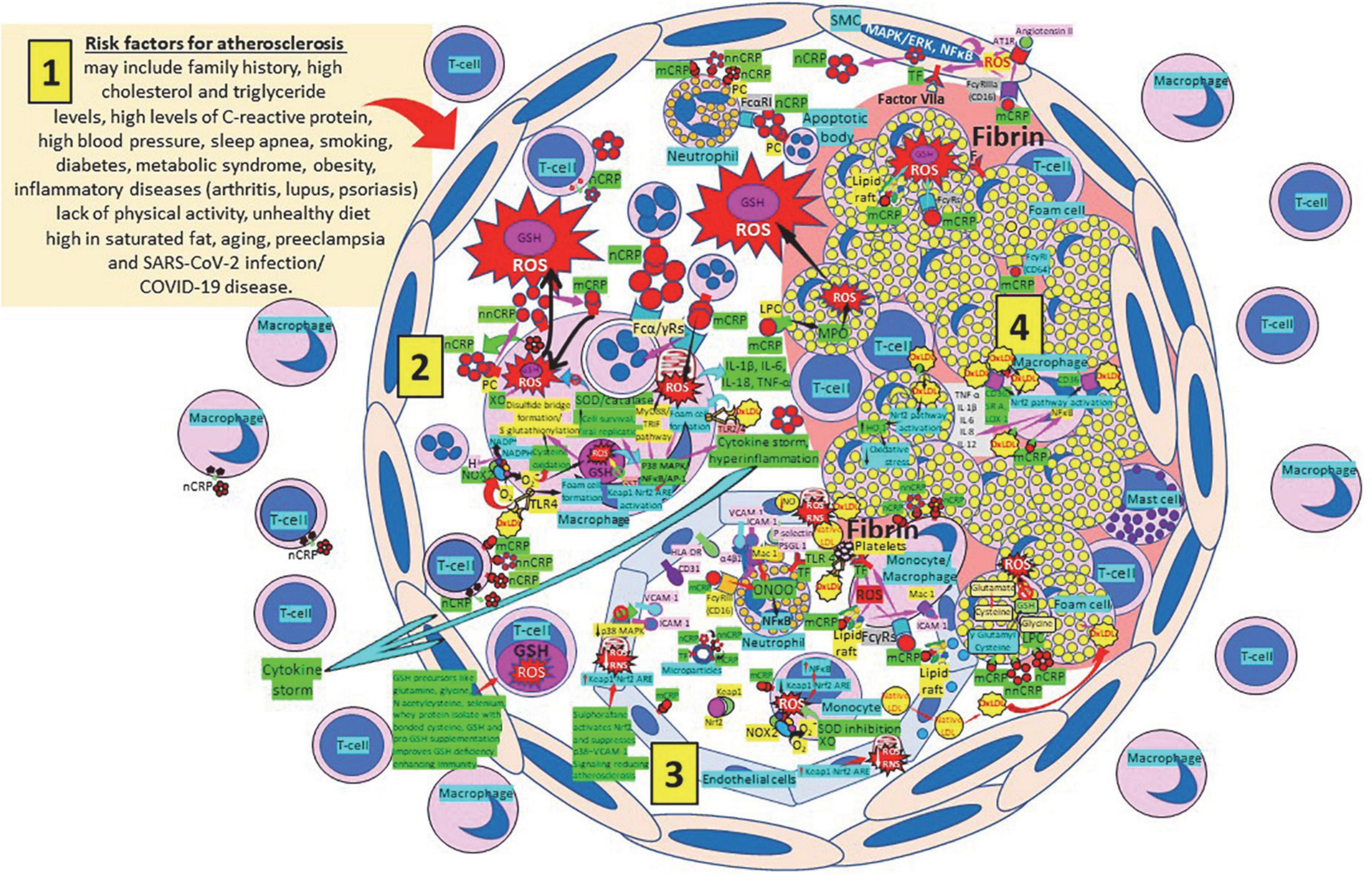
Figure 7. Oxidative stress, reduced glutathione (GSH) and atherosclerosis. (1) Risk factors for atherosclerosis. (2) Atherosclerosis risk factors facilitate oxidative stress and inflammation in the arterial intima. Native C-reactive protein (nCRP), a pattern recognition receptor produced in the liver, macrophages, lymphocytes, smooth muscle cells (SMC), and other cells, promotes inflammation through monomeric CRP (mCRP) enhancing intimal oxidative stress. Oxidized (ox) LDL binds macrophage toll-like receptor (TLR) 4 and facilitates nicotinamide adenine dinucleotide phosphate (NADP)H oxidase 2 (Nox2) activity and superoxide (O2–) production causing cysteine oxidation, disulfide bridge formation and S-glutathionylation. Xanthine oxidase (XO) and inhibition of superoxide dismutase (SOD)/catalase further facilitate O2– cellular activity. OxLDL bound to TLRs 2 and 4 promotes foam cell formation and activates transcription factors like nuclear factor (NF)-κB facilitating cytokine storm and hyperinflammation. Excessive mitochondrial reactive oxygen species (ROS) generation further enhances cytokine production. CRP (nCRP, mCRP) can facilitate macrophage and neutrophil uptake of apoptotic cells through Fcγ and Fcα receptors, respectively (FcRs). Oxidative stress also activates the Kelch-like ECH-associated protein 1 (Keap1)-Nuclear factor erythroid 2-related factor 2 (Nrf2)-antioxidant response element (ARE) redox regulator pathway in monocytes [see (3) and macrophages (2)], releasing Nrf2 to regulate the expression of genes that control antioxidant enzymes like glutathione S-transferase (GST), facilitating glutathione (GSH) activity. Macrophages, T-lymphocytes, neutrophils and SMCs can generate mCRP increasing inflammation. (3) Monocytes, macrophages, neutrophils, endothelial cells and microparticles can generate mCRP, increase O2– and ROS formation and reactive nitrogen species like peroxinitrite (ONOO–), and tissue factor (TF) expression enhancing oxidation, inflammation and thrombosis. TLR 4-mediated oxLDL-binding to platelets promotes thrombosis; mCRP binding to lipid rafts and FcγRs enhances inflammation; and endothelial activation allows intimal cell migration. GSH enhancement and Nrf2 activation augment immunity and reduce atherosclerosis. (4) Foam cells and smooth muscle cells associated with atherosclerotic plaques enhance ROS formation, cytokine release and tissue factor (TF)-mediated fibrin deposition. Abbreviations: MAPK/ERK, mitogen-activated protein kinases/extracellular signal-regulated kinases; AT1R, angiotensin II type 1 receptor; PC, phosphorylcholine; LPC, lysophosphatidylcholine; MPO, myeloperoxidase; nnCRP, non-native CRP; TNF, tumor necrosis factor; IL, interleukin; ACE, angiotensin converting enzyme; MyD88/TRIF, myeloid differentiation primary response 88/TIR-domain-containing adapter-inducing interferon-β; PI3K/Akt, phosphatidylinositol-3-kinase/protein kinase B; MAPK, mitogen-activated protein kinase; AP-1, activator protein 1; CD31, cluster of differentiation 31; ICAM-1, intercellular adhesion molecule-1; Mac-1, macrophage-1 antigen; PSGL-1, P-selectin glycoprotein ligand-1; HLA-DR, human leukocyte antigen–DR isotype.
The strong role of severe oxidative stress, reduced antioxidant defenses like GSH with increased lipid peroxidation and malondialdehyde generation (213), lipid, protein and DNA oxidation with increased apoptosis and necrosis in atherosclerosis as a major cause of cardiovascular diseases and stroke, supports the use of complementary and alternative medicines, dietary supplements, and antioxidants with hardly any adverse effect, able to restore homeostasis reversing oxidative stress (214). Enhancing GSH synthesis, selenium levels and redox-active selenoproteins, and activating Nrf2 and other antioxidant enzymes will strengthen the cardiovascular antioxidant defense. Phenolic compounds like phenolic acids, flavonoids, lignans and tannins can limit LDL oxidation and foam cell formation (215). Selenium is an essential micronutrient that modulates cardiovascular functions via its incorporation into selenoproteins as the amino acid selenocysteine (216, 217). Intravenous reduced GSH supplementation reverses endothelial dysfunction in patients with atherosclerosis enhancing NO activity and NO-mediated vasodilation (218). GSH stores and transports cysteine, and cysteine forms less diffusion-limited NO adducts that may transport NO to reach sites within vascular smooth muscle cells and platelets (218, 219). Since GSH is not carried inside the cell, exogenously administered GSH is most likely to act by increasing plasma GSH levels reducing luminal oxidative stress and increasing NO bioavailability in patients with endothelial dysfunction (220).
Administration of GSH precursors like cysteine/N-acetylcysteine, glycine and/or glutamic acid will facilitate the synthesis of GSH within each cell of the body including the atherosclerotic plaque, reducing ROS and LDL oxidation, enhancing NO production, and mitigating atherosclerosis and its complications (221–229). Considering the paramount importance of oxLDL in the pathogenesis of atherosclerosis, it is reasonable to evaluate the role of antioxidants in the treatment of the disease as adjuvant strategies to lipid-lowering or anti-inflammatory therapies designed to reduce the risk of cardiovascular disease (230). Since oxidation participates as an essential messenger of cellular signaling pathways, treatment of oxidative stress needs to consider maintaining that physiologic threshold (230, 231). The lack of standardized methods to evaluate total antioxidant capacity and the oxidation state and the use of inadequate antioxidants and/or improper concentrations of antioxidants lead to failure of numerous clinical trials directed to prevent or mitigate progression of atherosclerosis (230–233).
Nuclear factor erythroid 2-related factor 2 plays a fundamental role in the response to oxidative stress and xenobiotic metabolism and detoxification, and the Nrf2 signaling pathway is intimately associated with development of atherosclerosis. During development and progression of atherosclerosis, Nrf2 signaling modulates many physiological and pathophysiological processes, like regulation of lipid homeostasis, CD36 gene expression regulation, foam cell formation, macrophage polarization, immunity regulation (Th2 differentiation and inhibition of pro-inflammatory gene expression through NFκB down-regulation), redox regulation and inflammation, improvement of endothelial dysfunction, as well as GSH synthesis and utilization (234–245). Antioxidant pathways induced by NRF2 include enzymes for the reduced GSH synthesis, utilization, and regeneration. Glutamate-cysteine ligase catalytic and modulator subunits as well as GSH synthetase are the three NRF2 targets involved in the GSH synthesis (242). The redox cycling enzymes thioredoxin, thioredoxin reductase, sulfiredoxin, peroxiredoxin, GSH peroxidase, superoxide dismutase 1, and catalase, and several GSH S-transferases, which are the enzymes mediating the elimination of ROS, are all Nrf2 targets (242). Nrf2 displays both pro- and anti-atherogenic effects in experimental animal models, and the Nrf2 pathway becomes a promising target for atherosclerosis prevention (234). Macrophage Nrf2 activates genes encoding CD36, heme oxygenase-1 and other stress proteins in response to oxLDLs and other byproducts of lipid peroxidation (240). Nrf2 depletion in macrophages leads to increased foam cell formation, increases the M1 inflammatory phenotype with enhanced expression of pro-inflammatory monocyte chemoattractant protein-1 and interleukin-6, and aggravates atherosclerosis (244, 245). Nrf2 improves endothelial function by resisting oxidative stress and mitochondrial damage, thereby delaying atherosclerosis (245); and treatment with sulforaphane, a dietary antioxidant, activates Nrf2 and suppresses p38–VCAM-1 signaling, and may provide a novel therapeutic strategy to prevent or reduce atherosclerosis (237).
Glutathione and severe acute respiratory syndrome coronavirus 2
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection targets primarily the respiratory and cardiovascular systems causing COVID-19 disease identified largely by a respiratory tract infection (246, 247); sadly, many patients develop severe fatal outcomes because of overwhelming inflammation known as “cytokine storm” (248, 249), that leads to ROS-mediated cell death and tissue damage typical of RNA viruses (250). This intense inflammation is associated with damaging systemic events like oxidative stress, dysregulation of iron homeostasis, hypercoagulability and thrombus formation, acute respiratory distress syndrome, uncontrolled inflammation and organ failure (251–257) (Figure 8). Several viral infections, and the progression of virus-induced diseases, especially those associated with COVID-19, are characterized by an alteration in the intracellular redox balance (6). This imbalance disallows reactive intermediate detoxification by the cell biological systems. ROS production and associated inflammation are closely related to aging and numerous chronic diseases as diabetes, cardiovascular atherosclerosis-related diseases (144, 145) and respiratory diseases, known risk factors for developing severe illness and death in patients with SARS-CoV-2 and COVID-19 disease.
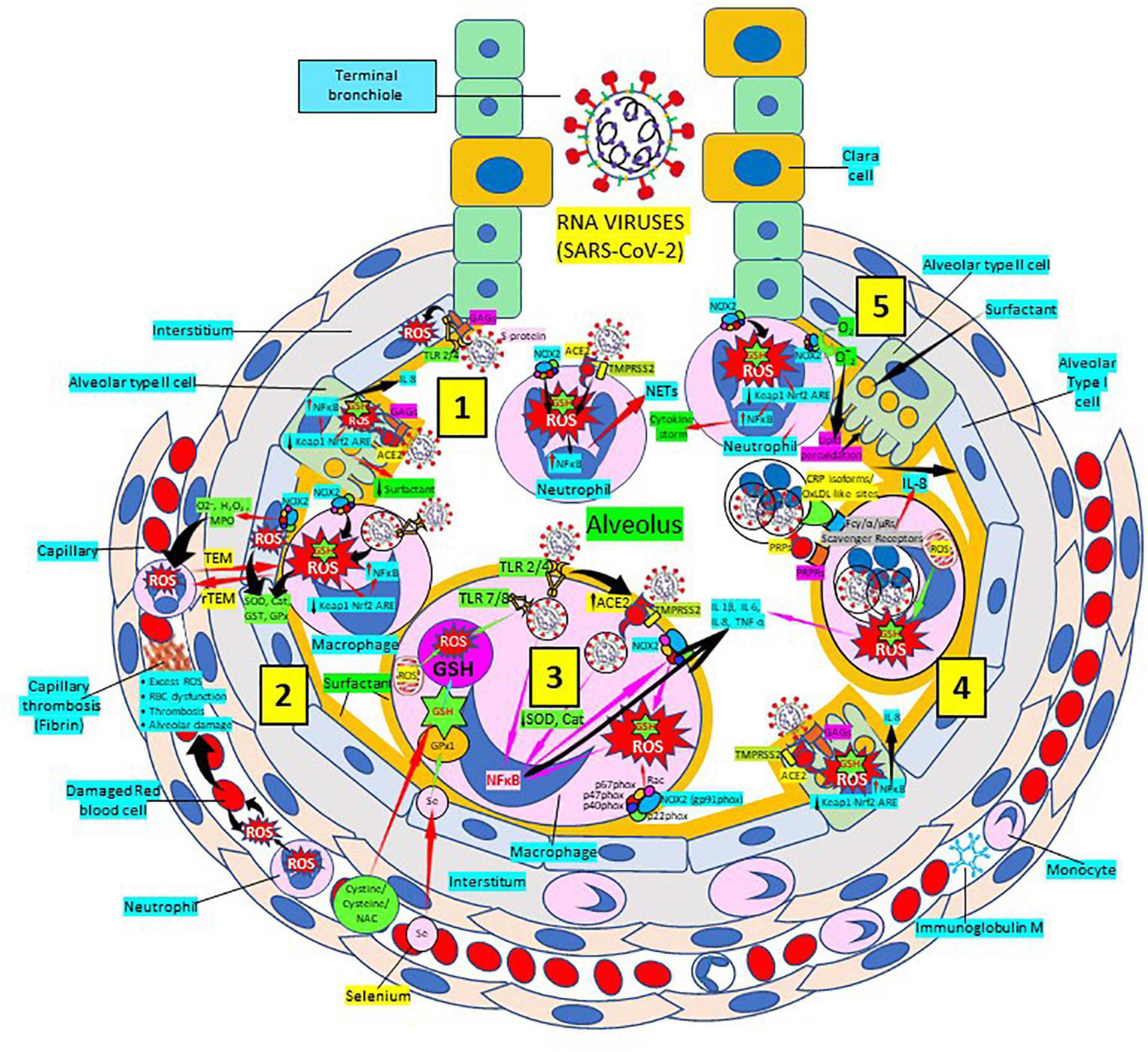
Figure 8. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) pulmonary infection, oxidative stress and antioxidant defenses. (1) After entry of SARS-CoV-2 into the alveolus, viruses invade type II alveolar cells through angiotensin-converting enzyme 2 receptors (ACE2) and glycosaminoglycans (GAGs) [see (4)], and infected cells increase reactive oxygen species (ROS) production, reduce Kelch-like ECH-associated protein 1 (Keap1)-Nuclear factor erythroid 2-related factor 2 (Nrf2)-antioxidant response element (ARE) redox regulator pathway and become defective for surfactant production. Infected cells activate nuclear factor (NF)-κB and release cytokines like interleukin (IL)-8. Alveolar type I cells augment ROS production via toll-like receptors (TLRs) 1 and 2. SARS-CoV-2 enhances neutrophil extracellular trap (NET) release and increases ROS production (2) SARS-CoV-2 augments macrophage’s ROS production, inhibiting Nrf2 activation and enhancing NF-κB upregulation. ROS are counterbalanced by enzymes like superoxide dismutase (SOD), catalase (Cat), glutathione S-transferase (GST), and glutathione peroxidase (GPx) to protect cells from oxidative damage caused by nicotinamide adenine-dinucleotide phosphate (NADPH) oxidase 2 (NOX2), superoxide (O2–), hydrogen peroxide (H2O2), and myeloperoxidase (MPO). Capillary neutrophils migrate to and from alveoli by trans-endothelial (TEM) and reverse transmigration (rTEM), respectively. SARS-CoV-2 infection can cause excessive ROS production in capillaries, red blood cell (RBC) dysfunction, thrombosis and alveolar damage. (3) Activated alveolar macrophages release increased levels of IL-1β, IL-6, IL-8, and tumor necrosis factor (TNF)-α. SARS-CoV-2-infected macrophages (via ACE2 and TLRs) reduce enzymes like SOD and Cat, among others, and activate NF-κB. NOX2 activation increases ROS production that enhance NF-κB activation. Glutathione (GSH) precursors (Cystine, cysteine, N-acetyl cysteine, NAC), and selenium (Se) restore GSH and GPx, respectively, to counteract the effects of ROS. (4) Alveolar macrophages engulf SARS-CoV-2-infected apoptotic cells via Fc (γ/α/μ) and scavenger receptors and/or pattern recognition protein receptors (PRPRs) leading to increased ROS production and cytokine release. (5) Neutrophils contribute to O2– production, lipid peroxidation and increased oxidative stress to promote the cytokine storm. Abbreviations: TMPRSS2, Transmembrane protease Serine 2; PRPs, pattern recognition proteins. Reprinted from Labarrere and Kassab (335).
Atherosclerosis, a chronic inflammatory disease, may be an ideal environment for the high viral replication capabilities of SARS-CoV-2 in human cells, enhancing hyper-inflammation secondary to immune system dysregulation (Figure 9) that leads to adverse outcomes, as shown in patients with cardiovascular risk factors (258, 259). In a vicious circle, feeding itself, SARS-CoV-2 may aggravate the evolution of atherosclerosis as a result of excessive and aberrant plasmatic concentration of cytokines (258–260). Atherosclerosis progression, as a chronic inflammatory mechanism, is characterized by immune system dysregulation associated with increased pro-inflammatory cytokine production, including interleukin 6 (IL-6), tumor necrosis factor-α (TNF-α), and IL-1β, as well as pattern recognition receptor proteins like CRP (170, 261–267). CRP, an active regulator of host innate immunity, is a biomarker of chronic inflammatory conditions and severe COVID-19 disease, including lung and atherosclerotic disease progression; strongly predicts the need for mechanical ventilation; and may guide intensification of treatment of COVID-19-associated uncontrolled inflammation (99, 183, 261, 262, 265, 267–269). Macrophage activation and foam cell formation may explain the elevated CRP serum levels and contribute to disease progression (Figure 9). CRP-mediated inflammation in atherosclerosis during SARS-CoV-2 infection may be related to the presence of mCRP in the lesions (188, 198, 204, 263, 267–269). The affinity of SARS-CoV-2 for ACE2 receptors makes the virus prone to cause vascular infection that could explain atherosclerosis progression and arterial and venous thrombosis (270, 271). Endothelial injury generated directly by intracellular viral replication and by ACE2 downregulation, exposing cells to angiotensin II in the absence of the modulator effects of angiotensin 1–7 (270, 271), and vascular chronic inflammation promoting the development of tissue macrophages overloaded by cholesterol (foam cells), both increase the possibility of acquiring a severe COVID-19 infection (170, 209, 258, 259, 272, 273).
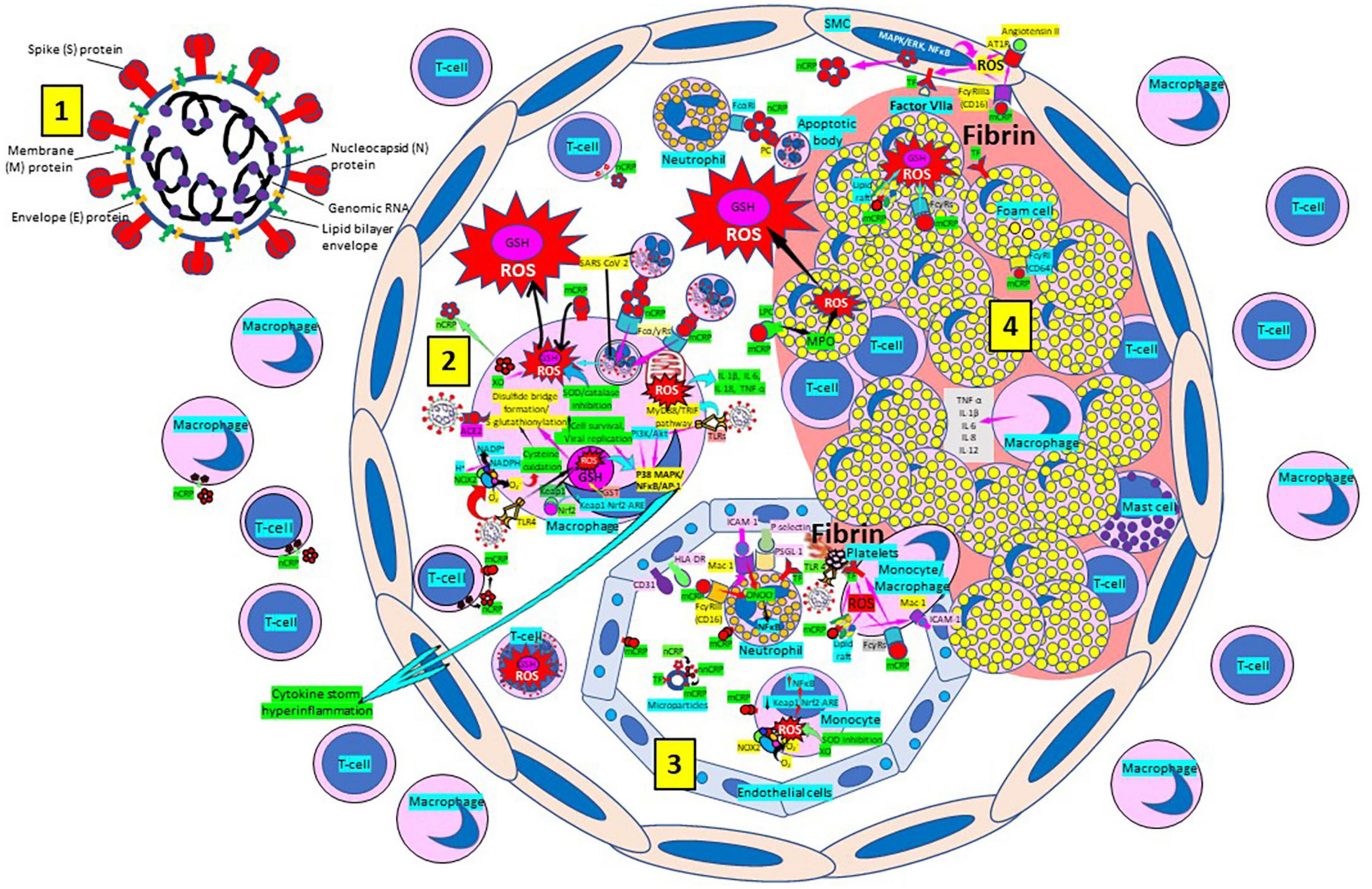
Figure 9. Severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) enhances oxidative stress and atherosclerosis progression. (1) SARS-CoV-2 structure. (2) SARS-CoV-2 viruses facilitate oxidative stress and inflammation in the arterial intima. Native C-reactive protein (nCRP), a marker of severe SARS-CoV-2 produced in liver, macrophages, lymphocytes, smooth muscle cells (SMC) and other cells, promotes inflammation through monomeric CRP (mCRP) enhancing intimal oxidative stress. SARS-CoV-2 binds macrophage toll-like receptor (TLR) 4 and facilitates nicotinamide adenine dinucleotide phosphate (NADP)H oxidase 2 (Nox2) activity and superoxide (O2–) production causing cysteine oxidation, disulfide bridge formation and S-glutathionylation. Xanthine oxidase (XO) and inhibition of superoxide dismutase (SOD)/catalase further facilitate O2– cellular activity. SARS-CoV-2 can bind TLRs 2 and 4 and activate transcription factors like nuclear factor (NF)-κB facilitating cytokine storm and hyperinflammation. Excessive mitochondrial reactive oxygen species (ROS) generation further enhances cytokine production. CRP (nCRP, mCRP) can facilitate macrophage and neutrophil uptake of SARS-CoV-2-infected apoptotic cells through Fcγ and Fcα receptors, respectively (FcRs). Oxidative stress also activates the Kelch-like ECH-associated protein 1 (Keap1)-Nuclear factor erythroid 2-related factor 2 (Nrf2)-antioxidant response element (ARE) redox regulator pathway in monocytes [see (3) and macrophages (2)], releasing Nrf2 to regulate the expression of genes that control antioxidant enzymes like glutathione S-transferase (GST), facilitating glutathione (GSH) activity. Macrophages, T-lymphocytes, neutrophils and SMCs can generate mCRP increasing inflammation. (3) Monocytes, macrophages, neutrophils, endothelial cells and microparticles can generate mCRP, increase superoxide (O2–) and ROS formation and reactive nitrogen species like peroxinitrite (ONOO–), and tissue factor (TF) expression enhancing oxidation, inflammation and thrombosis. TLR 4-mediated SARS-CoV-2-binding to platelets promotes thrombosis, mCRP binding to lipid rafts and FcγRs enhances inflammation and endothelial activation allows intimal cell migration. (4) Foam cells and smooth muscle cells associated with atherosclerotic plaques enhance ROS formation, cytokine release and tissue factor (TF)-mediated fibrin deposition. Abbreviations: MAPK/ERK, mitogen-activated protein kinases/extracellular signal-regulated kinases; AT1R, angiotensin II type 1 receptor; PC, phosphorylcholine; LPC, lysophosphatidylcholine; MPO, myeloperoxidase; nnCRP, non-native CRP; TNF, tumor necrosis factor; IL, interleukin; ACE, angiotensin converting enzyme; MyD88/TRIF, myeloid differentiation primary response 88/TIR-domain-containing adapter-inducing interferon-β; PI3K/Akt: phosphatidylinositol-3-kinase/protein kinase B; MAPK, mitogen-activated protein kinase; AP-1, activator protein 1; CD31, cluster of differentiation 31; ICAM-1, intercellular adhesion molecule-1; Mac-1, macrophage-1 antigen; PSGL-1, P-selectin glycoprotein ligand-1; HLA-DR, human leukocyte antigen–DR isotype. Reprinted from Labarrere and Kassab (335).
Summary and conclusions: Glutathione and early (premature) inflammaging
Chronic inflammatory diseases especially those compromising the lower respiratory system (chronic obstructive pulmonary disease, lower respiratory infections, cystic fibrosis, idiopathic pulmonary fibrosis, acute respiratory distress syndrome), diseases compromising the cardiovascular system (atherosclerosis, ischemic heart disease, stroke, and others), many other systemic inflammatory diseases like diabetes (274, 275); and actually SARS-CoV-2 causing COVID-19, all are characterized by persistent inflammation, continuous production of reactive oxygen/nitrogen species and oxidative stress that predominate over the antioxidant defenses (GSH and free radical scavenger enzymes) resulting in cell/tissue/organ aging associated with early (premature) chronic inflammation/”inflammaging.” We propose that, although inflammaging was introduced for the aging process (276–289), it could also apply to early (premature) tissue and organ aging associated with cell and tissue damage caused by excessive oxidative stress and lack of adequate antioxidant defenses, especially low GSH levels, in younger individuals. Indeed, inflammaging is associated with cell ROS over-production leading to oxidation/damage of cellular components, enhanced inflammation, and activation of cell death pathways; and oxidative stress and reduced antioxidant defenses contribute to progression of practically all diseases (290–297). In most diseases, ROS appear to have a direct connection with inflammaging and cell senescence, and oxidative stress and inflammaging increase the aging-related phenotype, and induce and aggravate the inflammatory response, creating a chronic state of systemic inflammation (290–297). Then, as proposed above, inflammaging can also be involved in aged cell/tissue processes in younger individuals. All chronic diseases, including COVID-19 with the long-COVID-19-syndrome (271), are characterized by the presence of persistent chronic inflammation and sustained generation of reactive oxygen and nitrogen species that when confronted with inadequate antioxidant defenses (likely leading components of anti-inflammaging) precipitate excessive oxidative stress. The demand for detailed analysis of the pathogenesis and clinical course of chronic diseases and viral diseases like COVID-19, as well as the use of efficacious therapies with minimal or no side effects are paramount. Here we present the antioxidant GSH as a potential unexplored way for further investigation as intervention to counteract inflammaging, premature inflammaging, inflammatory diseases and long-COVID-19-syndrome, since GSH levels are correlated with tissue and organ damage, disease severity and progression, and disease outcome (294–296, 298, 299). Enhancing GSH, mainly through NAC, GSH precursors rich in cysteine (whey protein, whey protein isolate rich in cysteine) or pro-GSH compound administration, becomes a potential treatment option for inflammatory diseases by reducing oxidative stress and cytokine expression especially in diabetic patients that also are at risk of more severe COVID-19 disease (299). GSH dysregulation might cause global immune cell autophagy decline with increased generation of proinflammatory cytokines in aging, further provoked by mitochondrial ROS signaling (293). Whey protein concentrate ameliorates lung damage and inhibits lung furin activity targeting SARS-CoV-2 S1/S2 site cleavage and SARS CoV-2 spike protein-angiotensin converting enzyme binding and could be used to protect against COVID-19 inhibiting SARS-CoV-2 cell entry (300). Glutamine, glycine, N-acetylcysteine, selenium, whey protein isolates with bonded cysteine, GSH and pro-GSH supplementation improves GSH deficiency, oxidative stress, mitochondrial dysfunction, inflammation, insulin resistance, endothelial dysfunction, genotoxicity, muscle strength, cognition and surfactant regeneration (301–307). A combination of vitamin D and L-cysteine administration significantly augmented GSH levels and lowered oxidative stress and inflammation (308, 309). Maintaining an adequate GSH redox status and 25-hydroxy-vitamin D levels will have the potential to reduce oxidative stress, enhance immunity and diminish the adverse clinical consequences of COVID-19 especially in African American communities having glucose-6-phosphate dehydrogenase (G6PD) deficiency, enzyme necessary to prevent GSH exhaustion and depletion (6, 213, 310). In normal red blood cells, pentose phosphate pathway and glycolysis are enhanced and G6PD is sufficient to produce NADPH efficiently for GSSG reduction and maintenance of GSH pool (311). G6PD-deficient cells are unable to generate enough NADPH under the condition of severe thiol depletion and GSH biosynthesis and methionine cycle are upregulated at the expense of ATP but fail to compensate for GSH depletion (311).
Severe acute respiratory syndrome coronavirus 2 can sequester mitochondria and replicate within them aging those vital organelles weakening immunity; facilitating over-stimulated or sustained inflammatory responses with interferon and cytokine release, influencing ROS production, iron storage, platelet coagulability, cytokine production stimulation, regulation of fission and fusion, mitochondrial biogenesis, and interference of apoptosis and mitophagy (312–319). By affecting all these cellular functions already impaired in aging individuals it could explain why older, comorbid patients have the most severe outcomes with COVID-19 (312) and stimulate the use of GSH and Nrf2 enhancers as well as develop new therapies to protect mitochondria. We propose that enhancement of the reduced form of GSH will reduce the body’s oxidation and inflammation associated with chronic inflammatory diseases and SARS-CoV-2 infection and COVID-19 disease (320–324). Maintaining GSH levels using therapies that do not deplete the body’s GSH (324) would be the best choice. In a patient that is overloaded with cytokine storm, the best way to fortify the immune system would be to supply it with reduced GSH, since reduced GSH is already able to provide reducing equivalents from its thiol group. This is particularly relevant when we consider GSH pathways, as well as their transcriptional regulator Nrf2, for proliferation, survival and function of T cells, B cells and macrophages (325, 326). The value of GSH and nutritional strategies like amino acids, vitamins, minerals, phytochemicals, sulforaphane to enhance cellular Nrf2, and other supplements used to restore GSH levels (327–330) as adjunct treatments for all inflammatory diseases including SARS-CoV-2 infection needs to be further emphasized. Reducing the levels of proinflammatory molecules like mCRP and nnCRP (331, 332) will further reduce the detrimental effects of inflammaging. Reestablishing the cellular metabolic homeostasis in inflammatory diseases as well as SARS-CoV-2 infection and COVID-19 disease especially in the lungs and cardiovascular system, could become paramount to balance altered innate and adaptive immunity and cell function and reduce morbimortality (333–335). Treatment of chronic inflammatory diseases and now COVID-19 appears to be complex and may resist finding a single silver bullet intervention (247) supporting the use of combination therapies (170); especially in COVID-19 bearing in mind that “no one is safe until everyone is safe” (336).
Author contributions
CL and GK participated in the design, writing, and final corrections of the manuscript. Both authors contributed to the article and approved the submitted version.
Acknowledgments
This is an open-access article distributed under the terms of the Creative Commons Attribution (CC-BY) License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Meister A, Anderson ME. Glutathione. Annu Rev Biochem. (1983) 52:711–60. doi: 10.1146/annurev.bi.52.070183.003431
2. Sastre J, Pallardo FV, Viña J. Glutathione. Handb Environ Chem. (2005) 2:91–108. doi: 10.1007/b101148
3. Lu SC. Glutathione synthesis. Biochim Biophys Acta. (2013) 1830:3143–53. doi: 10.1016/j.bbagen.2012.09.008
4. Meister A. On the discovery of glutathione. Trends Biochem Sci. (1988) 13:185–8. doi: 10.1016/0968-000490148-x
5. Marí M, de Gregorio E, de Dios C, Roca-Agujetas V, Cucarull B, Tutusaus A, et al. Mitochondrial glutathione: recent insights and role in disease. Antioxidants. (2020) 9:909. doi: 10.3390/antiox9100909
6. Polonikov A. Endogenous deficiency of glutathione as the most likely cause of serious manifestations and death in COVID-19 patients. ACS Infect Dis. (2020) 6:1558–62. doi: 10.1021/acsinfecdis.0c00288
7. Silvagno F, Vernone A, Pescarmona GP. The role of glutathione in protecting against the severe inflammatory response triggered by COVID-19. Antioxidants. (2020) 9:624. doi: 10.3390/antiox9070624
8. Forman HJ, Zhang H, Rinna A. Glutathione: overview of its protective roles, measurement, and biosynthesis. Mol Aspects Med. (2009) 30:1–12. doi: 10.1016/j.mam.2008.08.006
9. De Rey-Pailhade MJ. Sur un corps d’origine organique hydrogénant le soufre á froid. C R Hebd Séances Acad Sci. (1888) 106:1683–4.
10. Hopkins FG. On an autoxidisable constituent of the cell. Biochem J. (1921) 15:286–305. doi: 10.1042/bj0150286
12. Kendall EC, Mason HL, McKenzie BF. A study of glutathione. IV. Determination of the structure of glutathione. J Biol Chem. (1930) 88:409–23.
13. Harington CR, Mead TH. Synthesis of glutathione. Biochem J. (1935) 29:1602–11. doi: 10.1042/bj0291602
14. Kosower EM, Kosower NS. Lest I forget thee, glutathione. Nature. (1969) 224:117–20. doi: 10.1038/224117a0
15. Ballatori N, Krance SM, Notenboom S, Shi S, Tieu K, Hammond CL. Glutathione dysregulation and the etiology and progression of human diseases. Biol Chem. (2009) 390:191–214. doi: 10.1515/BC.2009.033
16. Jones DP, Carlson JL, Mody VC, Cai J, Lynn MJ, Sternberg P. Redox state of glutathione in human plasma. Free Radic Biol Med. (2000) 28:625–35. doi: 10.1016/S0891-5849(99)00275-0
17. Koehler CM, Beverly KN, Leverich EP. Redox pathways of the mitochondrion. Antioxid Redox Signal. (2006) 8:813–22. doi: 10.1089/ars.2006.8.813
18. Han D, Hanawa N, Saberi B, Kaplowitz N. Mechanisms of liver injury. III. Role of glutathione redox status in liver injury. Am J Physiol Gastrointest Liver Physiol. (2006) 291:G1–7. doi: 10.1152/ajpgi.00001.2006
19. Owen JB, Butterfield DA. Measurement of oxidized/reduced glutathione ratio. In: Bross P, Gregersen N editors. Methods in molecular biology, protein misfolding and cellular stress in disease and aging: concepts and protocols. (Vol. 648), (2010). p. 269–77. doi: 10.1007/978-1-60761-756-3_18
20. Sreekumar PG, Ferrington DA, Kannan R. Glutathione metabolism and the novel role of mitochondrial GSH in retinal degeneration. Antioxidants. (2021) 10:661. doi: 10.3390/antiox10050661
21. Tomin T, Schittmayer M, Birner-Gruenberger R. Addressing glutathione redox status in clinical samples by two-step alkylation with N-ethylmaleimide isotopologues. Metabolites. (2020) 10:71. doi: 10.3390/metabo10020071
22. Zitka O, Skalickova S, Gumulec J, Masarik M, Adam V, Hubalek J, et al. Redox status expressed as GSH:GSSG ratio as a marker for oxidative stress in paediatric tumour patients. Oncol Lett. (2012) 4:1247–53. doi: 10.3892/ol.2012.931
23. Giustarini D, Tsikas D, Colombo G, Milzani A, Dalle-Donne I, Fanti P, et al. Pitfalls in the analysis of the physiological antioxidant glutathione (GSH) and its disulfide (GSSG) in biological samples: an elephant in the room. J Chromatogr B Analyt Technol Biomed Life Sci. (2016) 1019:21–8. doi: 10.1016/j.jchromb2016.02.015
24. Meredith MJ, Reed DJ. Status of the mitochondrial pool of glutathione in the isolated hepatocyte. J Biol Chem. (1982) 257:3747–53.
25. Fernández-Checa JC, Kaplowitz N, García-Ruiz C, Colell A, Miranda M, Marí M, et al. GSH transport in mitochondria: defense against TNF-induced oxidative stress and alcohol-induced defect. Am J Physiol Gastrointest Liver Physiol. (1997) 273:G7–17. doi: 10.1152/ajpgi.1997.273.1.G7
26. Hwang C, Sinskey AJ, Lodish HF. Oxidized redox state of glutathione in the endoplasmic reticulum. Science. (1992) 257:1496–502. doi: 10.1126/science.1523409
27. Yuan L, Kaplowitz N. Glutathione in liver diseases and hepatotoxicity. Mol Aspects Med. (2009) 30:29–41. doi: 10.1016/j.mam.2008.08.003
28. Scirè A, Cianfruglia L, Minnelli C, Bartolini D, Torquato P, Principato G, et al. Glutathione compartmentalization and its role in glutathionylation and other regulatory processes of cellular pathways. Biofactors. (2019) 45:152–68. doi: 10.1002/biof.1476
29. Schafer FQ, Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic Biol Med. (2001) 30:1191–212. doi: 10.1016/S0891-5849(01)00480-4
30. Jones DP. Redefining oxidative stress. Antioxid Redox Signal. (2006) 8:1865–79. doi: 10.1089/ars.2006.8.1865
31. Kemp M, Go Y-M, Jones DP. Nonequilibrium thermodynamics of thiol/disulfide redox systems: a perspective on redox systems biology. Free Radic Biol Med. (2008) 44:921–37. doi: 10.1016/j.freeradbiomed.2007.11.008
32. Esposito F, Agosti V, Morrone G, Morra F, Cuomo C, Russo T, et al. Inhibition of the differentiation of human myeloid cell lines by redox changes induced through glutathione depletion. Biochem J. (1994) 301:649–53. doi: 10.1042/bj3010649
33. Hansen JM, Carney EW, Harris C. Altered differentiation in rat and rabbit limb bud micromass cultures by glutathione modulating agents. Free Radic Biol Med. (2001) 31:1582–92. doi: 10.1016/s0891-5849(01)00751-1
34. Hansen JM, Harris C. Glutathione during embryonic development. Biochim Biophys Acta. (2015) 1850:1527–42. doi: 10.1016/j.bbagen.2014.12.001
35. Nkabyo YS, Ziegler TR, Gu LH, Watson WH, Jones DP. Glutathione and thioredoxin redox during differentiation in human colon epithelial (Caco-2) cells. Am J Physiol Gastrointest Liver Physiol. (2002) 283:G1352–9. doi: 10.1152/ajpgi.00183.2002
36. Kim J-M, Kim H, Kwon SB, Lee SY, Chung S-C, Jeong D-W, et al. Intracellular glutathione status regulates mouse bone marrow monocyte-derived macrophage differentiation and phagocytic activity. Biochem Biophys Res Commun. (2004) 325:101–8. doi: 10.1016/j.bbrc.2004.09.220
37. Huh Y-J, Kim J-M, Kim H, Song H, So H, Lee SY, et al. Regulation of osteoclast differentiation by the redox-dependent modulation of nuclear import of transcription factors. Cell Death Differ. (2006) 13:1138–46. doi: 10.1038/sj.cdd.4401793
38. Hammond CL, Lee TK, Ballatori N. Novel roles for glutathione in gene expression, cell death, and membrane transport of organic solutes. J Hepatol. (2001) 34:946–54. doi: 10.1016/s0168-8278(01)00037-x
39. Ballatori N, Hammond CL, Cunningham JB, Krance SM, Marchan R. Molecular mechanisms of reduced glutathione transport: role of the MRP/CFTR/ABCC and OATP/SLC21A families of membrane proteins. Toxicol Appl Pharmacol. (2005) 204:238–55. doi: 10.1016/j.taap.2004.09.008
40. Won J-S, Singh I. Sphingolipid signaling and redox regulation. Free Radic Biol Med. (2006) 40:1875–88. doi: 10.1016/j.freeradbiomed.2006.01.035
41. Garcia-Ruiz C, Fernández-Checa JC. Redox regulation of hepatocyte apoptosis. J Gastroenterol Hepatol. (2007) 22:S38–42. doi: 10.1111/j.1440-1746.2006.04644.x
42. Sykes MC, Mowbray AL, Jo H. Reversible glutathiolation of caspase-3 by glutaredoxin as a novel redox signaling mechanism in tumor necrosis factor-alpha-induced cell death. Circ Res. (2007) 100:152–4. doi: 10.1161/01.RES.0000258171.08020.72
43. Cerutti PA. Prooxidant states and tumor promotion. Science. (1985) 227:375–81. doi: 10.1126/science.2981433
44. Townsend DM, Tew KD, Tapiero H. The importance of glutathione in human disease. Biomed Pharmacother. (2003) 57:145–55. doi: 10.1016/s0753-3322(03)00043-x
45. Liu H, Wang H, Shenvi S, Hagen TM, Liu R-M. Glutathione metabolism during aging and in Alzheimer disease. Ann N Y Acad Sci. (2004) 1019:346–9. doi: 10.1196/annals.1297.059
46. Estrela JM, Ortega A, Obrador E. Glutathione in cancer biology and therapy. Crit Rev Clin Lab Sci. (2006) 43:143–81. doi: 10.1080/10408360500523878
47. Franco R, Schoneveld OJ, Pappa A, Panayiotidis MI. The central role of glutathione in the pathophysiology of human diseases. Arch Physiol Biochem. (2007) 113:234–58. doi: 10.1080/13813450701661198
48. Kinnula VL, Vuorinen K, Ilumets H, Rytilä P, Myllärniemi M. Thiol proteins, redox modulation and parenchymal lung disease. Curr Med Chem. (2007) 14:213–22. doi: 10.2174/092986707779313345
49. Reynolds A, Laurie C, Mosley RL, Gendelman HE. Oxidative stress and the pathogenesis of neurodegenerative disorders. Int Rev Neurobiol. (2007) 82:297–325. doi: 10.1016/S0074-7742(07)82016-2
50. Seifried HE. Oxidative stress and antioxidants: a link to disease and prevention? J Nutr Biochem. (2007) 18:168–71. doi: 10.1016/j.jnutbio.2006.12.003
51. Vali S, Mythri RB, Jagatha B, Padiadpu J, Ramanujan KS, Andersen JK, et al. Integrating glutathione metabolism and mitochondrial dysfunction with implications for Parkinson’s disease: a dynamic model. Neuroscience. (2007) 149:917–30. doi: 10.1016/j.neuroscience.2007.08.028
52. Valko M, Leibfritz D, Moncol J, Cronin MTD, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. (2007) 39:44–84. doi: 10.1016/j.biocel.2006.07.001
53. Khanfar A, Al Qaroot B. Could glutathione depletion be the trojan horse of COVID-19 mortality? Eur Rev Med Pharmacol Sci. (2020) 24:12500–9. doi: 10.26355/eurrev_202012_24046
54. Denzoin Vulcano LA, Soraci AL, Tapia MO. Homeostasis del glutatión. Acta Bioquím Clín Latinoam. (2013) 47:529–39.
55. Lu SC. Regulation of glutathione synthesis. Mol Aspects Med. (2009) 30:42–59. doi: 10.1016/j.mam.2008.05.005
56. Kaplowitz N, Aw TY, Ockhtens M. The regulation of hepatic glutathione. Ann Rev Pharmacol Toxicol. (1985) 25:715–44. doi: 10.1146/annurev.pa.25.040185.003435
57. Pastore A, Federici G, Bertini E, Piemonte F. Analyses of glutathione: implication in redox and detoxification. Clin Chim Acta. (2003) 333:19–39. doi: 10.1016/s0009-8981(03)00200-6
58. Camera E, Picardo M. Analytical methods to investigate glutathione and related compounds in biological and pathological processes. J Chromatogr B Analyt Technol Biomed Life Sci. (2002) 781:181–206. doi: 10.1016/s1570-0232(02)00618-9
59. Seelig GF, Meister A. Glutathione biosynthesis: γ-glutamylcysteine synthetase from rat kidney. Methods Enzymol. (1985) 113:379–90. doi: 10.1016/s0076-6879(85)13050-8
60. Huang CS, Anderson ME, Meister A. Amino acid sequence and function of the light subunit of rat kidney γ-glutamylcysteine synthetase. J Biol Chem. (1993) 268:20578–83.
61. Franklin CC, Backos DS, Mohar I, White CC, Forman HJ, Kavanagh TJ. Structure, function, and post-translational regulation of the catalytic and modifier subunits of glutamate cysteine ligase. Mol Aspects Med. (2009) 30:86–98. doi: 10.1016/j.mam.2008.08.009
62. Grant CM, Maciver FH, Dawes IW. Glutathione synthetase is dispensable for growth under both normal and oxidative stress conditions in the yeast Saccharomyces cerevisiae due to an accumulation of the dipeptide gamma-glutamylcysteine. Mol Biol Cell. (1997) 8:1699–707. doi: 10.1091/mbc.8.9.1699
63. Circu ML, Aw TY. Glutathione and apoptosis. Free Radic Res. (2008) 42:689–706. doi: 10.1080/10715760802317663
64. Marí M, Morales A, Colell A, García-Ruiz C, Fernández-Checa JC. Mitochondrial glutathione, a key survival antioxidant. Antioxid Redox Signal. (2009) 11:2685–700. doi: 10.1089/ars.2009.2695
65. Circu ML, Aw TY. Glutathione and modulation of cell apoptosis. Biochim Biophys Acta. (2012) 1823:1767–77. doi: 10.1016/j.bbamcr.2012.06.019
66. Markovic J, Borrás C, Ortega A, Sastre J, Viña J, Pallardó FV. Glutathione is recruited into the nucleus in early phases of cell proliferation. J Biol Chem. (2007) 282:20416–24. doi: 10.1074/jbc.M609582200
67. Markovic J, Mora NJ, Broseta AM, Gimeno A, de-la-Concepción N, Viña J, et al. The depletion of nuclear glutathione impairs cell proliferation in 3t3 fibroblasts. PLoS One. (2009) 4:e6413. doi: 10.1371/journal.pone.0006413
68. Diaz-Vivancos P, Wolff T, Markovic J, Pallardó FV, Foyer CH. A nuclear glutathione cycle within the cell cycle. Biochem J. (2010) 431:169–78. doi: 10.1042/BJ20100409
69. Chakravarthi S, Jessop CE, Bulleid NJ. The role of glutathione in disulphide bond formation and endoplasmic-reticulum-generated oxidative stress. EMBO Rep. (2006) 7:271–5. doi: 10.1038/sj.embor.7400645
70. Feige MJ, Hendershot LM. Disulfide bonds in ER protein folding and homeostasis. Curr Opin Cell Biol. (2011) 23:167–75. doi: 10.1016/j.ceb.2010.10.012
71. Ellgaard L, Sevier CS, Bulleid NJ. How are proteins reduced in the endoplasmic reticulum? Trends Biochem Sci. (2018) 43:32–43. doi: 10.1016/j.tibs.2017.10.006
72. García-Giménez JL, Markovic J, Dasí F, Queval G, Schnaubelt D, Foyer CH, et al. Nuclear glutathione. Biochim Biophys Acta. (2013) 1830:3304–16. doi: 10.1016/j.bbagen.2012.10.005
73. Pallardó FV, Markovic J, García JL, Viña J. Role of nuclear glutathione as a key regulator of cell proliferation. Mol Asp Med. (2009) 30:77–85. doi: 10.1016/j.mam.2009.01.001
74. Griffith OW, Bridges RJ, Meister A. Formation of gamma-glutamycyst(e)ine in vivo is catalyzed by gamma-glutamyl transpeptidase. Proc Natl Acad Sci USA. (1981) 78:2777–81. doi: 10.1073/pnas.78.5.2777
75. Viña JR, Palacin M, Puertes IR, Hernandez R, Viña J. Role of the y-glutamyl cycle in the regulation of amino acid translocation. Am J Physiol-Endocrinol Metab. (1989) 257:E916–22. doi: 10.1152/ajpendo.1989.257.6.E916
76. Meister A. On the enzymology of amino acid transport: transport in kidney and probably other tissues is mediated by a cycle of enzymic reactions involving glutathione. Science. (1973) 180:33–9. doi: 10.1126/science.180.4081.33
77. Zuo L, Wijegunawardana D. Redox role of ROS and inflammation in pulmonary diseases. Adv Exp Med Biol. (2021) 1304:187–204. doi: 10.1007/978-3-030-68748-9_11
78. McGuinness AJ, Sapey E. Oxidative stress in COPD: sources, markers, and potential mechanisms. J Clin Med. (2017) 6:21. doi: 10.3390/jcm6020021
79. Kirkham PA, Barnes PJ. Oxidative stress in COPD. Chest. (2013) 144:266–73. doi: 10.1378/chest.12-2664
80. Zinellu E, Zinellu A, Pau MC, Piras B, Fois AG, Mellino S, et al. Glutathione peroxidase in stable chronic obstructive pulmonary disease: a systematic review and meta-analysis. Antioxidants. (2021) 10:1745. doi: 10.3390/antiox10111745
81. Rahman I, MacNee W. Oxidative stress and regulation of glutathione in lung inflammation. Eur Respir J. (2000) 16:534–54. doi: 10.1034/j.1399-3003.2000.016003534.x
82. Rahman I. The role of oxidative stress in the pathogenesis of COPD: implications for therapy. Treat Respir Med. (2005) 4:175–200. doi: 10.2165/00151829-200504030-00003
83. Van Eeden SF, Sin DD. Oxidative stress in chronic obstructive pulmonary disease: a lung and systemic process. Can Respir J. (2013) 20:27–9. doi: 10.1155/2013/509130
84. Barnes PJ, Burney PG, Silverman EK, Celli BR, Vestbo J, Wedzicha JA, et al. Chronic obstructive pulmonary disease. Nat Rev Dis Primers. (2015) 1:15076. doi: 10.1038/nrdp.2015.76
85. Barnes PJ. Oxidative stress-based therapeutics in COPD. Redox Biol. (2020) 33:101544. doi: 10.1016/j.redox.2020.101544
86. Santos MC, Oliveira AL, Viegas-Crespo AM, Vicente L, Barreiros A, Monteiro P, et al. Systemic markers of the redox balance in chronic obstructive pulmonary disease. Biomarkers. (2004) 9:461–9. doi: 10.1080/13547500400024768
87. Dabo AJ, Ezegbunam W, Wyman AE, Moon J, Railwah C, Lora A, et al. Targeting c-Src reverses accelerated GPX-1 mRNA decay in chronic obstructive pulmonary disease airway epithelial cells. Am J Respir Cell Mol Biol. (2020) 62:598–607. doi: 10.1165/rcmb.2019-0177OC
88. Sotgia S, Paliogiannis P, Sotgiu E, Mellino S, Zinellu E, Fois AG, et al. Systematic review and meta-analysis of the blood glutathione redox state in chronic obstructive pulmonary disease. Antioxidants. (2020) 9:1146. doi: 10.3390/antiox9111146
89. Wang C, Zhou J, Wang J, Li S, Fukunaga A, Yodoi J, et al. Progress in the mechanism and targeted drug therapy for COPD. Sig Transduct Target Ther. (2020) 5:248. doi: 10.1038/s41392-020-00345-x
90. de Oliveira Rodrigues S, Coeli da Cunha CM, Valladão Soares GM, Leme Silva P, Ribeiro Silva A, Gonçalves-de-Albuquerque CF. Mechanisms, pathophysiology and currently proposed treatments of chronic obstructive pulmonary disease. Pharmaceuticals. (2021) 14:979. doi: 10.3390/ph14100979
91. Barnes PJ. Oxidative stress in chronic obstructive pulmonary disease. Antioxidants. (2022) 11:965. doi: 10.3390/antiox11050965
92. Takeda K, Akira S. Toll-like receptors in innate immunity. Int Immunol. (2005) 17:1–14. doi: 10.1093/intimm/dxh186
93. Kawasaki T, Kawai T. Toll-like receptor signaling pathways. Front Immunol. (2014) 5:461. doi: 10.3389/fimmu.2014.00461
94. Yu L, Wang L, Chen S. Endogenous toll-like receptor ligands and their biological significance. J Cell Mol Med. (2010) 14:2592–603. doi: 10.1111/j.1582-4934.2010.01127.x
95. Vidya MK, Kumar VG, Sejian V, Bagath M, Krishnan G, Bhatta R. Toll-like receptors: significance, ligands, signaling pathways, and functions in mammals. Int Rev Immunol. (2018) 37:20–36. doi: 10.1080/08830185.2017.1380200
96. El-Zayat SR, Sibaii H, Mannaa FA. Toll-like receptors activation, signaling, and targeting: an overview. Bull Natl Res Cent. (2019) 43:187. doi: 10.1186/s42269-019-0227-2
97. Farooq M, Batool M, Kim MS, Choi S. Toll-like receptors as a therapeutic target in the era of immunotherapies. Front Cell Dev Biol. (2021) 9:756315. doi: 10.3389/fcell.2021.756315
98. Li D, Wu M. Pattern recognition receptors in health and diseases. Signal Transduct Target Ther. (2021) 6:291. doi: 10.1038/s41392-021-00687-0
99. Pepys MB. C-reactive protein predicts outcome in COVID-19: is it also a therapeutic target? Eur Heart J. (2021) 42:2280–3. doi: 10.1093/eurheartj/ehab169
100. Zeller J, Bogner B, McFadyen JD, Kiefer J, Braig D, Pietersz G, et al. Transitional changes in the structure of C-reactive protein create highly pro-inflammatory molecules: therapeutic implications for cardiovascular diseases. Pharmacol Ther. (2022) 235:108165. doi: 10.1016/j.pharmthera.2022.108165
101. Drost EM, Skwarski KM, Sauleda J, Soler N, Roca J, Agusti A, et al. Oxidative stress and airway inflammation in severe exacerbations of COPD. Thorax. (2005) 60:293–300. doi: 10.1136/thx.2004.027946
102. Schaberg T, Klein U, Rau M, Eller J, Lode H. Subpopulations of alveolar macrophages in smokers and nonsmokers: relation to the expression of CD11/CD18 molecules and superoxide anion production. Am J Respir Crit Care Med. (1995) 151:1551–8. doi: 10.1164/ajrccm.151.5.7735614
103. Grandjean EM, Berthet P, Ruffmann R, Leuenberger P. Efficacy of oral long-term N-acetylcysteine in chronic bronchopulmonary disease: a meta-analysis of published double-blind, placebo-controlled clinical trials. Clin Ther. (2000) 22:209–21. doi: 10.1016/S0149-2918(00)88479-9
104. Decramer M, Rutten-van Mölken M, Dekhuijzen PN, Troosters T, van Herwaarden C, Pellegrino R, et al. Effects of N-acetylcysteine on outcomes in chronic obstructive pulmonary disease (bronchitis randomized on NAC cost-utility study, BRONCUS): a randomised placebo-controlled trial. Lancet. (2005) 365:1552–60. doi: 10.1016/S0140-6736(05)66456-2
105. Zheng JP, Wen FQ, Bai CX, Wan HY, Kang J, Chen P, et al. PANTHEON study group. Twice daily N-acetylcysteine 600 mg for exacerbations of chronic obstructive pulmonary disease (PANTHEON): a randomised, double-blind placebo-controlled trial. Lancet Respir Med. (2014) 2:187–94. doi: 10.1016/S2213-2600(13)70286-8
106. Soltan-Sharifi MS, Mojtahedzadeh M, Najafi A, Khajavi MR, Rouini MR, Moradi M, et al. Improvement by N-acetylcysteine of acute respiratory distress syndrome through increasing intracellular glutathione, and extracellular thiol molecules and anti-oxidant power: evidence for underlying toxicological mechanisms. Hum Exp Toxicol. (2007) 26:697–703. doi: 10.1177/0960327107083452
107. Pacht ER, Timerman AP, Lykens MG, Merola AJ. Deficiency of alveolar fluid glutathione in patients with sepsis and the adult respiratory distress syndrome. Chest. (1991) 100:1397–403. doi: 10.1378/chest.100.5.1397
108. Schmidt R, Luboeinski T, Markart P, Ruppert C, Daum C, Grimminger F, et al. Alveolar antioxidant status in patients with acute respiratory distress syndrome. Eur Respir J. (2004) 24:994–9. doi: 10.1183/09031936.04.00120703
109. Moradi M, Mojtahedzadeh M, Mandegari A, Soltan-Sharifi MS, Najafi A, Khajavi MR, et al. The role of glutathione-S-transferase polymorphisms on clinical outcome of ALI/ARDS patient treated with N-acetylcysteine. Respir Med. (2009) 103:434–41. doi: 10.1016/j.rmed.2008.09.013
110. Duca L, Ottolenghi S, Coppola S, Rinaldo R, Dei Cas M, Rubino FM, et al. Differential redox state and iron regulation in chronic obstructive pulmonary disease, acute respiratory distress syndrome and coronavirus disease 2019. Antioxidants. (2021) 10:1460. doi: 10.3390/antiox10091460
111. Kogan I, Ramjeesingh M, Li C, Kidd JF, Wang Y, Leslie EM, et al. CFTR directly mediates nucleotide-regulated glutathione flux. EMBO J. (2003) 22:1981–9. doi: 10.1093/emboj/cdg194
112. Cantin AM, North SL, Hubbard RC, Crystal RG. Normal alveolar epithelial lining fluid contains high levels of glutathione. J Appl Physiol. (1987) 63:152–7. doi: 10.1152/jappl.1987.63.1.152
113. Roum JH, Buhl R, McElvaney NG, Borok Z, Crystal RG. Systemic deficiency of glutathione in cystic fibrosis. J Appl Physiol. (1993) 75:2419–24. doi: 10.1152/jappl.1993.75.6.2419
114. Kettle AJ, Turner R, Gangell CL, Harwood DT, Khalilova IS, Chapman AL, et al. Oxidation contributes to low glutathione in the airways of children with cystic fibrosis. Eur Respir J. (2014) 44:122–9. doi: 10.1183/09031936.00170213
115. Piaggi S, Marchi E, Carnicelli V, Zucchi R, Griese M, Hector A, et al. Airways glutathione S-transferase omega-1 and its A140D polymorphism are associated with severity of inflammation and respiratory dysfunction in cystic fibrosis. J Cyst Fibros. (2021) 20:1053–61. doi: 10.1016/j.jcf.2021.01.010
116. Griese M, Kappler M, Eismann C, Ballmann M, Junge S, Rietschel E, et al. Glutathione study group. Inhalation treatment with glutathione in patients with cystic fibrosis: a randomized clinical trial. Am J Respir Crit Care Med. (2013) 188:83–9. doi: 10.1164/rccm.201303-0427OC
117. Galli F, Battistoni A, Gambari R, Pompella A, Bragonzi A, Pilolli F, et al. Working group on inflammation in cystic fibrosis. Oxidative stress and antioxidant therapy in cystic fibrosis. Biochim Biophys Acta. (2012) 1822:690–713. doi: 10.1016/j.bbadis.2011.12.012
118. Corti A, Franzini M, Cianchetti S, Bergamini G, Lorenzini E, Melotti P, et al. Contribution by polymorphonucleate granulocytes to elevated gamma-glutamyltransferase in cystic fibrosis sputum. PLoS One. (2012) 7:e34772. doi: 10.1371/journal.pone.0034772
119. Roum JH, Borok Z, McElvaney NG, Grimes GJ, Bokser AD, Buhl R, et al. Glutathione aerosol suppresses lung epithelial surface inflammatory cell-derived oxidants in cystic fibrosis. J Appl Physiol. (1999) 87:438–43. doi: 10.1152/jappl.1999.87.1.438
120. Corti A, Paolicchi A, Franzini M, Dominici S, Casini AF, Pompella A. The S-thiolating activity of membrane g-glutamyltransferase: formation of cysteinyl-glycine mixed disulfides with cellular proteins and in the cell microenvironment. Antioxid Redox Signal. (2005) 7:911–8. doi: 10.1089/ars.2005.7.911
121. Corti A, Pompella A, Bergamini G, Melotti P. Glutathione inhalation treatments in cystic fibrosis: the interference of airway γ-glutamyltransferase. Am J Respir Crit Care Med. (2014) 189:233–4. doi: 10.1164/rccm.201305-0908LE
122. Hector A, Griese M. Reply: glutathione inhalation treatments in cystic fibrosis: the interference of airway γ-glutamyltransferase. Am J Respir Crit Care Med. (2014) 189:234–5. doi: 10.1164/rccm.201307-1289LE
123. Zhao J, Huang W, Zhang S, Xu J, Xue W, He B, et al. Efficacy of glutathione for patients with cystic fibrosis: a meta-analysis of randomized-controlled studies. Am J Rhinol Allergy. (2020) 34:115–21. doi: 10.1177/1945892419878315
124. Whillier S, Raftos JE, Chapman B, Kuchel PW. Role of N-acetylcysteine and cystine in glutathione synthesis in human erythrocytes. Redox Rep. (2009) 14:115–24. doi: 10.1179/135100009X392539
125. Bozic M, Goss CH, Tirouvanziam RM, Baines A, Kloster M, Antoine L, et al. GROW study group. Oral glutathione and growth in cystic fibrosis: a multicenter, randomized, placebo-controlled, double-blind trial. J Pediatr Gastroenterol Nutr. (2020) 71:771–7. doi: 10.1097/MPG.0000000000002948
126. Gao L, Kim KJ, Yankaskas JR, Forman HJ. Abnormal glutathione transport in cystic fibrosis airway epithelia. Am J Physiol. (1999) 277:L113–8. doi: 10.1152/ajplung.1999.277.1.L113
127. Beeh KM, Beier J, Haas IC, Kornmann O, Micke P, Buhl R. Glutathione deficiency of the lower respiratory tract in patients with idiopathic pulmonary fibrosis. Eur Respir J. (2002) 19:1119–23. doi: 10.1183/09031936.02.00262402
128. McMillan DH, van der Velden JLJ, Lahue KG, Qian X, Schneider RW, Iberg MS, et al. Attenuation of lung fibrosis in mice with a clinically relevant inhibitor of glutathione-S-transferase π. JCI Insight. (2016) 1:e85717. doi: 10.1172/jci.insight.85717
129. He N, Bai S, Huang Y, Xing Y, Chen L, Yu F, et al. Evaluation of glutathione S-transferase inhibition effects on idiopathic pulmonary fibrosis therapy with a near-infrared fluorescent probe in cell and mice models. Anal Chem. (2019) 91:5424–32. doi: 10.1021/acs.analchem.9b00713
130. Estornut C, Milara J, Bayarri MA, Belhadj N, Cortijo J. Targeting oxidative stress as a therapeutic approach for idiopathic pulmonary fibrosis. Front Pharmacol. (2022) 12:794997. doi: 10.3389/fphar.2021.794997
131. Borok Z, Buhl R, Grimes GJ, Bokser AD, Hubbard RC, Holroyd KJ, et al. Effect of glutathione aerosol on oxidant-antioxidant imbalance in idiopathic pulmonary fibrosis. Lancet. (1991) 338:215–6. doi: 10.1016/0140-6736(91)90350-x
132. Prousky J. The treatment of pulmonary diseases and respiratory-related conditions with inhaled (nebulized or aerosolized) glutathione. eCAM. (2008) 5:27–35. doi: 10.1093/ecam/nem040
133. Holroyd KJ, Buhl R, Borok Z, Roum JH, Bokser AD, Grimes GJ, et al. Correction of glutathione deficiency in the lower respiratory tract of HIV seropositive individuals by glutathione aerosol treatment. Thorax. (1993) 48:985–9. doi: 10.1136/thx.48.10.985
134. Ghezzi P. Role of glutathione in immunity and inflammation in the lung. Int J Gen Med. (2011) 4:105–13. doi: 10.2147/IJGM.S15618
135. Murphy SL, Kochanek KD, Xu JQ, Arias E. Mortality in the united states, 2020 NCHS data brief, no 427. Hyattsville, MD: National Center for Health Statistics (2021). doi: 10.15620/cdc:112079
136. Bajic VP, Van Neste C, Obradovic M, Zafirovic S, Radak D, Bajic VB, et al. Glutathione “redox homeostasis” and its relation to cardiovascular disease. Oxid Med Cell Long. (2019) 2019:5028181. doi: 10.1155/2019/5028181
137. Matuz-Mares D, Riveros-Rosas H, Vilchis-Landeros MM, Vázquez-Meza H. Glutathione participation in the prevention of cardiovascular diseases. Antioxidants. (2021) 10:1220. doi: 10.3390/antiox10081220
138. Willis MS, Patterson C. Proteotoxicity and cardiac dysfunction —Alzheimer’s disease of the heart? N Engl J Med. (2013) 368:455–64. doi: 10.1056/NEJMra1106180
139. Ghosh R, Vinod V, Symons JD, Boudina S. Protein and mitochondria quality control mechanisms and cardiac aging. Cells. (2020) 9:933. doi: 10.3390/cells9040933
140. Ren J, Wang X, Zhang Y. Editorial: new drug targets for proteotoxicity in cardiometabolic diseases. Front Physiol. (2021) 12:745296. doi: 10.3389/fphys.2021.745296
141. McLendon PM, Robbins J. Proteotoxicity and cardiac dysfunction. Circ Res. (2015) 116:1863–82. doi: 10.1161/CIRCRESAHA.116.305372
142. Ranek MJ, Bhuiyan MS, Wang X. Editorial: targeting cardiac proteotoxicity. Front Physiol. (2021) 12:669356. doi: 10.3389/fphys.2021.669356
143. Sandri M, Robbins J. Proteotoxicity: an underappreciated pathology in cardiac disease. J Mol Cell Cardiol. (2014) 71:3–10. doi: 10.1016/j.yjmcc.2013.12.015
144. Kattoor AJ, Pothineni NVK, Palagiri D, Mehta JL. Oxidative stress in atherosclerosis. Curr Atheroscler Rep. (2017) 19:42. doi: 10.1007/s11883-017-0678-6
145. Yang X, Li Y, Li Y, Ren X, Zhang X, Hu D, et al. Oxidative stress-mediated atherosclerosis: mechanisms and therapies. Front Physiol. (2017) 8:600. doi: 10.3389/fphys.2017.00600
146. Nickenig G, Harrison DG. The AT1-type angiotensin receptor in oxidative stress and atherogenesis. Part I: oxidative stress and atherogenesis. Circulation. (2002) 105:393–6.
147. Nowak WN, Deng J, Ruan XZ, Xu Q. Reactive oxygen species generation and atherosclerosis. Arterioscler Thromb Vasc Biol. (2017) 37:e41–52. doi: 10.1161/ATVBAHA.117.309228
148. Förstermann U, Xia N, Li H. Roles of vascular oxidative stress and nitric oxide in the pathogenesis of atherosclerosis. Circ Res. (2017) 120:713–35. doi: 10.1161/CIRCRESAHA.116.309326
149. Marchio P, Guerra-Ojeda S, Vila JM, Aldasoro M, Victor VM, Mauricio MD. Targeting early atherosclerosis: a focus on oxidative stress and inflammation. Oxid Med Cell Longev. (2019) 2019:8563845. doi: 10.1155/2019/8563845
150. Perrotta I, Aquila S. The role of oxidative stress and autophagy in atherosclerosis. Oxid Med Cell Longev. (2015) 2015:130315. doi: 10.1155/2015/130315
151. Madamanchi NR, Hakim ZS, Runge MS. Oxidative stress in atherogenesis and arterial thrombosis: the disconnect between cellular studies and clinical outcomes. J Thromb Haemost. (2005) 3:254–67. doi: 10.1111/j.1538-7836.2004.01085.x
152. Lapenna D, de Gioia S, Ciofani G, Mezzetti A, Ucchino S, Calafiore AM, et al. Glutathione-related antioxidant defenses in human atherosclerotic plaques. Circulation. (1998) 97:1930–4. doi: 10.1161/01.cir.97.19.1930
153. Musthafa QA, Abdul Shukor MF, Ismail NAS, Mohd Ghazi A, Mohd Ali R, Nor IF, et al. Oxidative status and reduced glutathione levels in premature coronary artery disease and coronary artery disease. Free Radic Res. (2017) 51:787–98. doi: 10.1080/10715762.2017.1379602
154. Bastani A, Rajabi S, Daliran A, Saadat H, Karimi-Busheri F. Oxidant and antioxidant status in coronary artery disease. Biomed Rep. (2018) 9:327–32. doi: 10.3892/br.2018.1130
155. Song J, Park J, Oh Y, Lee JE. Glutathione suppresses cerebral infarct volume and cell death after ischemic injury: involvement of FOXO3 inactivation and Bcl2 expression. Oxid Med Cell Longev. (2015) 2015:426069. doi: 10.1155/2015/426069
156. Paterson PG, Juurlink BHJ. Nutritional regulation of glutathione in stroke. Neurotox Res. (1999) 1:99–112. doi: 10.1007/BF03033274
157. Maksimova MY, Ivanov AV, Virus ED, Nikiforova KA, Ochtova FR, Suanova ET, et al. Impact of glutathione on acute ischemic stroke severity and outcome: possible role of aminothiols redox status. Redox Rep. (2021) 26:117–23. doi: 10.1080/13510002.2021.1952819
158. Ivanov AV, Maksimova MY, Nikiforova KA, Ochtova FR, Suanova ET, Alexandrin VV, et al. Plasma glutathione as a risk marker for the severity and functional outcome of acute atherothrombotic and cardioembolic stroke. Egypt J Neurol Psychiatry Neurosurg. (2022) 58:15. doi: 10.1186/s41983-022-00452-2
159. Cacciapuoti F. N-acetyl-cysteine supplementation lowers high homocysteine plasma levels and increases glutathione synthesis in the transsulfuration pathway. Beneficial effects on several cardiovascular and neurodegenerative diseases. Ital J Med. (2019) 13:234–40. doi: 10.4081/itjm.2019.1192
160. Paul BD. Neuroprotective roles of the reverse transsulfuration pathway in Alzheimer’s disease. Front Aging Neurosci. (2021) 13:659402. doi: 10.3389/fnagi.2021.659402
161. Moretti R, Caruso P. The controversial role of homocysteine in neurology: from labs to clinical practice. Int J Mol Sci. (2019) 20:231. doi: 10.3390/ijms20010231
162. Higashi Y, Aratake T, Shimizu T, Shimizu S, Saito M. Protective role of glutathione in the hippocampus after brain ischemia. Int J Mol Sci. (2021) 22:7765. doi: 10.3390/ijms22157765
163. Won SJ, Yoo BH, Brennan AM, Shin BS, Kauppinen TM, Berman AE, et al. EAAC1 gene deletion alters zinc homeostasis and exacerbates neuronal injury after transient cerebral ischemia. J Neurosci. (2010) 30:15409–18. doi: 10.1523/JNEUROSCI.2084-10.2010
164. Aratake T, Higashi Y, Hamada T, Ueba Y, Shimizu T, Shimizu S, et al. The role of diurnal fluctuations in excitatory amino acid carrier 1 levels in post-ischemic hippocampal Zn2+ accumulation. Exp Neurol. (2021) 336:113538. doi: 10.1016/j.expneurol.2020.113538
165. Potempa LA, Rajab IM, Olson ME, Hart PC. C-reactive protein and cancer: interpreting the differential bioactivities of its pentameric and monomeric, modified isoforms. Front Immunol. (2021) 12:744129. doi: 10.3389/fimmu.2021.744129
166. Yasojima K, Schwab C, McGeer EG, McGeer PL. Generation of C-reactive protein and complement components in atherosclerotic plaques. Am J Pathol. (2001) 158:1039–51. doi: 10.1016/S0002-9440(10)64051-5
167. Jabs WJ, Theissing E, Nitschke M, Bechtel JFM, Duchrow M, Mohamed S, et al. Local generation of C-reactive protein in diseased coronary artery venous bypass grafts and normal vascular tissue. Circulation. (2003) 108:1428–31. doi: 10.1161/01.CIR.0000092184.43176.91
168. Diehl EE, Haines GKIII, Radosevich JA, Potempa LA. Immunohistochemical localization of modified C-reactive protein antigen in normal vascular tissue. Am J Med Sci. (2000) 319:79–83. doi: 10.1097/00000441-200002000-00002
169. Jialal I, Devaraj S, Venugopal SK. C-reactive protein: risk marker or mediator in atherothrombosis? Hypertension. (2004) 44:6–11. doi: 10.1161/01.HYP.0000130484.20501.df
170. Labarrere CA, Kassab GS. Pattern recognition proteins: first line of defense against coronaviruses. Front Immunol. (2021) 12:652252. doi: 10.3389/fimmu.2021.652252
171. Zhang Y, Cao H. Monomeric C-reactive protein affects cell injury and apoptosis through activation of p38 mitogen-activated protein kinase in human coronary artery endothelial cells. Bosn J Basic Med Sci. (2020) 20:487–94. doi: 10.17305/bjbms.2020.4711
172. Zhao M, Liu Y, Wang X, New L, Han J, Brunk UT. Activation of the P38 MAP kinase pathway is required for foam cell formation from macrophages exposed to oxidized LDL. APMIS. (2002) 110:458–68. doi: 10.1034/j.1600-0463.2002.100604.x
173. Howell KW, Meng X, Fullerton DA, Jin C, Reece TB, Cleveland JC. Toll-like receptor 4 mediates oxidized LDL-induced macrophage differentiation to foam cells. J Surg Res. (2011) 171:e27–31. doi: 10.1016/j.jss.2011.06.033
174. Yang K, Liu X, Liu Y, Wang X, Cao L, Zhang X, et al. DC-SIGN and toll-like receptor 4 mediate oxidized low-density lipoprotein-induced inflammatory responses in macrophages. Sci Rep. (2017) 7:3296. doi: 10.1038/s41598-017-03740-7
175. Boguslawski G, Labarrere CA. The role of C-reactive protein as a cardiovascular risk predictor. Kardiochir Torakochir Pol. (2006) 3:16–28. doi: 10.1201/b14635-16
176. Boncler M, Wu Y, Watala C. The multiple faces of C-reactive protein—physiological and pathophysiological implications in cardiovascular disease. Molecules. (2019) 24:2062. doi: 10.3390/molecules24112062
177. Mihlan M, Blom AM, Kupreishvili K, Lauer N, Stelzner K, Bergström F, et al. Monomeric C-reactive protein modulates classic complement activation on necrotic cells. FASEB J. (2011) 25:4198–210. doi: 10.1096/fj.11-186460
178. Agassandian M, Shurin GV, Ma Y, Shurin MR. C-reactive protein and lung diseases. Int J Biochem Cell Biol. (2014) 53:77–88. doi: 10.1016/j.biocel.2014.05.016
179. Thompson D, Pepys MB, Wood SP. The physiological structure of human c-reactive protein and its complex with phosphocholine. Structure. (1999) 7:169–77. doi: 10.1016/S0969-2126(99)80023-9
180. Chang M-K, Binder CJ, Torzewski M, Witztum JLC-. Reactive protein binds to both oxidized LDL and apoptotic cells through recognition of a common ligand: phosphorylcholine of oxidized phospholipids. Proc Natl Acad Sci USA. (2002) 99:13043–8. doi: 10.1073/pnas.192399699
181. Boguslawski G, McGlynn PW, Potempa LA, Filep JG, Labarrere CA. Conduct unbecoming: C-reactive protein interactions with a broad range of protein molecules. J Heart Lung Trans. (2007) 26:705–13. doi: 10.1016/j.healun.2007.04.006
182. Lv J-M, Lü S-Q, Liu Z-P, Zhang J, Gao B-X, Yao Z-Y, et al. Conformational folding and disulfide bonding drive distinct stages of protein structure formation. Sci Rep. (2018) 8:1494. doi: 10.1038/s41598-018-20014-y
183. Smilowitz NR, Kunichoff D, Garshick M, Shah B, Pillinger M, Hochman JS, et al. C-reactive protein and clinical outcomes in patients with COVID-19. Eur Heart J. (2021) 42:2270–9. doi: 10.1093/eurheartj/ehaa1103
184. Thiele JR, Zeller J, Bannasch H, Stark GB, Peter K, Eisenhardt SU. Targeting C-reactive protein in inflammatory disease by preventing conformational changes. Mediators Inflam. (2015) 2015:372432. doi: 10.1155/2015/372432
185. Black S, Kushner I, Samols DC-. Reactive protein. J Biol Chem. (2004) 279:48487–90. doi: 10.1074/jbc.R400025200
186. Lv J-M, Chen J-Y, Liu Z-P, Yao Z-Y, Wu Y-X, Tong C-S, et al. Cellular folding determinants and conformational plasticity of native C-reactive protein. Front Immunol. (2020) 11:583. doi: 10.3389/fimmu.2020.00583
187. Trial J, Cieslik KA, Entman ML. Phosphocholine-containing ligands direct CRP induction of M2 macrophage polarization independent of T cell polarization: implication for chronic inflammatory states. Immun Inflamm Dis. (2016) 4:274–88. doi: 10.1002/iid3.112
188. Sproston NR, Ashworth JJ. Role of C-reactive protein at sites of inflammation and infection. Front Immunol. (2018) 9:754. doi: 10.3389/fimmu.2018.00754
189. Zouki C, Haas B, Chan JSD, Potempa LA, Filep JG. Loss of pentameric symmetry of c-reactive protein is associated with promotion of neutrophil-endothelial cell adhesion. J Immunol. (2001) 167:5355–61. doi: 10.4049/jimmunol.167.9.5355
190. Torzewski M, Rist C, Mortensen RF, Zwaka TP, Bienek M, Waltenberger J, et al. C-reactive protein in the arterial intima. role of C-reactive protein receptor-dependent monocyte recruitment in atherogenesis. Arterioscler Thromb Vasc Biol. (2000) 20:2094–9. doi: 10.1161/01.atv.20.9.2094
191. Khreiss T, Joìzsef L, Hossain S, Chan JSD, Potempa LA, Filep JG. Loss of pentameric symmetry of C-reactive protein is associated with delayed apoptosis of human neutrophils. J Biol Chem. (2002) 277:40775–81. doi: 10.1074/jbc.M205378200
192. Braig D, Kaiser B, Thiele JR, Bannasch H, Peter K, Stark GB, et al. A conformational change of C-reactive protein in burn wounds unmasks its proinflammatory properties. Int Immunol. (2014) 26:467–78. doi: 10.1093/intimm/dxu056
193. Zwaka TP, Hombach V, Torzewski JC-. Reactive protein–mediated low densitylipoprotein uptake by macrophages. Implic Ather Circ. (2001) 103:1194–7. doi: 10.1161/01.CIR.103.9.1194
194. Thiele JR, Habersberger J, Braig D, Schmidt Y, Goerendt K, Maurer V, et al. Dissociation of pentameric to monomeric C-reactive protein localizes and aggravates inflammation. In vivo proof of a powerful proinflammatory mechanism and a new anti-inflammatory strategy. Circulation. (2014) 130:35–50. doi: 10.1161/CIRCULATIONAHA.113.007124
195. Molins B, Peña E, Vilahur G, Mendieta C, Slevin M, Badimon LC-. Reactive protein isoforms differ in their effects on thrombus growth. Arterioscler Thromb Vasc Biol. (2008) 28:2239–46. doi: 10.1161/ATVBAHA.108.174359
196. Ji SR, Wu Y, Potempa LA, Sheng FL, Lu W, Zhao J. Cell membranes and liposomes dissociate C-reactive protein (CRP) to form a new, biologically active structural intermediate mCRPm. FASEB J. (2007) 21:284–94. doi: 10.1096/fj.06-6722com
197. Fujii H, Li S-H, Szmitko PE, Fedak PWM, Verma S. C-reactive protein alters antioxidant defenses and promotes apoptosis in endothelial progenitor cells. Arterioscler Thromb Vasc Biol. (2006) 26:2476–82. doi: 10.1161/01.ATV.0000242794.65541.02
198. Eisenhardt SU, Thiele JR, Bannasch H, Stark GB, Peter K. C-reactive protein. How conformational changes influence inflammatory properties. Cell Cycle. (2009) 8:3885–92. doi: 10.4161/cc.8.23.10068
199. Eisenhardt SU, Habersberger J, Murphy A, Chen Y-C, Woollard KJ, Bassler N, et al. Dissociation of pentameric to monomeric C-reactive protein on activated platelets localizes inflammation to atherosclerotic plaques. Circ Res. (2009) 105:128–37. doi: 10.1161/CIRCRESAHA.108.190611
200. Slevin M, Krupinski J. A role for monomeric C-reactive protein in regulation of angiogenesis, endothelial cell inflammation and thrombus formation in cardiovascular/cerebrovascular disease? Histol Histopathol. (2009) 24:1473–8. doi: 10.14670/HH-24.1473
201. Singh SK, Thirumalai A, Pathak A, Ngwa DN, Agrawal A. Functional transformation of C-reactive protein by hydrogen peroxide. J Biol Chem. (2017) 292:3129–36. doi: 10.1074/jbc.M116.773176
202. Thiele JR, Zeller J, Kiefer J, Braig D, Kreuzaler S, Lenz Y, et al. A conformational change in C-reactive protein enhances leukocyte recruitment and reactive oxygen species generation in ischemia/reperfusion injury. Front Immunol. (2018) 9:675. doi: 10.3389/fimmu.2018.00675
203. Zeinolabediny Y, Kumar S, Slevin M. Monomeric C-reactive protein – a feature of inflammatory disease associated with cardiovascular pathophysiological complications? In Vivo. (2021) 35:693–7. doi: 10.21873/invivo.12309
204. McFadyen JD, Kiefer J, Braig D, Loseff-Silver J, Potempa LA, Eisenhardt SU, et al. Dissociation of C-reactive protein localizes and amplifies inflammation: evidence for a direct biological role of C-reactive protein and its conformational changes. Front Immunol. (2018) 9:1351. doi: 10.3389/fimmu.2018.01351
205. Schmitz G, Grandl M. Role of redox regulation and lipid rafts in macrophages during Ox-LDL–mediated foam cell formation. Antioxid Redox Signal. (2007) 9:1499–518. doi: 10.1089/ars.2007.1663
206. Witztum JL, Steinberg D. Role of oxidized low-density lipoprotein in atherogenesis. J Clin Invest. (1991) 88:1785–92. doi: 10.1172/JCI115499
207. Li R, Ren M, Luo M, Chen N, Zhang Z, Luo B, et al. Monomeric C-reactive protein alters fibrin clot properties on endothelial cells. Thromb Res. (2012) 129:e251–6. doi: 10.1016/j.thromres.2012.03.014
208. Badimon L, Peña E, Arderiu G, Padró T, Slevin M, Vilahur G, et al. C-reactive protein in atherothrombosis and angiogenesis. Front Immunol. (2018) 9:430. doi: 10.3389/fimmu.2018.00430
209. Obermayer G, Afonyushkin T, Binder CJ. Oxidized low-density lipoprotein in inflammation-driven thrombosis. J Thromb Haemost. (2018) 16:418–28. doi: 10.1111/jth.13925
210. Singh RK, Haka AS, Asmal A, Barbosa-Lorenzi VC, Grosheva I, Chin HF, et al. TLR4 (toll-like receptor 4)-dependent signaling drives extracellular catabolism of LDL (low-density lipoprotein) aggregates. Arterioscler Thromb Vasc Biol. (2020) 40:86–102. doi: 10.1161/ATVBAHA.119.313200
211. Rhoads JP, Major AS. How oxidized low-density lipoprotein activates inflammatory responses. Crit Rev Immunol. (2018) 38:333–42. doi: 10.1615/CritRevImmunol.2018026483
212. Yang K, Zhang XJ, Cao LJ, Liu XH, Liu ZH, Wang XQ, et al. Toll-like receptor 4 mediates inflammatory cytokine secretion in smooth muscle cells induced by oxidized low-density lipoprotein. PLoS One. (2014) 9:e95935. doi: 10.1371/journal.pone.0095935
213. Uppal N, Uppal V, Uppal P. Progression of coronary artery disease (CAD) from stable angina (SA) towards myocardial infarction (MI): role of oxidative stress. J Clin Diagn Res. (2014) 8:40–3. doi: 10.7860/JCDR/2014/7966.4002
214. Malekmohammad K, Sewell RDE, Rafieian-Kopaei M. Antioxidants and atherosclerosis: mechanistic aspects. Biomolecules. (2019) 9:301. doi: 10.3390/biom9080301
215. Amarowicz R. Natural phenolic compounds protect LDL against oxidation. Eur J Lipid Sci Technol. (2016) 118:677–9. doi: 10.1002/ejlt.201600077
216. Liu H, Xu H, Huang K. Selenium in the prevention of atherosclerosis and its underlying mechanisms. Metallomics. (2017) 9:21. doi: 10.1039/c6mt00195e
217. Handy DE, Joseph J, Loscalzo J. Selenium, a micronutrient that modulates cardiovascular health via redox enzymology. Nutrients. (2021) 13:3238. doi: 10.3390/nu13093238
218. Prasad A, Andrews NP, Padder FA, Husain M, Quyyumi AA. Glutathione reverses endothelial dysfunction and improves nitric oxide bioavailability. J Am Coll Cardiol. (1999) 34:507–14. doi: 10.1016/S0735-1097(99)00216-8
219. Scharfstein JS, Keaney JF Jr., Slivka A, Welch GN, Vita JA, Stamler JS, et al. In vivo transfer of nitric oxide between a plasma protein-bound reservoir and low-molecular weight thiols. J Clin Invest. (1994) 94:1432–9. doi: 10.1172/JCI117480
220. Ignarro LJ, Napoli C, Loscalzo J. Nitric oxide donors and cardiovascular agents modulating the bioactivity of nitric oxide. Circ Res. (2002) 90:21–8. doi: 10.1161/hh0102.102330
221. Rom O, Liu Y, Finney AC, Ghrayeb A, Zhao Y, Shukha Y, et al. Induction of glutathione biosynthesis by glycine-based treatment mitigates atherosclerosis. Redox Biol. (2022) 52:102313. doi: 10.1016/j.redox.2022.102313
222. Rom O, Villacorta L, Zhang J, Chen YE, Aviram M. Emerging therapeutic potential of glycine in cardiometabolic diseases: dual benefits in lipid and glucose. Metab Opin Lipidol. (2018) 29:428–32. doi: 10.1097/MOL.0000000000000543
223. Ding Y, Svingen GFT, Pedersen ER, Gregory JF, Ueland PM, Tell GS, et al. Plasma glycine and risk of acute myocardial infarction in patients with suspected stable angina pectoris. J Am Heart Assoc. (2016) 5:e002621. doi: 10.1161/JAHA.115.002621
224. Zaric BL, Radovanovic JN, Gluvic Z, Stewart AJ, Essack M, Motwalli O, et al. Atherosclerosis linked to aberrant amino acid metabolism and immunosuppressive amino acid catabolizing enzymes. Front Immunol. (2020) 11:551758. doi: 10.3389/fimmu.2020.551758
225. Wittemans LBL, Lotta LA, Oliver-Williams C, Stewart ID, Surendran P, Karthikeyan S, et al. Assessing the causal association of glycine with risk of cardio-metabolic diseases. Nat Commun. (2019) 10:1060. doi: 10.1038/s41467-019-08936-1
226. Chen J, Zhang S, Wu J, Wu S, Xu G, Wei D. Essential role of nonessential amino acid glutamine in atherosclerotic cardiovascular disease. DNA Cell Biol. (2020) 39:8–15. doi: 10.1089/dna.2019.50348
227. Andrews NP, Prasad A, Quyyumi AA. N-acetylcysteine improves coronary and peripheral vascular function. J Am Coll Cardiol. (2001) 37:117–23. doi: 10.1016/S0735-1097(00)01093-7
228. Cui Y, Narasimhulu CA, Liu L, Zhang Q, Liu PZ, Li X, et al. N-acetylcysteine inhibits in vivo oxidation of native low-density lipoprotein. Sci Rep. (2015) 5:16339. doi: 10.1038/srep16339
229. Shimada K, Murayama T, Yokode M, Kita T, Uzui H, Ueda T, et al. N-acetylcysteine reduces the severity of atherosclerosis in apolipoprotein E-deficient mice by reducing superoxide production. Circ J. (2009) 73:1337–41. doi: 10.1253/circj.CJ-08-1148
230. Toledo-Ibelles P, Mas-Oliva J. Antioxidants in the fight against atherosclerosis: is this a dead end? Curr Atheroscler Rep. (2018) 20:36. doi: 10.1007/s11883-018-0737-7
231. Méndez I, Vázquez-Martínez O, Hernández-Muñoz R, Valente-Godínez H, Díaz-Muñoz M. Redox regulation and pro-oxidant reactions in the physiology of circadian systems. Biochimie. (2016) 124:178–86. doi: 10.1016/j.biochi.2015.04.014
232. Ahmad F, Leake DS. Antioxidants inhibit low density lipoprotein oxidation less at lysosomal pH: a possible explanation as to why the clinical trials of antioxidants might have failed. Chem Phys Lipids. (2018) 213:13–24. doi: 10.1016/j.chemphyslip.2018.03.001
233. Mathur P, Ding Z, Saldeen T, Mehta JL. Tocopherols in the prevention and treatment of atherosclerosis and related cardiovascular disease. Clin Cardiol. (2015) 38:570–6. doi: 10.1002/clc.22422
234. Mimura J, Itoh K. Role of Nrf2 in the pathogenesis of atherosclerosis. Free Radic Biol Med. (2015) 88:221–32. doi: 10.1016/j.freeradbiomed.2015.06.019
235. da Costa RM, Rodrigues D, Pereira CA, Silva JF, Alves JV, Lobato NS, et al. Nrf2 as a potential mediator of cardiovascular risk in metabolic diseases. Front Pharmacol. (2019) 10:382. doi: 10.3389/fphar.2019.00382
236. Dai G, Vaughn S, Zhang Y, Wang ET, Garcia-Cardena G, Gimbrone MA. Biomechanical forces in atherosclerosis-resistant vascular regions regulate endothelial redox balance via phosphoinositol 3-kinase/akt-dependent activation of Nrf2. Circ Res. (2007) 101:723–33. doi: 10.1161/CIRCRESAHA.107.152942
237. Zakkar M, Van der Heiden K, Luong LA, Chaudhury H, Cuhlmann S, Hamdulay SS, et al. Activation of Nrf2 in endothelial cells protects arteries from exhibiting a proinflammatory state. Arterioscler Thromb Vasc Biol. (2009) 29:1851–7. doi: 10.1161/ATVBAHA.109.193375
238. Fiorelli S, Porro B, Cosentino N, Di Minno A, Manega CM, Fabbiocchi F, et al. Activation of Nrf2/HO-1 pathway and human atherosclerotic plaque vulnerability: an in vitro and in vivo study. Cells. (2019) 8:356. doi: 10.3390/cells8040356
239. Maruyama A, Tsukamoto S, Nishikawa K, Yoshida A, Harada N, Motojima K, et al. Nrf2 regulates the alternative first exons of CD36 in macrophages through specific antioxidant response elements. Arch Biochem Biophys. (2008) 477:139–45. doi: 10.1016/j.abb.2008.06.004
240. Ishii T, Itoh K, Ruiz E, Leake DS, Unoki H, Yamamoto M, et al. Role of Nrf2 in the regulation of CD36 and stress protein expression in murine macrophages. Activation by oxidatively modified ldl and 4-hydroxynonenal. Circ Res. (2004) 94:609–16. doi: 10.1161/01.RES.0000119171.44657.45
241. Araujo JA, Zhang M, Yin F. Heme oxygenase-1, oxidation, inflammation, and atherosclerosis. Front Pharmacol. (2012) 3:119. doi: 10.3389/fphar.2012.00119
242. He F, Ru X, Wen T. NRF2, a transcription factor for stress response and beyond. Int J Mol Sci. (2020) 21:4777. doi: 10.3390/ijms21134777
243. Ding Y, Zhang B, Zhou K, Chen M, Wang M, Jia Y, et al. Dietary ellagic acid improves oxidant-induced endothelial dysfunction and atherosclerosis: role of Nrf2 activation. Int J Cardiol. (2014) 175:508–14. doi: 10.1016/j.ijcard.2014.06.045
244. Ruotsalainen A-K, Inkala M, Partanen ME, Lappalainen JP, Kansanen E, Mäkinen PI, et al. The absence of macrophage Nrf2 promotes early atherogenesis. Cardiovasc Res. (2013) 98:107–15. doi: 10.1093/cvr/cvt008
245. Huang Z, Wu M, Zeng L, Wang D. The beneficial role of Nrf2 in the endothelial dysfunction of atherosclerosis. Cardiol Res Pract. (2022) 2022:4287711. doi: 10.1155/2022/4287711
246. Cascella M, Rajnik M, Aleem A, Dulebohn SC, Di Napoli R. Features, evaluation, and treatment of coronavirus (COVID-19). (2022). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK554776/ (accessed May 4, 2022).
247. Brosnahan SB, Jonkman AH, Kugler MC, Munger JS, Kaufman DA. COVID-19 and respiratory system disorders. current knowledge, future clinical and translational research questions. Arterioscler Thromb Vasc Biol. (2020) 40:2586–97. doi: 10.1161/ATVBAHA.120.314515
248. Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ. HLH across speciality collaboration, UK. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet. (2020) 395:1033–4. doi: 10.1016/S0140-6736(20)30628-0
249. Fajgenbaum DC, June CH. Cytokine storm. N Engl J Med. (2020) 383:2255–73. doi: 10.1056/NEJMra2026131
250. Reshi ML, Su Y-C, Hong J-R. RAN viruses: ROS-mediated cell death. Int J Cell Biol. (2014) 2014:467452. doi: 10.1155/2014/467452
251. Khomich OA, Kochetkov SN, Bartosch B, Ivanov AV. Redox biology of respiratory viral infections. Viruses. (2018) 10:392. doi: 10.3390/v10080392
252. Sallenave J-M, Guillot L. Innate immune signaling and proteolytic pathways in the resolution or exacerbation of SARS-CoV-2 in Covid-19: key therapeutic targets? Front Immunol. (2020) 11:1229. doi: 10.3389/fimmu.2020.01229
253. Beltrán-García J, Osca-Verdegal R, Pallardó FV, Ferreres J, Rodríguez M, Mulet S, et al. Sepsis and coronavirus disease 2019: common features and anti-inflammatory therapeutic approaches. Crit Care Med. (2020) 48:1841–4. doi: 10.1097/CCM.0000000000004625
254. Ghati A, Dam P, Tasdemir D, Kati A, Sellami H, Sezgin GC, et al. Exogenous pulmonary surfactant: a review focused on adjunctive therapy for severe acute respiratory syndrome coronavirus 2 including SP-A and SP-D as added clinical marker. Curr Opin Colloid Int Sci. (2021) 51:101413. doi: 10.1016/j.cocis.2020.101413
255. Kernan KF, Carcillo JA. Hyperferritinemia and inflammation. Int Immunol. (2017) 29:401–9. doi: 10.1093/intimm/dxx031
256. Naidu SAG, Clemens RA, Naidu AS. SARS-CoV-2 infection dysregulates host iron (Fe)-redox homeostasis (Fe-R-H): role of fe-redox regulators, ferroptosis inhibitors, anticoagulants, and iron-chelators in COVID-19 control. J Diet. (2022) 22:1–60. doi: 10.1080/19390211.2022.2075072
257. Moore JB, June CH. Cytokine release syndrome in severe COVID-19. Lessons from arthritis and cell therapy in cancer patients point to therapy for severe disease. Science. (2020) 368:473–4. doi: 10.1126/science.abb8925
258. Liu Y, Zhang H-G. Vigilance on new-onset atherosclerosis following SARS-CoV-2 infection. Front Med. (2021) 7:629413. doi: 10.3389/fmed.2020.629413
259. Vinciguerra M, Romiti S, Fattouch K, De Bellis A, Greco E. Atherosclerosis as pathogenetic substrate for sars-Cov2 cytokine storm. J Clin Med. (2020) 9:2095. doi: 10.3390/jcm9072095
260. Yang L, Xie X, Tu Z, Fu J, Xu D, Zhou Y. The signal pathways and treatment of cytokine storm in COVID-19. Signal Transduct Target Ther. (2021) 6:255. doi: 10.1038/s41392-021-00679-0
261. Luan Y, Yin C-H, Yao Y-M. Update advances on C-reactive protein in COVID-19 and other viral infections. Front Immunol. (2021) 12:720363. doi: 10.3389/fimmu.2021.720363
262. Potempa LA, Rajab IM, Hart PC, Bordon J, Fernandez-Botran R. Insights into the use of C-reactive protein as a diagnostic index of disease severity in COVID-19 infections. Am J Trop Med Hyg. (2020) 103:561–3. doi: 10.4269/ajtmh.20-0473
263. Mosquera-Sulbaran JA, Pedreañez A, Carrero Y, Callejas D. C-reactive protein as an effector molecule in covid-19 pathogenesis. Rev Med Virol. (2021) 31:e2221. doi: 10.1002/rmv.2221
264. Labarrere CA, Zaloga GP. C-reactive protein: from innocent bystander to pivotal mediator of atherosclerosis. Am J Med. (2004) 117:499–507. doi: 10.1016/j.amjmed.2004.03.039
265. Fazal M. C-reactive protein a promising biomarker of COVID-19 severity. Korean J Clin Lab Sci. (2021) 53:201–7. doi: 10.15324/kjcls.2021.53.3.201
266. Fendl B, Weiss R, Eichhorn T, Linsberger I, Afonyushkin T, Puhm F, et al. Extracellular vesicles are associated with C–reactive protein in sepsis. Sci Rep. (2021) 11:6996. doi: 10.1038/s41598-021-86489-4
267. Ahnach M, Zbiri S, Nejjari S, Ousti F, Elkettani C. C-reactive protein as an early predictor of COVID-19 severity. J Med Biochem. (2020) 39:500–7. doi: 10.5937/jomb0-27554
268. Torzewski M. C-reactive protein: friend or foe? Phylogeny from heavy metals to modified lipoproteins and SARS-CoV-2. Front Cardiovasc Med. (2022) 9:797116. doi: 10.3389/fcvm.2022.797116
269. Slevin M, Heidari N, Azamfirei L. Monomeric C-reactive protein: current perspectives for utilization and inclusion as a prognostic indicator and therapeutic target. Front Immunol. (2022) 13:866379. doi: 10.3389/fimmu.2022.866379
270. Poznyak AV, Bezsonov EE, Eid AH, Popkova TV, Nedosugova LV, Starodubova AV, et al. ACE2 is an adjacent element of atherosclerosis and COVID-19 pathogenesis. Int J Mol Sci. (2021) 22:4691. doi: 10.3390/ijms22094691
271. Martínez-Salazar B, Holwerda M, Stüdle C, Piragyte I, Mercader N, Engelhardt B, et al. COVID-19 and the vasculature: current aspects and long-term consequences. Front Cell Dev Biol. (2022) 10:824851. doi: 10.3389/fcell.2022.824851
272. Vinciguerra M, Romiti S, Sangiorgi GM, Rose D, Miraldi F, Greco E. SARS-CoV-2 and atherosclerosis: should COVID-19 be recognized as a new predisposing cardiovascular risk factor? J Cardiovasc Dev Dis. (2021) 8:130. doi: 10.3390/jcdd8100130
273. Grzegorowska O, Lorkowski J. Possible correlations between atherosclerosis. Acute coronary syndromes and covid-19. J Clin Med. (2020) 9:3746. doi: 10.3390/jcm9113746
274. Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev Immunol. (2011) 11:98–107. doi: 10.1038/nri2925
275. Tsalamandris S, Antonopoulos AS, Oikonomou E, Papamikroulis G-A, Vogiatzi G, Papaioannou S, et al. The role of inflammation in diabetes: current concepts and future perspectives. Eur Cardiol Rev. (2019) 14:50–9. doi: 10.15420/ecr.2018.33.1
276. Franceschi C, Bonafè M, Valensin S, Olivieri F, De Luca M, Ottaviani E, et al. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann NY Acad Sci. (2000) 908:244–54. doi: 10.1111/j.1749-6632.2000.tb06651.x
277. Franceschi C, Capri M, Monti D, Giunta S, Olivieri F, Sevini F, et al. Inflammaging and anti-inflammaging: a systemic perspective on aging and longevity emerged from studies in humans. Mech Ageing Dev. (2007) 128:92–105. doi: 10.1016/j.mad.2006.11.016
278. Calçada D, Vianello D, Giampieri E, Sala C, Castellani G, de Graaf A, et al. The role of low-grade inflammation and metabolic flexibility in aging and nutritional modulation thereof: a systems biology approach. Mech Ageing Dev. (2014) 2014:138–47. doi: 10.1016/j.mad.2014.01.004
279. Franceschi C, Campisi J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J Gerontol A Biol Sci Med Sci. (2014) 69:S4–9. doi: 10.1093/gerona/glu057
280. Frasca D, Blomberg BB. Inflammaging decreases adaptive and innate immune responses in mice and humans. Biogerontology. (2016) 17:7–19. doi: 10.1007/s10522-015-9578-8
281. Giuliani A, Prattichizzo F, Micolucci L, Ceriello A, Procopio AD, Rippo MR. Mitochondrial (Dys) function in inflammaging: do mitomirs influence the energetic. Oxidative, and inflammatory status of senescent cells? Mediators Inflamm. (2017) 2017:2309034. doi: 10.1155/2017/2309034
282. Balistreri CR. Anti-inflamm-ageing and/or anti-age-related disease emerging treatments: a historical alchemy or revolutionary effective procedures? Mediators Inflamm. (2018) 2018:3705389. doi: 10.1155/2018/3705389
283. Ferrucci L, Fabbri E. Inflammageing: chronic inflammation in ageing, cardiovascular disease, and frailty. Nat Rev Cardiol. (2018) 15:505–22. doi: 10.1038/s41569-018-0064-2
284. Franceschi C, Garagnani P, Parini P, Giuliani C, Santoro A. Inflammaging: a new immune–metabolic viewpoint for age- related diseases. Nat Rev Endocrinol. (2018) 14:576–90. doi: 10.1038/s41574-018-0059-4
285. Olivieri F, Prattichizzo F, Grillari J, Balistreri CR. Cellular senescence and inflammaging in age-related diseases. Mediators Inflamm. (2018) 2018:9076485. doi: 10.1155/2018/9076485
286. Fulop T, Larbi A, Dupuis G, Le Page A, Frost EH, Cohen AA, et al. Immunosenescence and inflamm-aging as two sides of the same coin: friends or foes? Front Immunol. (2018) 8:1960. doi: 10.3389/fimmu.2017.01960
287. Franceschi C, Capri M, Garagnani P, Ostan R, Santoro A, Monti D, et al. Inflammaging. In: T Fulop, C Franceschi, K Hirokawa, G Pawelec editors. Handbook of immunosenescence. Cham: Springer (2019). p. 1599–629. doi: 10.1007/978-3-319-99375-1_45
288. Pietrobon AJ, Teixeira FME, Sato MN. Immunosenescence and Inflammaging: risk factors of severe COVID-19 in older people. Front Immunol. (2020) 11:579220. doi: 10.3389/fimmu.2020.579220
289. Fulop T, Larbi A, Pawelec G, Khalil A, Cohen AA, Hirokawa K, et al. Immunology of aging: the birth of inflammaging. Clin Rev Allergy Immunol. (2021) 18:1–14. doi: 10.1007/s12016-021-08899-6
290. Zuo L, Prather ER, Stetskiv M, Garrison DE, Meade JR, Peace TI, et al. Inflammaging and oxidative stress in human diseases: from molecular mechanisms to novel treatments. Int J Mol Sci. (2019) 20:4472.
291. Xia S, Zhou C, Kalionis B, Shuang X, Ge H, Gao W. Combined antioxidant, anti-inflammaging and mesenchymal stem cell treatment: a possible therapeutic direction in elderly patients with chronic obstructive pulmonary disease. Aging Dis. (2020) 11:129–40. doi: 10.14336/AD.2019.0508
292. Draganidis D, Karagounis LG, Athanailidis I, Chatzinikolaou A, Jamurtas AZ, Fatouros IG. Inflammaging and skeletal muscle: can protein intake make a difference? J Nutr. (2016) 146:1940–52. doi: 10.3945/jn.116.230912
293. Bharath LP, Nikolajczyk BS. Next steps in mechanisms of inflammaging. Autophagy. (2020) 16:2285–6. doi: 10.1080/15548627.2020.1822089
294. Birben E, Sahiner UM, Sackesen C, Erzurum S, Kalayci O. Oxidative stress and antioxidant defense. World Allergy Organ J. (2012) 5:9–19. doi: 10.1097/WOX.0b013e3182439613
295. Sharifi-Rad M, Kumar NVA, Zucca P, Varoni EM, Dini L, Panzarini E, et al. Lifestyle, oxidative stress, and antioxidants: back and forth in the pathophysiology of chronic diseases. Front Physiol. (2020) 11:694. doi: 10.3389/fphys.2020.00694
296. Liu Z, Ren Z, Zhang J, Chuang C-C, Kandaswamy E, Zhou T, et al. Role of ROS and nutritional antioxidants in human diseases. Front Physiol. (2018) 9:477. doi: 10.3389/fphys.2018.00477
297. Cuhna LL, Perazzio SF, Azzi J, Cravedi P, Riella LV. Remodeling of the immune response with aging: immunosenescence and its potential impact on COVID-19 immune response. Front Immunol. (2020) 11:1748. doi: 10.3389/fimmu.2020.01748
298. Kryukov EV, Ivanov AV, Karpov VO, Vasil’evich Alexandrin V, Dygai AM, Kruglova MP, et al. Association of low molecular weight plasma aminothiols with the severity of coronavirus disease 2019. Oxid Med Cell Longev. (2021) 2021:9221693. doi: 10.1155/2021/9221693
299. Singh M, Barrera Adame O, Nickas M, Robison J, Khatchadourian C, Venketaraman V. Type 2 diabetes contributes to altered adaptive immune responses and vascular inflammation in patients with SARS-CoV-2 infection. Front Immunol. (2022) 13:833355. doi: 10.3389/fimmu.2022.833355
300. Tufan E, Göksun Sivas G, Gürel-Gökmen B, Yılmaz-Karaoğlu S, Ercan D, Özbeyli D, et al. Inhibitory effect of whey protein concentrate on SARS-CoV-2-targeted furin activity and spike protein-ACE2 binding in methotrexate-induced lung damage. J Food Biochem. (2022) 46:e14039. doi: 10.1111/jfbc.14039
301. Kumar P, Liu C, Hsu JW, Chacko S, Minard C, Jahoor F, et al. Glycine and N-acetylcysteine (GlyNAC) supplementation in older adults improves glutathione deficiency, oxidative stress, mitochondrial dysfunction, inflammation, insulin resistance, endothelial dysfunction, genotoxicity, muscle strength, and cognition: results of a pilot clinical trial. Clin Transl Med. (2021) 11:e372. doi: 10.1002/ctm2.372
302. Kumar P, Osahon O, Vides DB, Hanania N, Minard CG, Sekhar RV. Severe glutathione deficiency, oxidative stress and oxidant damage in adults hospitalized with COVID-19: implications for GlyNAC (glycine and N-acetylcysteine) supplementation. Antioxidants. (2022) 11:50. doi: 10.3390/antiox11010050
303. Obayan AOE. Overview of the rationale for l-glutamine treatment in moderate-severe COVID-19 infection. J Infect Dis Epidemiol. (2021) 7:187. doi: 10.23937/2474-3658/1510187
304. Seale LA, Torres DJ, Berry MJ, Pitts MW. A role for selenium-dependent GPX1 in SARS-CoV-2 virulence. Am J Clin Nutr. (2020) 112:447–8. doi: 10.1093/ajcn/nqaa177
305. Taylor EW, Radding W. Understanding selenium and glutathione as antiviral factors in COVID-19: does the viral mpro protease target host selenoproteins and glutathione synthesis? Front Nutr. (2020) 7:143. doi: 10.3389/fnut.2020.00143
306. Fraternale A, Zara C, De Angelis M, Nencioni L, Palamara AT, Retini M, et al. Intracellular redox-modulated pathways as targets for effective approaches in the treatment of viral infection. Int J Mol Sci. (2021) 22:3603. doi: 10.3390/ijms22073603
307. Ross EK, Gray JJ, Winter AN, Linseman DA. Immunocal® and preservation of glutathione as a novel neuroprotective strategy for degenerative disorders of the nervous system. Recent Pat CNS Drug Discov. (2012) 7:230–5. doi: 10.2174/157488912803252014
308. Jain SK, Parsanathan R, Achari AE, Kanikarla-Marie P, Bocchini JA. Glutathione stimulates vitamin D regulatory and glucose-metabolism genes, lowers oxidative stress and inflammation, and increases 25- hydroxy-vitamin D levels in blood: a novel approach to treat 25-hydroxyvitamin d deficiency. Antioxid Redox Signal. (2018) 29:1792–807. doi: 10.1089/ars.2017.7462
309. Jain SK, Parsanathan R. Can vitamin D and L-cysteine co-supplementation reduce 25(OH)-vitamin D deficiency and the mortality associated with COVID-19 in African Americans? J Am Coll Nutr. (2020) 39:694–9. doi: 10.1080/07315724.2020.1789518
310. Jain SK, Parsanathan R, Levine SN, Bocchini JA, Holick MF, Vanchiere JA. The potential link between inherited G6PD deficiency, oxidative stress, and vitamin D deficiency and the racial inequities in mortality associated with COVID-19. Free Radic Biol Med. (2020) 161:84–91. doi: 10.1016/j.freeradbiomed.2020.10.002
311. Tang HY, Ho HY, Wu PR, Chen SH, Kuypers FA, Cheng ML, et al. Inability to maintain GSH pool in G6PD-deficient red cells causes futile AMPK activation and irreversible metabolic disturbance. Antioxid Redox Signal. (2015) 22:744–59. doi: 10.1089/ars.2014.6142
312. Ganji R, Reddy PH. Impact of COVID-19 on mitochondrial-based immunity in aging and age-related diseases. Front Aging Neurosci. (2021) 12:614650. doi: 10.3389/fnagi.2020.614650
313. Mazumder S, Bindu S, De R, Debsharma S, Pramanik S, Bandyopadhyay U. Emerging role of mitochondrial DAMPs, aberrant mitochondrial dynamics and anomalous mitophagy in gut mucosal pathogenesis. Life Sci. (2022) 305:120753. doi: 10.1016/j.lfs.2022.120753
314. Singh SP, Amar S, Gehlot P, Patra SK, Kanwar N, Kanwal A. Mitochondrial modulations, autophagy pathways shifts in viral infections: consequences of COVID-19. Int J Mol Sci. (2021) 22:8180. doi: 10.3390/ijms22158180
315. Srinivasan K, Pandey AK, Livingston A, Venkatesh S. Roles of host mitochondria in the development of COVID-19 pathology: could mitochondria be a potential therapeutic target? Mol Biomed. (2021) 2:38. doi: 10.1186/s43556-021-00060-1
316. Burtscher J, Burtscher M, Millet GP. The central role of mitochondrial fitness on antiviral defenses: an advocacy for physical activity during the COVID-19 pandemic. Redox Biol. (2021) 43:101976. doi: 10.1016/j.redox.2021.101976
317. Li X, Hou P, Ma W, Wang X, Wang H, Yu Z, et al. SARS-CoV-2 ORF10 suppresses the antiviral innate immune response by degrading MAVS through mitophagy. Cell Mol Immunol. (2022) 19:67–78. doi: 10.1038/s41423-021-00807-4
318. Pacheco Y, Valeyre D, El Jammal T, Vallee M, Chevalier F, Lamartine J, et al. Autophagy and mitophagy-related pathways at the crossroads of genetic pathways involved in familial sarcoidosis and host-pathogen interactions induced by coronaviruses. Cells. (2021) 10:1995. doi: 10.3390/cells10081995
319. Shang C, Liu Z, Zhu Y, Lu J, Ge C, Zhang C, et al. SARS-CoV-2 causes mitochondrial dysfunction and mitophagy impairment. Front Microbiol. (2022) 12:780768. doi: 10.3389/fmicb.2021.780768
320. Lushchak VI. Glutathione homeostasis and functions: potential targets for medical interventions. J Amino Acids. (2012) 2012:736837. doi: 10.1155/2012/736837
321. Franco R, Cidlowski JA. Apoptosis and glutathione: beyond an antioxidant. Cell Death Differ. (2009) 16:1303–14. doi: 10.1038/cdd.2009.107
322. Bartolini D, Stabile AM, Bastianelli S, Giustarini D, Pierucci S, Busti C, et al. SARS-CoV2 infection impairs the metabolism and redox function of cellular glutathione. Redox Biol. (2021) 45:102041. doi: 10.1016/j.redox.2021.102041
323. Karkhanei B, Talebi Ghane E, Mehri F. Evaluation of oxidative stress level: total antioxidant capacity, total oxidant status and glutathione activity in patients with COVID-19. New Microbes New Infect. (2021) 42:100897. doi: 10.1016/j.nmni.2021.100897
324. Sestili P, Fimognari C. Paracetamol-induced glutathione consumption: is there a link with severe COVID-19 illness? Front Pharmacol. (2020) 11:579944. doi: 10.3389/fphar.2020.579944
325. Labarrere CA, Kassab GS. Response: commentary: pattern recognition proteins: first line of defense against coronaviruses. Front Immunol. (2022) 13:853015. doi: 10.3389/fimmu.2022.853015
326. Muri J, Kopf M. Redox regulation of immunometabolism. Nat Rev Immunol. (2021) 21:363–81. doi: 10.1038/s41577-020-00478-8
327. Grigoletto Fernandes I, Alves de Brito C, Silva Dos Reis VM, Sato MN, Zanete Pereira N. SARS-CoV-2 and other respiratory viruses: what does oxidative stress have to do with it? Oxid Med Cell Longev. (2020) 2020:8844280. doi: 10.1155/2020/8844280
328. Minich DM, Brown BI. A review of dietary (phyto)nutrients for glutathione support. Nutrients. (2019) 11:2073. doi: 10.3390/nu11092073
329. Hermel M, Sweeney M, Ni Y-M, Bonakdar R, Triffon D, Suhar C, et al. Natural supplements for COVID19—background, rationale, and clinical trials. J Evid Based Integr Med. (2021) 26:2515690X211036875. doi: 10.1177/2515690X211036875
330. Yamamoto M, Kensler TW, Motohashi H. The keap1-Nrf2 system: a thiol-based sensor-effector apparatus for maintaining redox homeostasis. Physiol Rev. (2018) 98:1169–203. doi: 10.1152/physrev.00023.2017
331. Torzewski J, Heigl F, Zimmermann O, Wagner F, Schumann C, Hettich R, et al. First-in-man: case report of selective c-reactive protein apheresis in a patient with SARS-CoV-2 infection. Am J Case Rep. (2020) 21:e925020. doi: 10.12659/AJCR.925020
332. Ringel J, Ramlow A, Bock C, Sheriff A. Case report: C-reactive protein apheresis in a patient with COVID-19 and fulminant CRP increase. Front Immunol. (2021) 12:708101. doi: 10.3389/fimmu.2021.708101
333. Li S, Zhao F, Ye J, Li K, Wang Q, Du Z, et al. Cellular metabolic basis of altered immunity in the lungs of patients with COVID-19. Med Microbiol Immunol. (2022) 211:49–69. doi: 10.1007/s00430-021-00727-0
334. Hsu R-J, Yu W-C, Peng G-R, Ye C-H, Hu SY, Chong PCT, et al. The role of cytokines and chemokines in severe acute respiratory syndrome coronavirus 2 infections. Front Immunol. (2022) 13:832394. doi: 10.3389/fimmu.2022.832394
335. Labarrere CA, Kassab GS. Glutathione deficiency in the pathogenesis of SARS-CoV-2 infection and its effects upon the host immune response in severe COVID-19 disease. Front Microbiol. (2022) 13:979719. doi: 10.3389/fmicb.2022.979719
Keywords: glutathione, oxidative stress, reactive oxygen species, nuclear factor erythroid 2-related factor 2, inflammaging, chronic obstructive pulmonary disease, atherosclerosis, COVID-19
Citation: Labarrere CA and Kassab GS (2022) Glutathione: A Samsonian life-sustaining small molecule that protects against oxidative stress, ageing and damaging inflammation. Front. Nutr. 9:1007816. doi: 10.3389/fnut.2022.1007816
Received: 31 July 2022; Accepted: 12 October 2022;
Published: 01 November 2022.
Edited by:
Yannan Jin, De Montfort University, United KingdomReviewed by:
Mostafa Waly, Sultan Qaboos University, OmanAthanasios Jamurtas, University of Thessaly, Greece
Tzortzis Nomikos, Harokopio University, Greece
Li Chu, Hebei Medical University, China
Copyright © 2022 Labarrere and Kassab. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carlos A. Labarrere, clabarrere@sbcglobal.net