 Ruiyang Zhang
Ruiyang Zhang Junhua Liu
Junhua Liu Linshu Jiang3
Linshu Jiang3 Xinfeng Wang
Xinfeng Wang Shengyong Mao
Shengyong Mao- 1College of Animal Science and Veterinary Medicine, Shenyang Agricultural University, Shenyang, China
- 2Laboratory of Gastrointestinal Microbiology, Jiangsu Key Laboratory of Gastrointestinal Nutrition and Animal Health, College of Animal Science and Technology, Nanjing Agricultural University, Nanjing, China
- 3Beijing University of Agriculture, Beijing, China
- 4College of Animal Science and Technology, Shihezi University, Shihezi, China
At present, research on high-concentrate (HC) diets mostly focused on the rumen, and there is a paucity of information on the hindgut microbiota of dairy cows. In the present study, a 2 × 2 crossover design with four healthy Holstein cows was used, and the metagenomics approach was adopted to reveal the remodeling effects of HC diets on hindgut microbiota and their metabolic functions. Results showed that, compared with the low-concentrate (LC) diets, HC diets have markedly decreased (p < 0.05) the abundance of cellulolytic bacteria (such as Fibrobacter, Ruminococcus, and Ruminiclostridium) and methanogens (such as Methanobrevibacter, Methanosarcina, and Methanosphaera); and correspondingly, HC diets have significantly reduced (p < 0.05) the abundance of carbohydrate-active enzymes (CAZy) related to hemicellulases (GH10, GH11, and GH54) and cellulases (GH1, GH44, and GH45) and increased the abundance of one oligosaccharide-degrading enzyme (GH32). Furthermore, 62 Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways of hindgut microbiota were affected (p < 0.05) by different dietary treatments, and the major pathways altered by HC diets were “Methane metabolism” (enriched in the LC group), “Lipid metabolism” (enriched in the HC group), and several sub-pathways in “Amino acid metabolism” (such as Phenylalanine metabolism, and Phenylalanine, tyrosine, and tryptophan biosynthesis). Also, the microbial genes involved in the pathways “Methane metabolism” (except 1 gene), “Tryptophan metabolism”, and “Phenylalanine metabolism” were all decreased (p < 0.05) in the present study. These findings suggested that HC diets caused the remodeling of hindgut microbiota and its potential functions, and these results may benefit in gaining a deeper understanding of the impact of HC diets on the hindgut microbiota of dairy cows.
Introduction
For ruminants, the gastrointestinal tract-dwelling microbiota is central to nutrient digestion, production performance, and host health (1, 2). To maintain the high milk yields or growth rate, the current ruminant industry imposes a high-concentrate (HC) feeding pattern; this also induces gastrointestinal microdysbiosis, thus resulting in some health problems, such as subacute ruminal acidosis (SARA) (3, 4). Being the initial and primary organ of microbial fermentation, alterations in the ruminal microbiome of dairy cows after HC feeding have received much research attention (5, 6). Our previous study and others showed that the undegraded starch escaped from the rumen and the small intestine could also reach the hindgut under the HC feeding condition, which enabled to affect the internal environment and microbial structure in the hindgut of dairy cows (7, 8). However, compared with the ruminal microbiome, there is relatively limited information about the effects of HC feeding on the hindgut microbiome.
Previous studies demonstrated the events that occurred in the rumen mirrored in the hindgut after HC feeding, which also resulted in an acidic environment with potential pathogenic factors in the hindgut, specifically manifested as low pH, high levels of volatile fatty acids (VFAs), and lipopolysaccharides in feces of dairy cows (8–10). However, as compared with the multilayer squamous epithelium of rumen, the single-layer columnar epithelium of the hindgut is more fragile and susceptible to this potentially dangerous environment, which further damaged the host health and production performance (11, 12). Thus, the above findings indicate that HC feeding may alter the microbial composition and its metabolic functions in the hindgut of dairy cows. In addition to the host health, the enteric microbiota of dairy cows is related to food safety, fecal pollution and is also identified as the source of various microbial contamination, such as soil and water reservoirs (13, 14). Hence, a better understanding of how HC diets affect the hindgut microbiome is important in reducing fecal pollution and the related subsequent environmental problems. Moreover, taking feces through the rectum is less damaging to dairy cows and easier to harvest than the rumen contents. Therefore, enriching the information about fecal microbiome and the remodeling effects of HC diets could contribute to host health and environmental optimization and provide the possibility to use the characteristic microbial changes to reflect the effects of HC diets on the host response of dairy cows in the future.
Nowadays, the application of metagenomics has largely advanced our knowledge of the gastrointestinal microbiome. However, published studies that investigated the effects of HC diets on the hindgut microbial changes in dairy cows were limited to the methods of quantitative PCR or 16S rRNA sequencing (15) and could not unravel the alterations in microbial structure and its potential metabolic functions synchronously. This study, therefore, aimed to reveal the effects of HC diets on the fecal microbiome and their overall metabolic functions using a metagenomics approach, and the results of the present study may benefit in gaining a deeper understanding of the impact of HC feeding on the hindgut microbiota of dairy cows.
Materials and Methods
Animals, Feeding Diets, and Experimental Design
The present study was part of a series of studies on the impact of HC diets on the gastrointestinal tract of dairy cows, and detailed and complete animal experimental protocols have been provided previously (16, 17). In brief, the 2 × 2 crossover experimental design (two treatments and two periods) was applied in the present study. Four healthy and lactating Holstein dairy cows (460 ± 16.4 kg body weight and 84 ± 25 days in milk) with long-established fistulas were randomly allocated to the following two different experimental diets: low-concentrate (LC) diets (40% concentrate feed, DM) or HC diets (70% concentrate feed, DM). The detailed ingredients and nutrition levels of experimental diets in the present study are presented in Supplementary Table S1. Before the experiment began, all cows were fed LC diets (30% concentrate feed, DM) for 3 weeks. For cows fed HC diets, the dietary concentrate level was gradually increased to 70% in the first 2 days. During the whole feeding period, a total mixed ration was available ad libitum for experimental dairy cows and free access to water. In each experimental period, the dairy cows were fed the corresponding treatment diets for 21 days, and fecal samples were harvested by the rectal method at 4 h after morning feeding on the last day of each period.
The collected samples were stored in an −80°C experimental refrigerator until DNA extraction was performed.
DNA Extraction, Pyrosequencing, and Functional Annotations
Metagenomic DNA was isolated utilizing a QIAamp DNA Mini Kit (Qiagen, Valencia, CA, USA) in accordance with the standard extraction protocol provided by the manufacturer. To obtain efficiently extracted quality DNA from fecal samples, the bead-beating process was adopted to mechanically break the microbial cell wall. The above-extracted DNA has measured the extracted quality and quantified concentration by a Nanodrop Spectrophotometer (Nyxor Biotech, Paris, France). Finally, DNA samples were stored at −80°C until subsequent assays.
Total genomic DNA was fragmented by the Covaris M220 Focused-ultrasonicator (Covaris Inc., Woburn, MA, USA), and the sequencing libraries were constructed using a TruSeq™ DNA Sample Prep Kit (Illumina, San Diego, CA, USA) and sequenced on an Illumina HiSeq PE 150 platform. To harvest the clean data, the quality control of original sequencing data was conducted to remove adapters and low-quality reads using Trimmomatic (18). The retained reads were then mapped to Bos taurus ARS-UCD1.2 to remove the host-genome contaminations through the Burrows–Wheeler Aligner (BWA, V0.7.12) (19). The metagenome assembles of clean data were conducted by Megahit (V1.1.1) (20). Only the assembled contigs longer than 500 bp were adopted for subsequent analysis. The open reading frame (ORF) prediction was performed using the software MetaGeneMark (V2.10) (21), and the CD-HIT (V4.5.8) (22) was adopted to remove redundancy and constructed the initial gene catalog. The species annotations were harvested by blasting the Unigenes with the Non-Redundant (NR) database in the National Center for Biotechnology Information (NCBI). For further analysis, the Unigenes were blasted with the Kyoto Encyclopedia of Genes and Genomes (KEGG) and the Carbohydrate-active Enzymes (CAZy) to harvest the functional annotations information.
Statistical Analysis
Statistics on microbial taxonomic data and the relative abundance of KEGG Orthology (KO) genes and CAZy were conducted using the general linear model (GLM) in IBM SPSS statistics V20.0.0 (IBM Corp., Armonk, NY, USA). The statistical model applied in the present study was as follows: yi = μ + S + P + SP + ei. Where μ represents the overall mean, S represents the dietary treatment, P represents the fixed effect of the experimental period, SP represents the interactions between the diet treatment and period, and ei represents the random residual. The correction of values of p obtained by the above statistical model was performed through the false discovery rate (FDR), and the corrected p < 0.05 was defined as significant.
The principal coordinate analysis (PCoA) and the analysis of molecular variance (AMOVA) were run within R software packages (23) to evaluate the sample distribution and statistical differences between the groups. To identify the characteristic changes at the species level in fecal samples from different dietary treatments, the linear discriminant analysis effect size (LEfSe) was conducted. The hierarchical analysis of KEGG pathways (level 3) was conducted with the Number Cruncher Statistical System (NCSS, version 12.0.2; Kaysville, UT, USA).
Results
Microbial Ecology of the Fecal Microbiota in Dairy Cows
At the domain level (Figure 1A), the fecal microbial community in the LC and HC groups was dominated by bacteria (95.73 vs. 98.55%), followed by archaea (3.41 vs. 0.94%) and viruses (0.79 vs. 0.49%), and eukaryote was the least with <0.1% relative abundance (0.07 vs. 0.02%). At the phylum level, the fecal microbial community in the LC and HC groups was dominated by Firmicutes (57.43 vs. 54.89%), Bacteroidetes (24.72 vs. 28.93%), Proteobacteria (2.83 vs. 3.17%), Spirochaetes (2.15 vs. 0.83%), and Tenericutes (1.63 vs. 1.87%). At the genus level (Figure 1B), unclassified Firmicutes (10.25 vs. 13.08%), Clostridium (11.18 vs. 11.62%), Bacteroides (9.46 vs. 11.12%), Prevotella (4.24 vs. 10.79%), unclassified Lachnospiraceae (2.85 vs. 3.36%), Ruminococcus (4.12 vs. 2.04%), Alistipes (3.73 vs. 2.30%), Eubacterium (2.62 vs. 1.93%), Methanobrevibacter (3.01 vs. 0.61%), and Roseburia (0.84 vs. 2.17%) were dominated in the fecal microbial community in the LC and HC groups.
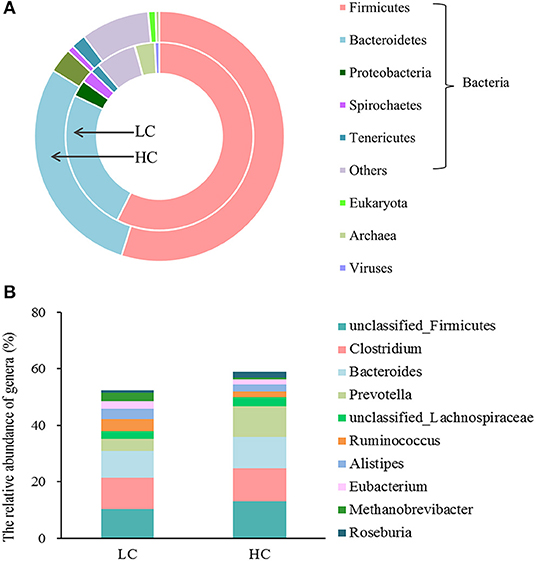
Figure 1. The overview of the fecal microbial composition of dairy cows at the level of the kingdom (A) and genus (B). Only the 5 most abundant phylum and the 10 most abundant genera in bacteria were shown in the figure.
The Fecal Microbial Community in HC-Fed Cows
Through the metagenomics analysis, we evaluated the effects of HC diets on the fecal microbial communities at different taxonomic levels. Of phyla (>0.05%; Figure 2A), a decrease in the relative abundance of Euryarchaeota (Archaea; p = 0.012), Candidatus Saccharibacteria (p = 0.014), Fibrobacteres (p = 0.021), Parcubacteria (p = 0.024), Synergistetes (p = 0.027), and Ascomycota (Eukaryota; p = 0.024) were found in HC-fed cows compared with LC-fed cows. Of genera (>0.1%) showing significant (Figure 2B), a decrease in the relative abundance of Ruminococcus (p = 0.033), Methanobrevibacter (Archaea; p = 0.028), Ruminiclostridium (p = 0.031), Bradyrhizobium (p = 0.033), Fibrobacter (p = 0.033), Candidatus Saccharimonas (p = 0.019), Dysgonomonas (p = 0.041), Paludibacter (p = 0.034), Candidatus Soleaferrea (p = 0.036), Butyricicoccus (p = 0.032), and 4 unclassified genera and an increase in unclassified Clostridiaceae (p = 0.049) were observed in HC-fed cows when compared with LC-fed cows. Besides, we also found the relative abundance of genera Methanosarcina and Methanosphaera (<0.1%, data not shown), belonging to Archaea, was also decreased (p < 0.05) by HC diets compared with LC diets.
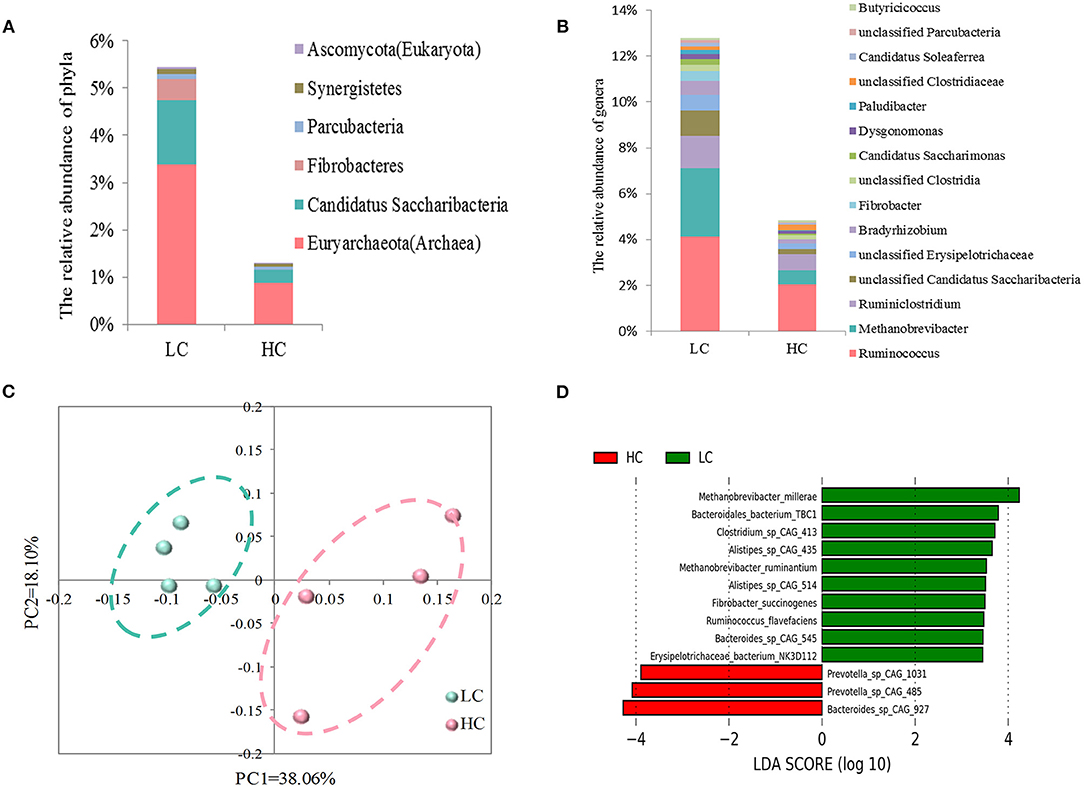
Figure 2. Effects of high-concentrate (HC) diets on the fecal microbial communities in dairy cows at different taxonomic levels. Comparison of the fecal microbial communities at the level of phylum (A) and genus (B). Only the phyla and genera that were significantly affected by dietary treatments were presented. (C) The principal coordinate analysis (PCoA) of fecal microbial functions at the species level. (D) Histogram of linear discriminant analysis (LDA) scores for abundant species between the LC and HC groups.
At the species level, the results of PCoA (Figure 2C) revealed that the LC samples were separated clearly from HC samples without any overlap, and the statistical analysis of AMOVA showed that this difference in LC and HC groups was significant (Fs = 3.164, p = 0.032). Then, LEfSe was adopted to identify the key species responsible for the differences between the LC and HC groups (Figure 2D). Bacteroides sp. CAG: 927, Prevotella sp. CAG: 485, and Prevotella sp. CAG: 1031, which were enriched in the HC group, and Methanobrevibacter millerae (Archaea), Methanobrevibacter ruminantium (Archaea), Bacteroidales bacterium TBC1, Bacteroides sp. CAG: 545, Clostridium sp. CAG: 413, Alistipes sp. CAG: 514, Alistipes sp. CAG: 435, Fibrobacter succinogenes, Ruminococcus flavefaciens, and Erysipelotrichaceae bacterium NK3D112, which were enriched in the LC group, were the dominant species that contributed to the microbial differences between the two groups.
Alterations in the Profiles of CAZy After HC Feeding
Due to the degradation process of carbohydrates requiring the participation of multiple enzymes, we focused on analyzing the differences in the profiles of CAZy between the LC- and HC-fed cows. At the class level (Figure 3A), the carbohydrate-binding module (CBM) was less abundant (p = 0.023) in the fecal microbiota of HC-fed cows compared with those in LC-fed cows. However, the dietary treatments did not affect the relative abundance of glycoside hydrolases (GH), glycosyltransferases (GT), carbohydrate esterases (CE), auxiliary activity (AA), polysaccharide lyases (PL), S-layer homology domain (SLH), dockerin, and cohesion.
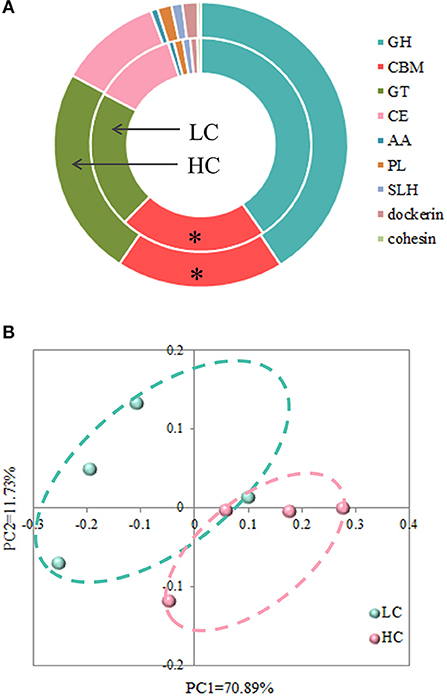
Figure 3. Effects of high-concentrate (HC) diets on profiles of carbohydrate-active enzymes at the class (A) and phylum (B) levels. *p < 0.05.
At the phylum level, the PCoA with Bray-Curtis distance (Figure 3B) suggested that the LC group was approximately separated from the HC group, and AMOVA analysis confirmed that these differences in the two groups were reached at statistical tendency (Fs = 3.26747, p = 0.085). For further understanding of the effects of dietary treatments on CAZy, we conducted a statistical analysis of the relative abundance of enzymes (Table 1). The results revealed that there were 24 abundant enzymes that (>0.01% relative abundance) were affected by HC diets, among them, the relative abundance of 1 oligosaccharide-degrading enzyme (GH32), 2 N-acetylglucosaminidase (GH85 and GH101), 1 mycodextranase (GH87), and 3 GTs (GT20, GT39, and GT83) were increased (p < 0.05) by HC diets when compared with LC diets. Meanwhile, the remaining 21 abundant enzymes, i.e., 1 oligosaccharide-degrading enzyme (GH17), 3 endo-hemicellulases (GH10, GH11, and GH54), 2 cellulases (GH1, GH44, and GH45), 1 N-acetylmuramidase (GH108), 3 CBMs (CBM16, CBM44, and CBM59), 3 CEs (CE12, CE13, and CE15), 2 PLs (PL9 and PL14), and 1 AA (AA4) were decreased (p < 0.05) by HC diets when compared with LC diets.
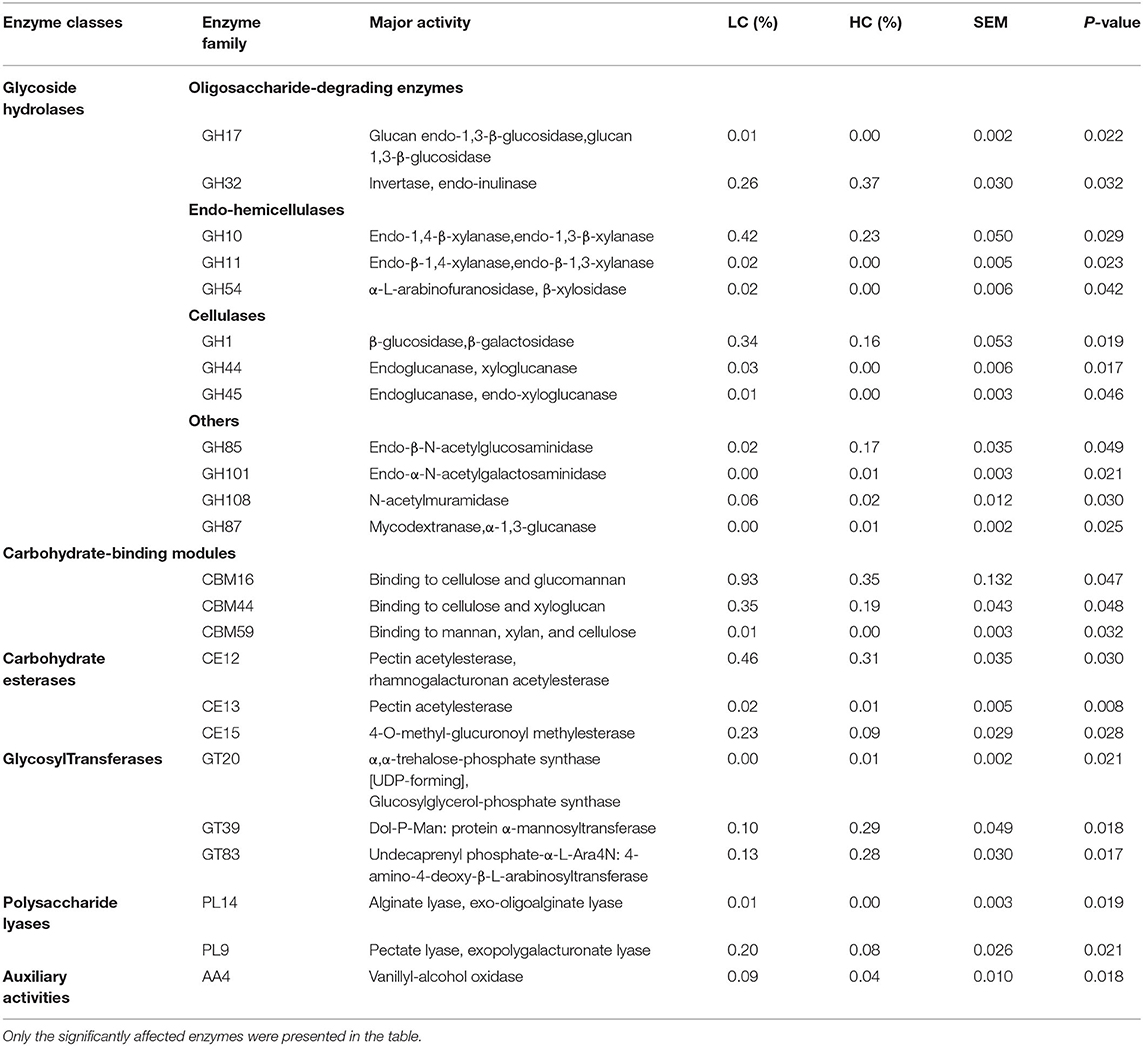
Table 1. The effects of high-concentrate (HC) diets on the relative abundance of abundant carbohydrate-active enzymes (>0.01% in at least one group) in the fecal samples of dairy cows.
Differences in KEGG Profiles of Fecal Microbiota After HC Feeding
For further understanding of how HC diets affected the microbial functions in the hindgut of dairy cows, the KEGG profiles (level 2 and level 3) were analyzed. At KEGG level 2 (Figure 4A), the results showed that the “Lipid metabolism” was enriched in the HC group, while the “Excretory system,” “Excretory system,” “Cardiovascular diseases,” “Transcription,” and “Translation” were enriched in the LC group. At the KEGG level 3, the results of PCoA and AMOVA (Supplementary Figure S1A) revealed that there were certain differences in the profiles of KEGG pathways between the samples of two groups in the present study (Fs = 7.00599, p = 0.067). There were 62 KEGG pathways affected by different dietary treatments (Supplementary Figure S1B), and after classification and re-carding, the main altered KEGG pathways are listed in Figure 4B. The results showed that two “Carbohydrate metabolism” pathways, three “Energy metabolism” pathways, five “Amino acid metabolism” pathways, eight “Lipid metabolism,” the pathway “Carbon metabolism,” and the pathway “Microbial metabolism in diverse environments” were affected by different dietary treatments in the present study. Among these main affected KEGG pathways, the pathways “Glycolysis/Gluconeogenesis,” “Carbon fixation pathways in prokaryotes,” “Methane metabolism,” “Tryptophan metabolism,” “Phenylalanine metabolism,” “Secondary bile acid biosynthesis”, and “Primary bile acid biosynthesis” were enriched in the LC group, and the other pathways were enriched in the HC group.
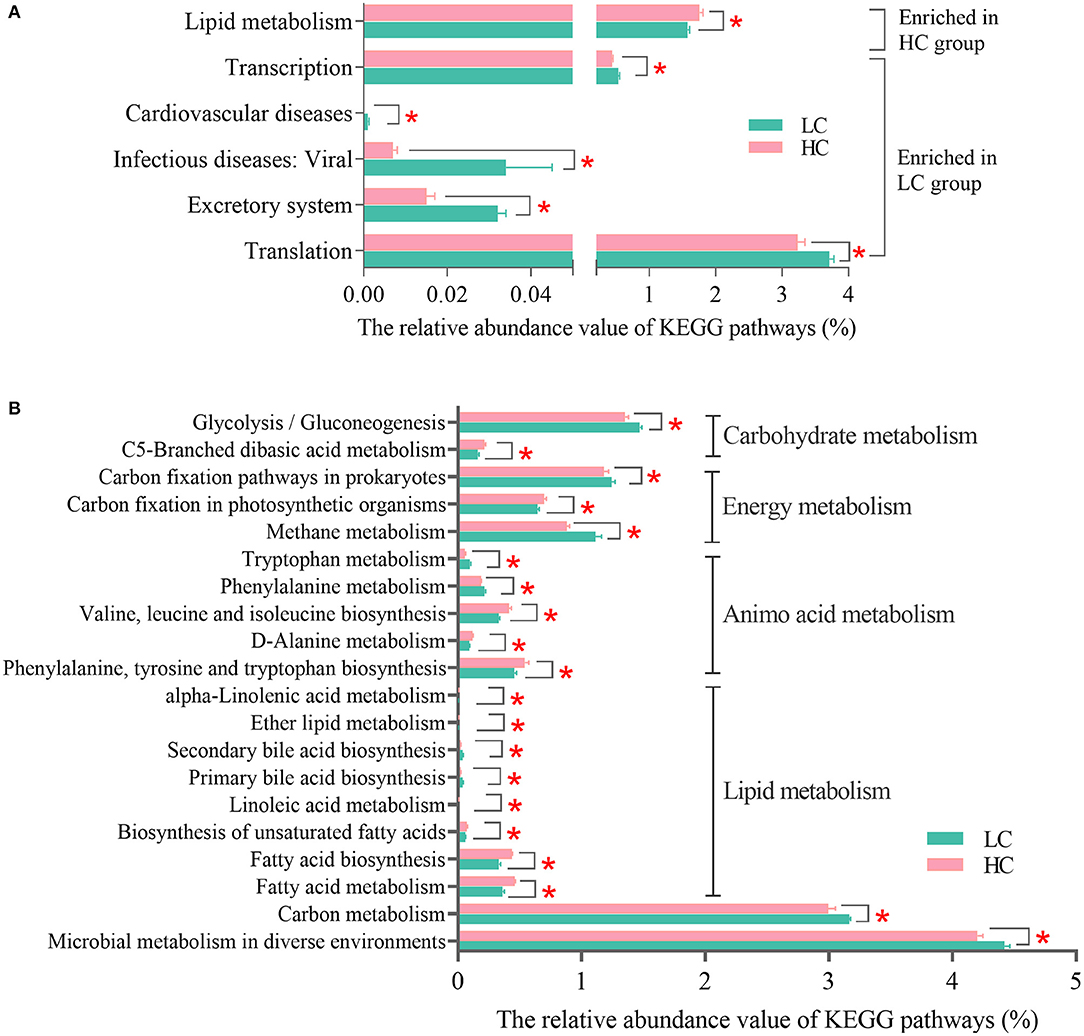
Figure 4. Effects of high-concentrate (HC) diets on KEGG profiles of the fecal microbiota of dairy cows at level 2 (A) and level 3 (B). KEGG, Kyoto Encyclopedia of Genes and Genomes. *p < 0.05.
Microbial Gene Abundance Analysis of Fecal Microbiota in HC-Fed Cows
In the present study, the KO genes significantly affected by diets were imported into the KEGG Mapper for visualization, and the results showed that 67 differential genes were related to methane metabolism. Since the methane in the gastrointestinal tract is mainly produced through the hydrogenotrophic route (predominant), the acetoclastic route, and the methylotrophic route (24), and hence, we separately screened the genes involved in these three routes. As for the hydrogenotrophic route (Figures 5A,B), the relative abundance of all genes encoding catalytic enzymes related to the reduction reactions of CO2/H2 to methane, such as formate dehydrogenase (Fdh, EC: 1.2.1.2), formylmethanofuran dehydrogenase (Fmd, EC: 1.2.99.5), formylmethanofuran-tetrahydromethanopterin N-formyltransferase (Ftr, EC: 2.3.1.101), methenyltetrahydromethanopterin cyclohydrolase (Mch, EC: 3.5.4.27), methylenetetrahydromethanopterin dehydrogenase (Mtd-Hmd, EC: 1.5.98.1), 5,10-methylenetetrahydromethanopterin reductase (Mer, EC: 1.5.98.2), tetrahydromethanopterin S-methyltransferase (Mtr, EC: 2.1.1.86), and methyl-coenzyme M reductase (Mcr, EC: 2.8.4.1), was decreased (p < 0.05) by HC diets when compared with LC diets. In addition, the relative abundance of genes encoding coenzyme F420 hydrogenase (Frh, EC: 1.12.98.1) and heterodisulfide reductase (Hdr, EC: 1.8.98.1) was also decreased (p < 0.05) after HC-diets feeding when compared with LC-diets feeding. As for the acetoclastic route and the methylotrophic route (Figures 5C,D), in addition to genes encoding Mtr and Mcr, which were also presented in the hydrogenotrophic route, only the relative abundance of genes encoding methanol-5-hydroxybenzimidazolylcobamide Co-methyltransferase (MtaB), methanol corrinoid protein (MtaC), and trimethylamine-corrinoid protein Co-methyltransferase (MttB) was decreased (p < 0.05), and the gene encoding trimethylamine corrinoid protein (MttC) was increased (p < 0.05) after HC-diets feeding when compared with LC-diets feeding.
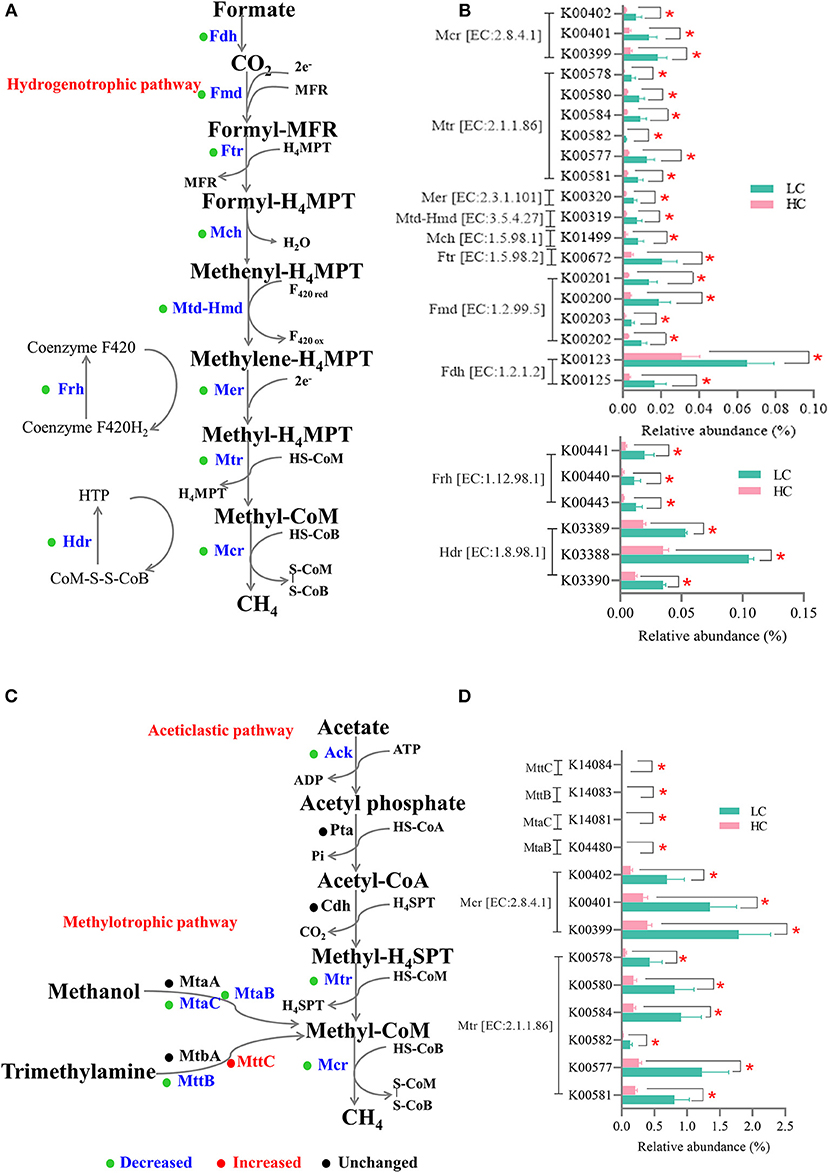
Figure 5. Effects of high-concentrate (HC) feeding on microbial genes involved in methanogenesis of dairy cows. The enzymes involved in the methanogenesis pathway—the hydrogenotrophic route (A) and the acetoclastic route, and the methylotrophic route (C). The relative abundance of microbial genes involved in the hydrogenotrophic route (B) and the acetoclastic and methylotrophic route (D). The blue font indicated that the relative abundance of this enzyme was significantly reduced in the HC group. *p < 0.05.
As for the microbial genes related to lipid metabolism (Figure 6), the relative abundance of genes encoding acyl-CoA dehydrogenase (Acd, EC: 1.3.8.7), enoyl-CoA hydratase (EchA, EC: 4.2.1.17), 3-hydroxyacyl-CoA dehydrogenase/enoyl-CoA hydratase/3-hydroxybutyryl-CoA epimerase (Fadj, EC: 1.1.1.35/4.2.1.17/5.1.2.3), and choloylglycine hydrolase (Cgh, EC: 3.5.1.24) was decreased (p < 0.05), whereas the relative abundance of genes encoding acetyl-CoA carboxylase, biotin carboxylase subunit (AccC, EC: 6.4.1.2), acetyl-CoA carboxylase carboxyl transferase subunit alpha (AccA, EC: 6.4.1.2), acetyl-CoA carboxylase biotin carboxyl carrier protein (AccB), and phospholipase A1 (PldA, EC: 3.1.1.32/3.1.1.4) was increased (p < 0.05) in the HC group when compared with the LC group.
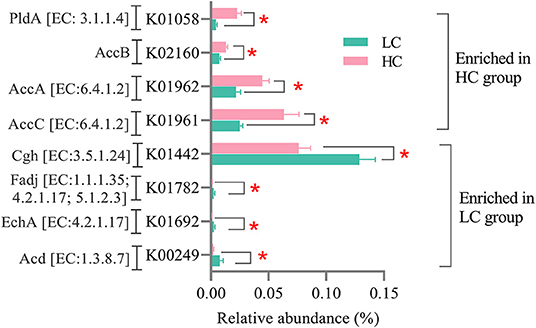
Figure 6. Effects of high-concentrate (HC) feeding on microbial genes involved in the lipid metabolism of dairy cows. *p < 0.05.
For the microbial genes related to amino acid metabolism (Supplementary Figure S2) and glycolysis/gluconeogenesis (Supplementary Figure S3), the relative abundance of affected microbial genes related to the pathways “Tryptophan metabolism” and “Phenylalanine metabolism” was all decreased (p < 0.05) in the HC group when compared with the LC group. The relative abundance of fructose-bisphosphate aldolase (Fba, EC: 4.1.2.13), cyclohexadienyl/prephenate dehydrogenase (TyrC, EC: 1.3.1.43), chorismate mutase (AroH, EC: 5.4.99.5), chorismate mutase (PheA, EC: 5.4.99.5), and 3-dehydroquinate synthase II (Dds, EC: 1.4.1.24) in the pathway “Phenylalanine, tyrosine, and tryptophan biosynthesis” and the leucine dehydrogenase (LeuDH, EC: 1.4.1.9) and branched-chain amino acid aminotransferase (IlvE, EC: 2.6.1.42) in the pathway “Valine, leucine and isoleucine biosynthesis” was decreased (p < 0.05), while the relative abundance of the other affected microbial genes in these two pathways was increased (p < 0.05) in the HC group when compared with the LC group.
Discussion
In the modern dairy farming industry, HC feeding was often used to maintain the high production performance of dairy cows, but some health problems ensued, and these problems were closely related to microbial fermentation and metabolism (3). Our previous study had confirmed that HC diets resulted in an increase in starch content in the hindgut of dairy cows, which in turn caused the accumulation of VFAs and pH decline (7). However, compared with the rumen, there is a paucity of information on the microbial composition and its metabolic functions in the hindgut of dairy cows after HC feeding. Hence, in the present study, a metagenomics approach was applied to ascertain the ability of HC diets to reshape the architecture (at the microbial phylum, genus, and species level) and functions (CAZy, KEGG pathways, and microbial genes) of hindgut microbiota in dairy cows. Our findings may provide a global understanding of the HC diets on microbial composition and functions in the hindgut of dairy cows.
The Hindgut Adapts to HC Diets by Remodeling the Microbial Composition, Especially Cellulolytic Bacteria and Methanogens
After long-term co-evolution, the animal gastrointestinal tract has formed a stable and complex micro-ecosystem in which bacteria, archaea, eukaryotes, and viruses coexist (25). Our results showed that similar to the rumen microbial structure, bacteria occupied an absolutely dominant position (>95%), and archaea were in a subordinate position of the total microbes (25). At lower taxonomic levels, Firmicutes, Bacteroidetes, Proteobacteria, Spirochaetes, and Tenericutes were the five most abundant phyla, and unclassified Firmicutes, Clostridium, Bacteroides, Prevotella, and unclassified Lachnospiraceae were the five most abundant genera in the fecal microbial community of dairy cows.
Various factors have the effect of remodeling the microbial structure, with the diet and particularly dietary carbohydrates are key determinants for the composition and activity of the gastrointestinal microbiome (26). As expected, our results of Bray-Curtis PCoA and AMOVA indicated that there were significant differences in the fecal microbial community between the LC and HC groups. Indeed, we found some differences in the specific microorganisms between two groups at the levels of phylum, genus, and species, respectively. At the phylum level, a decrease in the abundance of Fibrobacteres, Parcubacteria, Synergistetes, Candidatus Saccharibacteria, Euryarchaeota (Archaea), and Ascomycota (Eukaryota) was found in the HC group when compared with that in the LC group. The phylum Fibrobacteres, its only genus Fibrobacter, and its cultured species Fibrobacter succinogenes are known as major cellulolytic degraders in the herbivore gut (27). Unsurprising, consistent with a previous study (28), the relative abundance of Fibrobacteres, Fibrobacter, and Fibrobacter succinogenes (as mentioned below) was all decreased by HC feeding. This decrease may be due to the increased starch content, decreased fiber content, and increased acidity caused by the changes in fermentable substrates entered into the hindgut (12, 28). Ascomycota, the abundant fungus in the rumen, was reported to proliferate in the rumen of dairy cows after HC feeding (29), while its relative abundance was decreased in the feces of dairy cows after HC feeding in the present study. Due to the limited information, the reason for this inconsistent result is different regions of the gastrointestinal tract or others still need further study. Euryarchaeota was the dominant phylum in the fecal archaeal community and comprised methanogenic bacteria species (30); the decreased relative abundance of this phylum in the present study indicated that HC feeding may affect methane production, but further analysis at lower taxonomic levels is required.
At lower taxonomic levels, the genera Fibrobacter, Ruminococcus, and their important species F. succinogenes and R. flavefaciens, which are considered as representative cellulolytic bacteria in the herbivore gut (31), were decreased by the HC diets in the present study. As mentioned above, these cellulolytic bacteria are more sensitive to the low pH caused by increased starch, and hence, their proliferation in the hindgut during HC feeding is inhibited. Ruminiclostridium species was demonstrated to be able to secrete extracellular multi-enzymatic complexes, which further efficiently decomposed the cellulose (32). In the present study, a decrease in the relative abundance of genus Ruminiclostridium was observed in the HC group, and this decrease may be also caused by the substrate changes in the hindgut of dairy cows. Interestingly enough, we also found the genera Methanobrevibacter, Methanosarcina, and Methanosphaera (belonging to archaea), and the important species (M. millerae and M. ruminantium) in Methanobrevibacter, were also decreased by the HC feeding in the present study. Methanobrevibacter is the most dominant archaeal genus in the rumen and feces of dairy cows (33, 34). These archaea members are responsible for methane production in the gastrointestinal tract of livestock animals (35), and hence, the decreased methanogen populations in the present study may result in less methane production from the gastrointestinal tract of dairy cows. Although we did not measure methane production in the present study, several previous studies had confirmed that increasing the proportion of concentrate in the diets could reduce methane production (36–38). Besides, there is a symbiotic relationship between methanogens and bacteria in the animal gut, most cellulolytic bacteria produced hydrogen during their metabolic process, and a positive correlation was observed between the number of cellulolytic bacteria and methanogens in the rumen of various ruminant species (39). Hence, the reduction in the relative abundance of cellulolytic bacteria in the present study may limit the available quantity of hydrogens that were converted to methane.
The Relative Abundance of CAZy Associated With the Degradation of Cellulose and Hemicellulose Was Reduced by HC Feeding
The complex degradation process of carbohydrates in HC diets requires the participation of multiple enzymes, such as GH, PL, CE, GT, AA, and CBMs. The PCoA and AMOVA analysis of CAZy at the phylum level showed that HC diets had a certain effect on fecal microbial CAZy in dairy cows. Since the different content of cellulose and starch in dietary treatments in the present study, the degradation of cellulose, hemicellulose, and sugars attracted our special attention. GHs are a widespread and extremely important group of enzymes capable of hydrolyzing the glycosidic bonds that existed in carbohydrates, and CBMs possess the carbohydrate-binding activity and, hence, contribute to the degradation of carbohydrates (40). In the present study, the relative abundance of all GHs, which are classified as hemicellulases (GH10, GH11, and GH54) and cellulases (GH1, GH44, and GH45), were reduced after HC-diets feeding. Meanwhile, the relative abundance of all affected CBMs (CBM 16, CBM 44, and CBM59), which possessed cellulose-binding activity, was also reduced after HC-diets feeding. This decrease in the abundance of this hemicellulose- and cellulose-degradation-related enzymes may be caused by substrate dependence, corresponding to the low hemicellulose and cellulose in the LC diets. However, only one oligosaccharide-degrading enzyme—GH32—was more abundant in the HC group compared with the LC group. Since starch degrades faster than fiber, it seems that starch degradation is more dependent on substrate contents rather than drastically adjusting the microbial functions. This may be one of the reasons for the above phenomenon; on the other hand, the small sample size in the present study may also mask some differences.
HC Diets Altered Microbial Functions Related to the Metabolism of Methane, Lipid, and Amino Acids in the Hindgut of Dairy Cows
Corresponding to the changes in the abovementioned cellulolytic bacteria and methanogens, we found the KEGG pathway—methane metabolism—was enriched in the LC group when compared with the HC group. Subsequently, the visualization results of the KEGG mapper showed that 67 KOs significantly affected by HC diets were related to methane metabolism in the present study. The main methanogenesis pathways in the gastrointestinal tract of livestock animals include the hydrogenotrophic route (predominant), the acetoclastic route, and the methylotrophic route (24). Our results showed that the relative abundance of 10 main microbial genes (Fdh, Fmd, Ftr, Mch, Mtd-Hmd, Mer, Mtr, Mcr, Frh, and Hdr) related to the hydrogenotrophic route and 3 microbial genes related to the acetoclastic route (Ack, Mtr, and Mcr) were decreased by HC diets in the present study; as for the methylotrophic route, 4 microbial genes (MtaB, MtaC, MttB, and Mcr) were decreased, while 1 microbial gene (MttC) was increased by HC diets in the present study. Mcr, namely, methyl-coenzyme M reductase, the final and rate-limiting step enzyme for catalyzing the methane biogenesis, was shared with these three important methanogenesis routes (24). The decrease in the relative abundance of these microbial genes was involved in methanogenesis pathways further substantively proved that HC diets abled to reduce the methane production from dairy cows. In addition, among the abovementioned changed methanogens, Methanobrevibacter utilized hydrogen, and/or formate for methanogenesis, Methanosphaera utilized methyl-containing compounds (such as methanol and methylamine) to produce methane, and Methanosarcina harbored the abovementioned three methane methanogenesis pathways (41, 42). Hence, in the present study, HC feeding resulted in a decrease in the methanogens populations and gene expressions related to methanogenesis, which further resulted in a reduction in methane production.
Our results also found that the KEGG pathway—Lipid metabolism (level 2) and its sub-pathways included fatty acid metabolism, fatty acid biosynthesis, biosynthesis of unsaturated fatty acids, linoleic acid metabolism, primary bile acid biosynthesis, and secondary bile acid biosynthesis (level 3)—was all enriched in the HC group. Previous studies showed that the biohydrogenation process of poly-unsaturated fatty acids (PUFA) in the rumen was weakened when the HC diets were fed (43, 44). This response was considered to be related to a decrease in cellulolytic bacterial populations and pH value, further causing a reduction in the hydrogen supply of ruminal biohydrogenation (43, 45). Hence, these changes in the rumen may increase the amounts of unsaturated fatty acids entering the lower intestine. Furthermore, studies on pigs and mice confirmed that increasing the starch contents in the hindgut could regulate the lipid metabolism and increase the linoleic acid contents in the hindgut (46, 47). In addition, the reduction in the cellulolytic bacterial populations in hindgut in the present study may also exacerbate the above changes. For a deeper understanding, we screened the differential microbial genes involved in lipid metabolism. A decrease in the abundance of genes Acd, EchA, and Fadj and an increase in the abundance of genes AccA, AccB, AccC, and PldA were observed in the HC group when compared with the LC group in the present study. The microbial genes Acd, EchA, and Fadj were the main enzymes of the β-oxidation pathway in the mitochondrial matrix, and each cycle of this β-oxidation resulted in the production of acetyl-CoA (48); and the AccA, AccB, and AccC were the encoding genes of acetyl-CoA carboxylase's subunits, which abled to catalyze the carboxylation of acetyl-CoA to produce malonyl-CoA in the first committed step of the fatty acid synthesis pathway (49). In addition, the gene PldA involved in the linoleic acid metabolism and abled to catalyze the lecithin to produce linoleate; and the gene Cgh, namely, choloylglycine hydrolase, abled to catalyze the deconjugation of bile acids conjugated with glycine or taurine and further assisted in fatty acids metabolism (50, 51). According to the above analysis, it seems that fatty acid synthesis was enhanced and fatty acid metabolism was weakened, which may cause the accumulation of fatty acids in the hindgut of cows fed HC diets. However, because the fatty acids in the feces were not measured in the present study, this result still needs further confirmation in the future.
In addition, our results also revealed that several KEGG pathways belonging to “Carbohydrate metabolism” and “Amino acid metabolism” were also affected by dietary treatments in the present study. In particular, the pathways “Phenylalanine metabolism,” “Tryptophan metabolism”, and “Glycolysis/Gluconeogenesis” were enriched in the LC group, and the pathways “Phenylalanine, tyrosine, and tryptophan biosynthesis” and “Valine, leucine, and isoleucine biosynthesis” were enriched in the HC group. Previous studies demonstrated that increasing the contents of fermentable carbohydrates reached pig's hindgut, the microbes habited in hindgut tended to preferentially utilize the carbohydrates, and hence weaken the fermentation of amino acids (52, 53). Hence, the increased carbon source in the hindgut of dairy cows fed HC diets in the present study may explain the weakened pathways “Phenylalanine metabolism” and “Tryptophan metabolism”, and the enhanced pathways “Phenylalanine, tyrosine, and tryptophan biosynthesis” and “Valine, leucine, and isoleucine biosynthesis”. In addition, the aromatic amino acids phenylalanine and tryptophan are both glucogenic and ketogenic amino acids (54), and their weakened metabolism and sufficient glucose supply during HC feeding may partly contribute to weakening the pathway “Glycolysis/Gluconeogenesis” in the HC group in the present study.
Conclusion
In conclusion, a metagenomics approach was applied to reveal the changes in hindgut microbiota and their metabolic functions of dairy cows. The proposed schematic sketch of how HC diets affected hindgut microbiota and their metabolic functions of dairy cows is mapped in Figure 7. Overall, during HC feeding, hindgut microbiota, especially cellulolytic bacteria (such as Fibrobacter, and Ruminococcus) and methanogens (such as Methanobrevibacter, Methanosarcina, and Methanosphaera), was decreased and correspondingly resulted in a decrease in the relative abundance of CAZy related to hemicellulases (GH10, GH11, and GH54) and cellulases (GH1, GH44, and GH45). Furthermore, the KEGG pathways “Methane metabolism” (enriched in the LC group), “Lipid metabolism” (enriched in the HC group), and several sub-pathways in “Amino acid metabolism” (such as Phenylalanine metabolism, Phenylalanine, tyrosine, and tryptophan biosynthesis) of the hindgut microbiota of dairy cows were all affected by HC diets. Therefore, it may be reasonable to speculate that, HC diets caused the remodeling of hindgut microbiota, giving priority to utilize carbon sources, reducing the decomposition of amino acids and methane production, and increasing the accumulation of fatty acids. However, more studies on how HC diets affected hindgut microbiota with larger sample sizes and hindgut metabolites are required.
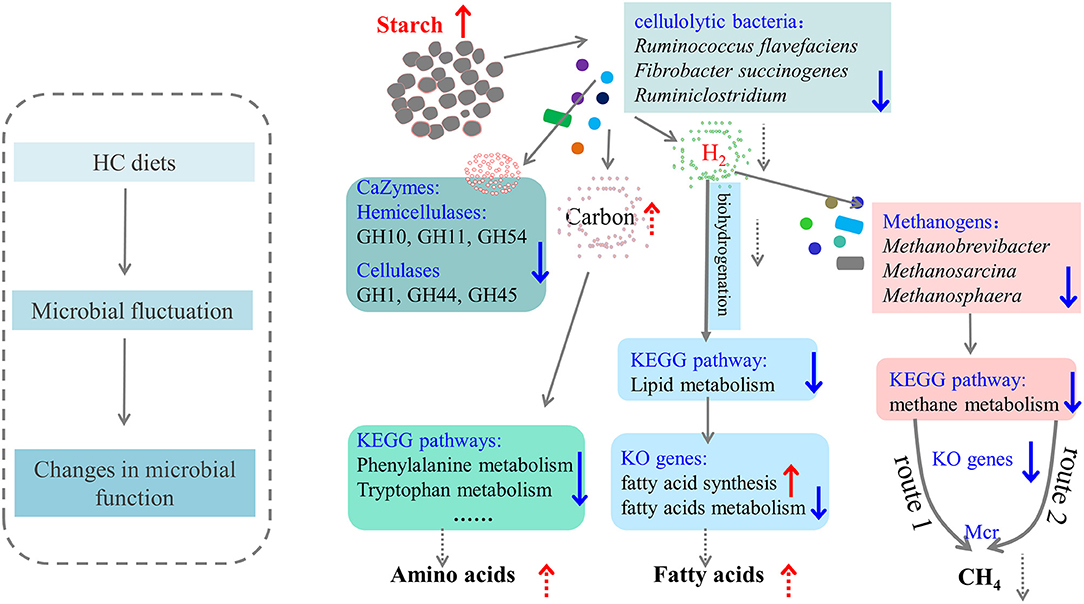
Figure 7. The proposed schematic diagram of how high-concentrate (HC) diets affected the hindgut microbiota and metabolic functions of dairy cows. The up arrows indicated this microbial taxa or function increased or enhanced in the HC group compared with the low-concentrate group. The dotted line was the speculated concept based on these results in the present study, which needed to be confirmed by future studies.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA641261.
Ethics Statement
The animal study was reviewed and approved by Nanjing Agricultural University Animal Care and Use Ethics Committee.
Author Contributions
SM and XW: designed the present experiment. RZ and JL: performed the experiment, analyzed the data, and wrote the manuscript. SM, XW, and LJ: revised the manuscript. All the authors had read and approved the final manuscript.
Funding
This study was funded by the Fundamental Research Funds for the Central Universities (JCQY201905), the Open Project of Beijing Key Laboratory of Dairy Cow Nutrition, Beijing University of Agriculture, China, the Fundamental Research Projects of Colleges and Universities of Liaoning Province of China (No. LSNQN201706), and the National Natural Science Foundation of China (No. 31360558).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnut.2021.809406/full#supplementary-material
References
1. Malmuthuge N, Guan LL. Gut microbiome and omics: a new definition to ruminant production and health. Animal Frontiers. (2016) 6:8–12. doi: 10.2527/af.2016-0017
2. O'Hara E, Neves AL, Song Y, Guan LL. The role of the gut microbiome in cattle production and health: driver or passenger? Ann Rev Animal Biosci. (2020) 8:199–220. doi: 10.1146/annurev-animal-021419-083952
3. Kleen J, Hooijer G, Rehage J, Noordhuizen J. Subacute ruminal acidosis (SARA): a review. J Vet Med Ser A. (2003) 50:406–14. doi: 10.1046/j.1439-0442.2003.00569.x
4. Plaizier J, Krause D, Gozho G, McBride B. Subacute ruminal acidosis in dairy cows: the physiological causes, incidence and consequences. Vet J. (2008) 176:21–31. doi: 10.1016/j.tvjl.2007.12.016
5. Petri R, Forster R, Yang W, McKinnon J, McAllister T. Characterization of rumen bacterial diversity and fermentation parameters in concentrate fed cattle with and without forage. J Appl Microbiol. (2012) 112:1152–62. doi: 10.1111/j.1365-2672.2012.05295.x
6. Khafipour E, Li S, Plaizier JC, Krause DO. Rumen microbiome composition determined using two nutritional models of subacute ruminal acidosis. Appl Environ Microbiol. (2009) 75:7115–24. doi: 10.1128/AEM.00739-09
7. Mao S, Zhang R, Wang D, Zhu W. The diversity of the fecal bacterial community and its relationship with the concentration of volatile fatty acids in the feces during subacute rumen acidosis in dairy cows. BMC Vet Res. (2012) 8:237. doi: 10.1186/1746-6148-8-237
8. Li S, Khafipour E, Krause DO, Kroeker A, Rodriguez-Lecompte JC, Gozho GN, et al. Effects of subacute ruminal acidosis challenges on fermentation and endotoxins in the rumen and hindgut of dairy cows. J Dairy Sci. (2012) 95:294–303. doi: 10.3168/jds.2011-4447
9. Diez-Gonzalez F, Callaway TR, Kizoulis MG, Russell JB. Grain feeding and the dissemination of acid-resistant Escherichia coli from cattle. Science. (1998) 281:1666–8. doi: 10.1126/science.281.5383.1666
10. Khafipour E, Li S, Tun H, Derakhshani H, Moossavi S, Plaizier J. Effects of grain feeding on microbiota in the digestive tract of cattle. Animal Front. (2016) 6:13–9. doi: 10.2527/af.2016-0018
11. Liu J, Xu T, Zhu W, Mao S. High-grain feeding alters caecal bacterial microbiota composition and fermentation and results in caecal mucosal injury in goats. Br J Nutr. (2014) 112:416–27. doi: 10.1017/S0007114514000993
12. Plaizier JC, Khafipour E, Li S, Gozho GN, Krause DO. Subacute ruminal acidosis (SARA), endotoxins and health consequences. Anim Feed Sci Technol. (2012) 172:9–21. doi: 10.1016/j.anifeedsci.2011.12.004
13. Romero-Pérez GA, Ominski KH, McAllister TA, Krause DO. Effect of environmental factors and influence of rumen and hindgut biogeography on bacterial communities in steers. Appl Environ Microbiol. (2011) 77:258–68. doi: 10.1128/AEM.01289-09
14. Shanks OC, Kelty CA, Archibeque S, Jenkins M, Newton RJ, McLellan SL, et al. Community structures of fecal bacteria in cattle from different animal feeding operations. Appl Environ Microbiol. (2011) 77:2992–3001. doi: 10.1128/AEM.02988-10
15. Plaizier JC Li S, Tun HM, Khafipour E. Nutritional models of experimentally-induced subacute ruminal acidosis (SARA) differ in their impact on rumen and hindgut bacterial communities in dairy cows. Front Microbiol. (2017) 7:2128. doi: 10.3389/fmicb.2016.02128
16. Mao S, Zhang R, Wang D, Zhu W. Impact of subacute ruminal acidosis (SARA) adaptation on rumen microbiota in dairy cattle using pyrosequencing. Anaerobe. (2013) 24:12–9. doi: 10.1016/j.anaerobe.2013.08.003
17. Zhang R, Zhu W, Jiang L, Mao S. Comparative metabolome analysis of ruminal changes in Holstein dairy cows fed low-or high-concentrate diets. Metabolomics. (2017) 13:74. doi: 10.1007/s11306-017-1204-0
18. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. (2014) 30:2114–20. doi: 10.1093/bioinformatics/btu170
19. Li H, Durbin R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics. (2010) 26:589–95. doi: 10.1093/bioinformatics/btp698
20. Li D, Liu CM, Luo R, Sadakane K, Lam TW, MEGAHIT. an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics. (2015) 31:1674–6. doi: 10.1093/bioinformatics/btv033
21. Zhu W, Lomsadze A, Borodovsky M. Ab initio gene identification in metagenomic sequences. Nucleic Acids Res. (2010) 38:e132. doi: 10.1093/nar/gkq275
22. Fu L, Niu B, Zhu Z, Wu S, Li W, CD-HIT. accelerated for clustering the next-generation sequencing data. Bioinformatics. (2012) 28:3150–2. doi: 10.1093/bioinformatics/bts565
23. Team RC. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria. (2013)
24. Shi W, Moon CD, Leahy SC, Kang D, Froula J, Kittelmann S, et al. Methane yield phenotypes linked to differential gene expression in the sheep rumen microbiome. Genome Res. (2014) 24:1517–25. doi: 10.1101/gr.168245.113
25. Jose VL, Appoothy T, More RP, Arun AS. Metagenomic insights into the rumen microbial fibrolytic enzymes in Indian crossbred cattle fed finger millet straw. AMB Expr. (2017) 7:13. doi: 10.1186/s13568-016-0310-0
26. Wang W, Hu H, Zijlstra RT, Zheng J, Gänzle MG. Metagenomic reconstructions of gut microbial metabolism in weanling pigs. Microbiome. (2019) 7:48. doi: 10.1186/s40168-019-0662-1
27. Ransom-Jones E, Jones DL, McCarthy AJ, McDonald JE. The fibrobacteres: an important phylum of cellulose-degrading bacteria. Microb Ecol. (2012) 63:267–81. doi: 10.1007/s00248-011-9998-1
28. Plaizier JC Li S, Danscher AM, Derakshani H, Andersen PH, Khafipour E. Changes in microbiota in rumen digesta and feces due to a grain-based subacute ruminal acidosis (SARA) challenge. Microb Ecol. (2017) 74:485–95. doi: 10.1007/s00248-017-0940-z
29. Han X, Li B, Wang X, Chen Y, Yang Y. Effect of dietary concentrate to forage ratios on ruminal bacterial and anaerobic fungal populations of cashmere goats. Anaerobe. (2019) 59:118–25. doi: 10.1016/j.anaerobe.2019.06.010
30. Zhang J, Shi H, Wang Y, Cao Z, Yang H, Li S. Effect of limit-fed diets with different forage to concentrate ratios on fecal bacterial and archaeal community composition in holstein heifers. Front Microbiol. (2018) 9:976. doi: 10.3389/fmicb.2018.00976
31. Koike S, Kobayashi Y. Fibrolytic rumen bacteria: their ecology and functions. Asian-Australas J Anim Sci. (2009) 22:131–8. doi: 10.5713/ajas.2009.r.01
32. Fosses A, Maté M, Franche N, Liu N, Denis Y, Borne R, et al. A seven-gene cluster in Ruminiclostridium cellulolyticum is essential for signalization, uptake and catabolism of the degradation products of cellulose hydrolysis. Biotechnol biofuels. (2017) 10:250. doi: 10.1186/s13068-017-0933-7
33. Pitta DW, Indugu N, Kumar S, Vecchiarelli B, Sinha R, Baker LD, et al. Metagenomic assessment of the functional potential of the rumen microbiome in Holstein dairy cows. Anaerobe. (2016) 38:50–60. doi: 10.1016/j.anaerobe.2015.12.003
34. Holman DB, Hao X, Topp E, Yang HE, Alexander TW. Effect of co-composting cattle manure with construction and demolition waste on the archaeal, bacterial, and fungal microbiota, and on antimicrobial resistance determinants. PLoS ONE. (2016) 11:e0157539. doi: 10.1371/journal.pone.0157539
35. Kumar S, Indugu N, Vecchiarelli B, Pitta DW. Associative patterns among anaerobic fungi, methanogenic archaea, and bacterial communities in response to changes in diet and age in the rumen of dairy cows. Front Microbiol. (2015) 6:781. doi: 10.3389/fmicb.2015.00781
36. Aguerre MJ, Wattiaux MA, Powell J, Broderick GA, Arndt C. Effect of forage-to-concentrate ratio in dairy cow diets on emission of methane, carbon dioxide, and ammonia, lactation performance, and manure excretion. J Dairy Sci. (2011) 94:3081–93. doi: 10.3168/jds.2010-4011
37. Mc Geough E. O'kiely P, Hart K, Moloney A, Boland T, Kenny D. Methane emissions, feed intake, performance, digestibility, and rumen fermentation of finishing beef cattle offered whole-crop wheat silages differing in grain content. J Anim Sci. (2010) 88:2703–16. doi: 10.2527/jas.2009-2750
38. Knapp J, Laur G, Vadas P, Weiss W, Tricarico J. Invited review: enteric methane in dairy cattle production: quantifying the opportunities and impact of reducing emissions. J Dairy Sci. (2014) 97:3231–61. doi: 10.3168/jds.2013-7234
39. Morvan B, Bonnemoy F, Fonty G, Gouet P. Quantitative determination of H 2-utilizing acetogenic and sulfate-reducing bacteria and methanogenic archaea from digestive tract of different mammals. Curr Microbiol. (1996) 32:129–33. doi: 10.1007/s002849900023
40. Suryawanshi PR, Badapanda C, Singh KM, Rathore A. Exploration of the rumen microbial diversity and carbohydrate active enzyme profile of black Bengal goat using metagenomic approach. Anim Biotechnol. (2019) 10:1–14 doi: 10.1080/10495398.2019.1609489
41. Xue F, Wang Y, Zhao Y, Nan X, Hua D, Sun F, et al. Ruminal methanogenic responses to the thiamine supplementation in high-concentrate diets. Animals. (2020) 10:935. doi: 10.3390/ani10060935
42. Lambie SC, Kelly WJ, Leahy SC Li D, Reilly K, McAllister TA, et al. The complete genome sequence of the rumen methanogen Methanosarcina barkeri CM1. Stand Genomic Sci. (2015) 10:57. doi: 10.1186/s40793-015-0038-5
43. Loor JJ, Ueda K, Ferlay A, Chilliard Y, Doreau M. Biohydrogenation, duodenal flow, and intestinal digestibility of trans fatty acids and conjugated linoleic acids in response to dietary forage:concentrate ratio and linseed oil in dairy cows. J Dairy Sci. (2004) 87:2472–85. doi: 10.3168/jds.S0022-0302(04)73372-X
44. Kalscheur KF, Teter BB, Piperova LS, Erdman RA. Effect of dietary forage concentration and buffer addition on duodenal flow of trans-C18:1 fatty acids and milk fat production in dairy cows. J Dairy Sci. (1997) 80:2104–14. doi: 10.3168/jds.S0022-0302(97)76156-3
45. Chilliard Y, Glasser F, Ferlay A, Bernard L, Rouel J, Doreau M. Diet, rumen biohydrogenation and nutritional quality of cow and goat milk fat. Eur J Lipid Sci Technol. (2007) 109:828–55. doi: 10.1002/ejlt.200700080
46. Yu M, Li Z, Chen W, Rong T, Wang G, Ma X. Microbiome-metabolomics analysis investigating the impacts of dietary starch types on the composition and metabolism of colonic microbiota in finishing pigs. Front Microbiol. (2019) 10:1143. doi: 10.3389/fmicb.2019.01143
47. Lee KY, Yoo S, Lee HG. The effect of chemically-modified resistant starch, RS type-4, on body weight and blood lipid profiles of high fat diet-induced obese mice. Starch-starke. (2012) 64:78–85. doi: 10.1002/star.201100057
48. Lionetti V, Stanley WC, Recchia FA. Modulating fatty acid oxidation in heart failure. Cardiovasc Res. (2011) 90:202–9. doi: 10.1093/cvr/cvr038
49. Polyak SW, Abell AD, Wilce MCJ, Zhang L, Booker GW. Structure, function and selective inhibition of bacterial acetyl-coa carboxylase. Appl Microbiol Biotechnol. (2012) 93:983–92. doi: 10.1007/s00253-011-3796-z
50. Chand D, Ramasamy S, Suresh CG, A. highly active bile salt hydrolase from Enterococcus faecalis shows positive cooperative kinetics. Process Biochem. (2016) 51:263–9. doi: 10.1016/j.procbio.2015.12.006
51. Marchesini MI, Connolly J, Delpino MV, Baldi PC, Mujer CV, DelVecchio VG, et al. Brucella abortus choloylglycine hydrolase affects cell envelope composition and host cell internalization. PLoS ONE. (2011) 6:e28480. doi: 10.1371/journal.pone.0028480
52. Sun Y, Zhou L, Fang L, Su Y, Zhu W. Responses in colonic microbial community and gene expression of pigs to a long-term high resistant starch diet. Front Microbiol. (2015) 6:877. doi: 10.3389/fmicb.2015.00877
53. Zhou L, Fang L, Sun Y, Su Y, Zhu W. Effects of a diet high in resistant starch on fermentation end-products of protein and mucin secretion in the colons of pigs. Starch. (2017) 69:1600032. doi: 10.1002/star.201600032
Keywords: high concentrate diets, hindgut microbiota, microbial function, dairy cows, metagenomics
Citation: Zhang R, Liu J, Jiang L, Wang X and Mao S (2022) The Remodeling Effects of High-Concentrate Diets on Microbial Composition and Function in the Hindgut of Dairy Cows. Front. Nutr. 8:809406. doi: 10.3389/fnut.2021.809406
Received: 05 November 2021; Accepted: 15 December 2021;
Published: 01 February 2022.
Edited by:
Yangchun Cao, Northwest A&F University, ChinaReviewed by:
Yuheng Luo, Sichuan Agricultural University, ChinaZhi Peng Li, Institute of Special Animal and Plant Sciences (CAAS), China
Copyright © 2022 Zhang, Liu, Jiang, Wang and Mao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xinfeng Wang, d3hmLTRAMTYzLmNvbQ==; Shengyong Mao, bWFvc2hlbmd5b25nQG5qYXUuZWR1LmNu