 Zhenhua Zeng1
Zhenhua Zeng1 Qiaobing Huang
Qiaobing Huang Sheng An
Sheng An Zhongqing Chen
Zhongqing Chen Weijin Zhang
Weijin Zhang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Nutr. , 14 December 2021
Sec. Nutrition and Metabolism
Volume 8 - 2021 | https://doi.org/10.3389/fnut.2021.783164
This article is part of the Research Topic Metabolic Flexibility View all 8 articles
Anaerobic glycolysis is the process by which glucose is broken down into pyruvate and lactate and is the primary metabolic pathway in sepsis. The pyruvate dehydrogenase complex (PDHC) is a multienzyme complex that serves as a critical hub in energy metabolism. Under aerobic conditions, pyruvate translocates to mitochondria, where it is oxidized into acetyl-CoA through the activation of PDHC, thereby accelerating aerobic oxidation. Both phosphorylation and acetylation affect PDHC activity and, consequently, the regulation of energy metabolism. The mechanisms underlying the protective effects of PDHC in sepsis involve the regulation on the balance of lactate, the release of inflammatory mediators, the remodeling of tricarboxylic acid (TCA) cycle, as well as on the improvement of lipid and energy metabolism. Therapeutic drugs that target PDHC activation for sepsis treatment include dichloroacetate, thiamine, amrinone, TNF-binding protein, and ciprofloxacin. In this review, we summarize the recent findings regarding the metabolic regulation of PDHC in sepsis and the therapies targeting PDHC for the treatment of this condition.
Sepsis is a major healthcare concern caused by an aberrant response to infection, which is easily complicated with multiple organ dysfunction syndromes (1–3). Despite the considerable progress for its clinical management, sepsis-related morbidity and mortality remain high (4), highlighting the importance of developing new strategies for sepsis prevention and treatment.
Sepsis is characterized by the release of a large number of inflammatory mediators, the imbalance in energy metabolism and the accumulation of lactic acid. The mechanisms underlying sepsis pathogenesis and development are complex, and involve inflammation, immunity, and metabolism, among other processes. Recent studies have shown that in the early stages of sepsis, high level inflammation relies on anaerobic glycolysis as the key energy source, while in the recovery stage, inflammation level was lowered and the cells switch to fat oxidation for their energy supply (5).
During the process of glycolysis, one molecule of glucose is broken down into two molecules of pyruvate. Under aerobic conditions, pyruvate translocates to mitochondria, where it is oxidized into acetyl-CoA through the activation of the pyruvate dehydrogenase complex (PDHC), thereby accelerating aerobic oxidation, whereas under hypoxic conditions, pyruvate produced through anaerobic glycolysis might result in the generation of lactic acid (Figure 1).
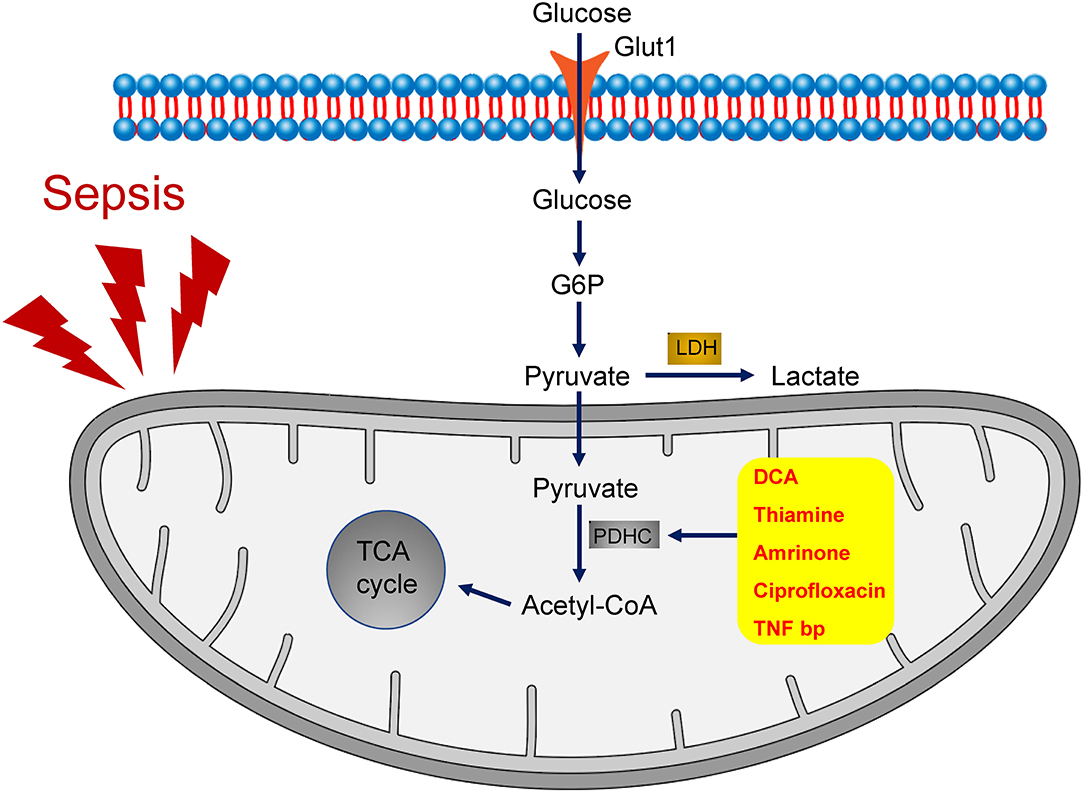
Figure 1. The role of PDHC in the metabolism of sepsis. During the process of glycolysis, one molecule of glucose is broken down into two molecules of pyruvate. Inactivation of PDHC results into anaerobic glycolysis, which is the primary metabolic pathway in sepsis. By contrast, activation of PDHC leads to pyruvate translocation to mitochondrial and the consequent acceleration of aerobic oxidation. A group of drugs that target PDHC activation, including dichloroacetate (DCA), thiamine, amrinone, ciprofloxacin, and TNF-binding protein (TNFbp), have been shown to ameliorate the symptoms of sepsis.
Although the amount of ATP produced by aerobic oxidation is significantly higher than that generated through anaerobic glycolysis, the speed of energy production via the former mechanism is significantly slower than that via the latter. Therefore, in the acute stage of sepsis, which is characterized by high energy consumption and hyper-inflammation, cells tend to generate energy to meet the significantly increased energy demands through anaerobic glycolysis, during which pyruvate converts into lactate. Furthermore, an absence of acetyl-CoA generated via pyruvate oxidation remodeled the tricarboxylic acid (TCA) cycle and lipid metabolism, with accumulation of two intermediates which could be produced through other pathways other than the physiological TCA cycle upon sepsis, namely succinate and citrate, both of which are pro-inflammatory mediators. In macrophages treated with lipopolysaccharide (LPS), succinate could be produced through anaplerosis of α-ketoglutarate into the TCA cycle and the subsequent increased glutamine metabolism. Moreover, LPS also increases γ-aminobutyric acid and its transporters, thus producing succinate. Citrate accumulates due to the shunting of cis-aconitate to itaconate through the enzymatic action of aconitase decarboxylase. Furthermore, free fatty acids and glycerol are released abundantly from adipose tissue into bloodstream, and are taken up by the liver to be oxidized into β-oxidation with the increase of acetyl-CoA, to produce more energy and ketone bodies, used as the source of energy by organs. However, this process is accompanied by the increased levels of palmitic acid and palmitoyl-carnitine, which might induce mitochondrial dysfunction. Together, lipotoxicity is also induced, characterized by the accumulation of malondialdehyde and 4-hydroxynonenal, which are the end products of lipid radical reactions and could cause cell death in liver and kidney (6–9). Following the acute stage of sepsis is the hypo-metabolic stage, which is characterized by low energy demand and hypo-inflammation. Cells are reprogrammed to low-energy state through aerobic oxidation, with the conversion of pyruvate to acetyl-CoA and the appropriate procession of TCA cycle and lipid metabolism (10). Switching metabolism to adjust energy supply and modulate inflammation in response to different stresses, including sepsis, is known as metabolic reprogramming. Early metabolic reprogramming in the acute phase of sepsis may be detrimental (11). Furthermore, early metabolic reprogramming in sepsis markedly alters lipid metabolism, resulting in lipotoxicity and glycerol accumulation (7, 9).
The generation of lactic acid, the major byproduct of anaerobic respiration, is the most frequently observed metabolic consequence of sepsis. Lactic acid can, in turn, activate Toll-like receptor 4 (TLR4) and promote the activation of nuclear factor-kappa B (NF-κB), as well as the further release of inflammatory mediators (12). Studies have shown that reducing lactic acid levels in the early stage of sepsis can improve the prognosis of patients (13, 14), suggesting that inhibiting early metabolic reprogramming, and subsequently decreasing subsequent lactate production and metabolic impairment, may represent a promising target for preventing the development of sepsis.
However, switching between these two metabolic pathways is necessary and might have beneficial effects. It is reported that, during the early stages of sepsis, metabolism in tubular epithelial cells can switch from aerobic oxidation to anaerobic glycolysis, suggestive of metabolic reprogramming (15). This alteration might enhance the capacity of cells to eliminate mitochondrial reactive oxygen species (ROS) accumulated due to aerobic oxidation in sepsis (16). Anaerobic glycolysis can also promote the pentose phosphate pathway and the production of NADPH, thus regenerating reduced glutathione, which favors the elimination of hydrogen peroxide (17). During sepsis, metabolic reprogramming helps to downregulate major energy sinks such as ion transport and fuel processes that are necessary for cell survival, thus contributing to energy maintenance (18). Furthermore, this switch from aerobic oxidation to anaerobic glycolysis is beneficial for the development of trained immunity, which helps build up the innate immune system to defend against future stresses (16). This study also demonstrated that a shift from oxidative phosphorylation to anaerobic glycolysis in glucose metabolism is the metabolic basis for trained immunity (19). In this context, early metabolic reprogramming and energy prioritization during sepsis might alleviate the extent of organ dysfunction, the progression to fibrosis, and the development of chronic kidney disease (16).
Concerning the wide-ranging effects of metabolic reprogramming, both beneficial and detrimental, further work is warranted for an improved understanding of the role of the different metabolic pathways in sepsis, as well as identifying strategies to improve outcome in septic patients through the regulation of metabolism.
A large number of studies have focused on PDHC, the key regulator of glucose metabolism, in exploring the underlying pathogenesis of sepsis (20). Accordingly, PDHC has been shown to regulate critical processes, including lactate production, the release of inflammatory mediators, TCA cycle, lipid metabolism and energy production. Here, we provide an overview of recent findings relating to how PDHC regulates metabolism in sepsis.
PDHC, a multienzyme complex composed of pyruvate dehydrogenase E1 (PDHE1), dihydrolipoamide transacetylase E2 (PDHE2), and dihydrolipoamide dehydrogenase E3 (PDHE3), serves as the critical hub in energy metabolism. Numerous studies have shown the positive regulatory role of PDHC in anaerobic glycolysis in sepsis. One study showed that PDHC activity was decreased by ~70% in skeletal muscle cells of septic rats, leading to cellular hypoxia and dysfunction (21). In terms of clinical research, Nuzzo et al. demonstrated that PDHC activity was significantly lower in peripheral blood mononuclear cells from sepsis patients than in those of healthy controls, and that this reduced activity may affect the prognosis of patients with sepsis (22). This suggests that PDHC activity may serve as a critical marker for the pathogenesis and development of sepsis.
PDHC is regulated via multiple mechanisms, including phosphorylation and acetylation. PDH kinase (PDK), consisting of four isoforms (PDK1–PDK4), acts as an upstream negative regulator of PDHC and is located in the mitochondrial matrix (23). PDK can decrease PDHC activity through the phosphorylation of PDHE1α on Ser293, Ser300, and Ser232, thereby inhibiting the TCA cycle and resulting in the accumulation of pyruvate and the production of lactic acid. In contrast, PDHC activity can be restored via PDH phosphatase (PDP)-mediated dephosphorylation (24). In skeletal muscle, the levels of the non-phosphorylated form of PDH and PDHC activity are both reduced following the induction of sepsis. Meanwhile, PDHC phosphorylation was reported to result in hyperlactatemia and disorder in energy metabolism (21). A different study showed that in LPS-stimulated mouse digital extensor muscle cells, the PDK mRNA expression level was upregulated 24-fold, while PDHC activity was decreased by 65%, resulting in an increase in lactic acid levels (25). These findings indicate that PDK is an important regulator of energy metabolism through its capacity to phosphorylate and inactivate PDHC.
Besides phosphorylation, studies have shown that PDHC acetylation also affects its activity and exerts a regulatory effect on energy metabolism (26). Notably, the acetylation level of PDHC is upregulated by acetyl-CoA acetyltransferase 1 (ACAT1), while PDHC deacetylation is catalyzed by the deacetylase SIRT3 (27). In skeletal muscle, an increase in PDHE1α acetylation levels reduces the activity of PDHC and causes metabolic disorder. That the switch from aerobic to anaerobic oxidation likely results in lactic acid accumulation indicates that PDHE1α acetylation plays a significant role in the regulation of PDHC activity and energy metabolism (28). Recent studies have also confirmed that increased levels of PDHC acetylation can lead to reductions in PDHC activity and total ATP synthesis, leading to mitochondrial dysfunction and myocardial damage (29). Consistent with these results, PDHE3 acetylation levels were sharply increased in a model of myocardial metabolic remodeling in obese mice, supporting the pivotal role of PDHE3 acetylation in mediating obesity-induced myocardial injury (30). However, relatively few studies have investigated the mechanism underlying the role of PDHC acetylation in sepsis.
Furthermore, there is crosstalk between the phosphorylation and acetylation of PDHC. The enhancement of PDHC acetylation through ACAT1 activation helps recruit PDK to the phosphorylation sites of PDH, thus inducing a decrease in PDHC activity, while the blockade of PDHC acetylation through SIRT3 activation may lead to its dephosphorylation and activation (31). Together, these results reveal that the direct targeting of PDHC acetylation or phosphorylation may be a promising strategy for sepsis management (Figure 2).
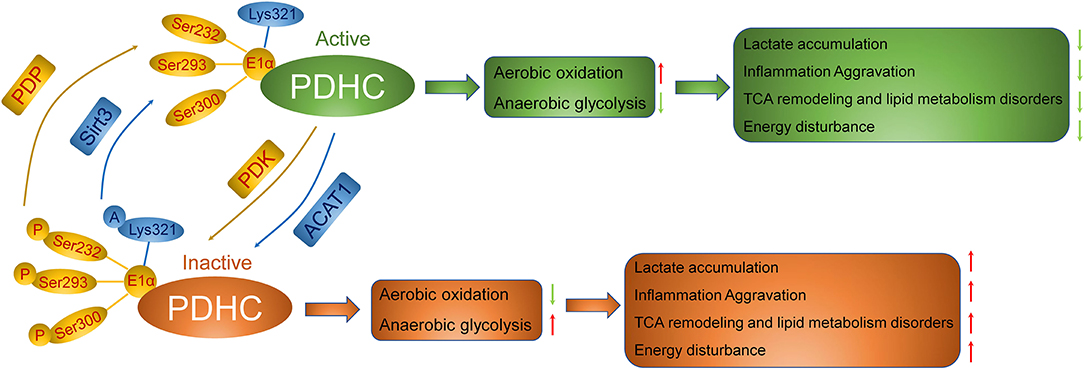
Figure 2. Regulation of PDHC in the metabolism of sepsis. The inactivation of PDHC through PDK-mediated phosphorylation and acetylation by ACAT1 results into anaerobic glycolysis, thereby inducing accumulation of lactic acid, the aggravation of inflammation, the remodeling of TCA cycle, the disorder of lipid metabolism, and the consequence of lower ATP synthesis relative to ATP demand. Activation of PDHC instead, via PDP-mediated de-phosphorylation and de-acetylation by Sirt3, leads to aerobic glycolysis, which is good for amelioration of sepsis.
As a critical node in metabolic regulation, PDHC converts pyruvate to acetyl-CoA in mitochondria, and its inhibition leads to early metabolic reprogramming in response to sepsis, resulting in the accumulation of lactic acid, the aggravation of inflammation, the remodeling of TCA cycle, and the disorder of lipid metabolism, and the consequence of lower ATP synthesis relative to ATP demand.
The inhibition of PDHC activation and the switch from aerobic oxidation to anaerobic glycolysis in sepsis is known to contribute to the formation of a large amount of lactic acid and intracellular acidosis, which can lead to intracellular Ca2+ overload, mitochondrial membrane damage (32, 33). The persistence of hyperlactatemia is responsible for arrhythmia, kidney injury, respiratory failure, central nervous system dysfunction, and damage to several other organs (34). There is evidence supporting that lactate triggers innate immune responses via TLR-mediated enhancement of NF-κB transcriptional activity and subsequent expression of pro-inflammatory genes in macrophages (12). Moreover, lactate has also been reported to promote HMGB1 acetylation in macrophages (35). From a clinical perspective, reducing lactic acid levels in the early stage of sepsis has been reported to improve the prognosis of patients (36). Collectively, these findings shed light on how inflammation and mitochondrial injury might be controlled through the regulation of lactate release in the treatment of sepsis.
As mentioned above, PDHC is a critical regulator of lactate levels. Bakalov et al. (20) confirmed that sepsis could lead to decreased PDHC activity and increased lactic acid levels in the gross tissues of Drosophila, while the upregulation of PDHC activity could reduce lactic acid levels and improve the survival rate of the flies. In a rat model of sepsis, inhibiting PDHC activity can lead to a significant increase in the level of lactate, illustrating the negative regulatory effect exerted by PDHC activity on the level of lactic acid (37). In addition, other studies have found that in septic mice, the administration of PDHC agonists can reduce the levels of lactate, thereby improving the disrupted metabolic regulation in the animals (38). These observations suggest that PDHC-mediated changes in lactate levels play a key role in sepsis-related metabolic changes. Accordingly, levels of PDHC activity and lactic acid may serve as biomarkers for the occurrence of sepsis.
Anaerobic glycolysis promotes the activation of a variety of inflammatory cells, including monocytes, dendritic cells, and macrophages (39, 40). Lactate, as a pro-inflammatory metabolite that could regulate macrophage polarization and increase the production of pro-inflammatory factors, is a marker for sepsis severity (41). Lactate has also been demonstrated to promote the expression of vascular endothelial growth factor, while lactate inhibition could block TLR4 signaling and attenuate the production of pro-inflammatory factors (12). Given that lactate is the major product of PDHC inactivation in anaerobic glycolysis, the targeting of PDHC may be a means for controlling lactate production and downstream inflammation pathways in sepsis.
Studies have confirmed that PDK is involved in the regulation of macrophage differentiation and glucose metabolism in bone marrow macrophages of septic mice. PDK1 knockout reduced the phosphorylation level of PDHE1α and upregulated the activity of PDHC, thus inhibiting LPS-induced anaerobic glycolysis and the polarization of macrophages from an M2 to an M1 phenotype and, consequently, reducing the levels of the inflammatory mediators interleukin (IL)-6, IL-12, and IL-1β. This finding highlights that PDK inhibition exerts positive regulatory effects in glycolysis via PDHC, allowing M1 macrophages to be polarized toward the M2 phenotype, thus tempering the release of inflammatory mediators (42). Bakalov et al. showed that PDHC activation led to the lowered level of cecropin-A and defensin, two releasing markers of pro-inflammatory factors, resulting in the improvement of life span of septic Drosophila (20). These results indicate that regulation of PDHC activity may represent a therapeutic target in the treatment of sepsis.
It is reported that in hepatocytes under septic condition, multiple TCA cycle-related metabolites, including citrate, cis-aconitate, and succinate, were markedly upregulated, indicating the remodeling of TCA (7). Citrate, exported from mitochondria via the mitochondrial citrate carrier (CIC), is a key molecule for the generation of energy. After citrate synthesis in the mitochondrial, citrate can enter the Krebs cycle and promote oxidative phosphorylation for energy production, which is required for the production of inflammatory molecules after LPS treatment. In the cytoplasm citrate is cleaved to acetyl-CoA which is the precursor for fatty acid, or cleaved to oxaloacetate which is converted to malate and then pyruvate (43). Excessive succinate accumulation following TCA remodeling in sepsis primes inflammation through succinate dehydrogenase (SDH)-mediated ROS generation and IL-1β production (44). In addition to TCA remodeling, lipid metabolism is also reprogrammed in sepsis. Studies showed that the amount of free fatty acids from the white adipose tissue into bloodstream was elevated under infection (45). Lipidomic analysis in livers of septic mice also demonstrated fatty acid uptake and β-oxidation are upregulated in sepsis to produce ketone bodies used as an energy source by the brain and other bodies, leading to excess free fatty acids and triglycerides, causing lipotoxicity (7, 9). Moreover, the hepatic levels of phospholipids, including phosphatidylcholine, phosphatidylethanolamine, and sphingomyelin, were also significantly elevated, all of which influence energy metabolism and have been linked with sepsis progression (46).
Given its critical role in the regulation of the TCA cycle and lipid metabolism, the impairment of which underlies the organ dysfunction observed in sepsis, PDHC may represent an effective target for sepsis treatment. The activation of PDHC significantly restored TCA metabolite levels to those of control and improved liver function in sepsis (7). PDHC activation also restored anabolic energy in inflammatory monocytes while also increasing the abundance of TCA cycle intermediates and the anaplerotic metabolism of branched-chain amino acids, thus promoting TCA-driven anabolic energetics (47). Furthermore, PDHC activation in septic mice reversed lipid disorder and mitochondrial dysfunction, indicative of the positive regulatory role of PDHC in lipid metabolism. Combined, these observations demonstrate that PDHC is a critical regulator of TCA cycling and lipid metabolism during sepsis.
PDHC is essential for glucose oxidation through its ability to promote a switch from the glycolytic to the oxidative pathway and the subsequent use of substrates through the respiratory chain in mitochondria. PDHC catalyzes the oxidative decarboxylation of pyruvate, yielding NADH and acetyl-CoA, key molecules for mitochondrial ATP generation (48). During sepsis, the downregulation of PDHC might contribute to energy metabolism disturbances by impairing the capacity of mitochondria for energy production. Furthermore, PDHC inactivation might help to explain the lower functional capacity of mitochondrial energy in premature neonates with sepsis compared with older children patients (49). Studies have shown that PDHC activation induced by norepinephrine could improve the respiratory function in mitochondria and attenuate the inflammation in septic rats (50). These data all support that enhancing PDHC activity and maintaining metabolism homeostasis can restore mitochondrial function and inhibit organ dysfunction in sepsis.
Given its critical role in the metabolism of sepsis, PDHC may represent an effective target for sepsis management. A study by McCall et al. (51) confirmed that the promotion of PDHC activity through PDK inhibition facilitates immune-metabolic adaptations in sepsis. A group of drugs that target PDHC activation, including dichloroacetate (DCA), thiamine, amrinone, ciprofloxacin, and TNF-binding protein (TNFbp), have been shown to ameliorate the symptoms of sepsis (Figure 1).
DCA is a small-molecule metabolism-regulating drug that is mainly used in the treatment of diseases related to mitochondrial defects and lactic acid accumulation (52). It is well-known as a PDK inhibitor and can reduce the level of PDHC phosphorylation by inhibiting the activity of PDK, upregulating the activity of PDHC, and promoting the entry of pyruvate into mitochondria for oxidative phosphorylation. Several studies have confirmed that DCA can activate PDHC, regulate glucose metabolism, and inhibit lactic acid accumulation in septic cells (53). The activation of PDHC with DCA significantly restored TCA metabolite levels to those of control and improved liver function in sepsis (7).
Furthermore, DCA administration to septic mice reversed lipid disorder and mitochondrial dysfunction, indicative of the positive regulatory role of PDHC in lipid metabolism. Similarly, compared with control septic animals, the activation of PDHC through DCA infusion led to decreases in plasma lactate concentrations and glycolytic activity, thus restoring normal glycolytic function (38).
Notably, clinical studies have also shown that DCA can promote a switch in glucose metabolism from anaerobic glycolysis to oxidative phosphorylation in patients with sepsis, thereby significantly inhibiting the occurrence of hyperlactatemia (54). These results suggest that DCA can effectively improve prognosis in septic patients. However, one randomized controlled study reported that DCA injection could significantly reduce lactate levels in sepsis patients, but did not significantly affect hemodynamics or mortality (55). Additionally, DCA has been associated with several side effects (56), highlighting that the clinical application of DCA requires further extensive verification.
Thiamine, also known as vitamin B1, consists of pyrimidine and thiazole moieties, and thiamine deficiency can lead to the disease beriberi (57). Studies have shown that thiamine, a coenzyme of PDHC, plays an important role in improving the activity of the latter in sepsis. Between 10 and 30% of patients with critically illness are deficient in thiamine (58). For patients with sepsis, thiamine deficiency can lead to the accumulation of pyruvate and induce the production of a large amount of lactate, thus significantly increasing the mortality of patients (59). Injecting thiamine is beneficial for mitigating increases in lactate levels and can also improve prognosis in sepsis patients (60, 61). Given the potential health-related significance of thiamine, current guidelines recommend that patients admitted to ICU should receive 100–300 mg of thiamine daily for the first 3 days to reduce the potential adverse prognosis of sepsis (62). Important though thiamine is in clinical drug application, further studies are required for thiamine dose determination and for identifying the causal relationship between PDHC activity regulation and the clinical effect of thiamine.
Amrinone, which was reported to inhibit TNF synthesis, could prevent alterations in muscle protein metabolism produced by sepsis. Burns et al. (63) found that in rats with septic shock, amrinone could induce a 2.5-fold increase in PDHC activity in cardiac tissue compared with that of control animals, and also increased the ATP level. Additionally, it has been shown that the injection of amrinone at a dose of 5 mg/(kg·day−1) for 5 days in septic rats helped to induce the activation of PDHC in skeletal muscle and to significantly reduce the level of lactate, suggesting that amrinone may be useful for the treatment of hyperlactatemia in septic patients (64). However, further evidence-based data are needed before amrinone can be administered clinically to septic patients.
Ciprofloxacin, a member of the quinolone family, can downregulate the levels of PDK1, which, in turn, activates PDHC, reverses the loss of ATP, and decreases the high mortality of mice exposed to ionizing radiation and trauma (65). It is imperative to determine whether ciprofloxacin provides therapeutic benefit for sepsis.
Studies have demonstrated that TNFbp injection can increase the activity of PDHC in skeletal muscle cells as well as reverse the increase in lactate levels in septic rats. Investigation of the effects of TNFbp in sepsis is ongoing (37).
Most studies to date have focused on the role of PDHC phosphorylation in sepsis, while reports on the effect of acetylation and the crosstalk between these two post-translational modifications are scarce. While we summarized the mechanisms underlying the role of PDHC in lactate production, inflammation, TCA cycle, and in lipid and energy metabolism (Table 1), whether other mechanisms are also involved remains to be determined. Regarding to the targeting treatment, although several drugs have been reported to improve the prognosis of patients with sepsis by activating PDHC, evidence-based data are still lacking. Many unsolved problem remain. Further exploration of the role of PDHC in sepsis is required for the understanding of septic pathogenesis and the management of sepsis.
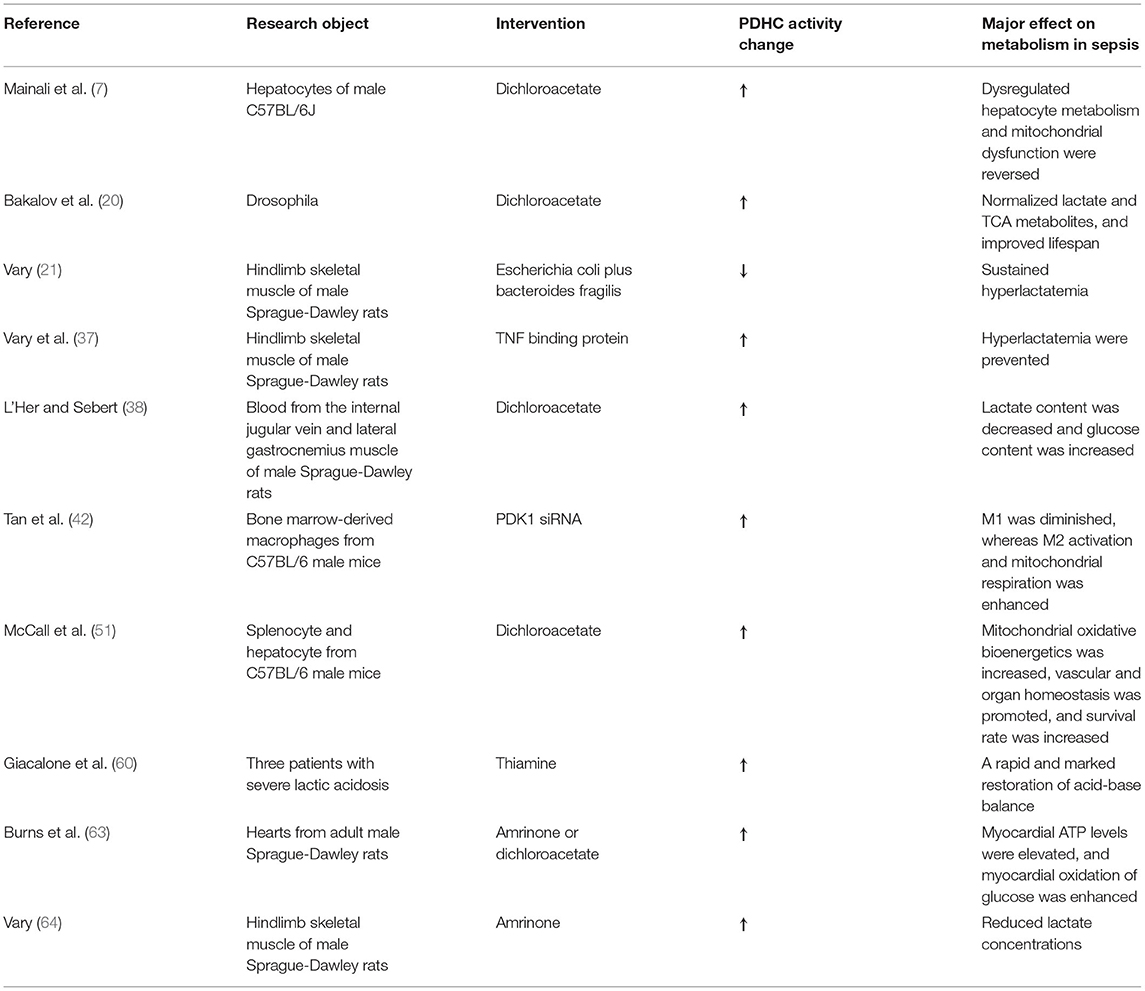
Table 1. Overview of included studies.
ZZ and QH contributed to the organization and writing of the manuscript. LM, JW, SA, and ZC wrote sections of the manuscript. WZ reviewed and edited the submitted version. All authors approved the manuscript for publication.
This work was supported by grants from National Natural Science Foundation of China (Grants 81900279).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Sterling SA, Puskarich MA, Glass AF, Guirgis F, Jones AE. The impact of the sepsis-3 septic shock definition on previously defined septic shock patients. Crit Care Med. (2017) 45:1436–42. doi: 10.1097/CCM.0000000000002512
2. Yu K, Wang Y, Wan T, Zhai Y, Cao S, Ruan W, et al. Tacrolimus nanoparticles based on chitosan combined with nicotinamide: enhancing percutaneous delivery and treatment efficacy for atopic dermatitis and reducing dose. Int J Nanomedicine. (2018) 13:129–42. doi: 10.2147/IJN.S150319
3. Peake SL, Delaney A, Bailey M, Bellomo R. Potential impact of the 2016 consensus definitions of sepsis and septic shock on future sepsis research. Ann Emerg Med. (2017) 70:553–61.e1. doi: 10.1016/j.annemergmed.2017.04.007
4. Seymour CW, Liu VX, Iwashyna TJ, Brunkhorst FM, Rea TD, Scherag A, et al. Assessment of clinical criteria for sepsis: for the third international consensus definitions for sepsis and septic shock (sepsis-3). JAMA. (2016) 315:762–74. doi: 10.1001/jama.2016.0288
5. Vachharajani VT, Liu T, Wang X, Hoth JJ, Yoza BK, McCall CE. Sirtuins link inflammation and metabolism. J Immunol Res. (2016) 2016:8167273. doi: 10.1155/2016/8167273
6. Mills E, O'Neill LA. Succinate: a metabolic signal in inflammation. Trends Cell Biol. (2014) 24:313–20. doi: 10.1016/j.tcb.2013.11.008
7. Mainali R, Zabalawi M, Long D, Buechler N, Quillen E, Key CC, et al. Dichloroacetate reverses sepsis-induced hepatic metabolic dysfunction. Elife. (2021) 10:e64611. doi: 10.7554/eLife.64611.sa2
8. Ayala A, Munoz MF, Arguelles S. Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid Med Cell Longev. (2014) 2014:360438. doi: 10.1155/2014/360438
9. Van Wyngene L, Vanderhaeghen T, Timmermans S, Vandewalle J, Van Looveren K, Souffriau J, et al. Hepatic PPARalpha function and lipid metabolic pathways are dysregulated in polymicrobial sepsis. EMBO Mol Med. (2020) 12:e11319. doi: 10.15252/emmm.201911319
10. Cheng SC, Scicluna BP, Arts RJ, Gresnigt MS, Lachmandas E, Giamarellos-Bourboulis EJ, et al. Broad defects in the energy metabolism of leukocytes underlie immunoparalysis in sepsis. Nat Immunol. (2016) 17:406–13. doi: 10.1038/ni.3398
11. Jha AK, Huang SC, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E, et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity. (2015) 42:419–30. doi: 10.1016/j.immuni.2015.02.005
12. Samuvel DJ, Sundararaj KP, Nareika A, Lopes-Virella MF, Huang Y. Lactate boosts TLR4 signaling and NF-kappaB pathway-mediated gene transcription in macrophages via monocarboxylate transporters and MD-2 up-regulation. J Immunol. (2009) 182:2476–84. doi: 10.4049/jimmunol.0802059
13. Bao L, Zhang M, Yan P, Wu X, Shao J, Zheng R. [Retrospective analysis of the value of arterial blood lactate level and its clearance rate on the prognosis of septic shock patients]. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue. (2015) 27:38–42. doi: 10.3760/cma.j.issn.2095-4352.2015.01.009
14. Kanashvili B, Saganelidze K, Ratiani L. The role of procalcitonin and blood lactic acid values in prognosis of sepsis and septic shock in polytrauma patients. Georgian Med News. (2018) 102–107.
15. Waltz P, Carchman E, Gomez H, Zuckerbraun B. Sepsis results in an altered renal metabolic and osmolyte profile. J Surg Res. (2016) 202:8–12. doi: 10.1016/j.jss.2015.12.011
16. Gomez H, Kellum JA, Ronco C. Metabolic reprogramming and tolerance during sepsis-induced AKI. Nat rev nephrol. (2017) 13:143–51. doi: 10.1038/nrneph.2016.186
17. Patra KC, Hay N. The pentose phosphate pathway and cancer. Trends Biochem Sci. (2014) 39:347–54. doi: 10.1016/j.tibs.2014.06.005
18. Carre JE, Singer M. Cellular energetic metabolism in sepsis: the need for a systems approach. Biochim Biophys Acta. (2008) 1777:763–71. doi: 10.1016/j.bbabio.2008.04.024
19. Cheng SC, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V, et al. mTOR- and HIF-1alpha-mediated aerobic glycolysis as metabolic basis for trained immunity. Science. (2014) 345:1250684. doi: 10.1126/science.1250684
20. Bakalov V, Reyes-Uribe L, Deshpande R, Maloy AL, Shapiro SD, Angus DC, et al. Dichloroacetate-induced metabolic reprogramming improves lifespan in a Drosophila model of surviving sepsis. PLoS ONE. (2020) 15:e0241122. doi: 10.1371/journal.pone.0241122
21. Vary TC. Sepsis-induced alterations in pyruvate dehydrogenase complex activity in rat skeletal muscle: effects on plasma lactate. Shock. (1996) 6:89–94. doi: 10.1097/00024382-199608000-00002
22. Nuzzo E, Berg KM, Andersen LW, Balkema J, Montissol S, Cocchi MN, et al. Pyruvate dehydrogenase activity is decreased in the peripheral blood mononuclear cells of patients with sepsis. A prospective observational trial. Ann Am Thorac Soc. (2015) 12:1662–6. doi: 10.1513/AnnalsATS.201505-267BC
23. Gudi R, Bowker-Kinley MM, Kedishvili NY, Zhao Y, Popov KM. Diversity of the pyruvate dehydrogenase kinase gene family in humans. J Biol Chem. (1995) 270:28989–94. doi: 10.1074/jbc.270.48.28989
24. Woolbright BL, Rajendran G, Harris RA, Taylor JR. Metabolic flexibility in cancer: targeting the pyruvate dehydrogenase kinase: pyruvate dehydrogenase axis. Mol Cancer Ther. (2019) 18:1673–81. doi: 10.1158/1535-7163.MCT-19-0079
25. Alamdari N, Constantin-Teodosiu D, Murton AJ, Gardiner SM, Bennett T, Layfield R, et al. Temporal changes in the involvement of pyruvate dehydrogenase complex in muscle lactate accumulation during lipopolysaccharide infusion in rats. J Physiol. (2008) 586:1767–75. doi: 10.1113/jphysiol.2007.149625
26. Ozden O, Park SH, Wagner BA, Song HY, Zhu Y, Vassilopoulos A, et al. SIRT3 deacetylates and increases pyruvate dehydrogenase activity in cancer cells. Free Radic Biol Med. (2014) 76:163–72. doi: 10.1016/j.freeradbiomed.2014.08.001
27. Fukushima A, Alrob OA, Zhang L, Wagg CS, Altamimi T, Rawat S, et al. Acetylation and succinylation contribute to maturational alterations in energy metabolism in the newborn heart. Am J Physiol Heart Circ Physiol. (2016) 311:H347–63. doi: 10.1152/ajpheart.00900.2015
28. Jing E, O'Neill BT, Rardin MJ, Kleinridders A, Ilkeyeva OR, Ussar S, et al. Sirt3 regulates metabolic flexibility of skeletal muscle through reversible enzymatic deacetylation. Diabetes. (2013) 62:3404–17. doi: 10.2337/db12-1650
29. Zhang X, Ji R, Liao X, Castillero E, Kennel PJ, Brunjes DL, et al. MicroRNA-195 regulates metabolism in failing myocardium via alterations in sirtuin 3 expression and mitochondrial protein acetylation. Circulation. (2018) 137:2052–67. doi: 10.1161/CIRCULATIONAHA.117.030486
30. Romanick SS, Ulrich C, Schlauch K, Hostler A, Payne J, Woolsey R, et al. Obesity-mediated regulation of cardiac protein acetylation: parallel analysis of total and acetylated proteins via TMT-tagged mass spectrometry. Biosci Rep. (2018) 38:BSR20180721. doi: 10.1042/BSR20180721
31. Fan J, Shan C, Kang HB, Elf S, Xie J, Tucker M, et al. Tyr phosphorylation of PDP1 toggles recruitment between ACAT1 and SIRT3 to regulate the pyruvate dehydrogenase complex. Mol Cell. (2014) 53:534–48. doi: 10.1016/j.molcel.2013.12.026
32. Nayler WG, Panagiotopoulos S, Elz JS, Daly MJ. Calcium-mediated damage during post-ischaemic reperfusion. J Mol Cell Cardiol. (1988) 20(Suppl. 2):41–54. doi: 10.1016/0022-2828(88)90331-8
33. Arulkumaran N, Deutschman CS, Pinsky MR, Zuckerbraun B, Schumacker PT, Gomez H, et al. Mitochondrial function in sepsis. Shock. (2016) 45:271–81. doi: 10.1097/SHK.0000000000000463
34. Perez RDGA, Kellum JA, Honigschnabel J, Kreymann B. Respiratory and metabolic acidosis correction with the ADVanced Organ Support system. Intensive Care Med Exp. (2019) 7:56. doi: 10.1186/s40635-019-0269-7
35. Yang L, Xie M, Yang M, Yu Y, Zhu S, Hou W, et al. PKM2 regulates the Warburg effect and promotes HMGB1 release in sepsis. Nat Commun. (2014) 5:4436. doi: 10.1038/ncomms5436
36. Bakker J, Nijsten MW, Jansen TC. Clinical use of lactate monitoring in critically ill patients. Ann Intensive Care. (2013) 3:12. doi: 10.1186/2110-5820-3-12
37. Vary TC, Hazen SA, Maish G, Cooney RN. TNF binding protein prevents hyperlactatemia and inactivation of PDH complex in skeletal muscle during sepsis. J Surg Res. (1998) 80:44–51. doi: 10.1006/jsre.1998.5324
38. L'Her E, Sebert P. Effects of dichloroacetate and ubiquinone infusions on glycolysis activity and thermal sensitivity during sepsis. J Lab Clin Med. (2004) 143:352–7. doi: 10.1016/j.lab.2004.03.004
39. Pearce EL, Poffenberger MC, Chang CH, Jones RG. Fueling immunity: insights into metabolism and lymphocyte function. Science. (2013) 342:1242454. doi: 10.1126/science.1242454
40. Chang CH, Curtis JD, Maggi LJ, Faubert B, Villarino AV, O'Sullivan D, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell. (2013) 153:1239–51. doi: 10.1016/j.cell.2013.05.016
41. Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. (2014) 513:559–63. doi: 10.1038/nature13490
42. Tan Z, Xie N, Cui H, Moellering DR, Abraham E, Thannickal VJ, et al. Pyruvate dehydrogenase kinase 1 participates in macrophage polarization via regulating glucose metabolism. J Immunol. (2015) 194:6082–9. doi: 10.4049/jimmunol.1402469
43. Infantino V, Iacobazzi V, Menga A, Avantaggiati ML, Palmieri F. A key role of the mitochondrial citrate carrier (SLC25A1) in TNFalpha- and IFNgamma-triggered inflammation. Biochim Biophys Acta. (2014) 1839:1217–25. doi: 10.1016/j.bbagrm.2014.07.013
44. Mills EL, Kelly B, Logan A, Costa A, Varma M, Bryant CE, et al. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell. (2016) 167:457–470.e13. doi: 10.1016/j.cell.2016.08.064
45. Wellhoener P, Vietheer A, Sayk F, Schaaf B, Lehnert H, Dodt C. Metabolic alterations in adipose tissue during the early phase of experimental endotoxemia in humans. Horm Metab Res. (2011) 43:754–9. doi: 10.1055/s-0031-1287854
46. van der Veen JN, Kennelly JP, Wan S, Vance JE, Vance DE, Jacobs RL. The critical role of phosphatidylcholine and phosphatidylethanolamine metabolism in health and disease. Biochim Biophys Acta Biomembr. (2017) 1859:1558–72. doi: 10.1016/j.bbamem.2017.04.006
47. Zhu X, Long D, Zabalawi M, Ingram B, Yoza BK, Stacpoole PW, et al. Stimulating pyruvate dehydrogenase complex reduces itaconate levels and enhances TCA cycle anabolic bioenergetics in acutely inflamed monocytes. J Leukoc Biol. (2020) 107:467–84. doi: 10.1002/JLB.3A1119-236R
48. Becherini P, Caffa I, Piacente F, Damonte P, Vellone VG, Passalacqua M, et al. SIRT6 enhances oxidative phosphorylation in breast cancer and promotes mammary tumorigenesis in mice. Cancer Metab. (2021) 9:6. doi: 10.1186/s40170-021-00240-1
49. Honzik T, Wenchich L, Bohm M, Hansikova H, Pejznochova M, Zapadlo M, et al. Activities of respiratory chain complexes and pyruvate dehydrogenase in isolated muscle mitochondria in premature neonates. Early Hum Dev. (2008) 84:269–76. doi: 10.1016/j.earlhumdev.2006.07.008
50. Dan W, Qi Z. Protective effects of continuous norepinephrine infusion in early septic rats. Chin Pediatr Emerg Med. (2013) 20:73–77. doi: 10.3760/cma.j.issn.1673-4912.2013.01.020
51. McCall CE, Zabalawi M, Liu T, Martin A, Long DL, Buechler NL, et al. Pyruvate dehydrogenase complex stimulation promotes immunometabolic homeostasis and sepsis survival. JCI Insight. (2018) 3:e99292. doi: 10.1172/jci.insight.99292
52. El SS, Baghdadi H, Ahmed NS, Almaramhy HH, Mahmoud AA, El-Sawy SA, et al. Dichloroacetate is an antimetabolite that antagonizes acetate and deprives cancer cells from its benefits: a novel evidence-based medical hypothesis. Med Hypotheses. (2019) 122:206–9. doi: 10.1016/j.mehy.2018.11.012
53. Lang CH, Bagby GJ, Blakesley HL, Spitzer JJ. Glucose kinetics and pyruvate dehydrogenase activity in septic rats treated with dichloroacetate. Circ Shock. (1987) 23:131–41.
54. Zhou ZH, McCarthy DB, O'Connor CM, Reed LJ, Stoops JK. The remarkable structural and functional organization of the eukaryotic pyruvate dehydrogenase complexes. Proc Natl Acad Sci USA. (2001) 98:14802–7. doi: 10.1073/pnas.011597698
55. Stacpoole PW, Wright EC, Baumgartner TG, Bersin RM, Buchalter S, Curry SH, et al. A controlled clinical trial of dichloroacetate for treatment of lactic acidosis in adults. The Dichloroacetate-Lactic Acidosis Study Group. N Engl J Med. (1992) 327:1564–9. doi: 10.1056/NEJM199211263272204
56. Chu QS, Sangha R, Spratlin J, Vos LJ, Mackey JR, McEwan AJ, et al. A phase I open-labeled, single-arm, dose-escalation, study of dichloroacetate (DCA) in patients with advanced solid tumors. Invest New Drugs. (2015) 33:603–10. doi: 10.1007/s10637-015-0221-y
57. Wilson RB. Pathophysiology, prevention, and treatment of beriberi after gastric surgery. Nutr Rev. (2020) 78:1015–29. doi: 10.1093/nutrit/nuaa004
58. Donnino MW, Carney E, Cocchi MN, Barbash I, Chase M, Joyce N, et al. Thiamine deficiency in critically ill patients with sepsis. J Crit Care. (2010) 25:576–81. doi: 10.1016/j.jcrc.2010.03.003
59. Sechi G, Serra A. Wernicke's encephalopathy: new clinical settings and recent advances in diagnosis and management. Lancet Neurol. (2007) 6:442–55. doi: 10.1016/S1474-4422(07)70104-7
60. Giacalone M, Martinelli R, Abramo A, Rubino A, Pavoni V, Iacconi P, et al. Rapid reversal of severe lactic acidosis after thiamine administration in critically ill adults: a report of 3 cases. Nutr Clin Pract. (2015) 30:104–10. doi: 10.1177/0884533614561790
61. Amrein K, Ribitsch W, Otto R, Worm HC, Stauber RE. Severe lactic acidosis reversed by thiamine within 24 hours. Crit Care. (2011) 15:457. doi: 10.1186/cc10495
62. Singer P, Berger MM, Van den Berghe G, Biolo G, Calder P, Forbes A, et al. ESPEN Guidelines on Parenteral Nutrition: intensive care. Clin Nutr. (2009) 28:387–400. doi: 10.1016/j.clnu.2009.04.024
63. Burns AH, Summer WR, Burns LA, Shepherd RE. Inotropic interactions of dichloroacetate with amrinone and ouabain in isolated hearts from endotoxin-shocked rats. J Cardiovasc Pharmacol. (1988) 11:379–86. doi: 10.1097/00005344-198804000-00001
64. Vary TC. Amrinone prevents the inhibition of muscle pyruvate dehydrogenase complex activity during sepsis. Shock. (1996) 5:229–32. doi: 10.1097/00024382-199603000-00011
Keywords: sepsis, glycolysis, metabolism, pyruvate dehydrogenase complex, therapy
Citation: Zeng Z, Huang Q, Mao L, Wu J, An S, Chen Z and Zhang W (2021) The Pyruvate Dehydrogenase Complex in Sepsis: Metabolic Regulation and Targeted Therapy. Front. Nutr. 8:783164. doi: 10.3389/fnut.2021.783164
Received: 25 September 2021; Accepted: 24 November 2021;
Published: 14 December 2021.
Edited by:
Julie-Anne Nazare, Université Claude Bernard Lyon 1, FranceReviewed by:
Bjørn Steen Skålhegg, University of Oslo, NorwayCopyright © 2021 Zeng, Huang, Mao, Wu, An, Chen and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Weijin Zhang, aTc5OTA3NzUzM0AxMjYuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.