 Yifan Wu
Yifan Wu Qian Zhang
Qian Zhang Xinhua Xiao
Xinhua Xiao- Key Laboratory of Endocrinology, Ministry of Health, Department of Endocrinology, Peking Union Medical College Hospital, Peking Union Medical College, Chinese Academy of Medical Sciences, Beijing, China
Diabetes has become the most common metabolic disease around the world. In addition to genetic and environmental factors in adulthood, the early life environment is critical to the progression of diabetes in adults, especially the environment during the fetal period; this concept is called “fetal programming.” Substantial evidence has illustrated the key role of early life macronutrient in programming metabolic diseases. Recently, the effect of maternal micronutrient intake on offspring glucose metabolism during later life has become an emerging field. This review focuses on updated human and animal evidence about the effect of maternal micronutrient status on offspring glucose metabolism and the underlying mechanism.
Introduction
Diabetes is a serious metabolic disease and a major health concern in modern society. The prevalence of diabetes is high and increasing worldwide. According to the latest statistics, in 2019, the number of patients (20–79 years old) with diabetes worldwide reached 463 million, and it is expected to reach 700 million in 2045 (1). It is also a serious chronic disease that leads to various types of complications and an increase in disability and death, hence causing a huge problem of medical financial cost.
Type 2 diabetes mellitus (T2DM) accounts for 90% of diabetes cases (1). However, the causes of the disease are not fully understood. There is a modern perception that it results from a combination of multigene pre-disposition and environmental triggers. This perception was first put forward by James Neel in 1962 in his “thrifty genotype hypothesis.” He proposed the “thrifty genotype” as a trigger for an increased efficacy in utilizing food and fat deposition promotion under better nutrition status after starvation (2). In a study on the Dutch famine of 1994–1945, individuals born during famine have increased blood glucose levels, which was related to their birth weight (3). Moreover, subjects born during powerful tropical storms in Puerto Rico during the 1920s and 1930s are prone to hypertension, high blood cholesterol, and diabetes (4). Nevertheless, the hypothesis fails to explain why the “diabesity” pandemic is still spreading rapidly and widely in modern times with adequate availability of nutrients. In 1992, Barker et al. raised the “thrifty phenotype” hypothesis which summarized the relationship of the early life environment and the prevalence of metabolic diseases in epidemiological findings. The hypothesis was spread as the “Barker hypothesis” or “fetal programming hypothesis,” which suggests that unfavorable environmental factors during early life (intrauterine and postnatal) lead to a high incidence of metabolic diseases, such as hypertension, impaired glucose tolerance, insulin resistance, and T2DM (5, 6). This concept gradually evolved into the Developmental Origins of Health and Disease (DOHaD).
DOHaD clearly demonstrates the link between the adverse intrauterine environment and the risk for chronic metabolic consequences, including obesity, insulin resistance and cardiovascular disease, and such an effect may persist for generations through epigenetic mechanisms (7–9), including DNA methylation, histone modifications and non-coding RNAs (8). Epigenetic modifications are easily affected by environmental factors, such as tobacco smoking (10, 11), environmental toxins (12, 13), low-dose radiation (14), lack of physical activity (15–17), psychological stress (18), and circadian dysregulation (19, 20).
In addition, maternal nutrition status also affects epigenetic modifications, including protein deprivation and caloric excess. The initial evidence of the link is from severe famines. A 50% increase in T2DM development was seen among the Ukrainian population born during the Holodomor famine in 1932–1933 (21). In other studies on the Dutch Hunger Winter of 1944–1945, exposure to famine during gestation and childhood had life-long health effects, especially an increased incidence of impaired glucose tolerance (22, 23), and had transgenerational effects on offspring health later in life (24). Offspring DNA methylation changes were reported in those exposed to the Dutch Hungry in the periconceptional period (25, 26). Exposure to the Chinese famine from 1959 to 1961 in early life has increased the incidence of overweight and T2DM, which may be one of the explanations of obesity and T2DM epidemics in China (27). An increase in methylation levels in the insulin-like growth factor 2 (IGF2) and insulin receptor (INSR) genes was also observed in those exposed to severe famine in their early life (28, 29). In recent years, many studies have reported that maternal undernutrition, especially protein restriction, and overnutrition in utero resulted in the effects of fetal programming, which can be observed in animal models. Our group and other scientists' research also show that maternal protein restriction leads to pancreatic β-cell dysfunction in mouse offspring with the regulation of microRNAs (miRNAs), which may lead to offspring that are pre-disposed to insulin resistance and T2DM (30, 31). In addition, exposure of mouse pups to a high-fat diet in the fetal period leads to hyperglycemia, high serum leptin, and methylation modification disorder of the hepatic insulin receptor substrate 2 (Irs2) gene, the mitogen-activated protein kinase kinase 4 (Map2k4) gene, the hypothalamic proopiomelanocortin (Pomc) gene, and the melanocortin 4 receptor (Mc4r) gene (32, 33).
Most studies have focused on maternal macronutrient deprivation and excess during early life and its effects on later-life health. Maternal micronutrient malnutrition is also a threatening problem worldwide. Although micronutrients are needed in small amounts, they are essential to life activity and homeostasis. They are classified into minerals, trace elements and vitamins, which play an important role in biochemical reactions and function. Micronutrient deficiency usually leads to health problems (34, 35). Widespread global micronutrient deficiency poses a threat to people's health, especially pregnant women and their children's health, and results in poor growth, perinatal complications, and an increased risk of metabolic disorders later in life (36). Animal studies on the effects of maternal micronutrient deficiency or surplus on insulin resistance in offspring can be traced back to 2004, when Raghunath et al. found that maternal multimineral (Fe, Zn, Mg, and Ca) or multivitamin restriction led to early growth retardation, body composition alteration, and insulin resistance in offspring. Increased oxidative stress and/or decreased antioxidant defense in these offspring were also observed (37, 38). Since then, animal and clinical studies on the effects of single micronutrients, including minerals (Zn, Cr, Ca, Fe, Mg, Se, Zn) and vitamins (vitamins A, D, B12, and folic acid), on offspring metabolic status have been carried out successively. The possible mechanisms behind this phenomenon include increased oxidative stress and inflammation, insulin signaling pathways, eating reflex, etc. have been explored. In recent years, discussions on epigenetic mechanisms (such as DNA methylation and miRNAs) have gradually increased. In 2007, Sinclair et al. carried out a study in a sheep model, in which restricted dietary methyl donor (i.e., methionine and vitamin B12) provision up to day 6 following insemination in sheep altered DNA methylation and led to hypertensive and insulin-resistant offspring (39). This review summarizes the previous studies linking maternal micronutrient surplus and deficiency and its adverse impacts on glucose metabolism in offspring and the mechanisms involved in this process.
Minerals
Calcium
Ca is a key micronutrient in bone construction, blood clotting, and muscle contraction (40). The best natural sources of Ca are milk and dairy products. In addition, we can also obtain Ca from leafy green vegetables, nuts or fortified foods, such as flour and dairy alternatives (41). A study reported consistently low Ca intake in pregnant subjects across Asian, African, and Latin American countries due to the deficiency of dairy products consumption (intake between 200 and 500 mg/day) (42, 43). The recommended dietary allowance (RDA) of Ca set by the USA is 1,200 mg per day for men and non-pregnant women aged 31–50 (44). In pregnancy and lactation, women, particularly women with low dietary Ca intake, require an increase in their Ca intake (1,000–2,000 mg/day) to meet the Ca demands of both mothers and fetuses/infants (45, 46). Therefore, strategies of Ca supplement to achieve Ca recommendations should be targeted at those groups.
Ca restriction in pregnant women is significantly linked with abnormal offspring glucose metabolism. Several human studies have demonstrated that serum Ca levels are positively correlated with serum glucose levels and insulin secretion in subjects whose fast serum glucose <7.0 mmol/L (47) and changed oxidative stress is related with Ca dysregulation around T2DM patients (48). However, there is still a lack of robust clinical evidence on the association of maternal Ca intake and offspring glucose metabolism. Furthermore, relevant animal studies showed Ca deficiency is linked with the progress of offspring insulin resistance. Takaya et al. found that in rat models, a maternal Ca-deficient diet increased insulin resistance in the adulthood of offspring, especially in male offspring. This alteration may be involved in the influenced hepatic hydroxysteroid 11-beta dehydrogenase 1 (Hsd11b1) expression (49) and the altered osteocalcin, a bone formation biomarker, that acts directly on pancreatic β-cells and increases insulin secretion (50). The undercarboxylated osteocalcin (Glu-osteocalcin) was increased to 76.7 ± 48.4 ng/mL in female offspring fed on a maternal low Ca diet, compared with 19.6 ± 9.9 ng/mL in control group (50). Moreover, the influences of maternal Ca deficiency on insulin secretion were observed in the next three generations (51).
Magnesium
Mg is an essential ion for protein production, muscle contraction, nerve transmission, and immune system health. Food sources of Mg usually include green leafy vegetables, nuts, legumes, and whole grains (52). Daily Mg intake at a dose of 310–360 mg/day (RDA) for girls/women aged 14 through 50 is recommended and the RDA for pregnant women for Mg increases to 350–400 mg/day. The range varies from adolescents to adults (53). The World Health Organization reported that the frequency of Mg deficiency is still problematic in general population in both developed and developing countries (54). A high percentage of pregnant women with low serum ionized Mg levels is found in the USA (55). Thus, pregnant women are still recommended to include good sources of Mg in their diets (56).
In glucose metabolism, Mg could improve insulin resistance. Widespread clinical evidence has proven that Mg intake deficiency and low serum Mg levels are related to T2DM, insulin resistance and metabolic syndrome (57). But the effect of maternal Mg restriction on offspring glucose metabolism currently lacks clinical evidence partly because the serum Mg levels cannot reflect the Mg stored in the body and the gold standard of Mg deficiency for pregnant women has not yet determined (56). Moreover, in rat models, Venu et al. found that maternal restriction of Mg in the diet resulted in an increase in body fat, induced insulin resistance, and impaired glucose tolerance in male pups and that rehabilitation with Mg supplementation can partially correct body composition and low-birth weight by 6 months of age (58). However, in another study, rehabilitation of Mg in the diet of the male pups of rats showed no correction of body composition at 18 months (59). The inconsistency between the two studies is likely to result from the different ages of the offspring.
Abnormal Mg balance is associated with the pathogenesis of T2DM. However, the plausible molecular mechanisms involved are still not fully understood. In carbohydrate metabolism, Mg serves as a critical cofactor of many enzymes (60). First, Mg deficiency impairs glucose intake in the endothelia of target tissues, including liver and adipose tissue, by inhibiting glucose transporter 4 (GLUT4) expression. In addition, low serum Mg affects insulin secretion and signaling because Mg acts as an insulin sensor to induce insulin receptor phosphorylation and regulate reporter tyrosine kinase activity. Furthermore, Mg deficiency is also associated with an impaired antioxidant system of endothelial cells, oxidative stress, and lipid metabolism disorder (61, 62). In an animal study on maternal Mg deficiency, the expression of fatty acid synthase (FASN) and fatty acyl transport protein 1 (FATP 1) in liver and adipose tissue was increased in offspring at 18 months of age (59). Additionally, chronic Mg intake deficiency in WNIN female rats not only increased serum corticosterone, leptin, and proinflammatory cytokine levels but also led to lipid metabolism disorder and insulin resistance in their offspring (63).
Chromium
Cr is an essential trace element that is important in carbohydrate metabolism. The adequate intake (AI) is 25 and 30 mcg/day for non-pregnant women and pregnant women, respectively (64). The data on Cr intakes from dietary supplements are very limited; thus, the frequency and degree of Cr restriction remain unclear (65). It is found as a cofactor in insulin signaling. Diabetic and prediabetic patients usually have low serum Cr concentrations (66).
The potential mechanisms of the link between Cr and glucose metabolism are as follows. The Cr-nicotinic acid complex has been proven to be a glucose tolerance factor (GTF) that potentiates the action of insulin in vitro and in vivo (67, 68). Cr also plays a potential anti-inflammatory role in glucose metabolism (69). Cr restriction leads to glucose and lipid metabolism disorders (70). Cr supplementation intake moderates fasting blood glucose levels in diabetic patients (71, 72) and diabetic rat models (73). The involved mechanism is mainly insulin signaling moderation, such as insulin binding, insulin receptor number, insulin internalization, pancreatic β cell sensitivity and insulin receptor enzymes (74).
Additionally, Cr intake in pregnant women can also affect glucose metabolism in offspring later in life (65). In a clinical study (n = 76), Cr levels in hair and blood samples of infants of diabetic mothers were significantly lower than those of infants from healthy mothers (age ranged 30–40) (75). Several animal studies have reached the same conclusion that maternal Cr restriction contributes to pre-disposal to diabetes and obesity in offspring, and the underlying mechanisms are complex. A study in 2010 reported that maternal Cr restriction increased body fat of the offspring at 12 months of age in both genders and was associated with impaired glucose uptake by muscle in WNIN rat offspring (76). Increased oxidative stress is also an underlying mechanism (77). Additionally, maternal Cr restriction inhibits hepatic insulin signaling and activates Wnt signaling (78). Our group data also found miRNA dysfunction and alteration of the methylation status of hepatic genes in animal models (79, 80).
Iron
Fe is an essential mineral functioning as a coenzyme in a variety of physiological processes, including oxygen transportation, red blood cell growth, energy metabolism and fat metabolism, immune response, and DNA synthesis and repair (81–83). Fe restriction leads to adverse outcomes during pregnancy. It is also a worldwide health problem, especially for infants, nursing mothers, and women of reproductive age (84, 85). The physiologic demand for Fe considerably increases during pregnancy compared with the average amount of Fe intake, changing from 0.8 mg to at most 7.5 mg absorbed Fe per day (86), increasing the risk of anemia in pregnant women. The adverse pregnancy outcomes of Fe deficiency include intrauterine growth retardation, pre-maturity, fetoplacental miss ratio, and higher risk for peripartum blood transfusion (87).
However, excess Fe, stored in the liver, influences glucose metabolism and leads to insulin resistance and T2DM (88). Plasma Fe overload is positively correlated with the risk of insulin resistance and T2DM, although it does not typically feature T2DM (89). Pregnant women with increased Fe stores are at higher risk of developing gestational diabetes mellitus (90). Transferrin saturation was 3–4-fold higher in patients with T2DM (91). Elevated Fe stores may be induced through multiple pathways, such as pancreatic β cell oxidative damage, hepatic insulin extraction impairment, and hepatic glucose production suppression (92, 93). Psyrogiannis et al. indicated that offspring from T2DM parents have relative Fe “overload,” which may be a major reason why insulin resistance worsens in offspring (94, 95). Furthermore, in rodent models, Zein et al. found that excessive Fe intake in pregnant mice on a high-fructose diet increased blood sugar in male offspring mice and body weight in female offspring mice, which appeared to be associated with decreased activities of the antioxidant enzyme glutathione peroxidase (GPx) (96). Thus, a high Fe intake increases the risk of glucose metabolism disorder and oxidative damage in newborns, indicating that we should take a more cautious attitude toward Fe supplementation during gestation. According to the Food and Nutrition Board (FNB), daily Fe intake range is 27–45 mg [RDA and upper intake level (UL)] for pregnant women in order to reduce the risk of both Fe restriction and excess due to the aforementioned reasons (64).
Selenium
Se, another trace element, binds to proteins to make Se-dependent glutathione peroxidases and other selenoprotein complexes to defend against oxidative stress (97). Se intakes and serum concentrations vary somewhat by geographical region because the amounts of Se in soil and in local foods differ from each other regions (98). Se deficiency diseases, Keshan disease, have been exclusively reported in low-Se rural areas in China (99). Se has a dual effect on glucose metabolism: Se serves as an antioxidant to prevent the progression of T2DM and its complications, while excessively high serum Se levels are related to a high prevalence of diabetes (100–102). Administration of Se (0.2 μmol/μL) in drinking water in non-obese diabetic mice for 3 weeks reduced fasting blood glucose and moderate lipid metabolism (103). In a follow-up study over two decades, individuals with high serum Se levels had a 24% reduction in diabetes risk, compared with those with low serum Se levels (104). In one cross-sectional analysis of diabetic patients and healthy people, GPx activity declined significantly, and a slight decrease was observed in the concentration of total Se, indicating the association of Se levels and the progression of diabetes (105). Therefore, Se supplementation is an efficient way to prevent glucose metabolism disorder because of the connection of its counteractive oxidative properties and the onset of metabolic diseases, such as T2DM. Conversely, a cross-sectional analysis of American adults discovered that high serum Se is positively correlated with the prevalence of diabetes (101). Additionally, a meta-analysis published in 2018 comes to the same conclusion (106). The potential explanation behind this difference is that serum Se reflects only the amount of Se intake and not the activity of selenoprotein, the form participating in the biologic function of Se. More studies are needed to explain the association and mechanism of excessive Se intake and glucose metabolism disorder.
During pregnancy, serum Se levels decrease significantly in the first few months of life. This reduction in the pregnant mother initiates oxidative stress in the fetus (107). Adequate ingestion of Se is basic nutrition, as its deficiency results in pregnancy-related complications, metabolic disorders, and other disorders in offspring (108, 109). Melo et al. found that in rats, maternal Se increases insulin secretion and glucose tolerance at 80 days of age (110). However, Zeng et al. found that excessive Se in the mother's diet led to insulin resistance in offspring at 112 days of age (111). The different conclusion between the two studies may arise from the different dosages in the maternal diet, as both the low-Se and high-Se diets during pregnancy can induce insulin resistance with different mechanisms (112).
The recommended Se intake by the FNB is stipulated at levels of 60 mg/day (RDA), changing from 55 mg/day for non-pregnant women to assure the optimal concentration of GPx in blood serum; the UL of Se intake is set at 400 mcg per day (113). A report published by the German, Austrian and Swiss nutrition societies suggested that as persons from Se-deficient regions (China) can achieve selenoprotein P (SelP) saturation by taking a daily intake of 49 mcg of Se, they recommend a daily Se intake of 70 mcg for men and 60 mcg for women, whether pregnant or not (114). Moreover, People living in areas with low Se soil concentration are recommended to increase Se intake (114).
Zinc
Zn directly affects the physiology and action of insulin (115), the regulation of cytokine expression, the suppression of inflammation, and the activation of antioxidant enzymes (116). Zn is important in the functions of epigenetic enzymes; thus, lack of Zn may disrupt the bioactivities through epigenetics mechanisms (117). Zn can also increase glucose transport into cells and potentiate insulin-induced glucose transport to moderate glucose and lipid metabolism (118).
Inadequate Zn intake is fairly common globally (119), especially in some select population subgroups, including lower socioeconomic groups, vegetarians, the elderly, and pregnant and lactating women (64, 120, 121). Pregnant women require an increased Zn intake, changing from the recommended amount of 18–27 mg/day (RDA), thus the risk of Zn deficiency around pregnant women is increased (64). Low serum Zn levels in pregnant women are related to several pregnancy complications (122). Adequate Zn supplementation during pregnancy reduces the risk of pre-term birth (123, 124). Zn deficiency is likely a reflection of poor nutrition, thus the best method to improve Zn deficiency is overall maternal nutrition supplementation (125). A randomized clinical study in Peru in 2017 did not reveal any evidence of the effect of maternal Zn restriction on insulin resistance (126). In contrast, many rodent studies have shown the link between Zn deficiency in pregnant rats and insulin resistance in their offspring. The first animal study was conducted by Rosario et al., in which they found that maternal Zn deficiency played a key role in the metabolic status of the offspring (127). In addition, decreased expression of placental hydroxysteroid 11-beta dehydrogenase 2 (HSD11B2) was observed in pregnant women having a Zn restriction diet, which increased fetal exposure to maternal cortisol and may lead to insulin resistance and hypertension in the offspring (128). In addition, Padmavathi et al. suggested that maternal Zn restriction not only induces insulin resistance, but also alters body composition and adipose profile (129). The alteration is associated with serum leptin levels and sex-specific changes in the insulin signaling pathway. Additionally, the effect of maternal Zn restriction is related to the nutrient intake of the offspring, as offspring fed an excessive nutrient diet had higher weight and insulin-like growth factor-1 (IGF1) levels (130, 131). Therefore, Zn restriction in rats in early life impairs downstream insulin signaling with sex differences, and Zn supplementation at weaning reverses the adverse effect (132).
Vitamins
Vitamin A
Vitamin A is the general term for retinol and its natural metabolites, including retinal and retinoic acid. About 10–20% of pregnant women develop vitamin A deficiency in developing countries, and subclinical vitamin A deficiency affects more individuals, particularly in Africa and Asia (36). Carotenoids such as B-carotene can be converted into retinol in the human body. These substances are collectively called carotenoids (133). In the past, the physiological functions of vitamin A and carotenoids were widely studied. They play an important role in reproduction, embryogenesis, vision, growth, cell differentiation and proliferation, maintenance of epithelial cell integrity and immune function (134–136). Studies on the link between vitamin A and metabolic diseases, such as obesity and T2DM, are receiving increasing attention (137, 138).
In a Denmark birth cohort study, high prenatal vitamin A exposure was related to a low risk of developing T2DM (139). Retinol binding protein 4 (RBP4) is a novel type of adipokine that affects vitamin A delivery to tissues. In 2005, Kahn et al. found high serum RBP4 levels in insulin-resistant mice and obese and T2DM individuals, which may participate in the pathogenesis of T2DM (140, 141). In addition, vitamin A is important in the development of the pancreas, especially the differentiation and growth of pancreatic β-cells (138, 142, 143). Impaired fetal pancreas function was seen in rodent models of vitamin A deficiency (144, 145). However, Guo et al. found that excessive supplementation of β-carotene during pregnancy disturbed the lipid metabolism of the offspring and induced impaired glucose tolerance in rat models (146). Thus, the proper intake of vitamin A and carotenoids in pregnancy is needed to avoid both maternal excess and deficiency intake. The recommended amount of vitamin A intake was given as retinol activity equivalents (RAE). The range of vitamin A intake is 700–3,000 mcg RAE/day for non-pregnant women, 770–3,000 mcg RAE/day for pregnant women, and 1,300–3,000 mcg RAE/day for lactation women (64).
Vitamin D
Vitamin D is a lipid-soluble steroid and an anti-inflammatory factor. The major physiological action of vitamin D is bone health. Its main resources include dietary intake and skin synthesis through sun exposure. After two hydrogenation conversions, vitamin D transforms into its active form, 1,25-dihydroxyvitamin (OH)2D, and then binds to vitamin D receptor (VDR) in the nuclear receptor and affects gene transcription, including calcium binding protein (CaBP), epithelial calcium channel (ECaC), 25(OH)D 24-hydroxylase (24-OHase), receptor activator nuclear factor-κB ligand (RANKL), alkaline phosphatase (alk PASE), prostate-specific antigen (PSA), and parathyroid hormone (PTH) (147, 148). As vitamin D can be obtained by receiving regular exposure to sunlight, it is recommended that pregnant women do not need extra vitamin D supplementation; thus, women, whether pregnant or not, are recommended to intake 5 mcg per day (53). Vitamin D enables the intestine to absorb more Ca and phosphorus and moderates the renal tubules reabsorbing Ca. The existence of VDR in the heart, liver, blood vessels, and central nervous system and other organs means that vitamin D also functions in these tissues (149). Vitamin D deficiency is associated with the risk of both type 1 diabetes mellitus and T2DM (148, 150, 151). In a cross-sectional study of 808 non-diabetic participants of the Framingham Offspring Study, plasma 25(OH)D concentrations were inversely associated with markers of the insulin resistant phenotype (152). The mechanism of the effects of vitamin D on glucose metabolism includes regulating insulin production and secretion, enhancing insulin sensitivity, and reducing inflammation (147, 153). In terms of epigenetics, vitamin D participates in the increased expression of DNA demethylases that inhibit hypermethylation of multiple gene promoter regions of many diabetes-related genes (154). High vitamin D intake during pregnancy results in an increase in maternal and infant 25(OH)D concentrations and engages in maternal glucose metabolism and fetal growth (155).
In recent decades, many studies have shown that vitamin D deficiency during pregnancy may potentially affect chronic disease susceptibility of fetuses later in life (156). Many clinical studies have proven that maternal vitamin D deficiency is linked with increased insulin resistance in offspring in early childhood (157–159). Rehabilitation of maternal and offspring vitamin D reduces fasting glucose levels, as shown in rat models (160). The underlying mechanism may be involved in the epigenetic alterations of special genes. In animal models, vitamin D deficiency during pregnancy contributed to insulin resistance and impaired glucose tolerance in male offspring, associated with increased DNA methylation of the nuclear factor κB inhibitor α (Iκbα) gene, which decreased luciferase activity (161).
Vitamin B12 and Folate
Vitamin B12 is a micronutrient essential for one-carbon metabolism. Lack of vitamin B12 may result in decreased RBC production, dysfunction of the nervous system, and metabolic disorders. During pregnancy, vitamin B12 is essential for fetal growth and development. The metabolic demands of fetuses for vitamin B12 are increasing during pregnancy (162). Vitamin B12 and folate deficiency mainly result from poor dietary intake, increased demand for maternal biological activity and fetal growth, and the increased loss of folate (163). Recommended vitamin B12 intake for pregnant women is 2.6 mcg per day, rising from 2.4 mcg per day for non-pregnant female adults (164). The FNB introduced dietary folate equivalents (DFEs) when calculating different absorption of folate. Individuals who are pregnant should consume 600 mcg DFE daily, increasing from 400 mcg DEF per day for non-pregnant women (164). However, maternal vitamin B12 deficiency has become increasingly common (165).
Vitamin B12 and folate deficiency in pregnant women are more likely to cause insulin resistance. In a clinical study in Pune, low maternal vitamin B12 and high folate status may increase the prevalence of obesity and T2DM in India (166). Other studies, one from Nepal in 2011 and one from India in 2014, obtained consistent findings that maternal vitamin B 12 deficiency is related to a high risk of insulin resistance in school-aged offspring (167, 168), but folate supplementation showed no significant improvement in insulin resistance among offspring at 6–8 years of age (167). Additionally, in a study in the UK, such changes were not observed in the cord blood of neonatal Caucasian patients (169). These findings indicated that the effects of maternal vitamin B12 restriction on offspring may not be observed immediately in early life. Additionally, in a systematic review including 46 articles in India, low maternal vitamin B12 status was associated with a high risk of adverse health outcomes (high adiposity, insulin resistance, and low offspring B12) in offspring (170).
Apart from many human studies, many animal models also provide substantial evidence and reveal the potential mechanisms of the relationship between maternal vitamin B12 and folate status and glucose metabolism in offspring. The first animal study is conducted by Sinclair et al. They found an association of folate reduction in maternal diet and alterations to DNA methylation and increased insulin resistance in offspring (39). The effect of vitamin B12 and folate is related to the offspring diet. Several animal studies found that in female offspring, increased body fat percentage, fasting hyperglycemia, and insulin resistance in later life associated with a maternal high folic acid and low vitamin B12 intake; though dependent on offspring diet intake (171, 172). Furthermore, vitamin B12 supplementation may partially reverse the adverse effect, but the timing is critical. In another study, rehabilitation at weaning did not show any improvement in the onset of adverse outcomes, but rehabilitation at parturition did (173). In another rodent study, altered DNA methylation in metabolic-related genes in the offspring of maternal vitamin B12 restriction was reported, and rehabilitation at conception and parturition partly reversed the methylation status (174).
Conclusion and Future Perspective
It is obvious that both maternal excess and deficiency of micronutrients affect glucose metabolism and induce insulin resistance in offspring later in life. Micronutrient deficiency largely results from poverty. Other possible causes of undernutrition and overnutrition include sociocultural factors, economic factors, inadequate care and feeding practices, improper dietary intake and diseases (36). The mechanisms are summarized in Figure 1. Recently, robust evidence from human and animal studies has convincingly shown that improper micronutrient intake in pregnant women induces epigenetic alterations in the placentae and fetuses that can alter the expression of genes in offspring, influence fetal growth and alter glucose metabolism in adulthood. These epigenetic factors are heritable for several generations. A list of human studies is summarized in Table 1 and a list of animal studies is in Table 2. Therefore, it should be seen as equally significant compared with genetic factors in the discussion of the risk of T2DM. However, it is still unclear how much micronutrient intake is proper in different gestational stages to avoid adverse effects on glucose metabolism and how many generations it induces epigenetic alterations. In addition to micronutrient intake, other environmental factors, including parental obesity/diabetes, macronutrients, toxins, stress, and physical activities, should also be taken into consideration because they also result in the inherited traits and contribute to diabetic offspring later in life.
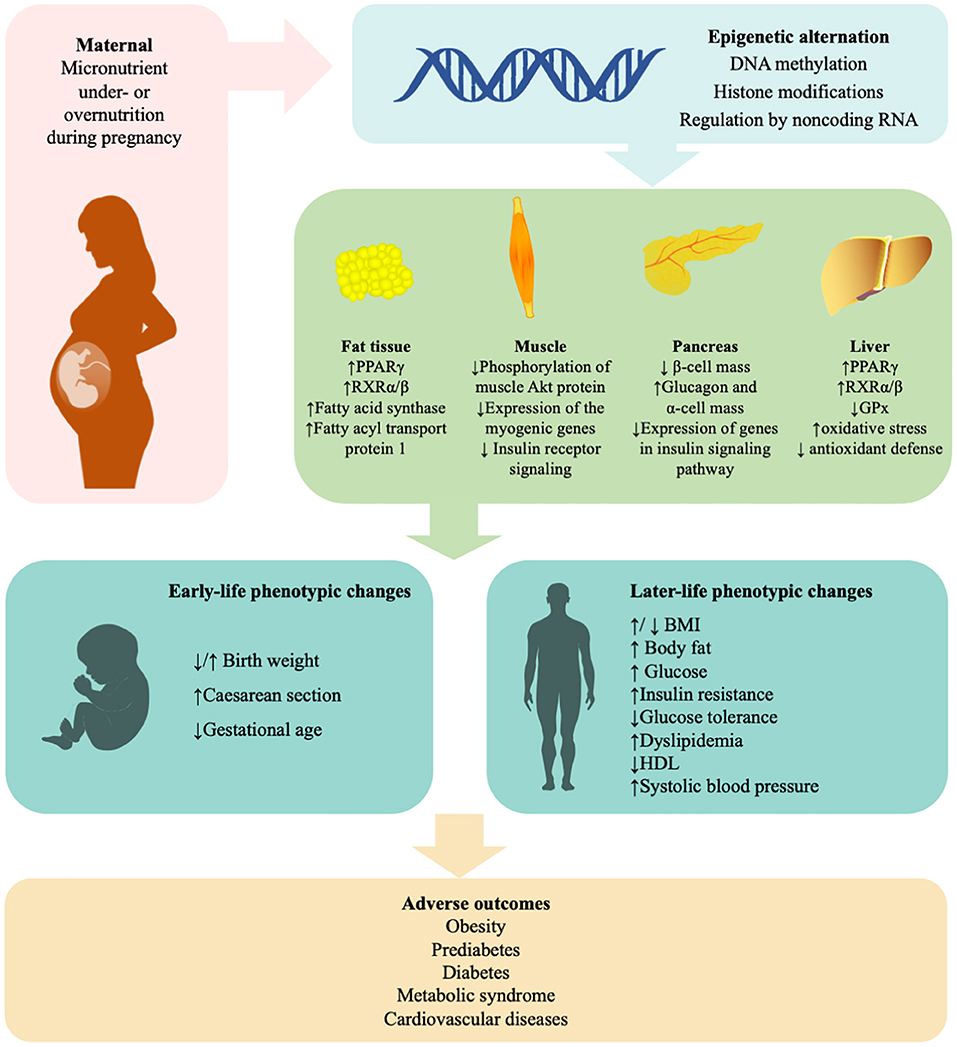
Figure 1. Mechanisms in the effects of maternal micronutrient on glucose metabolism of offspring. “Fetal programming” suggests that maternal micronutrient is linked with the susceptibility of insulin resistance, glucose intolerance, obesity, and type 2 diabetes in offspring. Epigenetic mechanisms, including DNA methylation, histone modifications, and microRNA, play a critical role in the process. Supplement of minerals and vitamins under medical guides will be beneficial to subsequent generations. BMI, body mass index; GPx, glutathione peroxidase; HDL, high-density lipoprotein; PPARγ, peroxisome proliferator-activated receptor gamma; RXR, retinoid X receptors. ↑, increased; ↓, decreased.
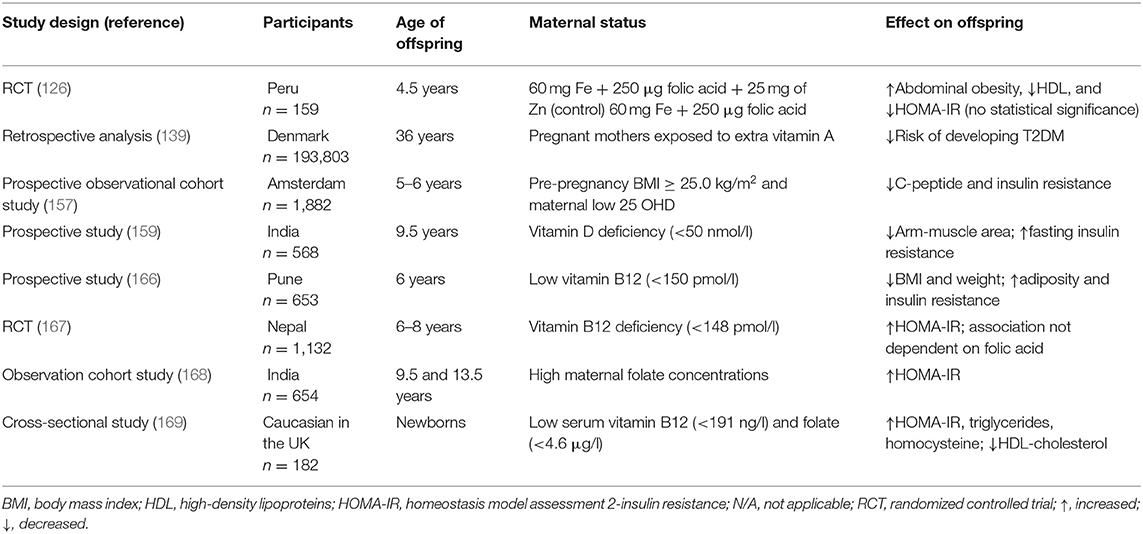
Table 1. Charting the maternal micronutrients status and the effect on glucose metabolism in offspring.
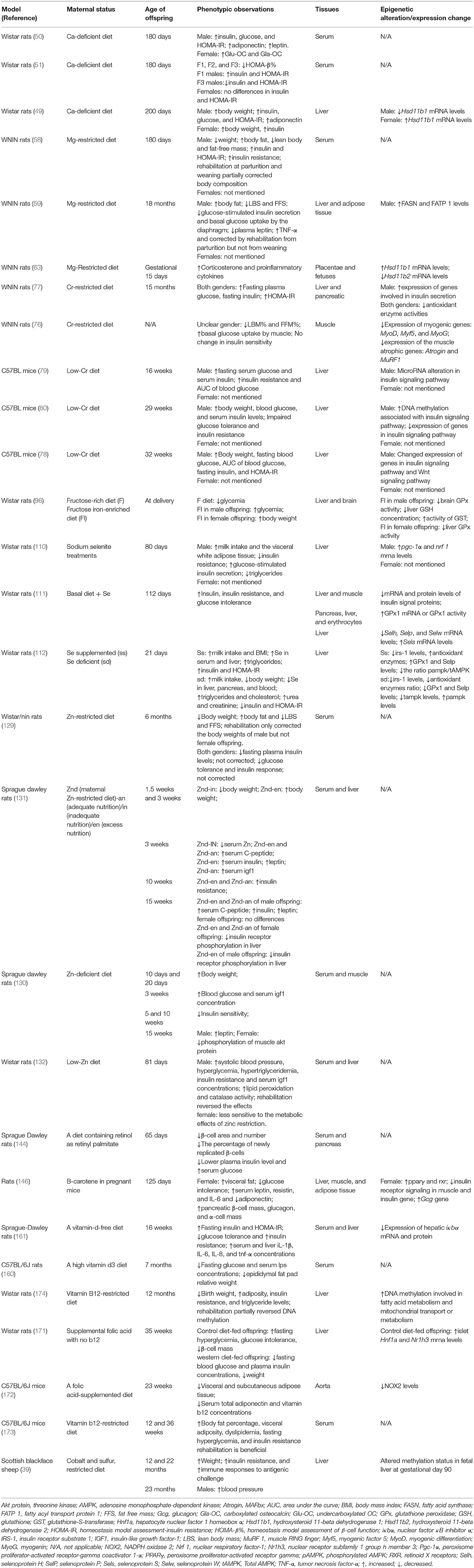
Table 2. Summary of animal studies investigating the effects of maternal micronutrients on glucose metabolism in offspring.
However, some limitations should be noted. Due to the lack of clinical studies, we largely focus on rodent experiments. Currently, clinical studies on micronutrients are relevantly difficult to conduct because of the difficulties in excluding possible confounding factors, controlling daily micronutrient intake, measuring dietary micronutrients intake at the microgram level and other methodologic challenges. The small numbers of participants involved in studies on certain micronutrient and limited follow-up are also disadvantages for further analysis. Therefore, the discussion on clinical studies is limited and lacks quantitative, detailed analysis.
In the process of our learning how epigenetic factors participate in the development of T2DM and its rapid expansion, there remains much more to be explored. How are DNA methylation, histone codes or non-coding RNAs involved in the process? When is the key period to develop epigenetic alterations? When should interventions be conducted during gestation to target specific epigenetic markers? Moreover, considering the period prior to conception or a certain time when pregnancy is not yet known, but nutrient requirements are essential, more questions require answers. The questions are given below. What is the relationship between the micronutrient status in these stages and offspring glucose metabolism later in life? How does micronutrient status affect offspring glucose metabolism during these periods? And what strategies can be adopted to optimize micronutrient status at that time? Finally, today, there are many limitations of existing epigenetic studies. Interventions for obesity and diabetes can be carried out by supplementing corresponding nutrients in the perinatal period, but more explicit guidelines are needed. To detect the adverse effects of maternal micronutrient status, whether relevant epigenetic indicators can be utilized in obstetric examinations also needs more research. Thus, studies have great potential to understand the causes of the current diabetic epidemic and reduce the occurrence of diabetes worldwide.
Author Contributions
YW drafted the manuscript. QZ and XX revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by National Natural Science Foundation of China (Nos. 81870545, 81870579, 82170854, 81570715, and 81170736), Beijing Natural Science Foundation (7202163), Beijing Natural Science Foundation (7202163), Beijing Municipal Science and Technology Commission (Z201100005520011), National Key R&D Program of China (2017YFC1309603), National Key Research and Development Program of China (2016YFA0101002 and 2018YFC2001100), Scientific Activities Foundation for Selected Returned Overseas Professionals of Human Resources and Social Security Ministry, Beijing Dongcheng District Outstanding Talent Funding Project (2019DCT-M-05), Medical Epigenetics Research Center, Chinese Academy of Medical Sciences (2017PT31036 and 2018PT31021), the Non-profit Central Research Institute Fund of Chinese Academy of Medical Sciences (Nos. 2017PT32020 and 2018PT32001), and Chinese Academy of Medical Sciences Innovation Fund for Medical Sciences (CIFMS2017-I2M-1-008).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Saeedi P, Petersohn I, Salpea P, Malanda B, Karuranga S, Unwin N, et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: results from the international diabetes federation diabetes atlas, 9 edition. Diabetes Res Clin Pract. (2019) 157:107843. doi: 10.1016/j.diabres.2019.107843
2. Neel JV. Diabetes mellitus: a “thrifty” genotype rendered detrimental by “progress”? Am J Hum Genet. (1962) 14:353–62.
3. Roseboom TJ, van der Meulen JH, Ravelli AC, Osmond C, Barker DJ, Bleker OP. Effects of prenatal exposure to the Dutch famine on adult disease in later life: an overview. Mol Cell Endocrinol. (2001) 185:93–8. doi: 10.1016/S0303-7207(01)00721-3
4. Sotomayor O. Fetal and infant origins of diabetes and ill health: evidence from Puerto Rico's 1928 and 1932 hurricanes. Econ Hum Biol. (2013) 11:281–93. doi: 10.1016/j.ehb.2012.02.009
5. Hales CN, Barker DJ. Type 2 (non-insulin-dependent) diabetes mellitus: the thrifty phenotype hypothesis. Diabetologia. (1992) 35:595–601. doi: 10.1007/BF00400248
6. Hales CN, Barker DJ. The thrifty phenotype hypothesis. Br Med Bull. (2001) 60:5–20. doi: 10.1093/bmb/60.1.5
7. Oestreich AK, Moley KH. Developmental and transmittable origins of obesity-associated health disorders. Trends Genet. (2017) 33:399–407. doi: 10.1016/j.tig.2017.03.008
8. Goyal D, Limesand SW, Goyal R. Epigenetic responses and the developmental origins of health and disease. J Endocrinol. (2019) 242:T105–19. doi: 10.1530/JOE-19-0009
9. Safi-Stibler S, Gabory A. Epigenetics and the developmental origins of health and disease: parental environment signalling to the epigenome, critical time windows and sculpting the adult phenotype. Semin Cell Dev Biol. (2020) 97:172–80. doi: 10.1016/j.semcdb.2019.09.008
10. Rogers JM. Smoking and pregnancy: epigenetics and developmental origins of the metabolic syndrome. Birth Defects Res. (2019) 111:1259–69. doi: 10.1002/bdr2.1550
11. Nobre JL, Lisboa PC, Santos-Silva AP, Lima NS, Manhães AC, Nogueira-Neto JF, et al. Calcium supplementation reverts central adiposity, leptin, and insulin resistance in adult offspring programed by neonatal nicotine exposure. J Endocrinol. (2011) 210:349–59. doi: 10.1530/JOE-11-0172
12. Gong Y, Xue Y, Li X, Zhang Z, Zhou W, Marcolongo P, et al. Inter- and transgenerational effects of paternal exposure to inorganic arsenic. Adv Sci (Weinh). (2021) 8:2002715. doi: 10.1002/advs.202002715
13. Xi Z, Fang L, Xu J, Li B, Zuo Z, Lv L, et al. Exposure to aroclor 1254 persistently suppresses the functions of pancreatic β-cells and deteriorates glucose homeostasis in male mice. Environ Pollut. (2019) 249:822–30. doi: 10.1016/j.envpol.2019.03.101
14. Martín L, Kamstra JH, Hurem S, Lindeman LC, Brede DA, Aanes H, et al. Altered non-coding RNA expression profile in F progeny 1 year after parental irradiation is linked to adverse effects in zebrafish. Sci Rep. (2021) 11:4142. doi: 10.1038/s41598-021-83345-3
15. Laker RC, Lillard TS, Okutsu M, Zhang M, Hoehn KL, Connelly JJ, et al. Exercise prevents maternal high-fat diet-induced hypermethylation of the Pgc-1α gene and age-dependent metabolic dysfunction in the offspring. Diabetes. (2014) 63:1605–11. doi: 10.2337/db13-1614
16. Antoun E, Kitaba NT, Titcombe P, Dalrymple KV, Garratt ES, Barton SJ, et al. Maternal dysglycaemia, changes in the infant's epigenome modified with a diet and physical activity intervention in pregnancy: Secondary analysis of a randomised control trial. PLoS Med. (2020) 17:e1003229. doi: 10.1371/journal.pmed.1003229
17. Mathias PCF, Elmhiri G, de Oliveira JC, Delayre-Orthez C, Barella LF, Tófolo LP, et al. Maternal diet, bioactive molecules, and exercising as reprogramming tools of metabolic programming. Eur J Nutr. (2014) 53:711–22. doi: 10.1007/s00394-014-0654-7
18. Balasubramanian P, Varde PA, Abdallah SL, Najjar SM, MohanKumar PS, MohanKumar SMJ. Differential effects of prenatal stress on metabolic programming in diet-induced obese and dietary-resistant rats. Am J Physiol Endocrinol Metab. (2015) 309:E582–E8. doi: 10.1152/ajpendo.00167.2015
19. Ding L, Xiao X-H. Gut microbiota: closely tied to the regulation of circadian clock in the development of type 2 diabetes mellitus. Chin Med J (Engl). (2020) 133:817–25. doi: 10.1097/CM9.0000000000000702
20. de Almeida Faria J, de Araújo TMF, Mancuso RI, Meulman J, da Silva Ferreira D, Batista TM, et al. Day-restricted feeding during pregnancy and lactation programs glucose intolerance and impaired insulin secretion in male rat offspring. Acta Physiol (Oxf). (2016) 217:240–53. doi: 10.1111/apha.12684
21. Lumey LH, Khalangot MD, Vaiserman AM. Association between type 2 diabetes and prenatal exposure to the Ukraine famine of 1932-33: a retrospective cohort study. Lancet Diabetes Endocrinol. (2015) 3:787–94. doi: 10.1016/S2213-8587(15)00279-X
22. Kyle UG, Pichard C. The Dutch Famine of 1944-1945: a pathophysiological model of long-term consequences of wasting disease. Curr Opin Clin Nutr Metab Care. (2006) 9:388–94. doi: 10.1097/01.mco.0000232898.74415.42
23. Painter RC, Roseboom TJ, Bleker OP. Prenatal exposure to the Dutch famine and disease in later life: an overview. Reprod Toxicol. (2005) 20:345–52. doi: 10.1016/j.reprotox.2005.04.005
24. Painter RC, Osmond C, Gluckman P, Hanson M, Phillips DIW, Roseboom TJ. Transgenerational effects of prenatal exposure to the Dutch famine on neonatal adiposity and health in later life. BJOG. (2008) 115:1243–9. doi: 10.1111/j.1471-0528.2008.01822.x
25. Heijmans BT, Tobi EW, Stein AD, Putter H, Blauw GJ, Susser ES, et al. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc Natl Acad Sci USA. (2008) 105:17046–9. doi: 10.1073/pnas.0806560105
26. Tobi EW, Goeman JJ, Monajemi R, Gu H, Putter H, Zhang Y, et al. DNA methylation signatures link prenatal famine exposure to growth and metabolism. Nat Commun. (2014) 5:5592. doi: 10.1038/ncomms6592
27. Li C, Lumey LH. Exposure to the Chinese famine of 1959-61 in early life and long-term health conditions: a systematic review and meta-analysis. Int J Epidemiol. (2017) 46:1157–70. doi: 10.1093/ije/dyx013
28. Shen L, Li C, Wang Z, Zhang R, Shen Y, Miles T, et al. Early-life exposure to severe famine is associated with higher methylation level in the IGF2 gene and higher total cholesterol in late adulthood: the Genomic Research of the Chinese Famine (GRECF) study. Clin Epigenetics. (2019) 11:88. doi: 10.1186/s13148-019-0676-3
29. Wang Z, Song J, Li C, Li Y, Shen L, Dong B, et al. DNA methylation of the INSR gene as a mediator of the association between prenatal exposure to famine and adulthood waist circumference. Sci Rep. (2020) 10:12212. doi: 10.1038/s41598-020-69120-w
30. Alejandro EU, Jo S, Akhaphong B, Llacer PR, Gianchandani M, Gregg B, et al. Maternal low-protein diet on the last week of pregnancy contributes to insulin resistance and β-cell dysfunction in the mouse offspring. Am J Physiol Regul Integr Comp Physiol. (2020) 319:R485–R96. doi: 10.1152/ajpregu.00284.2019
31. Zheng J, Xiao X, Zhang Q, Wang T, Yu M, Xu J. Maternal low-protein diet modulates glucose metabolism and hepatic microRNAs expression in the early life of offspring. Nutrients. (2017) 9:205. doi: 10.3390/nu9030205
32. Zhang Q, Xiao X, Zheng J, Li M, Yu M, Ping F, et al. A maternal high-fat diet induces DNA methylation changes that contribute to glucose intolerance in offspring. Front Endocrinol (Lausanne). (2019) 10:871. doi: 10.3389/fendo.2019.00871
33. Zheng J, Xiao X, Zhang Q, Yu M, Xu J, Wang Z, et al. Maternal and post-weaning high-fat, high-sucrose diet modulates glucose homeostasis and hypothalamic POMC promoter methylation in mouse offspring. Metab Brain Dis. (2015) 30:1129–37. doi: 10.1007/s11011-015-9678-9
34. Dubey P, Thakur V, Chattopadhyay M. Role of minerals and trace elements in diabetes and insulin resistance. Nutrients. (2020) 12:1864. doi: 10.3390/nu12061864
35. Young VR. Trace element biology: the knowledge base and its application for the nutrition of individuals and populations. J Nutr. (2003) 133(Suppl. 1):1581s−7s. doi: 10.1093/jn/133.5.1581S
36. Bailey RL, West KP, Black RE. The epidemiology of global micronutrient deficiencies. Ann Nutr Metab. (2015) 66(Suppl. 2):22–33. doi: 10.1159/000371618
37. Venu L, Harishankar N, Krishna TP, Raghunath M. Does maternal dietary mineral restriction per se predispose the offspring to insulin resistance? Eur J Endocrinol. (2004) 151:287–94. doi: 10.1530/eje.0.1510287
38. Venu L, Harishankar N, Prasanna Krishna T, Raghunath M. Maternal dietary vitamin restriction increases body fat content but not insulin resistance in WNIN rat offspring up to 6 months of age. Diabetologia. (2004) 47:1493–501. doi: 10.1007/s00125-004-1506-4
39. Sinclair KD, Allegrucci C, Singh R, Gardner DS, Sebastian S, Bispham J, et al. DNA methylation, insulin resistance, and blood pressure in offspring determined by maternal periconceptional B vitamin and methionine status. Proc Natl Acad Sci USA. (2007) 104:19351–6. doi: 10.1073/pnas.0707258104
40. Ross AC, Taylor CL, Yaktine AL, Del Valle HB, editors. Dietary Reference Intakes for Calcium and Vitamin D. The National Academies Collection: Reports funded by National Institutes of Health. Washington, DC: National Academies Press (US) (2011).
41. Wimalawansa SJ, Razzaque MS, Al-Daghri NM. Calcium and vitamin D in human health: hype or real? J Steroid Biochem Mol Biol. (2018) 180:4–14. doi: 10.1016/j.jsbmb.2017.12.009
42. Merialdi M, Mathai M, Ngoc NTN, Purwar M, Campodonico L, Abdel-Aleem H, et al. World Health Organization systematic review of the literature and multinational nutritional survey of calcium intake during pregnancy. Fetal Matern Med Rev. (2005) 16:1506. doi: 10.1017/S0965539505001506
43. Lee SE, Talegawkar SA, Merialdi M, Caulfield LE. Dietary intakes of women during pregnancy in low- and middle-income countries. Public Health Nutr. (2013) 16:1340–53. doi: 10.1017/S1368980012004417
44. Godswill AG, Somtochukwu IV, Ikechukwu AO, Kate EC. Health benefits of micronutrients (vitamins and minerals) and their associated deficiency diseases: a systematic review. Int J Food Sci. (2020) 3:1–32. doi: 10.47604/ijf.1024
45. Buppasiri P, Lumbiganon P, Thinkhamrop J, Ngamjarus C, Laopaiboon M. Calcium supplementation (other than for preventing or treating hypertension) for improving pregnancy and infant outcomes. Cochrane Database Syst Rev. (2011) 10:CD007079. doi: 10.1002/14651858.CD007079.pub2
46. Prentice A. Maternal calcium requirements during pregnancy and lactation. Am J Clin Nutr. (1994) 59(Suppl. 2):477S−82S. doi: 10.1093/ajcn/59.2.477S
47. Sun G, Vasdev S, Martin GR, Gadag V, Zhang H. Altered calcium homeostasis is correlated with abnormalities of fasting serum glucose, insulin resistance, and beta-cell function in the Newfoundland population. Diabetes. (2005) 54:3336–9. doi: 10.2337/diabetes.54.11.3336
48. Ly LD, Xu S, Choi S-K, Ha C-M, Thoudam T, Cha S-K, et al. Oxidative stress and calcium dysregulation by palmitate in type 2 diabetes. Exp Mol Med. (2017) 49:e291. doi: 10.1038/emm.2016.157
49. Takaya J, Yamanouchi S, Kaneko K. A calcium-deficient diet in rat dams during gestation and nursing affects hepatic 11β-hydroxysteroid dehydrogenase-1 expression in the offspring. PLoS ONE. (2014) 9:e84125. doi: 10.1371/journal.pone.0084125
50. Takaya J, Yamanouchi S, Kino J, Tanabe Y, Kaneko K. A calcium-deficient diet in dams during gestation increases insulin resistance in male offspring. Nutrients. (2018) 10:1745. doi: 10.3390/nu10111745
51. Takaya J, Yamanouchi S, Tanabe Y, Kaneko K. A calcium-deficient diet in rat dams during gestation decreases HOMA-β% in 3 generations of offspring. J Nutrigenet Nutrigenomics. (2016) 9:276–86. doi: 10.1159/000456025
52. Guerrera MP, Volpe SL, Mao JJ. Therapeutic uses of magnesium. Am Fam Physician. (2009) 80:157–62.
53. Institute of Medicine Standing Committee on the Scientific Evaluation of Dietary Reference I. The National Academies Collection: Reports funded by National Institutes of Health. Dietary Reference Intakes for Calcium, Phosphorus, Magnesium, Vitamin D, and Fluoride. Washington, DC: National Academies Press (US) Copyright 1997, National Academy of Sciences (1997).
54. Bartram J. Calcium and Magnesium in Drinking-water: Public Health Significance Geneva: World Health Organization Press (2009).
55. Altura BM, Li W, Zhang A, Zheng T, Shah NC, Shah GJ, et al. Sudden cardiac death in infants, children and young adults: possible roles of dietary magnesium intake and generation of platelet-activating factor in coronary arteries. J Heart Health. (2016) 2. doi: 10.16966/2379-769X.121
56. Dalton LM, Ní Fhloinn DM, Gaydadzhieva GT, Mazurkiewicz OM, Leeson H, Wright CP. Magnesium in pregnancy. Nutr Rev. (2016) 74:549–57. doi: 10.1093/nutrit/nuw018
57. Volpe SL. Magnesium, the metabolic syndrome, insulin resistance, and type 2 diabetes mellitus. Crit Rev Food Sci Nutr. (2008) 48:293–300. doi: 10.1080/10408390701326235
58. Venu L, Kishore YD, Raghunath M. Maternal and perinatal magnesium restriction predisposes rat pups to insulin resistance and glucose intolerance. J Nutr. (2005) 135:1353–8. doi: 10.1093/jn/135.6.1353
59. Venu L, Padmavathi IJN, Kishore YD, Bhanu NV, Rao KR, Sainath PB, et al. Long-term effects of maternal magnesium restriction on adiposity and insulin resistance in rat pups. Obesity (Silver Spring). (2008) 16:1270–6. doi: 10.1038/oby.2008.72
60. Tosiello L. Hypomagnesemia and diabetes mellitus. A review of clinical implications. Arch Intern Med. (1996) 156:1143–8. doi: 10.1001/archinte.156.11.1143
61. Kostov K. Effects of magnesium deficiency on mechanisms of insulin resistance in type 2 diabetes: focusing on the processes of insulin secretion and signaling. Int J Mol Sci. (2019) 20:1351. doi: 10.3390/ijms20061351
62. Feng J, Wang H, Jing Z, Wang Y, Cheng Y, Wang W, et al. Role of magnesium in type 2 diabetes mellitus. Biol Trace Elem Res. (2020) 196:74–85. doi: 10.1007/s12011-019-01922-0
63. Thomas AE, Inagadapa PJN, Jeyapal S, Merugu NM, Kalashikam RR, Manchala R. Maternal magnesium restriction elevates glucocorticoid stress and inflammation in the placenta and fetus of WNIN rat dams. Biol Trace Elem Res. (2018) 181:281–7. doi: 10.1007/s12011-017-1058-3
64. Trumbo P, Yates AA, Schlicker S, Poos M. Dietary reference intakes: vitamin A, vitamin K, arsenic, boron, chromium, copper, iodine, iron, manganese, molybdenum, nickel, silicon, vanadium, and zinc. J Am Diet Assoc. (2001) 101:294–301. doi: 10.1016/S0002-8223(01)00078-5
65. Adams JBB, Sorenson JCC, Pollard ELL, Kirby JKK, Audhya T. Evidence-based recommendations for an optimal prenatal supplement for women in the U.S., part two: minerals. Nutrients. (2021) 13:1849. doi: 10.3390/nu13061849
66. Rafiei R, Habyby Z, Fouladi L, Najafi S, Asgary S, Torabi Z. Chromium level in prediction of diabetes in pre-diabetic patients. Adv Biomed Res. (2014) 3:235. doi: 10.4103/2277-9175.145737
67. Mertz W. Chromium in human nutrition: a review. J Nutr. (1993) 123:626–33. doi: 10.1093/jn/123.4.626
68. Vincent JB. Elucidating a biological role for chromium at a molecular level. Acc Chem Res. (2000) 33:503–10. doi: 10.1021/ar990073r
69. Moradi F, Maleki V, Saleh-Ghadimi S, Kooshki F, Pourghassem Gargari B. Potential roles of chromium on inflammatory biomarkers in diabetes: a Systematic. Clin Exp Pharmacol Physiol. (2019) 46:975–83. doi: 10.1111/1440-1681.13144
70. Mahdi GS. Chromium deficiency might contribute to insulin resistance, type 2 diabetes mellitus, dyslipidaemia, and atherosclerosis. Diabet Med. (1996) 13:389–90. doi: 10.1002/(SICI)1096-9136(199604)13:4<389::AID-DIA65>3.0.CO;2-J
71. Anderson RA, Polansky MM, Bryden NA, Roginski EE, Mertz W, Glinsmann W. Chromium supplementation of human subjects: effects on glucose, insulin, and lipid variables. Metabolism. (1983) 32:894–9. doi: 10.1016/0026-0495(83)90203-2
72. Sharma S, Agrawal RP, Choudhary M, Jain S, Goyal S, Agarwal V. Beneficial effect of chromium supplementation on glucose, HbA1C and lipid variables in individuals with newly onset type-2 diabetes. J Trace Elem Med Biol. (2011) 25:149–53. doi: 10.1016/j.jtemb.2011.03.003
73. Feng W, Mao G, Li Q, Wang W, Chen Y, Zhao T, et al. Effects of chromium malate on glycometabolism, glycometabolism-related enzyme levels and lipid metabolism in type 2 diabetic rats: a dose-response and curative effects study. J Diabetes Investig. (2015) 6:396–407. doi: 10.1111/jdi.12350
74. Anderson RA. Nutritional factors influencing the glucose/insulin system: chromium. J Am Coll Nutr. (1997) 16:404–10. doi: 10.1080/07315724.1997.10718705
75. Afridi HI, Kazi TG, Kazi N, Baig JA, Jamali MK, Arain MB, et al. Status of essential trace metals in biological samples of diabetic mother and their neonates. Arch Gynecol Obstet. (2009) 280:415–23. doi: 10.1007/s00404-009-0955-x
76. Padmavathi IJN, Rao KR, Venu L, Ismail A, Raghunath M. Maternal dietary chromium restriction programs muscle development and function in the rat offspring. Exp Biol Med (Maywood). (2010) 235:349–55. doi: 10.1258/ebm.2009.009199
77. Padmavathi IJN, Rao KR, Raghunath M. Impact of maternal chromium restriction on glucose tolerance, plasma insulin and oxidative stress in WNIN rat offspring. J Mol Endocrinol. (2011) 47:261–71. doi: 10.1530/JME-11-0010
78. Zhang Q, Sun X, Xiao X, Zheng J, Li M, Yu M, et al. Maternal chromium restriction leads to glucose metabolism imbalance in mice offspring through insulin signaling and wnt signaling pathways. Int J Mol Sci. (2016) 17:1767. doi: 10.3390/ijms17101767
79. Zhang Q, Sun X, Xiao X, Zheng J, Li M, Yu M, et al. Maternal chromium restriction induces insulin resistance in adult mice offspring through miRNA. Int J Mol Med. (2018) 41:1547–59. doi: 10.3892/ijmm.2017.3328
80. Zhang Q, Sun X, Xiao X, Zheng J, Li M, Yu M, et al. Dietary chromium restriction of pregnant mice changes the methylation status of hepatic genes involved with insulin signaling in adult male offspring. PLoS ONE. (2017) 12:e0169889. doi: 10.1371/journal.pone.0169889
81. Musallam KM, Taher AT. Iron deficiency beyond erythropoiesis: should we be concerned? Curr Med Res Opin. (2018) 34:81–93. doi: 10.1080/03007995.2017.1394833
82. Ward RJ, Crichton RR, Taylor DL, Della Corte L, Srai SK, Dexter DT. Iron and the immune system. J Neural Transm (Vienna). (2011) 118:315–28. doi: 10.1007/s00702-010-0479-3
83. Rees WD, Hay SM, Hayes HE, Stevens VJ, Gambling L, McArdle HJ. Iron deficiency during pregnancy and lactation modifies the fatty acid composition of the brain of neonatal rats. J Dev Orig Health Dis. (2020) 11:264–72. doi: 10.1017/S2040174419000552
84. Vucic V, Berti C, Vollhardt C, Fekete K, Cetin I, Koletzko B, et al. Effect of iron intervention on growth during gestation, infancy, childhood, and adolescence: a systematic review with meta-analysis. Nutr Rev. (2013) 71:386–401. doi: 10.1111/nure.12037
85. Organization WH. Weekly Iron and Folic Acid Supplementation as an Anaemia-Prevention Strategy in Women and Adolescent Girls: Lessons Learnt from Implementation of Programmes Among Non-pregnant Women of Reproductive Age. Geneva: World Health Organization Press (2018).
86. Beard JL. Effectiveness and strategies of iron supplementation during pregnancy. Am J Clin Nutr. (2000) 71(Suppl. 5):1288S−94S. doi: 10.1093/ajcn/71.5.1288s
87. Breymann C. Iron deficiency anemia in pregnancy. Semin Hematol. (2015) 52:339–47. doi: 10.1053/j.seminhematol.2015.07.003
88. Fernández-Real JM, McClain D, Manco M. Mechanisms linking glucose homeostasis and iron metabolism toward the onset and progression of type 2 diabetes. Diabetes Care. (2015) 38:2169–76. doi: 10.2337/dc14-3082
89. Eshed I, Elis A, Lishner M. Plasma ferritin and type 2 diabetes mellitus: a critical review. Endocr Res. (2001) 27:91–7. doi: 10.1081/ERC-100107172
90. Lao TT, Chan PL, Tam KF. Gestational diabetes mellitus in the last trimester - a feature of maternal iron excess? Diabet Med. (2001) 18:218–23. doi: 10.1046/j.1464-5491.2001.00453.x
91. Thomas MC, MacIsaac RJ, Tsalamandris C, Jerums G. Elevated iron indices in patients with diabetes. Diabet Med. (2004) 21:798–802. doi: 10.1111/j.1464-5491.2004.01196.x
92. Fernández-Real JM, Ricart-Engel W, Arroyo E, Balançá R, Casamitjana-Abella R, Cabrero D, et al. Serum ferritin as a component of the insulin resistance syndrome. Diabetes Care. (1998) 21:62–8. doi: 10.2337/diacare.21.1.62
93. Wilson JG, Lindquist JH, Grambow SC, Crook ED, Maher JF. Potential role of increased iron stores in diabetes. Am J Med Sci. (2003) 325:332–9. doi: 10.1097/00000441-200306000-00004
94. Psyrogiannis A, Kyriazopoulou V, Symeonidis A, Leotsinidis M, Vagenakis AG. Relative iron “overload” in offspring of patients with type 2 diabetes mellitus: a new component in the conundrum of insulin resistance syndrome? Hormones (Athens). (2003) 2:161–8. doi: 10.14310/horm.2002.1196
95. Zafar U, Qureshi HJ, Imran M. Comparison of iron status and insulin resistance between non-diabetic offsprings of type. 2 diabetics and non-diabetics. J Ayub Med Coll Abbottabad. (2015) 27:307–11.
96. Zein S, Sitti F, Osman M, Arnaud J, Batandier C, Gauchez A-S, et al. Middle iron-enriched fructose diet on gestational diabetes risk and on oxidative stress in offspring rats. Biol Trace Elem Res. (2017) 175:405–13. doi: 10.1007/s12011-016-0791-3
97. Burk RF. Selenium, an antioxidant nutrient. Nutr Clin Care. (2002) 5:75–9. doi: 10.1046/j.1523-5408.2002.00006.x
98. Mistry HD, Broughton Pipkin F, Redman CWG, Poston L. Selenium in reproductive health. Am J Obstet Gynecol. (2012) 206:21–30. doi: 10.1016/j.ajog.2011.07.034
99. Moreno-Reyes R, Mathieu F, Boelaert M, Begaux F, Suetens C, Rivera MT, et al. Selenium and iodine supplementation of rural Tibetan children affected by Kashin-Beck osteoarthropathy. Am J Clin Nutr. (2003) 78:137–44. doi: 10.1093/ajcn/78.1.137
100. Mueller AS, Pallauf J. Compendium of the antidiabetic effects of supranutritional selenate doses. In vivo and in vitro investigations with type II diabetic db/db mice. J Nutr Biochem. (2006) 17:548–60. doi: 10.1016/j.jnutbio.2005.10.006
101. Bleys J, Navas-Acien A. Guallar E. Serum selenium and diabetes in US adults. Diabetes Care. (2007) 30:829–34. doi: 10.2337/dc06-1726
102. Pallauf K, Rivas-Gonzalo JC, Del Castillo M, Cano MP, de Pascual-Teresa S. Characterization of the antioxidant composition of strawberry tree (Arbutus unedo L.) fruits. J Food Comp Anal. (2008) 21:273–81. doi: 10.1016/j.jfca.2007.11.006
103. Hwang D, Seo S, Kim Y, Kim C, Shim S, Jee S, et al. Selenium acts as an insulin-like molecule for the down-regulation of diabetic symptoms via endoplasmic reticulum stress and insulin signalling proteins in diabetes-induced non-obese diabetic mice. J Biosci. (2007) 32:723–35. doi: 10.1007/s12038-007-0072-6
104. Park K, Rimm EB, Siscovick DS, Spiegelman D, Manson JE, Morris JS, et al. Toenail selenium and incidence of type 2 diabetes in US men and women. Diab Care. (2012) 35:1544–51. doi: 10.2337/dc11-2136
105. González de Vega R, Fernández-Sánchez ML, Fernández JC, Álvarez Menéndez FV, Sanz-Medel A. Selenium levels and Glutathione peroxidase activity in the plasma of patients with type II diabetes mellitus. J Trace Elem Med Biol. (2016) 37:44–9. doi: 10.1016/j.jtemb.2016.06.007
106. Vinceti M, Filippini T, Rothman KJ. Selenium exposure and the risk of type 2 diabetes: a systematic review and meta-analysis. Eur J Epidemiol. (2018) 33:789–810. doi: 10.1007/s10654-018-0422-8
107. Zachara BA, Wasowicz W, Gromadzińska J, Skłodowska M, Krasomski G. Glutathione peroxidase activity, selenium, and lipid peroxide concentrations in blood from a healthy Polish population: I. Maternal and cord blood. Biol Trace Elem Res. (1986) 10:175–87. doi: 10.1007/BF02795616
108. Nogales F, Ojeda ML, Del Valle PM, Serrano A, Murillo ML, Carreras Sánchez O. Metabolic syndrome and selenium during gestation and lactation. Eur J Nutr. (2017) 56:819–30. doi: 10.1007/s00394-015-1129-1
109. Hofstee P, McKeating DR, Bartho LA, Anderson ST, Perkins AV, Cuffe JSM. Maternal selenium deficiency in mice alters offspring glucose metabolism and thyroid status in a sexually dimorphic manner. Nutrients. (2020) 12:267. doi: 10.3390/nu12010267
110. Laureano-Melo R, Império GE, Kluck GEG, da Conceição RR, de Souza JS, Marinho BG, et al. Selenium supplementation during pregnancy and lactation promotes metabolic changes in Wistar rats' offspring. Clin Exp Pharmacol Physiol. (2020) 47:1272–82. doi: 10.1111/1440-1681.13268
111. Zeng M-S, Li X, Liu Y, Zhao H, Zhou J-C, Li K, et al. A high-selenium diet induces insulin resistance in gestating rats and their offspring. Free Radic Biol Med. (2012) 52:1335–42. doi: 10.1016/j.freeradbiomed.2012.01.017
112. Ojeda ML, Nogales F, Membrilla A, Carreras O. Maternal selenium status is profoundly involved in metabolic fetal programming by modulating insulin resistance, oxidative balance and energy homeostasis. Eur J Nutr. (2019) 58:3171–81. doi: 10.1007/s00394-018-1861-4
113. Institute of Medicine Panel on Dietary A Related C. Dietary Reference Intakes for Vitamin C, Vitamin E, Selenium, and Carotenoids. Washington, DC: National Academies Press (US) Copyright 2000 by the National Academy of Sciences. All rights reserved (2000).
114. Kipp AP, Strohm D, Brigelius-Flohé R, Schomburg L, Bechthold A, Leschik-Bonnet E, et al. Revised reference values for selenium intake. J Trace Elem Med Biol. (2015) 32:195–9. doi: 10.1016/j.jtemb.2015.07.005
115. Taylor CG. Zinc, the pancreas, and diabetes: insights from rodent studies and future directions. Biometals. (2005) 18:305–12. doi: 10.1007/s10534-005-3686-x
116. Olechnowicz J, Tinkov A, Skalny A, Suliburska J. Zinc status is associated with inflammation, oxidative stress, lipid, and glucose metabolism. J Physiol Sci. (2018) 68:19–31. doi: 10.1007/s12576-017-0571-7
117. Sanusi KO, Ibrahim KG, Abubakar B, Malami I, Bello MB, Imam MU, et al. Effect of maternal zinc deficiency on offspring health: the epigenetic impact. J Trace Elem Med Biol. (2021) 65:126731. doi: 10.1016/j.jtemb.2021.126731
118. Tang X, Shay NF. Zinc has an insulin-like effect on glucose transport mediated by phosphoinositol-3-kinase and Akt in 3T3-L1 fibroblasts and adipocytes. J Nutr. (2001) 131:1414–20. doi: 10.1093/jn/131.5.1414
119. Wessells KR, Singh GM, Brown KH. Estimating the global prevalence of inadequate zinc intake from national food balance sheets: effects of methodological assumptions. PLoS ONE. (2012) 7:e50565. doi: 10.1371/journal.pone.0050565
120. Wang Y, Jia X-F, Zhang B, Wang Z-H, Zhang J-G, Huang F-F, et al. Dietary zinc intake and its association with metabolic syndrome indicators among Chinese adults: an analysis of the china nutritional transition cohort survey 2015. Nutrients. (2018) 10:572. doi: 10.3390/nu10050572
121. Keen CL, Gershwin ME. Zinc deficiency and immune function. Annu Rev Nutr. (1990) 10:415–31. doi: 10.1146/annurev.nu.10.070190.002215
122. Wilson RL, Grieger JA, Bianco-Miotto T, Roberts CT. Association between maternal zinc status, dietary zinc intake and pregnancy complications: a systematic review. Nutrients. (2016) 8:641. doi: 10.3390/nu8100641
123. Terrin G, Berni Canani R, Di Chiara M, Pietravalle A, Aleandri V, Conte F, et al. Zinc in early life: a key element in the fetus and preterm neonate. Nutrients. (2015) 7:10427–46. doi: 10.3390/nu7125542
124. Ota E, Mori R, Middleton P, Tobe-Gai R, Mahomed K, Miyazaki C, et al. Zinc supplementation for improving pregnancy and infant outcome. Cochrane Datab Syst Rev. (2015) (2):CD000230. doi: 10.1002/14651858.CD000230.pub5
125. Mousa A, Naqash A, Lim S. Macronutrient and micronutrient intake during pregnancy: an overview of recent evidence. Nutrients. (2019) 11:443. doi: 10.3390/nu11020443
126. Mispireta ML, Caulfield LE, Zavaleta N, Merialdi M, Putnick DL, Bornstein MH, et al. Effect of maternal zinc supplementation on the cardiometabolic profile of Peruvian children: results from a randomized clinical trial. J Dev Orig Health Dis. (2017) 8:56–64. doi: 10.1017/S2040174416000568
127. Rosario JF, Gomez MP, Anbu P. Does the maternal micronutrient deficiency (copper or zinc or vitamin E) modulate the expression of placental 11 beta hydroxysteroid dehydrogenase-2 per se predispose offspring to insulin resistance and hypertension in later life? Indian J Physiol Pharmacol. (2008) 52:355–65.
128. Jansson T, Powell TL. Role of the placenta in fetal programming: underlying mechanisms and potential interventional approaches. Clin Sci (Lond). (2007) 113:339. doi: 10.1042/CS20060339
129. Padmavathi IJN, Kishore YD, Venu L, Ganeshan M, Harishankar N, Giridharan NV, et al. Prenatal and perinatal zinc restriction: effects on body composition, glucose tolerance and insulin response in rat offspring. Exp Physiol. (2009) 94:761–9. doi: 10.1113/expphysiol.2008.045856
130. Jou M-Y, Philipps AF, Lönnerdal B. Maternal zinc deficiency in rats affects growth and glucose metabolism in the offspring by inducing insulin resistance postnatally. J Nutr. (2010) 140:1621–7. doi: 10.3945/jn.109.119677
131. Jou M-Y, Lönnerdal B, Philipps AF. Maternal zinc restriction affects postnatal growth and glucose homeostasis in rat offspring differently depending upon adequacy of their nutrient intake. Pediatr Res. (2012) 71:228–34. doi: 10.1038/pr.2011.44
132. Mendes Garrido Abregú F, Gobetto MN, Castañón A, Lucero D, Caniffi C, Elesgaray R, et al. Fetal and postnatal zinc restriction: sex differences in metabolic alterations in adult rats. Nutrition. (2019) 65:18–26. doi: 10.1016/j.nut.2019.01.022
133. Bar-El Dadon S, Reifen R. Vitamin A and the epigenome. Crit Rev Food Sci Nutr. (2017) 57:2404–11. doi: 10.1080/10408398.2015.1060940
134. Duester G. Retinoic acid synthesis and signaling during early organogenesis. Cell. (2008) 134:921–31. doi: 10.1016/j.cell.2008.09.002
135. Stephensen CB. Vitamin A, infection, and immune function. Annu Rev Nutr. (2001) 21:167–92. doi: 10.1146/annurev.nutr.21.1.167
136. Cañete A, Cano E, Muñoz-Chápuli R, Carmona R. Role of vitamin A/retinoic acid in regulation of embryonic and adult hematopoiesis. Nutrients. (2017) 9:159. doi: 10.3390/nu9020159
137. Blaner WS. Vitamin A signaling and homeostasis in obesity, diabetes, and metabolic disorders. Pharmacol Ther. (2019) 197:153–78. doi: 10.1016/j.pharmthera.2019.01.006
138. Brun P-J, Wongsiriroj N, Blaner WS. Retinoids in the pancreas. Hepatobiliary Surg Nutr. (2016) 5:1–4. doi: 10.3978/j.issn.2304-3881.2015.09.03
139. Keller A, Ängquist L, Jacobsen R, Vaag A, Heitmann BL. A retrospective analysis of a societal experiment among the Danish population suggests that exposure to extra doses of vitamin A during fetal development may lower type 2 diabetes mellitus (T2DM) risk later in life. Br J Nutr. (2017) 117:731–6. doi: 10.1017/S000711451700037X
140. Graham TE, Yang Q, Blüher M, Hammarstedt A, Ciaraldi TP, Henry RR, et al. Retinol-binding protein 4 and insulin resistance in lean, obese, and diabetic subjects. N Engl J Med. (2006) 354:2552–63. doi: 10.1056/NEJMoa054862
141. Yang Q, Graham TE, Mody N, Preitner F, Peroni OD, Zabolotny JM, et al. Serum retinol binding protein 4 contributes to insulin resistance in obesity and type 2 diabetes. Nature. (2005) 436:356–62. doi: 10.1038/nature03711
142. Oström M, Loffler KA, Edfalk S, Selander L, Dahl U, Ricordi C, et al. Retinoic acid promotes the generation of pancreatic endocrine progenitor cells and their further differentiation into beta-cells. PLoS ONE. (2008) 3:e2841. doi: 10.1371/journal.pone.0002841
143. Pérez RJ, Benoit YD, Gudas LJ. Deletion of retinoic acid receptor β (RARβ) impairs pancreatic endocrine differentiation. Exp Cell Res. (2013) 319:2196–204. doi: 10.1016/j.yexcr.2013.05.032
144. Matthews KA, Rhoten WB, Driscoll HK, Chertow BS. Vitamin A deficiency impairs fetal islet development and causes subsequent glucose intolerance in adult rats. J Nutr. (2004) 134:1958–63. doi: 10.1093/jn/134.8.1958
145. Chertow BS, Blaner WS, Baranetsky NG, Sivitz WI, Cordle MB, Thompson D, et al. Effects of vitamin A deficiency and repletion on rat insulin secretion in vivo and in vitro from isolated islets. J Clin Invest. (1987) 79:163–9. doi: 10.1172/JCI112778
146. Guo J, Li B, Zuo Z, Chen M, Wang C. Maternal supplementation with β-carotene during pregnancy disturbs lipid metabolism and glucose homoeostasis in F1 female mice. Mol Nutr Food Res. (2019) 63:e1900072. doi: 10.1002/mnfr.201900072
147. Nakashima A, Yokoyama K, Yokoo T, Urashima M. Role of vitamin D in diabetes mellitus and chronic kidney disease. World J Diabetes. (2016) 7:89–100. doi: 10.4239/wjd.v7.i5.89
148. Holick MF. Sunlight and vitamin D for bone health and prevention of autoimmune diseases, cancers, and cardiovascular disease. Am J Clin Nutr. (2004) 80(Suppl. 6):1678S−88S. doi: 10.1093/ajcn/80.6.1678S
149. Stumpf WE, Sar M, Reid FA, Tanaka Y, DeLuca HF. Target cells for 1,25-dihydroxyvitamin D3 in intestinal tract, stomach, kidney, skin, pituitary, and parathyroid. Science. (1979) 206:1188–90. doi: 10.1126/science.505004
150. Isaia G, Giorgino R, Adami S. High prevalence of hypovitaminosis D in female type 2 diabetic population. Diabetes Care. (2001) 24:1496. doi: 10.2337/diacare.24.8.1496
151. Ishida H, Seino Y, Matsukura S, Ikeda M, Yawata M, Yamashita G, et al. Diabetic osteopenia and circulating levels of vitamin D metabolites in type 2 (noninsulin-dependent) diabetes. Metabolism. (1985) 34:797–801. doi: 10.1016/0026-0495(85)90101-5
152. Liu E, Meigs JB, Pittas AG, McKeown NM, Economos CD, Booth SL, et al. Plasma 25-hydroxyvitamin d is associated with markers of the insulin resistant phenotype in nondiabetic adults. J Nutr. (2009) 139:329–34. doi: 10.3945/jn.108.093831
153. Palomer X, González-Clemente JM, Blanco-Vaca F, Mauricio D. Role of vitamin D in the pathogenesis of type 2 diabetes mellitus. Diabetes Obes Metab. (2008) 10:185–97. doi: 10.1111/j.1463-1326.2007.00710.x
154. Berridge MJ. Vitamin D deficiency and diabetes. Biochem J. (2017) 474:1321–32. doi: 10.1042/BCJ20170042
155. Gallo S, McDermid JM, Al-Nimr RI, Hakeem R, Moreschi JM, Pari-Keener M, et al. Vitamin D supplementation during pregnancy: an evidence analysis center systematic review and meta-analysis. J Acad Nutr Diet. (2020) 120:898–924.e4. doi: 10.1016/j.jand.2019.07.002
156. Lapillonne A. Vitamin D deficiency during pregnancy may impair maternal and fetal outcomes. Med Hypotheses. (2010) 74:71–5. doi: 10.1016/j.mehy.2009.07.054
157. Hrudey EJ, Reynolds RM, Oostvogels AJJM, Brouwer IA, Vrijkotte TGM. The Association between maternal 25-hydroxyvitamin D concentration during gestation and early childhood cardio-metabolic outcomes: is there interaction with pre-pregnancy BMI? PLoS ONE. (2015) 10:e0133313. doi: 10.1371/journal.pone.0133313
158. Roomi MA, Lone KP, Madassar A. Vitamin D and cardiometabolic risk factors in adult non-diabetic offspring of type 2 diabetic parents. J Pak Med Assoc. (2014) 64:1229–34.
159. Krishnaveni GV, Veena SR, Winder NR, Hill JC, Noonan K, Boucher BJ, et al. Maternal vitamin D status during pregnancy and body composition and cardiovascular risk markers in Indian children: the Mysore Parthenon Study. Am J Clin Nutr. (2011) 93:628–35. doi: 10.3945/ajcn.110.003921
160. Villa CR, Chen J, Wen B, Sacco SM, Taibi A, Ward WE, et al. Maternal vitamin D beneficially programs metabolic, gut and bone health of mouse male offspring in an obesogenic environment. Int J Obes (Lond). (2016) 40:1875–83. doi: 10.1038/ijo.2016.177
161. Zhang H, Chu X, Huang Y, Li G, Wang Y, Li Y, et al. Maternal vitamin D deficiency during pregnancy results in insulin resistance in rat offspring, which is associated with inflammation and Iκbα methylation. Diabetologia. (2014) 57:2165–72. doi: 10.1007/s00125-014-3316-7
162. Knight BA, Shields BM, Brook A, Hill A, Bhat DS, Hattersley AT, et al. Lower circulating B12 is associated with higher obesity and insulin resistance during pregnancy in a non-diabetic white british population. PLoS ONE. (2015) 10:e0135268. doi: 10.1371/journal.pone.0135268
163. Antony AC. Evidence for potential underestimation of clinical folate deficiency in resource-limited countries using blood tests. Nutr Rev. (2017) 75:600–15. doi: 10.1093/nutrit/nux032
164. Institute of Medicine Standing Committee on the Scientific Evaluation of Dietary Reference I its Panel on Folate OBV Choline. The National Academies Collection: Reports funded by National Institutes of Health. Dietary Reference Intakes for Thiamin, Riboflavin, Niacin, Vitamin B(6), Folate, Vitamin B(12), Pantothenic Acid, Biotin, and Choline. Washington, DC: National Academies Press (US) Copyright 1998, National Academy of Sciences (1998).
165. Sukumar N, Rafnsson SB, Kandala N-B, Bhopal R, Yajnik CS, Saravanan P. Prevalence of vitamin B-12 insufficiency during pregnancy and its effect on offspring birth weight: a systematic review and meta-analysis. Am J Clin Nutr. (2016) 103:1232–51. doi: 10.3945/ajcn.115.123083
166. Yajnik CS, Deshpande SS, Jackson AA, Refsum H, Rao S, Fisher DJ, et al. Vitamin B12 and folate concentrations during pregnancy and insulin resistance in the offspring: the Pune Maternal Nutrition Study. Diabetologia. (2008) 51:29–38. doi: 10.1007/s00125-007-0793-y
167. Stewart CP, Christian P, Schulze KJ, Arguello M, LeClerq SC, Khatry SK, et al. Low maternal vitamin B-12 status is associated with offspring insulin resistance regardless of antenatal micronutrient supplementation in rural Nepal. J Nutr. (2011) 141:1912–7. doi: 10.3945/jn.111.144717
168. Krishnaveni GV, Veena SR, Karat SC, Yajnik CS, Fall CHD. Association between maternal folate concentrations during pregnancy and insulin resistance in Indian children. Diabetologia. (2014) 57:110–21. doi: 10.1007/s00125-013-3086-7
169. Adaikalakoteswari A, Vatish M, Lawson A, Wood C, Sivakumar K, McTernan PG, et al. Low maternal vitamin B12 status is associated with lower cord blood HDL cholesterol in white Caucasians living in the UK. Nutrients. (2015) 7:2401–14. doi: 10.3390/nu7042401
170. Behere RV, Deshmukh AS, Otiv S, Gupte MD, Yajnik CS. Maternal VItamin B12 status during pregnancy and its association with outcomes of pregnancy and health of the offspring: a systematic review and implications for policy in India. Front Endocrinol (Lausanne). (2021) 12:619176. doi: 10.3389/fendo.2021.619176
171. Henderson AM, Tai DC, Aleliunas RE, Aljaadi AM, Glier MB, Xu EE, et al. Maternal folic acid supplementation with vitamin B deficiency during pregnancy and lactation affects the metabolic health of adult female offspring but is dependent on offspring diet. FASEB J. (2018) 32:5039–50. doi: 10.1096/fj.201701503RR
172. Aleliunas RE, Aljaadi AM, Laher I, Glier MB, Green TJ, Murphy M, et al. Folic acid supplementation of female mice, with or without vitamin B-12, before and during pregnancy and lactation programs adiposity and vascular health in adult male offspring. J Nutr. (2015) 146:688–96. doi: 10.3945/jn.115.227629
173. Ghosh S, Sinha JK, Muralikrishna B, Putcha UK, Raghunath M. Chronic transgenerational vitamin B12 deficiency of severe and moderate magnitudes modulates adiposity-probable underlying mechanisms. Biofactors. (2017) 43:400–14. doi: 10.1002/biof.1350
Keywords: DOHaD, diabetes mellitus, micronutrient, glucose metabolism, offspring, maternal, epigenetics
Citation: Wu Y, Zhang Q and Xiao X (2021) The Effect and Potential Mechanism of Maternal Micronutrient Intake on Offspring Glucose Metabolism: An Emerging Field. Front. Nutr. 8:763809. doi: 10.3389/fnut.2021.763809
Received: 24 August 2021; Accepted: 27 September 2021;
Published: 22 October 2021.
Edited by:
Faidon Magkos, University of Copenhagen, DenmarkReviewed by:
Nina Geiker, University of Copenhagen, DenmarkJasper Most, Zuyderland Medical Centre, Netherlands
Copyright © 2021 Wu, Zhang and Xiao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qian Zhang, emhhbmdxaWFuNjA4OEBwdW1jaC5jbg==; Xinhua Xiao, eGlhb3hoMjAxNEB2aXAuMTYzLmNvbQ==