 Tim Vanuytsel1,2
Tim Vanuytsel1,2 Jan Tack
Jan Tack Ricard Farre
Ricard Farre- 1Department of Chronic Diseases, Translational Research Center for Gastrointestinal Disorders, Metabolism and Ageing, Catholic University Leuven, Leuven, Belgium
- 2Division of Gastroenterology and Hepatology, Leuven University Hospital, Leuven, Belgium
An increased intestinal permeability has been described in various gastrointestinal and non-gastrointestinal disorders. Nevertheless, the concept and definition of intestinal permeability is relatively broad and includes not only an altered paracellular route, regulated by tight junction proteins, but also the transcellular route involving membrane transporters and channels, and endocytic mechanisms. Paracellular intestinal permeability can be assessed in vivo by using different molecules (e.g., sugars, polyethylene glycols, 51Cr-EDTA) and ex vivo in Ussing chambers combining electrophysiology and probes of different molecular sizes. The latter is still the gold standard technique for assessing the epithelial barrier function, whereas in vivo techniques, including putative blood biomarkers such as intestinal fatty acid-binding protein and zonulin, are broadly used despite limitations. In the second part of the review, the current evidence of the role of impaired barrier function in the pathophysiology of selected gastrointestinal and liver diseases is discussed. Celiac disease is one of the conditions with the best evidence for impaired barrier function playing a crucial role with zonulin as its proposed regulator. Increased permeability is clearly present in inflammatory bowel disease, but the question of whether this is a primary event or a consequence of inflammation remains unsolved. The gut-liver axis with a crucial role in impaired intestinal barrier function is increasingly recognized in chronic alcoholic and metabolic liver disease. Finally, the current evidence does not support an important role for increased permeability in bile acid diarrhea.
Introduction
The concept of the “leaky gut” also known as increased intestinal permeability has been described many years ago and is currently receiving increasing attention in the scientific literature but also in the media because of its proposed associations with numerous conditions that do not necessarily affect the gastrointestinal (GI) tract, such as asthma, Alzheimer's disease, diabetes among others. This is the reason why different interventions such as probiotics, and dietary modifications intended to ameliorate the leaky gut, are suggested as potential treatments.
The principle behind the “leaky gut” theory is that endogenous (e.g., psychological stress, intestinal inflammation) and exogenous factors (e.g., diet, alcohol intake) can increase intestinal permeability, allowing the entrance of food antigens, commensal or pathogenic bacteria and bacterial components into the lamina propria and later on into the systemic circulation, provoking the systemic inflammation described in different disease conditions. The vast majority of studies evaluate the intestinal permeability of the paracellular route, despite the presence of other routes of transepithelial transport that are potentially more relevant in gastrointestinal disorders.
To better comprehend the role of an increased intestinal permeability in GI diseases, it is crucial to understand some basic concepts on how different molecules cross the intestinal epithelium and how intestinal permeability can be evaluated in humans.
Routes of Transport in the Epithelium: Paracellular, Transcellular, Transporter Mediated and Endocytosis
There are several routes for luminal products to cross the intestinal epithelium. They depend of the size, hydrophobicity and other physico-chemical properties (Figure 1). Overall there are four different pathways: (a) the transcellular route used by small hydrophilic and lipophilic compounds; (b) the paracellular route used by ions, water and larger hydrophilic compounds from ~400–600 Da up to 10–20 kDa and that cannot cross transcellularly [regulated by the tight junction (TJ) proteins]; (c) transcellular active transport used by nutrients as sugars, amino acids and vitamins that require specific transporters and energy and (d) endocytosis and basolateral exocytosis via vesicles are used by larger peptides and proteins, large bacterial components or even whole bacteria (1, 2). From a physiological point of view, the paracellular route and the transcellular endocytic route are the two relevant pathways in the context of the leaky gut. Nevertheless, most of the in vivo studies assessing human intestinal permeability (oral administration of sugars or other probes later detected in blood or urine) mainly target the paracellular pathway.
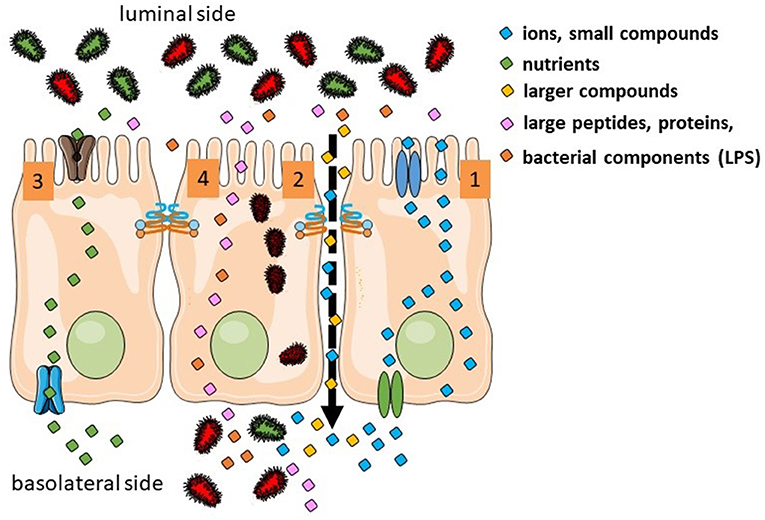
Figure 1. Transport routes in the intestinal epithelium. Transcellular transport of ions is controlled by transporters in the apical and basolateral surfaces (1). Ion, water, and larger hydrophilic compounds use the paracellular pathway (2). Sugars, amino acids, and vitamins use active transport (3). Large molecules and whole bacteria are endocytosed into vesicles (4).
The transcellular endocytic pathways is then understudied but has been shown very relevant in several diseases GI and non-GI disorders. A very illustrative example is the increased bacterial translocation (BT) observed in numerous pathologies and that is defined as the migration of viable micro-organisms and bacterial components from the gut lumen toward the mesenteric lymph nodes (MLNs) and extra-intestinal sites such as the peritoneal cavity, liver, spleen and kidney (3) among others. Importantly, in some pathological conditions as intestinal ischemia and advanced liver disease, BT can have fatal consequences. Lipopolysaccharide (LPS) is by far the most studied bacterial protein, located on the surface of Gram-negative bacteria (size of 10–20 kDa). LPS has been found in higher concentrations in the systemic circulation of patients with cirrhosis (4) and other diseases as inflammatory bowel disease (IBD) (5) and even in patients with diarrhea-predominant irritable bowel syndrome (IBS-D) (6).
The Paracellular Route
Evidences for the role of TJs as key regulators in the epithelial barrier are based historically on observation from two very different fields: electron microscopy (EM) and electrophysiology. In the most apical region of the epithelium there is an intercellular gap (around 90 Å) named zonula occludens or tight junctions, followed by the zonula adherens (gap of 200 Å), followed by the macula adherens or desmosomes (gap of ~240 Å) (7). More than 50 year ago, Hans Ussing initially described by performing electrophysiology in the frog skin that the TJ structure is a dynamic and permeant barrier that in physiological conditions is also permeable to ions and plays a role in processes like water absorption and secretion (8). Moreover, early experiments in rat kidney tubules using transmission EM showed that the flux of large molecules across the paracellular route is selectively regulated by complex tight junction strands (7, 9). More recently, knowledge has been accumulating on the complexity, selectivity, and dynamic character of the intestinal epithelial barrier. Most studies on the paracellular route of transport describe at least two populations of pores regulated by TJs: (1) a high capacity charge selective pore permeable to small ions and small uncharged molecules (referred to as the “pore” pathway), and (2) a much larger low-capacity pore (commonly referred to as the “leak” pathway) permeable to large ions and molecules irrespective of charge. At the molecular level, the first pore is mainly regulated by claudins and the latter by the TJ proteins occludin and the zonula occludens (ZO) family.
Role of Tight Junctions in Crucial Physiological Processes
The paracellular pathway includes several important aspects: (1) the ability of TJ to restrict paracellular diffusion processes, acting as a paracellular barrier; (2) the ability of the TJ to selectively allow paracellular transport of certain ion species acting as ion channel and (3) the ability of the TJs to maintain cell polarity, by blocking the free diffusion of proteins and lipids between the apical and basolateral domains of the plasma membrane. Moreover, TJ play a crucial role in other relevant physiological processes such as absorption and secretion of water and Ca2+ absorption.
Absorption of Glucose and Water
When luminal glucose concentrations are low, its absorption involves the transcellular route and the apical Na+-glucose co-transporter SGLT1 and the basolateral Na+-independent glucose transporter GLUT2 (6). In this environment, absorption of glucose is coupled to an electrochemical gradient; provided by the activity of Na+/K+-ATPase located at the basolateral membrane (10). Nevertheless, when the luminal concentrations are high as occurs after a meal (concentration range 50–300 mM), about 30% of glucose absorption is mediated via the paracellular route (leak pathway). This pathway includes the phosphorylation of the myosin light chain (MLC) by myosin light chain kinase (MLCK), followed by contraction of the cytoskeleton and the opening of the TJs (11). The osmotic gradient created by transcellular movement of Na+ and glucose enhances the paracellular flux of water. This, in combination with the increased paracellular permeability allows paracellular absorption of glucose and other nutrients (12). The Na+ absorption is in turn the driving force of the so-called voltage dependent Cl− absorption but it is uncertain whether this route is transcellular or paracellular. It believed that in physiologic conditions when the membrane transporters are saturated, the leak pathway complements the transcellular pathway (active transport) in absorbing water and nutrients (13).
Secretion of Water
The entire intestinal epithelium remains hydrated to assure its accurate physiological function. To assure this, there is a constant water secretion in the entire GI tract from the duodenum to the distal colon. The submucosal plexus precisely regulates the secretion of Cl− and consequently Na+ and water though by the TJs. The release of vasoactive intestinal peptide (VIP) and acetylcholine (Ach) by submucosal neurons stimulates the enterocyte and Cl− enters by co-transport with sodium and potassium through the basolateral Na+-K+-2Cl− channel. Then, the Na+-K+ ATPase pumps Na+ back out, and K+ is exported via K+ channels. The activation of VIP receptors stimulates the adenylyl cyclase to generate cytosolic cAMP that activate the cystic fibrosis transmembrane conductance regulator (CFTR), resulting in secretion of Cl− into the lumen. The activation of muscarinic receptors by Ach induces an increase in intracellular Ca2+ concentration that activates calcium-activated chloride channels (CaCC) inducing also Cl− secretion. Accumulation of Cl− in the lumen creates an electric potential that attracts Na+ through the TJ into the lumen. NaCl creates an osmotic gradient across the tight junction and water is drawn into the lumen. Some pathogenic bacteria produce toxins (for instance cholera toxin) that permanently activate the adenylate cyclase of the enterocyte. This leads to elevated levels of cAMP, causing the continuous opening of the CFTR channel. As a consequence, there is a massive secretion of water, provoking severe diarrhea (14).
Absorption of Calcium
Intestinal Ca2+ absorption is a critical physiological process for maintaining Ca2+ homeostasis in the entire human body and involves both the transcellular and paracellular pathways. In the proximal small intestine, luminal absorption of low Ca2+ concentrations involve mainly the transcellular route (vitamin D-dependent) and for high concentrations, the paracellular passive route (vitamin D-independent) is the predominant source for calcium absorption. In contrast, in the distal small intestine Ca2+ absorption mainly involves the paracellular route. Despite the physiological relevance of Ca2+ absorption, the mechanisms involved are poorly understood. These mechanisms involve different steps which are currently not well-characterized: (1) entrance of Ca2+ into the enterocytes through the Ca2+-selective transient receptor potential (TRP) channel TRPV6 and the voltage-gated calcium channel Cav1.3, a member of the L-type calcium channel family; (2) movement of Ca2+ to the basolateral membranes by binding to the high Ca2+ affinity protein calbindin-D 9 k; (3) basolateral extrusion of Ca2+ via plasma membrane Ca2+-ATPase PMCA1b and (4) Ca2+ extrusion into the blood (15, 16). The pore-forming claudin (CLDN)-2 and barrier forming CLDN-12 seem to be crucial in the Ca2+ transport through tight junction proteins (17). Research to elucidate whether other proteins of the pore or the leak pathways are involved in Ca2+ absorption is needed.
The Transcellular Pathway: Transcytosis and Posterior Exocytosis
An increased intestinal transcellular permeability found in different disease conditions has been postulated to be critical for the migration of bacteria and bacterial components such as endotoxins across the gut wall. Based on the molecular weight, isolated LPS can potentially cross the epithelium through the paracellular pathway as has been suggested in some studies (18). Nevertheless, the exact mechanism(s) by which endotoxins (LPS) cross the intestinal barrier remain to be elucidated. In contrast to the general belief, recent research in healthy tissue from animals suggests that LPS is not crossing the epithelium via the paracellular pathway (19). Instead, LPS can potentially cross the enterocyte via the different mechanisms involved in endocytosis: (1) clathrin-mediated endocytosis; (2) lipid-raft caveolae- mediated endocytosis; and (3) via the chylomicrons pathway (only in the small intestine) (19, 20). If these findings can be confirmed in disease conditions, they will strongly challenge the broadly extended concept of “leaky gut” (21).
Contrary to the widely held assumption that whole bacteria can cross the intestinal epithelium via the paracellular pathway, basic research strongly supports that endocytosis is the mechanism used for bacteria to translocate to the lamina propria (22). Whole bacterial translocation through the epithelium occurs not only in the Peyer patches but also in the normal epithelium and involves endocytosis. Nevertheless, the specific pathway(s) of endocytosis that might be involved are not well-characterized. Recent research indicates the role of clathrin-dependent endocytosis in bacterial entry (23). Moreover, Pai et al. (24) recently showed that commensal bacteria endocytosis is also involving MLCK activation and MLC phosphorylation (25), followed by contraction of the actomyosin ring. Then, the perijunctional actomyosin ring contraction produces a change in the configuration of the microvilli called brush border fanning that facilitates the contact of the bacteria with the enterocytes. Remarkable is the fact that this mechanism of bacterial internalization is modulated by tumor necrosis factor-like 1A and interferon (IFN)-γ at low concentrations. IFN-γ at high concentrations is a cytokine that also increases intestinal permeability to large molecules (24).
Despite the increased LPS translocation into the blood described in numerous pathologies, it is not clear whether blood endotoxin levels are the result of LPS from the lumen or whether LPS is released by whole bacteria translocated in the lamina propria by the wall breakdown from effective host defense mechanisms and by autolysis (26).
Methods to Assess Intestinal Permeability
Ex vivo Assessment of Intestinal Permeability With the Ussing Chambers Technique
In 1951, the Danish scientist Hans Ussing performed for the first time electrophysiology in the frog skin (27). Later, this methodology was adapted and used to study other epithelia of different organs including the GI tract.
Transepithelial Electrical Resistance and Paracellular Permeability
Currently, there are different commercially available Ussing chamber equipments that are designed to accommodate multiple epithelial sources such as cell cultures grown in membranes, animal tissue and human tissue from resection specimens or endoscopic biopsies. In such a way, each side of the epithelium is perfectly isolated in each hemi-chamber. Epithelia develop ex vivo a potential difference (PD) because they have two differential characteristics compared with other types of tissue: polarity and tightness. They are polarized tissues due to the differential expression of ion channels, pumps and transporters in the apical and the basolateral sides: (a) epithelial sodium channels (ENaC), Ca2+ activated chloride channels and the cystic fibrosis transmembrane conductance regulator (CFTR) are present in the apical plasma membrane and (b) the Na+-K+-ATPase ion pump and the Na+-K+-2Cl− cotransporter in the basolateral side. Epithelia are tight tissues due exclusively to the presence of TJ that selectively regulate the paracellular flux of ions (see Role of Tight Junctions in Crucial Physiological Processes). The presence of polarity and tightness generate a basal PD across epithelial tissue that is measured in the Ussing chamber using voltage electrodes. Then TEER is measured by (a) applying a constant electric current (open-circuit conditions) in the epithelium every couple of seconds with two current electrodes and (b) measuring then the new generated potential PD. TEER (resistance to the current flow) can then be calculated by using Ohm's law (V = I × R, V: voltage or PD, I: intensity of the current, R: resistance) and expressed as Ω × cm2. Low TEER values are indicative of increased permeability to ions. Permeability of the epithelium to ions can be also express in milliSiemens (mS) as the conductance (G) of the epithelium, the opposite of the TEER. Then, an increased epithelial permeability to ions is translated as an increase in conductance. TEER measures the net flux of all ions (cations and anions) across the epithelium and reflects the contribution of the paracellular resistance (Rpara) that reflects the resistance of the TJs, the transcellular resistance (Rtrans) that reflects the resistance to ions of the apical (Rapi) and basolateral (Rbas) membranes and finally the sub-epithelial resistance (Rsub). In other words, TEER is the reflection of the resistance that the epithelium is exerting against ions to cross from the luminal to the basolateral side. The use of more complex techniques as the one-path and the two-path impedance spectroscopy is able to measure the two components of the Repi, the Rpara and the Rtrans (Repi = Rpara + Rtrans) on top of the Rbas (28). Unfortunately, these complex impedance methodologies are only available in a few research groups and total TEER measurements are the most commonly used.
Importantly, TEER does not discriminate between the above-mentioned pore and leak pathways since an increased permeability of both pathways will reduce TEER. The permeability of the leak pathway can be better assessed ex vivo by measuring the flux of molecules from the luminal to the basolateral side such as EDTA, mannitol, sucrose, inulin and polyethylene glycols (PEG) or dextran molecules of variable sizes (ranging from 4 up to 20 KDa). For that, labeled molecules with radioactive isotopes or with fluorophores applied at the luminal side are commonly used. Samples from the basolateral side are taken at different time points during ~2 h (29).
The short circuit current (Isc) is also often calculated during electrophysiological measurements and refers to the amount of current needed to force the PD to 0 mV. This amount of current is continuously adjusted to clamp the epithelium at 0 mV. Cl− and Na+, the most common ions in physiological solutions, mainly generate the PD of the epithelium. Isc is the reflection of the net ionic transport through the epithelium and is expressed as μA/cm2. During Isc the voltage is clamped to values different from 0 mV enabling to estimate the TEER.
From the simple equation V = I × R, it is obvious that Isc can also be calculated in open-circuit conditions when R and PD are known. This value is often referred as the equivalent short-circuit current (Ieq).
Assessment of the Transcellular Route
The transcellular pathway involving endocytic vesicles and posterior exocytosis can also be assessed in Ussing chambers by adding probes of large molecular weight (MW) that are known to cross the intestinal epithelium using this route. Then, samples from the basolateral side are taken at different time points. Probes most commonly used are horseradish peroxidase (HRP, MW: 44 kDa) and dextran-labeled molecules of a molecular weight between 40 and 80 kDa.
The transcellular route is not only used by large peptides and proteins but also for the uptake of whole commensal bacteria into the lamina propria as part of the BT process. This process can be assessed ex vivo by luminal addition of commercially available fluorescently labeled, heat- or chemically killed bacteria such us Escherichia coli (K-12 strain) and Staphylococcus aureus (30). Alternatively, live green fluorescent protein-incorporated E. coli or to other bacteria can be used (31).
In vivo Assessment of the Intestinal Permeability
Directly: Mainly Assessing the Paracellular Pathway
Virtually all in vivo methods to assess paracellular intestinal permeability rely on the urinary excretion of orally ingested probes. Several markers, including different sizes of PEG, 51CrEDTA, and especially sugars such as sucrose, sucralose, lactulose, mannitol, and rhamnose, have been used, each with advantages and disadvantages. In general, the probe should be absorbed exclusively via the paracellular pathway, be metabolically inert and freely filtered at the glomerulus without tubular reabsorption (32).
The most commonly used test evaluates the differential urinary excretion of the disaccharide lactulose and the sugar alcohol mannitol. Many studies have used the urinary ratio of lactulose to mannitol (LMR) in order to correct for differences in absorption kinetics through gastrointestinal motility and alterations in renal excretion. The LMR in a urine collection of 0–2 h after ingestion is generally accepted as a read-out of small intestinal permeability (13) (Figure 2). A study using radio-isotopes has confirmed that a large proportion of the ingested probe is in the small intestine during this period (33). During 2–8 h, a mixed small intestinal and colonic presence is detected while the 8–24 h collection is most representative for the colonic permeability. However, lactulose and mannitol will be degraded by the colonic microbiota, and hence are not suited to evaluate colonic permeability (34). Other markers including sucralose, PEG, and 51CrEDTA are resistant to fermentation and can be used to measure total gastrointestinal permeability with longer urine collections up to 24 h (32). Sucrose is quickly hydrolyzed to fructose and glucose by the digestive enzyme sucrase in the small intestine, which provides the opportunity to estimate gastric and proximal duodenal permeability by measuring urinary sucrose excretion (35). The quantification of urinary sugars is not a standard lab test and should be carefully validated before implementation (36). Especially for lactulose, the urinary recovery can be close to the limit of quantification. Increasing the ingested dose above 5 g should be avoided since this will alter intestinal motility and water handling through its osmotic effect.
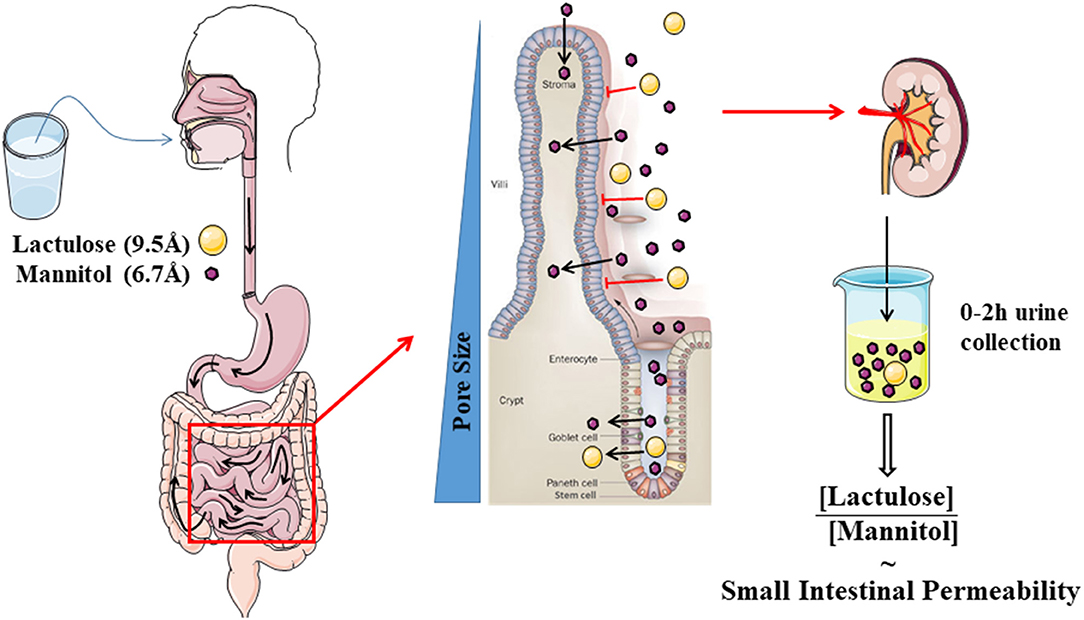
Figure 2. Schematic cartoon of the evaluation of small intestinal permeability by using the lactulose- mannitol test.
The Mayo clinic group has proposed the fractional excretion of mannitol, rather than the LMR to evaluate small intestinal permeability (33, 37, 38). Moreover, 13C mannitol has been suggested as an alternative to the common 12C mannitol because of the contamination with the latter from food sources in which mannitol is used as a sweetener (e.g., chewing gum) (37–39). However, until now, the test has only been used in healthy volunteers and patients with diarrhea-predominant IBS in whom an increased fractional mannitol excretion but no increased LMR was demonstrated in the 0–2 h collection (33). The effect of treatment on the mannitol excretion has only been evaluated in one study in which gluten induced a higher mannitol excretion and a higher LMR (40). The performance of the mannitol excretion test in other disease conditions and treatments will need to be evaluated and compared against the multitude of studies using a combined assessment of lactulose and mannitol excretion (13).
The truth of the matter is that the permeability pathways of many of the probes are still unclear. The hypothesis behind the LMR is that the smaller mannitol (6.7 Å) can pass via the pore pathway and can be considered as a marker for the total small intestinal surface, while the larger disaccharide lactulose (9.5 Å) will only pass the larger pores of the leak pathway in the crypt region and at sites where the barrier is damaged (13). However, the evidence supporting this hypothesis is scarce, leading to controversy on the validity and especially the interpretation of the lactulose-mannitol test (21). Some authors have suggested that mannitol can also pass transcellularly, although this is not backed up by experimental evidence and is not generally accepted (32). Nevertheless, the urinary recovery of mannitol is 100-fold higher than lactulose, which cannot be sufficiently explained by the size difference (21, 41). Intriguingly, when using the same probes ex vivo in Ussing chambers, the difference is much less pronounced with a LMR of ~0.8 ex vivo vs. 0.03 in vivo (42). The authors hypothesized that this difference is related to an increased absorption of mannitol in vivo through solvent drag at the tip of the villi.
Confocal laser endomicroscopy (CLE) is a more recent method to directly assess intestinal barrier function. After intravenous administration of fluorescein, enhancement of the gaps between epithelial cells (1), leakage of fluorescein into the bowel lumen (2), and cell shedding (3) can be directly visualized using a confocal probe equipped with a 488 nm laser (43, 44). Using these three features, increased leakiness has been reported in a variety of conditions, including IBD (45, 46), IBS (47), and acute pancreatitis (48). Moreover, two studies from the same group demonstrated increased duodenal permeability, assessed by CLE, in IBS patients upon perfusion of diluted food antigens (49, 50). However, it is important to keep in mind that CLE visualizes a basolateral (subepithelial) to apical (luminal) flux of a small molecule (fluorescein) of which the correlation to luminal to subepithelial passage of luminal compounds, which is hypothesized to trigger a mucosal immune response and symptoms, is still unclear. Only one study in patients with gastro-esophageal reflux disease (GERD) took the effort of comparing CLE to classical Ussing chamber experiments, which showed no differentiation between GERD and controls by probe-based CLE and no correlation of CLE findings and ex vivo permeability (51).
Finally, mucosal impedance testing is another tool to directly evaluate mucosal integrity, although most studies are limited to the esophagus. Impedance is the resistance—or inverse of conductivity—of an alternating electrical current between two adjacent electrodes on a luminal probe. Ten years ago, our group validated this technique to assess esophageal mucosal integrity by showing a correlation to ex vivo measurements and a decrease after esophageal perfusion with acidic solutions (52). Since then, it has become clear that baseline impedance, measured by multichannel intraluminal impedance (MII) testing is lower in GERD patients compared to healthy volunteers and improves with medical and surgical therapy (53, 54). More recently, a lower duodenal and jejunal baseline impedance was also demonstrated in patients with functional dyspepsia, corroborating previous ex vivo studies (55). The technique has recently been incorporated on a balloon that can be inserted through the biopsy channel of a standard endoscope. By inflating the balloon, the electrodes are brought into contact with the mucosa, and impedance is registered for 90 s as a read-out for mucosal integrity (56, 57). Studies in other segments of the GI tract and comparison of this short measurement to ex vivo permeability areawaited.
Indirect Assessment of Mucosal Integrity by Potential Blood Biomarkers
Both ex vivo and the direct in vivo tests for intestinal permeability are time and labor-intensive and are not readily available in most labs. Therefore, there is a growing interest in the validation of blood or urinary biomarkers for intestinal permeability. Strategies include the detection in the blood of molecules normally present in the intestinal lumen as a sign of impaired barrier function (e.g., LPS) or the detection of increased levels of proteins which are components of the intestinal barrier [intestinal fatty-acid binding protein (I-FABP) or tight-junction molecules] signaling damage to the intestinal wall or finally higher blood concentration of barrier-regulating proteins, i.e., zonulin. Unfortunately, with the possible exception of LPS and I-FABP, validation of these assays against standard permeability measurements and proper clinical validation studies in large groups of patients are missing and can therefore not be recommended to replace the direct permeability tests described above.
LPS is a 10–20 kDa protein located in the outer membrane of gram-negative bacteria, which is mainly transported across the epithelium via the transcellular route (58). Increased levels of LPS as a sign of barrier dysfunction have been reported in a variety of conditions, including cirrhosis (4), IBD (5), and IBS-D (6). To circumvent the pitfall of LPS-contamination of common lab equipment, LPS-binding protein (LBP) has been proposed as alternative non-invasive markers indicating increased transepithelial uptake of LPS (59). Even if LBP most likely evaluates the transcellular rather than paracellular pathway, a recent study demonstrated a significant correlation between LBP levels and the LMR in normal weight and obese subjects (60). Similarly, soluble CD14 (sCD14) can also bind LPS in the systemic circulation and is also used as a proxy readout of LPS-activity, although they are not always correlated (61).
I-FABP is a cytosolic protein present in differentiated enterocytes of the small intestine and to a lesser extent in the colon. In normal conditions, this protein is present in low amounts in the circulation, but upon damage to the intestinal barrier, it is released into the bloodstream (62). Increased I-FABP levels have been demonstrated in intestinal ischemia (63, 64), celiac disease (65), necrotizing enterocolitis (66), etc.
A limited number of studies have reported increased systemic levels of claudins as a marker for impaired intestinal barrier function, e.g., increased serum claudin-5 in children with attention deficit hyperactivity disorder (67) and increased urinary claudin-3 in patients with active Crohn's disease which was associated with decreased claudin-3 staining in intestinal biopsies (68). However, validation of these markers against the gold standard, i.e., Ussing chambers, is still lacking.
Finally, zonulin has become a widely used biomarker for intestinal permeability. In 2000, the Fasano lab discovered zonulin as the analog of the prokaryotic zonula-occludens toxin (Zot) in primates (69), and was later identified as prehaptoglobin-2 (70). Zonulin was shown to reversibly open tight-junctions in a protease-activated receptor 2 (PAR2) and epidermal growth factor receptor (EGFR) dependent manner resulting in dislocation of ZO-1 from the tight junction (71). Increased zonulin concentrations have been reported in many conditions, including celiac disease, type 1 diabetes, IBD, obesity, schizophrenia, etc. (72). However, it has become clear that the most widely used enzyme-linked immunosorbent assays (ELISA) do not detect zonulin but complement C3 and possibly properdin, a related molecule from the same family with unclear functional effects (73, 74). A more recent study also confirms that some zonulin ELISA kits are not specific and that increased blood zonulin levels in patients with IBS are not associated with colonic paracellular permeability assessed in Ussing chambers (75). Therefore, the available literature on zonulin as a non-invasive marker for disease characterized by barrier dysfunction should be interpreted with extreme caution.
Table 1 summarizes the different methods described above to evaluate intestinal permeability.
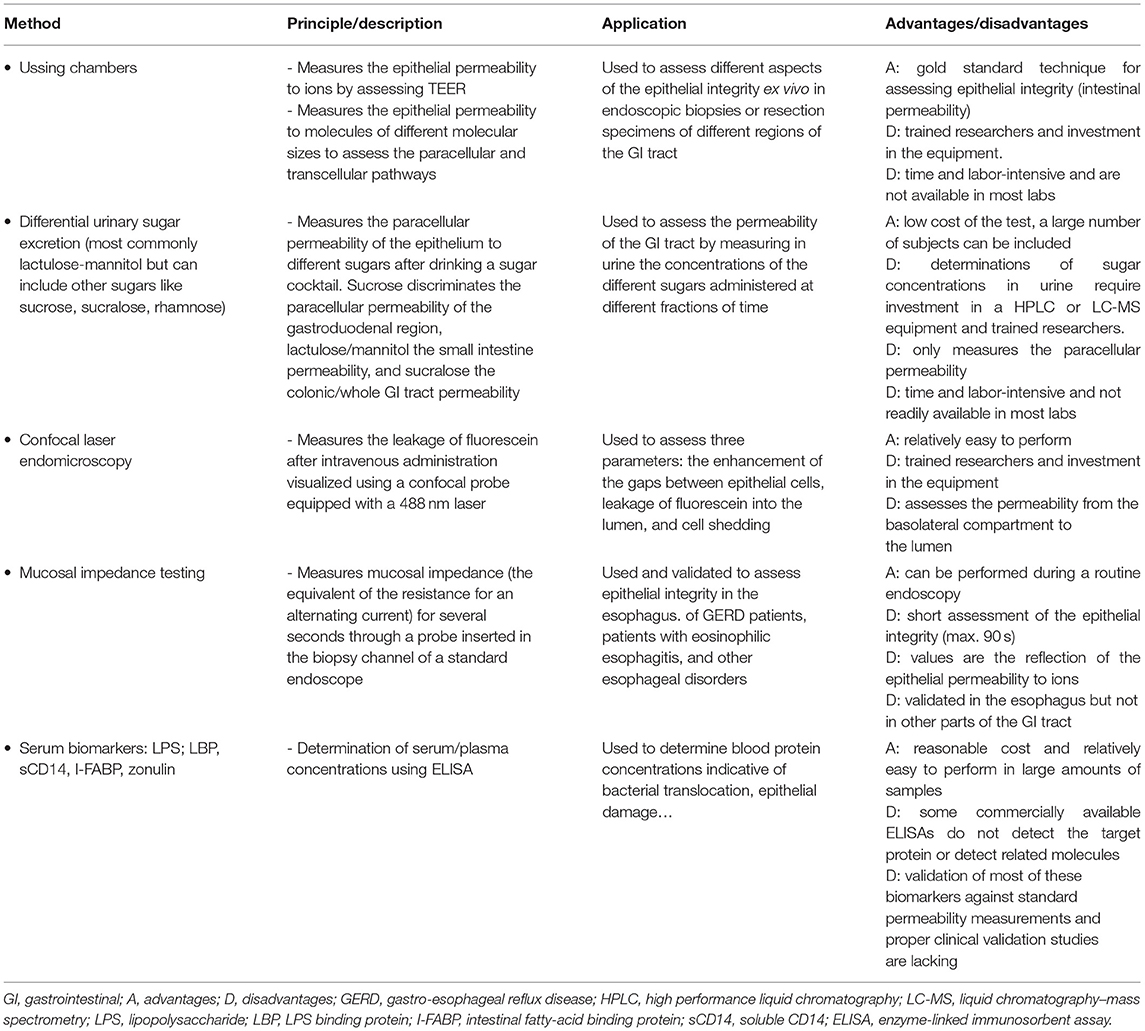
Table 1. Summary table of the different methods to assess intestinal permeability.
Intestinal Permeability in Selected GI Diseases
Celiac Disease
Celiac disease (CeD) is an immune-mediated enteropathy, triggered by dietary gluten—an alcohol-soluble protein fraction rich in glutamine and proline in wheat, barley, and rye—in genetically predisposed individuals (76). To date, celiac disease is the condition in which the pathogenic role of an impaired barrier function is best established and where steps are being made to translate this finding to a therapeutic target. In the late seventies, Cobden et al. found increased cellobiose excretion and decreased mannitol excretion in a 5 h urine collection in patients with active celiac disease (77). The decreased absorption was attributed to the reduced intestinal absorptive surface of the atrophic mucosa and forms the basis of using mannitol as a marker for intestinal surface area in the LMR. Similar data were presented in a study of 13 untreated CeD patients using a lactulose/rhamnose test (78). Gastroduodenal (sucrose excretion) (79) and whole gut permeability (51Cr-EDTA) (80) were also increased in patients with active CeD. These data were confirmed by a lower TEER in Ussing chambers (81, 82) and a decreased number of tight-junction strands shown by freeze-fracture electron microscopy (83). At the molecular level, lower levels of the sealing claudins-3,−5, and−7 and more pore-forming claudin-2 were reported (82). Moreover, in celiac disease claudin-5,−15, ZO-1 and occludin were displaced from their normal position at the tight junction (82, 84).
Gluten ingestion is causally related to the impaired intestinal barrier function in CeD. A single 30 g dose of gluten caused a transient increase in cellobiose excretion in well-controlled patients with normalization within 1 week (85). Moreover, with a gluten-free diet a fast improvement of the cellobiose excretion and a slower recovery of the mannitol excretion—supposedly a marker of regeneration of the intestinal villi—was observed (85). However, in several studies only a partial improvement but not normalization of the barrier was found on a long-term gluten-free diet (79–81), suggesting either that inadvertent dietary mistakes are made or that a barrier defect is a primary event in the pathophysiology of CeD. The latter hypothesis is also suggested by an intermediate (between healthy controls and CeD patients) small intestinal barrier function (LMR) in relatives of CeD patients (86).
The barrier defect in CeD is hypothesized to contribute to diarrhea by a paracellular flux of water and solutes (87). The controversy remains whether the impaired paracellular barrier plays a role in the transepithelial passage of gliadin. Recent data suggest a transcellular passage, dependent on binding to secretory immunoglobulin A (sIgA) and the transferrin receptor CD71 (88).
Two decades ago, increased zonulin protein concentration was shown in patients with active CeD by the group of Fasano (89). Gliadin induced release of zonulin by binding to CXCR3 and elevated permeability ex vivo in biopsies of healthy volunteers and patients with quiescent CeD (90, 91). However, both the baseline permeability and permeability after the addition of gliadin were higher and the luminal zonulin release was more pronounced and prolonged in patients with CeD. It is important to notice that these original studies were not performed with the previously mentioned commercial flawed ELISA (73, 74).
Comparison of the amino-terminal end of zonulin and the active fragment of Zot (92) revealed a conserved common motif (69, 93). A synthetic octapeptide (GGVLVQPG), named FZI/0 (92, 94), AT1001 (95, 96), and most recently larazotide (97, 98), corresponding to the amino acids 8–15 of this fragment, did not affect permeability, but pretreatment offered significant protection against the effect of subsequent treatment with purified Zot or zonulin (69, 92). To confirm the contribution of zonulin to early permeability changes in celiac disease, 20 patients on a gluten-free diet were randomized to a 3-day larazotide (12 mg once daily) or placebo treatment with a 2.5 g oral gluten challenge on the second day (96). Intestinal permeability was tested daily by a lactulose/mannitol urinary excretion test. In the placebo group, a significant 70% increase in intestinal permeability was observed after the gluten challenge in contrast to the AT1001 treated patients in whom no changes in permeability were observed. However, the difference between both groups failed to reach statistical significance. Leffler et al. evaluated a 2-week treatment with different doses of larazotide, ranging from 0.5 to 8 mg t.i.d., with daily gluten challenge in 86 celiac disease patients on a gluten-free diet (97). The primary endpoint, a difference in intestinal permeability, was not met in this study with large variability in the LMR. However, significantly fewer gastrointestinal symptoms were observed in patients in the active treatment arm (97). In a second study from the same group, evaluating 1, 4, and 8 mg of larazotide t.i.d. vs. placebo in 184 patients with daily gluten challenge for 6 weeks, gastrointestinal symptoms and anti-tissue transglutaminase IgA antibodies were lower in the active treatment groups. In contrast, and similar to the previous study, the urinary LMR was similar in both groups (98). Finally, 0.5 mg of larazotide t.i.d., but not the higher doses, improved symptoms in CeD patients with persistent symptoms despite following a gluten-free diet (99).
Inflammatory Bowel Disease
Inflammatory bowel disease (IBD) includes two chronic intestinal inflammatory conditions: Crohn's disease (CD) and ulcerative colitis (UC), characterized by an overactivation and dysregulation of the immune system with a relapsing-remitting pattern. Inflammation in UC is restricted to the superficial layers of the colon whereas, in CD, inflammation is transmural and can affect different regions of the GI tract including the upper part. The exact IBD pathogenesis is unknown, but mounting pieces of evidences indicate a complex interaction between the genome, the exposome, the microbiome, and the immunome (100).
The first attempts to measure intestinal permeability in IBD were performed in the late seventies by rectal instillation of radioiodine-labeled albumin and by measuring later the radioactivity in the plasma in healthy subjects and patients with UC (101). Intestinal permeability using sugar excretion tests (102) and 51Cr-EDTA (103) started to be used in the eighties mainly with small groups of patients. From this period, two relevant studies using a larger amount of subjects clearly showed that patients with IBD have an altered intestinal barrier. Total gut permeability using the oral administration of 99mTc-DTPA was increased in patients with CD and UC with both active and inactive disease. Interestingly, the authors found that the degree of intestinal and colonic inflammation was associated with permeability measurements (104). Jenkins et al. showed that the permeability to the oral administration of 51Cr-EDTA was increased in patients with small bowel disease and patients with colonic disease. In contrast, only patients with colonic disease had an increased permeability when 51Cr-EDTA was administered in the rectum. These findings suggest that the inflamed colon is the site of increased intestinal permeation (105). Since then, a large number of studies have been shown an increased gut permeability in IBD but discrepant findings are reported in different studies. These differences can be explained, at least in part, by the different probes used for the assessment (106). It has been stated and assumed by the majority of the research community that in vivo permeability is also increased in first-degree relatives of patients with IBD. Nevertheless, most of the studies do not support this. An increased intestinal PEG-400 permeability in first-degree healthy relatives of Crohn's disease patients was described for the first time in 1986 (107), whereas three other studies could not reproduce these findings by using the same probe (108–110). In addition, no differences were found between healthy relatives and control subjects when the LMR (110–112) or other sugars (110) were used. Interestingly, in vivo studies also show that the L/rhamnose ratio was restored with the induction of remission with an elemental diet in patients with active CD (113) and the urinary excretion of 51Cr-EDTA was normalized by an anti-TNF-α therapy (114). Moreover, some studies indicate that the increased intestinal permeability assessed in patients in clinical remission predicts a high risk of early relapse (115).
The first study ex vivo in Ussing chambers (1999) evaluating the epithelial barrier function in colonic tissue from UC patients with mild or moderate macroscopic disease activity showed a 50% decrease of the total electrical wall resistance (assessed by transmural impedance analysis) when compared to healthy subjects. This reduction was concomitant with increased paracellular permeability of mannitol. Moreover, the strand numbers of the TJs were decreased in UC suggesting a down regulation (116). Interestingly, Söderholm's group in Sweden showed that the TEER and the paracellular passage of 51Cr-EDTA and FD4 are not altered in the sigmoid colon of UC patients in remission but they have an increased transepithelial flux of the protein antigen HRP. Nevertheless, the same group recently showed that UC patients in remission have a reduced TEER and an increased paracellular passage of 51Cr-EDTA (117). The reason for this discrepancy is not known but could be related to differences in patient cohorts. Electrophysiological and molecular data discussed here contribute to a better understanding of the mechanisms for the increased permeability found in vivo and highlight the relevance of the transcellular route in UC.
Studies in CD have been performed in both affected and macroscopically non-inflamed tissues. The total electrical resistance, TEER, in the colon of CD patients with mild or moderate inflammation did not differ between patients with active and inactive disease (remission) when compared to control subjects. In contrast, the epithelial resistance, Rep, evaluated by transmural impedance analysis, was reduced in CD patients with active disease only (118). Occludin, claudin-5, and claudin-8 were found to be downregulated and redistributed, whereas claudin-2 (pore-forming TJ protein) was strongly upregulated. In the same direction, Söderholm et al. showed that the TEER of ileal inflamed tissue from CD patients is not altered but the paracellular permeability to 51Cr-EDTA is increased (119). The epithelium of the non-inflamed ileum shows a normal permeability to ions and a normal paracellular permeability to 51Cr-EDTA as compared to controls (119, 120) but is more susceptible to a challenge with sodium caprate indicating that TJs in the non-inflamed ileum of CD are more reactive to luminal stimuli contributing to the development of mucosal inflammation (120). Moreover, the non-inflamed ileal epithelium has an increased transcellular passage to protein antigens (119, 121) and increased transcellular uptake of non-pathogenic E. coli in the follicle-associated epithelium (FAE) in CD but not in UC (30, 122). Both transcellular mechanisms seem to be mediated by tumor necrosis factor alpha (121, 123).
An elegant study by Pastor-Rojo et al. showed that markers of an impaired barrier function associated with bacterial translocation as LPS and LBP are increased in the serum of CD patients with active and inactive disease whereas they are only increased in UC patients with active disease. Moreover, serum sCD14 levels are only increased in active CD and UC. Serum levels of these markers recovered after treatment to normal levels, although less completely in Crohn's disease (5). Hence, different treatment strategies in IBD patients do not only ameliorate the paracellular permeability (114) but also the transcellular uptake of luminal bacteria and bacterial components (5, 123). A more recent study has shown that the potential intestinal permeability biomarker I-FABP did not differ between endoscopic active disease and remission in both CD and UC (124).
In summary, the inflamed colonic and ileal epithelia from CD patients have a dysregulation of both the pore and the leak pathway. Remarkably, an altered epithelial barrier function has been found in non-inflamed ileal regions in CD. This alteration is characterized by an increased transcellular permeability to protein antigens and whole bacteria translocation in the FAE. The latter may lead to the increased load of commensal bacteria observed in inflamed and non-inflamed mucosa (125) and to the increased concentrations of LPS and LBP found in the blood of CD patients (5).
Chronic Alcoholic and Metabolic Liver Disease
Metabolic dysfunction-associated liver disease (MALFD), previously known as non-alcoholic liver disease (NAFLD) is the most common cause of chronic liver disease in the Western world with a global prevalence of 25% in the adult population (126). MAFLD covers a spectrum of disease stages ranging from simple steatosis over non-alcoholic steatohepatitis (NASH) to fibrosis and cirrhosis. Chronic and excessive alcohol use causes alcoholic liver disease with a similar progression from steatosis to alcoholic steatohepatitis (ASH) and cirrhosis.
Even if the pathophysiology of both conditions is not fully elucidated and not identical, the role of an impaired intestinal barrier function and altered microbiota in the pathogenesis and progression of chronic liver disease is increasingly recognized as a key player. This close and bidirectional interaction between the liver and the intestinal tract is termed the “gut-liver axis” (127–129). The central hypothesis of the gut-liver axis is that translocation of luminal microbiota or microbial products (pathogen-associated molecular patterns or PAMPs) through an impaired intestinal barrier function is an early step in the pathogenesis of chronic liver disease (4). A hepatic immune response will be triggered by the interaction of PAMPs (e.g., LPS and bacterial RNA) with pathogen recognition receptors such as Toll-like receptor (TLR)4 on hepatic Kuppfer cells, the resident macrophages in the liver sinusoids (130). This immune response will then lead to progressive liver injury and ultimately to fibrosis by activation of hepatic stellate cells (131–133).
Several groups have investigated in vivo intestinal permeability in MAFLD: small intestinal permeability by lactulose/mannitol (134–140) or lactulose/rhamnose (141) and whole gut permeability by 51Cr-EDTA (142) or sucralose (140). A recent systematic review and meta-analysis demonstrated increased small intestinal and whole gut permeability in MAFLD patients, even if this was not confirmed in all individual studies (134, 140, 141). All described studies evaluating small intestinal permeability used a 5 or 6 h urinary collection to assess small intestinal permeability, which may have confounded the results since a large amount of the sugars were probably already localized in the colon by the end of the urine collection (33). Increased zonulin levels were also reported in five pediatric and adult MAFLD studies, but because of the flawed commercial zonulin ELISA, these reports do not advance the field (73, 74). Taking into account these limitations, one of the more informative studies was performed by Miele et al. who showed increased 51Cr-EDTA in a 24 h urine collection in biopsy proven MAFLD vs. healthy controls (142). Moreover, higher intestinal permeability was present in patients with moderate or severe steatosis vs. patients with mild steatosis but was not associated to hepatic inflammation (NASH). Interestingly, patients with increased intestinal permeability were also more likely to have a pathological glucose breath test, which is indicative of small intestinal bacterial overgrowth (SIBO) (88.8 vs. 29.4% in MAFLD patients with increased vs. normal permeability; P < 0.001). Conversely, patients with SIBO also had a higher intestinal permeability. The authors also reported a correlation between impaired permeability and lower nuclear ZO-1 staining, the importance of which is difficult to interpret because its normal localization should be at the level of the tight-junctions (142). In a recent study in liver cirrhosis of any cause, not only urinary 51Cr-EDTA excretion was elevated in about half the patients and associated with Child-Pugh state, it was also present in all ascites samples from patients with spontaneous bacterial peritonitis (SBP) (143). Nevertheless, even if 85% of patients with cirrhosis in another study had a pathological LMR, the test failed to predict the development of SBP or survival (144).
In 1984, Bjarnason et al. were the first to describe increased whole gut permeability in patients with alcohol abuse without significant liver disease, measured by a 24 h urinary excretion of 51Cr-EDTA, which was confirmed by increased uptake of macromolecules (inulin, EDTA and cyanocobalamin) ex vivo in jejunal biopsies (145). Since then, increased whole gut permeability to larger molecular weight PEG molecules (MW 1,500 and 4,000) (146) and increased urinary excretion of sucrose in a 5–12 h collection (147) was reported in chronic alcoholic liver disease patients. Chronic alcohol use can by itself damage the intestinal barrier and increases plasma LPS levels, both of which normalized after a longer period of abstinence (145, 148). However, in a recent study in 106 patients with alcohol use disorder with variable degrees of liver disease, intestinal permeability normalized after a 3-week detoxification program, while markers of bacterial translocation (LBP) remained elevated (149). Moreover, baseline LBP and sCD14 levels were similar in patients with normal (2/3 of patients) and increased intestinal permeability (urinary 51Cr-EDTA excretion), highlighting again that bacterial translocation is not a paracellular phenomenon and that one should be careful of connecting clinical outcomes to paracellular permeability tests (149).
Very few studies have directly evaluated intestinal permeability in Ussing chambers. Du Plessis et al. described increased passage of labeled 4 kDa dextrans and a lowered TEER in duodenal biopsies in patients with compensated and decompensated cirrhosis from different etiologies (4). At the molecular level, an increased protein level of claudin-2, a pore-forming claudin, was demonstrated in comparison to healthy controls, although these pores are of insufficient size to allow diffusion of bacterial products such as LPS in decompensated cirrhosis (4). Decreased expression of occludin and claudin-1 in the duodenal mucosa have also been reported in cirrhosis and their expression levels correlated with LPS concentrations, severity of liver disease and portal hypertension (esophageal varices) (150). In a very recent study Haderer et al. reported a decreased mucus thickness in the colon of cirrhotic patients and a breakdown of E-cadherin and occludin by endogenous and newly identified bacterial proteases respectively, which could be future therapeutic targets (151).
Several etiological factors are most likely involved in the pathogenesis of the impaired barrier function in chronic liver disease, but the four most important ones are direct toxicity of alcohol, portal hypertension, alterations in the intestinal microbiome and altered bile acid signaling.
Alcohol and its metabolite acetaldehyde exert a direct toxic effect to the intestinal tight junctions with a redistribution of ZO-1 and occludin (152, 153). These data were also confirmed in vivo by duodenal perfusion of 20 g ethanol vs. placebo in healthy volunteers (154). Moreover, increased gene expression of MLCK pointed toward an activation of the leak pathway through contraction of the cytoskeleton. These effects were dependent on the activation of the mitogen activated protein kinase (MAPK) pathway (154). However, since small intestinal permeability was significantly higher in patients with alcoholic liver diseases than in patients with chronic alcohol abuse in the absence of liver disease, the direct barrier toxicity of alcohol does not fully account for the permeability defect (155).
Congestion of the intestinal microcirculation in portal hypertension may cause intestinal and vascular barrier dysfunction. A causal role for portal hypertension in the pathogenesis or further progression of the barrier defect in chronic liver disease is suggested by an improvement of intestinal permeability and LBP levels with non-selective beta-blockers and transjugular intrahepatic portosystemic shunting (TIPS) (156, 157). However, macrophage activation status (sCD163 levels) remained elevated even after normalization of the portal-venous pressure gradient with TIPS (157).
Several studies have shown microbial alterations in MALFD, alcoholic liver disease and acute-on-chronic liver failure which also evolve with disease severity (158–162). However, most studies are based on the analysis of stool microbiota which may not be fully representative of the small intestinal microbiota which can translocate through the intestinal barrier. Recently, Raj et al. investigated the mucosa-associated microbiota in duodenal biopsies of 35 patients with chronic liver disease of different etiologies. Patients displayed a lower microbial diversity and a significantly different composition of the duodenal microbiome with a lower abundance of the genera Moryella, Porphyromonas and Veillonella (163). The lower alpha diversity—but not serum LPS levels—also correlated with increased intestinal permeability, measured by a plasma lactulose/rhamnose ratio 90 min after ingestion of the sugar solution, which is a less validated way to assess intestinal permeability because this one sampling point does not take into account renal clearance. A similar correlation between intestinal barrier function and fecal microbial alterations has also been shown in alcohol-dependent patients (149, 164). How dysbiosis affects intestinal barrier function is largely unknown, but one mechanism is through decreased production of short-chain fatty acids (SCFA) such as butyrate, although this is largely based on preclinical data and would be mainly relevant for colonic barrier function (165).
Finally, the altered microbiome can change the bile acid pool by the conversion of primary to secondary bile acids. The altered bile acid pool is associated with impaired intestinal barrier function (166). Conversely, bile acids also affect the gut microbiota through activation of the innate immune system and production of antimicrobial peptides (127). Finally, decreased activation of the farnesoid X receptor (FXR) in the distal small intestine by bile acids may also contribute to impaired integrity of the intestinal barrier (127).
At this moment, treatments targeting the intestinal barrier in chronic liver disease are still lacking. Preclinical data demonstrate a restoration of intestinal barrier function upon stimulation with FXR agonists, e.g., obeticholic acid (128). However, to the best of our knowledge, clinical data of FXR agonists on barrier function in human chronic liver disease are lacking. In a recent study in 21 patients with MAFLD, fecal microbiota transfer (FMT), administered in the duodenum via endoscopy, improved the LMR in contrast to those who received an autologous FMT. However, there was no effect on insulin resistance or steatosis (167).
Bile Acid Malabsorption
Bile acids are synthesized in the liver and released into the duodenum to facilitate, through their detergent properties, the solubilization and absorption of fatty acid and monoglycerides in the small intestine. Under normal circumstances, more than 90% of bile acids are reabsorbed in the small intestine and transported back to the liver, a process referred to as enterohepatic circulation. The bile acid pool, estimated at 2–4 grams, circulates six to 10 dimes daily through the enterohepatic circulation. The reuptake of bile acids in the ileum occurs through active transport, for conjugated bile acids, or by passive absorption, for unconjugated bile acids.
Active transport of bile acids from the lumen into the enterocyte occurs through an apical sodium-dependent bile salt transporter in the brush border. Inside the enterocyte, ileal bile acid binding protein allows transfer through the cell and delivery at the basolateral side through organic solute transporters α and β. From there, the bile acids reach the portal circulation and the liver where they are recycled. Passage of bile acids through the enterocyte activates FXR, which promotes the synthesis of fibroblast growth factor 19 (FGF19), which also circulates through the portal vein to the liver where it activates fibroblast growth factor-receptor 4 resulting in inhibition of bile acid synthesis. Binding of FGF to FGFR4 is facilitated by the transmembrane protein Klotho β, and genetic variants in this protein have been associated with colonic transit in IBS with diarrhea. In case of low bile acid absorption, FXR is not activated, FGF19 stays low and bile acid synthesis is stimulated. Bile acids are synthesized from cholesterol and serum levels of 7α-hydroxy-4-cholesten-3-one (C4), an intermediate product of this process, reflect the activity of bile acid synthesis (168).
In case of organic disease, that affects the terminal ileum (e.g., Crohn's disease, ileal resection, etc.), bile acids are not absorbed and enter the colon, where they induce diarrhea. This is referred to as bile acid diarrhea type 1. In case of mutations affecting the function of the bile salt transporter, but especially when the FGF19-driven negative feedback pathway is defective, this leads to bile acid diarrhea type 2 (168). Patients with this type of bile acid diarrhea have significantly lower FGF19 and higher 7a-hydroxy-4-cholesten-3-one (C4) levels than healthy controls (169), which supports the hypothesis that excess synthesis of bile acids may overflow the reabsorption capacity in the ileum and hence allow bile acids to enter the colon in larger quantities.
Bile acids in the colon may facilitate the occurrence diarrhea in a number of ways. Through activation of the G protein-coupled receptor TGR5, expressed on enterocytes, entero-endocrine cells and on enteric neurons, bile acids may stimulate colonic motility (170). In man, colonic or rectal instillation of bile acid solution stimulates propulsive motility (171, 172). Administration of chenodeoxycholic acid accelerated colonic transit in healthy controls (173). Bile acids also promote colonic water secretion, through TGR5-mediated activation of adenylate cyclase, which stimulates chloride secretion through the cystic fibrosis transmembrane conductance regulator (CFTR) (174). In addition, bile acids also inhibit apical chloride/bicarbonate exchange in enterocytes (175).
Several ex vivo and in vivo studies have demonstrated that bile acids may affect mucosal integrity, in the esophagus and in the small intestine. TGR5-deficient mice have increased colonic mucosal permeability, through disruption of epithelial tight junctions (176). Hence, it has been suggested that bile acid diarrhea also involves increased colonic mucosal permeability. In the rabbit colon ex vivo, mucosal permeability was increased by deoxycholic and chenodeoxycholic acid (177). In a mixed cohort of controls and IBS patients, total fecal bile acid content was significantly correlated with mucosal permeability (178).
In IBS-D patients with elevated serum C4 a borderline increased colonic permeability was found based on urinary mannitol excretion (178). However, C4 levels in IBS-D are not significantly correlated to mucosal permeability as assessed by urinary excretion of lactulose/mannitol, and in 23 patients with IBS-D, treatment with colesevelam tended to improve stool consistency but did not alter mucosal permeability as assessed by lactulose/mannitol excretion (173). In a controlled colesevelam trial in 30 patients with IBS-D, colesevelam did not alter mucosal permeability (179). Taken together, these studies do not confirm increased mucosal permeability as a key mechanism involved in bile acid diarrhea.
Conclusion and Knowledge Gaps
The intestinal barrier function is a complex integrated system composed of several pathways. However, evaluation of the intestinal barrier in clinical studies is usually limited to the assessment of the paracellular permeability. Moreover, not all methodologies utilized in these studies are properly validated against the gold standard, i.e., the Ussing chamber, and therefore caution is needed in the interpretation of these permeability data. In the clinical conditions presented in the review, increased intestinal permeability has been demonstrated, but the central question of whether the barrier dysfunction is a primary event in the pathophysiology or a consequence of the disease is still not resolved in most diseases. Until therapies specifically correcting barrier function, which could demonstrate a causal role for intestinal permeability, become available, the controversy will remain and diagnosis of “leaky gut” has no role in diagnosis or treatment of human disease.
Author Contributions
TV, JT, and RF: contributed on writing and review the original draft. RF: editing. All authors contributed to the article and approved the submitted version.
Funding
TV is a senior clinical investigator of the Flanders Research Foundation (FWO Vlaanderen) and JT was supported by a Methusalem grant of the KU Leuven. This research was funded in part by Centro de Investigación Biomédica en Red de Enfermedades Hepáticas y Digestivas (CIBEREHD), Instituto de Salud Carlos III, Subdirección General de Investigación Sanitaria, Ministerio de Economía y Competitividad (Madrid): EHD16PI02 (RF).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Farre R, Vicario M. Abnormal barrier function in gastrointestinal disorders. Handb Exp Pharmacol. (2017) 239:193–217. doi: 10.1007/164_2016_107
2. Wallon C, Yang PC, Keita AV, Ericson AC, McKay DM, Sherman PM, et al. Corticotropin-releasing hormone (CRH) regulates macromolecular permeability via mast cells in normal human colonic biopsies in vitro. Gut. (2008) 57:50–8. doi: 10.1136/gut.2006.117549
3. Berg RD, Garlington AW. Translocation of certain indigenous bacteria from the gastrointestinal tract to the mesenteric lymph nodes and other organs in a gnotobiotic mouse model. Infect Immun. (1979) 23:403–11. doi: 10.1128/iai.23.2.403-411.1979
4. Du Plessis J, Vanheel H, Janssen CE, Roos L, Slavik T, Stivaktas PI, et al. Activated intestinal macrophages in patients with cirrhosis release NO and IL-6 that may disrupt intestinal barrier function. J Hepatol. (2013) 58:1125–32. doi: 10.1016/j.jhep.2013.01.038
5. Pastor Rojo O, Lopez San Roman A, Albeniz Arbizu E, de la Hera Martinez A, Ripoll Sevillano E, Albillos Martinez A. Serum lipopolysaccharide-binding protein in endotoxemic patients with inflammatory bowel disease. Inflamm Bowel Dis. (2007) 13:269–77. doi: 10.1002/ibd.20019
6. Dlugosz A, Nowak P, D'Amato M, Mohammadian Kermani G, Nystrom J, Abdurahman S, et al. Increased serum levels of lipopolysaccharide and antiflagellin antibodies in patients with diarrhea-predominant irritable bowel syndrome. Neurogastroenterol Motil. (2015) 27:1747–54. doi: 10.1111/nmo.12670
7. Farquhar MG, Palade GE. Junctional complexes in various epithelia. J Cell Biol. (1963) 17:375–412. doi: 10.1083/jcb.17.2.375
8. Lindemann B. Hans Ussing, experiments and models. J Membr Biol. (2001) 184:203–10. doi: 10.1007/s00232-001-0103-4
9. Claude P, Goodenough DA. Fracture faces of zonulae occludentes from “tight” and “leaky” epithelia. J Cell Biol. (1973) 58:390–400. doi: 10.1083/jcb.58.2.390
10. Shirazi-Beechey SP, Moran AW, Batchelor DJ, Daly K, Al-Rammahi M. Glucose sensing and signalling; regulation of intestinal glucose transport. Proc Nutr Soc. (2011) 70:185–93. doi: 10.1017/S0029665111000103
11. Turner JR, Rill BK, Carlson SL, Carnes D, Kerner R, Mrsny RJ, et al. Physiological regulation of epithelial tight junctions is associated with myosin light-chain phosphorylation. Am J Physiol. (1997) 273:C1378–85. doi: 10.1152/ajpcell.1997.273.4.C1378
12. Loo DD, Wright EM, Zeuthen T. Water pumps. J Physiol. (2002) 542:53–60. doi: 10.1113/jphysiol.2002.018713
13. Odenwald MA, Turner JR. Intestinal permeability defects: is it time to treat? Clin Gastroenterol Hepatol. (2013) 11:1075–83. doi: 10.1016/j.cgh.2013.07.001
14. Thiagarajah JR, Verkman AS. Chloride channel-targeted therapy for secretory diarrheas. Curr Opin Pharmacol. (2013) 13:888–94. doi: 10.1016/j.coph.2013.08.005
15. Kopic S, Geibel JP. Gastric acid, calcium absorption, and their impact on bone health. Physiol Rev. (2013) 93:189–268. doi: 10.1152/physrev.00015.2012
16. Lieben L, Carmeliet G. The involvement of TRP channels in bone homeostasis. Front Endocrinol. (2012) 3:99. doi: 10.3389/fendo.2012.00099
17. Fujita H, Sugimoto K, Inatomi S, Maeda T, Osanai M, Uchiyama Y, et al. Tight junction proteins claudin-2 and−12 are critical for vitamin D-dependent Ca2+ absorption between enterocytes. Mol Biol Cell. (2008) 19:1912–21. doi: 10.1091/mbc.e07-09-0973
18. Watson CJ, Hoare CJ, Garrod DR, Carlson GL, Warhurst G. Interferon-gamma selectively increases epithelial permeability to large molecules by activating different populations of paracellular pores. J Cell Sci. (2005) 118:5221–30. doi: 10.1242/jcs.02630
19. Akiba Y, Maruta K, Takajo T, Narimatsu K, Said H, Kato I, et al. Lipopolysaccharides transport during fat absorption in rodent small intestine. Am J Physiol Gastrointest Liver Physiol. (2020) 318:G1070–87. doi: 10.1152/ajpgi.00079.2020
20. Hollander D, Kaunitz JD. The “leaky gut”: tight junctions but loose associations? Dig Dis Sci. (2020) 65:1277–87. doi: 10.1007/s10620-019-05777-2
21. Camilleri M. Leaky gut: mechanisms, measurement and clinical implications in humans. Gut. (2019) 68:1516–26. doi: 10.1136/gutjnl-2019-318427
22. Bonazzi M, Cossart P. Bacterial entry into cells: a role for the endocytic machinery. FEBS Lett. (2006) 580:2962–7. doi: 10.1016/j.febslet.2006.04.010
23. Veiga E, Cossart P. The role of clathrin-dependent endocytosis in bacterial internalization. Trends Cell Biol. (2006) 16:499–504. doi: 10.1016/j.tcb.2006.08.005
24. Pai YC, Weng LT, Wei SC, Wu LL, Shih DQ, Targan SR, et al. Gut microbial transcytosis induced by tumor necrosis factor-like 1A-dependent activation of a myosin light chain kinase splice variant contributes to IBD. J Crohns Colitis. (2020) 15:258–72. doi: 10.1093/ecco-jcc/jjaa165
25. Wu LL, Peng WH, Kuo WT, Huang CY, Ni YH, Lu KS, et al. Commensal bacterial endocytosis in epithelial cells is dependent on myosin light chain kinase-activated brush border fanning by interferon-gamma. Am J Pathol. (2014) 184:2260–74. doi: 10.1016/j.ajpath.2014.05.003
26. Richter W, Vogel V, Howe J, Steiniger F, Brauser A, Koch MH, et al. Morphology, size distribution, and aggregate structure of lipopolysaccharide and lipid A dispersions from enterobacterial origin. Innate Immun. (2011) 17:427–38. doi: 10.1177/1753425910372434
27. Ussing HH, Zerahn K. Active transport of sodium as the source of electric current in the short-circuited isolated frog skin. Acta Physiol Scand. (1951) 23:110–27. doi: 10.1111/j.1748-1716.1951.tb00800.x
28. Krug SM, Fromm M, Gunzel D. Two-path impedance spectroscopy for measuring paracellular and transcellular epithelial resistance. Biophys J. (2009) 97:2202–11. doi: 10.1016/j.bpj.2009.08.003
29. Vanheel H, Vicario M, Vanuytsel T, Van Oudenhove L, Martinez C, Keita AV, et al. Impaired duodenal mucosal integrity and low-grade inflammation in functional dyspepsia. Gut. (2014) 63:262–71. doi: 10.1136/gutjnl-2012-303857
30. Keita AV, Salim SY, Jiang T, Yang PC, Franzen L, Soderkvist P, et al. Increased uptake of non-pathogenic E. coli via the follicle-associated epithelium in longstanding ileal Crohn's disease. J Pathol. (2008) 215:135–44. doi: 10.1002/path.2337
31. Keita AV, Gullberg E, Ericson AC, Salim SY, Wallon C, Kald A, et al. Characterization of antigen and bacterial transport in the follicle-associated epithelium of human ileum. Lab Invest. (2006) 86:504–16. doi: 10.1038/labinvest.3700397
32. Bjarnason I, MacPherson A, Hollander D. Intestinal permeability: an overview. Gastroenterology. (1995) 108:1566–81. doi: 10.1016/0016-5085(95)90708-4
33. Rao AS, Camilleri M, Eckert DJ, Busciglio I, Burton DD, Ryks M, et al. Urine sugars for in vivo gut permeability: validation and comparisons in irritable bowel syndrome-diarrhea and controls. Am J Physiol Gastrointest Liver Physiol. (2011) 301:G919–28. doi: 10.1152/ajpgi.00168.2011
34. Meddings JB, Gibbons I. Discrimination of site-specific alterations in gastrointestinal permeability in the rat. Gastroenterology. (1998) 114:83–92. doi: 10.1016/S0016-5085(98)70636-5
35. Smecuol E, Bai JC, Sugai E, Vazquez H, Niveloni S, Pedreira S, et al. Acute gastrointestinal permeability responses to different non-steroidal anti-inflammatory drugs. Gut. (2001) 49:650–5. doi: 10.1136/gut.49.5.650
36. Houben E, Vanuytsel T, Farre R, Tack J, Verbeke K. Validation of a GC-MS and HPLC-ELSD method to study intestinal permeability. Trends Chromatogr. (2013) 8:83–96.
37. Grover M, Camilleri M, Hines J, Burton D, Ryks M, Wadhwa A, et al. (13) C mannitol as a novel biomarker for measurement of intestinal permeability. Neurogastroenterol Motil. (2016) 28:1114–9. doi: 10.1111/nmo.12802
38. Khoshbin K, Khanna L, Maselli D, Atieh J, Breen-Lyles M, Arndt K, et al. Development and validation of test for “leaky gut” small intestinal and colonic permeability using sugars in healthy adults. Gastroenterology. (2021) 161:463–75.e13. doi: 10.1053/j.gastro.2021.04.020
39. Laker MF Bull HJ Menzies IS. Evaluation of mannitol for use as a probe marker of gastrointestinal permeability in man. Eur J Clin Invest. (1982) 12:485–91. doi: 10.1111/j.1365-2362.1982.tb02230.x
40. Vazquez-Roque MI, Camilleri M, Smyrk T, Murray JA, Marietta E, O'Neill J, et al. A controlled trial of gluten-free diet in patients with irritable bowel syndrome-diarrhea: effects on bowel frequency and intestinal function. Gastroenterology. (2013) 144:903–11 e3. doi: 10.1053/j.gastro.2013.01.049
41. Vanuytsel T, van Wanrooy S, Vanheel H, Vanormelingen C, Verschueren S, Houben E, et al. Psychological stress and corticotropin-releasing hormone increase intestinal permeability in humans by a mast cell-dependent mechanism. Gut. (2014) 63:1293–9. doi: 10.1136/gutjnl-2013-305690
42. Bijlsma PB, Peeters RA, Groot JA, Dekker PR, Taminiau JA, Van Der Meer R. Differential in vivo and in vitro intestinal permeability to lactulose and mannitol in animals and humans: a hypothesis. Gastroenterology. (1995) 108:687–96. doi: 10.1016/0016-5085(95)90440-9
43. Buchner AM. Confocal laser endomicroscopy in the evaluation of inflammatory Bowel disease. Inflamm Bowel Dis. (2019) 25:1302–12. doi: 10.1093/ibd/izz021
44. Chang J, Ip M, Yang M, Wong B, Power T, Lin L, et al. The learning curve, interobserver, and intraobserver agreement of endoscopic confocal laser endomicroscopy in the assessment of mucosal barrier defects. Gastrointest Endosc. (2016) 83:785–91 e1. doi: 10.1016/j.gie.2015.08.045
45. Chang J, Leong RW, Wasinger VC, Ip M, Yang M, Phan TG. Impaired intestinal permeability contributes to ongoing bowel symptoms in patients with inflammatory bowel disease and mucosal healing. Gastroenterology. (2017) 153:723–31 e1. doi: 10.1053/j.gastro.2017.05.056
46. Queneherve L, David G, Bourreille A, Hardouin JB, Rahmi G, Neunlist M, et al. Quantitative assessment of mucosal architecture using computer-based analysis of confocal laser endomicroscopy in inflammatory bowel diseases. Gastrointest Endosc. (2019) 89:626–36. doi: 10.1016/j.gie.2018.08.006
47. Turcotte JF, Kao D, Mah SJ, Claggett B, Saltzman JR, Fedorak RN, et al. Breaks in the wall: increased gaps in the intestinal epithelium of irritable bowel syndrome patients identified by confocal laser endomicroscopy (with videos). Gastrointest Endosc. (2013) 77:624–30. doi: 10.1016/j.gie.2012.11.006
48. Sun L, Yue M, Dai Y, Yu C, Chen C. Confocal laser endomicroscopy reveals alterations in duodenal permeability in patients with acute pancreatitis. J Int Med Res. (2019) 47:1279–87. doi: 10.1177/0300060518820430
49. Fritscher-Ravens A, Schuppan D, Ellrichmann M, Schoch S, Rocken C, Brasch J, et al. Confocal endomicroscopy shows food-associated changes in the intestinal mucosa of patients with irritable bowel syndrome. Gastroenterology. (2014) 147:1012–20 e4. doi: 10.1053/j.gastro.2014.07.046
50. Fritscher-Ravens A, Pflaum T, Mosinger M, Ruchay Z, Rocken C, Milla PJ, et al. Many patients with irritable bowel syndrome have atypical food allergies not associated with immunoglobulin E. Gastroenterology. (2019) 157:109–18 e5. doi: 10.1053/j.gastro.2019.03.046
51. Mutha PR, Fasullo M, Chu S, Schubert ML, Zfass A, Cooper P, et al. Correlation of probe-based confocal laser endomicroscopy (pCLE) and mucosal integrity testing (MIT) with epithelial barrier function and presence of gastroesophageal reflux disease (GERD). Dig Dis Sci. (2021). doi: 10.1007/s10620-021-06980-w. [Epub ahead of print].
52. Farre R, Blondeau K, Clement D, Vicario M, Cardozo L, Vieth M, et al. Evaluation of oesophageal mucosa integrity by the intraluminal impedance technique. Gut. (2011) 60:885–92. doi: 10.1136/gut.2010.233049
53. Frazzoni M, Savarino E, de Bortoli N, Martinucci I, Furnari M, Frazzoni L, et al. Analyses of the post-reflux swallow-induced peristaltic wave index and nocturnal baseline impedance parameters increase the diagnostic yield of impedance-pH monitoring of patients with reflux disease. Clin Gastroenterol Hepatol. (2016) 14:40–6. doi: 10.1016/j.cgh.2015.06.026
54. Rinsma NF, Farre R, Bouvy ND, Masclee AA, Conchillo JM. The effect of endoscopic fundoplication and proton pump inhibitors on baseline impedance and heartburn severity in GERD patients. Neurogastroenterol Motil. (2015) 27:220–8. doi: 10.1111/nmo.12468
55. Nakagawa K, Hara K, Fikree A, Siddiqi S, Woodland P, Masamune A, et al. Patients with dyspepsia have impaired mucosal integrity both in the duodenum and jejunum: in vivo assessment of small bowel mucosal integrity using baseline impedance. J Gastroenterol. (2020) 55:273–80. doi: 10.1007/s00535-019-01614-5
56. Vaezi MF, Choksi Y. Mucosal impedance: a new way to diagnose reflux disease and how it could change your practice. Am J Gastroenterol. (2017) 112:4–7. doi: 10.1038/ajg.2016.513
57. Patel DA, Higginbotham T, Slaughter JC, Aslam M, Yuksel E, Katzka D, et al. Development and validation of a mucosal impedance contour analysis system to distinguish esophageal disorders. Gastroenterology. (2019) 156:1617–26 e1. doi: 10.1053/j.gastro.2019.01.253
58. Drewe J, Beglinger C, Fricker G. Effect of ischemia on intestinal permeability of lipopolysaccharides. Eur J Clin Invest. (2001) 31:138–44. doi: 10.1046/j.1365-2362.2001.00792.x
59. Schumann RR, Leong SR, Flaggs GW, Gray PW, Wright SD, Mathison JC, et al. Structure and function of lipopolysaccharide binding protein. Science. (1990) 249:1429–31. doi: 10.1126/science.2402637
60. Seethaler B, Basrai M, Neyrinck AM, Nazare JA, Walter J, Delzenne NM, et al. Biomarkers for assessment of intestinal permeability in clinical practice. Am J Physiol Gastrointest Liver Physiol. (2021) 321:G11–G17. doi: 10.1152/ajpgi.00113.2021
61. Nier A, Engstler AJ, Maier IB, Bergheim I. Markers of intestinal permeability are already altered in early stages of non-alcoholic fatty liver disease: studies in children. PLoS ONE. (2017) 12:e0183282. doi: 10.1371/journal.pone.0183282
62. Wells JM, Brummer RJ, Derrien M, MacDonald TT, Troost F, Cani PD, et al. Homeostasis of the gut barrier and potential biomarkers. Am J Physiol Gastrointest Liver Physiol. (2017) 312:G171–93. doi: 10.1152/ajpgi.00048.2015
63. Kanda T, Fujii H, Tani T, Murakami H, Suda T, Sakai Y, et al. Intestinal fatty acid-binding protein is a useful diagnostic marker for mesenteric infarction in humans. Gastroenterology. (1996) 110:339–43. doi: 10.1053/gast.1996.v110.pm8566578
64. Ding CM, Wu YH, Liu XF. Diagnostic accuracy of intestinal fatty acid binding protein for acute intestinal ischemia: a systematic review and meta-analysis. Clin Lab. (2020) 66. doi: 10.7754/Clin.Lab.2019.191139
65. Adriaanse MP, Tack GJ, Passos VL, Damoiseaux JG, Schreurs MW, van Wijck K, et al. Serum I-FABP as marker for enterocyte damage in coeliac disease and its relation to villous atrophy and circulating autoantibodies. Aliment Pharmacol Ther. (2013) 37:482–90. doi: 10.1111/apt.12194
66. Heida FH, Hulscher JB, Schurink M, Timmer A, Kooi EM, Bos AF, et al. Intestinal fatty acid-binding protein levels in Necrotizing Enterocolitis correlate with extent of necrotic bowel: results from a multicenter study. J Pediatr Surg. (2015) 50:1115–8. doi: 10.1016/j.jpedsurg.2014.11.037
67. Aydogan Avsar P, Isik U, Aktepe E, Kilic F, Doguc DK, Buyukbayram HI. Serum zonulin and claudin-5 levels in children with attention-deficit/hyperactivity disorder. Int J Psychiatry Clin Pract. (2021) 25:49–55. doi: 10.1080/13651501.2020.1801754
68. Thuijls G, Derikx JP, de Haan JJ, Grootjans J, de Bruine A, Masclee AA, et al. Urine-based detection of intestinal tight junction loss. J Clin Gastroenterol. (2010) 44:e14–9. doi: 10.1097/MCG.0b013e31819f5652
69. Wang W, Uzzau S, Goldblum SE, Fasano A. Human zonulin, a potential modulator of intestinal tight junctions. J Cell Sci 113 Pt. (2000) 24:4435–40. doi: 10.1242/jcs.113.24.4435
70. Tripathi A, Lammers KM, Goldblum S, Shea-Donohue T, Netzel-Arnett S, Buzza MS, et al. Identification of human zonulin, a physiological modulator of tight junctions, as prehaptoglobin-2. Proc Natl Acad Sci USA. (2009) 106:16799–804. doi: 10.1073/pnas.0906773106
71. Clemente MG, De Virgiliis S, Kang JS, Macatagney R, Musu MP, Di Pierro MR, et al. Early effects of gliadin on enterocyte intracellular signalling involved in intestinal barrier function. Gut. (2003) 52:218–23. doi: 10.1136/gut.52.2.218
72. Fasano A. All disease begins in the (leaky) gut: role of zonulin-mediated gut permeability in the pathogenesis of some chronic inflammatory diseases. F1000Res. (2020) 9:F1000. doi: 10.12688/f1000research.20510.1
73. Scheffler L, Crane A, Heyne H, Tonjes A, Schleinitz D, Ihling CH, et al. Widely used commercial ELISA does not detect precursor of haptoglobin2, but recognizes properdin as a potential second member of the Zonulin Family. Front Endocrinol. (2018) 9:22. doi: 10.3389/fendo.2018.00022
74. Ajamian M, Steer D, Rosella G, Gibson PR. Serum zonulin as a marker of intestinal mucosal barrier function: may not be what it seems. PLoS ONE. (2019) 14:e0210728. doi: 10.1371/journal.pone.0210728
75. Meira de-Faria F, Bednarska O, Strom M, Soderholm JD, Walter SA, Keita AV. Colonic paracellular permeability and circulating zonulin-related proteins. Scand J Gastroenterol. (2021) 56:424–31. doi: 10.1080/00365521.2021.1879247
76. Fasano A, Catassi C. Clinical practice. Celiac disease. N Engl J Med. (2012) 367:2419–26. doi: 10.1056/NEJMcp1113994
77. Cobden I, Dickinson RJ, Rothwell J, Axon AT. Intestinal permeability assessed by excretion ratios of two molecules: results in coeliac disease. Br Med J. (1978) 2:1060. doi: 10.1136/bmj.2.6144.1060
78. Menzies IS Laker MF Pounder R Bull J Heyer S Wheeler PG. Abnormal intestinal permeability to sugars in villous atrophy. Lancet. (1979) 2:1107–9. doi: 10.1016/S0140-6736(79)92507-8
79. Smecuol E, Bai JC, Vazquez H, Kogan Z, Cabanne A, Niveloni S, et al. Gastrointestinal permeability in celiac disease. Gastroenterology. (1997) 112:1129–36. doi: 10.1016/S0016-5085(97)70123-9
80. Bjarnason I, Peters TJ, Veall N. A persistent defect in intestinal permeability in coeliac disease demonstrated by a 51Cr-labelled EDTA absorption test. Lancet. (1983) 1:323–5. doi: 10.1016/S0140-6736(83)91628-8
81. Schulzke JD, Schulzke I, Fromm M, Riecken EO. Epithelial barrier and ion transport in coeliac sprue: electrical measurements on intestinal aspiration biopsy specimens. Gut. (1995) 37:777–82. doi: 10.1136/gut.37.6.777
82. Schumann M, Gunzel D, Buergel N, Richter JF, Troeger H, May C, et al. Cell polarity-determining proteins Par-3 and PP-1 are involved in epithelial tight junction defects in coeliac disease. Gut. (2012) 61:220–8. doi: 10.1136/gutjnl-2011-300123
83. Schulzke JD, Bentzel CJ, Schulzke I, Riecken EO, Fromm M. Epithelial tight junction structure in the jejunum of children with acute and treated celiac sprue. Pediatr Res. (1998) 43:435–41. doi: 10.1203/00006450-199804000-00001
84. Ciccocioppo R, Finamore A, Ara C, Di Sabatino A, Mengheri E, Corazza GR. Altered expression, localization, and phosphorylation of epithelial junctional proteins in celiac disease. Am J Clin Pathol. (2006) 125:502–11. doi: 10.1309/DTYRA91G8R0KTM8M
85. Hamilton I, Cobden I, Rothwell J, Axon AT. Intestinal permeability in coeliac disease: the response to gluten withdrawal and single-dose gluten challenge. Gut. (1982) 23:202–10. doi: 10.1136/gut.23.3.202
86. van Elburg RM, Uil JJ, Mulder CJ, Heymans HS. Intestinal permeability in patients with coeliac disease and relatives of patients with coeliac disease. Gut. (1993) 34:354–7. doi: 10.1136/gut.34.3.354
87. Schumann M, Siegmund B, Schulzke JD, Fromm M. Celiac disease: role of the epithelial barrier. Cell Mol Gastroenterol Hepatol. (2017) 3:150–62. doi: 10.1016/j.jcmgh.2016.12.006
88. Lebreton C, Menard S, Abed J, Moura IC, Coppo R, Dugave C, et al. Interactions among secretory immunoglobulin A, CD71, and transglutaminase-2 affect permeability of intestinal epithelial cells to gliadin peptides. Gastroenterology. (2012) 143:698–707 e4. doi: 10.1053/j.gastro.2012.05.051
89. Fasano A, Not T, Wang W, Uzzau S, Berti I, Tommasini A, et al. Zonulin, a newly discovered modulator of intestinal permeability, and its expression in coeliac disease. Lancet. (2000) 355:1518–9. doi: 10.1016/S0140-6736(00)02169-3
90. Drago S, El Asmar R, Di Pierro M, Grazia Clemente M, Tripathi A, Sapone A, et al. Gliadin, zonulin and gut permeability: effects on celiac and non-celiac intestinal mucosa and intestinal cell lines. Scand J Gastroenterol. (2006) 41:408–19. doi: 10.1080/00365520500235334
91. Lammers KM, Lu R, Brownley J, Lu B, Gerard C, Thomas K, et al. Gliadin induces an increase in intestinal permeability and zonulin release by binding to the chemokine receptor CXCR3. Gastroenterology. (2008) 135:194–204 e3. doi: 10.1053/j.gastro.2008.03.023
92. Di Pierro M, Lu R, Uzzau S, Wang W, Margaretten K, Pazzani C, et al. Zonula occludens toxin structure-function analysis. Identification of the fragment biologically active on tight junctions and of the zonulin receptor binding domain. J Biol Chem. (2001) 276:19160–5. doi: 10.1074/jbc.M009674200
93. Uzzau S, Cappuccinelli P, Fasano A. Expression of Vibrio cholerae zonula occludens toxin and analysis of its subcellular localization. Microb Pathog. (1999) 27:377–85. doi: 10.1006/mpat.1999.0312
94. Watts T, Berti I, Sapone A, Gerarduzzi T, Not T, Zielke R, et al. Role of the intestinal tight junction modulator zonulin in the pathogenesis of type I diabetes in BB diabetic-prone rats. Proc Natl Acad Sci USA. (2005) 102:2916–21. doi: 10.1073/pnas.0500178102
95. Arrieta MC, Madsen K, Doyle J, Meddings J. Reducing small intestinal permeability attenuates colitis in the IL10 gene-deficient mouse. Gut. (2009) 58:41–8. doi: 10.1136/gut.2008.150888
96. Paterson BM, Lammers KM, Arrieta MC, Fasano A, Meddings JB. The safety, tolerance, pharmacokinetic and pharmacodynamic effects of single doses of AT-1001 in coeliac disease subjects: a proof of concept study. Aliment Pharmacol Ther. (2007) 26:757–66. doi: 10.1111/j.1365-2036.2007.03413.x
97. Leffler DA, Kelly CP, Abdallah HZ, Colatrella AM, Harris LA, Leon F, et al. A randomized, double-blind study of larazotide acetate to prevent the activation of celiac disease during gluten challenge. Am J Gastroenterol. (2012) 107:1554–62. doi: 10.1038/ajg.2012.211
98. Kelly CP, Green PH, Murray JA, Dimarino A, Colatrella A, Leffler DA, et al. Larazotide acetate in patients with coeliac disease undergoing a gluten challenge: a randomised placebo-controlled study. Aliment Pharmacol Ther. (2013) 37:252–62. doi: 10.1111/apt.12147
99. Leffler DA, Kelly CP, Green PH, Fedorak RN, DiMarino A, Perrow W, et al. Larazotide acetate for persistent symptoms of celiac disease despite a gluten-free diet: a randomized controlled trial. Gastroenterology. (2015) 148:1311–9 e6. doi: 10.1053/j.gastro.2015.02.008
100. Fiocchi C. Inflammatory bowel disease pathogenesis: where are we? J Gastroenterol Hepatol. (2015) 30(Suppl. 1):12–8. doi: 10.1111/jgh.12751
101. Dalmark M. Plasma radioactivity after rectal instillation of radioiodine-labelled human albumin in normal subjects and in patients with ulcerative colitis. Scand J Gastroenterol. (1968) 3:490–6. doi: 10.3109/00365526809179908
102. Cobden I, Rothwell J, Axon AT. Intestinal permeability and screening tests for coeliac disease. Gut. (1980) 21:512–8. doi: 10.1136/gut.21.6.512
103. Bjarnason I, O'Morain C, Levi AJ, Peters TJ. Absorption of 51chromium-labeled ethylenediaminetetraacetate in inflammatory bowel disease. Gastroenterology. (1983) 85:318–22. doi: 10.1016/0016-5085(83)90317-7
104. Casellas F, Aguade S, Soriano B, Accarino A, Molero J, Guarner L. Intestinal permeability to 99mTc-diethylenetriaminopentaacetic acid in inflammatory bowel disease. Am J Gastroenterol. (1986) 81:767–70.
105. Jenkins RT, Ramage JK, Jones DB, Collins SM, Goodacre RL, Hunt RH. Small bowel and colonic permeability to 51Cr-EDTA in patients with active inflammatory bowel disease. Clin Invest Med. (1988) 11:151–5.
106. Howden CW, Robertson C, Duncan A, Morris AJ, Russell RI. Comparison of different measurements of intestinal permeability in inflammatory bowel disease. Am J Gastroenterol. (1991) 86:1445–9.
107. Hollander D, Vadheim CM, Brettholz E, Petersen GM, Delahunty T, Rotter JI. Increased intestinal permeability in patients with Crohn's disease and their relatives. A possible etiologic factor. Ann Intern Med. (1986) 105:883–5. doi: 10.7326/0003-4819-105-6-883
108. Ruttenberg D, Young GO, Wright JP, Isaacs S. PEG-400 excretion in patients with Crohn's disease, their first-degree relatives, healthy volunteers. Dig Dis Sci. (1992) 37:705–8. doi: 10.1007/BF01296426
109. Teahon K Smethurst P Levi AJ Menzies IS Bjarnason I. Intestinal permeability in patients with Crohn's disease and their first degree relatives. Gut. (1992) 33:320–3. doi: 10.1136/gut.33.3.320
110. Munkholm P, Langholz E, Hollander D, Thornberg K, Orholm M, Katz KD, et al. Intestinal permeability in patients with Crohn's disease and ulcerative colitis and their first degree relatives. Gut. (1994) 35:68–72. doi: 10.1136/gut.35.1.68
111. May GR, Sutherland LR, Meddings JB. Is small intestinal permeability really increased in relatives of patients with Crohn's disease? Gastroenterology. (1993) 104:1627–32. doi: 10.1016/0016-5085(93)90638-S
112. Katz KD, Hollander D, Vadheim CM, McElree C, Delahunty T, Dadufalza VD, et al. Intestinal permeability in patients with Crohn's disease and their healthy relatives. Gastroenterology. (1989) 97:927–31. doi: 10.1016/0016-5085(89)91499-6
113. Sanderson IR, Boulton P, Menzies I, Walker-Smith JA. Improvement of abnormal lactulose/rhamnose permeability in active Crohn's disease of the small bowel by an elemental diet. Gut. (1987) 28:1073–6. doi: 10.1136/gut.28.9.1073
114. Suenaert P, Bulteel V, Lemmens L, Noman M, Geypens B, Van Assche G, et al. Anti-tumor necrosis factor treatment restores the gut barrier in Crohn's disease. Am J Gastroenterol. (2002) 97:2000–4. doi: 10.1111/j.1572-0241.2002.05914.x
115. Wyatt J, Vogelsang H, Hubl W, Waldhoer T, Lochs H. Intestinal permeability and the prediction of relapse in Crohn's disease. Lancet. (1993) 341:1437–9. doi: 10.1016/0140-6736(93)90882-H
116. Schmitz H, Barmeyer C, Fromm M, Runkel N, Foss HD, Bentzel CJ, et al. Altered tight junction structure contributes to the impaired epithelial barrier function in ulcerative colitis. Gastroenterology. (1999) 116:301–9. doi: 10.1016/S0016-5085(99)70126-5
117. Katinios G, Casado-Bedmar M, Walter SA, Vicario M, Gonzalez-Castro AM, Bednarska O, et al. Increased colonic epithelial permeability and mucosal eosinophilia in ulcerative colitis in remission compared with irritable Bowel syndrome and health. Inflamm Bowel Dis. (2020) 26:974–84. doi: 10.1093/ibd/izz328
118. Zeissig S, Burgel N, Gunzel D, Richter J, Mankertz J, Wahnschaffe U, et al. Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn's disease. Gut. (2007) 56:61–72. doi: 10.1136/gut.2006.094375
119. Soderholm JD, Peterson KH, Olaison G, Franzen LE, Westrom B, Magnusson KE, et al. Epithelial permeability to proteins in the noninflamed ileum of Crohn's disease? Gastroenterology. (1999) 117:65–72. doi: 10.1016/S0016-5085(99)70551-2
120. Soderholm JD, Olaison G, Peterson KH, Franzen LE, Lindmark T, Wiren M, et al. Augmented increase in tight junction permeability by luminal stimuli in the non-inflamed ileum of Crohn's disease. Gut. (2002) 50:307–13. doi: 10.1136/gut.50.3.307
121. Soderholm JD, Streutker C, Yang PC, Paterson C, Singh PK, McKay DM, et al. Increased epithelial uptake of protein antigens in the ileum of Crohn's disease mediated by tumour necrosis factor alpha. Gut. (2004) 53:1817–24. doi: 10.1136/gut.2004.041426
122. Keita AV, Soderholm JD. Barrier dysfunction and bacterial uptake in the follicle-associated epithelium of ileal Crohn's disease. Ann N Y Acad Sci. (2012) 1258:125–34. doi: 10.1111/j.1749-6632.2012.06502.x
123. Yakymenko O, Schoultz I, Gullberg E, Strom M, Almer S, Wallon C, et al. Infliximab restores colonic barrier to adherent-invasive E. coli in Crohn's disease via effects on epithelial lipid rafts. Scand J Gastroenterol. (2018) 53:677–84. doi: 10.1080/00365521.2018.1458146
124. Bodelier AG, Pierik MJ, Lenaerts K, de Boer E, Olde Damink SW, Hameeteman WM, et al. Plasma intestinal fatty acid-binding protein fails to predict endoscopic disease activity in inflammatory bowel disease patients. Eur J Gastroenterol Hepatol. (2016) 28:807–13. doi: 10.1097/MEG.0000000000000616
125. Tiveuung A, Soderholm JD, Olaison G, Jonasson J, Monstein HJ. Presence of eubacteria in biopsies from Crohn's disease inflammatory lesions as determined by 16S rRNA gene-based PCR. J Med Microbiol. (1999) 48:263–8. doi: 10.1099/00222615-48-3-263
126. Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology. (2016) 64:73–84. doi: 10.1002/hep.28431
127. Martin-Mateos R, Albillos A. The role of the gut-liver axis in metabolic dysfunction-associated fatty liver disease. Front Immunol. (2021) 12:660179. doi: 10.3389/fimmu.2021.660179
128. Simbrunner B, Mandorfer M, Trauner M, Reiberger T. Gut-liver axis signaling in portal hypertension. World J Gastroenterol. (2019) 25:5897–917. doi: 10.3748/wjg.v25.i39.5897
129. Szabo G. Gut-liver axis in alcoholic liver disease. Gastroenterology. (2015) 148:30–6. doi: 10.1053/j.gastro.2014.10.042
130. Chopyk DM, Grakoui A. Contribution of the intestinal microbiome and gut barrier to hepatic disorders. Gastroenterology. (2020) 159:849–63. doi: 10.1053/j.gastro.2020.04.077
131. Brandl K, Kumar V, Eckmann L. Gut-liver axis at the frontier of host-microbial interactions. Am J Physiol Gastrointest Liver Physiol. (2017) 312:G413–9. doi: 10.1152/ajpgi.00361.2016
132. Racanelli V, Rehermann B. The liver as an immunological organ. Hepatology. (2006) 43:S54–62. doi: 10.1002/hep.21060
133. Heymann F, Tacke F. Immunology in the liver–from homeostasis to disease. Nat Rev Gastroenterol Hepatol. (2016) 13:88–110. doi: 10.1038/nrgastro.2015.200
134. Pierri L, Saggese P, Guercio Nuzio S, Troisi J, Di Stasi M, Poeta M, et al. Relations of gut liver axis components and gut microbiota in obese children with fatty liver: a pilot study. Clin Res Hepatol Gastroenterol. (2018) 42:387–90. doi: 10.1016/j.clinre.2018.03.015
135. Troisi J, Pierri L, Landolfi A, Marciano F, Bisogno A, Belmonte F, et al. Urinary metabolomics in pediatric obesity and NAFLD identifies metabolic pathways/metabolites related to dietary habits and gut-liver axis perturbations. Nutrients. (2017) 9:485. doi: 10.3390/nu9050485
136. Guercio Nuzio S, Di Stasi M, Pierri L, Troisi J, Poeta M, Bisogno A, et al. Multiple gut-liver axis abnormalities in children with obesity with and without hepatic involvement. Pediatr Obes. (2017) 12:446–52. doi: 10.1111/ijpo.12164
137. Giorgio V, Miele L, Principessa L, Ferretti F, Villa MP, Negro V, et al. Intestinal permeability is increased in children with non-alcoholic fatty liver disease, and correlates with liver disease severity. Dig Liver Dis. (2014) 46:556–60. doi: 10.1016/j.dld.2014.02.010
138. Nobili V, Alisi A, Cutrera R, Carpino G, De Stefanis C, D'Oria V, et al. Altered gut-liver axis and hepatic adiponectin expression in OSAS: novel mediators of liver injury in paediatric non-alcoholic fatty liver. Thorax. (2015) 70:769–81. doi: 10.1136/thoraxjnl-2015-206782
139. Volynets V, Kuper MA, Strahl S, Maier IB, Spruss A, Wagnerberger S, et al. Nutrition, intestinal permeability, and blood ethanol levels are altered in patients with nonalcoholic fatty liver disease (NAFLD). Dig Dis Sci. (2012) 57:1932–41. doi: 10.1007/s10620-012-2112-9
140. Farhadi A, Gundlapalli S, Shaikh M, Frantzides C, Harrell L, Kwasny MM, et al. Susceptibility to gut leakiness: a possible mechanism for endotoxaemia in non-alcoholic steatohepatitis. Liver Int. (2008) 28:1026–33. doi: 10.1111/j.1478-3231.2008.01723.x
141. Wigg AJ, Roberts-Thomson IC, Dymock RB, McCarthy PJ, Grose RH, Cummins AG. The role of small intestinal bacterial overgrowth, intestinal permeability, endotoxaemia, and tumour necrosis factor alpha in the pathogenesis of non-alcoholic steatohepatitis. Gut. (2001) 48:206–11. doi: 10.1136/gut.48.2.206
142. Miele L, Valenza V, La Torre G, Montalto M, Cammarota G, Ricci R, et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology. (2009) 49:1877–87. doi: 10.1002/hep.22848
143. Scarpellini E, Valenza V, Gabrielli M, Lauritano EC, Perotti G, Merra G, et al. Intestinal permeability in cirrhotic patients with and without spontaneous bacterial peritonitis: is the ring closed? Am J Gastroenterol. (2010) 105:323–7. doi: 10.1038/ajg.2009.558
144. Vogt A, Reuken PA, Stengel S, Stallmach A, Bruns T. Dual-sugar tests of small intestinal permeability are poor predictors of bacterial infections and mortality in cirrhosis: a prospective study. World J Gastroenterol. (2016) 22:3275–84. doi: 10.3748/wjg.v22.i11.3275
145. Bjarnason I, Peters TJ, Wise RJ. The leaky gut of alcoholism: possible route of entry for toxic compounds. Lancet. (1984) 1:179–82. doi: 10.1016/S0140-6736(84)92109-3
146. Parlesak A, Schafer C, Schutz T, Bode JC, Bode C. Increased intestinal permeability to macromolecules and endotoxemia in patients with chronic alcohol abuse in different stages of alcohol-induced liver disease. J Hepatol. (2000) 32:742–7. doi: 10.1016/S0168-8278(00)80242-1
147. Farhadi A, Keshavarzian A, Kwasny MJ, Shaikh M, Fogg L, Lau C, et al. Effects of aspirin on gastroduodenal permeability in alcoholics and controls. Alcohol. (2010) 44:447–56. doi: 10.1016/j.alcohol.2010.05.004
148. Leclercq S, Cani PD, Neyrinck AM, Starkel P, Jamar F, Mikolajczak M, et al. Role of intestinal permeability and inflammation in the biological and behavioral control of alcohol-dependent subjects. Brain Behav Immun. (2012) 26:911–8. doi: 10.1016/j.bbi.2012.04.001
149. Maccioni L, Gao B, Leclercq S, Pirlot B, Horsmans Y, De Timary P, et al. Intestinal permeability, microbial translocation, changes in duodenal and fecal microbiota, and their associations with alcoholic liver disease progression in humans. Gut Microbes. (2020) 12:1782157. doi: 10.1080/19490976.2020.1782157
150. Assimakopoulos SF, Tsamandas AC, Tsiaoussis GI, Karatza E, Triantos C, Vagianos CE, et al. Altered intestinal tight junctions' expression in patients with liver cirrhosis: a pathogenetic mechanism of intestinal hyperpermeability. Eur J Clin Invest. (2012) 42:439–46. doi: 10.1111/j.1365-2362.2011.02609.x
151. Haderer M, Neubert P, Rinner E, Scholtis A, Broncy L, Gschwendtner H, et al. Novel pathomechanism for spontaneous bacterial peritonitis: disruption of cell junctions by cellular and bacterial proteases. Gut. (2021). doi: 10.1136/gutjnl-2020-321663. [Epub ahead of print].
152. Rao R. Endotoxemia and gut barrier dysfunction in alcoholic liver disease. Hepatology. (2009) 50:638–44. doi: 10.1002/hep.23009
153. Elamin E, Jonkers D, Juuti-Uusitalo K, van Ijzendoorn S, Troost F, Duimel H, et al. Effects of ethanol and acetaldehyde on tight junction integrity: in vitro study in a three dimensional intestinal epithelial cell culture model. PLoS ONE. (2012) 7:e35008. doi: 10.1371/journal.pone.0035008
154. Elamin E, Masclee A, Troost F, Pieters HJ, Keszthelyi D, Aleksa K, et al. Ethanol impairs intestinal barrier function in humans through mitogen activated protein kinase signaling: a combined in vivo and in vitro approach. PLoS ONE. (2014) 9:e107421. doi: 10.1371/journal.pone.0107421
155. Keshavarzian A, Holmes EW, Patel M, Iber F, Fields JZ, Pethkar S. Leaky gut in alcoholic cirrhosis: a possible mechanism for alcohol-induced liver damage. Am J Gastroenterol. (1999) 94:200–7. doi: 10.1111/j.1572-0241.1999.00797.x
156. Reiberger T, Ferlitsch A, Payer BA, Mandorfer M, Heinisch BB, Hayden H, et al. Hepatic hemodynamic, non-selective betablocker therapy decreases intestinal permeability and serum levels of LBP and IL-6 in patients with cirrhosis. J Hepatol. (2013) 58:911–21. doi: 10.1016/j.jhep.2012.12.011
157. Holland-Fischer P, Gronbaek H, Sandahl TD, Moestrup SK, Riggio O, Ridola L, et al. Kupffer cells are activated in cirrhotic portal hypertension and not normalised by TIPS. Gut. (2011) 60:1389–93. doi: 10.1136/gut.2010.234542
158. Aron-Wisnewsky J, Vigliotti C, Witjes J, Le P, Holleboom AG, Verheij J, et al. Gut microbiota and human NAFLD: disentangling microbial signatures from metabolic disorders. Nat Rev Gastroenterol Hepatol. (2020) 17:279–97. doi: 10.1038/s41575-020-0269-9
159. Lee G, You HJ, Bajaj JS, Joo SK, Yu J, Park S, et al. Distinct signatures of gut microbiome and metabolites associated with significant fibrosis in non-obese NAFLD. Nat Commun. (2020) 11:4982. doi: 10.1038/s41467-020-18754-5
160. Loomba R, Seguritan V, Li W, Long T, Klitgord N, Bhatt A, et al. Gut microbiome-based metagenomic signature for non-invasive detection of advanced fibrosis in human nonalcoholic fatty liver disease. Cell metabolism. (2017) 25:1054–62 e5. doi: 10.1016/j.cmet.2017.04.001
161. Wijarnpreecha K, Lou S, Watthanasuntorn K, Kroner PT, Cheungpasitporn W, Lukens FJ, et al. Small intestinal bacterial overgrowth and nonalcoholic fatty liver disease: a systematic review and meta-analysis. Eur J Gastroenterol Hepatol. (2020) 32:601–8. doi: 10.1097/MEG.0000000000001541
162. Sole C, Guilly S, Da Silva K, Llopis M, Le-Chatelier E, Huelin P, et al. Alterations in gut microbiome in cirrhosis as assessed by quantitative metagenomics: relationship with acute-on-chronic liver failure and prognosis. Gastroenterology. (2021) 160:206–18 e13. doi: 10.1053/j.gastro.2020.08.054
163. Raj AS, Shanahan ER, Tran CD, Bhat P, Fletcher LM, Vesey DA, et al. Dysbiosis of the duodenal mucosal microbiota is associated with increased small intestinal permeability in chronic liver disease. Clin Transl Gastroenterol. (2019) 10:e00068. doi: 10.14309/ctg.0000000000000068
164. Leclercq S, Matamoros S, Cani PD, Neyrinck AM, Jamar F, Starkel P, et al. Intestinal permeability, gut-bacterial dysbiosis, and behavioral markers of alcohol-dependence severity. Proc Natl Acad Sci USA. (2014) 111:E4485–93. doi: 10.1073/pnas.1415174111
165. Schroeder BO, Birchenough GMH, Stahlman M, Arike L, Johansson MEV, Hansson GC, et al. Bifidobacteria or fiber protects against diet-induced microbiota-mediated colonic mucus deterioration. Cell Host Microbe. (2018) 23:27–40 e7. doi: 10.1016/j.chom.2017.11.004
166. Ridlon JM, Alves JM, Hylemon PB, Bajaj JS. Cirrhosis, bile acids and gut microbiota: unraveling a complex relationship. Gut Microbes. (2013) 4:382–7. doi: 10.4161/gmic.25723
167. Craven L, Rahman A, Nair Parvathy S, Beaton M, Silverman J, Qumosani K, et al. Allogenic fecal microbiota transplantation in patients with nonalcoholic fatty liver disease improves abnormal small intestinal permeability: a randomized control trial. Am J Gastroenterol. (2020) 115:1055–65. doi: 10.14309/ajg.0000000000000661
168. Camilleri M. Bile acid diarrhea: prevalence, pathogenesis, and therapy. Gut Liver. (2015) 9:332–9. doi: 10.5009/gnl14397
169. Walters JR, Tasleem AM, Omer OS, Brydon WG, Dew T, le Roux CW. A new mechanism for bile acid diarrhea: defective feedback inhibition of bile acid biosynthesis. Clin Gastroenterol Hepatol. (2009) 7:1189–94. doi: 10.1016/j.cgh.2009.04.024
170. Poole DP, Godfrey C, Cattaruzza F, Cottrell GS, Kirkland JG, Pelayo JC, et al. Expression and function of the bile acid receptor GpBAR1 (TGR5) in the murine enteric nervous system. Neurogastroenterol Motil. (2010) 22:814–25, e227–8. doi: 10.1111/j.1365-2982.2010.01487.x
171. Coremans G, Tack J, Vantrappen G, Janssens J, Annese V. Is the irritable bowel really irritable? Ital J Gastroenterol. (1991) 23:39–40.
172. Dinning PG, Bampton PA, Kennedy ML, Lubowski DZ, King D, Cook IJ. Impaired proximal colonic motor response to rectal mechanical and chemical stimulation in obstructed defecation. Dis Colon Rectum. (2005) 48:1777–84. doi: 10.1007/s10350-005-0087-8
173. Odunsi-Shiyanbade ST, Camilleri M, McKinzie S, Burton D, Carlson P, Busciglio IA, et al. Effects of chenodeoxycholate and a bile acid sequestrant, colesevelam, on intestinal transit and bowel function. Clin Gastroenterol Hepatol. (2010) 8:159–65. doi: 10.1016/j.cgh.2009.10.020
174. Ao M, Sarathy J, Domingue J, Alrefai WA, Rao MC. Chenodeoxycholic acid stimulates Cl(-) secretion via cAMP signaling and increases cystic fibrosis transmembrane conductance regulator phosphorylation in T84 cells. Am J Physiol Cell Physiol. (2013) 305:C447–56. doi: 10.1152/ajpcell.00416.2012
175. Alrefai WA, Saksena S, Tyagi S, Gill RK, Ramaswamy K, Dudeja PK. Taurodeoxycholate modulates apical Cl-/OH- exchange activity in Caco2 cells. Dig Dis Sci. (2007) 52:1270–8. doi: 10.1007/s10620-006-9090-8
176. Cipriani S, Mencarelli A, Chini MG, Distrutti E, Renga B, Bifulco G, et al. The bile acid receptor GPBAR-1 (TGR5) modulates integrity of intestinal barrier and immune response to experimental colitis. PLoS ONE. (2011) 6:e25637. doi: 10.1371/journal.pone.0025637
177. Chadwick VS, Gaginella TS, Carlson GL, Debongnie JC, Phillips SF, Hofmann AF. Effect of molecular structure on bile acid-induced alterations in absorptive function, permeability, and morphology in the perfused rabbit colon. J Lab Clin Med. (1979) 94:661–74.
178. Camilleri M, Busciglio I, Acosta A, Shin A, Carlson P, Burton D, et al. Effect of increased bile acid synthesis or fecal excretion in irritable bowel syndrome-diarrhea. Am J Gastroenterol. (2014) 109:1621–30. doi: 10.1038/ajg.2014.215
179. Vijayvargiya P, Camilleri M, Carlson P, Nair A, Nord SL, Ryks M, et al. Effects of colesevelam on bowel symptoms, biomarkers, and colonic mucosal gene expression in patients with bile acid diarrhea in a randomized trial. Clin Gastroenterol Hepatol. (2020) 18:2962–70 e6. doi: 10.1016/j.cgh.2020.02.027
Keywords: intestinal permeability, GI diseases, coeliac disease, chronic liver disease, inflammatory bowel disease, bile acid malabsorption, paracellular route, transcellular route
Citation: Vanuytsel T, Tack J and Farre R (2021) The Role of Intestinal Permeability in Gastrointestinal Disorders and Current Methods of Evaluation. Front. Nutr. 8:717925. doi: 10.3389/fnut.2021.717925
Received: 31 May 2021; Accepted: 04 August 2021;
Published: 26 August 2021.
Edited by:
Francisco Javier Santos Vicente, Vall d'Hebron University Hospital, SpainReviewed by:
Anandh Babu Pon Velayutham, The University of Utah, United StatesClaudio Nicoletti, University of Florence, Italy
Copyright © 2021 Vanuytsel, Tack and Farre. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ricard Farre, cmljYXJkLmZhcnJlQGt1bGV1dmVuLmJl