 Montserrat Esteve
Montserrat Esteve
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Nutr., 02 September 2020
Sec. Clinical Nutrition
Volume 7 - 2020 | https://doi.org/10.3389/fnut.2020.00111
This article is part of the Research TopicDietary Antioxidants and Metabolic DiseasesView all 7 articles
An inverse correlation between vegetable consumption and the incidence of cancer has long been described. This protective effect is stronger when cruciferous vegetables are specifically consumed. The beneficial properties of vegetables are attributed to their bioactive components like fiber, antioxidants vitamins, antioxidants, minerals, and phenolic compounds. Cruciferous vegetables contain all these molecules; however, what makes them different are their sulfurous components, called glucosinolates, responsible for their special smell and taste. Glucosinolates are inactive biologically in the organism but are hydrolyzed by the enzyme myrosinase released as a result of chewing, leading to the formation of active derivatives such as isothiocyanates and indoles. A considerable number of in vitro and in vivo studies have reported that isothiocyanates and indoles elicit chemopreventive potency through multiple mechanisms that include modulation of phases I and II detoxification pathway enzymes, regulation of cell cycle arrest, and control of cell growth, induction of apoptosis, antioxidant activity, anti-angiogenic effects, and epigenetic regulation. Nuclear erythroid 2-related factor 2 (Nrf2) and Nuclear factor-κB (NF-κB) are key and central regulators in all these processes with a main role in oxidative stress and inflammation control. It has been described that isothiocyanates and indoles regulate their activity directly and indirectly. Today, the metabolic syndrome (central obesity, insulin resistance, hyperlipidemia, and hypertension) is responsible for a majority of deaths worldwide. All components of metabolic syndrome are characterized by chronic inflammation with deregulation of the PI3K/AKT/mTOR, MAPK/EKR/JNK, Nrf2, and NF-κB signaling pathways. The effects of GLSs derivatives controlling these pathways have been widely described in relation to cancer. Changes in food consumption patterns observed in the last decades to higher consumption of ultra-processed foods, with elevation in simple sugar and saturated fat contents and lower consumption of vegetables and fruits have been directly correlated with metabolic syndrome prevalence. In this review, it is summarized the knowledge regarding the mechanisms by which cruciferous glucosinolate derivatives (isothiocyanates and indoles) directly and indirectly regulate these pathways. However, the review places a special focus on the knowledge of the effects of glucosinolates derivatives in metabolic syndrome, since this has not been reviewed before.
The aim of the present review was to assess the current knowledge about the mechanisms through which isothiocyanates (ITCs) and indoles derived from glucosinolates (GLSs) yield their biological effects. The effects to GLSs derivatives related to cancer have been extensively reviewed but have not been reviewed in relation to metabolic syndrome (MetS), which is what the review is focused on. Presented first are the factors that influence the amount of GLSs consumed, such as plant concentrations conditioned by growing, storage, and cooking conditions. Further is explained the process by which they are absorbed and metabolized. Then, the known biological effects and mechanisms through which ITCs and indoles act are assessed. It should be noted that the effects of ITCs and indoles have been studied mainly in relation to cancer, both in cancer cell lines and in cancer studies of animal models. However, it is necessary to point out that not only the anticancer effects have been analyzed; their effects in other situations are also mentioned because the purpose was to highlight the biological mechanisms they regulate, which can also be altered in other pathologies. Finally, it is focus on the metabolic MetS (which is greatly influenced by diet and is a major cause of death today) and analyzed the protective effects of ITCs and indoles brought about by the regulation of pathways previously described in relation to cancer prevention.
High consumption of vegetables and fruit has been recommended widely for the primary prevention of major chronic diseases such as type 2 diabetes (1, 2), coronary heart disease (3, 4), and some cancers such as those of the esophagus, larynx, stomach, colon and rectum, breast, lung, and bladder (5, 6). However, not all studies have yielded consistent results (7, 8). Some data show that not all vegetables have equal protective efficiency. Schulz et al. (9) found no evidence of an inverse linear association between total fruit and vegetable intake and the risk of ovarian cancer, but found that the consumption of garlic/onion might exert a protective effect on the risk of ovarian cancer. In a large study of men and women in the United States, Bhupathiraju et al. (4) suggested that the absolute quantity of fruits and vegetable consumed (rather than the variety) is associated with a significantly lower risk of coronary heart disease. However, the analysis of the correlation between the variety and quantity showed that higher intakes of specific fruits (such as citrus fruits), vegetables rich in β-carotene or vitamin C, and green leafy vegetables were associated with the lowest risk of coronary heart disease. In another study, Bazzano et al. (1) found that the consumption of green leafy vegetables and whole fruit was associated with a lower hazard of diabetes. Thus, the diverse protective effects of vegetables in relation to chronic diseases could be attributed to the differences in the composition of micronutrients and phytochemical profiles, cooking methods, and individual genetic variability.
The consumption of specific cruciferous vegetables is more strongly associated with protection against cancer than that of other vegetables in general (10). The beneficial properties of vegetables are attributed to their bioactive components such as fiber, antioxidant vitamins (vitamin C and β-carotene), antioxidant minerals (selenium), and phenolic compounds. Cruciferous vegetables contain all these molecules. However, what makes them different is that they also contain some sulfur compounds called GLSs (11). Cruciferous vegetables are members of the Brassicaceae or Cruciferae family (the alternative name is due to the shape of their flowers whose four petals resemble a cross) consumed commonly, such as broccoli, cauliflower, cabbage, kale, Brussels sprouts, Chinese cabbage, radish, wasabi, mustard, and watercress. GLSs are responsible for their characteristic pungent odor and bitter taste. More than 120 different GLSs have been identified from several plants with a profile and quantity that vary depending on the cultivars and growing conditions, since these compounds are vital for plants' defense against biotic and abiotic stress (12–14). All GLSs share a basic chemical structure that contains a β-D-thioglucose group, a sulfonate oxime group, and a side chain derived from a branched chain amino acid, methionine, alanine, phenylalanine, tyrosine, or tryptophan (14, 15). According to their structure, they can be classified into aliphatic (derived from methionine, alanine, leucine, isoleucine, or valine), aromatic (derived from phenylalanine or tyrosine), and indolic (derived from tryptophan) GLSs (16). GLSs are biologically inactive and need to be hydrolyzed by the enzyme myrosinase to become active (12). GLSs and myrosinase are located at different compartments in intact plant cells. When the structure is damaged, both molecules come into contact and the reaction occurs. This usually occurs when the vegetables are cut or damaged (during harvesting and processing) or during chewing (17). Myrosinase removes β-D-thioglucose from GLS, leading to the formation of unstable compounds that finally become bioactive molecules such as thiocyanates, ITCs, indoles, and nitriles in a process influenced by the pH. At neutral pH, ITC formation is favored, while an acidic pH favors those of nitriles (13, 18) which do not have anticarcinogenic properties. In addition, in some plants and specific GLSs, specific proteins (different from myrosinase) could also play a role in these chemical changes. These include epithiospecifier protein (ESP) which drives the reaction of nitrile formation in plants that contain alkenyl-GLSs but not those that contain GLSs with a terminal alkene (13). Figure 1 shows a scheme of GLSs derivatives formation. The amount of GLSs ingested, and their active compounds (the ITCs) which can eventually reach the tissues, is determined by several factors including the content of GLSs determined by the variety of cruciferous vegetables consumed, the lability of GLSs to environmental conditions and cooking techniques, their bioavailability, and their metabolism (19). Table 1 contains a summary of the most abundant cruciferous GSLs, their derivative ITCs/indoles, and plants in which they are found.
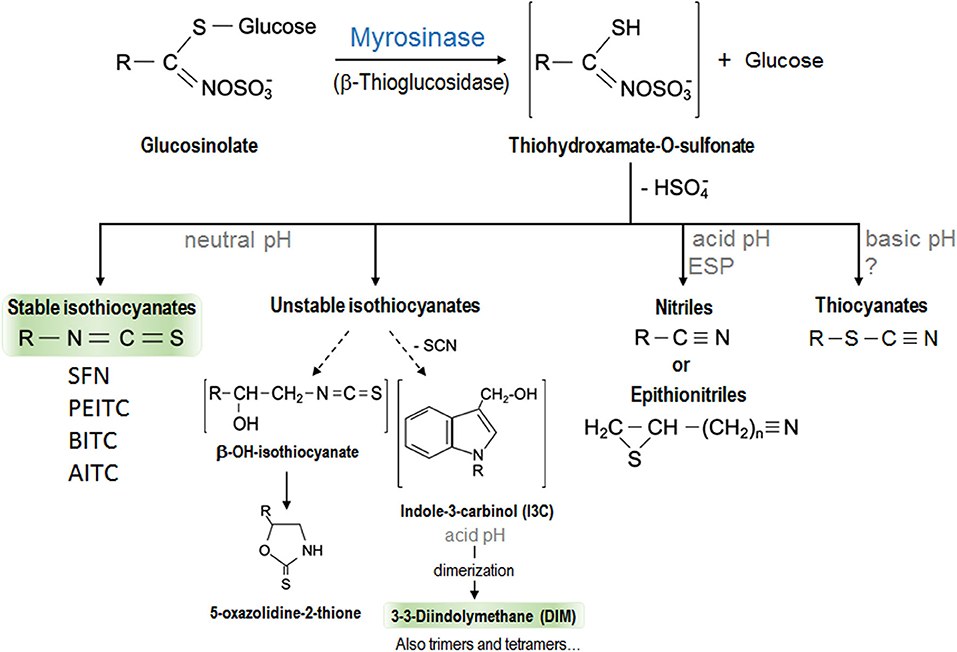
Figure 1. Scheme of the hydrolysis of glucosinolates by myrosinase enzyme with the resulted isothiocyanates and other derivatives. The biologically active hydrolysis products of glucosinolates, better studied and discussed in this review have been highlighted. EPS, epithiospecifer protein.
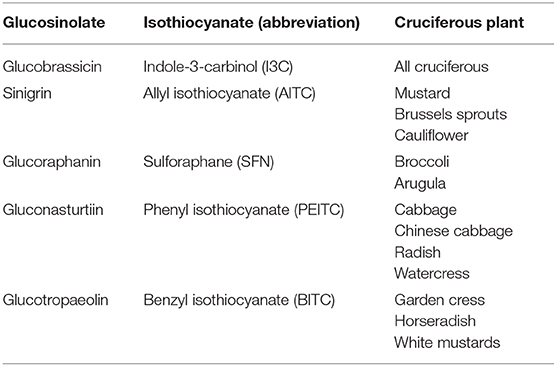
Table 1. The most abundant and studied GSLs, their ITCs derivatives and representative cruciferous-where found.
The content of GLSs varies widely depending on the cruciferous variety and is conditioned by the place and conditions of cultivation (12). Thus, it has been reported that cultivation under low temperatures decreased plant GLS levels; thus, plants harvested in the winter or autumn contain lower GLS levels than those harvested in the spring or summer (20, 21). In addition, the post-harvest and packaging conditions also modify the concentrations of GLSs in cruciferous vegetables (22). Rodrigues et al. (23) found a 79% reduction in the total GLS concentration in freshly harvested broccoli inflorescences stored at room temperature for 5 days, while those refrigerated at 4°C showed a 16% decrease. Therefore, a refrigerated mode of transport as well as the sale period could imply a significant loss of these compounds, which emphasizes the importance of consuming local vegetables. On the other hand, the cooking technique utilized may also cause more or less leakage of GLSs; however, the data about this are controversial as many variables need to be taken into account, such as whether the vegetable is chopped, the amount of water, cooking temperature, and duration of cooking (22, 24–26). Data emanating from diverse studies show that boiling leads to the largest loss of GLSs (by >50%), depending on the time and volume of water more than the temperature. During boiling, GLSs are mostly lost through leaching into the cooking water, and is favored by the convection movements of boiling water (27). The data led to the conclusion that boiling with greater amounts of water and for longer periods increases the losses of GLSs. Microwave cooking also leads to loss of GLSs although the increased osmotic pressure causes a breakdown of the cell structure which brings the GLSs and myrosinase into contact. The losses described are very variable (15–75%) and depend on factors such as the amount of water, the intensity of microwave radiation, and the duration of cooking (22, 25). Stir frying leads to an increased loss of GLSs (by >60%) (25, 28). In this case, temperature is the principal feature. It was observed that stir fried broccoli cooked for 3–5 min with oil preheated at 200°C did not lead to a significant change in the GLS content (27). However, cooking in preheated oil and maintenance at 140°C for 5 min led to losses of about 60% (25). Lastly, data showed that steaming was the best mode of cooking that ensured the preservation of GLSs. Most studies found little or no effect (26, 28), and some studies reported that an increase in GLS content (24, 28) was favored perhaps because steaming strongly limits their leach into the water.
There is evidence that very little of intact GLSs are absorbed (29–31) and it is their hydrolyzed products (the ITCs and indoles) that are absorbed (31). Chopping and chewing breaks the plant structure and promotes the hydrolyzation of GLSs to ITCs by myrosinase; however, the cooking temperature can inhibit the activity of myrosinase and limit the formation of ITCs (22). Therefore, it has been found that after ingestion of cruciferous vegetables, the myrosinase from gut microbiota has relevant influence in the conversion of inert GLSs to bioactive ITCs (32–36). Consequently, intact GLSs may reach the large intestine where they can be degraded by the resident gut microbiota leading to the release of ITCs which will then be absorbed (31, 32, 37, 38). Recently, it has been found that the consumption of broccoli modifies the gastrointestinal microbiota to a healthier profile (39), which coincides with that of bacterial genera that have myrosinase activity (33–35). Finally, ITCs must be absorbed and distributed by the body to reach the tissues where they will affect their biological mechanisms. The precise mechanism by which ITCs are absorbed has not been elucidated completely; however, numerous studies conducted using human (37, 40–42) and animal models (43–46) have described the mechanism through which ITCs are absorbed. Most studies conducted among humans have assessed absorption indirectly through the determination of their metabolites in urine or by the cyclo-condensation method (37), or by high performance liquid chromatography with tandem mass spectrometry (HPLC MS/MS) to measure plasma ITCs (42) after an oral dose. In studies conducted among humans after intake of a single dose of fresh broccoli, the peak plasma concentration of ITCs (mainly sulforaphane [SFN]) was found at about 3 h and disappeared at 24 h while the peak concentration in urine was recorded around 6 h and disappeared at 24 h (41, 42). Among rats, studies mostly assessed the fate of an oral dose of 14C-labeled ITCs. In general, rapid absorption of ITCs was observed but with some differences between individual compounds as the structure may affect the liposolubility (46, 47). For example, among animals given a 14C-phenethyl ITC (PEITC), the radioactivity in plasma peaked at 2.9 h while among those who received 14C-PHITC (phenylhexyl ITC), the peak plasma level was recorded at 8.9 h. Nevertheless, those that received 14C-PHITC excreted only about 7% in urine and 47% in feces in contrast to those that received 14C-PEITC who eliminated the majority in urine (about 89%) and only 10% was recovered in feces. These different absorption rates could perhaps be attributed to a greater lipophilicity of PHITC (46). All fractions of ITCs—those that are already present in previously consumed food, those that are formed during chewing, and those that result from microbiota activity—are absorbed by the epithelial cells of the small intestine or colon (31, 48). Available data indicate that the entry of ITCs into enterocytes through the apical membrane by passive diffusion, which is facilitated by rapid conjugation with reduced glutathione (GSH) by means of the enzyme glutathione-S-transferase (GST), results in the maintenance of the concentration gradient and favors rapid internal accumulation of GSH-ITCs (49, 50). GSH-ITCs are released through the basolateral membrane via multidrug-resistance-associated protein 1 (MRP1) and P-glycoprotein (P-gp) as occurs in other cell lines (51). In addition, some authors revealed that a proportion of absorbed ITCs and GSH conjugates (GSH-ITCs) effluxed back into the lumen as GSH-ITCs (49). Once absorbed, peak plasma concentrations of GSH-ITCs decline rapidly, signifying a rapid distribution (37, 52, 53). In blood, GSH-ITCs maintain an equilibrium with free ITCs due to the low concentrations of GSH in plasma and they are taken up by tissue through a similar mechanism within the enterocytes, passive diffusion, and formation of GSH-ITCs (49–51, 54). The capacity to synthesize GSH-ITCs is determined by the intracellular concentration of GSH and GST activity and could be related to the specific effects of ITCs in different tissues (31). Kim et al. confirmed that GSH-AITC accumulated to a greater extent in the liver within the first hour after an oral dose of AITC (25 mg/kg of body weight) followed by the kidneys, spleen, lungs, and heart (53). On the other hand, unstable ITC derivatives such as I3C (the major product of hydrolysis of indole-GLSs) in the acidic pH of the stomach are converted into condensed products (mainly, DIM [diindolylmethane] which is the most active) (55). De Kruif et al. in rats given 13C orally found DIM in tissues extracts from the stomach, small intestine and liver, proving that they are absolved by the small intestine (56). Anderton et al. determined the tissue distribution of DIM in mice after an oral load of I3C, and found that the liver retained higher levels (129 μg/g) of DIM, followed by the lungs (70.5 μg/g), kidneys (61.8 μg/g), heart (54.2 μg/g), and brain and plasma (19 μg/g). In addition, the peak plasma concentration of DIM occurred at 2 h after I3C administration (52, 57). In human studies, following I3C administration, the peak plasma concentration of DIM was detected after ~3 h (58); the lymphocyte activity of GST had increased (59) and urinary DIM was detected after ingestion of brussels sprouts (60).
After the initial conjugation with GSH catalyzed by the enzyme GST, the conjugates are metabolized to a mercapturic acid derivate (N-acetyl-cysteine-ITC [NAC-ITC]), which is excreted in the urine (53). Thus, the liver plays a relevant role in xenobiotic detoxification since it contains high levels of GSH and has the highest GST activity in the organism, and supports the formation and accumulation of high levels of GSH-ITC conjugates (52, 53). On the other hand, the kidney is the major organ implicated in the conversion of GSH-ITCs to the corresponding mercapturic acid derivate since it has elevated N-acetyltransferase activity (NAT) (61). Thus, it appears from experimental data (62) that the GSH-ITCs initially formed in the liver would be secreted as such and those formed in the kidney would be acetylated and excreted. In support of this, Kim et al. found that the level of GSH-AITC was higher in the liver but the level of NAC-AITC was more elevated in the kidney (53). In addition, the GSH-ITCs in the liver can also be metabolized to NAC-ITC and then excreted into the bile (62). The enterohepatic cycling was supported by evidence from human data that the excretion of dihydrocarbonates (such as GSH-ITCs) showed a biphasic curve after ingestion of horseradish (29). Data show that the specific structure of ITCs could affect their affinity to the enzyme GST, act as a phase I enzyme substrate, and be secreted into the bile and excreted in feces. For example, phenyl-, benzyl-, and allyl-ITCs such as PEITC are principally conjugated with GSH and metabolized via the mercapturic acid pathway to NAC-PEITC and excreted in urine. In contrast, a majority of PHITC (with an alkyl side chain) is excreted in the bile and recuperated in feces (46, 62, 63). Thus, phase I metabolism could contribute to the biotransformation and bioavailability of active ITCs. Figure 2 shows a schematic representation of the factors influencing the amount of GLSs ingested, their absorption, distribution, metabolism, and excretion.
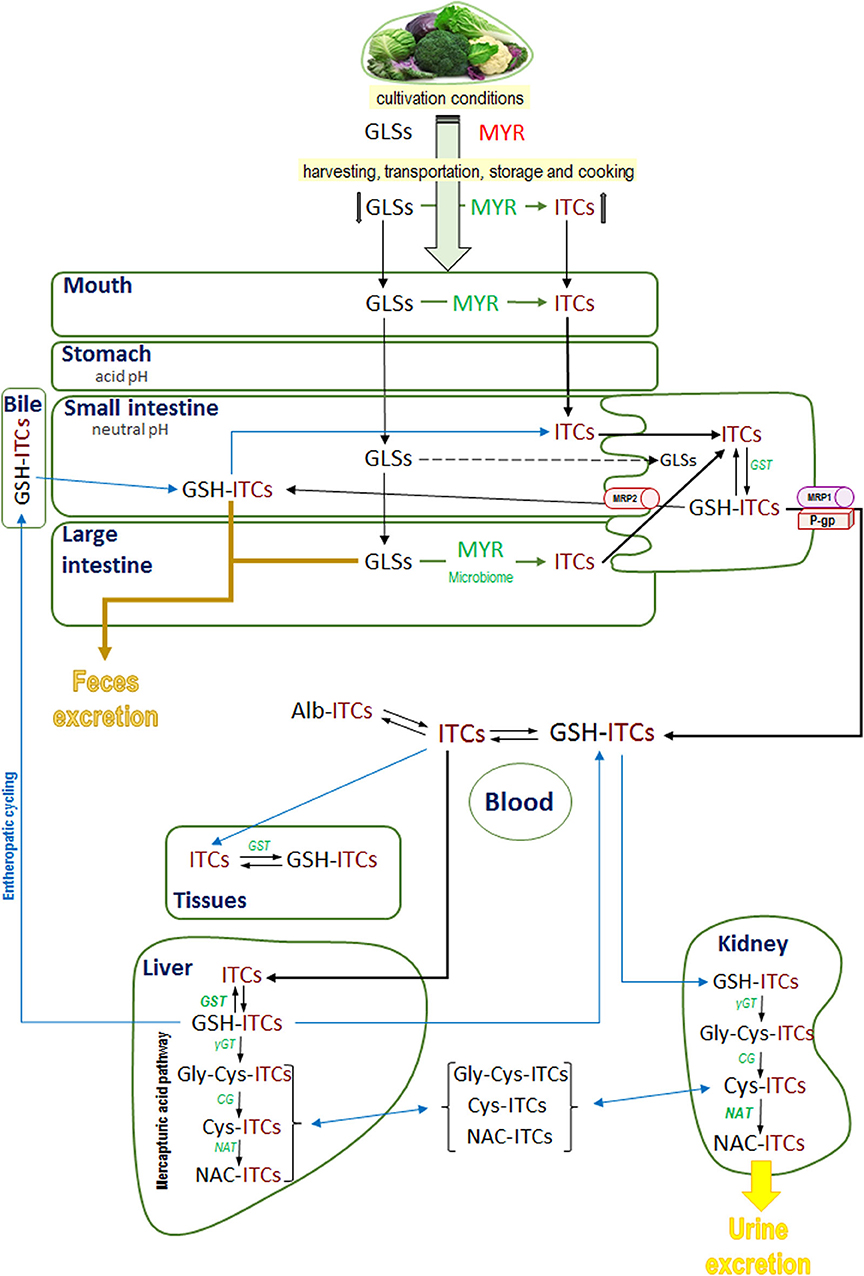
Figure 2. General overview of ingested glucosinolates and isothiocyanates fate: mouth hydrolysis, microbiota hydrolysis, absorption, metabolism, and excretion. GLSs, glucosinolates; ITCs, isothiocyanates; MYR, myrosinase; GSH-ITCs, glutathione isothiocyanates conjugates; Alb-ITCs, albumin isothiocyanates conjugates; Gly-Cys-ITCs, glycine-cysteine isothiocyanates conjugates; Cys-ITCs, cysteine isothiocyanates conjugates; NAC-ITC, N-acetylcysteine isothiocyanates conjugates; GST, glutathione-S-transferase; γGT, γglutamyltranspeptidase; CG, cysteinylglycinase; NAT, N-acetyltransferase.
As mentioned above, numerous epidemiological studies that reported a lower risk of suffering various types of cancers when cruciferous vegetables are consumed in the diet in a regular form have been published over the years. From these observations, abundant efforts have been made to understand the mechanisms through which hydrolyzed products derived from GLSs reduce the risk of cancer with the aim of arriving at potential cancer therapies. The most studied products of hydrolysis of GLSs, due to the abundance of their precursors in consumed cruciferous vegetables, have been the stable ITCs PEITC, SFN, BITC, AITC, and the unstable ITC derivatives (the indoles I3C/DIM). This section of the review focuses on the biological effects of ITCs and indoles that have been described in the regulation of key cell signaling pathways. These effects have been firstly and widely studied in relation to cancer. However, these pathways are not only altered in cancer but also in other conditions such as obesity, diabetes, and arteriosclerosis (MetS). This is why most of the cited studies will refer to cancer (though not all) and we reserve the final section of the review to focus on how ITCs and indoles can prevent MetS by modulating the common pathways described in cancer. It has been found that ITCs and indoles modulate the activity of enzymes involved in phases I and II of the detoxification pathway; modulate the cell cycle, oxidative stress, and inflammation; angiogenesis; as well as epigenetic effects.
Phases I and II of the detoxification pathway contain the most important enzyme group involved in xenobiotic biotransformation (64). The phase I enzymes are represented by the cytochrome P450 family (CYP450) that modify the molecule via oxidation, reduction, or hydration and, as a result, the xenobiotic (e.g., drugs, procarcinogens) may be activated or inactivated. The phase I metabolism generally increases the polarity of molecules and facilitates the excretion. Phase I reactions can be followed by phase II reactions to increase solubility, but phase I reactions are not a requirement to be substrate of Phase II enzymes. Phase II consists of a sulfation, glucuronidation, acylation, methylation or conjugation with GSH or amino acids, facilitating its excretion in urine or bile (64). However, some potential carcinogenic molecules become active when they are biotransformed through phase I enzymes (65). Inhibition of specific CYP enzymes involved in carcinogen activation inhibited cancer development in animal models (65, 66).
Stable ITCs (such as SFN, AITC, PEITC, and BITC), have been found to inhibit carcinogen activation through the inhibition of CYP enzymes in animal studies and cell lines (67–70). Leclercq et al. found that in human volunteers, a single ingestion of watercress homogenate (rich in the GLS precursor of PEITC) increased the area under the curve of time course plasma chlorzoxazone, suggesting that CYP2E1 activity was inhibited (71). Yoshigae et al. finally described a possible mechanism underlying the inactivation of human CYP2E1 by PEITC (72). Later, Nakajima et al., in an in vitro model of cells expressing specific human CYP isoforms, proved that PEITC yielded chemopreventive effects against nitrosamine-induced carcinogenesis through CYP450 inhibition (67). On the other hand, indole-GLS derivatives (such as I3C) and their condensed products (such as DIM) have a different effect than stable ITCs since they increase the activity of certain CYP enzymes (68, 69, 73). The mechanism described would be that DIM binds to the aryl hydrocarbon receptor, which then recognizes and binds to the xenobiotic response element (XRE) sequences in the DNA of genes of a large number of CYP enzymes, and increases their expression (74, 75). Increased expression of phase I enzymes in general could be considered beneficial to the elimination of the possible adverse effects of activated procarcinogenic agents. In breast cancer (characterized by altered CYP expression) I3C/DIM demonstrated a protective effect (73). In some breast cancer cell lines, CYP1A1 and CY1A2 are expressed at low levels; they are responsible for the production of estrogen metabolites that are protective against cancer, and it has been found that DIM increases their expression, favoring a protective role (76). Therefore, in a phase I clinical trial of healthy women, it was observed that the administration of I3C promotes CYP1A2 activity (59).
Numerous stable ITCs and indole derivatives of GLSs are potent inducers of phase II enzymes (49, 64, 77–79). The genes that encode phase II enzymes contain antioxidant response element (ARE) domains; ITCs recognize and bind to them, increasing their expression (68). A study conducted among smokers showed that watercress consumption promotes a higher excretion of glucuronidated nicotine in urine indicating activation of the phase II enzyme UDP-glucuronosyltransferase (UGT) (80). Most recently, Yuan et al., in a randomized, placebo-controlled, double-blind study conducted among smokers found a greater excretion of the mercapturic acids of benzene and acrolein (substances of tobacco smoke) in urine after oral administration of PEITC for 5 days (81). In addition, human polymorphisms for the GST gene that affect their activity have been described (82). Lower or null GST activity could affect the susceptibility to diseases due to reduced excretion of toxic molecules or could promote increased efficiency of cancer chemotherapy owing to a higher drug half-life (83). In the same sense, lower or null GST activity could result in a slower elimination and longer exposure to ITCs in cruciferous vegetables, maintaining their biological actions for a longer time. Several epidemiological studies found a higher cancer protective effect of cruciferous vegetable consumption among individuals carrying GSTM1-null and GSTT1-null than among those with normal GST activity (84–87). In the same vein, Yuan et al. (81) determined urine excretion of mercapturic acids of benzene and acrolein in cigarette smokers who received oral supplementation of PEITC, and observed a remarkably stronger effect of PEITC in subjects with GSTM1 and GSTT1-null genotype with a significant higher excretion than in those with a normal genotype (88). In Hep G2-C8 cells, Saw et al. described that I3C/DIM induced the expression of ARE-mediated phase II genes such as GSTM2, UGT1A1, and NQO1(NAD(P)H:quinone oxidoreductase 1, as well as a synergic effect with SFN and PEITC (89). In summary, the main mechanism of carcinogenesis inhibition described for ITCs appears to occur through two levels of the detoxification pathway: selective P450 enzyme inhibition (stable ITCs) or activation (I3C/DIM) and an induction of phase II enzymes, preventing the activation of procarcinogens and increasing their excretion (Figure 3). Although these activities are attributed exclusively to ITCs, an in vitro study described that intact GLSs modulate hepatic CYP450 and phase II enzymes (78).
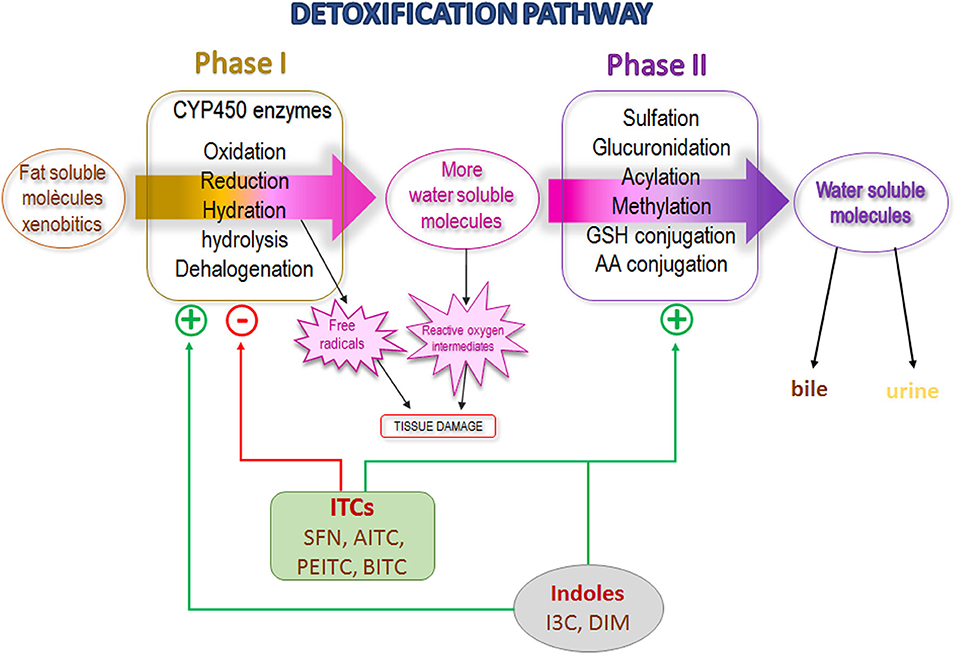
Figure 3. Detoxification pathway scheme showing isothiocyanates and indoles effect. SFN, Sulforaphane; AITC, Allyl isothiocyanate; PEITC, Phenyl isothiocyanate; BITC, Benzyl isothiocyanate; I3C, Indole-3-carbinol; DIM, diindolylmethane.
The cell cycle comprises a series of tightly regulated stages that include growth and division to eventually result in two new daughter cells. Multiple checkpoints are implicated in the regulation of the cell cycle such as growth signals, availability of nutrients, and the integrity of DNA. If the DNA is damaged, the cell cycle can be transiently arrested to repair it or the apoptosis pathway can be activated, leading to cell death. When cell cycle regulation is disturbed, the DNA is not repaired correctly and the mutation can propagate, contributing to the development of cancer (19). The effects of ITCs/I3C on cell cycle regulation have been studied in a wide variety of cancer cell lines and in animal models (55, 69, 90, 91). Numerous in vitro experiments in an extensive variety of cancer cell line models of the bladder, lung, prostate, osteosarcoma, adenocarcinoma, and colon (UM-UC3, J82, RT4, LTEP-A2, PC-3, LM8, and HT-29) have investigated the effects of SFN on cell cycle regulation. The results showed an extensive coincidence in that SFN arrests the cell cycle in the G2/M phase (92–100). Some of these studies also showed that its effect was mediated through induction of the expression of p21 and p53, which are cyclin-dependent kinase (CDK) inhibitors (94–96). Cyclins and CDK are important promoters of mitosis initiation as a consequence of cellular proliferation. In a manner similar to SFN, PEITC arrests the cell cycle in the G2/M phase in HeLa, Caco-2, and PC-3 cell lines (101–103), but in the G0/G1 phase in human prostate cancer DU-14 and LNCaP cell lines (104). In most of these studies, an increase in expression of p21 and a decrease in expression of the cyclins CDK and cdc25c was found (102, 103). Other studies regarding the effects of BITC and AITC on cell proliferation also showed their ability to arrest the cell cycle generally in G2/M by increasing p21 and/or p53 expression and decreasing the expressions of CDK and cyclins (cdk1, cyclin B) and cdc factors (cdc2, cdc25b, cdc25c) (93, 101, 105–111). The effects on cell proliferation have also been described for I3C/DIM; in MCF7 breast cancer cell lines, they arrest the cell cycle in G1 (112–114). Moreover, I3C/DIM increase the expressions of p53 (114), p21, and p27 and decrease that of CDK6 (112). Similar effects were observed in PC-3 prostate cancer cells treated with I3C (115).
In addition, the mechanisms through which ITCs inhibit the proliferation of cancer cells include the activation of apoptosis, as well as both the mitochondrial/intrinsic pathway and death receptor/extrinsic pathway (91). All the most studied ITCs (such as PEITC, SNF, BITC, AITC, and I3C/DIM) have been found to cause inhibition of the antiapoptotic factor Bcl-2 in different cell lines and mice (93, 94, 110, 116, 117). However, different mechanisms through which ITCs activate apoptosis have been described. Hu et al. (118) described that PEITC induces apoptosis via the intrinsic pathway through MAPK signaling, via activation of JNK through p38 that leads to cytochrome c release, and subsequent activation of caspase-9 and caspase-3 in HT-29 cells. In another study, Wang et al. (119) showed that induction of apoptosis by PEITC in PC-3 cells is mediated by ERK. Later, Xu et al. (120) demonstrated that ITC-induced apoptosis (by SFN, PEITC, and AITC) is tightly coupled to ERK and JNK signaling in human prostate PC-3 cells. Furthermore, in the bladder cancer cell lines HTB9 and RT112, Islam et al. (121) found that SFN inhibits proliferation of cells by decreasing the expressions of PI3K/Akt/mTOR. Other studies of 5637 and T24 human bladder cancer cells showed that treatment with SFN induces mitochondrial caspase dependent apoptosis due to reactive oxygen species (ROS) accumulation and activation of endoplasmic reticulum stress with activation of Nrf2 (nuclear erythroid 2-related factor 2) signaling pathway (121, 122). Other researchers have confirmed that the increase in ROS levels is a way by which BITC and PEITC induce apoptosis in cell lines of pancreatic cancer, chronic myeloid leukemia, and chronic lymphocytic leukemia (123–125). Nrf2 is a transcriptional factor that binds to ARE regions in DNA to induce the expression of major antioxidant enzymes that detoxify ROS (126, 127) and can inhibit the nuclear factor kappa B (NF-κB) transcriptional factor. NF-κB activity is linked to cellular processes that promote cancer, such as inflammation, cell proliferation, and angiogenesis and facilitate tumor growth and metastasis (128, 129); thus, its inhibition could yield positive effects in the prevention or treatment of cancer. The evidences that SFN and BITC decrease NF-κB activity have been described in the bladder and colon cancer cell lines T24 and T29, respectively (130, 131). Chen et al. showed that DIM also promotes apoptosis through AMPK activation, resulting in suppression of mTOR in C4-2B and LNCaP prostate cancer cells (132). In addition, in PC-3 prostate cancer cells, it was found that I3C/DIM induces apoptosis through inhibition of Akt/PI3K signaling with decreases in the expressions of Bcl-xL and BAD (133). Thus, I3C/DIM induce apoptosis via modulation of the PI3K/Akt/mTOR/NF-κB pathway (115, 134). Finally, it has been reported that ITCs and indoles play an important role in controlling the cell cycle, diminishing the proliferative capacity, and activating the apoptosis pathway; however, the specific underlying mechanism could differ according to the ITC or cell type.
Usually, oxidative stress is accompanied by an inflammatory response that is part of the responses to combat this oxidative damage. However, persistence of an oxidative stimulus (even if mild) will also lead to the maintenance of a chronic inflammatory state that can be more or less severe. Therefore, the organism fights this oxidative damage first to eliminate the causative agent and then to eliminate the inflammation to restore normalcy (135). As mentioned above, the genes that encode proteins relevant to the elimination of oxidative agents contain ARE sequences, as do phase II enzymes and stress-responsive antioxidant genes, such as GST, UGT, NAD(P)H:quinone oxidoreductase 1 (NQO1) and heme oxygenase-1 (HO-1) (136, 137). The central role of Nrf2 transcription factor in the upregulation of antioxidant genes in the oxidative stress response has been described (138). NF-κB plays a key role in immune response regulation to activate mediators (such as pro-inflammatory cytokines) to favor the attack of the inciting agent in the first step of the immune response, in which it also produces oxidation agents (135). However, in most normal cells NF-κB is in inactive form, and a constitutive activation of NF-κB has been noted in almost all cancers (128) and chronic physiological inflammatory conditions such as MetS (139). It has been reported that ITCs and indoles increase Nrf2 activity and inhibit NF-κB (140–145). In Raw 264.7 macrophage cells, it was found that BITC diminished the lipopolysaccharide (LPS) induced inflammatory response in a dose-dependent manner through the inhibition of NF-κB translocation to the nucleus as well as that of its target genes Ilβ1, IL6, TNFα, nitric oxide synthase (iNOS), and cyclooxygenase-2 (COX2) (145). In mice, topical application of BITC also ameliorated the TPA-induced swelling in the ears (model for inflammation diseases, including psoriasis) diminishing the expressions of iNOS and COX2 (145). The authors concluded that the anti-inflammatory effects of BITC could be mediated via the inhibition of Akt and ERK and the subsequent downregulation of NF-κB signaling. Zhou et al. recently confirmed that AITC attenuated oxidative stress in mice with chronic obstructive pulmonary disease via Nrf2 signaling (140). In a traumatic brain injury model of mice, AITC reduced infarct volume and brain swelling by decreasing the levels of pro-inflammatory molecules, IL1β, IL6 through the downregulation of NF-κB, and ameliorated some neuronal plasticity markers and increased the antioxidative mechanism by upregulating the Nrf2 pathway, evidenced by the double effect of AITC (141). Similar results have been described for PEITC, BITC, and SNF, in that they reduce the endothelial damage induced by oxidized LDL (146) or in NIH3T3 fibroblasts (142). Kim et al. described the protective effect against inflammation induced by DIM, but not for I3C, in LPS-treated BV-2 microglial cells by decreasing the expressions of iNOS, COX2, and NF-κB (147). In addition, DIM reduces the hippocampus inflammation caused by LPS administration in mice, thereby reducing the infiltration of pro-inflammatory macrophages (147). In mice, it has been found that DIM also attenuates colonic inflammation and tumorigenicity in a mouse colitis model and an azoxymethane (AOM)/DSS induced colon cancer model (148).
In the stages of cancer development, progression refers to the capacity for tumors to extend to other parts of the body (also referred to as metastasis). Angiogenesis denotes the construction of new blood vessels from existing ones to facilitate the delivery of nutrients and oxygen to tumor cells. Therefore, angiogenesis plays a fundamental role in tumor progression, and thus metastasis, which constitutes the most difficult phase for cancer control and treatment and is normally the cause of death (149–151). Metastasis is a complex process whereby cells lose their adhesive capacity and migrate through the circulation to other tissues. Thus, the new vascular network facilitates migration of cells; however, extracellular matrix degradation is an essential aspect of the loss of cell adhesion capacity. In this process, enzymes called metalloproteinases (MMPs) are important. There are pieces of evidence that SNF, PEITC, BITC, AITC, PITC, and I3C downregulate MMP2 and MMP9 in various cell lines (152–154). The angiogenic process is regulated by numerous molecules, in addition to MMPs, such as growth factors (vascular endothelial growth factor [VEGF], bFGF, angiopoietins, EGF, and TGF), integrins, and interleukins (IL1, IL2, IL6, IL8, IL12, and IL17) (149). The most studied among them is VEGF and there is evidence that ITCs and I3C inhibit angiogenesis by downregulating VEGF (152, 155). Treatment of HUVEC and PC3 cells with PEITC inhibited neovascularization and cell migration with suppression of VEGF secretion, downregulation of VEGF receptor 2 protein levels, and inactivation of Akt (156). Treatment of human microvascular endothelial cells (HMEC-1) with SFN decreases the formation of new microcapillaries with reduction in VEGF levels (152). This was also observed later by Kim et al. in HCT116 human colon cancer cells (157). Thejass et al. showed that AITC and PEITC treatment downregulated VEGF and pro-inflammatory IL1β, IL6, and TNFα, but increased the anti-inflammatory effects of IL2 and inhibition of metalloproteinase (TIMP1) (158). Among mice, Hajra et al. reported that I3C blocked angiogenesis by inhibiting VEGF-A and MMP-9 (155). One important factor that stimulates angiogenesis is hypoxia that results from tumor growth or due to other situations such as increase in adipose tissue caused by obesity, leading to the activation of hypoxia inducible factor-1α (HIF-1α) which in turn stimulates VEGF (152). In accordance with this, it was found that SFN dramatically decreases the expressions of HIF-1α and VEGF in HCT116 cells exposed to hypoxia (157). Similar results were found in MCF-7 cancer cells treated with PEITC (159, 160). PC-3 and C4-2B prostate cancer cells treated with DIM demonstrated decreases in HIF-1α and NF-κB expression and an increase in radiation efficacy (161). Gupta et al. described that in human glioma cells the inhibitory effect of PEITC on angiogenesis depended on PI3K/Akt and ERK/MAPK (160). In addition, it is known that IL1β upregulates HIF-1α through a classical inflammatory signaling pathway involving NF-κB and COX-2, ending in upregulation of VEGF. Thus, HIF-1α is recognized as a pivotal transcription factor linking the inflammatory and oncogenic pathways (127). The inhibition of NF-κB appears to be important in reducing cell proliferation, inflammatory status, and metastasis, and as discussed previously, there is abundant evidence that ITCs/indoles downregulate NF-κB activity (144).
The main mechanisms underlying epigenetic processes are post-translational histone modifications and DNA methylation, which are modulated by histone deacetylases (HDACs) and DNMTs (methyltranspherases), respectively. Histone acetylation results in an open chromatin conformation which facilitates the transcription process. HDACs remove the acetyl group from histones, leading to chromatin condensation which hinders transcription. The overexpression of HDACs are a common hallmark of cancer and their inhibition is considered to be therapeutic against cancer tumor progression (162, 163). ITCs and indoles have been found to exert a potent effect as HDAC inhibitors in various cancer lines (164–174). The most studied is SFN and the most common effect found is the inhibition of HDAC3 associated with an increase in p21and Bax expression and a decrease in cyclin D1 expression, which together results in cell cycle arrest and apoptosis activation (167, 168, 171, 173). In addition, in TPA-induced neoplastic transformation of mouse skin cells, Su et al. described that SFN promotes a decrease in HDAC expression with an increase in Nrf2 expression and their downstream target genes (such as HO-1 and NQO1) of phase II enzymes (169). PEITC in LNCaP prostate cancer cells inhibits HDAC3 with an increase in p21and p27 expression, leading to cell arrest (170). In another experiment, the inhibition of HDAC1, 2, 4, and 6 protein expression was observed and related to activation of the tumor suppressor factor RASSF1A (Ras-association domain family 1 isoform A) (175). In addition, Beaver et al. showed that DIM significantly decreased HDAC activity and was correlated with increased expression of p21 in PC-3 and LNCaP prostate cancer cells (166). In human pancreatic cancer cells, Sanjay et al. showed that BITC incubation promotes a decrease in the expression and activity of HDAC1 and HDAC3 with decreases in the expression and activity of cyclin D1 and NF-κB (172). In accordance, I3C and DIM decrease the expression of HDAC1, 2, 3, and 8 in T cells activated by staphylococcal enterotoxin B with a decreased secretion of pro-inflammatory cytokines (174). DNA methylation is an important process by which gene transcription is regulated (176). Changes in the pattern of methylation of genes have been related to the expression of oncogenes and cancer development (176). It has also been described that ITCs and indoles regulate DNMTs and mediate the process of carcinogenesis (175, 177–184). SFN is the ITC that has been studied the most, and in general, the research shows that SFN promotes a decrease in the DNA methylation of certain genes with anti-tumor action through the inhibition of DNMT1, DNMT3A, and DNMT3B. This leads to decreased expression of HDACs resulting in reactivation of genes [such as p21 (177), cyclin D2 (179) or Nrf2] and the downregulation of HO-1 and NQO1 (169, 181) depending on the nature of the study and the type of cell utilized. Similar results were obtained in DIM-treated TRAMP-C1 cells assessed by Wu et al., in which DNMT expression diminished and the methylation status of Nrf2 was reversed, resulting in its enhanced expression (184). Wong et al. described that in normal prostate cell (PeRC) and cancer cell lines (LNCaP and PC3), SFN and DIM altered promoter methylation in distinct sets of genes with similar targets within a single cell line. Further, they showed that SFN and DIM reversed many of the cancer-associated methylation alterations (178). PEITC in LNCaP cell lines also showed a reduction in the activity of RASSF1A promoter methylation through inhibition of DNMT3A and 3B (175). In leukemia T cells, the effects of PHITC were an inhibition of DNMT1 and DNMT3B, an increase in acetylation of histones 3 and 4, reversal of hypermethylation of the tumor suppressor gene p15, and reactivation of its transcription (180).
miRNAs are non-coding RNAs that are most abundant in animal cells. They have a length of 20–24 nucleotides and they play a pivotal role in regulatory pathways including differentiation, proliferation, and apoptosis, and regulate approximately one third of human genes (185, 186). During carcinogenesis, various miRNAs are deregulated (186). There is evidence that ITCs and indoles exert some of their effects by modulating specific miRNAs (187). It has been found that SFN in epithelial cancer cells (NCM460 and NC356) modulated 18 miRNAs (15 upregulated and 3 downregulated). Among the upregulated genes were miR-23b and miR-27b which are tumor suppressors, while the pro-oncogenic miR-155 was downregulated (188). PEITC in PC3 prostate cancer cells causes overexpression of miR-194 which decreases the expression of MMPs and hindering metastasis (189). I3C increases miR-34a expression in MCF-7 human breast cancer cells using p53 as a direct transcriptional target (190). In rats exposed to environmental cigarette smoke, I3C and PEITC showed a synergic effect by restoring the lung miRNA profile (191). In general, there is an increase in expression of those miRNAs related to decreased angiogenesis and proliferation or increased apoptosis and a decrease in expression of those miRNA implicated in the opposite direction (187, 192–195). Figure 4 summarizes the overall effects described for ITCs and indoles in the regulation of cell proliferation, apoptosis, inflammation, and angiogenesis.
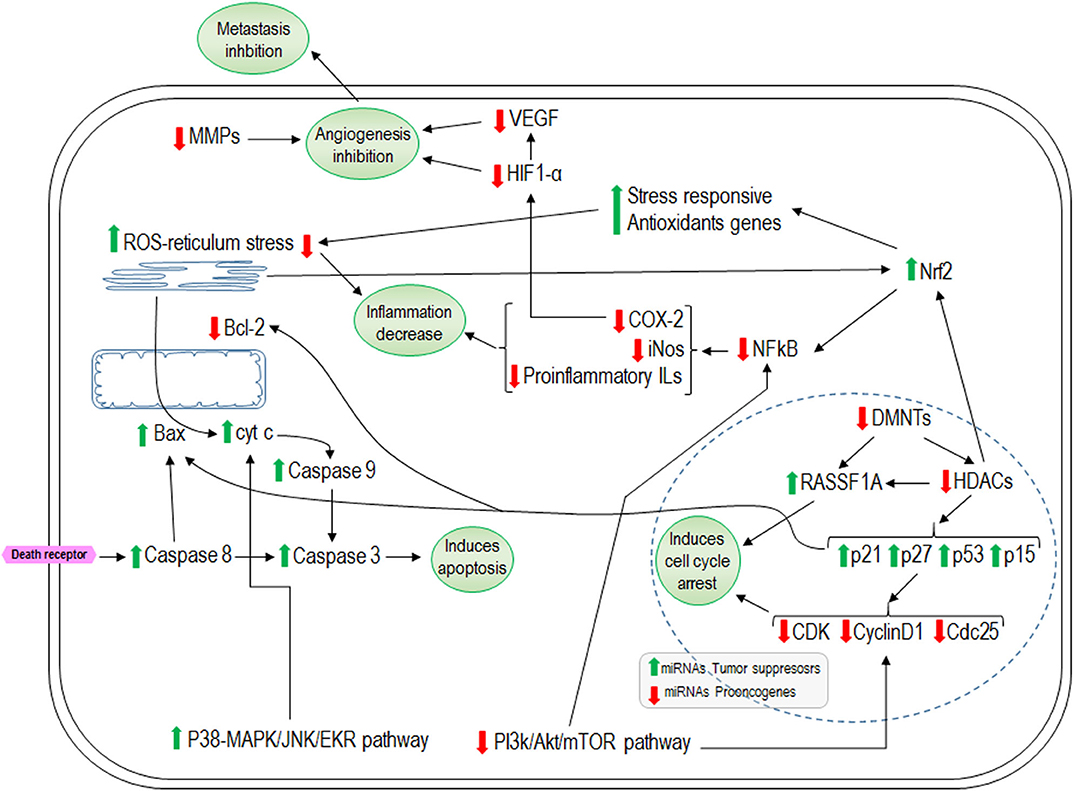
Figure 4. Representative effects of isothiocyanates on cellular cycle arrest, apoptosis, inflammation, and angiogenesis. Green arrows indicate the pathways that isothiocyanates increase and the red arrows the inhibited ones. HDACs, histone deacetylases; DNMTs, methyltranspherases MMPs; RASSF1A, Ras-association domain family 1 isoform A; ROS, reactive oxygen species; COX-2, ciclooxigenasa 2; HIF-1α, hypoxia inducible factor-1α; VEGF, Vascular Endothelial Growth Factor.
Obesity is characterized by excess body weight due to fat excess (196). The expansibility of white adipose tissue (WAT) in obesity entails deregulation between cellular proliferation and apoptosis, pro-inflammatory macrophage infiltration with increased pro-inflammatory cytokine secretion, and stimulation of angiogenesis. Obesity is frequently associated with other pathologies, mainly type 2 diabetes (insulin resistance), atherosclerosis (dyslipidemia), and hypertension. This cluster of associated pathologies is known as the MetS (197) and is currently a leading cause of death worldwide (198).
Several health organizations have provided definitions for MetS. The United Kingdom National Health Service (199) defines MetS as “… the medical term for a combination of diabetes, high blood pressure (hypertension), and obesity” and adds that “…it increases the risk of coronary heart disease, stroke, and other conditions that affect the blood vessels.” The International Diabetes Federation (IDF) (200), the American Heart Association (201) and the National Heart, Lung and Blood Institute (202) define MetS as a group of risk factors for heart attack or stroke. Specifically, the IDF posits that MetS “… is a cluster of the most dangerous heart attack risk factors: diabetes and raised fasting plasma glucose, abdominal obesity, high cholesterol, and high blood pressure.” According to data from the World Health Organization (2016), the leading cause of death worldwide was ischemic heart disease, followed by stroke and chronic obstructive pulmonary disease (198) all of which are related to MetS. Scientists have agreed on a set of criteria for the diagnosis of MetS. According to the IDF, for a person to be diagnosed with MetS, they must have: central obesity (defined according to the waist circumference with values specific to the ethnicity) (203) plus any two of the following four factors: raised triglyceride levels, reduced high density lipoprotein cholesterol level, raised blood pressure, and increased fasting plasma glucose concentration (200).
Obesity, the condition central to MetS, is characterized by an excess body weight due to fat excess (196). Today, obesity is defined as a mild but chronic inflammatory state (204). Animal studies have shown that the sequence of obesity establishment resembles the stages of the typical inflammatory response, only that it does not resolve in this case (205–207). In mice fed a high fat diet (HFD), it has been observed that in the first week, there already exists an increased infiltration of neutrophils in WAT (206). Then, around 5 weeks lymphocyte infiltration is observed (207) and finally after 16 weeks, when obesity is well-established, macrophages become the main infiltrating cells (206, 207). The lymphocyte and macrophage infiltrates in WAT of obese individuals (mice and humans) are mainly pro-inflammatory, while those in the WAT of lean individuals are anti-inflammatory (207–210). Thus, this inflammation has been interpreted as a response of the organism to an insult, which in this case is nutritional energetic overload, and whereby the stimulus persists and inflammation becomes chronic (211, 212). In addition, the diets that are usually consumed in middle- and high-income countries, where there is higher prevalence of obesity, are often unbalanced with very low fruit and vegetable consumption and with excessive meats and ultra-processed products that impose a burden of saturated fats and refined sugars (196, 213–216). Thus, the disturbed energy balance in favor of fat accumulation, that is obesity, leads to a WAT growth with hypertrophy and death of adipocytes promoting macrophage infiltration to phagocytize the remains; these in turn produce pro-inflammatory cytokines that favor further infiltration (217, 218). Saturated fatty acids directly stimulate differentiation to a pro-inflammatory macrophage profile while monounsaturated and polyunsaturated fatty acids promote an anti-inflammatory profile (219). Thus, if the stimulus persists, that is, if the diet is still rich in saturated fatty acids and energy, a vicious circle is established with continuous infiltration of pro-inflammatory macrophages, which maintains the inflammation chronically (220). In addition, while eicosanoids derived from ω-6 polyunsaturated fatty acids, prostaglandins, and leukotrienes, facilitate the initiation of the inflammatory response, the eicosanoids and docosahexanoids derived from ω-3 polyunsaturated fatty acids, resolvins, and protectins, are essential to finishing it (139, 221). It has been found that the diets of obese individuals are usually deficient in ω-3 fatty acids but not in ω-6 fatty acids, and this could contribute to maintaining the chronic inflammation (205, 221, 222). Besides, the hypertrophy of WAT also elicits tissue hypoxia, which favors inflammation and angiogenesis that are needed to expand WAT (209, 223). Therefore, to enable the adipocytes to hypertrophy, the MMPs must break down the extracellular matrix. Accordingly, in a mouse MMP14 null model, researchers observed a reduction in adipose tissue mass and smaller adipocytes (224). Moreover, in obese mice fed an HFD, increased HIF-1α and VEGF levels have been found, and HIF-1α overexpression was causally implicated in obesity-induced insulin resistance (221). Related to inflammation, the NF-κB signaling pathway is also activated in obesity and is responsible for the production of pro-inflammatory cytokines such as TNF-α that induce insulin resistance (139).
Type 2 diabetes is characterized by insulin resistance and is prevalent among people older than 40 years and normally associated with obesity. However, type 2 diabetes has been increasingly described among children and adolescents, related to the change in lifestyle and nutrition (222, 225, 226). Insulin resistance could be promoted through WAT inflammation generated in obesity and liver lipid accumulation (197), which is associated with nutritional energetic overload (225, 227). In addition, the diabetic state also increases liver inflammation with a pro-inflammatory profile of Kupffer cells and the secretion of pro-inflammatory cytokines that aggravate insulin resistance (228). Elevated circulating glucose levels interact with proteins and lipids which are glycated. The process occurs through the Maillard reaction, a complex sequence of chain reactions that leads to the formation of stable advanced glycation end products (AGEs) which are oxidants and can damage tissues (229, 230). AGEs may produce ROS, bind to specific cell surface receptors (RAGE) and contribute to an inflammatory state in diabetes. The interaction between AGE and endothelial RAGE leads to NF-κB upregulation, which transcribes its target genes such as vascular cell adhesion molecule-1 (VCAM-1), intercellular adhesion molecule-1 (ICAM-1), and VEGF; pro-inflammatory cytokines as IL1α, IL6, and TNFα; and RAGE itself (229, 231). The insulin resistance present in diabetes also promotes an increase in the level of circulating lipids (232) due to slow VLDL emptying since the level of lipoprotein lipase is diminished. In addition, there is reduced LDL clearance; consequently, their circulation time is increased, leading to hyperlipidemia and increasing their susceptibility to oxidative modifications (233). In LDL samples obtained from diabetic individuals, there have been observed significantly elevated levels of both apoB and lipid linked to AGE (233). Besides, it was found that macrophages take up the glycated LDL to a greater extent than native LDL, resulting in the formation of foam cells as is characteristic of the early atherosclerotic lesion (234). Thus, the formation of AGE in a hyperglycemic environment contributes to the vascular pathophysiology, promoting the development of atherosclerosis in diabetes (235). The atherosclerotic process also involves an inflammatory process, in which NF-κB plays a major role, contributing to the overall incidence of inflammation in MetS (236).
In addition to comorbidities that exist among obesity, type 2 diabetes, and atherosclerosis, obesity is associated with a higher risk of certain cancers, such as those of the pancreas, liver, colon and rectum, kidney, endometrium, and postmenopausal breast (237). Another condition that also occurs as a comorbidity to MetS is Alzheimer's disease [characterized by chronic brain inflammation in which diet is the risk factor (238–240) and represents the third leading cause of death in the highest income countries] (198).
Diverse studies have put in evidence regarding the relationship between alimentary habits and obesity prevalence and, in turn, their comorbidities in various countries such as Brazil, Canada, United States, Australia, and various European countries (241–246). These studies showed a trend in the last decades toward a decrease in the consumption of fresh food (mainly vegetables and fruits) in favor of ultra-processed foods (247) and how this change correlates with the increase in obesity prevalence worldwide. Other studies have correlated the consumption of ultra-processed foods with the increase in sugars, saturated and trans-fat, and Na intake, that in addition, correlate with an associated higher energy intake and obesity prevalence (248–251). In relation to the increase in cancer incidence associated with MetS, Fiolet et al. found that a 10% increase in the proportion of ultra-processed foods in the diet was associated with a significant increase (>10%) in the risk of overall cancer and more specifically of postmenopausal breast cancer, the most commonly related to obesity (237). In summary, all the scientific and epidemiological data lead us to a common point. That is, when diet is unbalanced with energy excess due to high intake of sugar and saturated fat, and with a shortage of fruit and vegetables, the MetS and comorbidities occur.
The effects of ITCs and indoles on MetS components have also been studied (252–258). The oxidative stress that exists in MetS, as a consequence of an overload of the homeostatic system, results in pro-inflammatory adipokine secretion, activation of the immune system, and chronic inflammation. The cellular mechanisms of defense against oxidative stress are orchestrated by the transcription factor Nrf2, as stated previously, making it a key target for the amelioration of MetS. Previous data have shown that ITCs and indoles activate Nrf2; thus, they are good candidates for further study. In mice fed an HFD supplemented with glucoraphanin (a GLS precursor of SFN), Nagata et al. (259) found a mitigated weight gain, attenuated fat depot (WAT), decreased hepatic steatosis, and improved glucose tolerance and insulin sensitivity. All these changes occurred without changes in intake but with an increase in energy expenditure. In addition, the increased energy expenditure occurred through WAT browning. The researchers also showed that decreased activation of NF-κB and JNK increased pAkt expression, together with diminished pro-inflammatory macrophage infiltration in WAT. Finally, they also found changes in microbiota with decreased relative abundance of Gram-negative bacteria that was correlated with diminished circulating LPS levels. Glucoraphanin supplementation had no effect in HFD fed Nrf2 knockout mice, suggesting that the anti-obesity effects of glucoraphanin (SFN) are due to the activation of the Nrf2 pathway. Similarly, using an adipocyte (3T3-L1)-macrophage (RAW264.7) co-culture system, Kang et al. described that brassinin (an indole) suppressed inflammation through Nrf2-HO-1 signaling pathway activation (260).
Several other authors described that SFN, AITC, BITC, PEITC, and indoles inhibited adipocyte differentiation in 3T3-L1 cells (261–265) and in mice (265–268). However, recently, Yang et al. described that the same doses of SFN and SFEN (sulforaphene) yielded stronger effects than AITC, BITC, or PEITC in inhibiting the differentiation of 3T3-L1 adipocytes. SFEN was also efficient in inhibiting adipogenesis in human adipose tissue-derived stem cells. The SFEN effect occurred when added early in differentiation and was mediated by decreased C/EBPβ stability, lowering PPARγ and C/EBPα expression (261). These results are in accordance with those reported previously by Chen et al. who showed that adipogenesis inhibition occurred in 3T3-L1 adipocytes through uptake of SFEN via the Hedgehog signaling pathway (263). Earlier, in 3T3-L1 preadipocytes, Choi et al. found that SFN arrested the cell cycle in the G0/G1 phase by increasing the expression of p27 and decreasing the expressions of cyclin D1, CDK4, cyclin A, and CDK2 (262). In the same study, the authors also found that SFN decreased the phosphorylation of ERK1/2 and Akt. Similar results were obtained by Choi et al. when 3T3-L1 preadipocytes were exposed to I3C; in this case, the cells were arrested in the transition from the S to the G2/M phase together with an increase in p27 expression and a diminished cyclin A expression (269). They also found that inhibition of adipogenesis, similar to that observed with SFEN, was achieved at an early stage of differentiation, and I3C regulates lipid synthesis via AMPKa signaling with diminished ERK and Akt phosphorylation. Yan et al. found that DIM arrested 3T3-L1 proliferation at the G0/G1 phase, but no effect on I3C was found. Moreover, the researchers also concluded that the inhibition of adipogenesis is mediated by targeting USP2 activity (265). In addition, the activation of apoptosis in 3T3-L1 adipocytes was found for SFN through downregulation of the Akt/p70s6k1/Bad pathway and upregulation of the ERK pathway as reported by Yao et al. (270).
In mice fed with an HFD to induce obesity, Choi et al. found that SFN prevents an increase in body weight without changes in food intake. SFN treatment also decreased circulating levels of leptin and cholesterol and increased the level of adiponectin. Fat deposition decrease in WAT takes place through inhibition of C/EBPα and PPARγ and the suppression of lipogenesis via activation of the AMPKa pathway (268). In HFD fed mice, Chuang et al. described that BITC and PEITC prevent body weight gain in a dose-dependent manner with diminishing adipogenesis and prevention of hepatosteatosis through inhibition of the lipogenic regulatory transcription factors PPARγ, LXRα, and SREBP1c, and a decrease in their regulated downstream enzymes (266). They also found that BITC and PEITC had similar effects and that 3T3-L1 cells were arrested in the G0/G1 phase. Similar results have been described for AITC also in HFD fed mice; in this case, the authors found prevention of body weight gain, decreased fat deposition in WAT and liver, and diminished inflammation. Diminished liver lipogenesis was mediated through activation of Sirt/AMPKa signaling, downregulation of SREBP1c, and upregulation of PPARα (271). They also investigated the decreasing liver inflammation and found diminished levels of TNFα, IL1β, and IL6 together with the activation of NF-κB signaling. Treatment of HFD fed mice with I3C (267) or DIM (265) also resulted in a decrease in body weight consequent to fat mass reduction and lipogenesis inhibition. Yagi et al. described that PEITC reduces food intake by activating leptin signaling via hypothalamic leptin receptors (Ob-Rb) and the Janus kinase 2 signal transducer (272). Furthermore, among mice, it has been found through the intraperitoneal glucose tolerance test that AITC reduces hyperglycemia and increases exogenous glucose consumption, ameliorating insulin sensibility (273). Ameliorated insulin sensibility was also found for BITC, PEITC, and AITC in HFD fed mice (266, 274). Jayakumar et al. found that I3C and DIM treatment of HFD fed mice induces decreases in the levels of glucose, insulin, and glycated hemoglobin and ameliorated overall oxidative stress (275). It has recently been reported that SFN could prevent Alzheimer progression and cerebral ischemia (276). Thus, all these pieces of evidence highlight that the use of GLS derivatives from cruciferous vegetables might be considered for the treatment of MetS.
Human studies aimed at investigating the effect of ITCs and indoles on MetS are scarce. Treatment of 40 hypertensive individuals (without diabetes and with normal levels of cholesterol) with 10 g of dried broccoli sprouts during a 4-week period failed to improve endothelial function (277). Another clinical trial examined 81 patients randomized to three groups: one to consume 10 g/d of broccoli sprouts powder (BSP), the other to consume 5 g/d of BSP, and another to consume the placebo for 4 weeks. The results showed that only the consumption of 10 g/d BSP resulted in a significant decrease in serum insulin concentration and HOMA-IR with improved insulin resistance (278). Japanese males with hepatic abnormalities treated with broccoli sprout extract capsules for 2 months showed significantly decreased serum levels of liver function markers (ALT, γGTP, and alkaline phosphatase activity); no changes were observed in the placebo group (279). A controlled trial conducted among healthy men analyzed the potential effect of 10 g freeze-dried nasturtium leaf (rich in BITC) administration on the levels of certain gut hormones that regulate food intake and satiety. The patients avoided the intake of cruciferous vegetables for a 1 week before the intervention. The results showed that the levels of peptide YY (PYY), an anorexigenic gut hormone, were increased after intake of freeze-dried nasturtium leaf during 6 h, and their effect could be more or less depending on whether they carried a polymorphism of the bitter taste receptor TAS2R38. Obese individuals frequently have lower levels of PYY and bariatric by-pass surgery results in elevated PYY levels. Therefore, administration of extract nasturtium (or BITC) or special diets containing nasturtium might be considered in the treatment of obesity (280). A randomized trial of 11 healthy individuals (www.controlled-trials.com ISRCTN19147515) assessed the effect of a dose of mustard (rich in AITC) on energy expenditure during 150 min. The results failed to show any relevant thermogenic response at the highest tolerable dose (281). Finally, López-Chillón et al. conducted a clinical trial of 40 healthy overweight subjects treated with broccoli sprouts over a long period (ClinicalTrials.gov ID NCT 03390855). The treatment phase consisted of 30 g/day of broccoli sprouts consumed for 10 weeks followed by a phase of 10 weeks of normal diet without consumption of these broccoli sprouts. The results showed a positive effect on inflammatory parameters with a significant decrease in IL6 levels that was maintained after treatment. In addition, the levels of GLSs, ITCs, and their metabolites (GRA, IB, SFN, SFN-GSH, SFN-NAC, SFN-CYS, I3C, and 3,3-DIM) were determined in urine as a control of broccoli intake, increased significantly during the treatment period, and decreased thereafter (282).
Figure 5 is a schema of how the increase in WAT size results in increased infiltration of pro-inflammatory macrophages and secretion of pro-inflammatory cytokines. High energy intake also promotes lipid deposition and inflammation in the liver. Both contribute to systemic inflammation that results in insulin resistance, dyslipidemia, and hypertension (i.e., MetS) which are the main risk factors for cardiac diseases. It shows also the described effects of ITCs and indoles that improve MetS.
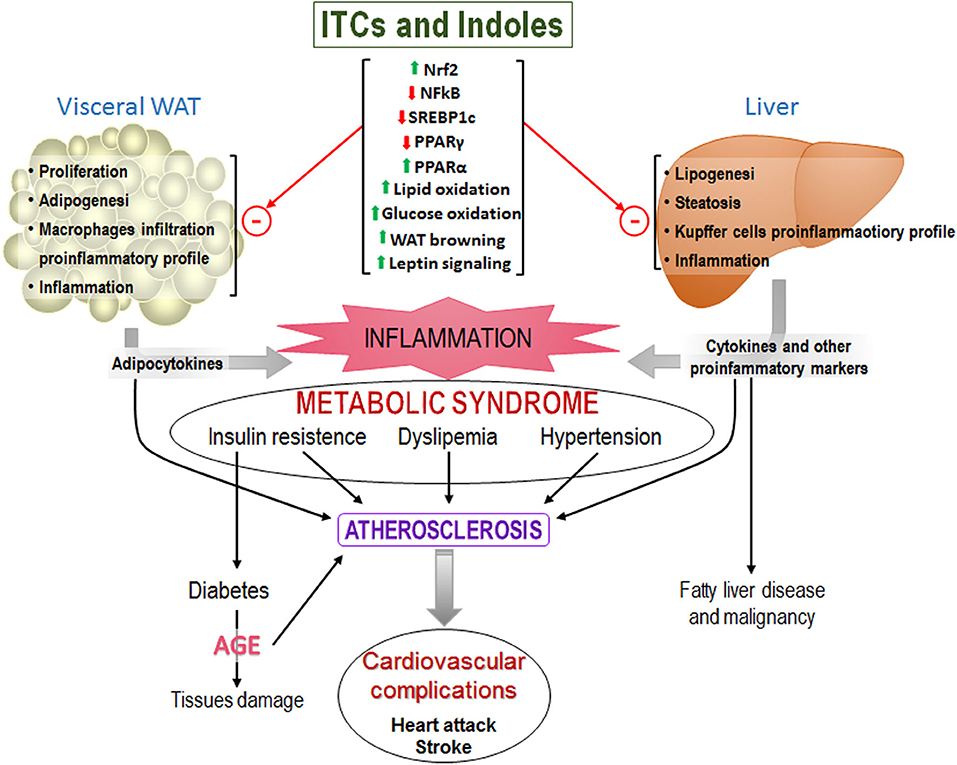
Figure 5. Representative scheme of the main components of the MetS that ultimately lead to cardiovascular problems and others. Upper of the figure are shown the effects of isothiocyanates that have been found and that could prevent MetS development.
In this review, the important regulatory biological role of cruciferous GLSs derivatives (ITCs and indoles) has been highlighted. Many scientific studies have put in evidence that GLSs derivatives play a key role in regulating central cell pathways and have epigenetic effects. For this reason, GLSs derivatives have been proposed as a possible co-therapy for the treatment of certain cancers, given their activity in the cell cycle, apoptosis, and angiogenesis regulation. More recently, they have been proposed as a possible mechanism in the prevention and/or therapy of MetS or disorders afflicting the central nervous system due to their anti-inflammatory effects. The results in animal and cell studies in relation to the MetS have shown that GLSs derivatives (ITCs and indoles) treatment reduces fat deposition in WAT and liver, decreases proliferation and differentiation of adipocytes, promotes WAT browning with an increase of energy expenditure, improves insulin sensitivity, reduces inflammation and decreases food intake. These results are promising to GLSs derivatives as a possible treatment to prevent MetS, however, there are few human trials. In addition, caution should be exercised since certain toxic effects have also been described (276). When other vegetable components with a high antioxidant capacity, like β-carotenes and vitamin E, have been administered as co-treatment or preventive agents for certain cancers, adverse effects have been found, reaching the point of having to stop the study because cancer progressed faster than among those that did not receive the supplement (283, 284). It is for this reason (without discarding the therapeutic application and the given scientific evidence regarding the important biological role of GLS derivatives in regulating key cellular pathways) that the consumption of cruciferous vegetables, and vegetables in general, in the diet should be claimed. There is a need to return to a healthier diet in which the pyramid base must constitute vegetable foods, of which cruciferous vegetables are essential.
The author confirms being the sole contributor of this work and has approved it for publication.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
I thank the CRAI of the University of Barcelona for the access to scientific journals and other resources.
1. Bazzano LA, Li TY, Joshipura KJ, Hu FB. Intake of fruit, vegetables, and fruit juices and risk of diabetes in women. Diabetes Care. (2008) 31:1311–7. doi: 10.2337/dc08-0080
2. Cooper AJ, Khaw KT, Sharp SJ, Wareham NJ, Lentjes MAH, Forouhi NG, et al. A prospective study of the association between quantity and variety of fruit and vegetable intake and incident type 2 diabetes. Diabetes Care. (2012) 35:1293–300. doi: 10.2337/dc11-2388
3. He FJ, Nowson CA, Lucas M, MacGregor GA. Increased consumption of fruit and vegetables is related to a reduced risk of coronary heart disease: meta-analysis of cohort studies. J Hum Hypertens. (2007) 21:717–28. doi: 10.1038/sj.jhh.1002212
4. Bhupathiraju SN, Wedick NM, Pan A, Manson JE, Rexrode KM, Willett WC, et al. Quantity and variety in fruit and vegetable intake and risk of coronary heart disease. Am J Clin Nutr. (2013) 98:1514–23. doi: 10.3945/ajcn.113.066381
5. Riboli E, Norat T. Epidemiologic evidence of the protective effect of fruit and vegetables on cancer risk. Am J Clin Nutr. (2003) 78(3 Suppl.):559S−69S. doi: 10.1093/ajcn/78.3.559s
6. Farvid MS, Chen WY, Michels KB, Cho E, Willett WC, Eliassen AH. Fruit and vegetable consumption in adolescence and early adulthood and risk of breast cancer: population based cohort study. BMJ. (2016) 353:i2343. doi: 10.1136/bmj.i2343
7. Van Gils CH, Peeters PHM, Bueno-de-Mesquita HB, Boshuizen HC, Lahmann PH, Clavel-Chapelon F, et al. Consumption of vegetables and fruits and risk of breast cancer. J Am Med Assoc. (2005) 293:183–93. doi: 10.1001/jama.293.2.183
8. Ma L, Liu G, Sampson L, Willett WC, Hu FB, Sun Q. Dietary glucosinolates and risk of type 2 diabetes in 3 prospective cohort studies. Am J Clin Nutr. (2018) 107:617–25. doi: 10.1093/ajcn/nqy003
9. Schulz M, Lahmann PH, Boeing H, Hoffmann K, Allen N, Key TJA, et al. Fruit and vegetable consumption and risk of epithelial ovarian cancer: the European prospective investigation into cancer and nutrition. Cancer Epidemiol Biomarkers Prev. (2005) 14:2531–5. doi: 10.1158/1055-9965.EPI-05-0159
10. Bayat Mokhtari R, Baluch N, Homayouni TS, Morgatskaya E, Kumar S, Kazemi P, et al. The role of Sulforaphane in cancer chemoprevention and health benefits: a mini-review. J Cell Commun Signal. (2018) 12:91–101. doi: 10.1007/s12079-017-0401-y
11. Liu RH. Potential synergy of phytochemicals in cancer prevention: mechanism of action. J Nutr. (2004) 134:3479S−85S. doi: 10.1093/jn/134.12.3479s
12. Fenwick GR, Heaney RK, Mullin WJ. Glucosinolates and their breakdown products in food and food plants. C R C Crit Rev Food Sci Nutr. (1982) 18:123–201. doi: 10.1080/10408398209527361
13. Lambrix V, Reichelt M, Mitchell-Olds T, Kliebenstein DJ, Gershenzon J. The arabidopsis epithiospecifier protein promotes the hydrolysis of glucosinolates to nitriles and influences Trichoplusia ni herbivory. Plant Cell. (2001) 13:2793–807. doi: 10.1105/tpc.13.12.2793
14. Kliebenstein DJ, Kroymann J, Brown P, Figuth A, Pedersen D, Gershenzon J, et al. Genetic control of natural variation in arabidopsis glucosinolate accumulation. Plant Physiol. (2001) 126:811–25. doi: 10.1104/pp.126.2.811
15. Finley JW. Proposed criteria for assessing the efficacy of cancer reduction by plant foods enriched in carotenoids, glucosinolates, polyphenols and selenocompounds. Ann Bot. (2005) 95:1075–96. doi: 10.1093/aob/mci123
16. Fahey JW, Zalcmann AT, Talalay P. The chemical diversity and distribution of glucosinolates and isothiocyanates among plants. Phytochemistry. (2001) 56:5–51. doi: 10.1016/S0031-9422(00)00316-2
17. Seo MS, Jin M, Chun JH, Kim SJ, Park BS, Shon SH, et al. Functional analysis of three BrMYB28 transcription factors controlling the biosynthesis of glucosinolates in Brassica rapa. Plant Mol Biol. (2016) 90:503–16. doi: 10.1007/s11103-016-0437-z
18. Tanii H. Allyl nitrile: toxicity and health effects. J Occup Health. (2017) 59:104–11. doi: 10.1539/joh.16-0147-RA
19. Stewart ZA, Westfall MD, Pietenpol JA. Cell-cycle dysregulation and anticancer therapy. Trends Pharmacol Sci. (2003) 24:139–45. doi: 10.1016/S0165-6147(03)00026-9
20. Kushad MM, Brown AF, Kurilich AC, Juvik JA, Klein BP, Wallig MA, et al. Variation of glucosinolates in vegetable crops of Brassica oleracea. J Agric Food Chem. (1999) 47:1541–8. doi: 10.1021/jf980985s
21. Cartea ME, Velasco P, Obregón S, Padilla G, de Haro A. Seasonal variation in glucosinolate content in Brassica oleracea crops grown in Northwestern Spain. Phytochemistry. (2008) 69:403–10. doi: 10.1016/j.phytochem.2007.08.014
22. Rungapamestry V, Duncan AJ, Fuller Z, Ratcliffe B. Effect of cooking brassica vegetables on the subsequent hydrolysis and metabolic fate of glucosinolates. Proc Nutr Soc. (2007) 66:69–81. doi: 10.1017/S0029665107005319
23. Rodrigues AS, Rosa EA. Effect of post-harvest treatments on the level of glucosinolates in broccoli. J Sci Food Agric. (1999) 79:1028–32. doi: 10.1002/(SICI)1097-0010(19990515)79:7<1028::AID-JSFA322>3.0.CO
24. Pellegrini N, Chiavaro E, Gardana C, Mazzeo T, Contino D, Gallo M, et al. Effect of different cooking methods on color, phytochemical concentration, and antioxidant capacity of raw and frozen brassica vegetables. J Agric Food Chem. (2010) 58:4310–21. doi: 10.1021/jf904306r
25. Yuan GF, Sun B, Yuan J, Wang QM. Effects of different cooking methods on health-promoting compounds of broccoli. J Zhejiang Univ Sci B. (2009) 10:580–8. doi: 10.1631/jzus.B0920051
26. Palermo M, Pellegrini N, Fogliano V. The effect of cooking on the phytochemical content of vegetables. J Sci Food Agric. (2014) 94:1057–70. doi: 10.1002/jsfa.6478
27. Song L, Thornalley PJ. Effect of storage, processing and cooking on glucosinolate content of Brassica vegetables. Food Chem Toxicol. (2007) 45:216–24. doi: 10.1016/j.fct.2006.07.021
28. Miglio C, Chiavaro E, Visconti A, Fogliano V, Pellegrini N. Effects of different cooking methods on nutritional and physicochemical characteristics of selected vegetables. J Agric Food Chem. (2008) 56:139–47. doi: 10.1021/jf072304b
29. Shapiro TA, Fahey JW, Wade KL, Stephenson KK, Talalay P. Human metabolism and excretion of cancer chemoprotective glucosinolates and isothiocyanates of cruciferous vegetables. Cancer Epidemiol Biomarkers Prev. (1998) 7:1091–100.
30. Sørensen JC, Frandsen HB, Jensen SK, Kristensen NB, Sørensen S, Sørensen H. Bioavailability and in vivo metabolism of intact glucosinolates. J Funct Foods. (2016) 24:450–60. doi: 10.1016/j.jff.2016.04.023
31. Holst B, Williamson G. A critical review of the bioavailability of glucosinolates and related compounds. Nat Prod Rep. (2004) 21:425. doi: 10.1039/b204039p
32. Krul C, Humblot C, Philippe C, Vermeulen M, van Nuenen M, Havenaar R, et al. Metabolism of sinigrin (2-propenyl glucosinolate) by the human colonic microflora in a dynamic in vitro large-intestinal model. Carcinogenesis. (2002) 23:1009–16. doi: 10.1093/carcin/23.6.1009
33. Palop MLl, Smiths JP, ten Brink B. Degradation of sinigrin by Lactobacillus agilis strain R16. Int J Food Microbiol. (1995) 26:219–29. doi: 10.1016/0168-1605(95)00123-2
34. Mullaney JA, Kelly WJ, McGhie TK, Ansell J, Heyes JA. Lactic acid bacteria convert glucosinolates to nitriles efficiently yet differently from enterobacteriaceae. J Agric Food Chem. (2013) 61:3039–46. doi: 10.1021/jf305442j
35. Cheng DL, Hashimoto K, Uda Y. In vitro digestion of sinigrin and glucotropaeolin by single strains of Bifidobacterium and identification of the digestive products. Food Chem Toxicol. (2004) 42:351–7. doi: 10.1016/j.fct.2003.09.008
36. Elfoul L, Rabot S, Khelifa N, Quinsac A, Duguay A, Rimbault A. Formation of allyl isothiocyanate from sinigrin in the digestive tract of rats monoassociated with a human colonic strain of Bacteroides thetaiotaomicron. FEMS Microbiol Lett. (2006) 197:99–103. doi: 10.1111/j.1574-6968.2001.tb10589.x
37. Ye L, Dinkova-Kostova AT, Wade KL, Zhang Y, Shapiro TA, Talalay P. Quantitative determination of dithiocarbamates in human plasma, serum, erythrocytes and urine: pharmacokinetics of broccoli sprout isothiocyanates in humans. Clin Chim Acta. (2002) 316:43–53. doi: 10.1016/s0009-8981(01)00727-6
38. Lai RH, Miller MJ, Jeffery E. Glucoraphanin hydrolysis by microbiota in the rat cecum results in sulforaphane absorption. Food Funct. (2010) 1:161–6. doi: 10.1039/c0fo00110d
39. Liu X, Wang Y, Hoeflinger J, Neme B, Jeffery E, Miller M. Dietary broccoli alters rat cecal microbiota to improve glucoraphanin hydrolysis to bioactive isothiocyanates. Nutrients. (2017) 9:262. doi: 10.3390/nu9030262
40. Mennicke WH, Görler K, Krumbiegel G, Lorenz D, Rittmann N. Studies on the metabolism and excretion of benzyl isothiocyanate in man. Xenobiotica. (1988) 18:441–7. doi: 10.3109/00498258809041680
41. Conaway CC, Getahun SM, Liebes LL, Pusateri DJ, Topham DKW, Botero-Omary M, et al. Disposition of glucosinolates and sulforaphane in humans after ingestion of steamed and fresh broccoli. Nutr Cancer. (2000) 38:168–78. doi: 10.1207/S15327914NC382_5
42. Atwell LL, Hsu A, Wong CP, Stevens JF, Bella D, Yu TW, et al. Absorption and chemopreventive targets of sulforaphane in humans following consumption of broccoli sprouts or a myrosinase-treated broccoli sprout extract. Mol Nutr Food Res. (2015) 59:424–33. doi: 10.1002/mnfr.201400674
43. Brüsewitz G, Cameron BD, Chasseaud LF, Görler K, Hawkins DR, Koch H, et al. The metabolism of benzyl isothiocyanate and its cysteine conjugate. Biochem J. (1977) 162:99–107. doi: 10.1042/bj1620099
44. Mennicke WH, Görler K, Krumbiegel G. Metabolism of some naturally occurring isothiocyanates in the rat. Xenobiotica. (1983) 13:203–7. doi: 10.3109/00498258309052256
45. Mennicke WH, Kral T, Krumbiegel G, Rittmann N. Determination of N-acetyl-S-(N-alkylthiocarbamoyl)-l-cysteine, a principal metabolite of alkyl isothiocyanates, in rat urine. J Chromatogr B Biomed Sci Appl. (1987) 414:19–24. doi: 10.1016/0378-4347(87)80020-8
46. Conaway CC, Jiao D, Kohri T, Liebes L, Chung FL. Disposition and pharmacokinetics of phenethyl isothiocyanate and 6-phenylhexyl isothiocyanate in F344 rats. Drug Metab Dispos. (1999) 27:13–20.
47. Conaway C, Yang Y, Chung F. Isothiocyanates as cancer chemopreventive agents: their biological activities and metabolism in rodents and humans. Curr Drug Metab. (2002) 3:233–55. doi: 10.2174/1389200023337496
48. Oliviero T, Verkerk R, Dekker M. Isothiocyanates from brassica vegetables—effects of processing, cooking, mastication, and digestion. Mol Nutr Food Res. (2018) 62:1701069. doi: 10.1002/mnfr.201701069
49. Petri N, Tannergren C, Holst B, Mellon FA, Bao Y, Plumb GW, et al. Absorption/metabolism of sulforaphane and quercetin, and regulation of phase II enzymes, in human jejunum in vivo. Drug Metab Dispos. (2003) 31:805–13. doi: 10.1124/dmd.31.6.805
50. Zhang Y. Role of glutathione in the accumulation of anticarcinogenic isothiocyanates and their glutathione conjugates by murine hepatoma cells. Carcinogenesis. (2000) 21:1175–82. doi: 10.1093/carcin/21.5.175
51. Zhang Y, Callaway EC. High cellular accumulation of sulphoraphane, a dietary anticarcinogen, is followed by rapid transporter-mediated export as a glutathione conjugate. Biochem J. (2002) 364:301–7. doi: 10.1042/bj3640301
52. Anderton MJ, Manson MM, Verschoyle R, Gescher A, Steward WP, Williams ML, et al. Physiological modeling of formulated and crystalline 3,3′ -diindolylmethane pharmacokinetics following oral administration in mice. Drug Metab Dispos. (2004) 32:632–8. doi: 10.1124/dmd.32.6.632
53. Kim YJ, Lee DH, Ahn J, Chung WJ, Jang YJ, Seong KS, et al. Pharmacokinetics, tissue distribution, and anti-lipogenic/adipogenic effects of allyl-isothiocyanate metabolites. PLoS ONE. (2015) 10:e0132151. doi: 10.1371/journal.pone.0132151
54. Zhang Y. Molecular mechanism of rapid cellular accumulation of anticarcinogenic isothiocyanates. Carcinogenesis. (2001) 22:425–31. doi: 10.1093/carcin/22.3.425
55. Licznerska B, Baer-Dubowska W. Indole-3-carbinol and its role in chronic diseases. In: Gupta SC, Sahdeo P, Aggarwal BB, editors. Anti-inflammatory Nutraceuticals and Chronic Diseases. Advances in Experimental Medicine and Biology. New York, NY: Springer LLC (2016). p. 131–54. doi: 10.1007/978-3-319-41334-1_6
56. De Kruif CA, Marsman JW, Venekamp JC, Falke HE, Noordhoek J, Blaauboer BJ, et al. Structure elucidation of acid reaction products of indole-3-carbinol: detection in vivo and enzyme induction in vitro. Chem Biol Interact. (1991) 80:303–15. doi: 10.1016/0009-2797(91)90090-T
57. Anderton MJ, Manson MM, Verschoyle RD, Gescher A, Lamb JH, Farmer PB, et al. Pharmacokinetics and tissue disposition of indole-3-carbinol and its acid condensation products after oral administration to mice. Clin Cancer Res. (2004) 10:5233–41. doi: 10.1158/1078-0432.CCR-04-0163
58. Reed GA, Arneson DW, Putnam WC, Smith HJ, Gray JC, Sullivan DK, et al. Single-dose and multiple-dose administration of indole-3-carbinol to women: pharmacokinetics based on 3,3′-diindolylmethane. Cancer Epidemiol Biomarkers Prev. (2006) 15:2477–81. doi: 10.1158/1055-9965.EPI-06-0396
59. Reed GA, Peterson KS, Smith HJ, Gray JC, Sullivan DK, Mayo MS, et al. A Phase I study of indole-3-carbinol in women : tolerability and effects. Cancer Epidemiol Biomarkers Prev. (2005) 14:1953–61. doi: 10.1158/1055-9965.EPI-05-0121
60. Fujioka N, Ainslie-waldman CE, Upadhyaya P, Carmella SG. Urinary 3,3'-diindolylmethane: a biomarker of glucobrassicin exposure and indole-3-carbinol uptake in humans. Cancer Epidemiol Biomarkers Prev. (2015) 23:282–7. doi: 10.1158/1055-9965.EPI-13-0645.Urinary
61. Lock EA, Reed CJ. Xenobiotic metabolizing enzymes of the kidney. Toxicol Pathol. (1998) 26:18–25. doi: 10.1177/019262339802600102
62. Kassahun K, Davis M, Hu P, Martin B, Baillie T. Biotransformation of the naturally occurring isothiocyanate sulforaphane in the rat: identification of phase I metabolites and glutathione conjugates. Chem Res Toxicol. (1997) 10:1228–33. doi: 10.1021/tx970080t
63. Jiao D, Eklind KI, Choi CI, Desai DH, Amin SG, Chung FL. Structure-activity relationships of isothiocyanates as mechanism-based inhibitors of 4-(Methylnitrosamino)-l-(3-pyridyl)-1-butanone-induced lung tumorigenesis in A/J mice. Cancer Res. (1994) 54:4327–33.
64. Iyanagi T. Molecular mechanism of phase I and phase II drug-metabolizing enzymes: implications for detoxification. Int Rev Cytol. (2007) 260:35–112. doi: 10.1016/S0074-7696(06)60002-8
65. Sharma R, Williams IS, Gatchie L, Sonawane VR, Chaudhuri B, Bharate SB. Khellinoflavanone, a semisynthetic derivative of khellin, overcomes benzo[a]pyrene toxicity in human normal and cancer cells that express CYP1A1. ACS Omega. (2018) 3:8553–66. doi: 10.1021/acsomega.8b01088
66. Hecht SS. Inhibition of carcinogenesis by isothiocyanates. Drug Metab Rev. (2000) 32:395–411. doi: 10.1081/dmr-100102342
67. Nakajima M, Yoshida R, Shimada N, Yamazaki H, Yokoi T. Inhibition and inactivation of human cytochrome P450 isoforms by phenethyl isothiocyanate. Drug Metab Dispos. (2001) 29:1110–3.
68. Higdon J, Delage B, Williams D, Dashwood R. Cruciferous vegetables and human cancer risk: epidemiologic evidence and mechanistic basis. Pharmacol Res. (2007) 55:224–36. doi: 10.1016/j.phrs.2007.01.009
69. Abbaoui B, Lucas CR, Riedl KM, Clinton SK, Mortazavi A. Cruciferous vegetables, isothiocyanates, and bladder cancer prevention. Mol Nutr Food Res. (2018) 62:e1800079. doi: 10.1002/mnfr.201800079
70. Plate AYA, Gallaher DD. Effects of indole-3-carbinol and phenethyl isothiocyanate on colon carcinogenesis induced by azoxymethane in rats. Carcinogenesis. (2006) 27:287–92. doi: 10.1093/carcin/bgi210
71. Leclercq I, Desager JP, Horsmans Y. Inhibition of chlorzoxazone metabolism, a clinical probe for CYP2E1, by a single ingestion of watercress. Clin Pharmacol Ther. (1998) 64:144–9. doi: 10.1016/S0009-9236(98)90147-3
72. Yoshigae Y, Sridar C, Kent UM, Hollenberg PF. The inactivation of human CYP2E1 by phenethyl isothiocyanate, a naturally occurring chemopreventive agent, and its oxidative bioactivation. Drug Metab Dispos. (2013) 41:858–69. doi: 10.1124/dmd.112.050609
73. Thomson CA, Ho E, Strom MB. Chemopreventive properties of 3,30-diindolylmethane in breast cancer: evidence from experimental and human studies. Nutr Rev. (2016) 74:432–43. doi: 10.1093/nutrit/nuw010
74. Guo Z, Smith TJ, Wang E, Sadrieh N, Ma Q, Thomas PE, et al. Effects of phenethyl isothiocyanate, a carcinogenesis inhibitor, on xenobiotic-metabolizing enzymes and nitrosamine metabolism in rats. Carcinogenesis. (1992) 13:2205–10. doi: 10.1093/carcin/13.12.2205
75. Chen I, McDougal A, Wang F, Safe S. Aryl hydrocarbon receptor-mediated antiestrogenic and antitumorigenic activity of diindolylmethane. Carcinogenesis. (1998) 19:1631–9. doi: 10.1093/carcin/19.9.1631
76. Szaefer H, Licznerska B, Krajka-Kuniak V, Bartoszek A, Baer-Dubowska W. Modulation of CYP1A1, CYP1A2 and CYP1B1 expression by cabbage juices and indoles in human breast cell lines. Nutr Cancer. (2012) 64:879–88. doi: 10.1080/01635581.2012.690928
77. Leoncini E, Malaguti M, Angeloni C, Motori E, Fabbri D, Hrelia S. Cruciferous vegetable phytochemical sulforaphane affects phase II enzyme expression and activity in rat cardiomyocytes through modulation of Akt signaling pathway. J Food Sci. (2011) 76:H175–1. doi: 10.1111/j.1750-3841.2011.02311.x
78. Abdull Razis AF, Bagatta M, De Nicola GR, Iori R, Ioannides C. Intact glucosinolates modulate hepatic cytochrome P450 and phase II conjugation activities and may contribute directly to the chemopreventive activity of cruciferous vegetables. Toxicology. (2010) 277:74–85. doi: 10.1016/j.tox.2010.08.080
79. Nho CW, Jeffery E. The synergistic upregulation of phase II detoxification enzymes by glucosinolate breakdown products in cruciferous vegetables. Toxicol Appl Pharmacol. (2001) 174:146–52. doi: 10.1006/taap.2001.9207
80. Hecht SS, Carmella SG, Murphy SE. Effects of watercress consumption on urinary metabolites of nicotine in smokers. Cancer Epidemiol Biomarkers Prev. (1999) 8:907–13.
81. Yuan JM, Murphy SE, Stepanov I, Wang R, Carmella SG, Nelson HH, et al. 2-phenethyl isothiocyanate, glutathione S-transferase M1 and T1 polymorphisms, and detoxification of volatile organic carcinogens and toxicants in tobacco smoke. Cancer Prev Res. (2016) 9:598–606. doi: 10.1158/1940-6207.CAPR-16-0032
82. Coles BF, Kadlubar FF. Detoxification of electrophilic compounds by glutathione S-transferase catalysis: determinants of individual response to chemical carcinogens and chemotherapeutic drugs? Biofactors. 17:115–30. doi: 10.1002/biof.5520170112
83. Coles BF, Kadlubar FF. Human alpha class glutathione S-transferases: genetic polymorphism, expression, and susceptibility to disease. Methods Enzymol. (2005) 401:9–42. doi: 10.1016/S0076-6879(05)01002-5
84. London SJ, Yuan JM, Chung FL, Gao YT, Coetzee GA, Ross RK, et al. Isothiocyanates, glutathione S-transferase M1 and T1 polymorphisms, and lung-cancer risk: a prospective study of men in Shanghai, China. Lancet. (2000) 356:724–9. doi: 10.1016/S0140-6736(00)02631-3
85. Spitz MR, Duphorne CM, Detry MA, Pillow PC, Amos CI, Lei L, et al. Dietary intake of isothiocyanates: evidence of a joint effect with glutathione S-transferase polymorphisms in lung cancer risk. Cancer Epidemiol Biomarkers Prev. (2000) 9:1017–20.
86. Lewis S, Brennan P, Nyberg F, Ahrens W, Constantinescu V, Mukeria A, et al. Cruciferous vegetable intake, GSTM1 genotype and lung cancer risk in a non-smoking population. IARC Sci Publ. (2002) 156:507–8.
87. Zhao B, Seow A, Lee EJ, Poh WT, Teh M, Eng P, et al. Dietary isothiocyanates, glutathione S-transferase -M1, -T1 polymorphisms and lung cancer risk among Chinese women in Singapore. Cancer Epidemiol Biomarkers Prev. (2001) 10:1063–7.
88. Boldry EJ, Yuan JM, Carmella SG, Wang R, Tessier K, Hatsukami DK, et al. Effects of 2-phenethyl isothiocyanate on metabolism of 1,3-butadiene in smokers. Cancer Prev Res. (2020) 13:91–100. doi: 10.1158/1940-6207.CAPR-19-0296
89. Saw CL-L, Cintron M, Wu T-Y, Guo Y, Huang Y, Jeong W-S, et al. Pharmacodynamics of dietary phytochemical indoles I3C and DIM: Induction of Nrf2-mediated Phase II drug metabolizing and antioxidant genes and synergism with isothiocyanates. Biopharm Drug Dispos. (2011) 32:289–300. doi: 10.1002/bdd.759.Pharmacodynamics
90. Soundararajan P, Kim JS. Anti-carcinogenic glucosinolates in cruciferous vegetables and their antagonistic effects on prevention of cancers. Molecules. (2018) 23:E2983. doi: 10.3390/molecules23112983
91. Singh D, Arora R, Bhatia A, Singh H, Singh B, Arora S. Molecular targets in cancer prevention by 4-(methylthio)butyl isothiocyanate - A comprehensive review. Life Sci. (2020) 241:117061. doi: 10.1016/j.lfs.2019.117061
92. Tang L, Zhang Y, Jobson HE, Li J, Stephenson KK, Wade KL, et al. Potent activation of mitochondria-mediated apoptosis and arrest in S and M phases of cancer cells by a broccoli sprout extract. Mol Cancer Ther. (2006) 5:935–44. doi: 10.1158/1535-7163.MCT-05-0476
93. Abbaoui B, Riedl KM, Ralston RA, Thomas-Ahner JM, Schwartz SJ, Clinton SK, et al. Inhibition of bladder cancer by broccoli isothiocyanates sulforaphane and erucin: characterization, metabolism, and interconversion. Mol Nutr Food Res. (2012) 56:1675–87. doi: 10.1002/mnfr.201200276
94. Fimognari C, Nüsse M, Berti F, Iori R, Cantelli-Forti G, Hrelia P. Cyclin D3 and p53 mediate sulforaphane-induced cell cycle delay and apoptosis in non-transformed human T lymphocytes. Cell Mol Life Sci. (2002) 59:2004–12. doi: 10.1007/pl00012523
95. Fimognari C, Nüsse M, Cersari R, Iori R, Cantelli-Forti G, Hrelia P. Growth inhibition, cell-cycle arrest and apoptosis in human T-cell leukemia by the isothiocyanate sulforaphane. Carcinogenesis. (2002) 23:581–6. doi: 10.1093/carcin/23.4.581
96. Matsui T-A, Murata H, Sakabe T, Sowa Y, Horie N, Nakanishi R, et al. Sulforaphane induces cell cycle arrest and apoptosis in murine osteosarcoma cells in vitro and inhibits tumor growth in vivo. Oncol Rep. (2007) 18:1263–8.
97. Liang H, Lai B, Yuan Q. Sulforaphane induces cell-cycle arrest and apoptosis in cultured human lung adenocarcinoma LTEP-A2 cells and retards growth of LTEP-A2 xenografts in vivo. J Nat Prod. (2008) 71:1911–4. doi: 10.1021/np800233q
98. Gamet-Payrastre L, Li P, Lumeau S, Cassar G, Dupont MA, Chevolleau S, et al. Sulforaphane, a naturally occurring isothiocyanate, induces cell cycle arrest and apoptosis in HT29 human colon cancer cells. Cancer Res. (2000) 60:1426–33.
99. Singh AV, Xiao D, Lew KL, Dhir R, Singh SV. Sulforaphane induces caspase-mediated apoptosis in cultured PC-3 human prostate cancer cells and retards growth of PC-3 xenografts in vivo. Carcinogenesis. (2004) 25:83–90. doi: 10.1093/carcin/bgg178
100. Chiao JW, Chung FL, Kancherla R, Ahmed T, Mittelman A, Conaway CC. Sulforaphane and its metabolite mediate growth arrest and apoptosis in human prostate cancer cells. Int J Oncol. (2002) 20:631–6. doi: 10.3892/ijo.20.3.631
101. Hasegawa T, Nishino H, Iwashima A. Isothiocyanates inhibit cell cycle progression of HeLa cells at G2/M phase. Anticancer Drugs. (1993) 4:273–9. doi: 10.1097/00001813-199304000-00021
102. Visanji JM, Duthie SJ, Pirie L, Thompson DG, Padfield PJ. Dietary isothiocyanates inhibit Caco-2 cell proliferation and induce G2/M phase cell cycle arrest, DNA damage, and G2/M checkpoint activation. J Nutr. (2004) 134:3121–6. doi: 10.1093/jn/134.11.3121
103. Xiao D, Johnson CS, Trump DL, Singh SV. Proteasome-mediated degradation of cell division cycle 25C and cyclin-dependent kinase 1 in phenethyl isothiocyanate-induced G2-M-phase cell cycle arrest in PC-3 human prostate cancer cells. Mol Cancer Ther. (2004) 3:567–75.
104. Chiao JW, Chung F, Krzeminski J, Amin S, Arshad R, Ahmed TCC. Modulation of growth of human prostate cancer cells by the N-acetylcysteine conjugate of phenethyl isothiocyanate. Int J Oncol. (2000) 16:1215–9. doi: 10.3892/ijo.16.6.1215
105. Zhang R, Loganathan S, Humphreys I, Srivastava SK. Benzyl isothiocyanate-induced DNA damage causes G2/M cell cycle arrest and apoptosis in human pancreatic cancer cells. J Nutr. (2006) 136:2728–34. doi: 10.1093/jn/136.11.2728
106. Miyoshi N, Uchida K, Osawa T, Nakamura Y. Benzyl isothiocyanate modifies expression of the G2/M arrest-related genes. Biofactors. (2008) 21:23–6. doi: 10.1002/biof.552210106
107. Melchini A, Costa C, Traka M, Miceli N, Mithen R, De Pasquale R, et al. Erucin, a new promising cancer chemopreventive agent from rocket salads, shows anti-proliferative activity on human lung carcinoma A549 cells. Food Chem Toxicol. (2009) 47:1430–6. doi: 10.1016/j.fct.2009.03.024
108. Zhang Y, Tang L, Gonzalez V. Selected isothiocyanates rapidly induce growth inhibition of cancer cells. Mol Cancer Ther. (2003) 2:1045–52.
109. Tang L, Zhang Y. Dietary Isothiocyanates inhibit the growth of human bladder carcinoma cells. J Nutr. (2004) 134:2004–10. doi: 10.1093/jn/134.8.2004
110. Xiao D, Srivastava SK, Lew KL, Zeng Y, Hershberger P, Johnson CS, et al. Allyl isothiocyanate, a constituent of cruciferous vegetables, inhibits proliferation of human prostate cancer cells by causing G2/M arrest and inducing apoptosis. Carcinogenesis. (2003) 24:891–7. doi: 10.1093/carcin/bgg023
111. Smith TK, Lund EK, Parker ML, Clarke RG, Johnson IT. Allyl-isothiocyanate causes mitotic block, loss of cell adhesion and disrupted cytoskeletal structure in HT29 cells. Carcinogenesis. (2004) 25:1409–15. doi: 10.1093/carcin/bgh149
112. Cover CM, Hsieh SJ, Tran SH, Hallden G, Kim GS, Bjeldanes LF, et al. Indole-3-carbinol inhibits the expression of cyclin-dependent kinase-6 and induces a G1 cell cycle arrest of human breast cancer cells independent of estrogen receptor signaling. J Biol Chem. (1998) 273:3838–47. doi: 10.1074/jbc.273.7.3838
113. Marconett CN, Singhal AK, Sundar SN, Firestone GL. Indole-3-carbinol disrupts estrogen receptor-alpha dependent expression of insulin-like growth factor-1 receptor and insulin receptor substrate-1 and proliferation of human breast cancer cells. Mol Cell Endocrinol. (2012) 363:74–84. doi: 10.1016/j.mce.2012.07.008
114. Brew CT, Aronchik I, Hsu JC, Sheen JH, Dickson RB, Bjeldanes LF, et al. Indole-3-carbinol activates the ATM signaling pathway independent of DNA damage to stabilize p53 and induce G1 arrest of human mammary epithelial cells. Int J Cancer. (2006) 118:857–68. doi: 10.1002/ijc.21445
115. Chinni SR, Li Y, Upadhyay S, Koppolu PK, Sarkar FH. Indole-3-carbinol (I3C) induced cell growth inhibition, G1 cell cycle arrest and apoptosis in prostate cancer cells. Oncogene. (2001) 20:2927–36. doi: 10.1038/sj.onc.1204365
116. Chen D, Carter TH, Auborn KJ. Apoptosis in cervical cancer cells: implications for adjunct anti-estrogen therapy for cervical cancer. Anticancer Res. (2004) 24:2649–56.
117. Chen D-Z, Qi M, Auborn KJ, Carter TH. Indole-3-carbinol and diindolylmethane induce apoptosis of human cervical cancer cells and in murine HPV16-transgenic preneoplastic cervical epithelium. J Nutr. (2001) 131:3294–302. doi: 10.1093/jn/131.12.3294
118. Hu R, Kim BR, Chen C, Hebbar V, Kong A-NT. The roles of JNK and apoptotic signaling pathways in PEITC-mediated responses in human HT-29 colon adenocarcinoma cells. Carcinogenesis. (2003) 24:1361–7. doi: 10.1093/carcin/bgg092
119. Xiao D, Singh SV. Phenethyl isothiocyanate-induced apoptosis in p53-deficient PC-3 human prostate cancer cell line is mediated by extracellular signal-regulated kinases. Cancer Res. (2002) 62:3615–9.
120. Xu C, Shen G, Yuan X, Kim JH, Gopalkrishnan A, Keum YS, et al. ERK and JNK signaling pathways are involved in the regulation of activator protein 1 and cell death elicited by three isothiocyanates in human prostate cancer PC-3 cells. Carcinogenesis. (2006) 27:437–45. doi: 10.1093/carcin/bgi251
121. Islam SS, Mokhtari RB, Akbari P, Hatina J, Yeger H, Farhat WA. Simultaneous targeting of bladder tumor growth, survival, and epithelial-to-mesenchymal transition with a novel therapeutic combination of acetazolamide (AZ) and Sulforaphane (SFN). Target Oncol. (2016) 11:209–27. doi: 10.1007/s11523-015-0386-5
122. Jo GH, Kim GY, Kim WJ, Park KY, Choi YH. Sulforaphane induces apoptosis in T24 human urinary bladder cancer cells through a reactive oxygen species-mediated mitochondrial pathway: The involvement of endoplasmic reticulum stress and the Nrf2 signaling pathway. Int J Oncol. (2014) 45:1497–506. doi: 10.3892/ijo.2014.2536
123. Wang Y, Wei S, Wang J, Fang Q, Chai Q. Phenethyl isothiocyanate inhibits growth of human chronic myeloid leukemia K562 cells via reactive oxygen species generation and caspases. Mol Med Rep. (2014) 10:543–9. doi: 10.3892/mmr.2014.2167
124. Trachootham D, Zhang H, Zhang W, Feng L, Du M, Zhou Y, et al. Effective elimination of fludarabine-resistant CLL cells by PEITC through a redox-mediated mechanism. Blood. (2008) 112:1912–22. doi: 10.1182/blood-2008-04-149815
125. Kasiappan R, Jutooru I, Karki K, Hedrick E, Safe S. Benzyl isothiocyanate (BITC) induces reactive oxygen species-dependent repression of STAT3 protein by down-regulation of specificity proteins in pancreatic cancer. J Biol Chem. (2016) 291:27122–33. doi: 10.1074/jbc.M116.746339
126. Morimitsu Y, Nakagawa Y, Hayashi K, Fujii H, Kumagai T, Nakamura Y, et al. A sulforaphane analogue that potently activates the Nrf2-dependent detoxification pathway. J Biol Chem. (2002) 277:3456–63. doi: 10.1074/jbc.M110244200
127. Sturm C, Wagner AE. Brassica-derived plant bioactives as modulators of chemopreventive and inflammatory signaling pathways. Int J Mol Sci. (2017) 18:1890. doi: 10.3390/ijms18091890
128. Aggarwal BB, Sung B. NF-κB in cancer: a matter of life and death. Cancer Discov. (2011) 1:469–471. doi: 10.1158/2159-8290.CD-11-0260
129. Monteillier A, Allard PM, Gindro K, Wolfender JL, Cuendet M. Lung cancer chemopreventive activity of patulin isolated from penicillium vulpinum. Molecules. (2018) 23:636. doi: 10.3390/molecules23030636
130. Shan Y, Zhang L, Bao Y, Li B, He C, Gao M, et al. Epithelial-mesenchymal transition, a novel target of sulforaphane via COX-2/MMP2, 9/Snail, ZEB1 and miR-200c/ZEB1 pathways in human bladder cancer cells. J Nutr Biochem. (2013) 24:1062–9. doi: 10.1016/j.jnutbio.2012.08.004
131. Lai KC, Huang ANC, Hsu SC, Kuo CL, Yang JS, Wu SH, et al. Benzyl Lsothiocyanate (BITC) inhibits migration and invasion of human colon cancer HT29 cells by inhibiting matrix metalloproteinase-2/-9 and urokinase plasminogen (uPA) through PKC and MAPK signaling pathway. J Agric Food Chem. (2010) 58:2935–42. doi: 10.1021/jf9036694
132. Chen D, Banerjee S, Cui QC, Kong D, Sarkar FH, Dou QP. Activation of AMP-activated protein kinase by 3,3′-Diindolylmethane (DIM) is associated with human prostate cancer cell death in vitro and in vivo. PLoS ONE. (2012) 7:e47186. doi: 10.1371/journal.pone.0047186
133. Chinni SR, Sarkar FH. Akt inactivation is a key event in indole-3-carbinol-induced apoptosis in PC-3 cells. Clin Cancer Res. (2002) 8:1228–36.
134. Ahmad A, Biersack B, Li Y, Kong D, Bao B, Schobert R, et al. Targeted regulation of PI3K/Akt/mTOR/NF-κB signaling by indole compounds and their derivatives: mechanistic details and biological implications for cancer therapy. Anticancer Agents Med Chem. (2013) 13:1002–13. doi: 10.2174/18715206113139990078
135. Medzhitov R, Horng T. Transcriptional control of the inflammatory response. Nat Rev Immunol. (2009) 9:692–703. doi: 10.1038/nri2634
136. Keum Y-S, Owuor ED, Kim B-R, Hu R, Kong ANT. Involvement of Nrf2 and JNK1 in the activation of antioxidant responsive element (ARE) by chemopreventive agent phenethyl isothiocyanate (PEITC). Pharm Res. (2003) 20:1351–6. doi: 10.1023/a:1025737622815
137. Xu C, Yuan X, Pan Z, Shen G, Kim JH, Yu S, et al. Mechanism of action of isothiocyanates: the induction of ARE-regulated genes is associated with activation of ERK and JNK and the phosphorylation and nuclear translocation of Nrf2. Mol Cancer Ther. (2006) 5:1918–26. doi: 10.1158/1535-7163.MCT-05-0497
138. Kobayashi M, Yamamoto M. Nrf2-Keap1 regulation of cellular defense mechanisms against electrophiles and reactive oxygen species. Adv Enzyme Regul. (2006) 46:113–40. doi: 10.1016/j.advenzreg.2006.01.007
139. Catrysse L, van Loo G. Inflammation and the metabolic syndrome: the tissue-specific functions of NF-κB. Trends Cell Biol. (2017) 27:417–29. doi: 10.1016/j.tcb.2017.01.006
140. Zhou Y, Xu X, Wu J, Xu L, Zhang M, Li Z, et al. Allyl isothiocyanate treatment alleviates chronic obstructive pulmonary disease through the Nrf2-Notch1 signaling and upregulation of MRP1. Life Sci. (2020) 243:117291. doi: 10.1016/j.lfs.2020.117291
141. Caglayan B, Kilic E, Dalay A, Altunay S, Tuzcu M, Erten F, et al. Allyl isothiocyanate attenuates oxidative stress and inflammation by modulating Nrf2/HO-1 and NF-κB pathways in traumatic brain injury in mice. Mol Biol Rep. (2019) 46:241–50. doi: 10.1007/s11033-018-4465-4
142. Ernst IMA, Wagner AE, Schuemann C, Storm N, Höppner W, Döring F, et al. Allyl-, butyl- and phenylethyl-isothiocyanate activate Nrf2 in cultured fibroblasts. Pharmacol Res. (2011) 63:233–40. doi: 10.1016/j.phrs.2010.11.005
143. Aggarwal BB, Ichikawa H. Molecular targets and anticancer potential of indole-3-carbinol and its derivatives. Cell Cycle. (2005) 4:1201–15. doi: 10.4161/cc.4.9.1993
144. Prawan A, Saw CLL, Khor TO, Keum YS, Yu S, Hu L, et al. Anti-NF-κB and anti-inflammatory activities of synthetic isothiocyanates: effect of chemical structures and cellular signaling. Chem Biol Interact. (2009) 179:202–11. doi: 10.1016/j.cbi.2008.12.014
145. Lee YM, Seon MR, Cho HJ, Kim JS, Park JHY. Benzyl isothiocyanate exhibits anti-inflammatory effects in murine macrophages and in mouse skin. J Mol Med. (2009) 87:1251–61. doi: 10.1007/s00109-009-0532-6
146. Huang CS, Lin AH, Liu CT, Tsai CW, Chang IS, Chen HW. Isothiocyanates protect against oxidized LDL-induced endothelial dysfunction by upregulating Nrf2-dependent antioxidation and suppressing NFκB activation. Mol Nutr Food Res. (2013) 57:1918–30. doi: 10.1002/mnfr.201300063
147. Kim HW, Kim J, Kim J, Lee S, Choi BR, Han JS, et al. 3,3'-Diindolylmethane inhibits lipopolysaccharide-induced microglial hyperactivation and attenuates brain inflammation. Toxicol Sci. (2014) 137:158–67. doi: 10.1093/toxsci/kft240
148. Kim YH, Kwon HS, Kim DH, Shin EK, Kang YH, Park JHY, et al. 3,3′-Diindolylmethane attenuates colonic inflammation and tumorigenesis in mice. Inflamm Bowel Dis. (2009) 15:1164–73. doi: 10.1002/ibd.20917
149. Benazzi C, Al-Dissi A, Chau CH, Figg WD, Sarli G, de Oliveira JT, et al. Angiogenesis in spontaneous tumors and implications for comparative tumor biology. Sci World J. (2014) 2014:919570. doi: 10.1155/2014/919570
150. Singhal S, Vachani A, Antin-Ozerkis D, Kaiser LR, Albelda SM. Prognostic implications of cell cycle, apoptosis, and angiogenesis biomarkers in non-small cell lung cancer: a review. Clin Cancer Res. (2005) 11:3974–86. doi: 10.1158/1078-0432.CCR-04-2661
151. Moserle L, Casanovas O. Anti-angiogenesis and metastasis: a tumour and stromal cell alliance. J Intern Med. (2013) 273:128–37. doi: 10.1111/joim.12018
152. Bertl E, Bartsch H, Gerhäuser C. Inhibition of angiogenesis and endothelial cell functions are novel sulforaphane-mediated mechanisms in chemoprevention. Mol Cancer Ther. (2006) 5:575–85. doi: 10.1158/1535-7163.MCT-05-0324
153. Lee C-S, Cho H-J, Jeong Y-J, Shin J-M, Park K-K, Park Y-Y, et al. Isothiocyanates inhibit the invasion and migration of C6 glioma cells by blocking FAK/JNK-mediated MMP-9 expression. Oncol Rep. (2015) 34:2901–8. doi: 10.3892/or.2015.4292
154. Ho JN, Jun W, Choue R, Lee J. I3C and ICZ inhibit migration by suppressing the EMT process and FAK expression in breast cancer cells. Mol Med Rep. (2013) 7:384–8. doi: 10.3892/mmr.2012.1198
155. Hajra S, Patra AR, Basu A, Saha P, Bhattacharya S. Indole-3-Carbinol (I3C) enhances the sensitivity of murine breast adenocarcinoma cells to doxorubicin (DOX) through inhibition of NF-κβ blocking angiogenesis and regulation of mitochondrial apoptotic pathway. Chem Biol Interact. (2018) 290:19–36. doi: 10.1016/j.cbi.2018.05.005
156. Xiao D, Singh SV. Phenethyl isothiocyanate inhibits angiogenesis in vitro and ex vivo. Cancer Res. (2007) 67:2239–46. doi: 10.1158/0008-5472.CAN-06-3645
157. Kim DH, Sung B, Kang YJ, Hwang SY, Kim MJ, Yoon JH, et al. Sulforaphane inhibits hypoxia-induced HIF-1α and VEGF expression and migration of human colon cancer cells. Int J Oncol. (2015) 47:2226–32. doi: 10.3892/ijo.2015.3200
158. Thejass P, Kuttan G. Inhibition of endothelial cell differentiation and proinflammatory cytokine production during angiogenesis by allyl isothiocyanate and phenyl isothiocyanate. Integr Cancer Ther. (2007) 6:389–99. doi: 10.1177/1534735407309084
159. Wang X-H, Cavell BE, Syed Alwi SS, Packham G. Inhibition of hypoxia inducible factor by phenethyl isothiocyanate. Biochem Pharmacol. (2009) 78:261–72. doi: 10.1016/j.bcp.2009.04.010
160. Gupta B, Chiang L, Chae K, Lee D-H. Phenethyl isothiocyanate inhibits hypoxia-induced accumulation of HIF-1α and VEGF expression in human glioma cells. Food Chem. (2013) 141:1841–6. doi: 10.1016/j.foodchem.2013.05.006
161. Singh-Gupta V, Banerjee S, Yunker CK, Rakowski JT, Joiner MC, Konski AA, et al. B-DIM impairs radiation-induced survival pathways independently of androgen receptor expression and augments radiation efficacy in prostate cancer. Cancer Lett. (2012) 318:86–92. doi: 10.1016/j.canlet.2011.12.006
162. Fraga MF, Ballestar E, Villar-Garea A, Boix-Chornet M, Espada J, Schotta G, et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet. (2005) 37:391–400. doi: 10.1038/ng1531
163. Eckschlager T, Plch J, Stiborova M, Hrabeta J. Histone deacetylase inhibitors as anticancer drugs. Int J Mol Sci. (2017) 18:1414. doi: 10.3390/ijms18071414
164. Yehuda H, Soroka Y, Zlotkin-Frušić M, Gilhar A, Milner Y, Tamir S. Isothiocyanates inhibit psoriasis-related proinflammatory factors in human skin. Inflamm Res. (2012) 61:735–42. doi: 10.1007/s00011-012-0465-3
165. Ho E, Clarke JD, Dashwood RH. Dietary Sulforaphane, a Histone Deacetylase Inhibitor for Cancer Prevention. J Nutr. (2009) 139:2393–6. doi: 10.3945/jn.109.113332
166. Beaver LM, Yu TW, Sokolowski EI, Williams DE, Dashwood RH, Ho E. 3,3'-Diindolylmethane, but not indole-3-carbinol, inhibits histone deacetylase activity in prostate cancer cells. Toxicol Appl Pharmacol. (2012) 263:345–51. doi: 10.1016/j.taap.2012.07.007
167. Rajendran P, Kidane AI, Yu T-W, Dashwood W-M, Bisson WH, Löhr CV, et al. HDAC turnover, CtIP acetylation and dysregulated DNA damage signaling in colon cancer cells treated with sulforaphane and related dietary isothiocyanates. Epigenetics. (2013) 8:612–23. doi: 10.4161/epi.24710
168. Yuanfeng W, Gongnian X, Jianwei M, Shiwang L, Jun H, Lehe M. Dietary sulforaphane inhibits histone deacetylase activity in B16 melanoma cells. J Funct Foods. (2015) 18:182–9. doi: 10.1016/j.jff.2015.07.002
169. Su ZY, Zhang C, Lee JH, Shu L, Wu TY, Khor TO, et al. Requirement and epigenetics reprogramming of Nrf2 in suppression of tumor promoter TPA-induced mouse skin cell transformation by sulforaphane. Cancer Prev Res. (2014) 7:319–29. doi: 10.1158/1940-6207.CAPR-13-0313-T
170. Wang LG, Liu XM, Fang Y, Dai W, Chiao FB, Puccio GM, et al. De-repression of the p21 promoter in prostate cancer cells by an isothiocyanate via inhibition of HDACs and c-Myc. Int J Oncol. (2008) 33:375–80. doi: 10.3892/ijo_00000018
171. Abbaoui B, Telu KH, Lucas CR, Thomas-Ahner JM, Schwartz SJ, Clinton SK, et al. The impact of cruciferous vegetable isothiocyanates on histone acetylation and histone phosphorylation in bladder cancer. J Proteomics. (2017) 156:94–103. doi: 10.1016/j.jprot.2017.01.013
172. Batra S, Sahu RP, Kandala PK, Srivastava SK. Benzyl isothiocyanate-mediated inhibition of histone deacetylase leads to NF-κB turnoff in human pancreatic carcinoma cells. Mol Cancer Ther. (2010) 9:1596–608. doi: 10.1158/1535-7163.MCT-09-1146
173. Myzak MC, Hardin K, Wang R, Dashwood RH, Ho E. Sulforaphane inhibits histone deacetylase activity in BPH-1, LnCaP and PC-3 prostate epithelial cells. Carcinogenesis. (2006) 27:811–9. doi: 10.1093/carcin/bgi265
174. Busbee PB, Nagarkatti M, Nagarkatti PS. Natural indoles, indole-3-carbinol and 3,3′-diindolymethane, inhibit T cell activation by staphylococcal enterotoxin B through epigenetic regulation involving HDAC expression. Toxicol Appl Pharmacol. (2014) 274:7–16. doi: 10.1016/j.taap.2013.10.022
175. Boyanapalli SSS, Li W, Fuentes F, Guo Y, Ramirez CN, Gonzalez XP, et al. Epigenetic reactivation of RASSF1A by phenethyl isothiocyanate (PEITC) and promotion of apoptosis in LNCaP cells. Pharmacol Res. (2016) 114:175–84. doi: 10.1016/j.phrs.2016.10.021
176. Akhavan-Niaki H, Samadani AA. DNA methylation and cancer development: molecular mechanism. Cell Biochem Biophys. (2013) 67:501–13. doi: 10.1007/s12013-013-9555-2
177. Kaufman-Szymczyk A, Majewski G, Lubecka-Pietruszewska K, Fabianowska-Majewska K. The role of sulforaphane in epigenetic mechanisms, including interdependence between histone modification and DNA methylation. Int J Mol Sci. (2015) 16:29732–43. doi: 10.3390/ijms161226195
178. Wong CP, Hsu A, Buchanan A, Palomera-Sanchez Z, Beaver LM, Houseman EA, et al. Effects of sulforaphane and 3,3′-diindolylmethane on genome-wide promoter methylation in normal prostate epithelial cells and prostate cancer cells. PLoS ONE. (2014) 9:e86787. doi: 10.1371/journal.pone.0086787
179. Hsu A, Wong CP, Yu Z, Williams DE, Dashwood RH, Ho E. Promoter de-methylation of cyclin D2 by sulforaphane in prostate cancer cells. Clin Epigenetics. (2011) 3:3. doi: 10.1186/1868-7083-3-3
180. Jiang S, Ma X, Huang Y, Xu Y, Zheng R, Chiao J-W. Reactivating aberrantly hypermethylated p15 gene in leukemic T cells by a phenylhexyl isothiocyanate mediated inter-active mechanism on DNA and chromatin. J Hematol Oncol. (2010) 3:48. doi: 10.1186/1756-8722-3-48
181. Zhang C, Su ZY, Khor TO, Shu L, Kong ANT. Sulforaphane enhances Nrf2 expression in prostate cancer TRAMP C1 cells through epigenetic regulation. Biochem Pharmacol. (2013) 85:1398–404. doi: 10.1016/j.bcp.2013.02.010
182. Meeran SM, Patel SN, Li Y, Shukla S, Tollefsbol TO. Bioactive dietary supplements reactivate ER expression in ER-negative breast cancer cells by active chromatin modifications. PLoS ONE. (2012) 7:e37748. doi: 10.1371/journal.pone.0037748
183. Meeran SM, Patel SN, Tollefsbol TO. Sulforaphane causes epigenetic repression of hTERT expression in human breast cancer cell lines. PLoS ONE. (2010) 5:e11457. doi: 10.1371/journal.pone.0011457
184. Wu TY, Khor TO, Su ZY, Saw CLL, Shu L, Cheung KL, et al. Epigenetic modifications of Nrf2 by 3,3′-diindolylmethane in vitro in TRAMP C1 cell line and in vivo TRAMP prostate tumors. AAPS J. (2013) 15:864–74. doi: 10.1208/s12248-013-9493-3
185. Farazi TA, Juranek SA, Tuschl T. The growing catalog of small RNAs and their association with distinct Argonaute/Piwi family members. Development. (2008) 135:1201–14. doi: 10.1242/dev.005629
186. Jansson MD, Lund AH. MicroRNA and cancer. Mol Oncol. (2012) 6:590–610. doi: 10.1016/j.molonc.2012.09.006
187. Pan JH, Abernathy B, Kim YJ, Lee JH, Kim JH, Shin EC, et al. Cruciferous vegetables and colorectal cancer prevention through microRNA regulation: a review. Crit Rev Food Sci Nutr. (2018) 58:2026–38. doi: 10.1080/10408398.2017.1300134
188. Slaby O, Sachlova M, Brezkova V, Hezova R, Kovarikova A, Bischofová S, et al. Identification of microRNAs regulated by isothiocyanates and association of polymorphisms inside their target sites with risk of sporadic colorectal cancer. Nutr Cancer. (2013) 65:247–54. doi: 10.1080/01635581.2013.756530
189. Zhang C, Shu L, Kim H, Khor TO, Wu R, Li W, et al. Phenethyl isothiocyanate (PEITC) suppresses prostate cancer cell invasion epigenetically through regulating microRNA-194. Mol Nutr Food Res. (2016) 60:1427–36. doi: 10.1002/mnfr.201500918
190. Hargraves KG, He L, Firestone GL. Phytochemical regulation of the tumor suppressive microRNA, miR-34a, by p53-dependent and independent responses in human breast cancer cells. Mol Carcinog. (2016) 55:486–98. doi: 10.1002/mc.22296
191. Izzotti A, Calin GA, Steele VE, Cartiglia C, Longobardi M, Croce CM, et al. Chemoprevention of cigarette smoke-induced alterations of microRNA expression in rat lungs. Cancer Prev Res. (2010) 3:62–72. doi: 10.1158/1940-6207.CAPR-09-0202
192. Melkamu T, Zhang X, Tan J, Zeng Y, Kassie F. Alteration of microRNA expression in vinyl carbamate-induced mouse lung tumors and modulation by the chemopreventive agent indole-3-carbinol. Carcinogenesis. (2010) 31:252–8. doi: 10.1093/carcin/bgp208
193. Paik WH, Kim HR, Park JK, Song BJ, Lee SH, Hwang JH. Chemosensitivity induced by down-regulation of MicroRNA-21 in gemcitabine-resistant pancreatic cancer cells by indole-3-carbinol. Anticancer Res. (2013) 33:1473–82.
194. Lan F, Pan Q, Yu H, Yue X. Sulforaphane enhances temozolomide-induced apoptosis because of down-regulation of miR-21 via Wnt/β-catenin signaling in glioblastoma. J Neurochem. (2015) 134:811–8. doi: 10.1111/jnc.13174
195. Wang DX, Zou YJ, Zhuang XB, Chen SX, Lin Y, Li WL, et al. Sulforaphane suppresses EMT and metastasis in human lung cancer through miR-616-5p-mediated GSK3β/β-catenin signaling pathways. Acta Pharmacol Sin. (2017) 38:241–51. doi: 10.1038/aps.2016.122
196. About Obesity | World Obesity Federation. Available online at: https://www.worldobesity.org/about/about-obesity
197. Virtue S, Vidal-Puig A. Adipose tissue expandability, lipotoxicity and the Metabolic Syndrome - An allostatic perspective. Biochim Biophys Acta. (2010) 1801:338–49. doi: 10.1016/j.bbalip.2009.12.006
198. The Top 10 Causes of Death. Available online at: https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death
199. Metabolic syndrome - NHS. Available online at: https://www.nhs.uk/conditions/metabolic-syndrome/
200. The IDF consensus worldwide definition of the Metabolic Syndrome. Fed Int Diabetis. (2006). Available online at: https://www.idf.org/e-library/consensus-statements/60-idfconsensus-worldwide-definitionof-the-metabolic-syndrome.html
201. About Metabolic Syndrome | American Heart Association. Available online at: https://www.heart.org/en/health-topics/metabolic-syndrome/about-metabolic-syndrome
202. Metabolic Syndrome | National Heart Lung and Blood Institute (NHLBI). Available online at: https://www.nhlbi.nih.gov/health-topics/metabolic-syndrome
203. Mathieu P, Poirier P, Pibarot P, Lemieux I, Després J-P. Visceral obesity: the link among inflammation, hypertension, and cardiovascular disease. Hypertension. (2009) 53:577–84. doi: 10.1161/HYPERTENSIONAHA.108.110320
204. Trayhurn P. Adipose tissue in obesity - An inflammatory issue. Endocrinology. (2005) 146:1003–5. doi: 10.1210/en.2004-1597
205. Clària J, González-Périz A, López-Vicario C, Rius B, Titos E. New insights into the role of macrophages in adipose tissue inflammation and fatty liver disease: modulation by endogenous omega-3 fatty acid-derived lipid mediators. Front Immunol. (2011) 2:49. doi: 10.3389/fimmu.2011.00049
206. Elgazar-Carmon V, Rudich A, Hadad N, Levy R. Neutrophils transiently infiltrate intra-abdominal fat early in the course of high-fat feeding. J Lipid Res. (2008) 49:1894–903. doi: 10.1194/jlr.M800132-JLR200
207. Kintscher U, Hartge M, Hess K, Foryst-Ludwig A, Clemenz M, Wabitsch M, et al. T-lymphocyte infiltration in visceral adipose tissue: a primary event in adipose tissue inflammation and the development of obesity-mediated insulin resistance. Arterioscler Thromb Vasc Biol. (2008) 28:1304–10. doi: 10.1161/ATVBAHA.108.165100
208. Wu H, Ghosh S, Perrard XD, Feng L, Garcia GE, Perrard JL, et al. T-cell accumulation and regulated on activation, normal T cell expressed and secreted upregulation in adipose tissue in obesity. Circulation. (2007) 115:1029–38. doi: 10.1161/CIRCULATIONAHA.106.638379
209. Rausch ME, Weisberg S, Vardhana P, Tortoriello DV. Obesity in C57BL/6J mice is characterized by adipose tissue hypoxia and cytotoxic T-cell infiltration. Int J Obes. (2008) 32:451–63. doi: 10.1038/sj.ijo.0803744
210. Feuerer M, Herrero L, Cipolletta D, Naaz A, Wong J, Nayer A, et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med. (2009) 15:930–9. doi: 10.1038/nm.2002
211. Johnson AR, Justin Milner J, Makowski L. The inflammation highway: metabolism accelerates inflammatory traffic in obesity. Immunol Rev. (2012) 249:218–38. doi: 10.1111/j.1600-065X.2012.01151.x
212. Esteve Ràfols M. Adipose tissue: cell heterogeneity and functional diversity. Endocrinol Nutr. (2014) 61:100–12. doi: 10.1016/j.endonu.2013.03.011
213. Nour M, Lutze SA, Grech A, Allman-Farinelli M. The relationship between vegetable intake and weight outcomes: a systematic review of cohort studies. Nutrients. (2018) 10:1626. doi: 10.3390/nu10111626
214. Siervo M, Montagnese C, Mathers JC, Soroka KR, Stephan BCM, Wells JCK. Sugar consumption and global prevalence of obesity and hypertension: an ecological analysis. Public Health Nutr. (2014) 17:587–96. doi: 10.1017/S1368980013000141
215. Drewnowski A. The real contribution of added sugars and fats to obesity. Epidemiol Rev. (2007) 29:160–71. doi: 10.1093/epirev/mxm011
216. Wang Y, Beydoun MA. Meat consumption is associated with obesity and central obesity among US adults. Int J Obes. (2009) 33:621–8. doi: 10.1038/ijo.2009.45
217. Jernås M, Palming J, Sjöholm K, Jennische E, Svensson P-A, Gabrielsson BG, et al. Separation of human adipocytes by size: hypertrophic fat cells display distinct gene expression. FASEB J. (2006) 20:1540–2. doi: 10.1096/fj.05-5678fje
218. Cinti S, Mitchell G, Barbatelli G, Murano I, Ceresi E, Faloia E, et al. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J Lipid Res. (2005) 46:2347–55. doi: 10.1194/jlr.M500294-JLR200
219. Radzikowska U, Rinaldi AO, Sözener ZÇ, Karaguzel D, Wojcik M, Cypryk K, et al. The influence of dietary fatty acids on immune responses. Nutrients. (2019) 11:2990. doi: 10.3390/nu11122990
220. Heilbronn L, Campbell L. Adipose tissue macrophages, low grade inflammation and insulin resistance in human obesity. Curr Pharm Des. (2008) 14:1225–30. doi: 10.2174/138161208784246153
221. Lee YS, Kim JW, Osborne O, Oh DY, Sasik R, Schenk S, et al. Increased adipocyte O2 consumption triggers HIF-1α, causing inflammation and insulin resistance in obesity. Cell. (2014) 157:1339–52. doi: 10.1016/j.cell.2014.05.012
222. Xu H, Verre MC. Type 2 diabetes mellitus in children. Am Fam Physician. (2018) 98:590–4. doi: 10.5005/jp/books/14155_31
223. Strissel KJ, Stancheva Z, Miyoshi H, Perfield JW, Defuria J, Jick Z, et al. Adipocyte death, adipose tissue remodeling, and obesity complications. Diabetes. (2007) 56:2910–8. doi: 10.2337/db07-0767
224. Chun TH, Hotary KB, Sabeh F, Saltiel AR, Allen ED, Weiss SJ. A pericellular collagenase directs the 3-dimensional development of white adipose tissue. Cell. (2006) 125:577–91. doi: 10.1016/j.cell.2006.02.050
225. Pulgaron ER, Delamater AM. Obesity and type 2 diabetes in children: epidemiology and treatment. Curr Diab Rep. (2014) 14:508. doi: 10.1007/s11892-014-0508-y
226. Lascar N, Brown J, Pattison H, Barnett AH, Bailey CJ, Bellary S. Type 2 diabetes in adolescents and young adults. Lancet Diabetes Endocrinol. (2018) 6:69–80. doi: 10.1016/S2213-8587(17)30186-9
227. Kang K, Reilly SM, Karabacak V, Gangl MR, Fitzgerald K, Hatano B, et al. Adipocyte-derived Th2 cytokines and myeloid PPARdelta regulate macrophage polarization and insulin sensitivity. Cell Metab. (2008) 7:485–95. doi: 10.1016/j.cmet.2008.04.002
228. Odegaard JI, Ricardo-Gonzalez RR, Red Eagle A, Vats D, Morel CR, Goforth MH, et al. Alternative M2 activation of kupffer cells by PPARδ ameliorates obesity-induced insulin resistance. Cell Metab. (2008) 7:496–507. doi: 10.1016/j.cmet.2008.04.003
229. Yan SF, Ramasamy R, Naka Y, Schmidt AM. Glycation, inflammation, and RAGE. Circ Res. (2003) 93:1159–69. doi: 10.1161/01.RES.0000103862.26506.3D
230. Goldin A, Beckman JA, Schmidt AM, Creager MA. Advanced glycation end products: sparking the development of diabetic vascular injury. Circulation. (2006) 114:597–605. doi: 10.1161/CIRCULATIONAHA.106.621854
231. Li J, Schmidt AM. Characterization and functional analysis of the promoter of RAGE, the receptor for advanced glycation end products. J Biol Chem. (1997) 272:16498–506. doi: 10.1074/jbc.272.26.16498
232. Dunn FL. Hyperlipidemia and diabetes. Med Clin North Am. (1982) 66:1347–60. doi: 10.1016/s0025-7125(16)31368-2
233. Bucala R, Makita Z, Vega G, Grundy S, Koschinsky T, Cerami A, et al. Modification of low density lipoprotein by advanced glycation end products contributes to the dyslipidemia of diabetes and renal insufficiency. Proc Natl Acad Sci USA. (1994) 91:9441–5. doi: 10.1073/pnas.91.20.9441
234. Klein RL, Laimins M, Lopes-Virella MF. Isolation, characterization, and metabolism of the glycated and nonglycated subfractions of low-density lipoproteins isolated from type I diabetic patients and nondiabetic subjects. Diabetes. (1995) 44:1093–8. doi: 10.2337/diab.44.9.1093
235. Lindsey JB, De Lemos JA, Cipollone F, Ayers CR, Rohatgi A, Morrow DA, et al. Association between circulating soluble receptor for advanced glycation end products and atherosclerosis: observations from the Dallas Heart Study. Diabetes Care. (2009) 32:1218–20. doi: 10.2337/dc09-0053
236. Baker RG, Hayden MS, Ghosh S. NF-κB, inflammation, and metabolic disease. Cell Metab. (2011) 13:11–22. doi: 10.1016/j.cmet.2010.12.008
237. World Cancer Research Fund/American Institute for Cancer Research. Diet, Nutrition, Physical Activity and Cancer: a Global Perspective. In: Continuous Update Project Expert Report. (2018) p. 1–53. Available onlline at: https://www.wcrf.org/dietandcancer/resources-and-toolkit
238. Morris MC, Schneider JA, Li H, Tangney CC, Nag S, Bennett DA, et al. Brain tocopherols related to Alzheimer's disease neuropathology in humans. Alzheimer's Dement. (2015) 11:32–9. doi: 10.1016/j.jalz.2013.12.015
239. Bowman GL, Dodge HH, Guyonnet S, Zhou N, Donohue J, Bichsel A, et al. A blood-based nutritional risk index explains cognitive enhancement and decline in the multidomain Alzheimer prevention trial. Alzheimer's Dement Transl Res Clin Interv. (2019) 5:953–63. doi: 10.1016/j.trci.2019.11.004
240. Serhan CN, Petasis NA. Resolvins and protectins in inflammation resolution. Chem Rev. (2011) 111:5922–43. doi: 10.1021/cr100396c
241. Juul F, Martinez-Steele E, Parekh N, Monteiro CA, Chang VW. Ultra-processed food consumption and excess weight among US adults. Br J Nutr. (2018) 120:90–100. doi: 10.1017/S0007114518001046
242. Rauber F, Louzada ML da C, Steele EM, Millett C, Monteiro CA, et al. Ultra-processed food consumption and chronic non-communicable diseases-related dietary nutrient profile in the UK (2008–2014). Nutrients. (2018) 10:E587. doi: 10.3390/nu10050587
243. Nardocci M, Leclerc BS, Louzada ML, Monteiro CA, Batal M, Moubarac JC. Consumption of ultra-processed foods and obesity in Canada. Can J Public Heal. (2019) 110:4–14. doi: 10.17269/s41997-018-0130-x
244. Martínez Steele E, Juul F, Neri D, Rauber F, Monteiro CA. Dietary share of ultra-processed foods and metabolic syndrome in the US adult population. Prev Med.(2019) 125:40–8. doi: 10.1016/j.ypmed.2019.05.004
245. Srour B, Fezeu LK, Kesse-Guyot E, Allès B, Méjean C, Andrianasolo RM, et al. Ultra-processed food intake and risk of cardiovascular disease: prospective cohort study (NutriNet-Santé). BMJ. (2019) 365:1451. doi: 10.1136/bmj.l1451
246. Monteiro CA, Moubarac JC, Levy RB, Canella DS, Da Costa Louzada ML, Cannon G. Household availability of ultra-processed foods and obesity in nineteen European countries. Public Health Nutr. (2018) 21:18–26. doi: 10.1017/S1368980017001379
247. Monteiro CA, Cannon G, Levy RB, Moubarac JC, Louzada MLC, Rauber F, et al. Ultra-processed foods: What they are and how to identify them. Public Health Nutr. (2019) 22:936–41. doi: 10.1017/S1368980018003762
248. Neri D, Martinez-Steele E, Monteiro CA, Levy RB. Consumption of ultra-processed foods and its association with added sugar content in the diets of US children, NHANES 2009-2014. Pediatr Obes. (2019) 14:e12563. doi: 10.1111/ijpo.12563
249. Rauber F, Louzada MLDC, Martinez Steele E, De Rezende LFM, Millett C, Monteiro CA, et al. Ultra-processed foods and excessive free sugar intake in the UK: a nationally representative cross-sectional study. BMJ Open. (2019) 9:e027546. doi: 10.1136/bmjopen-2018-027546
250. Machado PP, Steele EM, Louzada ML da C, Levy RB, Rangan A, et al. Ultra-processed food consumption drives excessive free sugar intake among all age groups in Australia. Eur J Nutr. (2020) 59:2783–92. doi: 10.1007/s00394-019-02125-y
251. MacHado PP, Steele EM, Levy RB, Sui Z, Rangan A, Woods J, et al. Ultra-processed foods and recommended intake levels of nutrients linked to non-communicable diseases in Australia: Evidence from a nationally representative cross-sectional study. BMJ Open. (2019) 9:e029544. doi: 10.1136/bmjopen-2019-029544
252. Zhang Z, Zhou S, Jiang X, Wang YH, Li F, Wang YG, et al. The role of the Nrf2/Keap1 pathway in obesity and metabolic syndrome. Rev Endocr Metab Disord. (2015) 16:35–45. doi: 10.1007/s11154-014-9305-9
253. Vasileva LV, Savova MS, Amirova KM, Dinkova-Kostova AT, Georgiev MI. Obesity and NRF2-mediated cytoprotection: Where is the missing link? Pharmacol Res. (2020) 156:104760. doi: 10.1016/j.phrs.2020.104760
254. Seo HA, Lee IK. The role of NRF2: adipocyte differentiation, obesity, and insulin resistance. Oxid Med Cell Longev. (2013) 2013:184598. doi: 10.1155/2013/184598
255. Patel B, Mann GE, Chapple SJ. Concerted redox modulation by sulforaphane alleviates diabetes and cardiometabolic syndrome. Free Radic Biol Med. (2018) 122:150–60. doi: 10.1016/j.freeradbiomed.2018.02.004
256. Okulicz M. Multidirectional time-dependent effect of sinigrin and allyl isothiocyanate on metabolic parameters in rats. Plant Foods Hum Nutr. (2010) 65:217–24. doi: 10.1007/s11130-010-0183-3
257. Bai Y, Wang X, Zhao S, Ma C, Cui J, Zheng Y. Sulforaphane protects against cardiovascular disease via Nrf2 activation. Oxid Med Cell Longev. (2015) 2015:407580. doi: 10.1155/2015/407580
258. Xu L, Nagata N, Ota T. Glucoraphanin: a broccoli sprout extract that ameliorates obesity-induced inflammation and insulin resistance. Adipocyte. (2018) 7:218–25. doi: 10.1080/21623945.2018.1474669
259. Nagata N, Xu L, Kohno S, Ushida Y, Aoki Y, Umeda R, et al. Glucoraphanin ameliorates obesity and insulin resistance through adipose tissue browning and reduction of metabolic endotoxemia in mice. Diabetes. (2017) 66:1222–36. doi: 10.2337/db16-0662
260. Kang B, Kim CY, Hwang J, Suh HJ, Choi H. Brassinin, a phytoalexin in cruciferous vegetables, suppresses obesity-induced inflammatory responses through the Nrf2-HO-1 signaling pathway in an adipocyte-macrophage co-culture system. Phyther Res. (2019) 33:1426–37. doi: 10.1002/ptr.6333
261. Yang H, Kang MJ, Hur G, Lee TK, Park IS, Seo SG. Sulforaphene suppresses adipocyte differentiation via induction of post-translational degradation of CCAAT/Enhancer binding protein beta (C/EBPβ). Nutrients. (2020) 12:758. doi: 10.3390/nu12030758
262. Choi KM, Lee YS, Sin DM, Lee S, Lee MK, Lee YM, et al. Sulforaphane inhibits mitotic clonal expansion during adipogenesis through cell cycle arrest. Obesity. (2012) 20:1365–71. doi: 10.1038/oby.2011.388
263. Chen J, Bao C, Kim JT, Cho JS, Qiu S, Lee HJ. Sulforaphene inhibition of adipogenesis via hedgehog signaling in 3T3-L1 adipocytes. J Agric Food Chem. (2018) 66:11926–34. doi: 10.1021/acs.jafc.8b04330
264. Chae SY, Seo SG, Yang H, Yu JG, Suk SJ, Jung ES, et al. Anti-adipogenic effect of erucin in early stage of adipogenesis by regulating Ras activity in 3T3-L1 preadipocytes. J Funct Foods. (2015) 19:700–9. doi: 10.1016/j.jff.2015.09.060
265. Yang H, Seo SG, Shin SH, Min S, Kang MJ, Yoo R, et al. 3,3'-Diindolylmethane suppresses high-fat diet-induced obesity through inhibiting adipogenesis of pre-adipocytes by targeting USP2 activity. Mol Nutr Food Res. (2017) 61:1700119. doi: 10.1002/mnfr.201700119
266. Chuang WT, Liu YT, Huang CS, Lo CW, Yao HT, Chen HW, et al. Benzyl isothiocyanate and phenethyl isothiocyanate inhibit adipogenesis and hepatosteatosis in mice with obesity induced by a high-fat diet. J Agric Food Chem. (2019) 67:7136–46. doi: 10.1021/acs.jafc.9b02668
267. Chang HP, Wang ML, Hsu CY, Liu ME, Chan MH, Chen YH. Suppression of inflammation-associated factors by indole-3-carbinol in mice fed high-fat diets and in isolated, co-cultured macrophages and adipocytes. Int J Obes. (2011) 35:1530–8. doi: 10.1038/ijo.2011.12
268. Choi KM, Lee YS, Kim W, Kim SJ, Shin KO, Yu JY, et al. Sulforaphane attenuates obesity by inhibiting adipogenesis and activating the AMPK pathway in obese mice. J Nutr Biochem. (2014) 25:201–7. doi: 10.1016/j.jnutbio.2013.10.007
269. Choi HS, Jeon HJ, Lee OH, Lee BY. Indole-3-carbinol, a vegetable phytochemical, inhibits adipogenesis by regulating cell cycle and AMPKα signaling. Biochimie. (2014) 104:127–36. doi: 10.1016/j.biochi.2014.06.010
270. Yao A, Shen Y, Wang A, Chen S, Zhang H, Chen F, et al. Sulforaphane induces apoptosis in adipocytes via Akt/p70s6k1/Bad inhibition and ERK activation. Biochem Biophys Res Commun. (2015) 465:696–701. doi: 10.1016/j.bbrc.2015.08.049
271. Li CX, Gao JG, Wan XY, Chen Y, Xu CF, Feng ZM, et al. Allyl isothiocyanate ameliorates lipid accumulation and inflammation in nonalcoholic fatty liver disease via the Sirt1/AMPK and NF-κB signaling pathways. World J Gastroenterol. (2019) 25:5120–33. doi: 10.3748/wjg.v25.i34.5120
272. Yagi M, Nakatsuji Y, Maeda A, Ota H, Kamikubo R, Miyoshi N, et al. Phenethyl isothiocyanate activates leptin signaling and decreases food intake. PLoS ONE. (2018) 13:e0206748. doi: 10.1371/journal.pone.0206748
273. Mori N, Kurata M, Yamazaki H, Hosokawa H, Nadamoto T, Inoue K, et al. Intragastric administration of allyl isothiocyanate reduces hyperglycemia in intraperitoneal glucose tolerance test (IPGTT) by enhancing blood glucose consumption in mice. J Nutr Sci Vitaminol. (2013) 59:56–63. doi: 10.3177/jnsv.59.56
274. Ahn J, Lee H, Im SW, Jung CH, Ha TY. Allyl isothiocyanate ameliorates insulin resistance through the regulation of mitochondrial function. J Nutr Biochem. (2014) 25:1026–34. doi: 10.1016/j.jnutbio.2014.05.006
275. Jayakumar P, Pugalendi KV, Sankaran M. Attenuation of hyperglycemia-mediated oxidative stress by indole-3-carbinol and its metabolite 3, 3′-diindolylmethane in C57BL/6J mice. J Physiol Biochem. (2014) 70:525–34. doi: 10.1007/s13105-014-0332-5
276. Huang C, Wu J, Chen D, Jin J, Wu Y, Chen Z. Effects of sulforaphane in the central nervous system. Eur J Pharmacol. (2019) 853:153–68. doi: 10.1016/j.ejphar.2019.03.010
277. Christiansen B, Bellostas Muguerza N, Petersen AM, Kveiborg B, Madsen CR, Thomas H, et al. Ingestion of broccoli sprouts does not improve endothelial function in humans with hypertension. PLoS ONE. (2010) 5:e12461. doi: 10.1371/journal.pone.0012461
278. Bahadoran Z, Tohidi M, Nazeri P, Mehran M, Azizi F, Mirmiran P. Effect of broccoli sprouts on insulin resistance in type 2 diabetic patients: a randomized double-blind clinical trial. Int J Food Sci Nutr. (2012) 63:767–71. doi: 10.3109/09637486.2012.665043
279. Kikuchi M, Ushida Y, Shiozawa H, Umeda R, Tsuruya K, Aoki Y, et al. Sulforaphane-rich broccoli sprout extract improves hepatic abnormalities in male subjects. World J Gastroenterol. (2015) 21:12457–67. doi: 10.3748/wjg.v21.i43.12457
280. Schiess S, Platz S, Kemper M, Schreiner M, Mewis I, Rohn S, et al. Oral administration of nasturtium affects peptide YY secretion in male subjects. Mol Nutr Food Res. (2017) 61:1600886. doi: 10.1002/mnfr.201600886
281. Langeveld M, Tan CY, Soeters MR, Virtue S, Watson LPE, Murgatroyd PR, et al. No metabolic effects of mustard allyl-isothiocyanate compared with placebo in men. Am J Clin Nutr. (2017) 106:1197–205. doi: 10.3945/ajcn.116.148395
282. López-Chillón MT, Carazo-Díaz C, Prieto-Merino D, Zafrilla P, Moreno DA, Villaño D. Effects of long-term consumption of broccoli sprouts on inflammatory markers in overweight subjects. Clin Nutr. (2019) 38:745–52. doi: 10.1016/j.clnu.2018.03.006.This
283. Kaiser J. Antioxidants could spur tumors by acting on cancer gene. Science. (2014) 343:477. doi: 10.1126/science.343.6170.477
Keywords: cruciferous, glucosinolates, isothiocyanates, indoles, metabolic syndrome, inflammation, Nuclear erythroid 2-related factor (Nrf2), Nuclear Factor-κB (NF-κB)
Citation: Esteve M (2020) Mechanisms Underlying Biological Effects of Cruciferous Glucosinolate-Derived Isothiocyanates/Indoles: A Focus on Metabolic Syndrome. Front. Nutr. 7:111. doi: 10.3389/fnut.2020.00111
Received: 02 April 2020; Accepted: 16 June 2020;
Published: 02 September 2020.
Edited by:
Daniele Vergara, University of Salento, ItalyReviewed by:
Emily Ho, Oregon State University, United StatesCopyright © 2020 Esteve. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Montserrat Esteve, bWVzdGV2ZUB1Yi5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.