 Inez Trouwborst
Inez Trouwborst Suzanne M. Bowser
Suzanne M. Bowser Gijs H. Goossens
Gijs H. Goossens Ellen E. Blaak
Ellen E. Blaak- Department of Human Biology, NUTRIM School of Nutrition and Translational Research in Metabolism, Maastricht University Medical Center+, Maastricht, Netherlands
Cardiometabolic diseases are one of the leading causes for disability and mortality in the Western world. The prevalence of these chronic diseases is expected to rise even further in the next decades. Insulin resistance (IR) and related metabolic disturbances are linked to ectopic fat deposition, which is the storage of excess lipids in metabolic organs such as liver and muscle. Notably, a vicious circle exists between IR and ectopic fat, together increasing the risk for the development of cardiometabolic diseases. Nutrition is a key-determining factor for both IR and ectopic fat deposition. The macronutrient composition of the diet may impact metabolic processes related to ectopic fat accumulation and IR. Interestingly, however, the metabolic phenotype of an individual may determine the response to a certain diet. Therefore, population-based nutritional interventions may not always lead to the most optimal (cardiometabolic) outcomes at the individual level, and differences in the metabolic phenotype may underlie conflicting findings related to IR and ectopic fat in dietary intervention studies. Detailed metabolic phenotyping will help to better understand the complex relationship between diet and metabolic regulation, and to optimize intervention outcomes. A subgroup-based approach that integrates, among others, tissue-specific IR, cardiometabolic parameters, anthropometrics, gut microbiota, age, sex, ethnicity, and psychological factors may thereby increase the efficacy of dietary interventions. Nevertheless, the implementation of more personalized nutrition may be complex, costly, and time consuming. Future studies are urgently warranted to obtain insight into a more personalized approach to nutritional interventions, taking into account the metabolic phenotype to ultimately improve insulin sensitivity and reduce the risk for cardiometabolic diseases.
Introduction
Cardiometabolic diseases such as obesity, diabetes mellitus type 2 (T2DM), and cardiovascular diseases are the leading causes of death and disability in the Western world. In 2014, T2DM affected about 422 million adults worldwide, and its prevalence is expected to rise even further in the next decades (1), representing a large burden for society. Although different etiologies precede the development of cardiometabolic diseases, insulin resistance (IR) is a major contributing risk factor, and is therefore a relevant target for preventive healthcare.
IR represents a physiological state in which the action of the hormone insulin is impaired. As a consequence, the body is not effectively able to adapt to its metabolic or energy demands, also described as metabolic inflexibility (2, 3). IR can develop simultaneously in multiple organs and the IR severity may vary between different organs. More specifically, the regulation of metabolic processes such as glucose uptake and oxidation, glycogen synthesis and breakdown, lipid storage, and lipolysis may be disturbed. A wide range of literature has linked these metabolic disturbances to accumulation of excess lipids in organs such as the liver, skeletal muscle, pancreas, and heart, also referred to as ectopic fat deposition (4, 5). The link between ectopic fat, IR and metabolic inflexibility, however, is multifaceted. The type of tissue (e.g., liver or skeletal muscle), the accumulation of lipids (e.g., triacylglcyerol, diacylglycerol) and lipid metabolites (e.g., ceramides and acylcarnitines) as well as composition, turnover, and localization of lipids have all been shown to play important roles in the metabolic consequences of ectopic fat accumulation (6, 7). More globally, age, sex, lifestyle, and (epi)genetics, as well as microbial composition and functionality, may also play an important role in ectopic fat accumulation (5, 8, 9). These factors impact the development of distinct IR phenotypes, and should be considered when targeting ectopic fat and IR.
Diet and physical activity are key modifiable risk factors that play a significant role in the development, progression and reversal of IR and ectopic fat accumulation (5, 10). Related to the diet, not only caloric intake, but also the macronutrient quantity and quality of the diet may affect insulin sensitivity, which may be mediated by changes in ectopic fat accumulation (11–14). Interestingly, however, recent evidence has shown that the metabolic phenotype of an individual seems to contribute to the inter-individual differences in response to dietary intervention (13, 15). Therefore, interventions to prevent or reverse IR may be optimized using a more subgroup-based or personalized approach.
In this review, we will provide an overview of the mechanisms that play a role in ectopic fat storage and its relationship to distinct IR phenotypes. Additionally, it will be discussed how dietary macronutrient quality and quantity may affect ectopic fat and, consequently, IR in individuals with different metabolic phenotypes.
Ectopic Fat and Insulin Resistance: Pathophysiology and Inter-Relationships
Cellular Mechanism Leading to Ectopic Fat and IR
IR conditions are often accompanied by a systemic overflow of lipids, which is mainly the result of disturbances in adipose tissue lipid handling, in combination with a chronic excess energy intake (2). Disturbances in adipose tissue function are, beside an impaired lipid storage capacity, characterized by an increased infiltration of macrophages and other immune cells leading to a state of chronic low-grade inflammation in obesity (2). Adipose tissue inflammation may induce alterations in adipose tissue metabolism (i.e., lipid metabolism/lipolysis) and the storage capacity of dietary lipid in adipose tissue (2, 16). The excessive flux of lipids toward multiple oxidative tissues, including the liver and skeletal muscle, has marked effects on lipid uptake and metabolism in these tissues, thereby contributing to the development of ectopic fat deposition and IR. Furthermore, the increased secretion of inflammatory factors by adipose tissue may lead to systemic inflammation, which in turn may affect insulin signaling and metabolism of non-adipose tissues, like the liver and skeletal muscle, contributing to the accumulation of ectopic fat and development of IR (2). The mechanisms by which ectopic fat accumulates and how this accumulation affects IR are, however, different between tissues, and has been extensively studied for the skeletal muscle (17). Importantly, the link between ectopic fat and IR is not a one-directional pathway. Accumulation of lipids within the liver, for example, may also be driven by increased de novo lipogenesis due to hyperinsulinemia, as present in IR conditions, and by increased NEFA supply from the adipose tissue (18, 19). Additionally, a reduced capacity to oxidize fatty acids upon increased supply in skeletal muscle (metabolic inflexibility) is often observed in obese insulin resistant conditions, which may be accompanied by mitochondrial dysfunction (2). Hence, a vicious cycle exists between the progression of IR and ectopic fat deposition. In the following sections, we will provide an overview of the mechanisms underlying the accumulation of ectopic fat in the liver and skeletal muscle and its effect on IR.
Disturbances in Liver and Skeletal Muscle Lipid Uptake
IR individuals are characterized by an altered plasma lipid profile (20). In fasted and postprandial IR conditions, (slightly) elevated levels of non-esterified fatty acids (NEFA), very-low-density lipoprotein (VLDL)-TAG (21), and dietary-derived chylomicron-TAG (22) are observed, although data is not entirely consistent (23, 24). Notably, NEFA concentrations may not be elevated in proportion to the increased fat mass, since NEFA concentrations per unit fat mass are down regulated in individuals with excess adipose tissue mass (24). Elevated levels of circulating TAG concentrations may be attributed to an increased VLDL-TAG production by the liver or reduced clearance of TAG by adipose tissue (2). Indeed, the removal of TAG across adipose tissue was found to be impaired in obesity, insulin resistance and T2DM due to a reduced insulin-mediated stimulation of lipoprotein lipase (LPL) activity (23, 25–29), suggesting less efficient removal of dietary lipids by adipose tissue in these subjects. Notably, LPL in AT macrophages has been found to increase AT lipid storage in obese mice (30, 31). In line, studies in high-fat-diet-fed mice showed local lipid fluxes are central regulators of AT macrophage recruitment and that once recruited these macrophages can buffer local increases in lipid concentrations (30). These data suggest that AT macrophages may compensate for the insufficient lipid storage in adipocytes in the obese insulin resistant state, reversing thereby glucose intolerance and insulin resistance.
Despite slightly elevated plasma NEFA concentrations in T2DM as compared to healthy control subjects, a similar postprandial NEFA uptake in skeletal muscle was observed in both groups (32). Likewise, we have recently demonstrated a comparable skeletal muscle uptake of circulating NEFA in overweight and obese subjects with mild vs. more severe IR (33). Elevated plasma NEFA levels may, however, lead to increased uptake of NEFA into the liver (34). An increased supply of NEFA to the liver leads to an increased partitioning of NEFA toward VLDL-TAG synthesis, and may be accompanied by a decreased insulin-mediated suppression of VLDL-TAG synthesis (35). Both processes contribute to elevated postprandial VLDL-TAG levels in IR. Interestingly, skeletal muscle extraction of VLDL-TAG in postprandial conditions is higher in more severe vs. mild IR individuals, despite similar VLDL-TAG supply (7, 33). In accordance with these findings, IR subjects show higher skeletal muscle VLDL-TAG extraction than normal glucose tolerant (NGT) individuals (36). Together, these findings indicate that disturbances in lipid supply and/or uptake in both liver and skeletal muscle may contribute to the pathogenesis of ectopic fat deposition and IR.
Plasma lipids can be transported into the liver or skeletal muscle cell by receptor-mediated transport, as well as passive diffusion via the cell membrane (37). The rate of lipid uptake is dependent on the type of tissue, type, and activity of transporter proteins on the cell membrane, and is a key factor in lipid handling by the liver or skeletal muscle cells. LPL in skeletal muscle and the membrane carrier proteins (specifically CD36) and fatty acid transport proteins (FATP) in liver and skeletal muscle are, therefore, an integral part of this process. LPL is, next to its large abundance in adipose tissue, expressed in endothelial cells near skeletal muscle and is an important facilitator of chylomicron- and VLDL-TAG extraction across the endothelium into the skeletal muscle cell (38). LPL has the capacity to bind and hydrolyze TAG on the surface of the capillary lumen, thereby releasing NEFA which can be taken up by the tissue (39). FATPs are involved in the transport of long-chain fatty acids across the membrane. Different members of the FATP family are expressed in the liver (FATP3,5) and skeletal muscle (FATP3,4), but may have similar properties related to lipid uptake (40). CD36 is a well-characterized protein that is also responsible for the uptake of a variety of lipids in the cell. Expression of FATPs and CD36 in liver and skeletal muscle is often dependent on the metabolic condition. Overexpression of these transport proteins is often seen in response to hyperinsulinemia, and may lead to an increased uptake of lipids in IR conditions, which in turn contributes to ectopic fat accumulation in these tissues and associated metabolic impairments (38, 41–44). Interestingly, this may not be the case for LPL in skeletal muscle since insulin seems to stimulate LPL activity in adipose tissue, but not in skeletal muscle (45).
Altogether, human data suggest that merely TAG rather than NEFA uptake into the skeletal muscle may be a putative target in the prevention and/or treatment of ectopic fat deposition and IR in individuals with an impaired oxidative capacity. The role of disturbances in lipid uptake by the liver in relation to liver fat accumulation and hepatic IR is less established.
Disturbances in Intramyocellular Lipid Metabolism
In addition to lipid uptake, insulin sensitivity is strongly related to the oxidative capacity of key metabolic organs such as the liver and the skeletal muscle. The high oxidative capacity represents the ability of the body to adapt to the appropriate metabolic condition, also described as metabolic flexibility (46). Following uptake from the circulation, lipids can either be oxidized or stored for later use. In healthy, insulin sensitive individuals, lipid accumulation within the cell is limited, albeit not absent, as an elevated influx of lipids into the cell triggers the conversion of lipids to long-chain fatty acyl-CoA (LC-CoA), which is transported into the mitochondria and, consequently, degraded by beta-oxidation.
In contrast, IR conditions are characterized by disturbed skeletal muscle lipid metabolism (2). In obese individuals with more severe as compared to mild IR the following has been reported: a higher saturation of skeletal muscle NEFA and DAG (in particular membrane-bound DAG), a tendency toward a lower fractional synthesis of TAG, and reduced gene expression of oxidative metabolism markers (33, 36, 47). In line, in overweight and obese adults, more severe IR individuals showed lower re-esterification of intramuscular NEFA into TAG, and decreased expression of several genes encoding for proteins involved in TAG synthesis as compared to individuals with mild IR (7). Additionally, in individuals with long-term diagnosed T2DM, a blunted insulin-mediated suppression of skeletal muscle lipolysis was reported, which was related to accumulation of more saturated DAG at the skeletal muscle membrane as compared to normal glucose tolerant (NGT) individuals (47).
These disturbances reflect, at least partly, a decreased capacity of the skeletal muscle to oxidize lipids (48) and a decreased ability to switch from carbohydrate to fat oxidation (49). The decreased skeletal muscle oxidative capacity may be a result of reduced mitochondrial activity, content, and/or plasticity, leading to an imbalance between lipid supply, lipid oxidation, and conversion into TAG (50). Furthermore, decreased mitochondrial function may lead to overproduction of reactive oxygen species (ROS), which are reactive molecules and free radicals, further contributing to impaired mitochondrial function and oxidative stress (51). The reduced oxidative capacity may contribute to lipid peroxidation whereby lipotoxic compounds are formed in the skeletal muscle, including diacylglycerol (DAG) and ceramides (48, 52). DAG and ceramides may have lipotoxic effects on mitochondrial DNA, RNA, and proteins, thereby promoting a further decline in mitochondrial bioenergetics (48). In addition, DAG are responsible for the activation of protein kinase C (PKC) isoforms. PKCs are important signaling molecules involved in insulin-stimulated glucose uptake, since PKC inhibits several steps of the insulin signaling pathway (53). As a consequence, PKC activation by lipid-intermediates interferes with insulin signaling, resulting in a reduced skeletal muscle glucose uptake, as extensively reviewed elsewhere (5). The relevance of ceramide accumulation in muscle insulin resistance in humans is still unclear (17). These findings illustrate the important role of lipotoxic lipid-intermediates in the development and progression of IR in the skeletal muscle.
Disturbances in Intrahepatocellular Lipid Metabolism
As discussed, lipid accumulation within the liver has been linked to IR. The mechanisms within hepatocytes that link lipid metabolism to IR are, however, less understood. It has been shown that insulin increases hepatic (de novo) lipogenesis, TAG storage, as well as the production of VLDL-TAG in the liver in IR conditions (17). Moreover, similar to skeletal muscle, an impaired mitochondrial function in the liver has been found in obese IR individuals (17). The link between mitochondrial dysfunction in the liver and IR may be explained by excess lipid supply, as often seen in IR conditions. The chronic increased activation of beta-oxidation due to excess lipid supply may stimulate oxidative stress and lipotoxicity, in turn resulting in impairments in mitochondrial oxidative capacity (54). In line, in vivo MRS measurements showed that IR-individuals exhibited lower hepatic ATP levels, which related to liver-IR and partly accounted for hepatic lipid accumulation (55). However, a causal relationship between mitochondrial function and hepatic IR has yet to be confirmed.
Impaired mitochondrial function in the liver may lead to reduced oxidation of lipids, resulting in the formation of different lipid moieties such as ceramides (54). The accumulation of both total DAG and cytosolic DAG in the liver have been linked to HOMA-IR as well as to liver-IR, as determined by insulin-induced suppression of hepatic glucose output (56, 57). Peterson et al. (58) hypothesized that DAG mediates lipid-induced IR in the liver by inhibition of insulin signaling in a similar manner as occurs in skeletal muscle. As discussed earlier, DAG may activate several PKC isoforms, thereby inhibiting several steps of the insulin signaling pathway. Furthermore, DAG accumulation in the liver could result in decreased mitochondrial function, inflammation, and increased VLDL-TAG production, which may affect IR on a local (liver-IR) and systemic level. Consequently, DAG accumulation in the liver may contribute to liver-IR and, consequently, to decreased glycogen synthesis and increased hepatic gluconeogenesis (2, 17, 58). In humans, there is currently no evidence for a relationship between ceramide accumulation and impaired hepatic insulin action (17, 56, 57). It must be noted that the number of studies related to lipid metabolism in the liver and IR are limited, studies are predominantly cross-sectional, or in vitro models have been used, which makes it difficult to understand the molecular mechanisms involved in the putative relationship between disturbances in hepatic lipid metabolism and liver IR.
To summarize, cellular mechanisms responsible for IR and lipid accumulation in both liver and skeletal muscle are related to altered lipid uptake into the cell as well as to an impaired intracellular regulation of lipid metabolism. Together, these impairments may cause an excess accumulation of lipotoxic metabolites if oxidative capacity is insufficient, which in turn interferes with insulin signaling.
Ectopic Fat and Liver- and Muscle-IR
As mentioned earlier, IR can occur in multiple key metabolic organs such as the liver and skeletal muscle. Nevertheless, insulin sensitivity and lipid metabolism may substantially differ between organs within an individual (59). Moreover, the extent to which IR is present in these distinct organs may vary among individuals. These inter-individual differences in tissue-specific insulin sensitivity may partly be explained by the location where excess lipids are stored. Ectopic fat and lipotoxic intermediates affect metabolism on a local tissue level, as described above. Thus, intrahepatocellular lipids (IHCL) and intramyocellular lipids (IMCL) are therefore often linked to the development of liver-IR and muscle-IR, respectively (5). Nonetheless, ectopic fat is not only related to site-specific disturbances in glucose homeostasis and IR, but also to more systemic disturbances (40). It could be speculated that tissue specific-IR may lead to a redistribution of substrates from the IR tissue toward other tissues during postprandial conditions (60). For example, muscle-IR may promote postprandial glucose supply to the liver, leading to increased de novo lipogenesis and hepatic steatosis (61, 62). This highlights the complex relationship between ectopic fat deposition and tissue-specific IR. Also, it should be noted that liver- and muscle-IR often coexist, which may bi-directionally affect metabolic perturbations in both organs. However, irrespective of the relationship between liver and skeletal muscle fat accumulation and liver- and muscle-IR, both IHCL and IMCL seem to play an important role in the progression toward T2DM (2). Clearly, multiple factors play a role in the complex relationship between ectopic fat deposition and tissue specific-IR, and careful interpretation of research findings is therefore needed.
Interestingly, plasma lipid profiles and lipid handling seem to differ between various IR-states, contributing to ectopic fat accumulation in both liver and skeletal muscle (33, 63). More specifically, higher fasting and postprandial plasma TAG, FFA, and VLDL-TAG concentrations have been observed in individuals with IGT as compared to impaired fasting glucose (IFG) (63–66), and in T2DM vs. non-diabetic individuals (67). Furthermore, an early postprandial increase in TAG concentration and higher skeletal muscle uptake TAG was observed in both IFG and IGT as compared to NGT (63). Additionally, IGT individuals showed higher skeletal muscle uptake of VLDL-TAG, higher intramuscular TAG content and higher intramuscular saturation of FFA compared to IFG individuals (33).
Interestingly, although IGT and IFG are both IR-states, these seem to differ in their tissue-specific IR, as liver-IR and muscle-IR seem to be the primary disorders in IFG and IGT, respectively (10, 68–70). Consistently, IGT is characterized by a decreased peripheral glucose disposal (71), whereas IFG is characterized by impaired insulin-induced suppression of liver gluconeogenesis and glycogenolysis (69). Not surprisingly, both IFG and IGT have been linked to the accumulation of ectopic fat in liver and skeletal muscle (72–74). Interestingly however, liver-IR is often considered to be a more severe IR-state as compared to muscle-IR. To exemplify, liver-IR seems to be accompanied with elevated postprandial levels of TAG and VLDL compared to muscle-IR (67), and has been linked to more severe cardiometabolic complications (75). This seems opposite to the finding that IGT is accompanied by higher fasting and postprandial plasma TAG and VLDL-TAG levels (63–66). These findings emphasize that the relationship between ectopic fat deposition, glucometabolic status (i.e., NGT, IFG, IGT, T2DM) and IR is complex, and suggest that these inter-relationships are dependent on certain exogenous (e.g., sex, ethnicity, and age) and/or endogenous factors (e.g., physical activity and diet). A better understanding of the different factors involved in these metabolic perturbations is needed to develop more personalized strategies to prevent or reverse IR, as discussed below.
Endogenous and Exogenous Factors Affect Ectopic Fat and Liver- and Muscle-IR
Endogenous Factors
Sex-differences in health and disease is a well-recognized concept in literature (76–78). More specifically, sex differences have been linked to ectopic fat accumulation and IR (9). Difference between sexes may be partly related to differences in body fat distribution between men and women. Generally, women have a higher percentage of subcutaneous adipose tissue (SAT) as compared to BMI-matched men, who generally have more visceral adipose tissue (VAT) (79, 80). Interestingly, the expandability of subcutaneous adipose tissue seems to be a critical factor in the development of insulin resistance (81, 82). Lipids may be predominantly stored in SAT before marked VAT expansion occurs (81, 83). VAT has been linked to higher levels of inflammatory markers, insulin resistance and other cardiometabolic complications (83, 84). Moreover, in general women accumulate more adipose tissue in the gluteo-femoral as compared to the abdominal fat depot. Abdominal obesity is associated with an increased risk of developing type 2 diabetes and cardiovascular diseases (85). In contrast, lower body fat has protective properties that are associated with an improved cardiometabolic risk profile in men and women (86, 87). These metabolic differences in adipose tissue depots may partially explain why women are relatively protected against metabolic diseases compared to men with the same BMI (84).
Furthermore, although not completely elucidated, the metabolic differences between men and women are, at least partly, a consequence of differences in hormonal status. Estrogen has been shown to reduce IHCL accumulation and IR in both sexes (77). Until menopause, women have a lower risk for developing fatty liver, whereas post-menopausal women have a similar risk compared to age-matched males. The increased risk after menopause may be the result of the marked drop in estrogen concentrations during the menopausal phase (88). Indeed, estrogen treatment in the menopausal phase is protective for the development of non-alcoholic fatty liver (89). Furthermore, BMI was reported to be independently related to IHCL content in both men and women, whereas postprandial glucose level was independently related to IHCL content in women only (90). In contrast to IHCL content, some studies reported that IMCL content was significantly lower in both lean and overweight men as compared to BMI matched pre- and post-menopausal women (91–93), although conflicting data have also been reported (94). Noteworthy, for a given BMI, men generally have more muscle mass as compared to women, which may at least partly explain the latter findings (8). Interestingly, despite a higher IMCL content in women, insulin sensitivity and plasma lipid levels were similar between men and women, suggesting that women are relatively protected against lipid-induced IR (93). The relative protection against IMCL was, however, not observed in women using oral contraceptives (93). Interestingly, higher IMCL, lower fractional synthesis rate (FSR) and lower oxidative capacity was observed in prediabetic vs. NGT men, while no differences were observed in these parameters in prediabetes vs. NGT age and anthropometry matched post-menopausal obese women (95). Together, these findings suggest sex-specific differences in both IHCL and IMCL content. Whether these sex-specific differences in ectopic fat are dependent or independently related to IR, and which mechanisms are involved, is not yet fully understood and requires further investigation (59).
The relationship between ectopic fat storage, IR and sex seem to be largely dependent on the population. IMCL was significantly associated with IR in European Americans, while in African Americans, IMCL varied independent of IR (96). Another study reported a stronger association between IHCL and IR in individuals born in Iraq as compared to individuals born in Sweden (97). Moreover, it is well-established that South-Asians are more likely to develop IR and have more ectopic fat (both IHCL and IMCL) when compared to BMI-matched Caucasians (98–101). A possible explanation for this could be that South-Asians have a decreased lipid storage capacity in subcutaneous adipose tissue, leading to an excess flux of lipids toward other tissues such as visceral adipose tissue, the liver, pancreas, heart, and skeletal muscle (102). Interestingly, when IHCL and IMCL content in South-Asian and Caucasian men were adjusted for whole-body insulin sensitivity, IMCL was similar, while Asian men maintained a two-fold higher IHCL content, compared to Caucasian men (100). These findings suggest that the amount of IHCL, in contrast to IMCL content, varied independent of whole-body insulin sensitivity in South-Asians. In a study of Goedecke et al. (103), IHCL content was lower and IMCL content was higher in pre-menopausal black vs. white women. Interestingly, this same study reported that the association between ectopic fat (both IHCL and IMCL) and IR was only present in black women (103). Noteworthy, the total amount of lipids rather than the concentration of toxic lipid-intermediates was measured in these studies. Therefore, it remains uncertain whether the relationship between lipid-intermediates in liver and skeletal muscle and insulin sensitivity depends on ethnicity. Differences between ectopic fat and IR in these populations may be the result of certain genetic polymorphisms. For example, polymorphism of the ApoC3 and PNPLA3 gene have been linked to alterations in IHCL, and are more prevalent in South-Asian and Hispanic individuals, respectively (52, 104, 105). To conclude, the studies described above illustrate that the amount of ectopic fat may not always be directly related to IR, and seems to differ between populations.
Age seems to be a very relevant factor in the etiology and pathophysiology of IR, and seems to be positively related to ectopic fat accumulation (8, 106). Interestingly, there also appears to be an interaction between sex and age, especially related to the pre- or post-menopausal state of women, as already indicated above. More specific, pre-menopausal women are as insulin sensitive as age-matched men, despite higher IMCL content, while this relative protection against IR disappears after menopause (93). However, whether relationships between ectopic fat, IR, age and sex remain after adjustment for visceral fat or total fat mass is not fully known, but has been extensively reviewed elsewhere (8).
Exogenous Factors
Lifestyle factors, such as unhealthy diet and a lack of physical activity, are key factors for the accumulation of ectopic fat and the development of IR (5, 10). Physical activity, with its different modalities such as resistance, endurance, or concurrent exercise, exerts several metabolic advantages. For example, improvements not only in cardiovascular fitness but also ectopic fat and insulin sensitivity are often observed (5). Furthermore, dietary intake is an important exogenous factor in relation to ectopic fat and IR. Hypocaloric diets, with resulting weight loss, will lead to a loss of body weight and fat mass, which is accompanied by a decrease in ectopic fat storage (5, 107, 108); metabolic improvements are often observed (5). In addition, the individual macronutrients in the diet, fats, carbohydrates, and proteins, seem to determine metabolic adaptations that have been related to both ectopic fat and IR. In the next section, we will focus on the relationship between macronutrient composition of the diet, ectopic fat storage, and IR.
Macronutrient Quality and Quantity Impact Ectopic Fat and IR
Ample data is available on the effect of different types of isocaloric diets with varying macronutrient composition on IR. Nevertheless, fewer studies are available that report the effects of a specific diet on both IR and ectopic fat accumulation. As discussed, the accumulation of ectopic fat is a major factor in the development and progression of IR. To select relevant studies, a semi-systematic literature search was performed. A literature search was performed in January 2018 using the databases PubMed and Google Scholar. The search strategy consisted of a combination of the following search terms using the Boolean operator “AND” and “OR”:
“Insulin resistance”[MeSH], “choristoma”[MeSH], “fatty liver”[MeSH], “Non-alcoholic Fatty Liver Disease”[MeSH], “liver steatosis” [TIAB], “ectopic fat”[TIAB], “intramuscular fat”[TIAB], “intramuscular adipose tissue”[TIAB], “muscle fat”[TIAB], “Glucose tolerance”[TIAB], “impaired fasting glucose”[TIAB], “impaired glucose tolerance”[TIAB], “energy metabolism”[Mesh], “metabolic flexibility”[TIAB], “liver insulin resistance”[TIAB], “hepatic insulin resistance”[TIAB], “muscle insulin resistance”[TIAB], “peripheral insulin resistance”[TIAB], “liver insulin sensitivity”[TIAB], “hepatic insulin sensitivity”[TIAB], “muscle insulin sensitivity”[TIAB], “peripheral insulin sensitivity”[TIAB], “dietary proteins”[Mesh], “dietary carbohydrates”[Mesh], “dietary fiber”[Mesh], “dietary fats”[Mesh], “diet therapy”[Mesh], “diet”[Mesh].
Articles included in Table 1 are selected with the following inclusion and exclusion criteria: articles written in both English and Dutch languages were included. Articles were excluded if in vitro or animal models were used. Studies examining the effects of single meals (acute setting) rather than dietary interventions, studies investigating hypo- or hyper-caloric diets, and review articles were excluded. Endnote X7 was used for the management and selection of articles. The search yielded a total of 729 papers. The majority of the studies were excluded based on abstract (n = 694), and others after having read the full-text (n = 25), resulting in a final selection of 10 original articles (Table 1).
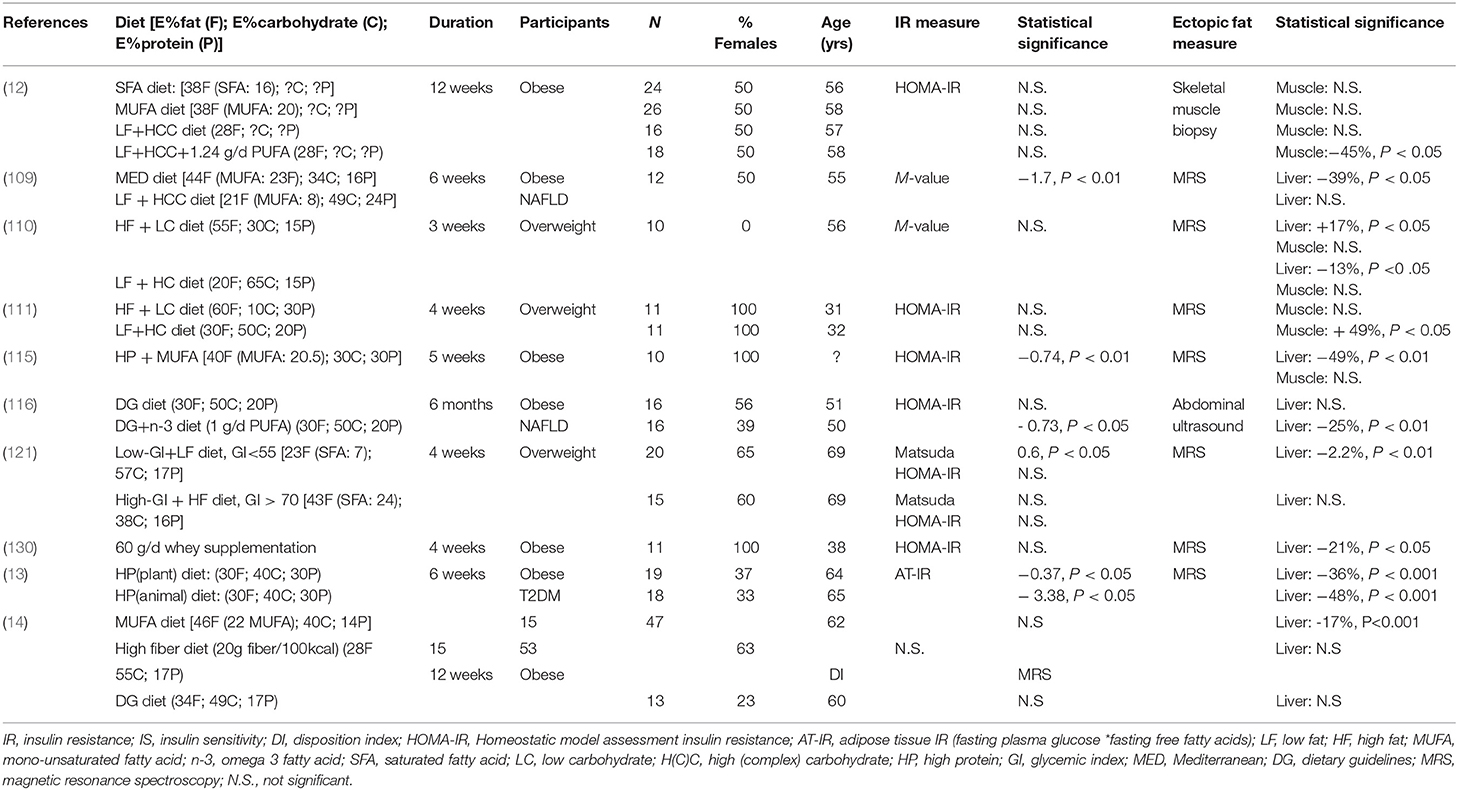
Table 1. Effect of isocaloric diets on insulin resistance and liver and muscle fat content in populations at risk for T2DM.
Carbohydrate/Fat Ratio of the Diet
Four different studies included in Table 1 investigated the effect of a low-fat high-complex carbohydrate (LFHCC) diet on ectopic fat and IR in overweight or obese individuals. The studies investigated the effect of diets that were slightly different in macronutrient composition and study duration (ranging between 3 and 12 weeks; Table 1). However, these studies consistently reported no significant effects of a LFHCC or a low-fat high- carbohydrate (LFHC) diet on IR, determined by either HOMA-IR or the M-value (12, 109–111). Study outcomes were less consistent with respect to ectopic fat content. The study of Van Herpen et al. (110) reported a significant decrease in IHCL following 3 weeks of a LFHC diet [21 energy percent (E%) fat, 49 E% carbohydrate, 24 E% protein], while Ryan et al. (109) did not report changes in IHCL after a 6 week LFHCC diet (20 E% fat, 65 E% carbohydrate, 15 E% protein). Two studies including LFHC or LFHCC diets did not report changes in IMCL in overweight and obese individuals after 3 or 12 weeks of dietary intervention (12, 110), while the study by Parente et al. (111) showed a 49% increase in IMCL content after a 4-week LFHC diet. It was not clear whether the carbohydrates were mainly complex or not. These discrepant findings may partly be explained by differences in the metabolic phenotype of the study participants. For example, it has been found that the intake of a carbohydrate-rich meal resulted in more than two–fold increase in de novo lipogenesis and TAG synthesis leading to a significant increase in IHCL content in the liver in IR individuals as compared to insulin sensitive individuals (61). Interestingly, in the latter study, no changes were observed in IMCL content between the groups. However, the proportion of saturated (SFA), mono-unsaturated (MUFA), and poly-unsaturated (PUFA) fatty acids in these diets was either not comparable or not described, making it difficult to compare these studies.
Dietary Fat Quality
There is substantial evidence that dietary fat quality is related to ectopic fat storage. In an in vivo study in rats and an in vitro study in myotubes, it was reported that SFA preferentially accumulate as DAG in skeletal muscle and may thereby potentially interfere with insulin signaling, whereas MUFA and PUFA more readily convert to TAG (112, 113). Also, after incubation of human skeletal muscle cells with either the MUFA oleic acid or the SFA palmitic acid, higher lipolytic rates were observed with oleic acid (114). Furthermore, the glucose and insulin responses to a high SFA meal was greater when compared to a high PUFA meal in IR men, indicating an impaired postprandial insulin sensitivity, which was accompanied by a decreased skeletal muscle lipid turnover and FSR of TAG and DAG after the high SFA meal (11).
These data are supported by longer-term human dietary intervention studies. The study of Jans et al. (12) reported no significant change in IMCL following a 12-week LFHCC diet in obese older adults, but reported a significant reduction in IMCL when this diet was supplemented with 1.24 g/day of PUFA omega-3 (LFHCC+n3 diet, Table 1). Interestingly, the latter study also reported that a 12-week diet rich in MUFA, defined as 20E%, had the tendency to decrease skeletal muscle FSR into DAG and TAG compared to pre-intervention (12). Both the MUFA diet and LFHCC+n-3 diet decreased the expression of lipogenic genes, although this was not reflected in IMCL changes in participants after completing the MUFA diet (12). Nonetheless, it shows that the LFHCC+n3 and MUFA diets have the potential to decrease skeletal muscle lipid (intermediate) accumulation, which in turn may affect insulin signaling. However, neither of the diets showed that this effect was related to changes in IR (12).
Clearly, dietary fat quality may have significant effects on ectopic fat and potentially also on insulin sensitivity. Significant reductions in IHCL content and improvements in insulin sensitivity were found following a MUFA diet (5 weeks) (115), a diet supplemented with omega-3 (6 months) (116) and a Mediterranean diet (high in MUFA and PUFA) (6 weeks) (109), see also Table 1. Interestingly however, excessive intake of fat (>55E% for 3 weeks) in the diet (including high PUFA and MUFA) resulted in a 17% increase in IHCL content in overweight men, but did not change IMCL (110). Whether these changes in IHCL content are also related to changes in insulin sensitivity remains to be elucidated. Several longer-term (12–24 weeks) large dietary intervention studies that investigated the effects of isoenergetic replacement of SFA with MUFA and/or PUFA on insulin sensitivity either did not report significant improvements in insulin sensitivity following the different diets (117, 118), or only reported significance when absolute intake of fat was not high (< 37E%) (119).
Carbohydrate Quality
The metabolic implications of dietary carbohydrates are largely dependent on the type of carbohydrate. The intake of simple carbohydrates have especially been linked to metabolic disorders (120). A diet low in glycemic index (GI), which is a relative ranking of foods and their effect on blood glucose values, may therefore be beneficial in relation to insulin sensitivity, and has also been linked to decreased IHCL accumulation (120). Indeed, an isocaloric low-GI diet for 4 weeks in overweight older adults led to a significant decrease in IHCL and improvement in insulin sensitivity (Matsuda index) compared to a 4-week isocaloric high-GI diet (121) (Table 1).
Individual carbohydrates and their effect on insulin sensitivity and ectopic fat have been studied. For example, excessive dietary fructose, as present in sugar-sweetened beverages, is thought to promote hepatic de novo lipogenesis, IHCL and IMCL content, and has accordingly been correlated to the development of cardiometabolic diseases (122–124), although This has been confirmed in animal models (125, 126). However, a 4-week isocaloric high-fructose diet (1.5 g/kg/body weight) in healthy men resulted in an increase in fasting plasma concentrations of TAG, VLDL-TAG, and glucose, but did not significantly alter ectopic fat content and insulin sensitivity in the liver and skeletal muscle (127). Furthermore, another study measured fat content in liver or skeletal muscle by computed tomography (CT) and found no differences between a 10-week isocaloric diet high in fructose and an isocaloric diet high in sucrose (123). Unfortunately, insulin sensitivity was not reported in the latter study. Finally, a study including both NGT as well as IGT individuals compared the effect of 50 g of added carbohydrate to the diet from either honey, sucrose, or high-fructose for 2 weeks (128). Insulin sensitivity and lipid levels were not different between the different types of carbohydrates after 2 weeks of dietary intervention (128). Although the complexity of dietary carbohydrates may be important in relation to insulin sensitivity and ectopic fat, simple carbohydrates, such as fructose and sucrose, when consumed in iso-energetic exchange for other CHO sources may not be directly related to differences in insulin sensitivity and ectopic fat (129).
Protein Intake
The amount of protein in the diet also seems to be important in relation to IR and ectopic fat. Markova et al. (13) showed that a 6-week diet high in protein (30 E%, either high in plant- or animal-source protein) reduced IHCL content in individuals with T2DM, as measured by MRS. Remarkably, these diets induced pronounced changes in IHCL, ranging from approximately −36% for the plant protein diet to −48% for the animal protein diet. Additionally, the reduction in IHCL content correlated with several parameters related to glucose homeostasis, including decreased fasting glucose levels, improved whole-body insulin sensitivity and decreased de novo lipogenesis. A decreased IHCL content following a high protein diet was also reported in a study supplementing whey protein (60 g/d) for 4 weeks in obese individuals (130). However, the latter study was not controlled. The effect on IMCL was not reported for either of the above-mentioned studies. The number of studies describing the effect of protein quality and quantity is limited; however, it seems that protein quality and quantity in the diet may have a significant effect on ectopic fat accumulation in the liver.
Overall, drawing conclusions on diets that are most effective in improving ectopic fat is highly challenging. Comparing dietary intervention studies is very difficult as characteristics between populations vary with age, sex, BMI, amongst others. Moreover, the duration of the intervention and macronutrient composition between studies differs considerably. Also, the majority of studies measure either IHCL or IMCL, but rarely do they assess both. Moreover, measures of IR (i.e., HOMA-IR, Matsuda Index, M-value) differ between studies and may reflect different etiologies of IR (131, 132). In addition, lipid-intermediates were not assessed in most of these studies, and the majority of the studies performed to date did not take liver- and muscle-IR into account when assessing the effectiveness of interventions. Finally, the baseline metabolic phenotype of individuals may be an important determinant on the effectiveness of dietary interventions, as discussed in more detail in the next section.
Differential Effect of Macronutrients On Ectopic Fat in Distinct IR Phenotypes; Toward Personalized Nutrition
For several decades, many studies have focused on diets either low in carbohydrate or in fat as the most effective approach to improve glucose homeostasis and manage T2DM (133). Interestingly, however, not all individuals may benefit from the same diet. Recent studies have shown large inter-individual variation in response to a meal (134), which may also explain why individuals respond differently to the same dietary intervention. Indeed, in recent years, several studies have demonstrated that the macronutrient quality and quantity in the diet can lead to a differential response, depending on the individual's metabolic phenotype (11, 12, 14, 75, 135, 136). More specifically, this response may be dependent on liver- or muscle-IR, ectopic fat content, age, ethnicity, sex, and likely other factors that have not been studied extensively thus far.
There has been a debate on different dietary approaches in the prevention of T2DM, one is the Mediterranean diet, rich in olive oil, which may provide cardiovascular benefits. Second, diets low in fat and high in complex carbohydrates with increased fiber content (within the context of a lifestyle intervention) may decrease the cumulative incidence of diabetes by more than 50% over 3–6 years (137–139). In the CORDIOPREV-DIAB study, the Mediterranean (high in MUFAs) and the LFHCC dietary patterns were compared with respect to tissue-specific insulin resistance and beta-cell function in cardiovascular patients not treated for diabetes (n = 642, analysis at baseline and at 2 years follow-up) (75). Interestingly, the change in disposition index (DI), which is a composite score of insulin sensitivity and secretion, after a long-term dietary intervention, was related to the tissue-specific IR phenotype of the participants at baseline. The tissue-specific IR phenotypes were defined as no-IR, muscle-IR, liver-IR or combined muscle and liver-IR, based on tertiles of the muscle insulin sensitivity index (MISI) and hepatic insulin resistance index (HIRI), which was modeled from an OGTT (140). Interestingly, the Mediterranean diet was reported to be most beneficial for individuals with muscle-IR, while the LFHCC diet was most beneficial for the liver-IR phenotype (75). In addition, a recent post-hoc analysis in the European project LIPGENE, which focused on the effects of dietary fat quantity and quality in the metabolic syndrome (MetS), found that IR MetS individuals were more sensitive to health effects from the substitution of a high-saturated fat diet by either high-MUFA or high (complex)-carbohydrates (with added n-3 PUFA) diets (15). On the contrary, the individuals with higher insulin sensitivity were more susceptible to the detrimental effects of SFA. Furthermore, Guess et al. (141) recently reported that 6 weeks of supplementation with the dietary fiber inulin (30 g/d) decreased HOMA-IR in IFG but not in IGT individuals. These results may indicate that inulin has beneficial effects in subjects with liver-IR rather than muscle-IR, as IFG individuals have often a more pronounced liver-IR.
Mediterranean or LFHC high-fiber diets may be healthy for all, however, these studies indicate that dietary prevention or treatment may require a more subgroup-based or personalized approach to optimize the effects of dietary interventions. Indeed, a recent study by Zeevi et al. (134) demonstrated that personalized diets seem more effective in lowering postprandial blood glucose responses compared to diets based on expert advice. In this study, a machine-learning approach was used to predict blood glucose responses, using parameters such as dietary habits, physical activity, gut microbiota composition, and anthropometrics, thereby developing the most optimal diet for each individual. The studies that are mentioned in this section are illustrative of the importance of personalized nutrition to optimize the effect of dietary interventions in IR individuals. Targeting tissue-specific IR phenotypes, such as liver- and muscle-IR, might prove to be an effective strategy to optimize metabolic outcomes of dietary interventions (Figure 1).
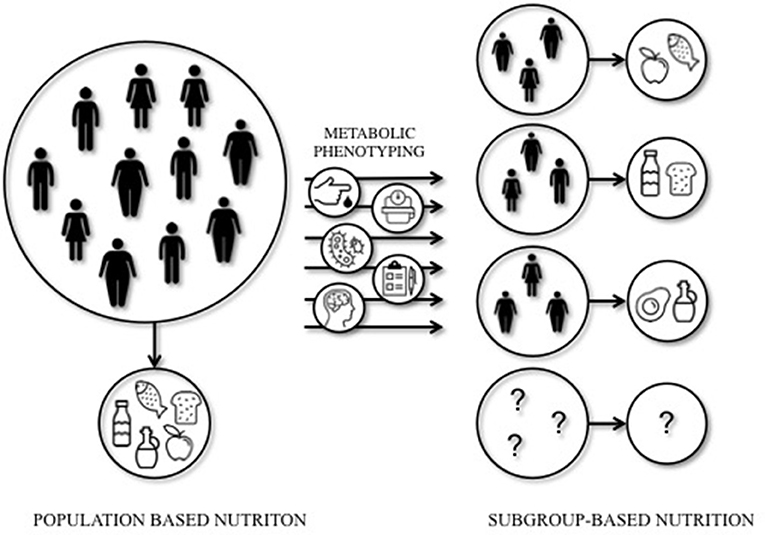
Figure 1. Population-based vs. subgroup-based nutrition. Large inter-individual variation can be seen in the response to dietary interventions. This implies that population-based nutritional interventions may not always lead to the most optimal (metabolic) outcomes for each individual. A subgroup-based approach that integrates among others cardiometabolic parameters, anthropometrics, gut microbiota, mental status, age, sex, and ethnicity, may increase the efficacy of dietary interventions. Future studies should elucidate the most optimal diet for a certain (metabolic) phenotype.
Nonetheless, the available literature to date does not provide sufficient evidence from prospective studies into the optimal quantity and quality of macronutrients in the diet based on metabolic phenotype. The tissue-specific IR phenotypes are related to a variety of different characteristics, such as ectopic fat, glucose control, gut microbiota, sex, age, and others, and are likely not isolated entities. Defining metabolic phenotypes is difficult and may therefore not always do justice to its complexity. Moreover, although not surprising, the studies mentioned in this review only focus on a specific phenotype, such as muscle- vs. liver-IR or mild- vs. severe-IR, and mainly focus on Caucasian, middle-aged populations. Therefore trying to understand an optimal diet for other ethnicities, age groups, or other metabolic (sub-)phenotypes such as IFG vs. IGT is difficult. Recommendations or conclusions on the optimization of diets for each metabolic phenotype are therefore not yet available (Figure 1).
Available evidence clearly indicates that there is great potential to optimize the effectiveness of dietary interventions on glucose homeostasis by, for example, targeting liver- and muscle-IR phenotypes. A first step toward the development of more personalized nutrition could be to investigate the role of tissue-specific IR and related ectopic fat content in the effectiveness of dietary interventions, in particular when studying the effect of manipulation of the macronutrient composition of the diet. Macronutrient composition, both quality and quality, is highly important and should be taken into account for these types of intervention studies. To develop effective personalized dietary interventions, it seems necessary to perform extensive phenotyping of individuals to understand the complexity of metabolic regulation and its target for different macronutrients, as illustrated in Figure 1. Here, a schematic overview is given of the concept of subgroup-based or more personalized nutrition. Based on the baseline metabolic phenotype, a specific dietary intervention may be initiated to optimize intervention outcomes.
In conclusion, to understand the complexity of the interaction of an individual's metabolic phenotype and the response to diet, a detailed physiological phenotyping including the use of advanced -omics methodologies like (epi)genomics, metagenomics, and metabolomics is required. Additionally, mobile apps and wearable devices may facilitate real-time assessment of dietary intake and physical activity, and may provide individual feedback to optimize personalization of advices. By integrating these technologies with big data analytics, personalized nutrition has the potential to provide targeted nutrition and lifestyle guidance for more effective prevention and management of type 2 diabetes and related chronic diseases. Despite all technological advances, the step toward implementation into a public health and clinical setting is still more remote and also includes other factors like dietary preferences, the socio-economic context and behavioral factors. Evidence is required to demonstrate the efficacy, cost-effectiveness as well as additional benefits of a personalized or subgroup-based approach beyond a traditional approach before these nutritional interventions can be implemented in daily practice in the future.
Author Contributions
IT wrote the manuscript; SB, GG, and EB conceptualized, reviewed, and edited the manuscript. All authors have read and approved the final version of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. World Health Organisation. Diabetes Factsheet (2017) (Accessed September 03,2018). Available online at: http://www.who.int/mediacentre/factsheets/fs312/en/
2. Stinkens R, Goossens GH, Jocken JW, Blaak EE. Targeting fatty acid metabolism to improve glucose metabolism. Obes Rev. (2015) 16:715–57. doi: 10.1111/obr.12298
3. Goodpaster BH, Sparks LM. Metabolic flexibility in health and disease. Cell Metab. (2017) 25:1027–36. doi: 10.1016/j.cmet.2017.04.015
4. Hocking S, Samocha-Bonet D, Milner KL, Greenfield JR, Chisholm DJ. Adiposity and insulin resistance in humans: the role of the different tissue and cellular lipid depots. Endocr Rev. (2013) 34:463–500. doi: 10.1210/er.2012-1041
5. Snel M, Jonker JT, Schoones J, Lamb H, de Roos A, Pijl H, et al. Ectopic fat and insulin resistance: pathophysiology and effect of diet and lifestyle interventions. Int J Endocrinol. (2012) 2012:983814. doi: 10.1155/2012/983814
6. Moro C, Bajpeyi S, Smith SR. Determinants of intramyocellular triglyceride turnover: implications for insulin sensitivity. Am J Physiol Endocrinol Metab. (2008) 294:E203–13. doi: 10.1152/ajpendo.00624.2007
7. van der Kolk BW, Goossens GH, Jocken JW, Blaak EE. Altered skeletal muscle fatty acid handling is associated with the degree of insulin resistance in overweight and obese humans. Diabetologia (2016) 59:2686–96. doi: 10.1007/s00125-016-4104-3
8. Kuk JL, Saunders TJ, Davidson LE, Ross R. Age-related changes in total and regional fat distribution. Ageing Res Rev. (2009) 8:339–48. doi: 10.1016/j.arr.2009.06.001
9. Bredella MA. Sex differences in body composition. Adv Exp Med Biol. (2017) 1043:9–27. doi: 10.1007/978-3-319-70178-3_2
10. Faerch K, Borch-Johnsen K, Holst JJ, Vaag A. Pathophysiology and aetiology of impaired fasting glycaemia and impaired glucose tolerance: does it matter for prevention and treatment of type 2 diabetes? Diabetologia (2009) 52:1714–23. doi: 10.1007/s00125-009-1443-3
11. Jans A, Konings E, Goossens GH, Bouwman FG, Moors CC, Boekschoten MV, et al. PUFAs acutely affect triacylglycerol-derived skeletal muscle fatty acid uptake and increase postprandial insulin sensitivity. Am J Clin Nutr. (2012) 95:825–36. doi: 10.3945/ajcn.111.028787
12. Jans A, van Hees AM, Gjelstad IM, Sparks LM, Tierney AC, Riserus U, et al. Impact of dietary fat quantity and quality on skeletal muscle fatty acid metabolism in subjects with the metabolic syndrome. Metabolism (2012) 61:1554–65. doi: 10.1016/j.metabol.2012.04.003
13. Markova M, Pivovarova O, Hornemann S, Sucher S, Frahnow T, Wegner K, et al. Isocaloric diets high in animal or plant protein reduce liver fat and inflammation in individuals with type 2 diabetes. Gastroenterology (2017) 152:571–85.e8. doi: 10.1053/j.gastro.2016.10.007
14. Errazuriz I, Dube S, Slama M, Visentin R, Nayar S, O'Connor H, et al. Randomized Controlled Trial of a MUFA or fiber-rich diet on hepatic fat in prediabetes. J Clin Endocrinol Metab. (2017) 102:1765–74. doi: 10.1210/jc.2016-3722
15. Yubero-Serrano EM, Delgado-Lista J, Tierney AC, Perez-Martinez P, Garcia-Rios A, Alcala-Diaz JF, et al. Insulin resistance determines a differential response to changes in dietary fat modification on metabolic syndrome risk factors: the LIPGENE study. Am J Clin Nutr. (2015) 102:1509–17.doi: 10.3945/ajcn.115.111286
16. Bluher M. The distinction of metabolically 'healthy' from 'unhealthy' obese individuals. Curr Opin Lipidol. (2010) 21:38–43. doi: 10.1097/MOL.0b013e3283346ccc
17. Gancheva S, Jelenik T, Alvarez-Hernandez E, Roden M. Interorgan metabolic crosstalk in human insulin resistance. Physiol Rev. (2018) 98:1371–415. doi: 10.1152/physrev.00015.2017
18. Otero YF, Stafford JM, McGuinness OP. Pathway-selective insulin resistance and metabolic disease: the importance of nutrient flux. J Biol Chem. (2014) 289:20462–9. doi: 10.1074/jbc.R114.576355
19. Qureshi K, Abrams GA. Metabolic liver disease of obesity and role of adipose tissue in the pathogenesis of nonalcoholic fatty liver disease. World J Gastroenterol. (2007) 13:3540–53. doi: 10.3748/wjg.v13.i26.3540
20. Murakami T, Yamada N. [Molecular mechanism of insulin resistance in hyperlipidemia]. Nihon Rinsho (1999) 57:2657–61.
21. Choi SH, Ginsberg HN. Increased very low density lipoprotein (VLDL) secretion, hepatic steatosis, and insulin resistance. Trends Endocrinol Metab. (2011) 22:353–63. doi: 10.1016/j.tem.2011.04.007
22. Bickerton AS, Roberts R, Fielding BA, Tornqvist H, Blaak EE, Wagenmakers AJ, et al. Adipose tissue fatty acid metabolism in insulin-resistant men. Diabetologia (2008) 51:1466–74. doi: 10.1007/s00125-008-1040-x
23. McQuaid SE, Hodson L, Neville MJ, Dennis AL, Cheeseman J, Humphreys SM, et al. Downregulation of adipose tissue fatty acid trafficking in obesity: a driver for ectopic fat deposition? Diabetes (2011) 60:47–55. doi: 10.2337/db10-0867
24. Karpe F, Dickmann JR, Frayn KN. Fatty acids, obesity, and insulin resistance: time for a reevaluation. Diabetes (2011) 60:2441–9. doi: 10.2337/db11-0425
25. Riemens SC, Sluiter WJ, Dullaart RP. Enhanced escape of non-esterified fatty acids from tissue uptake: its role in impaired insulin-induced lowering of total rate of appearance in obesity and Type II diabetes mellitus. Diabetologia (2000) 43:416–26. doi: 10.1007/s001250051324
26. Panarotto D, Remillard P, Bouffard L, Maheux P. Insulin resistance affects the regulation of lipoprotein lipase in the postprandial period and in an adipose tissue-specific manner. Eur J Clin Invest. (2002) 32:84–92. doi: 10.1046/j.1365-2362.2002.00945.x
27. Coppack SW, Evans RD, Fisher RM, Frayn KN, Gibbons GF, Humphreys SM, et al. Adipose tissue metabolism in obesity: lipase action in vivo before and after a mixed meal. Metabolism (1992) 41:264–72.
28. Potts JL, Coppack SW, Fisher RM, Humphreys SM, Gibbons GF, Frayn KN. Impaired postprandial clearance of triacylglycerol-rich lipoproteins in adipose tissue in obese subjects. Am J Physiol. (1995) 268(4 Pt 1):E588–94.
29. Annuzzi G, Giacco R, Patti L, Di Marino L, De Natale C, Costabile G, et al. Postprandial chylomicrons and adipose tissue lipoprotein lipase are altered in type 2 diabetes independently of obesity and whole-body insulin resistance. Nutr Metab Cardiovasc Dis. (2008) 18:531–8. doi: 10.1016/j.numecd.2007.12.003
30. Kosteli A, Sugaru E, Haemmerle G, Martin JF, Lei J, Zechner R, et al. Weight loss and lipolysis promote a dynamic immune response in murine adipose tissue. J Clin Invest. (2010) 120:3466–79. doi: 10.1172/JCI42845
31. Aouadi M, Vangala P, Yawe JC, Tencerova M, Nicoloro SM, Cohen JL, et al. Lipid storage by adipose tissue macrophages regulates systemic glucose tolerance. Am J Physiol Endocrinol Metab. (2014) 307:E374–83. doi: 10.1152/ajpendo.00187.2014
32. Labbe SM, Croteau E, Grenier-Larouche T, Frisch F, Ouellet R, Langlois R, et al. Normal postprandial nonesterified fatty acid uptake in muscles despite increased circulating fatty acids in type 2 diabetes. Diabetes (2011) 60:408–15. doi: 10.2337/db10-0997
33. Goossens GH, Moors CC, Jocken JW, van der Zijl NJ, Jans A, Konings E, et al. Altered skeletal muscle fatty acid handling in subjects with impaired glucose tolerance as compared to impaired fasting glucose. Nutrients (2016) 8:164. doi: 10.3390/nu8030164
34. Kawano Y, Cohen DE. Mechanisms of hepatic triglyceride accumulation in non-alcoholic fatty liver disease. J Gastroenterol. (2013) 48:434–41. doi: 10.1007/s00535-013-0758-5
35. Hodson L, Frayn KN. Hepatic fatty acid partitioning. Curr Opin Lipidol. (2011) 22:216–24. doi: 10.1097/MOL.0b013e3283462e16
36. van Hees AM, Jans A, Hul GB, Roche HM, Saris WH, Blaak EE. Skeletal muscle fatty acid handling in insulin resistant men. Obesity (2011) 19:1350–9. doi: 10.1038/oby.2011.10
37. Schwenk RW, Holloway GP, Luiken JJ, Bonen A, Glatz JF. Fatty acid transport across the cell membrane: regulation by fatty acid transporters. Prostaglandins Leukot Essent Fatty Acids (2010) 82:149–54. doi: 10.1016/j.plefa.2010.02.029
38. Goldberg IJ, Eckel RH, Abumrad NA. Regulation of fatty acid uptake into tissues: lipoprotein lipase- and CD36-mediated pathways. J Lipid Res. (2009) 50(Suppl.):S86–90. doi: 10.1194/jlr.R800085-JLR200
39. Mead JR, Irvine SA, Ramji DP. Lipoprotein lipase: structure, function, regulation, and role in disease. J Mol Med. (2002) 80:753–69. doi: 10.1007/s00109-002-0384-9
40. Kazantzis M, Stahl A. Fatty acid transport proteins, implications in physiology and disease. Biochim Biophys Acta (2012) 1821:852–7.doi: 10.1016/j.bbalip.2011.09.010
41. Kashyap SR, Ioachimescu AG, Gornik HL, Gopan T, Davidson MB, Makdissi A, et al. Lipid-induced insulin resistance is associated with increased monocyte expression of scavenger receptor CD36 and internalization of oxidized LDL. Obesity (2009) 17:2142–8. doi: 10.1038/oby.2009.179
42. Corpeleijn E, Pelsers MM, Soenen S, Mensink M, Bouwman FG, Kooi ME, et al. Insulin acutely upregulates protein expression of the fatty acid transporter CD36 in human skeletal muscle in vivo. J Physiol Pharmacol. (2008) 59:77–83.
43. Love-Gregory L, Abumrad NA. CD36 genetics and the metabolic complications of obesity. Curr Opin Clin Nutri Metab Care (2011) 14:527–34. doi: 10.1097/MCO.0b013e32834bbac9
44. Wu Q, Ortegon AM, Tsang B, Doege H, Feingold KR, Stahl A. FATP1 is an insulin-sensitive fatty acid transporter involved in diet-induced obesity. Mol Cell Biol. (2006) 26:3455–67. doi: 10.1128/MCB.26.9.3455-3467.2006
45. Farese RV Jr, Yost, TJ, Eckel RH. Tissue-specific regulation of lipoprotein lipase activity by insulin/glucose in normal-weight humans. Metabolism (1991) 40:214-6. doi: 10.1016/0026-0495(91)90178-Y
46. Kelley DE. Skeletal muscle fat oxidation: timing and flexibility are everything. J Clin Invest. (2005) 115:1699–702. doi: 10.1172/JCI25758
47. Jocken JW, Goossens GH, Boon H, Mason RR, Essers Y, Havekes B, et al. Insulin-mediated suppression of lipolysis in adipose tissue and skeletal muscle of obese type 2 diabetic men and men with normal glucose tolerance. Diabetologia (2013) 56:2255–65. doi: 10.1007/s00125-013-2995-9
48. Schrauwen P, Schrauwen-Hinderling V, Hoeks J, Hesselink MK. Mitochondrial dysfunction and lipotoxicity. Biochim Biophys Acta (2010) 1801:266–71. doi: 10.1016/j.bbalip.2009.09.011
49. Corpeleijn E, Mensink M, Kooi ME, Roekaerts PM, Saris WH, Blaak EE. Impaired skeletal muscle substrate oxidation in glucose-intolerant men improves after weight loss. Obesity (2008) 16:1025–32. doi: 10.1038/oby.2008.24
50. Szendroedi J, Phielix E, Roden M. The role of mitochondria in insulin resistance and type 2 diabetes mellitus. Nat Rev Endocrinol. (2011) 8:92–103. doi: 10.1038/nrendo.2011.138
51. Montgomery MK, Turner N. Mitochondrial dysfunction and insulin resistance: an update. Endocr Connect. (2015) 4:R1–15. doi: 10.1530/EC-14-0092
52. Shulman GI. Ectopic fat in insulin resistance, dyslipidemia, and cardiometabolic disease. New Engl J Med. (2014) 371:2237–8. doi: 10.1056/NEJMra1011035
53. Boucher J, Kleinridders A, Kahn CR. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb Perspect Biol. (2014) 6:a009191. doi: 10.1101/cshperspect.a009191
54. Koliaki C, Roden M. Alterations of mitochondrial function and insulin sensitivity in human obesity and diabetes mellitus. Ann Rev Nutr. (2016) 36:337–67. doi: 10.1146/annurev-nutr-071715-050656
55. Schmid AI, Szendroedi J, Chmelik M, Krssak M, Moser E, Roden M. Liver ATP synthesis is lower and relates to insulin sensitivity in patients with type 2 diabetes. Diabetes Care (2011) 34:448–53. doi: 10.2337/dc10-1076
56. Magkos F, Su X, Bradley D, Fabbrini E, Conte C, Eagon JC, et al. Intrahepatic diacylglycerol content is associated with hepatic insulin resistance in obese subjects. Gastroenterology (2012) 142:1444–6 e2. doi: 10.1053/j.gastro.2012.03.003
57. Ter Horst KW, Gilijamse PW, Versteeg RI, Ackermans MT, Nederveen AJ, la Fleur SE, et al. Hepatic diacylglycerol-associated protein kinase cepsilon translocation links hepatic steatosis to hepatic insulin resistance in humans. Cell Rep. (2017) 19:1997–2004. doi: 10.1016/j.celrep.2017.05.035
58. Petersen MC, and Shulman GI Roles of diacylglycerols and ceramides in hepatic insulin resistance. Trends Pharmacol Sci. (2017) 38:649–65. doi: 10.1016/j.tips.2017.04.004
59. Blaak EE. Characterisation of fatty acid metabolism in different insulin-resistant phenotypes by means of stable isotopes. Proc Nutr Soc. (2017) 19:1–7. doi: 10.1017/S0029665116003013
60. Kato K, Takeshita Y, Misu H, Zen Y, Kaneko S, Takamura T. Liver steatosis is associated with insulin resistance in skeletal muscle rather than in the liver in Japanese patients with non-alcoholic fatty liver disease. J Diabetes Invest. (2015) 6:158–63. doi: 10.1111/jdi.12271
61. Petersen KF, Dufour S, Savage DB, Bilz S, Solomon G, Yonemitsu S, et al. The role of skeletal muscle insulin resistance in the pathogenesis of the metabolic syndrome. Proc Natl Acad Sci USA. (2007) 104:12587–94. doi: 10.1073/pnas.0705408104
62. Flannery C, Dufour S, Rabol R, Shulman GI, Petersen KF. Skeletal muscle insulin resistance promotes increased hepatic de novo lipogenesis, hyperlipidemia, and hepatic steatosis in the elderly. Diabetes (2012) 61:2711–7. doi: 10.2337/db12-0206
63. Moors CC, van der Zijl NJ, Diamant M, Blaak EE, Goossens GH. Impaired insulin sensitivity is accompanied by disturbances in skeletal muscle fatty acid handling in subjects with impaired glucose metabolism. Int J Obes. (2012) 36:709–17. doi: 10.1038/ijo.2011.123
64. Lorenzo C, Hartnett S, Hanley AJ, Rewers MJ, Wagenknecht LE, Karter AJ, et al. Impaired fasting glucose and impaired glucose tolerance have distinct lipoprotein and apolipoprotein changes: the insulin resistance atherosclerosis study. J Clin Endocrinol Metab. (2013) 98:1622–30. doi: 10.1210/jc.2012-3185
65. Miyazaki Y, Akasaka H, Ohnishi H, Saitoh S, DeFronzo RA, Shimamoto K. Differences in insulin action and secretion, plasma lipids and blood pressure levels between impaired fasting glucose and impaired glucose tolerance in Japanese subjects. Hypertens Res. (2008) 31:1357–63. doi: 10.1291/hypres.31.1357
66. Chakarova N, Tankova T, Atanassova I, Dakovska L. Serum lipid and hsCRP levels in prediabetes–impaired fasting glucose (IFG) and impaired glucose tolerance (IGT). Diabetes Res Clin Pract. (2009) 86:56–60. doi: 10.1016/j.diabres.2009.04.005
67. Leon-Acuna A, Alcala-Diaz JF, Delgado-Lista J, Torres-Pena JD, Lopez-Moreno J, Camargo A, et al. Hepatic insulin resistance both in prediabetic and diabetic patients determines postprandial lipoprotein metabolism: from the CORDIOPREV study. Cardiovasc Diabetol. (2016) 15:68. doi: 10.1186/s12933-016-0380-y
68. Nathan DM, Davidson MB, DeFronzo RA, Heine RJ, Henry RR, Pratley R, et al. Impaired fasting glucose and impaired glucose tolerance: implications for care. Diabetes Care (2007) 30:753–9. doi: 10.2337/dc07-9920
69. Bock G, Chittilapilly E, Basu R, Toffolo G, Cobelli C, Chandramouli V, et al. Contribution of hepatic and extrahepatic insulin resistance to the pathogenesis of impaired fasting glucose: role of increased rates of gluconeogenesis. Diabetes (2007) 56:1703–11. doi: 10.2337/db06-1776
70. Meyer C, Pimenta W, Woerle HJ, Van Haeften T, Szoke E, Mitrakou A, et al. Different mechanisms for impaired fasting glucose and impaired postprandial glucose tolerance in humans. Diabetes Care (2006) 29:1909–14. doi: 10.2337/dc06-0438
71. Berrish TS, Hetherington CS, Alberti KG, Walker M. Peripheral and hepatic insulin sensitivity in subjects with impaired glucose tolerance. Diabetologia (1995) 38:699–704. doi: 10.1007/BF00401842
72. Borel AL, Nazare JA, Smith J, Aschner P, Barter P, Van Gaal L, et al. Visceral, subcutaneous abdominal adiposity and liver fat content distribution in normal glucose tolerance, impaired fasting glucose and/or impaired glucose tolerance. Int J Obes. (2015) 39:495–501. doi: 10.1038/ijo.2014.163
73. van der Zijl NJ, Goossens GH, Moors CC, van Raalte DH, Muskiet MH, Pouwels PJ, et al. Ectopic fat storage in the pancreas, liver, and abdominal fat depots: impact on beta-cell function in individuals with impaired glucose metabolism. J Clin Endocrinol Metab. (2011) 96:459–67. doi: 10.1210/jc.2010-1722
74. Komiya H, Mori Y, Yokose T, Kurokawa N, Horie N, Tajima N. Effect of intramuscular fat difference on glucose and insulin reaction in oral glucose tolerance test. J Atheroscler Thromb. (2006) 13:136–42. doi: 10.5551/jat.13.136
75. Blanco-Rojo R, Alcala-Diaz JF, Wopereis S, Perez-Martinez P, Quintana-Navarro GM, Marin C, et al. The insulin resistance phenotype (muscle or liver) interacts with the type of diet to determine changes in disposition index after 2 years of intervention: the CORDIOPREV-DIAB randomised clinical trial. Diabetologia (2015) 59:67–76. doi: 10.1007/s00125-015-3776-4
76. Regitz-Zagrosek V. Sex and gender differences in health. Science & society series on sex and science. EMBO Rep. (2012) 13:596–603. doi: 10.1038/embor.2012.87
77. Kautzky-Willer A, Harreiter J, Pacini G. Sex and gender differences in risk, pathophysiology and complications of type 2 diabetes mellitus. Endocr Rev. (2016) 37:278–316. doi: 10.1210/er.2015-1137
78. Mauvais-Jarvis F. Sex differences in metabolic homeostasis, diabetes, and obesity. Biol Sex Differ. (2015) 6:14. doi: 10.1186/s13293-015-0033-y
79. Karastergiou K, Smith SR, Greenberg AS, Fried SK. Sex differences in human adipose tissues - the biology of pear shape. Biol Sex Differ. (2012) 3:13. doi: 10.1186/2042-6410-3-13
80. White UA, Tchoukalova YD. Sex dimorphism and depot differences in adipose tissue function. Biochim Biophys Acta (2014) 1842:377–92. doi: 10.1016/j.bbadis.2013.05.006
81. Goossens GH. The metabolic phenotype in obesity: fat mass, body fat distribution, and adipose tissue function. Obes Facts (2017) 10:207–15. doi: 10.1159/000471488
82. Virtue S, Vidal-Puig A. Adipose tissue expandability, lipotoxicity and the metabolic syndrome–an allostatic perspective. Biochim Biophys Acta (2010) 1801:338–49. doi: 10.1016/j.bbalip.2009.12.006
83. Pellegrinelli V, Carobbio S, Vidal-Puig A. Adipose tissue plasticity: how fat depots respond differently to pathophysiological cues. Diabetologia (2016) 59:1075–88. doi: 10.1007/s00125-016-3933-4
84. Shah RV, Murthy VL, Abbasi SA, Blankstein R, Kwong RY, Goldfine AB, et al. Visceral adiposity and the risk of metabolic syndrome across body mass index: the MESA Study. JACC Cardiovasc Imaging (2014) 7:1221–35. doi: 10.1016/j.jcmg.2014.07.017
85. Bjorntorp P. Obesity: a chronic disease with alarming prevalence and consequences. J Intern Med. (1998) 244:267–9. doi: 10.1046/j.1365-2796.1998.00417.x
86. Manolopoulos KN, Karpe F, Frayn KN. Gluteofemoral body fat as a determinant of metabolic health. Int J Obes. (2010) 34:949–59. doi: 10.1038/ijo.2009.286
87. Manolopoulos KN, Karpe F, Frayn KN. Marked resistance of femoral adipose tissue blood flow and lipolysis to adrenaline in vivo. Diabetologia (2012) 55:3029–37. doi: 10.1007/s00125-012-2676-0
88. Brady CW. Liver disease in menopause. World J Gastroenterol. (2015) 21:7613–20. doi: 10.3748/wjg.v21.i25.7613
89. Hart-Unger S, Arao Y, Hamilton KJ, Lierz SL, Malarkey DE, Hewitt SC, et al. Hormone signaling and fatty liver in females: analysis of estrogen receptor alpha mutant mice. Int J Obes. (2017) 41:945–54. doi: 10.1038/ijo.2017.50
90. Lonardo A, Trande P. Are there any sex differences in fatty liver? A study of glucose metabolism and body fat distribution. J Gastroenterol Hepatol. (2000) 15:775–82. doi: 10.1046/j.1440-1746.2000.02226.x
91. Machann J, Thamer C, Schnoedt B, Stefan N, Stumvoll M, Haring HU, et al. Age and gender related effects on adipose tissue compartments of subjects with increased risk for type 2 diabetes: a whole body MRI/MRS study. MAGMA (2005) 18:128–37. doi: 10.1007/s10334-005-0104-x
92. Velan SS, Said N, Narasimhan K, Papan C, Bahu A, Vargo JA, et al. Gender differences in musculoskeletal lipid metabolism as assessed by localized two-dimensional correlation spectroscopy. Magn Reson Insights (2008) 2008:1–6. doi: 10.4137/MRI.S958
93. Perseghin G, Scifo P, Pagliato E, Battezzati A, Benedini S, Soldini L, et al. Gender factors affect fatty acids-induced insulin resistance in nonobese humans: effects of oral steroidal contraception. J Clin Endocrinol Metab. (2001) 86:3188–96. doi: 10.1210/jc.86.7.3188
94. Ortiz-Nieto F, Johansson L, Ahlstrom H, Weis J. Quantification of lipids in human lower limbs using yellow bone marrow as the internal reference: gender-related effects. Magn Reson Imaging (2010) 28:676–82. doi: 10.1016/j.mri.2010.03.014
95. Perreault L, Bergman BC, Hunerdosse DM, Eckel RH. Altered intramuscular lipid metabolism relates to diminished insulin action in men, but not women, in progression to diabetes. Obesity (2010) 18:2093–100. doi: 10.1038/oby.2010.76
96. Lara-Castro C, Garvey WT. Intracellular lipid accumulation in liver and muscle and the insulin resistance syndrome. Endocrinol Metab Clin North Am. (2008) 37:841–56. doi: 10.1016/j.ecl.2008.09.002
97. Bennet L, Groop L, Franks PW. Country of birth modifies the association of fatty liver index with insulin action in Middle Eastern immigrants to Sweden. Diabetes Res Clin Pract. (2015) 110:66–74. doi: 10.1016/j.diabres.2015.07.011
98. Yip WCY, Sequeira IR, Plank LD, Poppitt SD. Prevalence of pre-diabetes across ethnicities: a review of impaired fasting glucose (IFG) and impaired glucose tolerance (IGT) for classification of dysglycaemia. Nutrients (2017) 9:E1273. doi: 10.3390/nu9111273
99. Wulan SN, Westerterp KR, Plasqui G. Ethnic differences in body composition and the associated metabolic profile: a comparative study between Asians and Caucasians. Maturitas (2010) 65:315–9. doi: 10.1016/j.maturitas.2009.12.012
100. Petersen KF, Dufour S, Feng J, Befroy D, Dziura J, Dalla Man C, et al. Increased prevalence of insulin resistance and nonalcoholic fatty liver disease in Asian-Indian men. Proc Natl Acad Sci USA. (2006) 103:18273–7. doi: 10.1073/pnas.0608537103
101. Anand SS, Tarnopolsky MA, Rashid S, Schulze KM, Desai D, Mente A, et al. Adipocyte hypertrophy, fatty liver and metabolic risk factors in South Asians: the molecular study of health and risk in ethnic groups (mol-SHARE). PLoS ONE (2011) 6:e22112. doi: 10.1371/journal.pone.0022112102.
102. Sniderman AD, Bhopal R, Prabhakaran D, Sarrafzadegan N, Tchernof A. Why might South Asians be so susceptible to central obesity and its atherogenic consequences? The adipose tissue overflow hypothesis. Int J Epidemiol. (2007) 36:220–5. doi: 10.1093/ije/dyl245
103. Goedecke JH, Keswell D, Weinreich C, Fan J, Hauksson J, Victor H, et al. Ethnic differences in hepatic and systemic insulin sensitivity and their associated determinants in obese black and white South African women. Diabetologia (2015) 58:2647–52. doi: 10.1007/s00125-015-3720-7
104. Petersen KF, Dufour S, Hariri A, Nelson-Williams C, Foo JN, Zhang XM, et al. Apolipoprotein C3 gene variants in nonalcoholic fatty liver disease. New Engl J Med. (2010) 362:1082–9. doi: 10.1056/NEJMoa0907295
105. Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. (2008) 40:1461–5. doi: 10.1038/ng.257
106. Muller DC, Elahi D, Tobin JD, Andres R. The effect of age on insulin resistance and secretion: a review. Semin Nephrol. (1996) 16:289–98.
107. Dube JJ, Amati F, Toledo FG, Stefanovic-Racic M, Rossi A, Coen P, et al. Effects of weight loss and exercise on insulin resistance, and intramyocellular triacylglycerol, diacylglycerol and ceramide. Diabetologia (2011) 54:1147–56. doi: 10.1007/s00125-011-2065-0
108. Jazet IM, Schaart G, Gastaldelli A, Ferrannini E, Hesselink MK, Schrauwen P, et al. Loss of 50% of excess weight using a very low energy diet improves insulin-stimulated glucose disposal and skeletal muscle insulin signalling in obese insulin-treated type 2 diabetic patients. Diabetologia (2008) 51:309–19. doi: 10.1007/s00125-007-0862-2
109. Ryan MC, Itsiopoulos C, Thodis T, Ward G, Trost N, Hofferberth S, et al. The Mediterranean diet improves hepatic steatosis and insulin sensitivity in individuals with non-alcoholic fatty liver disease. J Hepatol. (2013) 59:138–43. doi: 10.1016/j.jhep.2013.02.012
110. van Herpen NA, Schrauwen-Hinderling VB, Schaart G, Mensink RP, Schrauwen P. Three weeks on a high-fat diet increases intrahepatic lipid accumulation and decreases metabolic flexibility in healthy overweight men. J Clin Endocrinol Metab. (2011) 96:E691–5. doi: 10.1210/jc.2010-2243
111. Parente EB, Pereira PHGR, Nunes VS, Lottenberg AM, Lima CSLM, Rochitte CE, et al. The effects of high-fat or high-carbohydrate diet on intramyocellular lipids. J Food Nutri Disord. (2014) 3:6. doi: 10.4172/2324-9323.1000158
112. Lee JS, Pinnamaneni SK, Eo SJ, Cho IH, Pyo JH, Kim CK, et al. Saturated, but not n-6 polyunsaturated, fatty acids induce insulin resistance: role of intramuscular accumulation of lipid metabolites. J Appl Physiol. (2006) 100:1467–74. doi: 10.1152/japplphysiol.01438.2005
113. Gaster M, Rustan AC, Beck-Nielsen H. Differential utilization of saturated palmitate and unsaturated oleate: evidence from cultured myotubes. Diabetes (2005) 54:648–56. doi: 10.2337/diabetes.54.3.648
114. Bakke SS, Moro C, Nikolic N, Hessvik NP, Badin PM, Lauvhaug L, et al. Palmitic acid follows a different metabolic pathway than oleic acid in human skeletal muscle cells; lower lipolysis rate despite an increased level of adipose triglyceride lipase. Biochim Biophys Acta (2012) 1821:1323–33. doi: 10.1016/j.bbalip.2012.07.001
115. Ryberg M, Sandberg S, Mellberg C, Stegle O, Lindahl B, Larsson C, et al. A Palaeolithic-type diet causes strong tissue-specific effects on ectopic fat deposition in obese postmenopausal women. J Internal Med. (2013) 274:67–76. doi: 10.1111/joim.12048
116. Spadaro L, Magliocco O, Spampinato D, Piro S, Oliveri C, Alagona C, et al. Effects of n-3 polyunsaturated fatty acids in subjects with nonalcoholic fatty liver disease. Dig Liver Dis. (2008) 40:194–9. doi: 10.1016/j.dld.2007.10.003
117. Jebb SA, Lovegrove JA, Griffin BA, Frost GS, Moore CS, Chatfield MD, et al. Effect of changing the amount and type of fat and carbohydrate on insulin sensitivity and cardiovascular risk: the RISCK (Reading, Imperial, Surrey, Cambridge, and Kings) trial. Am J Clin Nutr. (2010) 92:748–58. doi: 10.3945/ajcn.2009.29096
118. Tierney AC, McMonagle J, Shaw DI, Gulseth HL, Helal O, Saris WH, et al. Effects of dietary fat modification on insulin sensitivity and on other risk factors of the metabolic syndrome–LIPGENE: a European randomized dietary intervention study. Int J Obes. (2011) 35:800–9. doi: 10.1038/ijo.2010.209
119. Vessby B, Uusitupa M, Hermansen K, Riccardi G, Rivellese AA, Tapsell LC, et al. Substituting dietary saturated for monounsaturated fat impairs insulin sensitivity in healthy men and women: the KANWU Study. Diabetologia (2001) 44:312–9. doi: 10.1007/s001250051620
120. Le KA, Bortolotti M. Role of dietary carbohydrates and macronutrients in the pathogenesis of nonalcoholic fatty liver disease. Current Opin Clin Nutri Metab Care (2008) 11:477–82. doi: 10.1097/MCO.0b013e328302f3ec
121. Utzschneider KM, Bayer-Carter JL, Arbuckle MD, Tidwell JM, Richards TL, Craft S. Beneficial effect of a weight-stable, low-fat/low-saturated fat/low-glycaemic index diet to reduce liver fat in older subjects. Br J Nutr. (2013) 109:1096–104. doi: 10.1017/S0007114512002966
122. Tappy L. Fructose metabolism and noncommunicable diseases: recent findings and new research perspectives. Curr Opin Clin Nutr Metab Care (2018) 21:214–22. doi: 10.1097/MCO.0000000000000460
123. Bravo S, Lowndes J, Sinnett S, Yu Z, Rippe J. Consumption of sucrose and high-fructose corn syrup does not increase liver fat or ectopic fat deposition in muscles. Appl Physiol Nutr Metab. (2013) 38:681–8. doi: 10.1139/apnm-2012-0322
124. Lim JS, Mietus-Snyder M, Valente A, Schwarz JM, Lustig RH. The role of fructose in the pathogenesis of NAFLD and the metabolic syndrome. Nat Rev Gastroenterol Hepatol. (2010) 7:251–64. doi: 10.1038/nrgastro.2010.41
125. Wei Y, Wang D, Topczewski F, Pagliassotti MJ. Fructose-mediated stress signaling in the liver: implications for hepatic insulin resistance. J Nutr Biochem. (2007) 18:1–9. doi: 10.1016/j.jnutbio.2006.03.013
126. Roglans N, Vila L, Farre M, Alegret M, Sanchez RM, Vazquez-Carrera M, et al. Impairment of hepatic Stat-3 activation and reduction of PPARalpha activity in fructose-fed rats. Hepatology (2007) 45:778–88. doi: 10.1002/hep.21499
127. Le KA, Faeh D, Stettler R, Ith M, Kreis R, Vermathen P, et al. A 4-wk high-fructose diet alters lipid metabolism without affecting insulin sensitivity or ectopic lipids in healthy humans. Am J Clin Nutr. (2006) 84:1374–9. doi: 10.1093/ajcn/84.6.1374
128. Raatz SK, Johnson LK, Picklo MJ. Consumption of honey, sucrose, and high-fructose corn syrup produces similar metabolic effects in glucose-tolerant and -intolerant individuals. J Nutr. (2015) 145:2265–72. doi: 10.3945/jn.115.218016
129. Blaak EE. Carbohydrate quantity and quality and cardio-metabolic risk. Curr Opin Clin Nutr Metab Care (2016) 19:289–93. doi: 10.1097/MCO.0000000000000290
130. Bortolotti M, Maiolo E, Corazza M, Van Dijke E, Schneiter P, Boss A, et al. Effects of a whey protein supplementation on intrahepatocellular lipids in obese female patients. Clin Nutr. (2011) 30:494–8. doi: 10.1016/j.clnu.2011.01.006
131. DeFronzo RA, Tobin JD, Andres R. Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol. (1979) 237:E214–23. doi: 10.1152/ajpendo.1979.237.3.E214
132. Gutch M, Kumar S, Razi SM, Gupta KK, Gupta A. Assessment of insulin sensitivity/resistance. Indian J Endocrinol Metab. (2015) 19:160–4. doi: 10.4103/2230-8210.146874
133. Spritzler F. A low-carbohydrate, whole-foods approach to managing diabetes and prediabetes. Diabetes Spectr. (2012) 25:238–43. doi: 10.2337/diaspect.25.4.238
134. Zeevi D, Korem T, Zmora N, Israeli D, Rothschild D, Weinberger A, et al. Personalized Nutrition by prediction of glycemic responses. Cell (2015) 163:1079–94. doi: 10.1016/j.cell.2015.11.001
135. Baratta F, Pastori D, Polimeni L, Bucci T, Ceci F, Calabrese C, et al. Adherence to mediterranean diet and non-alcoholic fatty liver disease: effect on insulin resistance. Am J Gastroenterol. (2017) 112:1832–9. doi: 10.1038/ajg.2017.371
136. Lopez S, Bermudez B, Ortega A, Varela LM, Pacheco YM, Villar J, et al. Effects of meals rich in either monounsaturated or saturated fat on lipid concentrations and on insulin secretion and action in subjects with high fasting triglyceride concentrations. Am J Clin Nutr. (2011) 93:494–9. doi: 10.3945/ajcn.110.003251
137. Penn L, White M, Lindstrom J, den Boer AT, Blaak E, Eriksson JG, et al. Importance of weight loss maintenance and risk prediction in the prevention of type 2 diabetes: analysis of european diabetes prevention study RCT. PLoS ONE (2013) 8:e57143. doi: 10.1371/journal.pone.0057143
138. Tuomilehto J, Lindstrom J, Eriksson JG, Valle TT, Hamalainen H, Ilanne-Parikka P, et al. Prevention of type 2 diabetes mellitus by changes in lifestyle among subjects with impaired glucose tolerance. New Engl J Med. (2001) 344:1343–50. doi: 10.1056/NEJM200105033441801
139. Lindstrom J, Louheranta A, Mannelin M, Rastas M, Salminen V, Eriksson J, et al. The finnish diabetes prevention study (DPS): lifestyle intervention and 3-year results on diet and physical activity. Diabetes Care (2003) 26:3230–6. doi: 10.2337/diacare.26.12.3230
140. Abdul-Ghani MA, Matsuda M, Balas B, DeFronzo RA. Muscle and liver insulin resistance indexes derived from the oral glucose tolerance test. Diabetes Care (2007) 30:89–94. doi: 10.2337/dc06-1519
Keywords: insulin resistance, ectopic fat, personalized nutrition, type 2 diabetes mellitus, metabolic phenotype
Citation: Trouwborst I, Bowser SM, Goossens GH and Blaak EE (2018) Ectopic Fat Accumulation in Distinct Insulin Resistant Phenotypes; Targets for Personalized Nutritional Interventions. Front. Nutr. 5:77. doi: 10.3389/fnut.2018.00077
Received: 20 June 2018; Accepted: 15 August 2018;
Published: 04 September 2018.
Edited by:
Ondřej Šeda, Charles University, CzechiaReviewed by:
Michaela Tencerova, University of Southern Denmark Odense, DenmarkAkihiko Ando, Nishio Hospital, Japan
Copyright © 2018 Trouwborst, Bowser, Goossens and Blaak. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ellen E. Blaak, ZS5ibGFha0BtYWFzdHJpY2h0dW5pdmVyc2l0eS5ubA==