
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurosci., 15 July 2020
Sec. Neurodegeneration
Volume 14 - 2020 | https://doi.org/10.3389/fnins.2020.00674
This article is part of the Research TopicLRRK2 - Fifteen Years From Cloning to the ClinicView all 20 articles
 Steven P. Seegobin1,2,3
Steven P. Seegobin1,2,3 George R. Heaton1,2,3
George R. Heaton1,2,3 Dongxiao Liang1,2,3,4
Dongxiao Liang1,2,3,4 Insup Choi1,2,3
Insup Choi1,2,3 Marian Blanca Ramirez1,2,3
Marian Blanca Ramirez1,2,3 Beisha Tang4,5
Beisha Tang4,5 Zhenyu Yue1,2,3*
Zhenyu Yue1,2,3*Mutations in the leucine-rich repeat kinase 2 (LRRK2) gene are the most frequent cause of familial Parkinson’s disease (PD). Several genetic manipulations of the LRRK2 gene have been developed in animal models such as rodents, Drosophila, Caenorhabditis elegans, and zebrafish. These models can help us further understand the biological function and derive potential pathological mechanisms for LRRK2. Here we discuss common phenotypic themes found in LRRK2-associated PD animal models, highlight several issues that should be addressed in future models, and discuss emerging areas to guide their future development.
Parkinson’s disease (PD) is a common, age-dependent, neurodegenerative disease with a lifetime risk of approximately 1.5% (Lees et al., 2009). The defining diagnostic features of PD include bradykinesia, resting tremor, rigidity, cognitive impairment, and psychiatric dysfunction (Goetz et al., 2008). Neuropathologically, PD is characterized by degeneration of dopaminergic (DA) neurons in the substania nigra pars compacta (SNpc) and the presence of Lewy Bodies (LBs) throughout the central nervous system (CNS) (Gelb et al., 1999; Dickson et al., 2009). Extensive human genetic studies have led to the discovery of several PD-causing genes, including leucine-rich repeat kinase 2 (LRRK2) (Nalls et al., 2019). PD-causing mutations in LRRK2 occur in up to 41% of select patient populations and, as such, they represent the greatest known cause of heritable PD (Khan et al., 2005; Lesage and Brice, 2009). Mutations in LRRK2 are also found in sporadic PD, occurring at a rate of 1–3% (Gilks et al., 2005; Lesage et al., 2007; Paisán-Ruíz et al., 2008). Importantly, the clinical presentation of Parkinsonism in LRRK2 mutation carriers has been described as indistinguishable from sporadic PD patients (Adams et al., 2005; Kay et al., 2006). Given its importance in both sporadic and familial PD, understanding LRRK2 biology will assist in elucidation of common mechanisms of disease pathogenesis (for a more thorough review, see Kluss et al., 2019).
The LRRK2 gene encodes a large, 2,527-amino acid, 286-kDa, multi-domain protein belonging to the ROCO family (Zimprich et al., 2004). All ROCO proteins are characterized by a GTPase Ras-like G domain (Roc), followed in tandem by a C-terminal of Roc domain (COR) (Bosgraaf and Van Haastert, 2003; Marín, 2006). LRRK2 also contains a serine-threonine kinase domain, capable of phosphorylating both itself and a small group of substrates (West et al., 2005; Sheng et al., 2012; Steger et al., 2016). To date, most of the pathogenic mutations are clustered within the Roc, COR, or kinase domains and are found to alter LRRK2’s biochemical activity (Chen and Wu, 2018). Although LRRK2 activity has been linked to a diverse range of cellular processes (reviewed by Berwick et al., 2019), a large body of work suggests LRRK2 plays a key role in membrane trafficking along the endo-lysosomal pathway. These functions include synaptic vesicle endocytosis, degradation, and recycling of trans-membrane receptors, anterograde trafficking of receptors from the trans-golgi network to lysosomes, and retromer-mediated transmembrane recycling. LRRK2-dependent regulation of these cellular processes may be associated with LRRK2’s ability to bind and phosphorylate a cluster of Rab GTPases (Shin et al., 2008; Piccoli et al., 2011; MacLeod et al., 2013; Gómez-Suaga et al., 2014; Schreij et al., 2014; Pan et al., 2017; Steger et al., 2017; Pellegrini et al., 2018; Rivero-Ríos et al., 2019; Sheehan and Yue, 2019).
A diverse range of animal models have been developed which overexpress, knock-out, knock-down, or knock-in mutated and wildtype forms of the LRRK2 gene, for the characterization of LRRK2 biological and pathophysiological functions. In this review, we discuss common phenotypic themes found in LRRK2-associated rodent, Drosophila, Caenorhabditis elegans, and zebrafish animal models and highlight several new avenues toward development of future LRRK2 animal models.
Rodent models have been widely used in the study of LRRK2 biology due to their genetic and neuroanatomical similarities to humans. Both mice and rats possess a mammalian homolog of LRRK2 which shares approximately 86–88% sequence identity to human LRRK2. Importantly, all residues affected by pathogenic mutations in humans are conserved in rodent LRRK2 (Langston et al., 2016). Rodents also possess a LRRK1 homolog, which shares ankyrin repeats (ANK), leucine-rich repeats (LRR), and Roc, COR, and kinase domains with LRRK2, but may also contain a WD40 domain that is still contested in the literature (Biskup et al., 2007; Civiero et al., 2012; Sejwal et al., 2017; Xing et al., 2017). The dopaminergic neuroanatomical pathways of rodents and humans are also highly similar, leading to the development of an array of sensitive behavioral tests in rodents that may correlate to dopamine loss in human PD (Meredith and Kang, 2006; Redgrave et al., 2010). Therefore, rodents represent an ideal candidate for genetic manipulations to investigate LRRK2 biology toward investigation of PD pathogenesis.
LRRK2 knock-out (KO) models have been chiefly employed to investigate the physiological function of endogenous LRRK2 (Tables 1, 2). An emerging theme of LRRK2 KO rodent models is that the resulting phenotypes do not mimic LRRK2-assocated PD. Rodents born without LRRK2 exhibit no loss of dopaminergic (DA) neurons, demonstrate mild to no behavioral or locomotor defects, have limited neuropathology, and have unchanged dopamine synthesis (as measured by DOPAC and HVA) (Andres-Mateos et al., 2009; Lin et al., 2009; Tong et al., 2010b; Herzig et al., 2011; Hinkle et al., 2012; Daher et al., 2014).
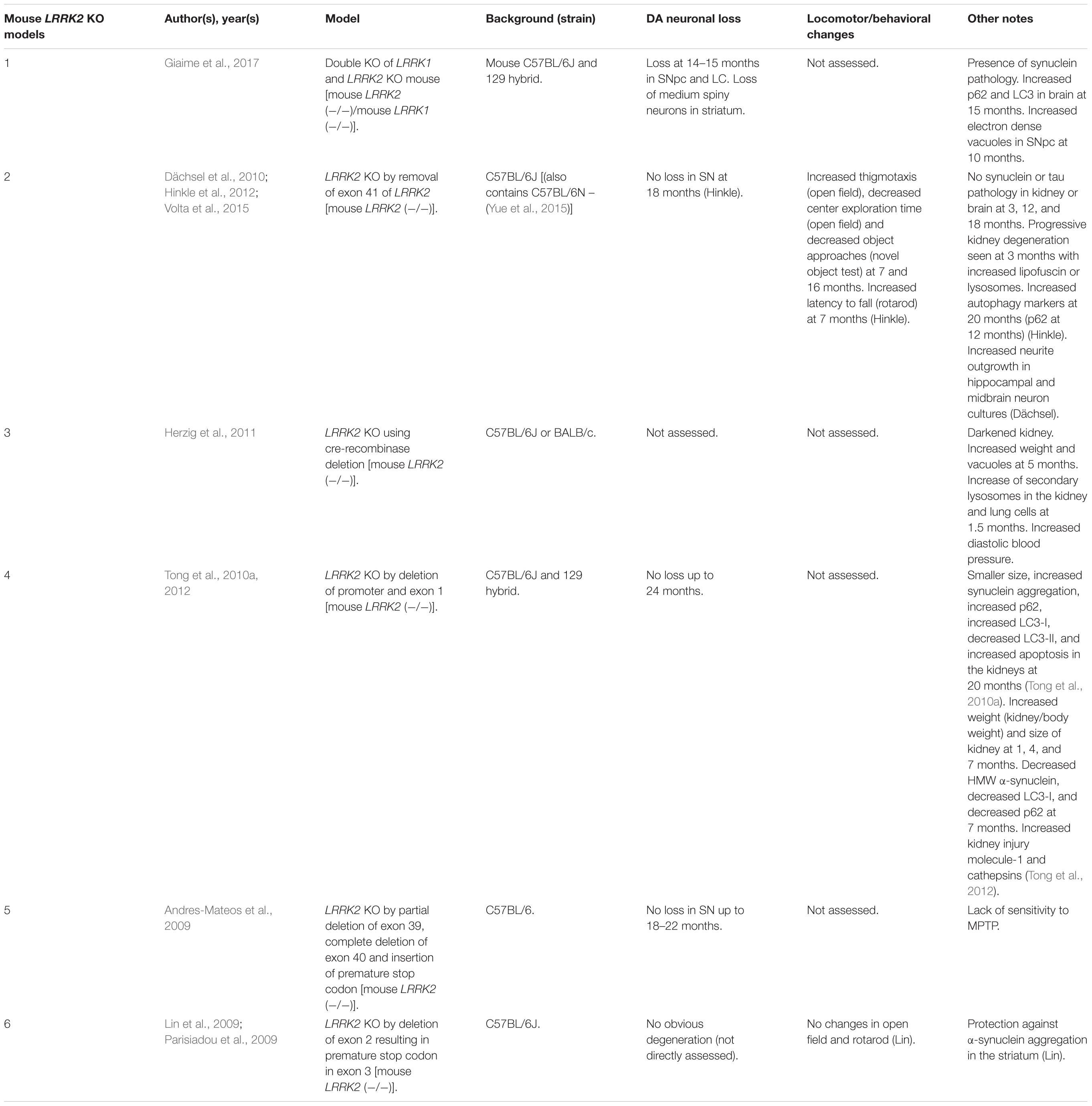
Table 1. Mouse LRRK2 KO models.
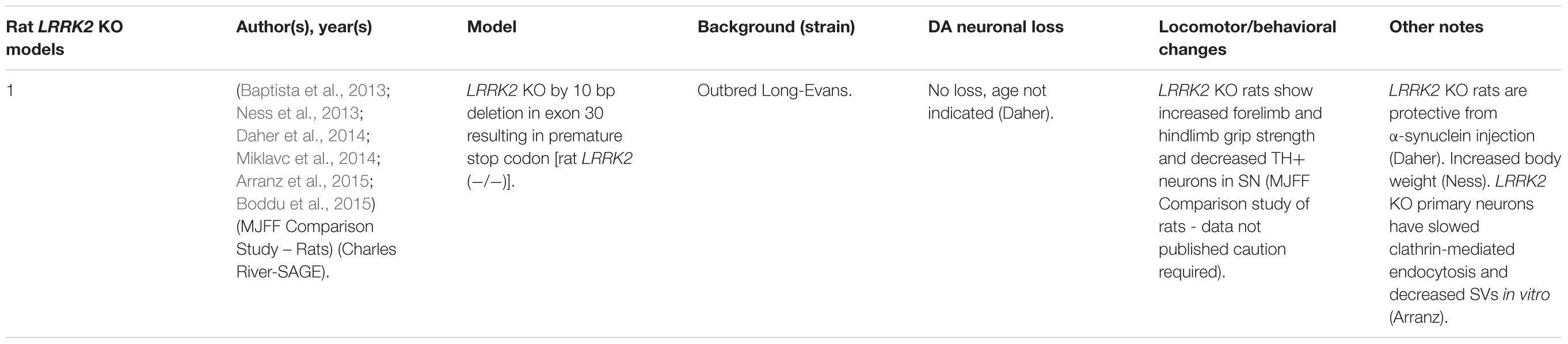
Table 2. Rat LRRK2 KO models.
Although no clear neuronal phenotypes have emerged, several studies have suggested that LRRK2 may fulfill key functions in peripheral tissues. Most notably, LRRK2 KO kidneys exhibit striking age-dependent changes in color and weight, along with ultrastructural abnormalities (Tong et al., 2010a, 2012; Herzig et al., 2011; Hinkle et al., 2012; Baptista et al., 2013). A darker coloration of LRRK2 KO kidneys has been observed as early as 2–3 months, while increases in weight are reported as early as 1–5 months in a sex-specific manner (Herzig et al., 2011; Hinkle et al., 2012; Tong et al., 2012; Ness et al., 2013; Boddu et al., 2015). Curiously, one group has reported a decrease in the weight of LRRK2 KO kidneys at 20–27 months in age (Tong et al., 2010b, 2012). Electron microscopy (EM) analysis of KO kidneys has further revealed an increase in the number and size of lysosomes in epithelial cells of the renal cortex, starting at 4 months of age (Herzig et al., 2011; Hinkle et al., 2012; Tong et al., 2012; Baptista et al., 2013). These studies also found increases in lipid-containing vesicles or droplets and lipofuscin (potentially contained within lysosomes). Alongside ultrastructural abnormalities, multiple groups have reported changes in autophagic markers, LC3, and p62 and lysosomal markers, LAMP-1 and Cathepsin D, consistent with alterations in the autophagy–lysosomal pathway (Tong et al., 2010a, 2012; Hinkle et al., 2012). Changes in homeostatic parameters regulated by the kidneys have also been reported in LRRK2 KO animals, including increased diastolic blood pressure, decreased plasma and serum chloride, and decreased specific gravity of urine (Herzig et al., 2011; Baptista et al., 2013; Ness et al., 2013; Boddu et al., 2015; Fuji et al., 2015). Kidney dysfunction may be detected using the biomarker of kidney health known as lipocalin-2 (NGAL), which has also been observed to be reduced in plasma and urine (Ness et al., 2013; Fuji et al., 2015).
Several additional abnormalities have been reported in other peripheral organs in LRRK2 KO animals. Lungs of LRRK2 KO rats exhibit an increased number and size of type II alveolar cell lamellar bodies, a lysosome-derived secretory vesicle that stores surfactant (Herzig et al., 2011; Baptista et al., 2013; Miklavc et al., 2014). Indications of liver dysfunction such as increased AST, ALT, and cholesterol levels have also been reported (Ness et al., 2013; Baptista et al., 2013). To a lesser degree, abnormalities in the spleen, such as decreased size and changes in cellular composition, were also found (Ness et al., 2013).
A less-clear phenotype seen has been the disruption of α-synuclein homeostasis in LRRK2 KO rodent models. While several studies have shown that LRRK2 KO or kinase inhibition is protective against α-synuclein aggregation in the brain (Lin et al., 2009; Herzig et al., 2011; Daher et al., 2014; Bae et al., 2018), others have reported that LRRK2 KO causes accumulation of α-synuclein in the kidneys (Tong et al., 2010a, 2012). These seemingly conflicting findings may indicate tissue or model specific differences in α-synuclein metabolism.
An exception to phenotypes seen in single LRRK2 KO models has been a recently developed LRRK1 and LRRK2 double-KO mouse that shows α-synuclein pathology, disruption of the autophagy–lysosomal pathway, and DA neurodegeneration in the CNS (Giaime et al., 2017). While the result suggests the possibility that LRRK1 compensates for the loss of LRRK2 in the brain, it raises the question as to whether PD mutations of LRRK2 could be implicated in the loss of LRRK1/LRRK2 functions. Future studies will have to further verify these findings and elucidate potential mechanisms.
Several rodent transgenic models have been generated to overexpress (OE) WT or pathogenic variants of LRRK2. These transgenic models have been developed using either insertions of LRRK2 cDNA or bacterial artificial chromosomes (BACs) (Tables 3–5).
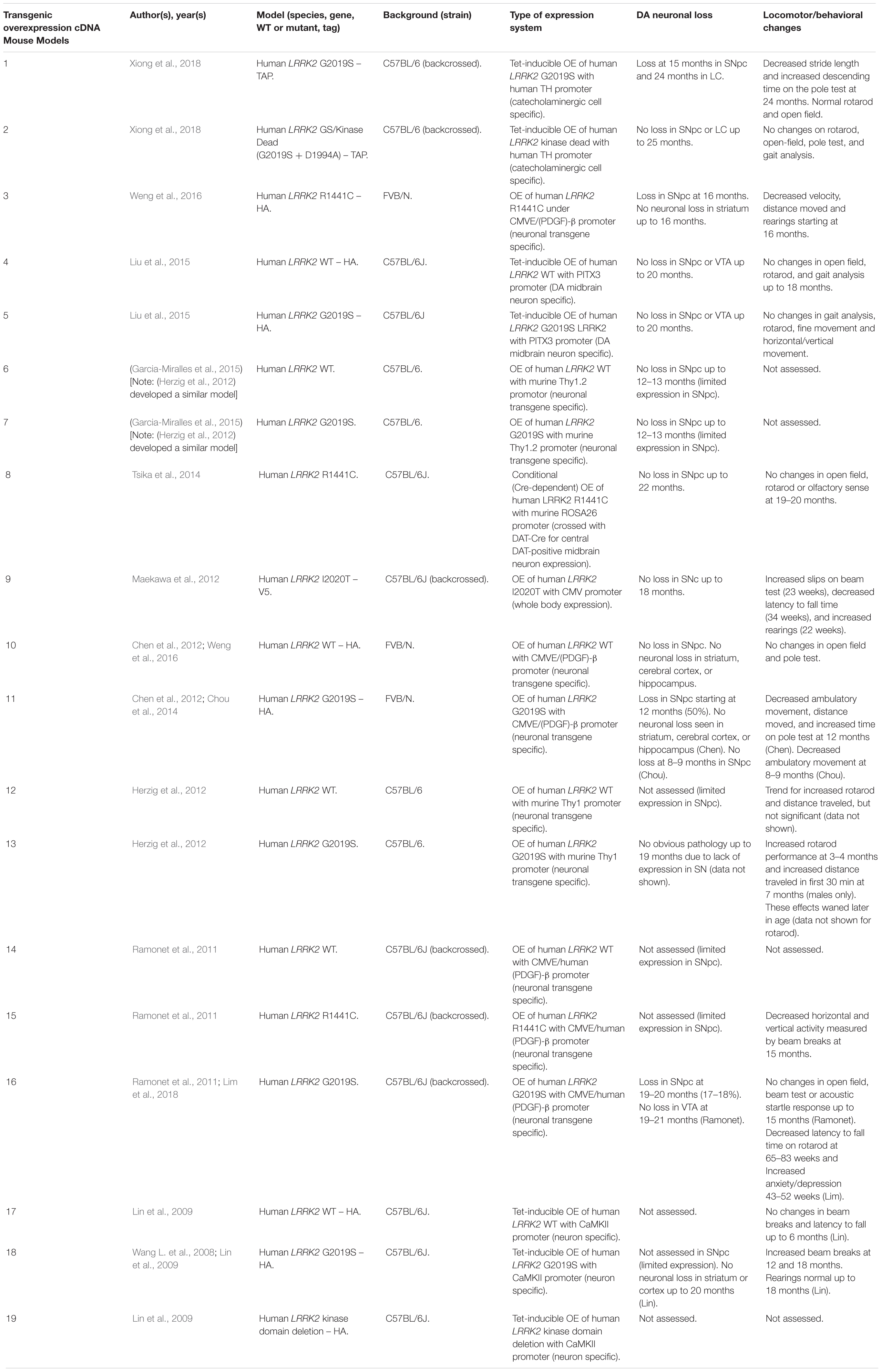
Table 3. Transgenic OE of LRRK2 using cDNA in mouse models.
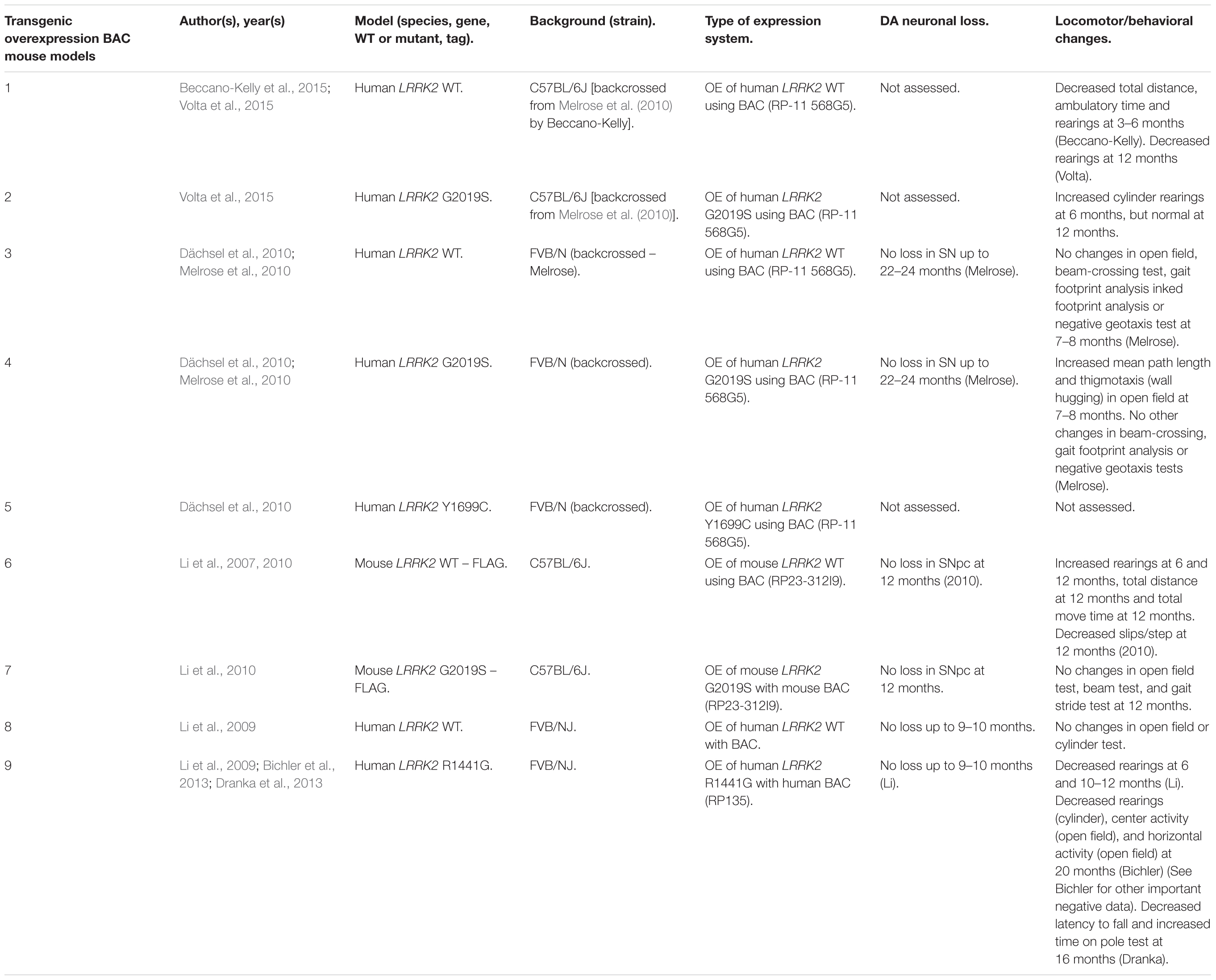
Table 4. Transgenic OE of LRRK2 using BAC in mouse models.
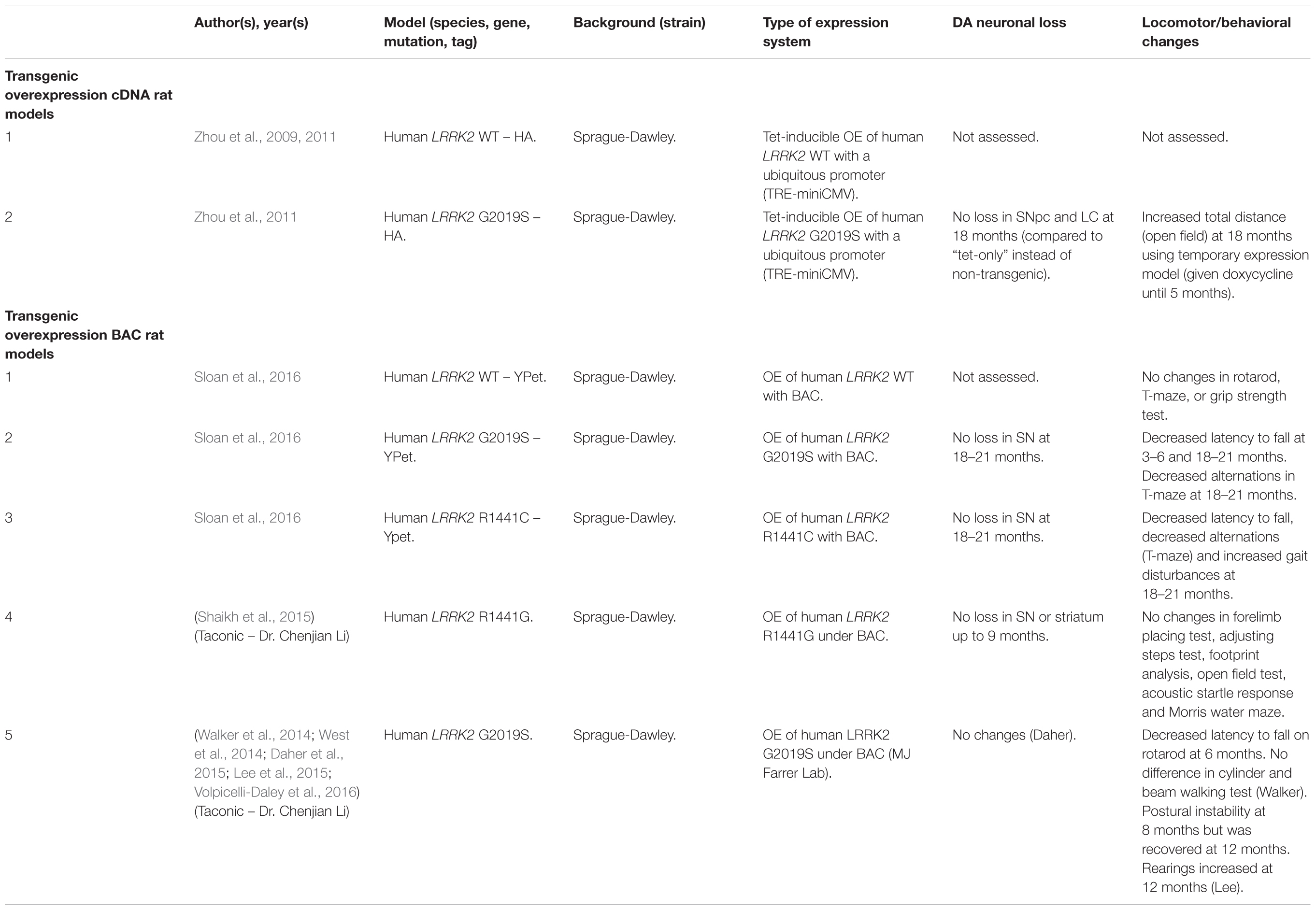
Table 5. Rat transgenic LRRK2 models.
An emerging theme in transgenic mouse models is that overexpression of pathogenic LRRK2 mutants, such as G2019S or R1441C, can induce PD-like phenotypes. Reported phenotypes include DA neuronal loss, disruption of dopamine homeostasis, ultrastructural changes, L-DOPA responsive locomotor defects, and pathological accumulations of tau and α-synuclein (Ramonet et al., 2011; Chen et al., 2012; Weng et al., 2016; Xiong et al., 2018). Several of these phenotypes have also been reproduced in rats, though without any obvious DA neuronal loss (Zhou et al., 2011; West et al., 2014; Sloan et al., 2016). In mice, DA neuron degeneration is typically observed at mid- to older age, occurring typically at 15–20 months in the SNpc (Ramonet et al., 2011; Weng et al., 2016; Xiong et al., 2018). Prior to cell death, DA neurons are frequently observed to exhibit abnormal morphology and reduced synaptic vesicles (Burke and O’Malley, 2013; Tsika et al., 2014; Liu et al., 2015; Weng et al., 2016). Decreased striatal dopamine content, dopamine metabolites, and evoked dopamine release also frequently occur in conjunction with DA neuronal death (Ramonet et al., 2011; Zhou et al., 2011; Liu et al., 2015; Sloan et al., 2016; Lim et al., 2018; Xiong et al., 2018). Consequently, these rodents exhibit locomotor defects, which are partially rescued with L-DOPA (Sloan et al., 2016; Weng et al., 2016; Xiong et al., 2018). To date, two studies using cDNA LRRK2-G2019S overexpression have been able to show both DA neurodegeneration and pathological inclusions (Chen et al., 2012; Xiong et al., 2018). In one such model, phosphorylated tau was increased at 12 months in the SNpc, in parallel with DA neuronal loss (Chen et al., 2012). The other reported an increase in phosphorylated and high molecular weight α-synuclein in the striatum and ventral medial body at 24 months of age (Xiong et al., 2018).
Although transgenic OE rodent models can capture many of the cardinal features of PD patients to various extents, there are several key caveats to bear in mind. For example, the question of whether DA neuronal loss occurs in a cell-autonomous or non-cell-autonomous manner has arisen, due to cell type-specific expression. Only mouse models which employ a DA neuron specific (TH) or neuronal transgene (CMV enhancer/PDGF-β) promoter has been able to induce DA neurodegeneration (Ramonet et al., 2011; Chen et al., 2012; Weng et al., 2016; Xiong et al., 2018). Of note, other neuronal transgene promoters, such as Thy1.2 and CaMKII, have failed to induce DA neuronal death, possibly owing to a lack of sufficient expression in midbrain DA neurons (Tsika et al., 2014; Garcia-Miralles et al., 2015). Alternative rodent models generated using a BAC approach, which use endogenous LRRK2 promoter to drive the expression has presented with more subtle phenotypes, such as changes in dopamine homeostasis and mild behavioral or locomotor dysfunction (Tables 4, 5).
One clear advantage of rodent OE models is that they have enabled the study of neurodegenerative mechanisms. One potential mechanism of action is LRRK2-mediated phosphorylation of synaptic proteins with known functions in vesicle endocytosis. Specifically, LRRK2 has been shown to phosphorylate the synaptic proteins synaptojanin 1 (SYNJ1), Endophilin A1 (SH3GL2) and auxilin (DNAJC6) (Matta et al., 2012; Arranz et al., 2015; Pan et al., 2017; Nguyen and Krainc, 2018). Broadly speaking, these studies have proposed that increased LRRK2-kinase activity disrupts the physiological function of these presynaptic trafficking proteins, resulting in defects in synaptic vesicle endocytosis (Pan et al., 2019).
Currently, the disease mutations of LRRK2 knock-in (KI) models have only been developed in mice (Table 6). Unlike OE models, KI models do not suffer from potential overexpression artifacts or interspecies differences. However, LRRK2 KI mouse models have failed to show DA neuron degeneration or α-synuclein pathology. Rather, these models exhibit neurophysiological changes, altered DA homeostasis, and modest behavioral abnormalities.
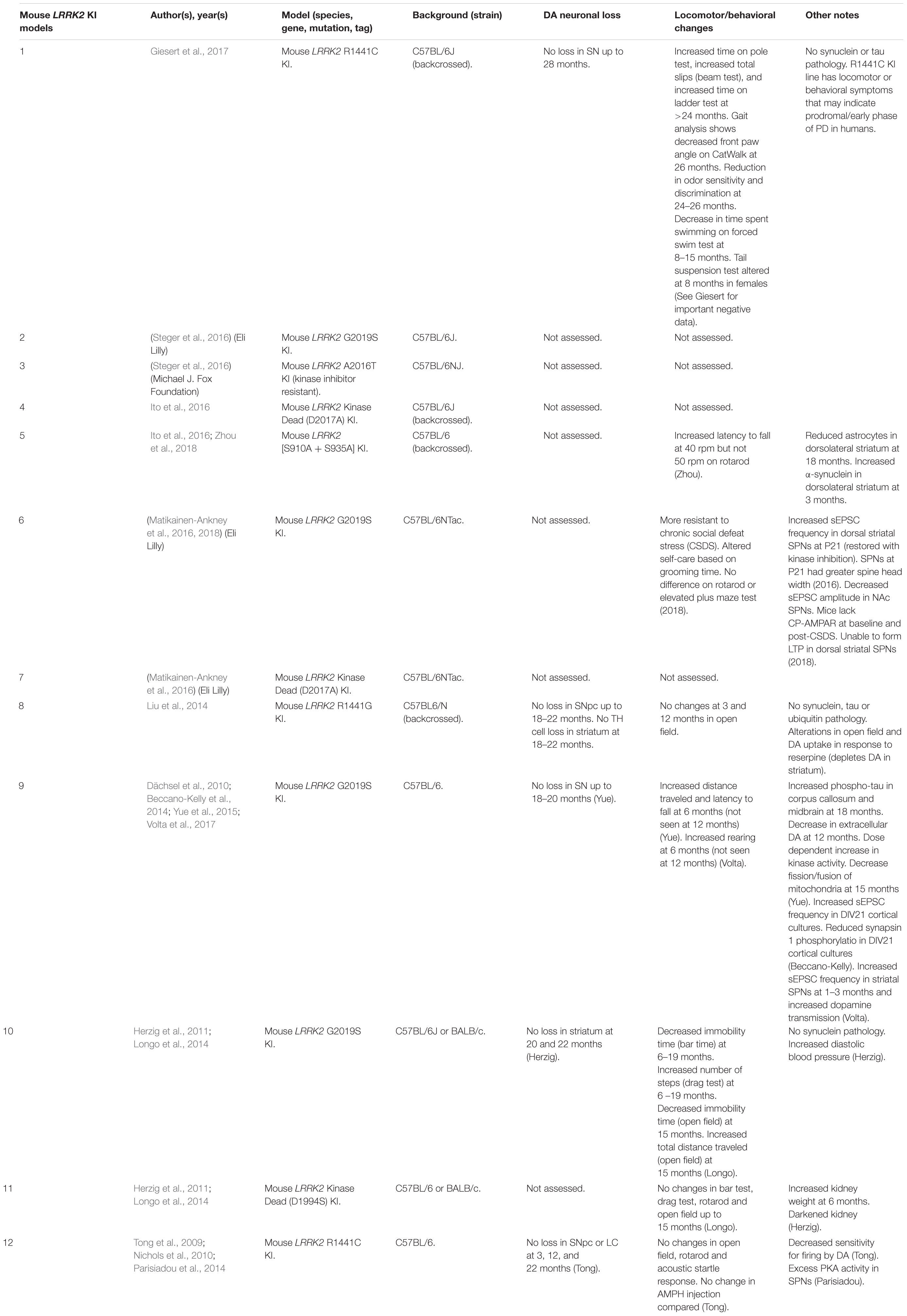
Table 6. Mouse LRRK2 KI models.
Two independent groups have reported that the LRRK2-G2019S KI mice exhibit increased frequency of spontaneous excitatory post-synaptic potentials (sEPSCs) in spiny projection neurons (SPNs) of the striatum (Matikainen-Ankney et al., 2016; Volta et al., 2017). Young (1–3 months) G2019S KI mice also exhibit elevated dopamine release upon repeated stimulation (Volta et al., 2017). These changes are mirrored by increases in total distance moved and rearings indicative of hyperkinesia at a young age (Longo et al., 2014; Yue et al., 2015; Volta et al., 2017). However, in older mice (>12 months), extracellular levels of dopamine appear decreased and hyperkinetic behavior is reduced (Yue et al., 2015; Volta et al., 2017). Perhaps surprisingly, a recent study has reported that young G2019S KI mice are more resilient to chronic social defeat stress (CSDS) (Matikainen-Ankney et al., 2018). The authors suggest that this may be due to the inability for calcium-permeable AMPA receptors (CP-AMPARs) to integrate into the synaptic membrane, blocking the formation of long-term potentiation (LTP). However, whether the synaptic changes at young adulthood in rodents confer altered non-motor or motor phenotypes in late-onset PD remains unclear.
Drosophila LRRK2 models can offer several advantages over rodents. Firstly, the relatively short lifespan of Drosophila (∼2 to 3 months) allows age-dependent changes in phenotypic variability to manifest quicker than in rodent models (e.g., synuclein pathology; Feany and Bender, 2000). In addition, the presence of the UAS-GAL4 system in several Drosophila lines can create a diverse range of genetic manipulations. Finally, the dopaminergic system in Drosophila consists of six well-defined neuronal clusters that can easily be quantified to assay DA-specific neuronal death.
Unlike rodents and humans, which possess both LRRK1 and LRRK2 genes, Drosophila only possesses a single LRRK2 ortholog, dLRRK. dLRRK is 2,445 amino acids in length and shares an overall sequence identity of 24% with human LRRK2 (Wang D. et al., 2008). Like LRRK2 KO rodent models, loss of dLRRK does not cause DA neurodegeneration (Table 7) (Imai et al., 2008; Wang D. et al., 2008; Tain et al., 2009). Several studies have, however, reported locomotor deficits and loss of fertility in dLRRK KOs (Lee et al., 2007; Imai et al., 2008; Tain et al., 2009). An unclear point of investigation is whether dLRRK KOs are sensitive or protective to treatment with reactive oxygen species (ROS) with two studies observing conflicting evidence for H2O2 on mortality (Imai et al., 2008; Wang D. et al., 2008). Future studies should evaluate reasons for this discrepancy and probe potential mechanisms for ROS sensitivity or protection in dLRRK KOs.
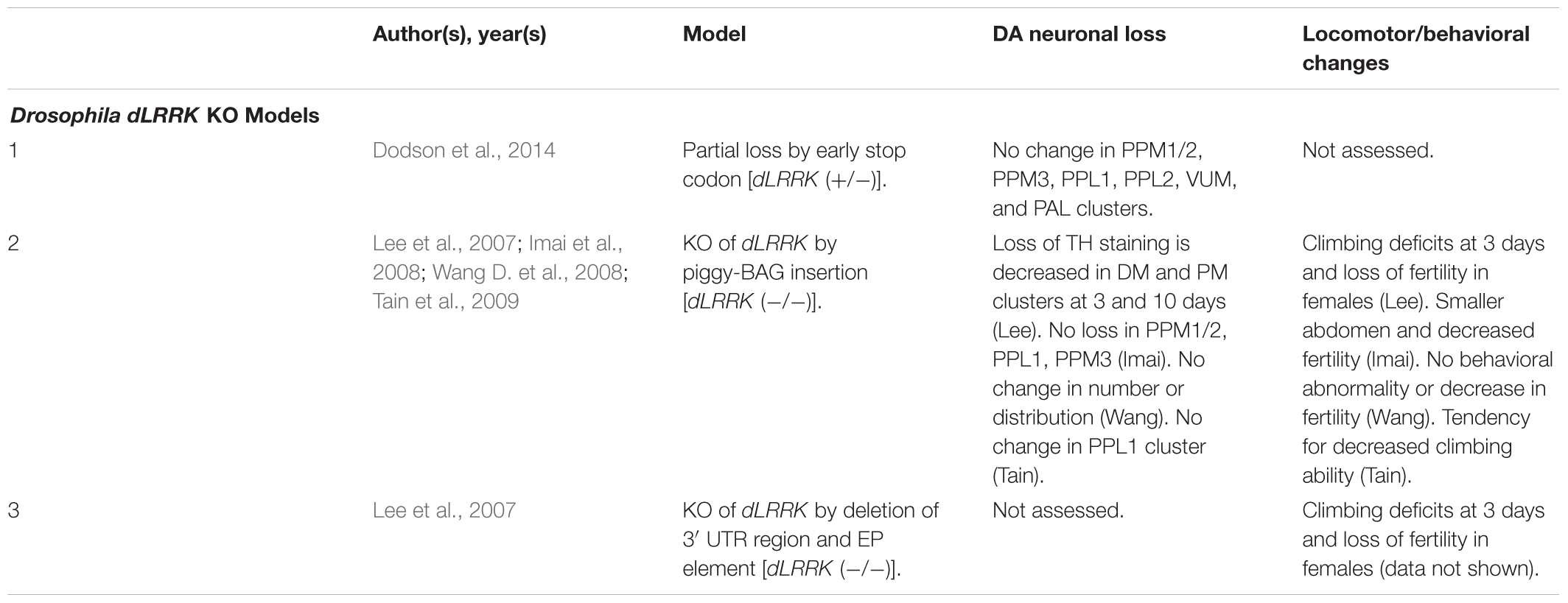
Table 7. Drosophila dLRRK KO models.
As with rodent OE models, Drosophila OE of human LRRK2 G2019S and R1441C has resulted in DA neuronal loss, disruptions of dopamine homeostasis, and L-DOPA responsive locomotor defects (Table 8). Interestingly, other variants such as I1122V, Y1699C, I2020T, and G2385R have also been shown to cause DA neuronal loss in Drosophila (Imai et al., 2008; Liu et al., 2008; Ng et al., 2009; Venderova et al., 2009; Lin et al., 2010; Godena et al., 2014). This loss occurs at mid- to older age (35–60 days) and is commonly seen in the PPL1 DA neuronal cluster (Imai et al., 2008; Liu et al., 2008; Ng et al., 2009; Venderova et al., 2009). In addition to cell loss, transgenic models also exhibit decreased DA content and reduced climbing ability starting at birth or at 10 days, respectively (Imai et al., 2008; Ng et al., 2009). Several studies have also reported sensitivity to ROS in flies expressing pathogenic LRRK2 mutations (Imai et al., 2008; Ng et al., 2009).
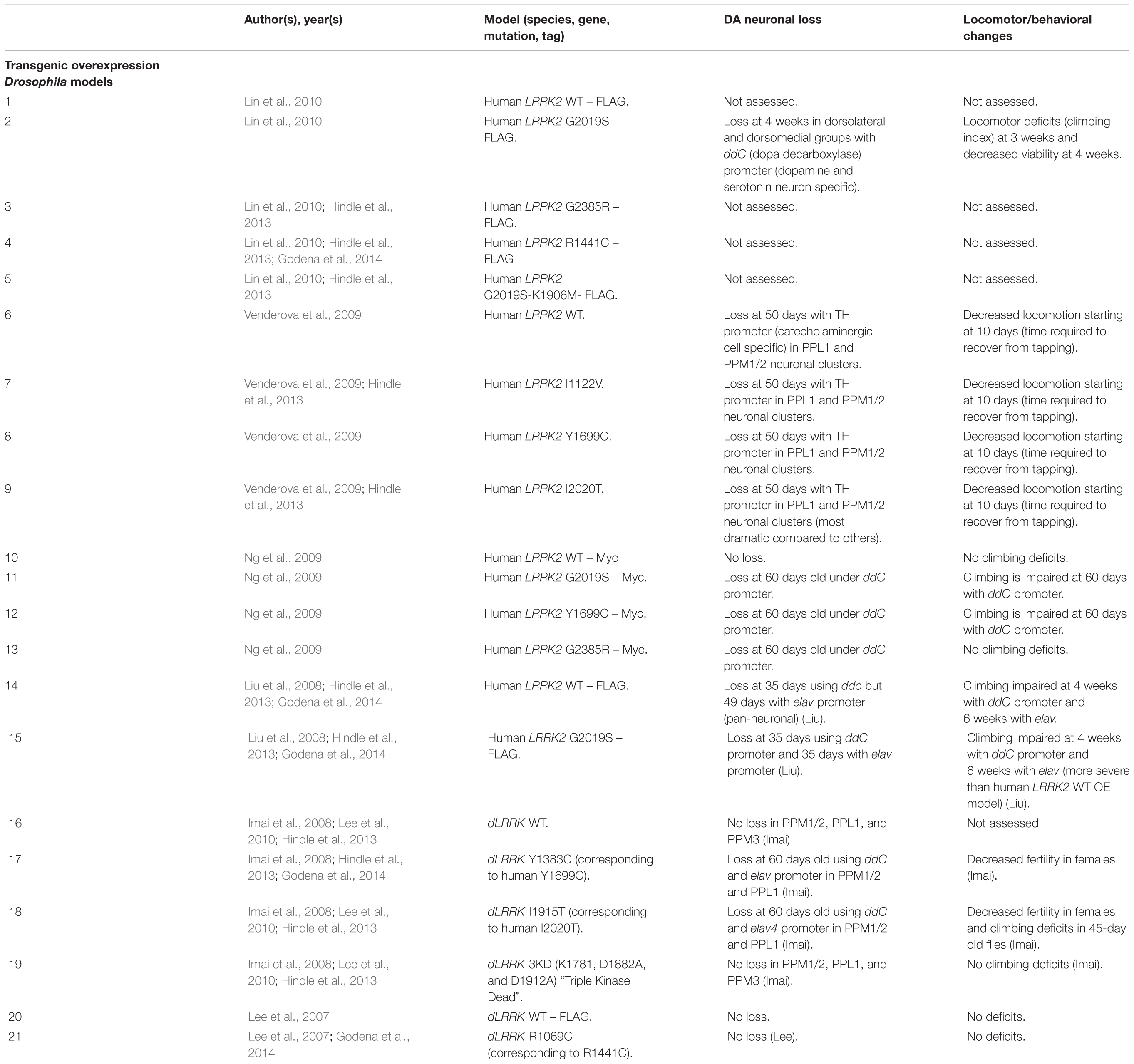
Table 8. Drosophila transgenic LRRK2 models.
Unlike rodent models, OE of human WT LRRK2 has also been reported to induce DA neurodegeneration (Liu et al., 2008). In addition, OE of Drosophila WT dLRRK (under a TH promoter) has also been shown to cause neurodegeneration (Imai et al., 2008). These models raise several questions, including whether DA neuronal loss may be caused by expression of a different species’ LRRK2 and whether cell-type specific expression can cause DA neuronal loss.
Caenorhabditis elegans have a well-characterized CNS with a total of 302 neurons, 8 of which are dopaminergic. Unlike rodents, the simplicity of the C. elegans’s nervous system has allowed for quantification and visualization of DA neuron morphology in vivo (Yao et al., 2010). In addition, its shorter lifespan, small size and cost-effectiveness have facilitated high-throughput drug screening (Maulik et al., 2017). Like Drosophila, C. elegans has one single ortholog of LRRK2, Lrk-1, which is broadly expressed in head and tail neurons, the hypodermis, intestine, and muscles (Sakaguchi-Nakashima et al., 2007). Both Lrk-1 KO and transgenic OE models of LRRK2 have been reported in C. elegans.
Like other LRRK2 KO models, loss of Lrk-1 does not lead to the degeneration of DA neurons or striking locomotor phenotypes (Sakaguchi-Nakashima et al., 2007; Saha et al., 2009; Sämann et al., 2009; Yao et al., 2010). C. elegans do, however, demonstrate a mis-localization of synaptic vesicles in DA neurons in vivo (Sakaguchi-Nakashima et al., 2007; Sämann et al., 2009). In addition, Lrk-1 KOs have been found to be more sensitive to ER stressors, such as tunicamycin, which has been recently observed to induce the recruitment of LRRK2 from the trans-Golgi to the lysosome in a Rab29-dependent manner (Sämann et al., 2009; Kuwahara et al., 2016; Eguchi et al., 2018). Both of these processes depend on the phosphorylation of Rab GTPases by LRRK2, and future studies in C. elegans may reveal more mechanistic insight in vivo.
The OE of human LRRK2 G2019S and R1441C in C. elegans has been shown to cause DA neurodegeneration, reduced dopamine levels, and locomotor dysfunction (Sämann et al., 2009; Yao et al., 2010; Liu et al., 2011). Interestingly, two conflicting studies suggest that LRRK2-mediated neurodegeneration may depend on either GTPase or kinase activity. One study suggests that OE of human LRRK2 K1347A (a defective GTP binding mutant) does not produce Parkinsonian phenotypes in C. Elegans, inferring that GTPase activity is essential for toxicity (Yao et al., 2010), while another indicates that the kinase dead human LRRK2 OE also does not produce loss of DA neurons (Liu et al., 2011). Furthermore, OE of human LRRK2 WT has also been shown to cause DA neurodegeneration (Yao et al., 2010). Future C. elegans transgenic OE models will have to clarify these discrepancies and use better GTPase inactive mutants in C. elegans to assess for toxicity.
Zebrafish (Danio rerio) is an attractive model due to its well-characterized neuronal circuitry, conserved neurobiochemical mechanisms between humans, optical transparency, small size, and ease of drug administration (Vaz et al., 2018). The lifespan of zebrafish is relatively long compared to other rodent models, with 71% survivability at 5 years, making it ideal for aging research (Vaz et al., 2018). Like Drosophila and C. elegans, zebrafish possess a sole ortholog to human LRRK2, called zLRRK2, sharing a 47% amino acid sequence identity with humans (Sheng et al., 2010).
Since an initial study reported that zLRRK2 KO is embryonically lethal, a targeted knock-down (KD) approach has been used as an alternative to reduce the expression of zLRRK2 (Sheng et al., 2010). This initial study saw that targeting the WD40 domain of zLRRK2 using morpholino oligonucleotides resulted in the loss of diencephalon DA neurons and locomotor defects (Sheng et al., 2010). A later study supported this finding and also observed α-synuclein aggregates in multiple brain regions (Prabhudesai et al., 2016). However, one study using a dosage of morpholinos in between these two studies achieved a greater KD of zLRRK2, but could not replicate these phenotypes (Ren et al., 2011). These studies may show conflicting results due to the reported off-target effects of morpholinos, and thus different approaches should be considered (Kok et al., 2015). As such, further study is required to establish a consistent theme in zebrafish zLRRK2 KD models.
Limited zebrafish mutant models have been developed, but a recent report using zinc finger nucleases (ZFNs) was able to introduce a mutation in the WD40 domain of zLRRK2 to generate a KI model (Sheng et al., 2018). They observed increases in locomotion in the adult stage and a weakened antibacterial response. Another report using transient overexpression of human LRRK2 WT and G2019S in zebrafish embryos was able to see disruptions in the ubiquitin-proteasome system (UPS) (Lichtenberg et al., 2011). Future studies will have to develop more mutant models of zebrafish models in order to gain more phenotypic insights.
Here we highlight several phenotypic themes found in LRRK2-associated PD animal models, with an emphasis on rodents.
LRRK2 KO models in mice, rats, Drosophila, and C. elegans have not produced DA neurodegeneration. Rather, phenotypic changes have been observed primarily in peripheral tissues such as the kidneys and lungs. Interestingly, primates treated with LRRK2 kinase inhibitors have also show disruptions in the kidneys and lungs with no obvious neuropathological changes (Fuji et al., 2015). Future models should work to uncover other novel phenotypes associated with LRRK2 KO models, which can be indicative of LRRK2 kinase inhibitor treatment, thereby facilitating the investigation of drug-based toxicity or efficacy. By contrast, transgenic OE of pathogenic LRRK2 in mice and Drosophila has been shown to cause robust DA neurodegeneration, tau and α-synuclein pathology, locomotor/behavioral deficits, alterations in DA homeostasis, and L-DOPA-responsive behavior. A recently reported mouse model was found to have many of these features including DA neurodegeneration, locomotor changes and α-synuclein pathology, and it may be useful for determining drug efficacy (Xiong et al., 2018). Other phenotypes, such as tau pathology and cognitive, behavioral, and peripheral organ changes have not been assessed in this model and could provide other important clinical measures. Finally, LRRK2 KI models used in mice and zebrafish have produced neurophysiological changes and modest behavioral or locomotor abnormalities. These phenotypes seen can be indicative of early or prodromal PD, and with careful behavioral analysis, such as one done by Giesert et al. (2017) in a R1441C KI mouse model, it can reveal other important phenotypes.
An emerging area in LRRK2 biology that may help guide future LRRK2 models is the role of Rab proteins and whether they can be used as an in vivo biomarker for LRRK2 activity. LRRK2 is capable of phosphorylating Rab3A/B/C/D, Rab8A/B, Rab10, Rab12, Rab29, Rab35, and Rab43 (Steger et al., 2016, 2017). Using LRRK2 G2019S, R1441C, kinase-dead, and phosphomimetic (S910A + S935A) KI rodent models, the endogenous phosphorylation of Rab proteins can be carefully evaluated in a tissue-specific manner. This should provide important clues for cell-type and region-specific readouts of LRRK2 activity (Ito et al., 2016; Lis et al., 2018). Furthermore, a recent study has highlighted LRRK2’s role at the lysosome, where LRRK2 is recruited onto stressed lysosomes by Rab29 and phosphorylates Rab8A and Rab10 (Eguchi et al., 2018). Interestingly, Rab8A KO mice have been developed, which may give important insights into LRRK2 biology at the lysosome (Sato et al., 2007). Furthermore, RAB29 was also observed to be a risk factor for the development of PD, and future studies should investigate a potential unifying mechanism for LRRK2 and Rab29 in PD pathogenesis (MacLeod et al., 2013; Beilina et al., 2014; Nalls et al., 2019).
Another emerging area in LRRK2 animal models is the role of LRRK2 in producing immune system abnormalities. A recent LRRK2 phosphomimetic (S910A + S935A) KI mouse model has shown a reduction in astrogliosis, while a transgenic OE of LRRK2-G2019S in mice exhibited an increase in astrogliosis (Xiong et al., 2018; Zhou et al., 2018). Another study using G2019S KI mice has exhibited astrogliosis using a sub-toxic dose of MPTP dose to cause DA neurodegeneration (Arbez et al., 2019). Investigation of LRRK2 and astrogliosis using these models may help elucidate potential pathogenic mechanisms for LRRK2 in the CNS.
LRRK2 animal models have thus facilitated our understanding of LRRK2 biology, have led us to determine PD pathogenic mechanisms, and have facilitated the discovery of novel phenotypes in LRRK2 pathogenesis in PD and therapeutic development.
SS, GH, and ZY wrote and edited the manuscript. DL had curated information on C. elegans and zebrafish models with assistance from BT. IC and MB provided topic specific knowledge on LRRK2 and guided sections in writing. All authors contributed to the article and approved the submitted version.
This work was supported by an NIH grant (R01NS06123, R01GM115844, and P50NS094733) to ZY. In addition, DL is supported by China Scholarship Council and MB is supported Fundación Ramón Areces.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling Editor HR declared a past co-authorship with the author ZY.
We would like to thank the Yue lab members for helpful discussions and our colleagues at the Icahn School of Medicine for their support.
Adams, J. R., Van Netten, H., Schulzer, M., Mak, E., Mckenzie, J., Strongosky, A., et al. (2005). PET in LRRK2 mutations: comparison to sporadic Parkinson’s disease and evidence for presymptomatic compensation. Brain 128, 2777–2785. doi: 10.1093/brain/awh607
Andres-Mateos, E., Mejias, R., Sasaki, M., Li, X., Lin, B. M., Biskup, S., et al. (2009). Unexpected lack of hypersensitivity in LRRK2 knock-out mice to MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine). J. Neurosci. 29, 15846–15850. doi: 10.1523/JNEUROSCI.4357-09.2009
Arbez, N., He, X., Huang, Y., Ren, M., Liang, Y., Nucifora, F., et al. (2019). G2019S-LRRK2 mutation enhances MPTP-linked parkinsonism in mice. Hum. Molec. Genet. 29, 580–590. doi: 10.1093/hmg/ddz271
Arranz, A. M., Delbroek, L., Kolen, K., Van Guimara, M. R., Daneels, G., Matta, S., et al. (2015). LRRK2 functions in synaptic vesicle endocytosis through a kinase- dependent mechanism. J. Cell. Sci. 128, 541–552. doi: 10.1242/jcs.158196
Bae, E. J., Kim, D. K., Kim, C., Mante, M., Adame, A., Rockenstein, E., et al. (2018). LRRK2 kinase regulates α-synuclein propagation via RAB35 phosphorylation. Nat. Commun. 9:18. doi: 10.1038/s41467-018-05958-z
Baptista, M. A. S., Dave, K. D., Frasier, M. A., Sherer, T. B., Greeley, M., Beck, M. J., et al. (2013). Loss of leucine-rich repeat kinase 2 (LRRK2) in rats leads to progressive abnormal phenotypes in peripheral organs. PLoS One 8:80705. doi: 10.1371/journal.pone.0080705
Beccano-Kelly, D. A., Kuhlmann, N., Tatarnikov, I., Volta, M., Munsie, L. N., Chou, P., et al. (2014). Synaptic function is modulated by LRRK2 and glutamate release is increased in cortical neurons of G2019S LRRK2 knock-in mice. Front. Cell. Neurosci. 8, 301. doi: 10.3389/fncel.2014.00301
Beccano-Kelly, D. A., Volta, M., Munsie, L. N., Paschall, S. A., Tatarnikov, I., Kimberly, C., et al. (2015). LRRK2 overexpression alters glutamergic presynaptic plasticity, striatal dopamine tone, postsynaptic signal transduction, motor activity and memory. Hum. Mol. Genet. 24, 1336–1349. doi: 10.1093/hmg/ddu543
Beilina, A., Rudenko, I. N., Kaganovich, A., Civiero, L., Chau, H., Kailia, S. K., et al. (2014). Unbiased screen for interactors of leucine-rich repeat kinase 2 supports a common pathway for sporadic and familial parkinson disease. Proc. Natl. Acad. Sci. U.S.A. 111, 2626–2631. doi: 10.1073/pnas.1318306111
Berwick, D. C., Heaton, G. R., Azeggagh, S., and Harvey, K. (2019). LRRK2 Biology from structure to dysfunction: research progresses, but the themes remain the same. Mol. Neurodegener. 14, 1–22. doi: 10.1186/s13024-019-0344-2
Bichler, Z., Lim, H. C., Zeng, L., and Tan, E. K. (2013). Non-motor and motor features in LRRK2 transgenic mice. PLoS One 8:e70249. doi: 10.1371/journal.pone.0070249
Biskup, S., Moore, D. J., Rea, A., Lorenz-Deperieux, B., Coombes, C. E., Dawson, V. L., et al. (2007). Dynamic and redundant regulation of LRRK2 and LRRK1 expression. BMC Neurosci. 8:102. doi: 10.1186/1471-2202-8-102
Boddu, R., Hull, T. D., Bolisetty, S., Hu, X., Moehle, M. S., Daher, J. P. L., et al. (2015). Leucine-rich repeat kinase 2 deficiency is protective in rhabdomyolysis-induced kidney injury. Hum. Mol. Genet. 24, 4078–4093. doi: 10.1093/hmg/ddv147
Bosgraaf, L., and Van Haastert, P. J. M. (2003). Roc, a Ras/GTPase domain in complex proteins. Biochim. Biophys. Acta Mol. Cell Res. 1643, 5–10. doi: 10.1016/j.bbamcr.2003.08.008
Burke, R. E., and O’Malley, K. (2013). Axon degeneration in Parkinson’s disease. Exp. Neurol. 246, 72–83. doi: 10.1016/j.expneurol.2012.01.011
Chen, C. Y., Weng, Y. H., Chien, K. Y., Lin, K. J., Yeh, T. H., Cheng, Y. P., et al. (2012). (G2019S) LRRK2 activates MKK4-JNK pathway and causes degeneration of SN dopaminergic neurons in a transgenic mouse model of PD. Cell Death Differ. 19, 1623–1633. doi: 10.1038/cdd.2012.42
Chen, M. L., and Wu, R. M. (2018). LRRK2 gene mutations in the pathophysiology of the ROCO domain and therapeutic targets for Parkinson’s disease: a review Julie Y.H. Chan. J. Biomed. Sci. 25, 1–11. doi: 10.1186/s12929-018-0454-0
Chou, J. S., Chen, C. Y., Chen, Y. L., Weng, Y. H., Yeh, T. H., Lu, C. S., et al. (2014). (G2019S) LRRK2 causes early-phase dysfunction of SNpc dopaminergic neurons and impairment of corticostriatal long-term depression in the PD transgenic mouse. Neurobiol. Dis. 68, 190–199. doi: 10.1016/j.nbd.2014.04.021
Civiero, L., Vancraenenbroeck, R., Belluzzi, E., Beilina, A., Lobbestael, E., Reyniers, L., et al. (2012). Biochemical characterization of highly purified leucine-rich repeat kinases 1 and 2 demonstrates formation of homodimers. PLoS One 7:43472. doi: 10.1371/journal.pone.0043472
Dächsel, J. C., Behrouz, B., Yue, M., Beevers, J. E., Melrose, H. L., and Farrer, M. J. (2010). A comparitive study of Lrrk2 function in primary neuronal cultures. Parkinsonism. Relat. Disord. 16, 650–655. doi: 10.1016/j.parkreldis.2010.08.018
Daher, J. P. L., Abdelmotilib, H. A., Hu, X., Volpicelli-Daley, L. A., Moehle, M. S., and Fraser, K. B. (2015). Leucine-rich repeat kinase 2 (LRRK2) pharmacological inhibition abates α-synuclein gene-induced neurodegeneration. J. Biol. Chem. 290, 19433–19444. doi: 10.1074/jbc.M115.660001
Daher, J. P. L., Volpicelli-daley, L. A., Blackburn, J. P., Moehle, M. S., and West, A. B. (2014). Abrogation of α -synuclein - mediated dopaminergic neurodegeneration in LRRK2-deficient rats. Proc. Natl. Acad. Sci. U.S.A. 111, 9289–9294. doi: 10.1073/pnas.1403215111
Dickson, D. W., Braak, H., Duda, J. E., Duyckaerts, C., Gasser, T., Halliday, G. M., et al. (2009). Neuropathological assessment of Parkinson’s disease: refining the diagnostic criteria. Lancet Neurol. 8, 1150–1157. doi: 10.1016/S1474-4422(09)70238-8
Dodson, M. W., Leung, L. K., Lone, M., Lizzio, M. A., and Guo, M. (2014). Novel ethyl methanesulfonate (EMS)-induced null allelles of the Drosophila homolog of LRRK2 reveal a crucial role in endolysosomal functions and autophagy in vivo. Dis. Models Mech. 7, 1351–1363. doi: 10.1242/dmm.017020
Dranka, B. P., Gifford, A., Ghosh, A., Zielonka, J., Jospeh, J., Kanthasamy, A. G., et al. (2013). Diapocynin prevents early parkinsons disease symptoms in the leucine-rich repeat kinase 2 (LRRK2R1441G) transgenic mouse. Neurosci. Lett. 549, 57–62. doi: 10.1016/j.neulet.2013.05.034
Eguchi, T., Kuwahara, T., Sakurai, M., Komori, T., Fujimoto, T., Ito, G., et al. (2018). LRRK2 and its substrate Rab GTPases are sequentially targeted onto stressed lysosomes and maintain their homeostasis. Proc. Natl. Acad. Sci. U.S.A. 115, E9115–E9124. doi: 10.1073/pnas.1812196115
Feany, M. B., and Bender, W. W. (2000). A Drosophila model of Parkinson’s disease. Nature 404, 394–398. doi: 10.1038/35006074
Fuji, R. N., Flagella, M., Baca, M., Baptista, M. A. S., Brodbeck, J., Chan, B. K., et al. (2015). Effect of selective LRRK2 kinase inhibition on nonhuman primate lung. Sci. Transl. Med. 7:273ra15. doi: 10.1126/scitranslmed.aaa3634
Garcia-Miralles, M., Coomaraswamy, J., Häbig, K., Herzig, M. C., Funk, N., Gillardon, F., et al. (2015). No dopamine cell loss or changes in cytoskeleton function in transgenic mice expressing physiological levels of wild type or G2019S mutant LRRK2 and in human fibroblasts. PLoS One 10:118947. doi: 10.1371/journal.pone.0118947
Gelb, D. J., Oliver, E., and Gilman, S. (1999). Diagnostic criteria for Parkinson disease. Arch. Neurol. 56, 33–39. doi: 10.1001/archneur.56.1.33
Giaime, E., Tong, Y., Wagner, L. K., Yuan, Y., Huang, G., and Shen, J. (2017). Age-dependent dopaminergic neurodegeneration and impairment of the autophagy-lysosomal pathway in LRRK-deficient mice. Neuron 96, 796–807. doi: 10.1016/j.neuron.2017.09.036
Giesert, F., Glasl, L., Zimprich, A., Ernst, L., Piccoli, G., Stautner, C., et al. (2017). The pathogenic LRRK2 R1441C mutation induces specific deficits modeling the prodromal phase of Parkinson’s disease in the mouse. Neurobiol. Dis. 105, 179–193. doi: 10.1016/j.nbd.2017.05.013
Gilks, W., Abousleiman, P., Gandhi, S., Jain, S., Singleton, A., Lees, A., et al. (2005). A common mutation in idiopathic Parkinson’s disease. Lancet 365, 415–416. doi: 10.1016/s0140-6736(05)70237-3
Godena, V. K., Brookes-Hocking, N., Moller, A., Shaw, G., Oswald, M., Sancho, R. M., et al. (2014). Increasing microtubule acetylation rescues axonal transport and locomotor deficits caused by LRRK2 Roc-COR domain mutations. Nat. Commun. 5:5245. doi: 10.1038/ncomms6245
Goetz, C. G., Tilley, B. C., Shaftman, S. R., Stebbins, G. T., Fahn, S., Martinez-Martin, P., et al. (2008). Movement disorder society-sponsored revision of the unified Parkinson’s disease rating scale (MDS-UPDRS): scale presentation and clinimetric testing results. Mov. Disord. 23, 2129–2170. doi: 10.1002/mds.22340
Gómez-Suaga, P., Rivero-Ríos, P., Fdez, E., Ramírez, M. B., Ferrer, I., Aiastui, A., et al. (2014). LRRK2 delays degradative receptor trafficking by impeding late endosomal budding through decreasing Rab7 activity. Hum. Mol. Genet. 23, 1–18. doi: 10.1093/hmg/ddu395
Herzig, M. C., Bidinosti, M., Schweizer, T., Hafner, T., Stemmelen, C., Wiess, A., et al. (2012). High LRRK2 levels fail to induce or exacerbate neuronal alpha-synucleinopathy in mouse brain. PLoS One 7:e36581. doi: 10.1371/journal.pone.0036581
Herzig, M. C., Kolly, C., Persohn, E., Theil, D., Schweizer, T., Hafner, T., et al. (2011). LRRK2 protein levels are determined by kinase function and are crucial for kidney and lung homeostasis in mice. Hum. Mol. Genet. 20, 4209–4223. doi: 10.1093/hmg/ddr348
Hindle, S., Afsari, F., Stark, M., Middleton, C. A., Evans, G. J. O., Sweeney, S. T., et al. (2013). Dopaminergic expression of the parkinsonian gene LRRK2-G2019S leads to non-autonomous visual neurodegeneration, accelerated by increased neural demands for energy. Hum. Mol. Genet. 22, 2129–2140. doi: 10.1093/hmg/ddt061
Hinkle, K. M., Yue, M., Behrouz, B., Dächsel, J. C., Lincoln, S. J., Bowles, E. E., et al. (2012). LRRK2 knockout mice have an intact dopaminergic system but display alterations in exploratory and motor co-ordination behaviors. Mol. Neurodegener. 7, 1–17. doi: 10.1186/1750-1326-7-25
Imai, Y., Gehrke, S., Wang, H., Takahashi, R., Hasegawa, K., Oota, E., et al. (2008). Phosphorylation of 4E-BP by LRRK2 affects the maintenance of dopaminergic neurons in Drosophila. EMBO J. 27, 2432–2443. doi: 10.1038/emboj.2008.163
Ito, G., Kasternonova, K., Tonelli, F., Lis, P., Baptista, M. A. S., and Shpiro, N. (2016). Phos-tag analysis of Rab10 phosphorylation by LRRK2: a powerful assay for assessing kinase function and inhibitors. J. Biochem. 473, 2671–2685. doi: 10.1042/BCJ20160557
Kay, D. M., Zabetian, C. P., Factor, S. A., Nut, J. G., Samii, A., Griffith, A., et al. (2006). Parkinson’s disease and LRRK2: frequency of a common mutation in U.S. movement disorder clinics. Mov. Disord. 21, 519–523. doi: 10.1002/mds.20751
Khan, N. L., Jain, S., Lynch, J. M., Pavese, N., Abou-Sleiman, P., Holton, J. L., et al. (2005). Mutations in the gene LRRK2 encoding dardarin (PARK8) cause familial Parkinson’s disease: clinical, pathological, olfactory and functional imaging and genetic data. Brain 128, 2786–2796. doi: 10.1093/brain/awh667
Kluss, J. H., Mamais, A., and Cookson, M. R. (2019). LRRK2 links genetic and sporadic Parkinson’s disease. Biochem. Soc. Trans. 47, 651–661. doi: 10.1042/BST20180462
Kok, F. O., Shin, M., Ni, C. W., Gupta, A., Grosse, A. S., van Impel, A., et al. (2015). Reverse genetic screening reveals poor correlation between morpholino-induced mutant phenotypes in zebrafish. Dev. Cell. 32, 97–108. doi: 10.1016/j.devcel.2014.11.018
Kuwahara, T., Inoue, K., D’Agati, V. D., Fujimoto, T., Eguchi, T., and Saha, S. (2016). LRRK2 and RAB7L1 cooardinately regulate axonal morphology and lysosome integrity in diverse cellular contexts. Sci. Rep. 6:22945. doi: 10.1038/srep29945
Langston, R. G., Rudenko, I. N., and Cookson, M. R. (2016). The function of orthologues of the human Parkinson’s disease gene LRRK2 across species: implications for disease modelling in preclinical research. Biochem. J. 473, 221–232. doi: 10.1042/BJ20150985
Lee, S., Liu, H. P., Lin, W. Y., Guo, H., and Lu, B. (2010). LRRK2 kinase regulates synaptic morphology through distinct substrates at the presynaptic and postsynaptic compartments of the Drosophila neuromuscular junction. J. Neurosci. 30, 16959–16969. doi: 10.1523/JNEUROSCI.1807-10.2010
Lee, S. B., Kim, W., Lee, S., and Chung, J. (2007). Loss of LRRK2/PARK8 induces degeneration of dopaminergic neurons in Drosophila. Biochem. Biophys. Res. Commun. 358, 534–539. doi: 10.1016/j.bbrc.2007.04.156
Lee, J. W., Tapias, V., Di Maio, R., Greenamyre, J. T., and Cannon, J. R. (2015). Behavioral, neurochemical, and pathogenic alterations in bacterial artificial chromosomes transgenic G2019S leucine-rich repeated kinase 2 rats. Neurobiol. Aging 36, 505–518. doi: 10.106/j.neurobiolaging.2014.07.011
Lees, A. J., Hardy, J., and Revesz, T. (2009). Parkinson’s disease. Lancet 373, 2055–2066. doi: 10.1016/S0140-6736(09)60492-X
Lesage, S., and Brice, A. (2009). Parkinson’s disease: from monogenic forms to genetic susceptibility factors. Hum. Mol. Genet. 18, 48–59. doi: 10.1093/hmg/ddp012
Lesage, S., Janin, S., Lohmann, E., Leutenegger, A. L., Leclere, L., Viallet, F., et al. (2007). LRRK2 exon 41 mutations in sporadic Parkinson disease in Europeans. Arch. Neurol. 64, 425–430. doi: 10.1001/archneur.64.3.425
Li, X., Nicholson, C., Yue, Z., Rice, M. E., Avshalumov, M. V., Patel, J. C., et al. (2010). Enhanced striatal dopamine transmission and motor performance with LRRK2 overexpression in mice is eliminated by familial Parkinson’s disease mutation G2019S. J. Neurosci. 30, 1788–1797. doi: 10.1523/jneurosci.5604-09.2010
Li, X., Tan, Y. C., Poulose, S., Olanow, C. W., Huang, X. Y., and Yue, Z. (2007). Leucine-rich repeat kinase 2 (LRRK2)/PARK8 possess GTPase activity that is altered in familial Parkinson’s disease R1441C/G mutants. J. Neurochem. 103, 238–247. doi: 10.1111/j.1471-5159.2007.04743.x
Li, Y., Liu, W., Oo, T. F., Wang, L., Tang, Y., Jackson-Lewis, V., et al. (2009). Mutant LRRK2R1441G BAC transgenic mice recapitulate cardinal features of Parkinson’s Disease. Nat. Neurosci. Brief. Commun. 12, 826–828. doi: 10.1038/nn.2349
Lichtenberg, M., Mansilla, A., Zecchini, V. R., Fleming, A., and Rubinsztein, D. C. (2011). The Parkinson’s disease protein LRRK2 impairs proteasome substrate clearance without affecting proteasome catalytic activity. Cell Death Dis. 2, 1–12. doi: 10.1038/cddis.2011.81
Lim, J., Bang, Y., Choi, J. H., Han, A., Kwon, M. S., Liu, K. H., et al. (2018). LRRK2 G2019S induces anxiety/depression-like behavior before the onset of motor dysfunction with 5-HT 1A receptor upregulation in mice. J. Neurosci. 38, 1611–1621. doi: 10.1523/JNEUROSCI.4051-15.2017
Lin, C. H., Tsai, P. I., Wu, R. M., and Chien, C. T. (2010). LRRK2 G2019S mutation induces dendrite degeneration through mislocalization and phosphorylation of tau by recruiting autoactivated GSK3β. J. Neurosci. 30, 13138–13149. doi: 10.1523/JNEUROSCI.1737-10.2010
Lin, X., Parisiadou, L., Gu, X. L., Wang, L., Shim, H., Sun, L., et al. (2009). Leucine-Rich repeat kinase 2 regulates the progression of neuropathology induced by parkinson’s-disease-related mutant α-synuclein. Neuron 64, 807–827. doi: 10.1016/j.neuron.2009.11.006
Lis, P., Burel, S., Steger, M., Mann, M., Brown, F., Diez, F., et al. (2018). Development of phospho-specific rab protein antibodies to monitor in vivo activity of the LRRK2 Parkinson’s Disease Kinase. J. Biochem. 475, 1–22. doi: 10.1042/BCJ20170802
Liu, Z., Hamamichi, S., Lee, B. D., Yang, D., Ray, A., Caldwell, G. A., et al. (2011). Inhibitors of LRRK2 kinase attenuate neurodegeneration and Parkinson-like phenotypes in Caenorhabditis elegans and Drosophila Parkinson’s disease models. Hum. Mol. Genet. 20, 3933–3942. doi: 10.1093/hmg/ddr312
Liu, H. F., Lu, S., Ho, P. W., Tse, H. M., Pang, S. Y., Kung, M. H., et al. (2014). LRRK2 R1441G mice are more liable to dopamine depletion and locomotor inactivity. Ann. Clin. Transl. Neurol. 1, 199–208. doi: 10.1002/acn3.45
Liu, G., Sgobio, C., Gu, X., Sun, L., Lin, X., Yu, J., et al. (2015). Selective expression of Parkinson’s disease-related Leucine-rich repeat kinase 2 G2019S missense mutation in midbrain dopaminergic neurons impairs dopamine release and dopaminergic gene expression. Hum. Mol. Genet. 24, 5299–5312. doi: 10.1093/hmg/ddv249
Liu, Z., Wang, X., Yu, Y., Li, X., Wang, T., Jiang, H., et al. (2008). A Drosophila model for LRRK2-linked parkinsonism. Proc. Natl. Acad. Sci. U.S.A. 105, 2693–2698. doi: 10.1073/pnas.0708452105
Longo, F., Russo, I., Shimshek, D. R., Greggio, E., and Morari, M. (2014). Genetic and pharmacological evidence that G2019S LRRK2 confers a hyperkinetic phenotype, resistant to motor decline associated with aging. Neurobiol. Dis. 71, 62–73. doi: 10.1016/j.nbd.2014.07.013
MacLeod, D. A., Rhinn, H., Kuwahara, T., Zolin, A., Di Paolo, G., McCabe, B. D., et al. (2013). RAB7L1 interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson’s disease risk. Neuron 77, 425–439. doi: 10.1016/j.neuron.2012.11.033
Maekawa, T., Mori, S., Sasaki, Y., Miyajima, T., Azuma, S., Ohta, E., et al. (2012). The I2020T leucine-rich repeat kinase 2 transgenic mouse exhibits impaired locomotive ability accompanied by dopaminergic neuron abnormalities. Mol. Neurodegener. 7:15. doi: 10.1186/1750-1326-7-15
Marín, I. (2006). The Parkinson disease gene LRRK2: evolutionary and structural insights. Mol. Biol. Evol. 23, 2423–2433. doi: 10.1093/molbev/msl114
Matikainen-Ankney, B. A., Kezunovic, N., Menard, C., Flanigan, M. E., Zhong, Y., Russo, S., et al. (2018). Parkinson’s disease-linked LRRK2-G2019S mutation alters synaptic plasticity and promotes resilience to chronic social stress in young adulthood. J. Neurosci. 38, 9700–9711. doi: 10.1523/JNEUROSCI.1457-18.2018
Matikainen-Ankney, B. A., Kezunovic, N., Mesias, R. E., Tian, Y., Williams, F. M., Huntley, G. W., et al. (2016). Altered development of synapse structure and function in striatum caused by Parkinson’s disease-linked LRRK2-G2019S mutation. J. Neurosci. 36, 7128–7141. doi: 10.1523/JNEUROSCI.3314-15.2016
Matta, S., Van Kolen, K., da Cunha, R., van den Bogaart, G., Mandemakers, W., Miskiewicz, K., et al. (2012). LRRK2 controls an endoa phosphorylation cycle in synaptic endocytosis. Neuron 75, 1008–1021. doi: 10.1016/j.neuron.2012.08.022
Maulik, M., Mitra, S., Bult-Ito, A., Taylor, B. E., and Vayndorf, E. M. (2017). Behavioral phenotyping and pathological indicators of Parkinson’s disease in C. elegans models. Front. Genet. 8:77. doi: 10.3389/fgene.2017.00077
Melrose, H. L., Dächsel, J. C., Behrouz, B., Lincoln, S. J., Yue, M., Hinkle, K. M., et al. (2010). Impaired dopaminergic transmission and microtubule-associated protein tau alterations in human LRRK2 transgenic mice. Neurobiol. Dis. 40, 503–517. doi: 10.1016/j.nbd.2010.07.010
Meredith, G. E., and Kang, U. J. (2006). Behavioral models of Parkinsons disease in rodents: a new look at an old problem. Mov. Disord. 21, 1595–1606. doi: 10.1002/mds.21010
Miklavc, P., Ehinger, K., Thompson, K. E., Hobi, N., Shimshek, D. R., and Frick, M. (2014). Surfactant secretion in LRRK2 knock-out rats: changes in lamellar body morphology and rate of exocytosis. PLoS One 9:84926. doi: 10.1371/journal.pone.0084926
Nalls, M. A., Blauwendraat, C., Vallerga, C. L., Heilbron, K., Bandres-Ciga, S., Chang, D., et al. (2019). Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol. 18, 1091–1102. doi: 10.1016/S1474-4422(19)30320-5
Ness, D., Ren, Z., Gardai, S., Sharpnack, D., Johnson, V. J., Brennan, R. J., et al. (2013). Leucine-rich repeat kinase 2 (LRRK2)-deficient rats exhibit renal tubule injury and perturbations in metabolic and immunological homeostasis. PLoS One 8:e066164. doi: 10.1371/journal.pone.0066164
Ng, C. H., Mok, S. Z. S., Koh, C., Ouyang, X., Fivaz, M. L., Tan, E. K., et al. (2009). Parkin protects against LRRK2 G2019S mutant-induced dopaminergic neurodegeneration in Drosophila. J. Neurosci. 29, 11257–11262. doi: 10.1523/JNEUROSCI.2375-09.2009
Nguyen, M., and Krainc, D. (2018). LRRK2 phosphorylation of auxilin mediates synaptic defects in dopaminergic neurons from patients with Parkinson’s disease. Proc. Natl. Acad. Sci. U.S.A. 115, 5576–5581. doi: 10.1073/pnas.1717590115
Nichols, R. J., Dzamko, N., Morrice, N. A., Campbell, D. G., Deak, M., Ordureau, A., et al. (2010). 14-3-3 binding to LRRK2 is disrupted by multiple Parkinson’s disease-associated mutations and regulates cytoplasmid localization. J. Biochem. 430, 393–404. doi: 10.1042/BJ20100483
Paisán-Ruíz, C., Nath, P., Washecka, N., Gibbs, J. R., and Singleton, A. B. (2008). Comprehensive analysis of LRRK2 in publicly available Parkinson’s disease cases and neurologically normal controls. Hum. Mutat. 29, 485–490. doi: 10.1002/humu.20668
Pan, P.-Y., Li, X., Wicinski, B., Yue, Z., Chen, Z., Powell, J., et al. (2017). Parkinson’s disease-associated LRRK2 hyperactive kinase mutant disrupts synaptic vesicle trafficking in ventral midbrain neurons. J. Neurosci. 37, 11366–11376. doi: 10.1523/jneurosci.0964-17.2017
Pan, P. Y., Zhu, Y., Shen, Y., and Yue, Z. (2019). Crosstalk between presynaptic trafficking and autophagy in Parkinson’s disease. Neurobiol. Dis. 122, 64–71. doi: 10.1016/j.nbd.2018.04.020
Parisiadou, L., Xie, C., Cho, H. J., Lin, X., Gu, X. L., and Long, C. X. (2009). Phosphorylation of Ezrin/Radixin/moesin proteins by LRRK2 promotes the rearrangement of actin cytoskeleton in neuronal morphogenesis. Neurbiol. Dis. 29, 13971–13980. doi: 10.1523/JNEUROSCI.3799-09.2009
Parisiadou, L., Yu, J., Sgobio, C., Xie, C., Liu, G., Sun, L., et al. (2014). LRRK2 regulates synaptogenesis and dopamine receptor activation through modulation of PKA activity. Nat. Neurosci. 17, 367–376. doi: 10.1038/nn.3636
Pellegrini, L., Hauser, D. N., Li, Y., Mamais, A., Beilina, A., Kumaran, R., et al. (2018). Proteomic analysis reveals co-ordinated alterations in protein synthesis and degradation pathways in LRRK2 knockout mice. Hum. Mol. Genet. 27, 3257–3271. doi: 10.1093/hmg/ddy232
Piccoli, G., Condliffe, S. B., Bauer, M., Giesert, F., Boldt, K., De Astis, S., et al. (2011). LRRK2 controls synaptic vesicle storage and mobilization within the recycling pool. J. Neurosci. 31, 2225–2237. doi: 10.1523/JNEUROSCI.3730-10.2011
Prabhudesai, S., Bensabeur, F. Z., Abdullah, R., Basak, I., Baez, S., Alves, G., et al. (2016). LRRK2 knockdown in zebrafish causes developmental defects, neuronal loss, and synuclein aggregation. J. Neurosci. Res. 94, 717–735. doi: 10.1002/jnr.23754
Ramonet, D., Daher, J. P. L., Lin, B. M., Stafa, K., Kim, J., Banerjee, R., et al. (2011). Dopaminergic neuronal loss, reduced neurite complexity and autophagic abnormalities in transgenic mice expressing G2019S mutant LRRK2. PLoS One 6:18568. doi: 10.1371/journal.pone.0018568
Redgrave, P., Rodriguez, M., Smith, Y., Rodriguez-Oroz, M. C., Lehericy, S., Bergman, H., et al. (2010). Goal-directed and habitual control in the basal ganglia: implications for Parkinson’s disease. Nat. Rev. Neurosci. 11, 760–772. doi: 10.1038/nrn2915
Ren, G., Xin, S., Li, S., Zhong, H., and Lin, S. (2011). Disruption of lrrk2 does not cause specific loss of dopaminergic neurons in zebrafish. PLoS One 6:20630. doi: 10.1371/journal.pone.0020630
Rivero-Ríos, P., Romo-Lozano, M., Madero-Pérez, J., Thomas, A. P., Biosa, A., Greggio, E., et al. (2019). The G2019S variant of leucine-rich repeat kinase 2 (LRRK2) alters endolysosomal trafficking by impairing the function of the GTPase RAB8A. J. Biol. Chem. 294, 4738–4758. doi: 10.1074/jbc.RA118.005008
Saha, S., Guillily, M. D., Ferree, A., Lanceta, J., Chan, D., Ghosh, J., et al. (2009). LRRK2 modulates vulnerability to mitochondrial dysfunction in Caenorhabditis elegans. J. Neurosci. 29, 9210–9218. doi: 10.1523/JNEUROSCI.2281-09.2009
Sakaguchi-Nakashima, A., Meir, J. Y., Jin, Y., Matsumoto, K., and Hisamoto, N. (2007). LRK-1, a C. elegans PARK8-related kinase, regulates axonal-dendritic polarity of SV proteins. Curr. Biol. 17, 592–598. doi: 10.1016/j.cub.2007.01.074
Sämann, J., Hegermann, J., von Gromoff, E., Eimer, S., Baumeister, R., and Schmidt, E. (2009). Caenorhabditits elegans LRK-1 and PINK-1 act antagonistically in stress response and neurite outgrowth. J. Biol. Chem. 284, 16482–16491. doi: 10.1074/jbc.M808255200
Sato, T., Mushiake, S., Kato, Y., Sato, K., Sato, M., Takeda, N., et al. (2007). The Rab8 GTPase regulates apical protein localization in intestinal cells. Nature 448, 366–369. doi: 10.1038/nature05929
Schreij, A. M., Chaineau, M., Ruan, W., Lin, S., Barker, P. A., Fon, E. A., et al. (2014). LRRK2 localizes to endosomes and interacts with clathrin-light chains to limit Rac1 activation. EMBO Rep. 16, 79–86. doi: 10.15252/embr.201438714
Sejwal, K., Chami, M., Rémigy, H., Vancraenenbroeck, R., Sibran, W., Sütterlin, R., et al. (2017). Cryo-EM analysis of homodimeric full-length LRRK2 and LRRK1 protein complexes. Sci. Rep. 7, 1–12. doi: 10.1038/s41598-017-09126-z
Shaikh, K. T., Yang, A., Youshin, E., and Schmid, S. (2015). Transgenic LRRK2 (R1441G) rats-a model for Parkinson disease? PeerJ. 3:e945. doi: 10.7717/peerj.945
Sheehan, P., and Yue, Z. (2019). Deregulation of autophagy and vesicle trafficking in Parkinson’s disease. Neurosci. Lett. 697, 59–65. doi: 10.1016/j.neulet.2018.04.013
Sheng, D., Qu, D., Kwok, K. H. H., Ng, S. S., Lim, A. Y. M., Aw, S. S., et al. (2010). Deletion of the WD40 domain of LRRK2 in zebrafish causes parkinsonism-like loss of neurons and locomotive defect. PLoS Genet. 6:e01000914. doi: 10.1371/journal.pgen.1000914
Sheng, D., See, K., Hu, X., Yu, D., Wang, Y., Liu, Q., et al. (2018). Disruption of LRRK2 in Zebrafish leads to hyperactivity and weakened antibacterial response. Biochem. Biophys. Res. Commun. 497, 1104–1109. doi: 10.1016/j.bbrc.2018.02.186
Sheng, Z., Zhang, S., Bustos, D., Kleinheinz, T., Le Pichon, C. E., Dominguez, S. L., et al. (2012). Ser1292 autophosphorylation is an indicator of LRRK2 kinase activity and contributes to the cellular effects of PD mutations. Sci. Transl. Med. 4:485. doi: 10.1126/scitranslmed.3004485
Shin, N., Jeong, H., Kwon, J., Heo, H. Y., Kwon, J. J., Yun, H. J., et al. (2008). LRRK2 regulates synaptic vesicle endocytosis. Exp. Cell Res. 314, 2055–2065. doi: 10.1016/j.yexcr.2008.02.015
Sloan, M., Alegre-Abarrategui, J., Potgieter, D., Kaufmann, A. K., Exley, R., Deltheil, T., et al. (2016). LRRK2 BAC transgenic rats develop progressive, L-DOPA-responsive motor impairment, and deficits in dopamine circuit function. Hum. Mol. Genet. 25, 951–963. doi: 10.1093/hmg/ddv628
Steger, M., Diez, F., Dhekne, H. S., Lis, P., Nirujogi, R. S., Karayel, O., et al. (2017). Systematic proteomic analysis of LRRK2-mediated rab GTPase phosphorylation establishes a connection to ciliogenesis. eLife 6, 1–22. doi: 10.7554/eLife.31012
Steger, M., Tonelli, F., Ito, G., Davies, P., Trost, M., Vetter, M., et al. (2016). Phosphoproteomics reveals that Parkinson’s disease kinase LRRK2 regulates a subset of Rab GTPases. eLife 5:e01. doi: 10.7554/eLife.12813.001
Tain, L. S., Mortiboys, H., Tao, R. N., Ziviani, E., Bandmann, O., and Whitworth, A. J. (2009). Rapamycin activation of 4E-BP prevents parkinsonian dopaminergic neuron loss. Nat. Neurosci. 12, 1129–1135. doi: 10.1038/nn.2372
Tong, Y., Boyle, S., Shen, J., Kelleher, R. J., Kopan, R., Giaime, E., et al. (2010a). Loss of leucine-rich repeat kinase 2 causes impairment of protein degradation pathways, accumulation of -synuclein, and apoptotic cell death in aged mice. Proc. Natl. Acad. Sci. U.S.A. 107, 9879–9884. doi: 10.1073/pnas.1004676107
Tong, Y., Giaime, E., Yamaguchi, H., Ichimura, T., Liu, Y., Si, H., et al. (2012). Loss of leucine-rich repeat kinase 2 causes age-dependent bi-phasic alterations of the autophagy pathway. Mol. Neurodegener. 7:2. doi: 10.1186/1750-1326-7-2
Tong, Y., Pisani, A., Martella, G., Karouani, M., Yamaguchi, H., Pothos, E. N., et al. (2009). R1441C mutation in LRRK2 impairs dopaminergic neurotransmission in mice. Proc. Natl. Acad. Sci. USA 106, 14622–14627. doi: 10.1073/pnas.0906334106
Tong, Y., Yamaguchi, H., Giaime, E., Boyle, S., Kopan, R., Kelleher, R. J., et al. (2010b). Loss of leucine-rich repeat kinase 2 causes impairment of protein degradation pathways, accumulation of α-synuclein, and apoptotic cell death in aged mice. Proc. Natl. Acad. Sci. U.S.A. 107, 9879–9884. doi: 10.1073/pnas.1004676107
Tsika, E., Kannan, M., Foo, C. S. Y., Dikeman, D., Glauser, L., Gellhaar, S., et al. (2014). Conditional expression of Parkinson’s disease-related R1441C LRRK2 in midbrain dopaminergic neurons of mice causes nuclear abnormalities without neurodegeneration. Neurobiol. Dis. 71, 345–358. doi: 10.1016/j.nbd.2014.08.027
Vaz, R. L., Outeiro, T. F., and Ferreira, J. J. (2018). Zebrafish as an animal model for drug discovery in Parkinson’s disease and other movement disorders: a systematic review. Front. Neurol. 9:347. doi: 10.3389/fneur.2018.00347
Venderova, K., Kabbach, G., Abdel-Messih, E., Zhang, Y., Parks, R. J., Imai, Y., et al. (2009). Leucine-rich repeat kinase 2 interacts with Parkin, DJ-1 and PINK-1 in a Drosophila melanogaster model of Parkinson’s disease. Hum. Mol. Genet. 18, 4390–4404. doi: 10.1093/hmg/ddp394
Volpicelli-Daley, L. A., Abdelmotilib, H., Liu, Z., Stokya, L., Daher, J. P. L., Milnerwood, A. J., et al. (2016). G2019S-LRRK2 expression augments α-synuclein sequestration into inclusions in neurons. J. Neurosci. 36, 7415–7427. doi: 10.1523/JNEUROSCI.3642-15.2016
Volta, M., Beccano-Kelly, D. A., Paschall, S. A., Cataldi, S., Macisaac, S. E., Kuhlmann, N., et al. (2017). Initial elevations in glutamate and dopamine neurotransmission decline with age, as does exploratory behavior, in LRRK2 G2019S knock-in mice. eLife 6:28377. doi: 10.7554/eLife.28377
Volta, M., Cataldi, S., Beccano-Kelly, D., Munsie, L., Tatarnikov, I., Chou, P., et al. (2015). Chronic and acute LRRK2 silencing has no long-term behavioral effects, whereas wild-type and mutant LRRK2 overexpression induce motor and cognitive deficits and altered regulation of dopamine release. Parkinsonism. Relat. Disord. 21, 1156–1163. doi: 10.1016/j.parkreldis.2015.07.025
Walker, M. D., Volta, M., Cataldi, S., Dinelle, K., Beccano-Kelly, D., Munsie, L., et al. (2014). Behavioral deficits and striatal DA signaling in LRK2 p. G2019S transgenic rats: a multimodal invesigation including PET neuroimaging. J. Park. Dis. 4, 483–498. doi: 10.3233/JPD-140344
Wang, D., Tang, B., Zhao, G., Pan, Q., Xia, K., Bodmer, R., et al. (2008). Dispensable role of Drosophila ortholog of LRRK2 kinase activity in survival of dopaminergic neurons. Mol. Neurodegener. 3:3. doi: 10.1186/1750-1326-3-3
Wang, L., Xie, C., Greggio, E., Parisadou, L., Shim, H., Sun, L., et al. (2008). The chaperone activity of heat shock protein 90 is critical for maintaining the stability of leucine-rich repeat kinase 2. J. Neurosci. 28, 3384–3391. doi: 10.1523/JNEUROSCI.0815-08.2008
Weng, Y. H., Chen, C. Y., Lin, K. J., Chen, Y. L., Yeh, T. H., Hsiao, I. T., et al. (2016). (R1441C) LRRK2 induces the degeneration of SN dopaminergic neurons and alters the expression of genes regulating neuronal survival in a transgenic mouse model. Exp. Neurol. 275, 104–115. doi: 10.1016/j.expneurol.2015.09.001
West, A. B., Cowell, R. M., Daher, J. P. L., Moehle, M. S., Hinkle, K. M., Melrose, H. L., et al. (2014). Differential LRRK2 expression in the cortex, striatum, and substantia nigra in transgenic and nontransgenic rodents. J. Comp. Neurol. 522, 2465–2480. doi: 10.1002/cne.23583
West, A. B., Moore, D. J., Biskup, S., Bugayenko, A., Smith, W. W., Ross, C. A., et al. (2005). Parkinson’s disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc. Natl. Acad. Sci. U.S.A. 102, 16842–16847. doi: 10.1073/pnas.0507360102
Xing, W., Goodluck, H., Zeng, C., and Mohan, S. (2017). Role and mechanism of action of leucine-rich repeat kinase 1 in bone. Bone Res. 5:17003. doi: 10.1038/boneres.2017.3
Xiong, Y., Neifert, S., Karuppagounder, S. S., Liu, Q., Stankowski, J. N., Lee, B. D., et al. (2018). Robust kinase- and age-dependent dopaminergic and norepinephrine neurodegeneration in LRRK2 G2019S transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 115, 1635–1640. doi: 10.1073/pnas.1712648115
Yao, C., El Khoury, R., Wang, W., Byrd, T. A., Pehek, E. A., Thacker, C., et al. (2010). LRRK2-mediated neurodegeneration and dysfunction of dopaminergic neurons in a Caenorhabditis elegans model of Parkinson’s disease. Neurobiol. Dis. 40, 73–81. doi: 10.1016/j.nbd.2010.04.002
Yue, M., Hinkle, K. M., Davies, P., Trushina, E., Fiesel, F. C., Christenson, T. A., et al. (2015). Progressive dopaminergic alterations and mitochondrial abnormalities in LRRK2 G2019S knock-in mice. Neurobiol. Dis. 78, 172–195. doi: 10.1016/j.nbd.2015.02.031
Zhou, H., Huang, C., Tong, J., Hong, W. C., Liu, Y. J., and Xia, X. G. (2011). Temporal expression of mutant LRRK2 in adult rats impairs dopamine reuptake. Int. J. Biol. Sci. 7, 753–761. doi: 10.7150/ijbs.7.753
Zhou, H., Huang, C., Yang, M., Landel, C. P., Xia, P. Y., Liu, Y. J., et al. (2009). Developing tTA transgenic rats for inducible and reverisble gene expression. Int. J. Biol. Sci. 5, 171–181. doi: 10.7150/ijbs.5.171
Zhou, Y., Keshiya, S., Atashrazm, F., Gao, J., Ittner, L. M., Allesi, D. R., et al. (2018). Nigrostriatal pathology with reduced astrocytes in LRRK2 S910/S935 phosphorylation deficient knockin mice. Neurbiol. Dis. 120, 76–87. doi: 10.106/j.nbd.2018.09.003
Keywords: LRRK2, models, rodent, Drosophila, C. elegans, zebrafish, phenotypes
Citation: Seegobin SP, Heaton GR, Liang D, Choi I, Blanca Ramirez M, Tang B and Yue Z (2020) Progress in LRRK2-Associated Parkinson’s Disease Animal Models. Front. Neurosci. 14:674. doi: 10.3389/fnins.2020.00674
Received: 03 March 2020; Accepted: 02 June 2020;
Published: 15 July 2020.
Edited by:
Hardy Rideout, Biomedical Research Foundation of the Academy of Athens (BRFAA), GreeceReviewed by:
Yulan Xiong, Kansas State University, United StatesCopyright © 2020 Seegobin, Heaton, Liang, Choi, Blanca Ramirez, Tang and Yue. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhenyu Yue, emhlbnl1Lnl1ZUBtc3NtLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.