 Xiaojuan Su
Xiaojuan Su Lingyi Huang
Lingyi Huang Dongqiong Xiao
Dongqiong Xiao Yi Qu
Yi Qu Dezhi Mu
Dezhi Mu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurosci., 09 October 2018
Sec. Neuroenergetics and Brain Health
Volume 12 - 2018 | https://doi.org/10.3389/fnins.2018.00697
Activin A belongs to the transforming growth factor superfamily and has a variety of biological functions. Studies have revealed that activin A can regulate the body's immune and inflammatory responses and participate in the regulation of cell death. In addition, activin A also has neurotrophic function and plays an important role in the repair of brain damage. This article summarizes recent advances in understanding the role and mechanism of action of activin A in brain injury and provides new hints into the application of activin A in the treatment of brain injury.
Brain injury is a typical functional disorder of the nervous system, with many types of pathogenic factors involved and a complex pathogenesis. The main pathogenesis involves massive cell death in injured brain areas and surrounding tissue, which leads to tissue damage and eventually destruction, mainly caused by systematic inflammation, including ischemia and traumatic brain injury (Chandra et al., 2018).
Activin A, a member of the transforming growth factor beta (TGF-β) superfamily, regulates the body's immune and inflammatory responses, and participates in the regulation of cell death. In addition, activin A has a neurotrophic function and plays an important role in repair following brain damage (Ageta and Tsuchida, 2011). For example, a transgenic mouse study showed that activin A exerts a critical role in neuroprotection following various types of brain damage, by regulating spine formation and adult neurogenesis (Müller et al., 2006).
In recent years, the role of activin A and its molecular mechanisms in brain injury have been studied extensively. Activin A may thus represent a promising therapeutic target, and knowledge about its function may provide some hints for clinical treatment and drug discovery. The present review discusses recent progress in the study of the role and mechanism of action of activin A in brain injury.
Activin A is a widely expressed homodimer that is composed of two β A chains. Sequence analysis showed that the β subunit has the typical structural features of the TGF-β superfamily, i.e., the C-terminal active portion of the molecule has nine conserved cysteine residues (Wang X. et al., 2016). In addition, the 14-kDa mature human β A chain of activin A has 100% amino acid sequence identity in cattle, cats, mice, pigs, etc., indicating its highly conserved structure (Tanimoto et al., 1991).
The receptor for activin A is a serine threonine protein kinase, and there are three types of receptors: type I (RI), type II (RII), and type III (RIII). RI and RII are mainly involved in the regulation of activin A activity, whereas RIII is not indispensable for its activity. RI is also known as activin receptor-like kinase 5 (ALK5) and can be phosphorylated by RII (de Kroon et al., 2015). Upon signal transduction, activin A first binds to RII, which in turn activates RI to form the receptor complex. After being activated, signaling of the formed receptor complex results in activation of the Smad signaling pathway, which includes phosphorylation of transcription factors Smad2 or Smad3 by ALK5 protein kinase, and leads to the transcriptional regulation of activin A response genes. Furthermore, phosphorylated Smad2 or Smad3 bind to Smad4, and the resulting complex translocates from the cytoplasm to the nucleus, where the Smad complex interacts in a cell-specific manner with various other transcription factors, thus exerting its biological activity (Peterson et al., 2012; Figure 1). In addition to the Smad-dependent signaling pathways, there are activin A pathways that are Smad-independent and mainly include the nuclear factor-κB pathway, extracellular signal-regulated kinase (ERK1/2) pathway, ubiquitin-proteolytic pathways, mitogen-activated protein kinase (MAPK) pathways, and other signal transduction pathways (Derynck and Zhang, 2003; Kim et al., 2015). The activin A/Smad molecular pathway is shown in Figure 1.
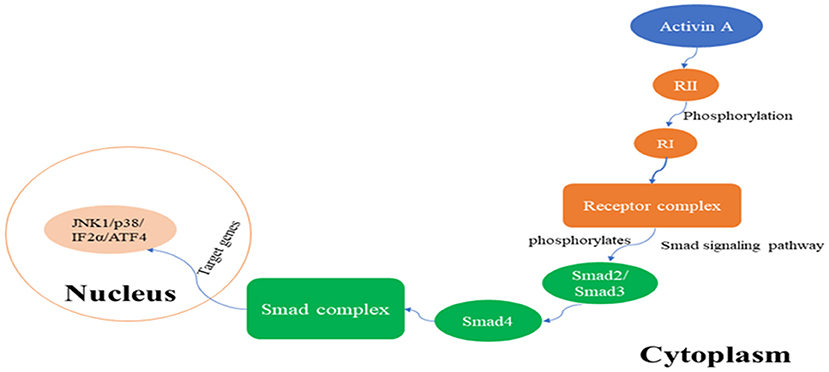
Figure 1. The activin A/Smad signaling pathway.
Studies have found that activin A activity is regulated by numerous factors. For example, it is negatively regulated by some factors, including the bone morphogenetic protein (BMP), BMP and activin membrane-bound inhibitor, glycan, and uterine natural killer, which limit its ability to induce the assembly of receptor complexes (Shi et al., 2009; Upton et al., 2009; Mai et al., 2014; Peng et al., 2015). Other factors, like 2-macroglobulin, follistatin, and follistatin-related genes, limit activin A bioavailability by binding to it (Asashima et al., 1991; Mather, 1996; de Kretser et al., 2012). In contrast, it has been reported that, in the early stages of tissue damage, TGF-β, epidermal growth factor, and platelet-derived growth factor are released from platelets, thus triggering the upregulation of activin A and leading to protection of the damaged nerve (Hübner et al., 1999; Wang and Ge, 2004; Protic et al., 2017). These factors were also found to promote the expression of the activin β A subunit in both cultured fibroblasts and keratinocytes (Huang et al., 2004). In vitro studies have shown that high levels of interleukin (IL)-1 and tumor necrosis factor alpha (TNF-α) are expressed in polymorphonuclear leucocytes and macrophages, both of which can also promote activin A secretion (Arai et al., 2011; Kelly et al., 2016). Moreover, recent studies have discovered a new group of intracellular proteins, termed activin A receptor-interacting proteins, which interact with activin A RII and regulate an activin A-dependent intracellular signaling process, influenced by activin A histological distribution and biological activity (Liu et al., 2009; Liu H. Y. et al., 2013; Qi et al., 2013; Table 1). The molecules that regulate activin A activity are summarized in Table 1.
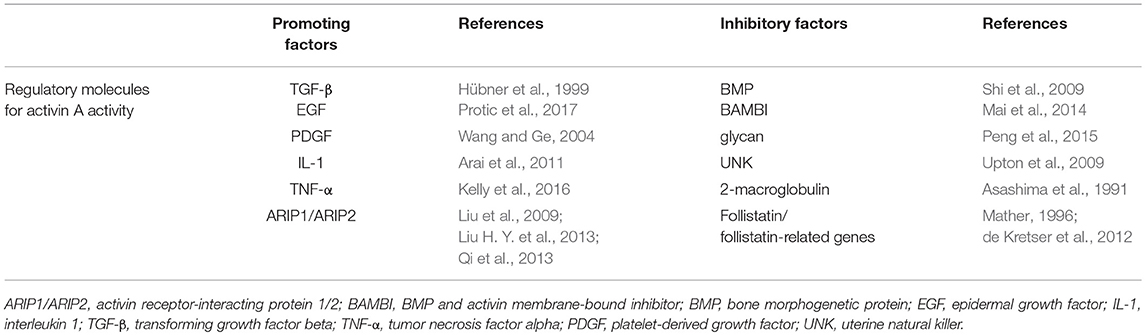
Table 1. Molecules regulating activin A activity.
Activin A and its receptors are widely expressed in brain tissue. Studies have shown that the expression of activin A is upregulated after nerve cells are subjected to acute injury from various sources (Mukerji et al., 2007). The neuroprotective effect of activin A after brain injury occurs mainly through its anti-inflammatory activity; however, the inhibition of the secretion of certain reactive proteins, reduction of cytotoxic brain edema, anti-oxidation, inhibition of free radical aggregation, upregulation of brain-derived neurotrophic factor and induction of its synthesis, as well as the antagonization of excitatory amino acid-induced neurotoxicity, among other functions, render activin A an important molecule with an endogenous protective role (Wang Q. et al., 2016). Moreover, a study found that activin A increases the number of synapses and the length of the dendritic aponeurosis neck in hippocampal neurons cultured in vitro, while it also increases the activity of neuronal voltage-gated Na+/K+ channels and triggers the maturation of synapses (Manickam and Tulsawani, 2014).
The rat model of cerebral ischemic and hypoxic injury induces the overexpression of follistatin, activin A, and BMP-4, depending on development and age, all of which are protective against nerve injury. As the rat grows older, the expression of follistatin and activin A decreases gradually, whereas BMP-4 expression decreases significantly in adulthood. (Tian et al., 2014). During ischemic and hypoxic injury, the exogenous use of activin A was shown to inhibit the expression of caspase-3 and apoptosis in neural cells (He et al., 2011). Besides, activin A also negatively regulate autophagy, which was found to inhibit the c-Jun N-terminal kinase 1 (JNK) and p38 MAPK signaling pathways during cerebral ischemia (Xue et al., 2017). In addition, a study found that activin A is upregulated in the early stage of acute ischemic brain injury, whereby it exerts its neuroprotective role by downregulating nitric oxide levels and increasing superoxide dismutase activity and neuronal tolerance to ischemic injury, through the activin A/Smad signaling pathway (Nakajima et al., 2014). In a mouse model of ischemia/reperfusion, it was found that intracerebroventricular injection of activin A inhibits neuronal apoptosis and significantly reduces the infarct size (Ma et al., 2016).
In a PC12-cell oxygen-glucose deprivation (OGD) and endoplasmic reticulum stress (ERS) lesion model, the protein levels of activin A and phosphorylated Smad3 (p-Smad3) were significantly upregulated (Guo et al., 2014). Treatment with exogenous activin A was shown to further increase activin A and p-Smad3 protein levels, as well as cell viability, and to significantly reduce the number of apoptotic nuclei and the levels of the C/EBP homologous protein and caspase-12. These findings indicate that activin A/Smad signaling exerts neuroprotective effects by inhibiting ERS-mediated apoptosis during OGD. The study also found that the expression of microtubule-associated protein light chain 3 (LC3) and of Beclin1 is significantly upregulated after OGD in PC12 cells and increases with the extension of OGD duration. Interestingly, application of exogenous activin A significantly inhibits LC3II and Beclin1 protein levels (Wang et al., 2016a). Together, the in vivo and in vitro study suggest that activin A exerts its neuroprotection role mainly through negatively regulate apoptotic and autophagic pathway.
When transient cerebral ischemia and hypoxia occurs, the expression of activin A, as a neuronal survival factor, as well as that of its effectors RII or Smad3, is significantly upregulated. It was found that activin A and Smad3 are mainly expressed in the cytoplasm and nucleus, whereas RII is mainly expressed in the cytoplasm and membrane of the cells. This change in expression levels occurs specifically in neurons, suggesting that the activin A/Smad pathway is activated after focal cerebral ischemia (Mukerji et al., 2009).
It was also reported that activin A, as a neuronal autocrine factor, may act on the neuron itself and mediate signal transduction through the activin A/Smad pathway after ischemia (Hiratochi et al., 2007). In addition, in PC12 OGD models, blockade of activin A RII site in the activin A transmembrane signal transduction pathway leads to aggravation of OGD-induced neuronal damage, and the expression of activin A and Smad3 is significantly downregulated (Xue et al., 2016). These results suggest that neuronal damage, induced by OGD, activates the activin A/Smad pathway, which exerts a neuroprotective role through the inhibition of apoptosis. Upregulation of RII may be the initiating factor in the activation of the activin A/Smad pathway induced by OGD injury, which may rely on an activin A positive feedback regulation mechanism (Table 2).

Table 2. Activin A targets in brain injury.
Sublethal ischemic damage induces increased anti-ischemic response in the late stages of brain injury. This phenomenon is called ischemic tolerance (IT) and is induced by ischemic pre-conditioning (IPC) (Nishio et al., 2000). Ischemic brain injury can induce high expression of activin A, which can then activate an endogenous neuroprotective signaling pathway (Wu et al., 1999). It was demonstrated that after IPC in PC12 cells, the expression of activin A RII is upregulated, suggesting that the IPC-induced IT is mediated by the signaling pathway involving activin A, RII, and downstream Smad proteins (Xue et al., 2016). The main mechanism of IPC involves JNK1 activation, which inhibits Smad3 phosphorylation and the entry of the downstream Smad4 complex into the nucleus (Wang et al., 2016b). Furthermore, pre-treatment with JNK1 inhibitors also induces IT in PC12 cells. Given the existing crosstalk between the intracellular JNK1 protein and activin A/Smad pathway, JNK1 inhibitors may represent potential therapeutic agents for drug-induced IT (Wang et al., 2016b; Table 2).
Studies have found that activin A is an immunosuppressive factor that induces cardiovascular and cerebrovascular diseases by inhibiting the activity of T lymphocytes in the body (Zipori and Barda-Saad, 2001; Ofstad et al., 2013; Yoon et al., 2013). Patients with cerebral hemorrhage show a significant increase in activin A and activin A binding protein levels, while patients with cerebral infarction show a significant increase in activin A (Ebert et al., 2006; Nicolas et al., 2017). Therefore, activin A and activin A binding protein levels in peripheral blood may be associated with cerebrovascular disease occurrence and development. Thus, detecting changes in these proteins may be helpful in guiding clinical diagnosis and treatment of cerebrovascular diseases, treatment decisions, and prognosis (Table 2).
Brain injury in pre-term infants is mainly restricted to white matter damage. Its primary neuropathological feature is damage of oligodendrocyte precursor cells (OPCs), which inhibits the formation of myelin (Yue et al., 2018). A study found that the activin A receptor subtype Acvr2a/Acvr2b signaling pathway is involved in the regulation of myelin repair after brain injury in pre-term infants (Dillenburg et al., 2018). Acvr2b competes with Acvr2a for binding to activin A, thus resulting in decreased levels of Acvr2a-bound activin A, which in turn hinders Acvr2a-driven oligodendrocyte (OL) differentiation and myelination. Therefore, the activin A receptor Acvr2a may serve as a novel therapeutic target for the repair of myelin damage (Dillenburg et al., 2018).
Activin A levels in the serum are significantly increased in pre-term infants during infection, which significantly inhibits the release of pro-inflammatory mediators from stimulated neonatal peripheral blood mononuclear cells in vitro and is associated with a dramatic increase in IL-10, an anti-inflammatory and immunoregulatory mediator (Petrakou et al., 2013; González-Domínguez et al., 2016). This suggests that activin A and IL-10 have strong anti-inflammatory and immunomodulatory effects in neonatal infection and are crucial for controlling the inflammatory response in neonates. Thus, activin A may be a target for the treatment of brain damage in prematurely born infants (Table 2).
One of the pathogenic mechanisms of sepsis encephalopathy is the activation of inflammation and apoptosis, for which TNF-α and IL-6 are the two most important inflammatory cytokines, produced in the early stages of this disease (Sun et al., 2017). activin A promotes the expression of TNF-α, IL-6, and IL-1, in inflammatory and immune reactions, and eventually promotes the occurrence of inflammatory responses (Tania et al., 2014). In addition, studies have shown that serum activin A is elevated during acute and chronic inflammation, which may further increase the uninhibited inflammatory response leading to multiple organ failure and even death (Lee et al., 2016). However, other studies have indicated that activin A inhibits the inflammatory response by inhibiting caspase-1, IL-1β, and IL-8, thus leading to the dramatic increase in the production of the anti-inflammatory mediator IL-10 (Sierra-Filardi et al., 2011; Petrakou et al., 2013). Therefore, activin A has both pro-inflammatory and anti-inflammatory functions, is associated with the severity of sepsis encephalopathy, and can be used as an early predictor of this pathogenesis (Table 2).
White matter damage is characterized by myelin injury, mainly affecting OLs (Liu X. B. et al., 2013). One study found that activin A, as a neurotrophic factor, plays a role in the repair of white matter damage (Dutta et al., 2014). Moreover, in an in vitro study, human recombinant activin A was added to cultures of primary OLs (no axons) or to neuro-glia co-cultures (Goebbels et al., 2017). After 3–5 days in culture, the number of OPCs in primary OL cultures that differentiated into mature OLs was significantly higher in treated versus untreated cells. Moreover, in the neuron-OL co-culture, the area of myelin was significantly higher in the activin A-treated cells. Thus, activin A seems to act as a myelination promoting factor. Researchers also found that activin A promotes the differentiation of OPCs into OLs by activating the ERK1/2 MAPK signaling pathway (Goebbels et al., 2017).
Using the hypoxia-ischemia brain damage model in neonatal rats, it was shown that pathological changes in the brain are significantly reduced by an intraperitoneal injection of activin A, confirming that the application of activin A reduces brain tissue damage induced by hypoxia-ischemia (An et al., 2005). In addition, in the OGD/reperfusion model in primary neuronal cultures, researchers used neuroprotector stress-induced phosphoprotein 1 (STI1) to treat neurons and found that STI1 is dependent on activin-A receptor 1 (ALK2) for the inhibition of neuronal apoptosis (Beraldo et al., 2018). Thus, since ALK2 acts as a downstream mediator of the STI1 neuroprotection pathway, it may be useful as a therapeutic target for ischemic brain injury (Beraldo et al., 2018). It was also shown that activin A reduces brain edema and the release of inflammatory cytokines in neonatal rats after hypoxic-ischemic brain damage and plays a role in nerve regeneration and functional repair, by increasing nestin protein expression, as well as the number and differentiation of neural stem cells (Peng et al., 2006; Zhang et al., 2016).
Focal cerebral ischemia and reperfusion lead to complex interactions between cells and molecules that eventually cause cells to either repair themselves or be destroyed (Cole et al., 1992; Li et al., 2004). Studies have found that activin A is an early response gene for cerebral ischemia and supports the survival of cortical neurons in vitro (Mukerji et al., 2009). In addition, activin A treatment protects neurons in the ischemic cerebral hemisphere, decreases the number of activated microglia, and reduces the activation of terminal kinases, like p38 and c-Jun N, involved in neuronal apoptosis after stroke. These findings suggest that activin A promotes tissue survival after focal cerebral ischemia/reperfusion.
One study demonstrated that administration of emodin in OGD-treated neuronal cultures significantly increases cell viability and activin A content in the culture medium, whereas it significantly decreases the expression of caspase-3 (Guo et al., 2013). Therefore, emodin promotes the up-regulation of activin A expression, increases the viability of neurons, and inhibits neuronal apoptosis in ischemic and hypoxic conditions, thus playing a neuroprotective role.
In summary, activin A, as a neuroprotective factor, maintains the survival of neurons in the central nervous system, protects neurons from neurotoxicity, and plays important roles in brain injury (Iwahori et al., 1997; Keelan et al., 2000). Therefore, it can be used as a clinical test index, which is of great significance for disease diagnosis and prognosis (Bergestuen et al., 2010). The protective effect of the activin A-mediated signaling pathway against brain injury and its application in the clinic have received increased attention. Future research on the molecular mechanisms involved in activin A neuroprotection will provide new insight for developing treatments against brain damage.
XS is the primary author who contributed to the conception and design of the review and the drafting of this article. LH and DX critically reviewed the article for some intellectual content. YQ and DM contributed to approving the final version of the manuscript submitted for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by the National Key Research Program of R & D (2017YFA0104200); National Natural Science Foundation of China (No.81330016, 81630038, 81771634); Science and Technology Project of Sichuan Province (2016TD0002); National Key Project of Neonatal Children (1311200003303). In addition, we would like to thank Editage [www.editage.cn] for English language editing.
Ageta, H., and Tsuchida, K. (2011). Multifunctional roles of activins in the brain. Vitam. Horm. 85, 185–206. doi: 10.1016/B978-0-12-385961-7.00009-3
An, L., Zhu, W. W., Wang, P., and Cao, Y. L. (2005). [Protective effects and mechanism of activin in neonatal rats with hypoxic ischemic brain damage]. Zhonghua Er Ke Za Zhi 43, 465–466.
Arai, K. Y., Ono, M., Kudo, C., Fujioka, A., Okamura, R., Nomura, Y., et al. (2011). IL-1 beta stimulates activin beta A mRNA expression in human skin fibroblasts through the MAPK pathways, the nuclear factor-kappa B pathway, and prostaglandin E-2. Endocrinology 152, 3779–3790. doi: 10.1210/en.2011-0255
Asashima, M., Nakano, H., Uchiyama, H., Sugino, H., Nakamura, T., Eto, Y., et al. (1991). Follistatin inhibits the mesoderm-inducing activity of activin A and the vegetalizing factor from chicken embryo. Roux. Arch. Dev. Biol. 200, 4–7. doi: 10.1007/BF02457635
Beraldo, F. H., Ostapchenko, V. G., Xu, J. Z., Di Guglielmo, G. M., Fan, J., Nicholls, P. J., et al. (2018). Mechanisms of neuroprotection against ischemic insult by stress-inducible phosphoprotein-1/prion protein complex. J. Neurochem. 145, 68–79. doi: 10.1111/jnc.14281
Bergestuen, D. S., Edvardsen, T., Aakhus, S., Ueland, T., Oie, E., Vatn, M., et al. (2010). activin A in carcinoid heart disease: a possible role in diagnosis and pathogenesis. Neuroendocrinology 92, 168–177. doi: 10.1159/000318014
Chandra, A., Geng, X., and Ding, Y. (2018). Brain and disease: an insight into new developments in the pathogenesis and novel therapies for neurological disorders. Neurol. Res. 40, 419–420. doi: 10.1080/01616412.2018.1451270
Cole, D. J., Schell, R. M., Drummond, J. C., Patel, P. M., and Marcantonio, S. (1992). Focal cerebral ischemia in rats: effect of phenylephrine-induced hypertension during reperfusion. J. Neurosurg. Anesthesiol. 4, 78–84. doi: 10.1097/00008506-199204000-00002
de Kretser, D. M., O'Hehir, R. E., Hardy, C. L., and Hedger, M. P. (2012). The roles of activin A and its binding protein, follistatin, in inflammation and tissue repair. Mol. Cell Endocrinol. 359, 101–106. doi: 10.1016/j.mce.2011.10.009
de Kroon, L. M., Narcisi, R., Blaney Davidson, E. N., Cleary, M. A., van Beuningen, H. M., Koevoet, W. J., et al. (2015). Activin receptor-like kinase receptors ALK5 and ALK1 are both required for TGFbeta-induced chondrogenic differentiation of human bone marrow-derived mesenchymal stem cells. PLoS ONE 10:e0146124. doi: 10.1371/journal.pone.0146124
Derynck, R., and Zhang, Y. E. (2003). Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 425, 577–584. doi: 10.1038/nature02006
Dillenburg, A., Ireland, G., Holloway, R. K., Davies, C. L., Evans, F. L., Swire, M., et al. (2018). Activin receptors regulate the oligodendrocyte lineage in health and disease. Acta Neuropathol. 135, 887–906. doi: 10.1007/s00401-018-1813-3
Dutta, D. J., Zameer, A., Mariani, J. N., Zhang, J., Asp, L., Huynh, J., et al. (2014). Combinatorial actions of Tgf beta and Activin ligands promote oligodendrocyte development and CNS myelination. Development 141, 2414–2428. doi: 10.1242/dev.106492
Ebert, S., Phillips, D. J., Jenzewski, P., Nau, R., O'Connor, A. E., and Michel, U. (2006). activin A concentration in human cerebrospinal fluid are age-dependent and elevated in meningitis. J. Neurol. Sci. 250, 50–57. doi: 10.1016/j.jns.2006.06.026
Goebbels, S., Wieser, G. L., Pieper, A., Spitzer, S., Weege, B., Yan, K., et al. (2017). A neuronal PI(3,4,5)P3-dependent program of oligodendrocyte precursor recruitment and myelination. Nat. Neurosci. 20, 10–15. doi: 10.1038/nn.4425
González-Domínguez, É., Domínguez-Soto, Á., Nieto, C., Flores-Sevilla, J. L., Pacheco-Blanco, M., Campos-Peña, V., et al. (2016). Atypical activin A and IL-10 production impairs human CD16+ monocyte differentiation into anti-inflammatory macrophages. J. Immunol. 196, 1327–1337. doi: 10.4049/jimmunol.1501177
Guo, H., Shen, X., Xu, Y., He, Y., and Hu, W. (2014). The effect of activin A on signal transduction pathways in PC12 cells subjected to oxygen and glucose deprivation. Int. J. Mol. Med. 33, 135–141. doi: 10.3892/ijmm.2013.1539
Guo, H., Shen, X., Xu, Y., Yuan, J., Zhao, D., and Hu, W. (2013). Emodin prevents hypoxic-ischemic neuronal injury: involvement of the activin A pathway. Neural Regen. Res. 8, 1360–1367. doi: 10.3969/j.issn.1673-5374.2013.15.002
He, J. T., Mang, J., Mei, C. L., Yang, L., Wang, J. Q., Xing, Y., et al. (2011). Neuroprotective effects of exogenous activin A on oxygen-glucose deprivation in PC12 cells. Molecules 17, 315–327. doi: 10.3390/molecules17010315
Hiratochi, M., Nagase, H., Kuramochi, Y., Koh, C. S., Ohkawara, T., and Nakayama, K. (2007). The Delta intracellular domain mediates TGF-beta/Activin signaling through binding to Smads and has an important bi-directional function in the Notch-Delta signaling pathway. Nucleic Acids Res. 35, 912–922. doi: 10.1093/nar/gkl1128
Huang, H. M., Chang, T. W., and Liu, J. C. (2004). Basic fibroblast growth factor antagonizes activin A-mediated growth inhibition and hemoglobin synthesis in K562 cells by activating ERK1/2 and deactivating p38 MAP kinase. Biochem. Biophys. Res. Commun. 320, 1247–1252. doi: 10.1016/j.bbrc.2004.06.083
Hübner, G., Alzheimer, C., and Werner, S. (1999). Activin: a novel player in tissue repair processes. Histol. Histopathol. 14, 295–304.
Iwahori, Y., Saito, H., Torii, K., and Nishiyama, N. (1997). Activin exerts a neurotrophic effect on cultured hippocampal neurons. Brain Res. 760, 52–58. doi: 10.1016/S0006-8993(97)00275-8
Keelan, J. A., Zhou, R. L., and Mitchell, M. D. (2000). activin A exerts both pro- and anti-inflammatory effects on human term gestational tissues. Placenta 21, 38–43. doi: 10.1053/plac.1999.0451
Kelly, E. A., Esnault, S., Johnson, S. H., Liu, L. Y., Malter, J. S., Burnham, M. E., et al. (2016). Human eosinophil activin A synthesis and mRNA stabilization are induced by the combination of IL-3 plus TNF. Immunol. Cell Biol. 94, 701–708. doi: 10.1038/icb.2016.30
Kim, Y. I., Park, S. W., Kang, I. J., Shin, M. K., and Lee, M. H. (2015). Activin suppresses LPS-induced Toll-like receptor, cytokine and inducible nitric oxide synthase expression in normal human melanocytes by inhibiting NF-kappaB and MAPK pathway activation. Int. J. Mol. Med. 36, 1165–1172. doi: 10.3892/ijmm.2015.2308
Lee, J. K., Choi, S. M., Lee, J., Park, Y. S., Lee, C. H., Yim, J. J., et al. (2016). Serum activin-A as a predictive and prognostic marker in critically ill patients with sepsis. Respirology 21, 891–897. doi: 10.1111/resp.12751
Li, J. S., Ren, X. Q., Liu, K., Liu, Z. G., and Kong, L. F. (2004). [Study on changes of neuron apoptosis after focal cerebral ischemia/reperfusion in the aged rats]. Zhongguo Wei Zhong Bing Ji Jiu Yi Xue 16, 151–154.
Liu, H. Y., Chen, F. F., Ge, J. Y., Wang, Y. N., Zhang, C. H., Cui, X. L., et al. (2009). Expression and localization of activin receptor-interacting protein 2 in mouse tissues. Gen. Comp. Endocrinol. 161, 276–282. doi: 10.1016/j.ygcen.2009.01.020
Liu, H. Y., Wang, Y. N., Ge, J. Y., Li, N., Cui, X. L., and Liu, Z. H. (2013). Localisation and role of activin receptor-interacting protein 1 in mouse brain. J. Neuroendocrinol. 25, 87–95. doi: 10.1111/j.1365-2826.2012.02371.x
Liu, X. B., Shen, Y., Plane, J. M., and Deng, W. (2013). Vulnerability of premyelinating oligodendrocytes to white-matter damage in neonatal brain injury. Neurosci. Bull. 29, 229–238. doi: 10.1007/s12264-013-1311-5
Ma, J., Zhang, L., He, G., Tan, X., Jin, X., and Li, C. (2016). Transcutaneous auricular vagus nerve stimulation regulates expression of growth differentiation factor 11 and activin-like kinase 5 in cerebral ischemia/reperfusion rats. J. Neurol. Sci. 369, 27–35. doi: 10.1016/j.jns.2016.08.004
Mai, Y., Zhang, Z., Yang, H., Dong, P., Chu, G., Yang, G., et al. (2014). BMP and activin membrane-bound inhibitor (BAMBI) inhibits the adipogenesis of porcine preadipocytes through Wnt/beta-catenin signaling pathway. Biochem. Cell Biol. 92, 172–182. doi: 10.1139/bcb-2014-0011
Manickam, M., and Tulsawani, R. (2014). Survival response of hippocampal neurons under low oxygen conditions induced by Hippophae rhamnoides is associated with JAK/STAT signaling. PLoS ONE 9:e87694. doi: 10.1371/journal.pone.0087694
Mather, J. P. (1996). Follistatins and alpha 2-macroglobulin are soluble binding proteins for inhibin and activin. Horm. Res. 45, 207–210. doi: 10.1159/000184789
Mukerji, S. S., Katsman, E. A., Wilber, C., Haner, N. A., Selman, W. R., and Hall, A. K. (2007). Activin is a neuronal survival factor that is rapidly increased after transient cerebral ischemia and hypoxia in mice. J. Cereb. Blood Flow Metab. 27, 1161–1172. doi: 10.1038/sj.jcbfm.9600423
Mukerji, S. S., Rainey, R. N., Rhodes, J. L., and Hall, A. K. (2009). Delayed activin A administration attenuates tissue death after transient focal cerebral ischemia and is associated with decreased stress-responsive kinase activation. J. Neurochem. 111, 1138–1148. doi: 10.1111/j.1471-4159.2009.06406.x
Müller, M. R., Zheng, F., Werner, S., and Alzheimer, C. (2006). Transgenic mice expressing dominant-negative activin receptor IB in forebrain neurons reveal novel functions of activin At glutamatergic synapses. J. Biol. Chem. 281, 29076–29084. doi: 10.1074/jbc.M604959200
Nakajima, A., Tsuji, M., Inagaki, M., Tamura, Y., Kato, M., Niiya, A., et al. (2014). Neuroprotective effects of propofol on ER stress-mediated apoptosis in neuroblastoma SH-SY5Y cells. Eur. J. Pharmacol. 725, 47–54. doi: 10.1016/j.ejphar.2014.01.003
Nicolas, N., Muir, J. A., Hayward, S., Chen, J. L., Stanton, P. G., Gregorevic, P., et al. (2017). Induction of experimental autoimmune orchitis in mice: responses to elevated circulating levels of the activin-binding protein, follistatin. Reproduction 154, 193–205. doi: 10.1530/REP-17-0010
Nishio, S., Yunoki, M., Chen, Z. F., Anzivino, M. J., and Lee, K. S. (2000). Ischemic tolerance in the rat neocortex following hypothermic preconditioning. J. Neurosurg. 93, 845–851. doi: 10.3171/jns.2000.93.5.0845
Ofstad, A. P., Gullestad, L., Orvik, E., Aakhus, S., Endresen, K., Ueland, T., et al. (2013). Interleukin-6 and activin A are independently associated with cardiovascular events and mortality in type 2 diabetes: the prospective Asker and Baerum Cardiovascular Diabetes (ABCD) cohort study. Cardiovasc. Diabetol. 12:126. doi: 10.1186/1475-2840-12-126
Peng, H., Zhou, H., and Xiong, Y. (2006). [Effect of basic fibral growth factor on nestin expression in neonatal rats following hypoxic-ischemic brain damage]. Zhongguo Dang Dai Er Ke Za Zhi 8, 235–238.
Peng, J., Monsivais, D., You, R., Zhong, H., Pangas, S. A., and Matzuk, M. M. (2015). Uterine activin receptor-like kinase 5 is crucial for blastocyst implantation and placental development. Proc. Natl. Acad. Sci. U.S.A. 112, E5098–E5107. doi: 10.1073/pnas.1514498112
Peterson, A. J., Jensen, P. A., Shimell, M., Stefancsik, R., Wijayatonge, R., Herder, R., et al. (2012). R-Smad competition controls activin receptor output in Drosophila. PLoS ONE 7:e36548. doi: 10.1371/journal.pone.0036548
Petrakou, E., Fotopoulos, S., Anagnostakou, M., Anatolitou, F., Samitas, K., Semitekolou, M., et al. (2013). Activin-A exerts a crucial anti-inflammatory role in neonatal infections. Pediatr. Res. 74, 675–681. doi: 10.1038/pr.2013.159
Protic, O., Islam, M. S., Greco, S., Giannubilo, S. R., Lamanna, P., Petraglia, F., et al. (2017). activin A in inflammation, tissue repair, and fibrosis: possible role as inflammatory and fibrotic mediator of uterine fibroid development and growth. Semin. Reprod. Med. 35, 499–509. doi: 10.1055/s-0037-1607265
Qi, Y., Ge, J. Y., Wang, Y. N., Liu, H. Y., Li, Y. M., Liu, Z. H., et al. (2013). Co-expression of activin receptor-interacting protein 1 and 2 in mouse nerve cells. Neurosci. Lett. 542, 53–58. doi: 10.1016/j.neulet.2013.03.012
Shi, J., Yoshino, O., Osuga, Y., Koga, K., Hirota, Y., Hirata, T., et al. (2009). Bone morphogenetic protein-6 stimulates gene expression of follicle-stimulating hormone receptor, inhibin/activin beta subunits, and anti-Mullerian hormone in human granulosa cells. Fertil. Steril. 92, 1794–1798. doi: 10.1016/j.fertnstert.2009.05.004
Sierra-Filardi, E., Puig-Kröger, A., Blanco, F. J., Nieto, C., Bragado, R., Palomero, M. I., et al. (2011). activin A skews macrophage polarization by promoting a proinflammatory phenotype and inhibiting the acquisition of anti-inflammatory macrophage markers. Blood 117, 5092–5101. doi: 10.1182/blood-2010-09-306993
Sun, W., Pei, L., and Liang, Z. (2017). mRNA and Long non-coding RNA expression profiles in rats reveal inflammatory features in sepsis-associated encephalopathy. Neurochem. Res. 42, 3199–3219. doi: 10.1007/s11064-017-2357-y
Tania, N. P., Schmidt, M., and Gosens, R. (2014). Activin-A: active in inflammation in COPD. Eur. Respir. J. 43, 954–955. doi: 10.1183/09031936.00011414
Tanimoto, K., Handa, S., Ueno, N., Murakami, K., and Fukamizu, A. (1991). Structure and sequence analysis of the human activin beta A subunit gene. DNA Seq. 2, 103–110. doi: 10.3109/10425179109039678
Tian, A. H., Zhao, W., Zhang, L., Dong, J. L., Li, W. W., and Jin, H. (2014). [Expression of follistatin, activin A and BMP-4 in rats with hypoxic-ischemic brain damage]. Sichuan Da Xue Xue Bao Yi Xue Ban 45, 772–776.
Upton, P. D., Davies, R. J., Trembath, R. C., and Morrell, N. W. (2009). Bone morphogenetic protein (BMP) and activin type II receptors balance BMP9 signals mediated by activin receptor-like kinase-1 in human pulmonary artery endothelial cells. J. Biol. Chem. 284, 15794–15804. doi: 10.1074/jbc.M109.002881
Wang, J. Q., Liang, W. Z., Cui, Y., He, J. T., Liu, H. Y., Wang, Y., et al. (2016a). Noncanonical activin A signaling in PC12 cells: a self-limiting feedback loop. Neurochem. Res. 41, 1073–1084. doi: 10.1007/s11064-015-1797-5
Wang, J. Q., Xu, Z. H., Liang, W. Z., He, J. T., Cui, Y., Liu, H. Y., et al. (2016b). Effects of c-Jun N-terminal kinase on activin A/Smads signaling in PC12 cell suffered from oxygen-glucose deprivation. Cell Mol. Biol. (Noisy-le-grand) 62, 81–86. doi: 10.14715/cmb/2016.62.2.14
Wang, Q., Yin, J., Wang, S., Cui, D., Lin, H., Ge, M., et al. (2016). Effects of activin A and its downstream ERK1/2 in oxygen and glucose deprivation after isoflurane-induced postconditioning. Biomed. Pharmacother. 84, 535–543. doi: 10.1016/j.biopha.2016.09.075
Wang, X., Fischer, G., and Hyvönen, M. (2016). Structure and activation of pro-activin A. Nat. Commun. 7:12052. doi: 10.1038/ncomms12052
Wang, Y., and Ge, W. (2004). Cloning of epidermal growth factor (EGF) and EGF receptor from the zebrafish ovary: evidence for EGF as a potential paracrine factor from the oocyte to regulate activin/follistatin system in the follicle cells. Biol. Reprod. 71, 749–760. doi: 10.1095/biolreprod.104.028399
Wu, D. D., Lai, M., Hughes, P. E., Sirimanne, E., Gluckman, P. D., and Williams, C. E. (1999). Expression of the activin Axis and neuronal rescue effects of recombinant activin A following hypoxic-ischemic brain injury in the infant rat. Brain Res. 835, 369–378. doi: 10.1016/S0006-8993(99)01638-8
Xue, L. X., Liu, H. Y., Cui, Y., Dong, Y., Wang, J. Q., Ji, Q. Y., et al. (2017). Neuroprotective effects of activin A on endoplasmic reticulum stress-mediated apoptotic and autophagic PC12 cell death. Neural Regen. Res. 12, 779–786. doi: 10.4103/1673-5374.206649
Xue, L. X., Xu, Z. H., Wang, J. Q., Cui, Y., Liu, H. Y., Liang, W. Z., et al. (2016). activin A/Smads signaling pathway negatively regulates oxygen glucose deprivation-induced autophagy via suppression of JNK and p38 MAPK pathways in neuronal PC12 cells. Biochem. Biophys. Res. Commun. 480, 355–361. doi: 10.1016/j.bbrc.2016.10.050
Yoon, J. H., Jung, S. M., Park, S. H., Kato, M., Yamashita, T., Lee, I. K., et al. (2013). Activin receptor-like kinase5 inhibition suppresses mouse melanoma by ubiquitin degradation of Smad4, thereby derepressing eomesodermin in cytotoxic T lymphocytes. EMBO Mol. Med. 5, 1720–1739. doi: 10.1002/emmm.201302524
Yue, Y., Zhang, L., Qu, Y., and Mu, D. Z. (2018). [Neuroprotective effects of oligodendrocyte precursor cells on white matter damage in preterm infants]. Zhongguo Dang Dai Er Ke Za Zhi 20, 326–331.
Zhang, X., Zhu, C., Luo, Q., Dong, J., Liu, L., Li, M., et al. (2016). Impact of siRNA targeting of beta-catenin on differentiation of rat neural stem cells and gene expression of Ngn1 and BMP4 following in vitro hypoxic-ischemic brain damage. Mol. Med. Rep. 14, 3595–3601. doi: 10.3892/mmr.2016.5667
Keywords: activin A, transforming growth factor β, activin A/Smad, brain injury, neuroprotection, target therapy
Citation: Su X, Huang L, Xiao D, Qu Y and Mu D (2018) Research Progress on the Role and Mechanism of Action of Activin A in Brain Injury. Front. Neurosci. 12:697. doi: 10.3389/fnins.2018.00697
Received: 20 June 2018; Accepted: 18 September 2018;
Published: 09 October 2018.
Edited by:
Sebastian Cerdan, Consejo Superior de Investigaciones Científicas (CSIC), SpainReviewed by:
Vladimir Parpura, University of Alabama at Birmingham, United StatesCopyright © 2018 Su, Huang, Xiao, Qu and Mu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yi Qu, cXV5aTcxMjAwMkAxNjMuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.