 Valentin Loser
Valentin Loser Alex Vicino
Alex Vicino- Department of Clinical Neurosciences, Nerve-Muscle Unit, Service of Neurology, Lausanne University Hospital and University of Lausanne, Lausanne, Switzerland
A great proportion of neuromuscular diseases are immune-mediated, included myasthenia gravis, Lambert-Eaton myasthenic syndrome, acute- and chronic-onset autoimmune neuropathies (anti-MAG neuropathy, multifocal motor neuropathy, Guillain-Barré syndromes, chronic inflammatory demyelinating polyradiculoneuropathy, CANDA and autoimmune nodopathies), autoimmune neuronopathies, peripheral nerve hyperexcitability syndromes and idiopathic inflammatory myopathies. The detection of autoantibodies against neuromuscular structures has many diagnostic and therapeutic implications and, over time, allowed a better understanding of the physiopathology of those disorders. In this paper, we will review the main autoantibodies described in neuromuscular diseases and focus on their use in clinical practice.
Introduction
Autoantibodies are antibodies (ab) directed against self-antigens. They may be the direct cause of a pathological process (through activation of the complement cascade for example) or be detected as an epiphenomenon. Their main usefulness is to confirm a diagnosis suspicion, but some of them can also be used as severity or treatment response biomarkers. In neuromuscular disorders, several antibodies are highly sensitive and/or specific. In this review, we will describe the main autoantibodies used in the diagnostic process of neuromuscular diseases.
Autoimmune neuromuscular junction disorder
Myasthenia gravis
Myasthenia gravis (MG) is the prototype of post-synaptic neuromuscular junction disorder. Its pathophysiology involves immunological dysregulation, with autoimmunization beginning in the thymus. It is a humoral-mediated disease, associated with the presence of antibodies directed against some epitopes of the neuromuscular junction. The most frequently described antibodies are directed against the nicotinic acetylcholine receptor (AChR) and the muscle kinase (MuSK). The disease is characterized by fluctuating muscle weakness, typically affecting the ocular and bulbar regions, and sometimes involving the cervical and proximal limb muscles.
Anti-AChR antibodies
Serum anti-AChR-ab are of IgG1 and IgG3 subtypes and impair neuromuscular transmission by two main mechanisms: antigenic modulation by inducing endocytosis of the acetylcholine receptor and its lysosomal proteolysis, and its degradation by activation of the complement cascade and membrane attack complex (1, 2). The current reference method for detecting these antibodies is a radioimmunoassay (RIA) (3), but detection can also be performed by ELISA, with a lower sensitivity, reason why this method is less widely used routinely (4). These antibodies are detected in approximately 85% of patients with generalized MG and 50–60% in ocular MG, with a very good specificity (5). In the recently published multicenter SCREAM study, the specificity of anti-AChR-ab was 97.8% by RIA and 94.8% by ELISA (6). False positives have been described in patients with peripheral neuropathies, Guillain-Barré syndrome, ophthalmoplegia externa, neuromyelitis optica spectrum disorder and even in healthy controls. There appears to be no correlation between antibody titer and clinical severity, although ocular forms tend to have lower titers. There is no consensus on the use of anti-AChR-ab during follow-up; previous studies reported conflicting results about an association between changes in anti-AChR-ab serum levels and clinical severity (7–14). Monitoring anti-AChR-ab titers is therefore currently not indicated. Detection of serum anti-AChR-ab has two therapeutic implications. First, 10–15% anti AChR+ MG are associated with a thymoma, which, on the contrary, has not been described in anti-AChR- MG patients (15). Accordingly, thymectomy is recommended in patients with thymoma and anti-AChR+ MG (16). More recently, the MGTX trial demonstrated that thymectomy was also beneficial in a subgroup of patients under 65 years of age with non-thymomatous anti-AChR+ generalized MG (17). Those results confirmed the strong implication of the thymus, even in the absence of thymoma, in the pathophysiology of anti-AChR+ MG, and have been integrated into the latest international consensus guidance for management of MG (18). Secondly, severe refractory generalized anti-AChR+ MG may benefit from second-line therapy with complement inhibitors (eculizumab, ravulizumab, zilucoplan) (19–21) or antagonist of the neonatal Fc receptor (efgartigimod, rozanolixizumab) (22, 23).
“Low-affinity” anti-AChR antibodies
“Low-affinity” anti-AChR-ab can be found in patients with seronegative MG. Unlike anti-AChR and MuSK antibodies, which are high-affinity antibodies, low-affinity anti-AChR-ab are detected only by cell-based assay (CBA), by binding to a high-density AChR cluster (24–28). This detection method is less widely available and usually performed only in research centers. The detection rate of these antibodies in patients with double seronegative MG varies between studies but is probably close to 20% (26, 27). Specificity seems to be close to 100% (25). These antibodies are IgG1 and activate the complement pathway. Detection of low-affinity anti-AChR-ab seems to be associated with a less severe phenotype and a better response to immunotherapy (25). Low-affinity anti-AChR+ MG should probably be managed in the same way as anti-AChR+ MG, although clear recommendations are lacking.
Anti-MuSK antibodies
Serum anti-MuSK-ab are present in 30–60% of patients seronegative for anti-AChR-ab, representing 5–8% of patients with MG (29). These antibodies are predominantly IgG4, and induce functional blockade of the MuSK protein, without inducing antigenic modulation or activation of the complement pathway (30). The clinical phenotype of anti-MuSK+ MG may be different from anti-AChR+ forms, dominated by oculo-bulbar involvement, sometimes with amyotrophy and lingual fasciculations that may mimic amyotrophic lateral sclerosis with bulbar onset (31). The preferential involvement of certain muscles in anti-MuSK+ MG may reflect a different composition of the endplates in these muscles. These antibodies can be detected by RIA or ELISA, with a specificity close to 100% (6, 32, 33). CBA methods appear to be more sensitive but are not yet routinely available (34). Anti-MuSK-ab titer may correlate with disease severity and response to immunotherapy (35). It is exceptional for anti-MuSK-ab to be detected in the presence of anti-AChR-ab (36). Detection of serum anti-MuSK-ab has also significant therapeutic implications. In anti-MuSK+ MG patients, classical treatment options, including acetylcholinesterase inhibitors, corticosteroid-sparing immunosuppressive treatments, and intravenous immunoglobulins (IVIg), are usually less effective than in anti AChR+ MG (37). On the other hand, anti-CD20 therapy with Rituximab has shown efficacy in a few studies in anti-MuSK+ MG (38–41). Consequently, according to the latest international recommendations, Rituximab should be considered as an early therapeutic option in anti-MuSK+ MG patients, who have an unsatisfactory response to initial immunotherapy (18). As anti-MuSK+ MG are not related to thymus disorders, thymectomy is not recommended in this population (42). Moreover, complement inhibitors are probably not effective in anti-MuSK+ MG, as antibodies are predominantly IgG4, unable to activate the complement pathway.
Anti-LRP4 antibodies
LRP4 (lipoprotein-related protein receptor 4) is an endplate protein that, along with MuSK, is an agrin receptor and is required for AChR clustering and normal neuromuscular junction function. Antibodies to LRP4 have been detected in 7–33% of patients with double seronegative MG (43). Most of these are IgG1 antibodies, which can activate the complement pathway. Anti LRP4-ab are identified in 8% of patients with anti-AChR-ab, 15% of patients with anti-MuSK-ab, 4% of patients with other neurological autoimmune pathologies and in 0–4% of healthy controls (44, 45). Although initially considered to be relatively specific to MG, these antibodies were surprisingly detected in 10–23% of patients with amyotrophic lateral sclerosis (46). Indeed, LRP4 antigen appears to be also expressed in motor neurons and muscle, which may explain the presence of this antibody in motor neuron diseases. In some unclear situations, anti-LRP4-ab - cannot therefore distinguish reliably MG from amyotrophic lateral sclerosis. Anti-LRP4+ MG are usually associated with a less severe phenotype (ocular or mild generalized form) at disease onset, and a similar response to immunotherapy as anti-AChR+ MG patients (43). However, double positive patients (AChR/LRP4 and MuSK/LRP4) may have a more severe phenotype at onset (43). The favored detection method is a CBA.
Anti-striated muscle antibodies
Other antibodies are directed against intracellular muscle epitopes, known as anti-striated (striational) muscle antibodies. They recognize intracellular muscle proteins (titin, myosin, actin, ryanodine receptor or RyR); since these antibodies have no direct access to the antigen, they are unlikely to be pathogenic. Those antibodies are generally associated with anti-AChR-ab, making them of little diagnostic value. However, anti-titin and anti-RyR antibodies are present in most patients with thymoma (46). In particular, anti-titin was found in 80% of patients with MG and thymoma, especially in patients younger than 60-year-old (47). This good sensitivity in the presence of thymoma may therefore make these antibodies interesting biomarkers of thymoma, albeit with rather low specificity (39–70%) (48). Those antibodies are not specific for MG and may occur in patients with other autoimmune disease and in patients with thymoma without MG. Anti-titin and anti-RyR titers also appear to correlate with disease severity (49). The usefulness of anti-striated muscle antibodies testing in clinical practice is yet unclear.
Diagnostic workup
There are no clear international recommendations on the serological diagnostic approach to MG. Current consensus, underlined in the 2019 Italian recommendations, is to test for anti-AChR-ab and, if negative, for anti-MuSK-ab, in every clinical suspicion of MG (50). It is also recommended to repeat the assay at 6–12 months if initially negative (as antibodies can be undetectable at onset and become positive afterwards) (50). In Ho Chan et al., 15.2% of initially seronegative AChR MG patients became seropositive at 12 months (51). In double seronegative MG patients, clear recommendations are lacking. It is generally suggested to test for low affinity anti-AChR and/or anti-MuSK antibodies by CBA (if available), then for anti-LRP4 antibodies (50).
Testing for anti-MuSK antibodies need also to be considered in patients with bulbar involvement, suspect of ALS. According to Huijbers et al., anti-LRP4-ab testing in ALS patients may be considered in patients with prominent bulbar weakness, prolonged disease course, minor fluctuations and absence of upper motor neuron involvement (49).
Lambert-Eaton myasthenic syndrome
Lambert-Eaton myasthenic syndrome (LEMS) is the prototype of pre-synaptic neuromuscular junction disorder. The disease results from the production of antibodies directed against voltage-gated calcium channels (VGCC), and is paraneoplastic in 50–60% of cases, mainly associated with small-cell lung cancer (SCLC) (52). Patient often present with a triad of lower extremity weakness, areflexia and autonomic dysfunction.
P/Q-type VGCC antibodies
P/Q-type (Cav2.1) anti-VGCC-ab are present in 85–90% of patients with LEMS, and nearly 100% of paraneoplastic LEMS, associated with SCLC (53, 54). VGCC are expressed at the presynaptic cleft in the neuromuscular junction, and are also expressed by SCLC, suggesting that autoimmunization by the tumor is the cause of paraneoplastic LEMS (55). The subtype of VGCC antibodies is unknown, probably IgG1 and IgG3, capable of activating the complement pathway. The preferred detection method is RIA, with a probably good specificity. Although initially considered to be very specific to LEMS, it is not the case and P/Q-type VGCC-ab may be encountered in other neuroinflammatory diseases, such as paraneoplastic cerebellar degeneration (54), autoimmune encephalitis, myelopathies or inflammatory neuropathies (56). In one study, P/Q-type and N-type anti-VGCC antibodies were found in 1.7% of healthy controls and 4% of neurologically asymptomatic patients with SCLC (57). A high titer of P/Q-type anti-VGCC-ab (>1 nmoL/L) seems to be strongly correlated with an autoimmune neurological diagnosis, while low titers (0.03–0.09 nmoL/L) can be seen in other neurological disorders (56). The positivity of these antibodies must therefore be correlated with clinical and electrophysiological data. There seems to be no correlation between antibody titers and clinical severity.
N-type VGCC antibodies
N-type VGCC antibodies have also been reported in 33–49% of LEMS patients, usually in association with P/Q-type VGCC antibodies (54). Similarly to P/Q-type VGCC antibodies, those antibodies are more frequent in paraneoplastic LEMS. However, according to a recent study, the additional assay of N-type to P/Q type anti-VGCC may not significantly improve diagnostic performance (58). Indeed, among 93 patients with LEMS, 25 (26.9%) were positive for both anti-VGCC-ab type, 67 (72%) were positive for P/Q-type anti-VGCC-ab only and only one (1.1%) was positive for N-type anti-VGCC-ab only.
SOX1 antibodies
SOX protein are immunogenic antigens expressed in SCLC (59). They belong to the Sry-like high mobility group superfamily of developmental transcription factors and may be important for neurogenesis (60). Antibodies against SOX proteins have been identified in SCLC patients. Anti-SOX1-ab are found in 64–67% of patients with LEMS and SCLC (60–62). These antibodies are specifically associated with the presence of a SCLC and are absent in almost all non-paraneoplastic LEMS (specificity 95–100%) (60–62). In Sabater et al., none of the 50 idiopathic LEMS had anti-SOX1-ab (61). However, anti-SOX1-ab are not specific to LEMS, and may be found in other paraneoplastic neurological syndromes, or in SCLC alone; anti-SOX1-ab are therefore a marker of paraneoplastic neurological syndromes (61, 63). In a patient with LEMS, the detection of anti-SOX1-ab should therefore imply an aggressive search for SCLC. Anti-SOX1-ab are generally part of onconeural antibodies panels, and detection is made by immunoblot, confirmed by indirect immunofluorescence on tissue (tissue-based assay or TBA). However, TBA may have a lower sensitivity to detect anti-SOX1-ab compared to other onconeural antibodies, and confirmation with CBA of HEK cells expressing SOX1 is probably the gold standard (64). Of note however, anti-SOX1-ab CBA is only available in specialized centers.
Any clinical suspicion of LEMS should therefore be confirmed with a dedicated electrophysiological assessment and testing for P/Q-type anti-VGCC-ab. The usefulness of anti-SOX1-abtesting in a patient with confirmed LEMS has yet to be determined, but could imply a more aggressive search for cancer, particularly SCLC.
Characteristic of antibodies associated with neuromuscular junction disorders is summarized in Table 1.
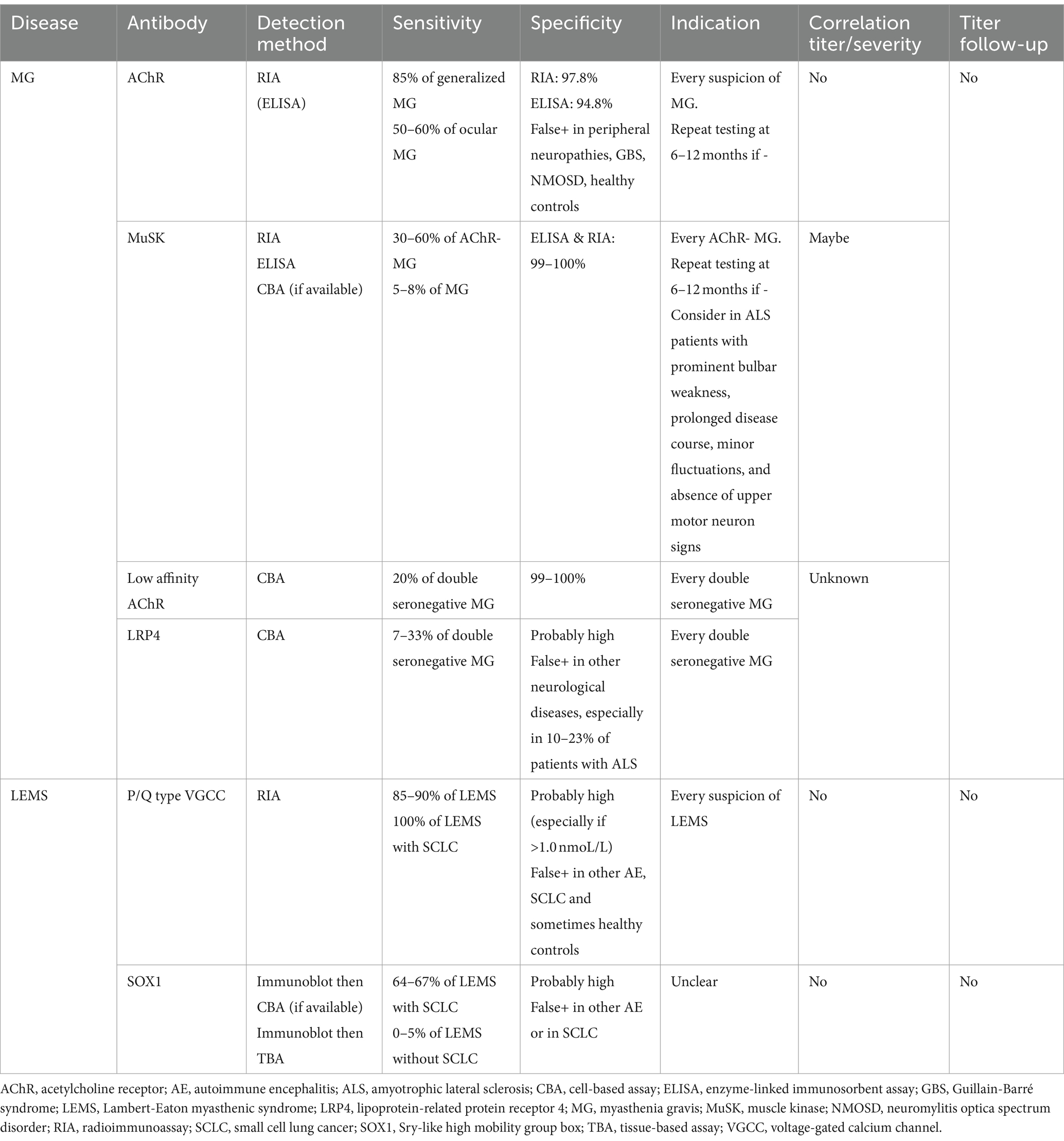
Table 1. Characteristics of antibodies associated with neuromuscular junction disorders.
Autoimmune peripheral neuropathies
Acute-onset autoimmune neuropathies: Guillain-Barré syndrome and variants
Guillain-Barré syndrome (GBS) is an acute-onset and monophasic inflammatory polyradiculoneuropathy, which includes different clinical variants et electrophysiologic patterns. The most frequent clinical phenotype is characterized by a proximo-distal tetraparesis, areflexia, and sensory involvement. It is characterized by an autoimmune attack on myelin, nodal/paranodal or axonal structures of the peripheral nerves and is usually post-infectious. The neural antigens targeted in this autoimmune process, particularly in “axonal” forms of GBS, are gangliosides, proteins expressed on axonal membranes, Schwann cells and myelin, where they participate in cell signaling and cell-to-cell communication (65). The main gangliosides are GM1, GD1a, Gd1b, GT1a and Gq1b. GM1 antigen is expressed mainly in the axolemma of nodes of Ranvier, the myelin of motor neurons and the dorsal ganglion (66), GD1a in the nodes of Ranvier of motor neurons (67), GT1a in the vagus and glossopharyngeal nerves (68), GQ1b in oculomotor nerves, neuromuscular spindles and possibly brainstem reticular matter (69) and GalNAc-GD1a in the axonal membrane of motor neurons and the axolemma of the sural nerve (70). Antibodies against gangliosides are detected in more than 50% of GBS patients (71). As each ganglioside has a different distribution in the peripheral nervous system, each antibody can be associated with a specific clinical phenotype.
Acute inflammatory demyelinating polyradiculoneuropathy
In acute inflammatory demyelinating polyradiculoneuropathy (AIDP), the classic form of GBS, there is no clear association with antiganglioside-ab, probably reflecting a rather cellular immune mechanism. It is therefore not recommended to test these antibodies in this subtype of GBS. Some studies have found anti-galactocerebroside (Gal-C), LM1 or GD1b antibodies in AIDP patients, but this association is unclear (72–74). A certain proportion of sera from AIDP patients binds to nodal and paranodal structure of the peripheral nerves, suggesting that yet unidentified antibodies may also play a role (75).
Acute motor axonal neuropathy
“Axonal” forms of GBS (in reality, nodopathies) classically includes acute axonal motor neuropathy (AMAN) and acute axonal motor and sensory neuropathy (AMSAN). AMAN is frequently associated with anti-GM1 and anti-GD1a IgG antibodies. In a recent Japanese-Italian collaborative study, 83% of patients with AMAN had anti-GM1, GD1a, GalNAc-GD1a or GM1b IgG (76). Pathogenesis of those antibodies, particularly anti-GM1 and anti-GD1a IgG, are the prototypic example of molecular mimicry (77). Almost half of patients with AMAN have a prior documented C. jejuni infection (78). C. jejuni contains in its lipo-oligosaccharides Gal(β1-3) GalNac, an epitope also present in GM1 and GD1a gangliosides; exposition to C. jejuni triggers an immune reaction with IgG1 and IgG3 production directed against gangliosides and causing the disease (79).
Acute ataxic neuropathies
Acute ataxic neuropathies include Miller-Fisher syndrome (MFS), Bickerstaff brainstem encephalitis (BBE) and acute sensory ataxic neuropathy (ASAN).
MFS is a variant of GBS characterized by ophtalmoparesis, ataxia and lower limb areflexia. Anti-GQ1b IgG antibodies were first described in 1992 in MFS patients (80). Few other studies confirmed the presence of anti-GQ1b in MFS, with a complete absence in normal and disease control groups, suggesting a high specificity (81). Anti-GQ1b-ab were later also associated with BBE (an overlap with MFS which includes a central nervous system involvement with disturbance of consciousness and corticospinal signs), acute ophthalmoparesis, or overlap syndromes between GBS and MFS/BBE (81). A comparative study found these antibodies in 83% patients with MFS and 68% patients with BBE (82). As for AMAN and AMSAN, cross-reactivity of anti-GQ1b IgG antibody with surface epitope on C. jejuni strains supports the hypothesis of molecular mimicry mechanism (83). Anti-GQ1b-ab cross-react with the structurally similar ganglioside GT1a.
ASAN is characterized by a sensory neuropathy with profound sensory ataxia and is often considered an incomplete form of MFS. It has been associated with anti-GD1b and, less frequently, anti-GQ1b IgG (84).
Pharyngeal-cervical-brachial variant
The pharyngeal-cervical-brachial (PCB) variant of GBS consists of an acute-onset bulbar paralysis and cervicobrachial weakness associated with upper limb areflexia. It is associated with anti-GT1a IgG in 50% of cases, some of these antibodies being able to cross-react with anti-GQ1b (85). Cross-reaction frequently occurs between GT1a and Gq1b-ab.
More recently, cases of severe GBS, with rapid tetraplegia, cranial nerve involvement, autonomic dysfunction and resistance to conventional treatments, have been associated with IgG1 antibodies to both neurofascin isoforms, known as anti-pan-neurofascin (86). Such antibodies, specific to the nodal regions, make these severe phenotypes of GBS enter the spectrum of autoimmune nodopathies, described in detail in the corresponding chapter.
Chronic sensory-ataxic autoimmune neuropathies
Anti-MAG neuropathy
Anti-MAG neuropathy is a paraproteinemic neuropathy, characterized by a distal predominant demyelinating polyneuropathy mainly affecting large sensory nerve fibers, a phenotype previously known as DADS (distal acquired demyelinating symmetric neuropathy), and is associated with an IgM monoclonal gammopathy. MAG is a transmembrane glycoprotein localized in the myelin of Schwann cells and oligodendrocytes and plays an important role in myelin formation and axon-myelin interaction (87). Up to 50–65% of patients with a “DADS phenotype” neuropathy and IgM monoclonal gammopathy have IgM antibodies with anti-MAG activity (88). Historically, these antibodies were detected by western blot, but this technique has now been replaced by ELISA, with an improved sensitivity (89). The main issue with ELISA technique is a low specificity in case of low antibody titers. Indeed, using the manufacturer’s recommended cut-off of 1,000 Bühlmann titer unit (BTU), specificity is imperfect, with false positives in cases of chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) for example (89). In a recent study, cut-offs of >1,000 BTU, >1,500 BUT and > 7,000 BTU were associated with a sensitivity of 100, 100 and 92.5% respectively, and a specificity of 90.99, 95.5 and 100%, respectively, to diagnose anti-MAG neuropathy (90). There is no clear correlation between antibody titer and disease severity. However, there seems to be a relationship between antibody titer and clinical response to treatment in responders; a > 50% titer reduction compared with its pre-treatment value might be a good indicator of therapeutic response (91). In patients with a clear electro-clinical diagnosis, anti-MAG titers can therefore be monitored, targeting a > 50% reduction in antibody levels (92).
A new ELISA technique, specifically targeting the HNK1 epitope of the MAG protein, has recently been developed, with a diagnostic sensitivity of 98% and specificity of 99% (93). With this technique, anti-HNK1 MAG antibody titers appear to correlate with the severity of neuropathy (93).
Anti-MAG IgM antibodies should be tested in any patients with a predominantly distal demyelinating neuropathy (DADS phenotype) and in the presence of IgM monoclonal gammopathy. According to EAN/PNS 2021 recommendations for CIDP, anti-MAG-ab should also be tested in all patients fulfilling CIDP diagnostic criteria and in presence of IgM monoclonal gammopathy (90, 94). The indication for anti-HNK1 MAG antibodies screening has yet to be clarified.
Chronic ataxic neuropathy with disialosyl antibodies
Chronic ataxic neuropathy with disialosyl antibodies (CANDA) is a rare syndrome characterized by a chronic sensory and ataxic, usually demyelinating neuropathy, occurring in the presence of an IgM monoclonal gammopathy reacting against gangliosides containing disialosyl epitopes (95). Some patients also present ophtalmoparesia, hence the term CANOMAD (chronic ataxic neuropathy with ophtalmoplegia, M-protein, cold agglutinins and disialosyl antibodies) (96). In CANDA/CANOMAD, IgM antibodies are directed against gangliosides harboring disialosyl groups, containing the sequence NeuNAc (α2-8)NeuNAc (α2-3)Gal, i.e., GD2, GD3, GD1b, GT1b, GT1a et GQ1b. The most frequently described antibodies are GD1b, GD3, GT1b and GQ1b, and most patients have antibodies reacting with multiple gangliosides (96–98). In the largest cohort of patients with CANDA/CANOMAD, 78% of cases had anti-GD1b autoantibodies, while other anti-disialosyl antibodies were each observed in less than 51% of patients (99). Those gangliosides are present in the neurons of the dorsal root ganglia and in oculomotor nerves, which explains the clinical symptoms.
Chronic motor autoimmune neuropathies: multifocal motor neuropathy
Multifocal motor neuropathy (MMN) is an inflammatory neuropathy with a purely motor, slowly progressive and asymmetric involvement, generally affecting the distal limbs. One of its electrophysiological features is the presence of motor conduction blocks, distinguishing it from motor neuron disease, with which it shares many clinical features (100). The disease can be associated with the presence of specific IgM antibodies against the GM1 ganglioside (101). GM1 antigen is mainly expressed in the axolemma of the nodes of Ranvier, the myelin of motor neurons and, to a lesser extent, the dorsal root ganglion. It is unclear why, in MMN, the injury is restricted to motor nerves, even though GM1 is present is motor and sensory nerves. These antibodies appear to be pathogenic, firstly by inducing direct functional alteration of paranodal regions, and secondly indirectly by activating the complement cascade (102).
Anti-GM1 IgM are detected in around 50% (20–85%) of patients with MMN (102–104). High levels of antibodies seem to be relatively specific for MMN, while low levels however can be found in other inflammatory neuropathies, motor neuron disease or even in healthy controls (104). Detection of high titers of IgM anti-GM1-ab has very useful diagnostic implications, mainly in the differential diagnosis with other lower motor neuron syndromes, including amyotrophic lateral sclerosis, when motor conduction blocks are not present. However, gangliosides antibodies, including anti-GM1 IgM, have also rarely been described in amyotrophic lateral sclerosis, usually at low titers (105). There seems to be no correlations between antiganglioside levels and disease severity, nor necessity in monitoring antibody levels.
Chronic inflammatory demyelinating polyradiculoneuropathy
CIDP is the chronic form of GBS, whose pathophysiology is less well understood, and may involve both humoral and cellular immunities. The presence of immunoglobulin and complement deposition in sural nerve biopsies from CIDP patients and the response to plasma exchanges and IVIg support the role of autoantibodies in CIDP pathogenesis (106). To date, no specific antibodies have been detected in patients with CIDP, apart from those directed against the nodal and paranodal regions of the nodes of Ranvier. However, patients carrying these antibodies are no longer classified as suffering from CIDP, but from autoimmune nodopathies (94).
Chronic axonal polyradiculoneuropathies
Several neural antibodies can cause a chronic axonal polyradiculoneuropathy, including antibodies directed to collapsin response mediator protein-5 (CRMP5 [anti-CV2]), antineuronal nuclear antibody type 1 (ANNA-1 [anti-Hu]), amphiphysin, Purkinje cell cytoplasmic antibody type 2 (PCA-2 [microtubule-associated protein 1B (MAP1B)]) and adaptor-related protein complex 3, beta 2 subunit (AP3B2) (107). CRMP5 and amphiphysin IgG usually cause an asymmetric and painful polyradiculoneuropathy, usually axonal or mixed axonal-demyelinating, which may mimic CIDP (108, 109). A coexisting myelopathy (myeloneuropathy) has also been reported in some patients with CRMP-5 [anti-CV-2], amphiphysin, ANNA-1 [anti-Hu] and AP3B2 IgG (107). CRMP5 has been shown to be involved in axon-Schwann cell interaction, and is expressed in immature Schwann cells as well as in unmyelinated Schwann cells (Remak cells) (110). Little is known about the expression and localization of other neural antigens in the peripheral nervous system.
Testing for those antibodies can be considered in suspected CIDP patients with unexpected poor response to treatment, severe and rapid evolution, prominent axonal loss and association with other neurological signs (110). Most of these antibodies (ANNA-1 [anti-Hu], CRMP-5 [anti-CV-2], amphiphysin and PCA-2) are strongly associated with cancer (111). Testing those antibodies must be performed simultaneously in the serum and in the CSF to improve specificity (112).
Antiganglioside antibodies testing: limitations and indications
Antigangliosides-ab are generally tested in a panel, containing the main gangliosides (GM1, GM2, GD1a, GD1b, GQ1b, GT1a, GT1b). The detection of antiganglioside-ab is difficult for several reasons: gangliosides are difficult to isolate based on their molecular weight or charge, making detection by western-blot complicated. Moreover, there is no reliable CBA for their expression. Finally, several gangliosides share the same backbone, leading to frequent cross-reactions. The gold standard technique for antigangliosides-ab detection is probably thin-layer chromatography, a technique not routinely available and requiring some expertise. Routinely detection is performed using ELISA commercial kits. Recommendations were published on how to best perform this ELISA (113). Immunodots or immunoblots are also used in certain settings, but their diagnostic performance is probably poorer (114).
According to the latest EAN/PNS 2023 guidelines, testing antigangliosides-ab is not recommended in most patient with typical sensory-motor GBS, because of their moderate diagnostic sensitivity and delay in result obtention (115). However, testing for those antibodies can be useful to confirm diagnostic suspicion in variant and atypical cases, and to rule out other disorders. For example, in pharyngeal-cervical-brachial variant, testing for anti-gangliosides-ab may be particularly useful in partial forms of the disease (e.g., acute bulbar palsy), in which a positive anti-GT1a may clarify the diagnosis. As nerve conduction studies cannot reliably distinguish between demyelinating and axonal form of GBS in the early phase of the disease, testing for antigangliosides-ab may help to make this distinction. Anti-GQ1b-ab testing is strongly recommended in MFS spectrum patients, because of their high sensitivity and specificity, and is integrated in the latest guidelines (115).
In the case of a chronic motor demyelinating neuropathy with conduction blocks on nerve conduction studies (suspicion of MMN), testing of anti-GM1 IgM antibodies is also required (116). Lastly, antigangliosides-ab screening is also indicated in CANOMAD/CANDA suspicion, i.e., in case of chronic ataxic sensory neuropathy with IgM monoclonal gammopathy, as the differential diagnosis of chronic sensory ataxic neuropathies are broad, including toxic, metabolic, mitochondrial or vitamin deficiencies (95). Testing for antigangliosides-ab is not recommended in CIDP (94).
Characteristics of antibodies associated with autoimmune neuropathies are summarized in Table 2.
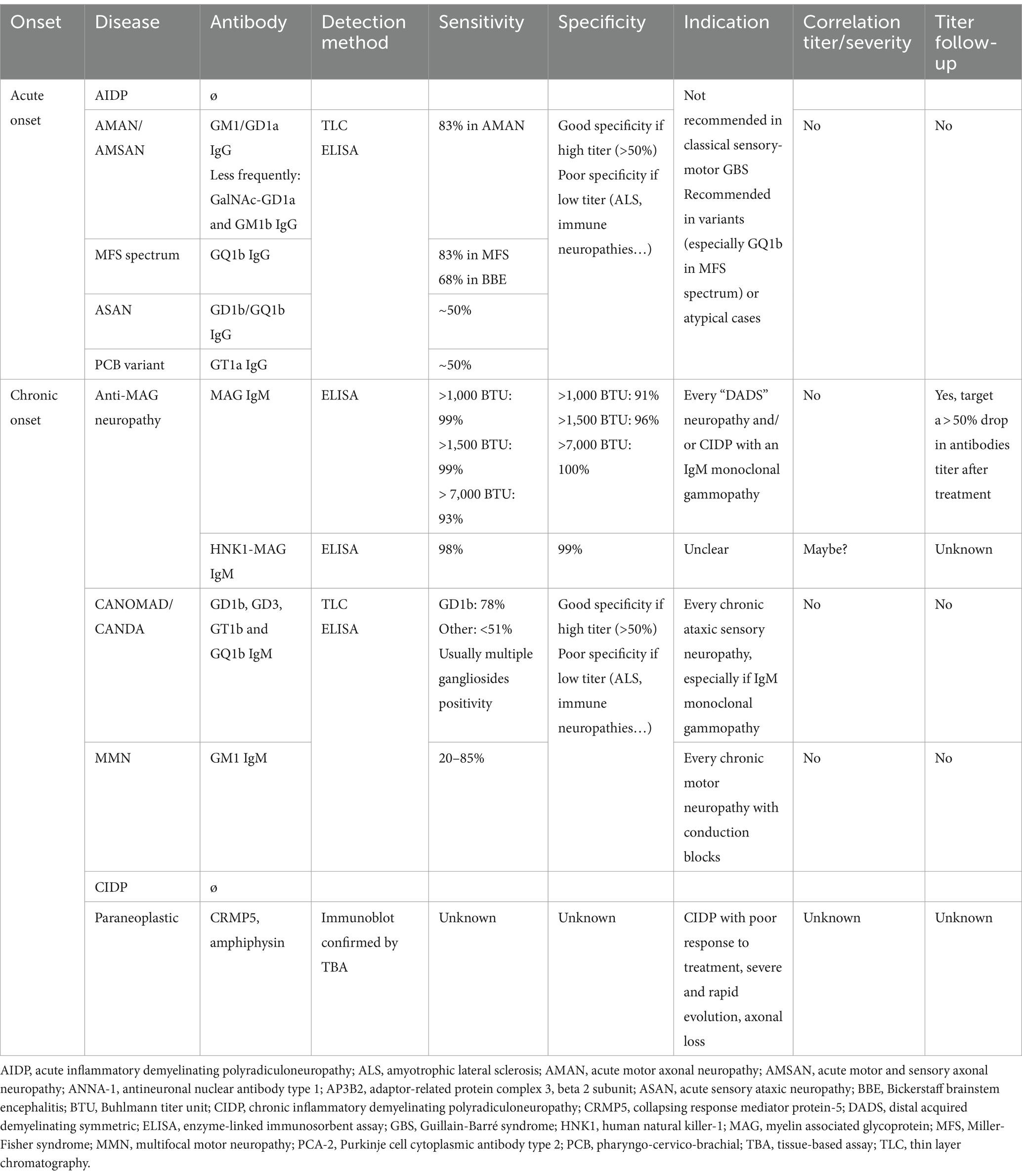
Table 2. Characteristics of antibodies associated with autoimmune neuropathies.
Autoimmune nodopathies
The recent discovery of antibodies directed against nodes of Ranvier led to the definition of a new group of immune-mediated neuropathies: autoimmune nodopathies (117). They usually manifest as GBS or acute CIDP, usually in an aggressive form. Autoimmune nodopathies are thought to account for 5–10% of patients fulfilling diagnostic criteria for CIDP, and response to IVIg is usually poor. Of note, according to the latest EAN/PNS recommendations, patients harboring those antibodies are not considered having CIDP anymore (94).
In these auto-immune neuropathies, antibodies target nodal and paranodal cell adhesion molecules; contactin 1 (CNTN1) (118), contactin-associated protein 1 (Caspr1) (119), neurofascin 155 (NF155) (120), neurofascin 140 (NF140) and 186 (NF186), also called pan-neurofascin when all isoforms of neurofascin are targeted (panNF) (121). In most cases, nodal/paranodal antibodies are predominantly of the IgG4 subclass, known for its non-inflammatory properties. This can explain why response to IVIg is poor, and response to plasmapheresis and B-cell-depleting therapy is better. More rarely, antibodies of the IgG3 subclass have been reported in acute-onset auto-immunes nodopathies, such as panNF neuropathy. IgG3 have a strong proinflammatory effect and activate the complement pathway. In this case, treatment with IVIg can be beneficial. Nerve biopsy samples from patients with autoimmune nodopathy show paranodal detachment, without evidence of overt inflammation or demyelination, confirming that this immune neuropathy is pathologically very different from CIDP (122). Each antibody is associated with a specific clinical phenotype.
Neurofascin 155
NF155 antibodies are the most common among autoimmune nodopathies. Patients with IgG4 antibodies against NF155 share a similar phenotype: younger age of onset (mean age 42.4 years), rapid progression, distal motor involvement, ataxia with cerebellar features and a prominent low-frequency tremor (123). This antibody has also been associated with central nervous system demyelination, in addition to peripheral nerve pathology; “combined central and peripheral demyelination” (124). Response to IVIg or steroids is usually poor, whereas most patients respond to Rituximab. Anti-NF155 titers may correlate with clinical severity within the same patient, and anti-NF155 titers decrease in all Rituximab-treated patients (123).
Contactin 1
Patients with IgG4 anti-CNTN 1 also share a similar phenotype: an aggressive and acute neuropathy mimicking GBS, old age at onset, axonal features in nerve conduction studies, poor response to IVIg and nephrotic syndrome (125, 126). Anti-CNTN1 IgG3 antibodies have also been detected during the early phase and may explain why some patients respond initially to IVIg (127).
Caspr 1
Antibodies targeting Caspr1 or the Caspr1/contactin 1 complex have been less frequently reported. Patients usually presented with acute-CIDP, severe pain, axonal involvement in nerve conduction studies, a poor response to IVIg and a good response to Rituximab (128). The main antibody subtype is IgG4, but IgG4 antibodies were also detected during the early phase in a few patients with GBS.
Neurofascin 186
Nodopathy with anti-NF186 has also been rarely reported. Patients typically presented with a severe acute or subacute CIDP phenotype, characterized by predominant distal motor and sensory impairment, along with mostly demyelinating features in nerve conduction studies, without axonal loss (129). In the initial description, none of the patients exhibited tremors (which were commonly seen in NF155 nodopathy), and concomitant glomerulonephritis or retroperitoneal fibrosis was also noted (121). The main antibodies subtypes are IgG3 and IgG4, and some patients may respond to IVIg (130). Patient with specific NF186 antibodies being rare, it is debatable whether these patients should be characterized as a “pan neurofascin” nodopathy, with predominant reactivity to NF186 (130).
Pan neurofascin
Some patients with autoimmune nodopathies harbor antibodies interacting with all neurofascin isoforms, NF140, NF155 and NF 186; they are described as panNF neuropathy (86, 131). Antibodies may be of the IgG1, IgG3 or IgG4 subtypes, and patients usually displayed an aggressive and fast clinical progression. These patients exhibit a fulminant disease course, with a severe sensorimotor tetraplegia, severe cranial nerve involvement, autonomic dysfunction, often requiring mechanical ventilation due to severe respiratory insufficiency. Most patient have a monophasic course and are diagnosed as an “explosive GBS.” Response to IVIg is usually poor, whereas most patients respond to Rituximab (86).
LGI4
IgG4 antibodies targeting leucine-rich repeat LGI family member 4 (LGI4) have been recently reported in 4 Japanese patients with a subacute sensory-motor CIDP phenotype (132). Those antibodies need further validation.
Detection of nodal/paranodal antibodies
Detection of nodal/paranodal-ab is of clinical relevance, as it can identify a specific subgroup of patients who share some clinical characteristics and response to treatment very different from CIDP patients. Indeed, response to IVIg treatment is usually poor whereas most patients respond well to Rituximab. Antibodies to nodal/paranodal antigens are detected in the serum. The screening and confirmation assays used mainly depend on the laboratory, but it is highly recommended to confirm the positivity of an antibody with a second assay, to ensure its specificity (93). According to EAN/PNS latest recommendations, CBA with plasmids encoding for human recombinant proteins, ELISA and teased-nerve immunohistochemistry are the recommended methods (94, 114). CBA and ELISA are good screening techniques, and teased-nerve immunohistochemistry is usually performed as a confirmatory test. Detection of nodal/paranodal antibodies need to be performed in expert centers.
According to the latest EAN/PNS 2021 recommendations, testing for antibodies to nodal/paranodal antigens are advised in CIDP patients with the following features (94):
• Resistance to standard therapy with IVIg and corticosteroids.
• Acute or subacute aggressive onset, previous diagnosis of GBS or acute CIDP.
• Low-frequency tremor, ataxia disproportionate to the sensory involvement or other cerebellar features or predominantly distal weakness.
• Respiratory failure and cranial nerve involvement.
• Associated nephrotic syndrome.
• Very high CSF protein levels.
Characteristics of nodal/paranodal antibodies are summarized in Table 3.
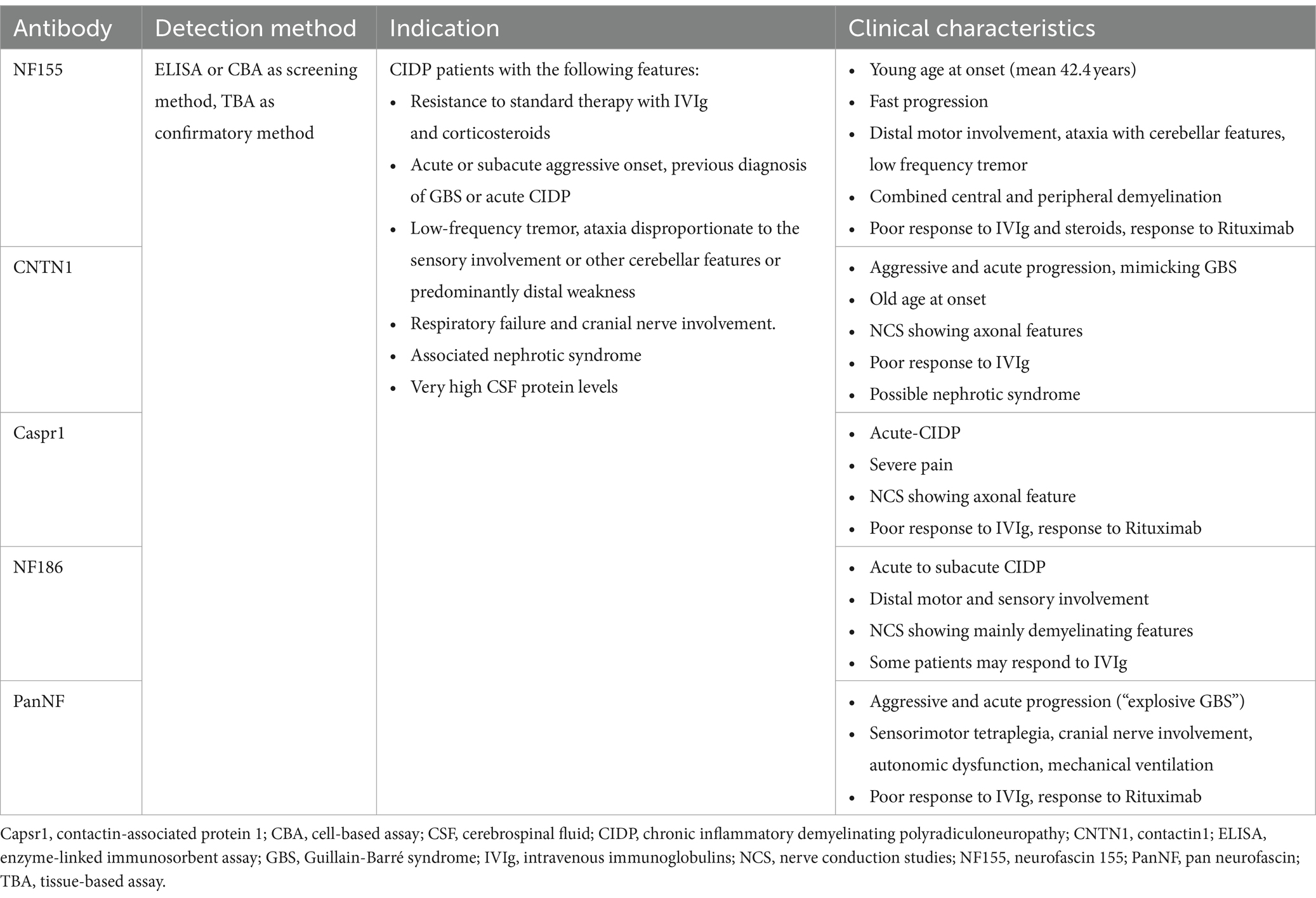
Table 3. Characteristics of antibodies associated with autoimmune nodopathies.
Autoimmune neuronopathies
Autoimmune autonomic ganglionopathy
Autoimmune autonomic ganglionopathy (AAG) is a severe immune mediated autonomic failure of acute-subacute onset, characterized by orthostatic hypotension, xerostomia, impaired pupil response, urinary retention, anhidrosis and gastrointestinal dysmotility. The disease is associated with the presence of ganglionic AChR type α3-ab, present in about 50% of patients (133, 134). Ganglionic AChR type α3 receptors are involved in synaptic transmission in autonomic ganglia. Those antibodies have pathogenic properties resulting in deterioration of synaptic transmission in sympathetic, parasympathetic, and enteric ganglia (134). High antibody titers >1.0 nmoL/L appear to be relatively specific for AAG (135), with a good correlation between antibody levels and the severity of autonomic involvement (133, 136, 137). Intermediate antibody levels are quite unspecific and may be found in cases of pure autonomic failure, postural orthostatic tachycardia syndrome (POTS) or gastrointestinal dysmotility (138). Low antibody levels (<0.2 nmoL/L) are non-specific and may be found in healthy controls (135). Three detection methods are available: RIA, Luciferase immunoprecipitation (LIP) and immunomodulation assay (139). RIA is the most widely established diagnostic method. A CBA method, which detects only potentially pathogenic antibodies, has recently been developed. In Karagiorgou et al., CBA was equally sensitive as the RIA, but was specific for AAG; all 15 patients with AAG and AChR type α3-ab were positive with CBA, and there was no false positive (140). This CBA assay should therefore replace RIA or LIP methods, but is still not yet widely available.
Autoimmune and paraneoplastic sensory neuronopathies
Sensory neuronopathies are characterized by degeneration of the dorsal root sensory ganglion, and manifests as a pure sensory non-length-dependent neuropathy. Acquired forms are due to a variety of dysimmune, toxic, paraneoplastic or idiopathic etiologies (141). Sjögren syndrome is the most frequent dysimmune cause of sensory neuronopathy, and sensory neuronopathy is the most frequent and typical neuropathy of Sjögren syndrome (142).
Anti-FGFR3 antibodies
In 2015, antibodies directed against the intracellular domain of fibroblast growth factor receptor 3 (FGFR3) have been described (143). In a multicenter prospective study, 15% of patients with a sensory neuropathy had FGFR3 antibodies; 2/3 of patients fulfilled the criteria for sensory neuronopathy, 17% for small-fiber neuropathy and 19% for other sensory neuropathies, the majority (89%) being non-length-dependent phenotypes (144). Patients with anti-FGFR3 sensory neuronopathy have usually a progressive disease-onset, symmetric involvement of 4 limbs, impairment of all sensory modalities with neuropathic pain, and a motor involvement in 25% of cases (144). IVIg are effective in 80% of cases (145). In adult rats, FGFR3 was expressed in small and large sensory neurons of the dorsal root ganglia, and activation of the receptor could play a role in regulation of many cell functions including survival (146). It is yet unknown whether those antibodies play a pathogenic role or are only biomarkers of a dysimmune process. However, FGFR3 antibodies recognize the intracellular domain of the protein, and should these antibodies be able to enter the neurons (which is yet unknown), they could interfere with signal transduction and dorsal root ganglia function. ELISA is used to detect those antibodies.
Anti-AGO1 antibodies
In 2021, antibodies to argonaute proteins 1 and 2 (AGO1 and 2) have been identified in immune diseases of the central and peripheral nervous systems, in particular sensory neuronopathies and limbic encephalitis (147). These antibodies are not specific to neuronopathies. The presence of these antibodies was strongly associated with an autoimmune context, particularly Sjögren’s syndrome. In a recent retrospective study, 13% of patients with sensory neuronopathy had anti-AGO1 antibody, which was associated with a more severe phenotype, but a better response to immunotherapy with IVIg (148). AGO are involved in degradation of RNA. The site of tissue expression of AGO proteins is not well known; it is unclear whether AGO antibodies have a pathogenic role directly in the dorsal root ganglia, or whether they are only a biomarker of dysimmune process (147, 148). AGO antibodies are detected by ELISA.
Paraneoplastic forms
Sensory neuronopathies are among the most common paraneoplastic neurological syndromes and have been associated with ANNA-1 [anti-Hu], CRMP-5 [anti-CV2], PCA-2 [MAP1B] and amphiphysin (108). Some paraneoplastic sensory neuronopathies are seronegative, especially those associated with hematologic malignancies (149). The most frequent paraneoplastic form of sensory neuronopathy is Denny-Brown syndrome, which is associated with ANNA-1 [anti-Hu] antibodies (150, 151). Anti-Hu paraneoplastic syndromes can manifest with various clinical syndromes. In a series of 27 patients with anti-Hu syndromes, 20 (74%) had a clinical neuropathy (most frequently a pure sensory neuropathy) (152). Anti-Hu recognizes a family of RNA-binding proteins expressed in the nuclei of neurons and SCLC cells; in neurons, they have a role in the development and maintenance of the neuronal phenotype (153). There is no evidence that the anti-Hu antibodies are pathogenic, they may rather be a marker of dysimmune reaction. Anti-Hu antibodies are usually identified in patients’ sera but can also be identified in the CSF. In patients with peripheral neuropathy, CSF anti-Hu titers are probably lower compared to patients with encephalomyelitis (154). The identification of anti-Hu antibodies strongly predicts an underlying cancer, most commonly a SCLC, with an estimated sensitivity of 82% and specificity of 99% (155). There is probably no correlation between Hu antibodies titers and neurological outcome; there is therefore no indication for monitoring antibody levels (156).
Autoimmune and paraneoplastic motor neuronopathies
Motor neuronopathies (or motor neuron diseases) manifest as a pure motor, symmetric or asymmetric, neuropathy. They are rarely of autoimmune origin and are frequently neurodegenerative (as in ALS) or, more rarely, of an infectious origin (as in poliomyelitis).
IgLON5
Ig-like cell adhesion molecule 5 (IgLON5) antibodies have been first described in 2014 in patients presenting with sleep apnea, REM-sleep or non-REM-sleep disorders (157). In 2021, 5 patients with a bulbar onset motor neuronopathy were found having serum IgLON5 antibodies, 4/5 patients having a coexisting sleep disorder (158). In another series, all patients had a coexisting neurologic finding, especially a vocal cord paresis, chorea or REM sleep behavior disorder (159). Most patients have IgLON5 antibodies in the serum, and a few also in the CSF. Anti-IgLON5 antibody testing is performed using a CBA (157). An early immunotherapy, prior to advanced neurodegeneration, seems to be efficient and associated with a better long-term clinical outcome (160). No paraneoplastic forms have yet been described.
Other antibodies
Paraneoplastic motor neuronopathies have also been rarely described in association with anti-Ma2 and ANNA-2 [anti-Ri] antibodies (161, 162). More recently, leucine zipper protein 4 (LUZP4) antibodies have been described in patients with neurological paraneoplastic syndromes, presenting as rhombencephalitis, limbic encephalitis, seizures and/or motor neuronopathy (163). Ma2 and ANNA-2 antibodies are included in screening panels for onconeuronal antibodies, detected usually by immunoblot, confirmed by TBA. LUZP4 antibodies testing is not routinely available.
Autoimmune small fiber neuropathies
Small fiber neuropathy (SFN) is characterized by isolated involvement of A δ and C small nerve fibers, causing neuropathic pain, reduced sensation to pain and temperature, and dysautonomia. Similarly to sensory neuropathies and neuronopathies, SFN can be caused by a variety of metabolic, toxic, infectious, inherited and dysimmune etiologies, and cryptogenic forms are frequent (164). Sjögren syndrome, less frequently systemic lupus erythematosus and sarcoidosis, may be responsible for autoimmune SFN (164). Paraneoplastic neurological syndromes with ANNA-1 [anti-Hu] and CRMP5 [anti-CV2] antibodies can also cause a small fiber involvement, but rarely isolated (109, 110). According to de Greef et al, up to 19% of patients with cryptogenic SFN may have underlying immunological abnormalities, suggesting an immune component (165). Diagnosing autoimmune SFN is important since immunotherapy may be beneficial in those patients.
Anti-FGFR3 and anti-TS-HDS antibodies
As mentioned previously, anti-FGFR3-ab have been described in patients with sensory neuronopathies, 17% of them fulfilling a SFN diagnosis (144). In a recent retrospective study, 17% of patients with SFN and dysautonomia had anti-FGFR3 antibodies (166). Those antibodies are not specific to SFN and are usually rather found in neuronopathies.
Trisulfated heparan disaccharide (TS-HDS) is a disaccharide component of the glycosylation moieties of heparan and heparan sulfate and is expressed in peripheral nerves surface. IgM antibodies binding to TS-HDS have been first described in patients with a painful, predominantly sensory neuropathy (167). In a recent retrospective cohort of cryptogenic SFN, 37% of patients had IgM anti-TS-HDS-ab, and 15% had IgG anti-FGFR3; the presence of anti-TS-HDS defined a subset of female patients with acute-onset, and non-length dependent SFN (168). In another study, 28% of patients with SFN and dysautonomia had anti-TS-HDS-ab; most of these patients presented with dysautonomia, but 30% of them had a normal intraepidermal nerve density in skin biopsy (166). The specificity of anti-TS-HDS antibodies for autoimmune SFN is still questioned. In a recent real-world study, among 77 patients with TS-HDS antibodies, 34% did not have evidence of neuropathy and 12% had another known cause of neuropathy (169).
Data about response to immunotherapy in TS-HDS/FGFR3+ SFN are conflicting. In two studies, most patients with those antibodies improved with immunotherapy, either IVIg or plasma exchange, with a significant reduction in pain scores (170, 171). In Chompoopong P et al. however, response to immunotherapy was observed only in 31% of TS-HDS positive patients and was not higher than in TS-HDS negative patients (169). More recently, a double-blind placebo-controlled pilot study did not show any benefit of IVIg treatment in SFN patients with anti-TS-HDS and/or FGFR-3 antibodies (172). In conclusion, anti-FGFR3/TS-HDS antibodies may not be specific to SFN, and their pathogenicity in neuropathies has yet to be proven in animal or cell culture models. Nevertheless, detection of such antibodies can give a clue about a potential autoimmune etiology/context and may prompt immunotherapy, even though response to immunotherapy is uncertain. Those antibodies can be detected with ELISA (144, 169).
Anti-plexin D1 antibodies
Anti-plexin D1 IgG are antibodies binding selectively to mouse unmyelinated C-fiber neuron in the dorsal root ganglia. Those antibodies have been initially described in a fraction of patients with neuropathic pain and neuroinflammatory diseases, and in patients with idiopathic trigeminal neuropathy (173, 174). Recently, plexin-D1 IgG were found in 12.7% (8/63) of patients with probable SFN and in 0% of healthy controls (175). Those antibodies seem to be pathogenic, according to their mechanism of action, and may be more specific to SFN than FGFR3 and TS-HDS antibodies. Response to immunotherapy of anti-plexin D1+ patients is currently unknown. Those antibodies can be detected with ELISA (175).
Other antibodies
Other novel antibodies have been recently described in idiopathic SFN; anti-MX1, anti-DBNL and anti-KRT8 (176). MX1 is an interferon-induced GTP-binding protein, DBNL (drebrin-like protein) is an adapter protein playing a role in endocytosis and synapse formations, and KRT8 (keratin type II cytoskeletal 8) is a contractile apparatus to dystrophin (176). Those antibodies need to be further validated and their pathogenicity is currently not known.
Peripheral hyperexcitability syndromes
Peripheral hyperexcitability syndromes can concern the nerve or rarely the muscle.
Peripheral nerve hyperexcitability syndromes
Peripheral nerve hyperexcitability syndromes (PNHS) are a group of rare pathologies characterized by spontaneous and continuous muscle activity, including muscle spasm, stiffness and pain. Primary forms include Isaacs syndrome, Morvan syndrome and cramp fasciculation syndrome. Cramp fasciculation syndrome is the least severe form of the spectrum, and is characterized by episodic cramping, fasciculations, stiffness and muscle pain. Morvan and Isaacs syndromes are characterized by relatively specific neuromyotonic discharges in needle myographies, but also cramps and myokymia. Morvan’s syndrome is also commonly associated with encephalopathy and dysautonomia.
PNHS are usually associated with anti-CASPR2-ab, more rarely LGI1-ab; these antibodies were historically described as targeting the voltage-gated potassium channels (VGKC) complex (177). They can be detected in the serum and the cerebrospinal fluid (CSF), but in case of peripheral nervous system presentation, sensitivity appears to be better in the serum. CASPR2/LGI1 antibodies are described in 0–24% of cramp fasciculation syndromes, 20–30% of Isaacs syndromes and 60–75% of Morvan syndromes (178). However, they are not specific for PNHS, and can be found in limbic encephalitis, movement disorders, epileptic seizures, etc. (179). CASPR2 is a cell-surface adhesion molecule, a critical component of the VGKC complex, present in myelinated axons (180). In mice models, antibodies against CASPR2 were shown to decrease expression of VGKC in dorsal root ganglion neurons and juxtaparanodes, causing pain-related hypersensitivity (180). Another animal model showed that CASPR2 antibodies can also alter CASPR2 protein function in the hippocamp, explaining central nervous system involvement (181). LGI1 is a neuronally secreted molecule, which has been proven to have an essential role in central nervous system neuronal hyperexcitability, through action on VGKC, and in synaptic transmission through AMPA receptors (182). It has been shown that LGI1 is also highly expressed in dorsal root ganglia and spinal cord dorsal horn neurons in mice and human (183). A considerable proportion of patient with Isaac and Morvan syndrome, especially the seropositive case for CASPR2 antibodies, have underlying thymoma (181). These antibodies are included in screening panels for antineural antibodies and are generally detected by CBA, then confirmed by TBA.
Immune-mediated rippling muscle disease
Rippling muscle disease is a generally benign, myotonic-like myopathy associated with rapid rolling contractions and percussion-induced contractions, caused by a muscle hyperexcitability. One of the main characteristics is that those visible contractions are electrically silent during an EMG recording (184). Genetic forms have been reported, caused by a pathogenic variant in caveolin-3 (CAV3) or less frequently cavin-1 (CAVIN1) genes (185). In 2022, immune-mediated forms of rippling muscle disease have been described, associated with cavin-4 IgG in 80% of the cases, most of them responding to immunotherapy (186). More recently, a paraneoplastic immune-mediating rippling muscle disease has been described, associated with thymoma (187).
Characteristics of antibodies associated with neuronopathies, small fiber neuropathies, motor neuron diseases and peripheral hyperexcitability syndromes are summarized in Table 4. The location, characteristics, and mechanism of action of the main antibodies and their antigens involved in neuromuscular junction and peripheral nerve disorders are summarized in Figure 1.
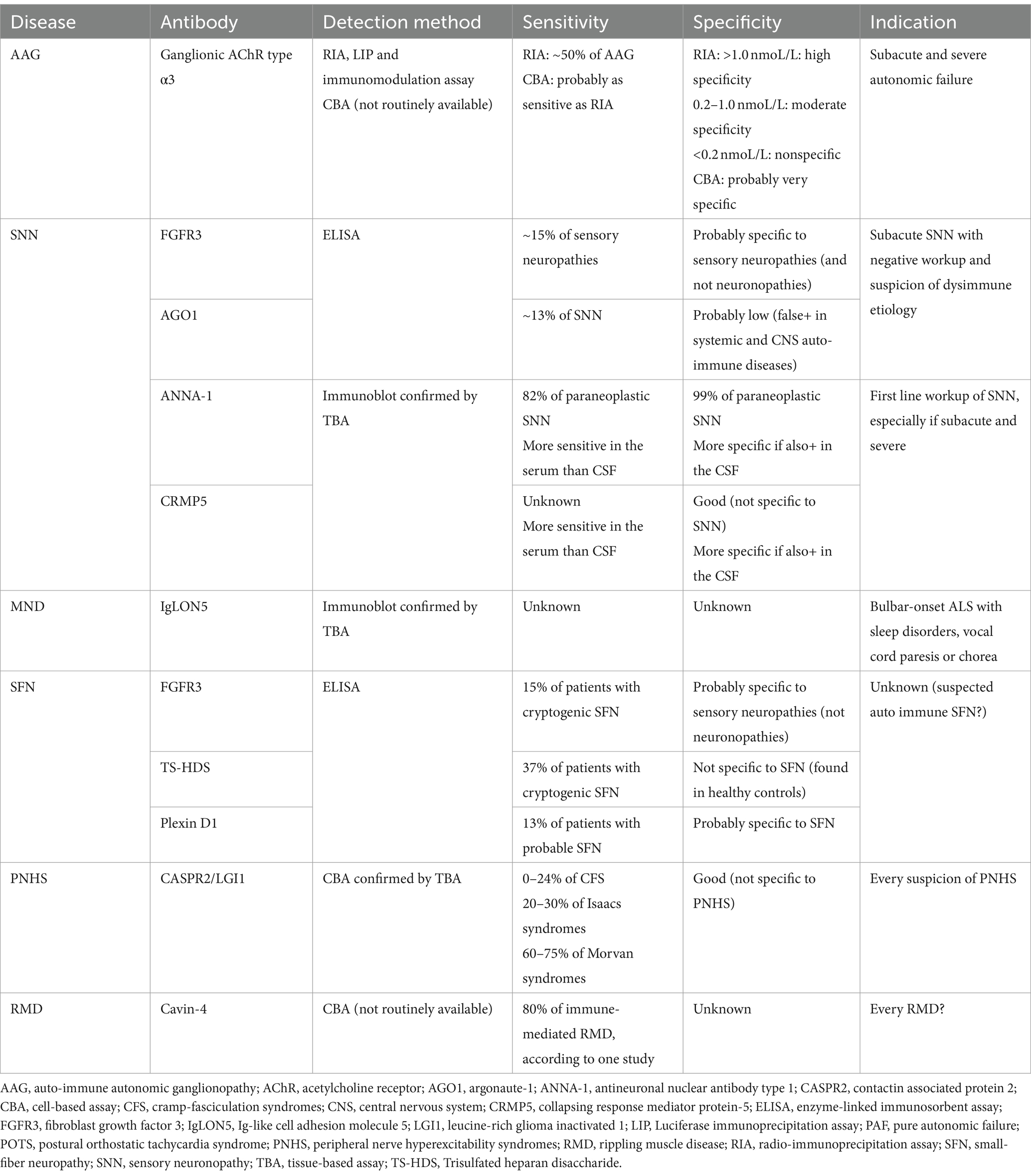
Table 4. Characteristics of antibodies associated with neuronopathies, small fiber neuropathies, motor neuron diseases, and peripheral hyperexcitability syndromes.
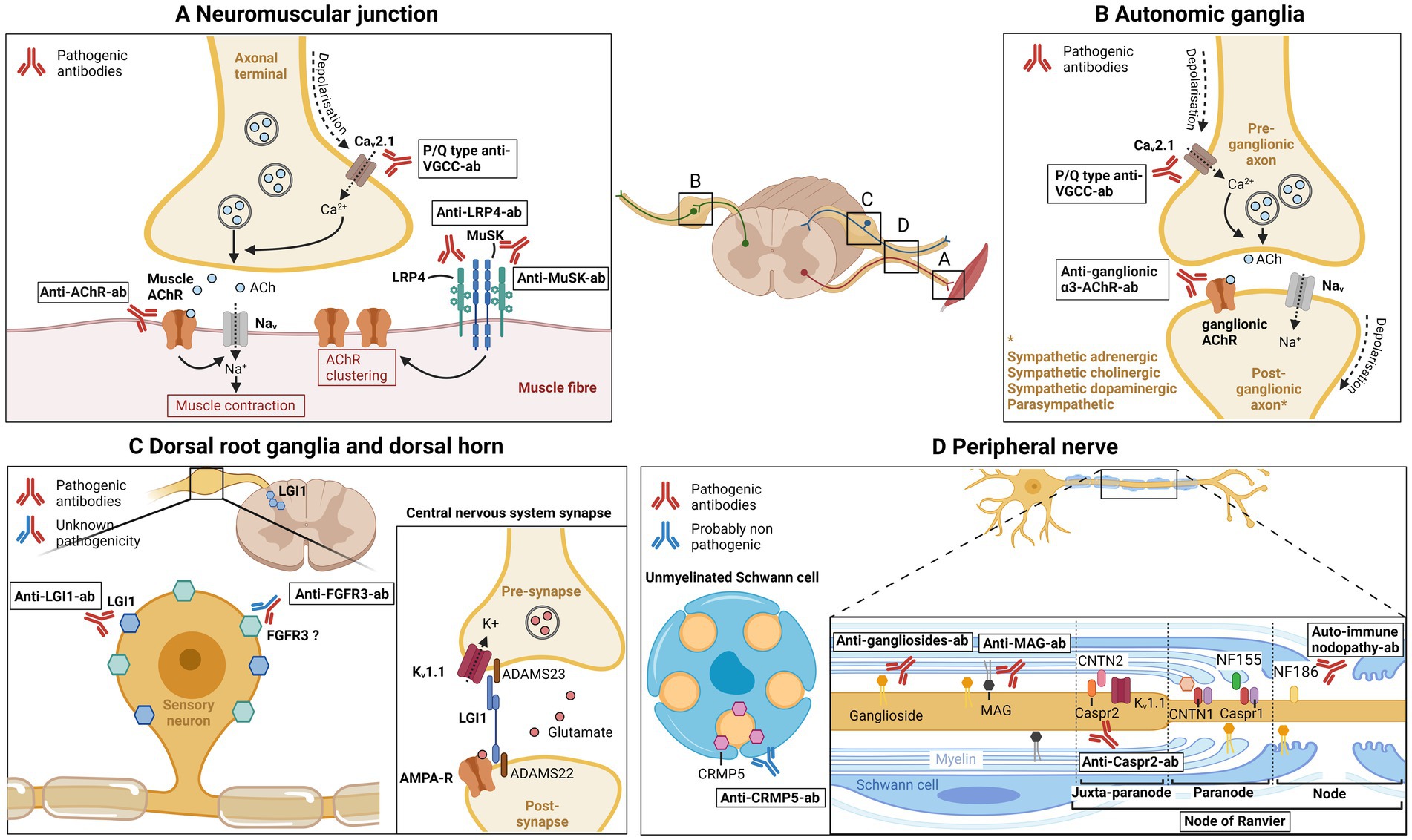
Figure 1. Main antibodies in neuromuscular disorders. (A) Neuromuscular junction. P/Q type anti-VGCC antibodies are directed against the voltage-gated calcium channel (VGCC or Cav2.1) in the axonal terminal, thus blocking calcium influx, acetylcholine vesicle fusion, and neuromuscular transmission. Anti-AChR antibodies target the nicotinic acetylcholine muscle receptor, preventing muscular membrane depolarization and generation of muscle action potential. Anti-LRP4 and anti-MuSK antibodies target the LRP4/MuSK complex, implicated in formation and maintenance of the neuromuscular junction and in acetylcholine receptor clustering. (B) Autonomic ganglia. Anti-ganglionic-α3-AChR antibodies are directed against the ganglionic acetylcholine receptor, preventing post-synaptic depolarization, blocking autonomic neurotransmission. P/Q type anti-VGCC antibodies also blocks the Cav2.1 present in presynaptic axonal terminal. (C) Dorsal root ganglia and dorsal horn. Leucin-rich glioma inactivated 1 (LGI1) was shown to be expressed in dorsal root ganglia and spinal cord dorsal horn, and is the target of anti-LGI1 antibodies. In central nervous system synapses, LGI1 binds to ADAM22/23 at the presynaptic side and modulate Kv1.1 (or voltage gated potassium channel; VGKC) channel; at the post-synaptic side, LGI1 binds to ADAM22 and modulate AMPA receptor. LGI1 therefore modulates central nervous system synaptic transmission. Anti-FGFR3 antibodies are directed against fibroblast growth factor receptor 3, which was shown to be expressed in small and large sensory neurons of the dorsal root ganglia in adult rats. Pathogenicity of anti-FGFR3 antibodies is currently unknown. (D) Peripheral nerve. Anti-gangliosides antibodies target gangliosides, glycoproteins that are expressed on neuronal membranes, Schwann cells and myelin. Anti-MAG antibodies recognizes myelin associated glycoprotein, a glycoprotein localized in periaxonal Schwann cells, which has function in glia-axon interaction. At the nodal and paranodal region, contactin 1, contactin-associated protein 1, neurofascin 155 and neurofascin 186, have different function such as axon-myelin and axon-Schwann cell binding and sodium channel clustering. Antibodies against paranodal/nodal proteins are responsible for auto-immune nodopathies. Contactin-associated protein 2 is expressed in the juxtaparanodal region (and also in dorsal root ganglia), connects to contactin 2 and organizes Kv1.1 (VGKC). Antibodies directed against Caspr2 cause peripheral nerve hyperexcitability. Collapsin response mediator protein-5 has been shown to be expressed in unmyelinated Schwann cells, and is involved in axon-Schwann cell interactions. Antibodies against CRMP5 are probably nonpathogenic, and a marker of paraneoplastic neurological syndrome. ACh, acetylcholine; AChR, Acetylcholine receptor; Cav2.1, voltage-dependent calcium channel 2.1; Caspr1, contactin-associated protein 1; Caspr2, contactin-associated protein 2; CNTN1, contactin 1; CNTN2, contactin 2; CRMP5, Collapsin response mediator protein-5; FGFR3, fibroblast growth factor 3; Kv1.1, voltage-gated potassium channel; LRP4, lipoprotein-related protein 4; MAG, myelin associated glycoprotein; MuSK, muscle kinase; NF155, neurofascin-155; NF186, neurofascin-186; VGCC, voltage gated calcium channel; VGKC, voltage-gated potassium channel.
Idiopathic inflammatory myopathies
Idiopathic inflammatory myopathies (IIM), also known as myositis, are a heterogeneous group of autoimmune diseases affecting multiples organs, including muscles, skin, lungs and joints (188, 189). Muscle involvement is typically characterized by subacute proximal weakness, sometimes associated with muscle pain. A major advance in the field of IIM is the discovery of myositis-specific autoantibodies, present in up to 60% of patients with IIM (190). Those antibodies are specific for myositis and are strongly associated with distinct clinical phenotypes. In addition, patients with IIM may be positive for autoantibodies that are present in other autoimmune disorders (systemic lupus erythematosus, systemic sclerosis etc.); they are often named myositis-associated auto-antibodies (189). Based on a combination of myositis-specific autoantibodies, clinical presentation, muscle MRI pattern and muscle biopsy features, patients can be classified in five distinct subtypes; dermatomyositis, antisynthetase syndrome, overlap myositis, immune-mediated necrotizing myopathy (IMNM) and inclusion-body myositis (IBM) (191, 192). The old and imprecise term “polymyositis” does not correspond to a precise entity, since more than 90% of polymyositis turn out to be IBM or IMNM upon follow-up.
Dermatomyositis
Dermatomyositis (DM) is defined by the presence of characteristic cutaneous manifestations (heliotrope rash, V sign rash, Gottron signs and papules etc.) and myositis, which can be inconstantly associated (189). Most patients with DM (~70%) have a myositis-specific antibody: anti-Mi-2, anti- MDA5, anti-TIF1, anti-NXP2 or anti-SAE (189). Each autoantibody is associated with a distinct clinical phenotype, leading to a subclassification of DM according to those antibodies (191). These associations have important clinical impact: patients with anti-TIF1 and -NXP2-ab have a higher risk of oncological disease, whereas anti-Mi-2 present with milder myositis. Anti-MDA-5 and -SAE are usually amyopathic but may have a dramatic course with severe interstitial lung disease or severe dysphagia, respectively.
Anti-synthetase syndrome
Anti-synthetase syndrome (ASyS) is a relatively homogeneous multisystem disease, usually classified as IIM although myositis is not always present. The most common manifestation is interstitial lung disease, which also determines the functional outcome and prognosis. ASyS is characterized by auto-antibodies against one of many amino-acyl transfer RNA (tRNA). Eight auto-antibodies have been identified: anti-Jo1, anti-Ha/YRS, anti-Zo, anti-EJ, anti-PL-7, anti-OJ, anti-KS and anti-PL-12 (190, 191).
Immune-mediated necrotizing myopathy
Immune-mediated necrotizing myopathy (IMNM) are characterized by a proximal symmetric and axial myopathy, usually extremely high muscle enzyme levels, and muscle biopsy showing necrosis or regeneration with minimal lymphocytic infiltrate (193). Patients rarely have prominent systemic manifestations. Patients with IMNM usually (60–70%) have auto-antibodies recognizing either 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR) or the signal recognition particle (SRP). According to those autoantibodies, IMNM can be classified as anti-HGMCR, anti-SRP and auto-antibody-negative IMNM, each subtype being a specific entity. Patients with anti-SRP usually have a more severe myopathy, extra-muscular manifestations (including cardiac involvement) and higher chance of dysphagia (194). The risk of developing anti-HMGCR IMNM is higher following statin exposure, and patients usually have a severe myopathy, without extra-muscular manifestations; the risk of developing cancer is higher in anti-HMGCR and seronegative IMNM (194).
Overlap myositis
Myositis can occur with other connective tissue disease such as systemic lupus erythematosus, systemic sclerosis, Sjögren syndrome or rheumatoid arthritis. Autoantibodies detected in overlap myositis include anti-U1RNP, anti-Ku, anti-PM-Scl, anti-RuvBL1, anti-RuvBL2, anti-Ro/SSA and anti-La/SSB (189, 195).
Inclusion-body myositis
Inclusion-body myositis (IBM) is characterized by asymmetrical weakness of both proximal and distal muscles that predominates on the quadriceps and long finger flexors, with a very slowly progressive course, and occurs mainly in individuals >50 years of age. As of the ENMC diagnostic criteria, typical clinical presentation and histological feature (endomysial infiltrate surrounding non necrotic fibers) are sufficient to make a diagnosis. In case of incomplete or atypical clinical picture, supportive criteria are required. These criteria include other histopathological features, imaging studies, and the positivity of autoantibodies recognizing cytosolic 5′-nucleotidase 1A (cN1a) antibodies (196). Anti-cN1a-ab have been described in 2013 (197) and may be detected in 33–80% of patients with IBM (198, 199). In initial studies, the specificity of those antibodies has been reported in the range of 92–100% (199). However, more recent studies showed that anti-cN1a-ab can also been found in patients with IIM (PM, DM, IMNM), SLE and Sjögren syndrome (198, 200). Various detection method can be used (ELISA, WB, CBA, immunoprecipitation), and the best method is currently unknown (199, 200).
Detection of IIM antibodies
Detection of myositis-specific antibodies and myositis-associated antibodies can be made with various detection methods. As many of those antibodies target cytoplasmic antigens, a weak or negative nuclear staining is achieved with indirect immunofluorescence using Hep2 cells. Indirect immunofluorescence has therefore limited utility as screening test. Immunoprecipitation is probably the gold-standard for most myositis autoantibodies detection, but is time-consuming, expensive and availability is limited to specialized centers (201). ELISA is a reliable method, but not all myositis-specific and myositis-associated antibodies are amenable to this testing (202). In the recent years, commercial line immunoblot assay and immunodot assay have been developed and have improved availability and diagnostic accuracy of myositis subtypes (203). Two of those commercial immune assays have been assessed and compared to immunoprecipitation in a recent study. Overall, those assays performed relatively well with sensitivity and specificity varying according to antibodies, with two notable exceptions: a poor detection rate of anti-TIF1γ (40% of false negative with line blot and 76% of false negative with dot blot) and a poor detection rate of rare anti-synthetase autoantibodies (200). In addition, a high false-positive rate was observed, in 13.7% of samples analyzed by line blot and 12.1% analyzed by dot blot, usually at low titer (201). Results of those commercial immunoassays must therefore be interpreted within the clinical context, and in selected cases, immunoprecipitation should be considered.
Myositis-specific and associated antibodies need to be tested in every suspicion of inflammatory myopathy. As detection of such antibody is part of the classification process of IMM, a positive antibody will help guide investigations (e.g., paraneoplastic workup) and guide treatment. It is important to note that the diagnostic value of myositis-specific and myositis-associated antibodies depends on pre-test probability, determined mainly by the clinical pictures and complementary exams like electromyography (204).
Characteristics of antibodies associated with IIM are summarized in Table 5.
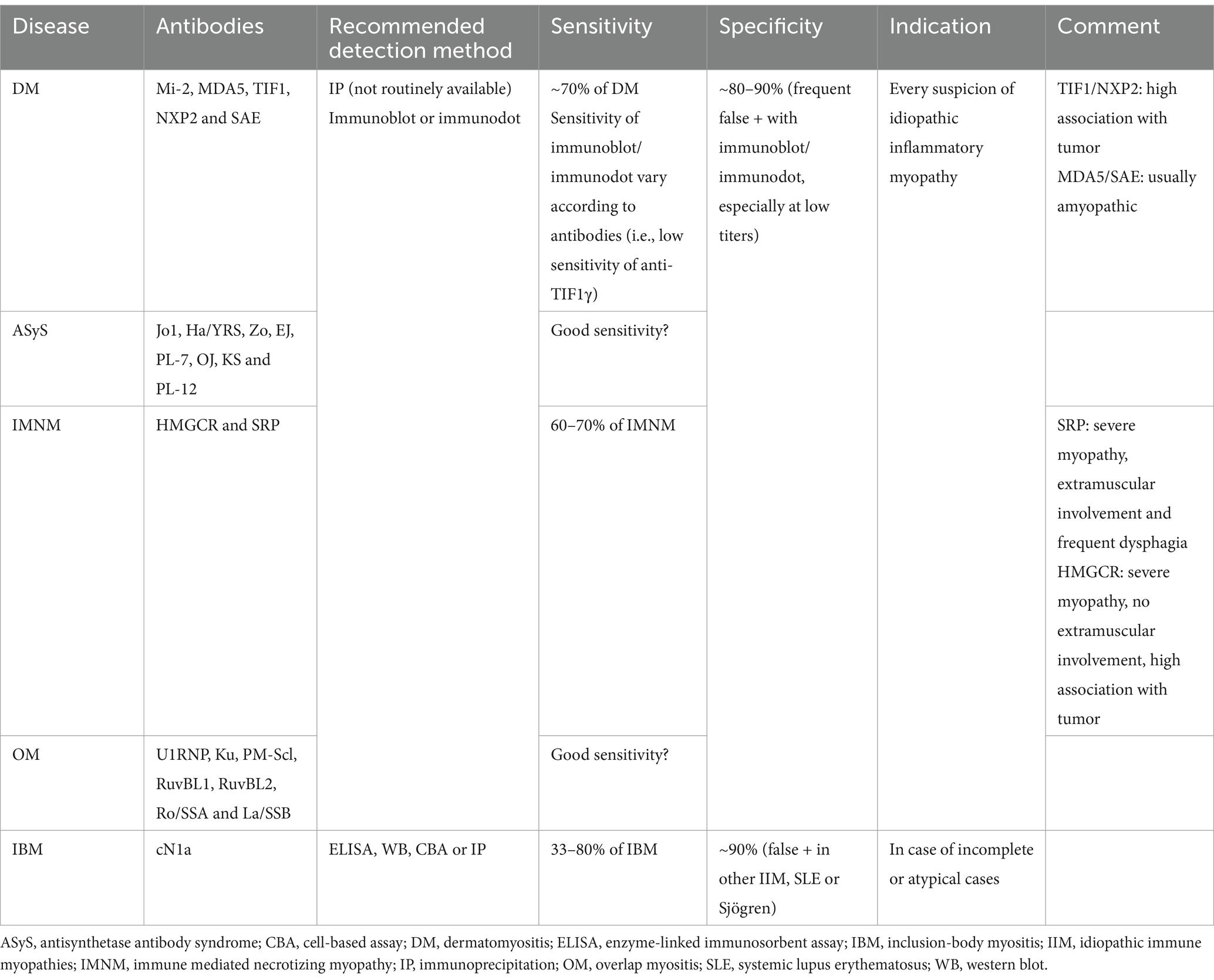
Table 5. Characteristics of antibodies associated with autoimmune myopathies.
Conclusion
Autoantibody testing is extremely useful in the management of a great number of patients with inflammatory neuromuscular disorders (205). In addition to confirming a diagnosis, the presence of such autoantibody can guide the paraneoplastic workup (e.g., the screening for thymoma in anti-AChR+ MG) and treatment (e.g., anti-C5 therapy in anti-AChR+ MG, anti-CD20 therapy in anti-MuSK+ MG and autoimmune nodopathy or IVIg therapy in anti-FGFR3+ sensory neuronopathy). Moreover, the recent discovery of new autoantibodies (such as anti-nodal and paranodal antibodies in auto-immune nodopathies, anti-FGFR3 and AGO1 in sensory neuronopathies or anti-cN1a in inclusion-body myositis) has led to a better understanding of the pathophysiology of immune-mediated neuromuscular diseases.
Despite their high clinical usefulness, clinicians should be aware of limitations related to such autoantibodies screening. First, their specificity is not always optimal. For example, anti-LRP4 antibodies are not specific to MG and may be found in patients with ALS. Anti-gangliosides antibodies are non-specific at low titers, and only some antibodies are associated with a specific clinical phenotype. False positives are frequent when testing for muscle-specific and muscle-associated autoantibodies. Finally, when testing for autoantibodies, clinicians should be aware of the used detection method. Indeed, sensitivity and specificity of detection techniques can vary widely, and the use of the reference method should be encouraged. However, some of these techniques (such as immunoprecipitation for the detection of muscle autoantibodies) are not widely available in clinical practice.
Author contributions
VL: Conceptualization, Investigation, Writing – original draft. AV: Writing – review & editing. MT: Conceptualization, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Noridomi, K, Watanabe, G, Hansen, MN, Han, GW, and Chen, L. Structural insights into the molecular mechanisms of myasthenia gravis and their therapeutic implications. eLife. (2017) 6:e23043. doi: 10.7554/eLife.23043
2. Howard, JF Jr. Myasthenia gravis: the role of complement at the neuromuscular junction. Ann N Y Acad Sci. (2018) 1412:113–28. doi: 10.1111/nyas.13522
3. Lindstrom, J. An assay for antibodies to human acetylcholine receptor in serum from patients with myasthenia gravis. Clin Immunol Immunopathol. (1977) 7:36–43. doi: 10.1016/0090-1229(77)90027-7
4. Oger, J, and Frykman, H. An update on laboratory diagnosis in myasthenia gravis. Clin Chim Acta. (2015) 449:43–8. doi: 10.1016/j.cca.2015.07.030
5. Benatar, M. A systematic review of diagnostic studies in myasthenia gravis. Neuromuscul Disord. (2006) 16:459–67. doi: 10.1016/j.nmd.2006.05.006
6. Li, Z, Zhang, C, Chang, T, Zhang, X, Yangd, H, Gao, F, et al. SCREAM study investigators. A multicentre, prospective, double-blind study comparing the accuracy of autoantibody diagnostic assays in myasthenia gravis: the SCREAM study. Lancet Reg Health West Pac. (2023) 38:100846. doi: 10.1016/j.lanwpc.2023.100846
7. Vincent, A, Newsom-Davis, J, Newton, P, and Beck, N. Acetylcholine re-ceptor antibody and clinical response to thymectomy in myasthenia gravis. Neurology. (1983) 33:1276–82. doi: 10.1212/WNL.33.10.1276
8. Oosterhuis, HJ, Limburg, PC, Hummel-Tappel, E, and The, TH. Anti-acetylcholine receptor antibodies in myasthenia gravis. Part 2.Clinical and serological follow-up of individual patients. J Neurol Sci. (1983) 58:371–85. doi: 10.1016/0022-510X(83)90096-5
9. Oosterhuis, HJ, Limburg, PC, Hummel-Tappel, E, Van den Burg, W, and The, TH. Anti- acetylcholine receptor antibodies in myasthenia gravis. Part 3. The effect of thymectomy. J Neurol Sci. (1985) 69:335–43. doi: 10.1016/0022-510X(85)90144-3
10. Heldal, AT, Eide, GE, Romi, F, Owe, JF, and Gilhus, NE. Repeated acetyl-choline receptor antibody- concentrations and association to clinical myasthenia gravis development. PLoS One. (2014) 9:e114060. doi: 10.1371/journal.pone.0114060
11. Roses, AD, Olanow, CW, McAdams, MW, and Lane, RJ. No direct correlation between serum antiacetylcholine receptor antibody levels and clinical state of individual patients with myasthenia gravis. Neurology. (1981) 31:220–4. doi: 10.1212/WNL.31.2.220
12. Olanow, CW, Wechsler, AS, and Roses, AD. A prospective study of thymectomy and serum acetylcholine receptor antibodies in myasthenia gravis. Ann Surg. (1982) 196:113–21. doi: 10.1097/00000658-198208000-00001
13. Sanders, DB, Burns, TM, Cutter, GR, Massey, JM, Juel, VC, Hobson-Webb, L, et al. Does change in acetylcholine receptor antibody level correlate with clinical change in myasthenia gravis? Muscle Nerve. (2014) 49:483–6. doi: 10.1002/mus.23944
14. Marcuse, F, Brandts, L, Moens, D, Damoiseaux, J, Hochstenbag, M, Hoeijmakers, JGJ, et al. The association between anti-acetylcholine receptor antibody level and clinical improvement in myasthenia gravis. Eur J Neurol. (2022) 29:1187–97. doi: 10.1111/ene.15238
15. Romi, F. Thymoma in myasthenia gravis: from diagnosis to treatment. Autoimmune Dis. (2011) 2011:474512:1–5. doi: 10.4061/2011/474512
16. Aydin, Y, Ulas, AB, Mutlu, V, Colak, A, and Eroglu, A. Thymectomy in myasthenia gravis. Eurasian J Med. (2017) 49:48–52. doi: 10.5152/eurasianjmed.2017.17009
17. Wolfe, GI, Kaminski, HJ, Aban, IB, Minisman, G, Kuo, HC, Marx, A, et al. Randomized trial of Thymectomy in myasthenia gravis. N Engl J Med. (2016) 375:511–22. doi: 10.1056/NEJMoa1602489
18. Narayanaswami, P, Sanders, DB, Wolfe, G, Benatar, M, Cea, G, Evoli, A, et al. International consensus guidance for Management of Myasthenia Gravis: 2020 update. Neurology. (2021) 96:114–22. doi: 10.1212/WNL.0000000000011124
19. Muppidi, S, Utsugisawa, K, Benatar, M, Murai, H, Barohn, RJ, Illa, I, et al. Long-term safety and efficacy of eculizumab in generalized myasthenia gravis. Muscle Nerve. (2019) 60:14–24. doi: 10.1002/mus.26447
20. Meisel, A, Annane, D, Vu, T, Mantegazza, R, Katsuno, M, Aguzzi, R, et al. Long-term efficacy and safety of ravulizumab in adults with anti-acetylcholine receptor antibody-positive generalized myasthenia gravis: results from the phase 3 CHAMPION MG open-label extension. J Neurol. (2023) 270:3862–75. doi: 10.1007/s00415-023-11699-x
21. Howard, JF Jr, Bresch, S, Genge, A, Hewamadduma, C, Hinton, J, Hussain, Y, et al. Safety and efficacy of zilucoplan in patients with generalised myasthenia gravis (RAISE): a randomised, double-blind, placebo-controlled, phase 3 study. Lancet Neurol. (2023) 22:395–406. doi: 10.1016/S1474-4422(23)00080-7
22. Howard, JF Jr, Bril, V, Vu, T, Karam, C, Peric, S, Margania, T, et al. Safety, efficacy, and tolerability of efgartigimod in patients with generalised myasthenia gravis (ADAPT): a multicentre, randomised, placebo-controlled, phase 3 trial. Lancet Neurol. (2021) 20:526–36. doi: 10.1016/S1474-4422(21)00159-9
23. Bril, V, Drużdż, A, Grosskreutz, J, Habib, AA, Mantegazza, R, Sacconi, S, et al. Safety and efficacy of rozanolixizumab in patients with generalised myasthenia gravis (MycarinG): a randomised, double-blind, placebo-controlled, adaptive phase 3 study. Lancet Neurol. (2023) 22:383–94. doi: 10.1016/S1474-4422(23)00077-7
24. Leite, MI, Jacob, S, Viegas, S, Cossins, J, Clover, L, Morgan, BP, et al. IgG1 antibodies to acetylcholine receptors in 'seronegative' myasthenia gravis. Brain. (2008) 131:1940–52. doi: 10.1093/brain/awn092
25. Rodríguez Cruz, PM, al-Hajjar, M, Huda, S, Jacobson, L, Woodhall, M, Jayawant, S, et al. Clinical features and diagnostic usefulness of antibodies to clustered acetylcholine receptors in the diagnosis of seronegative myasthenia gravis. JAMA Neurol. (2015) 72:642–9. doi: 10.1001/jamaneurol.2015.0203
26. Devic, P, Petiot, P, Simonet, T, Stojkovic, T, Delmont, E, Franques, J, et al. Antibodies to clustered acetylcholine receptor: expanding the phenotype. Eur J Neurol. (2014) 21:130–4. doi: 10.1111/ene.12270
27. Masi, G, Li, Y, Karatz, T, Pham, MC, Oxendine, SR, Nowak, RJ, et al. The clinical need for clustered AChR cell-based assay testing of seronegative MG. J Neuroimmunol. (2022) 367:577850. doi: 10.1016/j.jneuroim.2022.577850
28. Vincent, A, Huda, S, Cao, M, Cetin, H, Koneczny, I, Rodriguez-Cruz, P, et al. Serological and experimental studies in different forms of myasthenia gravis. Ann N Y Acad Sci. (2018) 1413:143–53. doi: 10.1111/nyas.13592
29. Guptill, JT, Sanders, DB, and Evoli, A. Anti-MuSK antibody myasthenia gravis: clinical findings and response to treatment in two large cohorts. Muscle Nerve. (2011) 44:36–40. doi: 10.1002/mus.22006
30. Klooster, R, Plomp, JJ, Huijbers, MG, Niks, EH, Straasheijm, KR, Detmers, FJ, et al. Muscle-specific kinase myasthenia gravis IgG4 autoantibodies cause severe neuromuscular junction dysfunction in mice. Brain. (2012) 135:1081–101. doi: 10.1093/brain/aws025
31. Evoli, A, Alboini, PE, Damato, V, Iorio, R, Provenzano, C, Bartoccioni, E, et al. Myasthenia gravis with antibodies to MuSK: an update. Ann N Y Acad Sci. (2018) 1412:82–9. doi: 10.1111/nyas.13518
32. Matthews, I, Chen, S, Hewer, R, McGrath, V, Furmaniak, J, and Rees Smith, B. Muscle-specific receptor tyrosine kinase autoantibodies-a new immunoprecipitation assay. Clin Chim Acta. (2004) 348:95–9. doi: 10.1016/j.cccn.2004.05.008
33. Kwon, YN, Woodhall, M, Sung, JJ, Kim, KK, Lim, YM, Kim, H, et al. Clinical pitfalls and serological diagnostics of MuSK myasthenia gravis. J Neurol. (2023) 270:1478–86. doi: 10.1007/s00415-022-11458-4
34. Huda, S, Waters, P, Woodhall, M, Leite, MI, Jacobson, L, de Rosa, A, et al. IgG-specific cell-based assay detects potentially pathogenic MuSK-abs in seronegative MG. Neurol Neuroimmunol Neuroinflamm. (2017) 4:e357. doi: 10.1212/NXI.0000000000000357
35. Bartoccioni, E, Scuderi, F, Minicuci, GM, Marino, M, Ciaraffa, F, and Evoli, A. Anti-MuSK antibodies: correlation with myasthenia gravis severity. Neurology. (2006) 67:505–7. doi: 10.1212/01.wnl.0000228225.23349.5d
36. Ohta, K, Shigemoto, K, Kubo, S, Maruyama, N, Abe, Y, Ueda, N, et al. MuSK antibodies in AChR ab-seropositive MG vs AChR ab-seronegative MG. Neurology. (2004) 62:2132–3. doi: 10.1212/01.WNL.0000129274.12702.92
37. Vakrakou, AG, Karachaliou, E, Chroni, E, Zouvelou, V, Tzanetakos, D, Salakou, S, et al. Immunotherapies in MuSK-positive myasthenia gravis; an IgG4 antibody-mediated disease. Front Immunol. (2023) 14:1212757. doi: 10.3389/fimmu.2023.1212757
38. Hehir, MK, Hobson-Webb, LD, Benatar, M, Barnett, C, Silvestri, NJ, Howard, JF Jr, et al. Rituximab as treatment for anti-MuSK myasthenia gravis: multicenter blinded prospective review. Neurology. (2017) 89:1069–77. doi: 10.1212/WNL.0000000000004341
39. Díaz-Manera, J, Martínez-Hernández, E, Querol, L, Klooster, R, Rojas-García, R, Suárez-Calvet, X, et al. Long-lasting treatment effect of rituximab in MuSK myasthenia. Neurology. (2012) 78:189–93. doi: 10.1212/WNL.0b013e3182407982
40. Beecher, G, Anderson, D, and Siddiqi, ZA. Rituximab in refractory myasthenia gravis: extended prospective study results. Muscle Nerve. (2018) 58:452–5. doi: 10.1002/mus.26156
41. Anderson, D, Phan, C, Johnston, WS, and Siddiqi, ZA. Rituximab in refractory myasthenia gravis: a prospective, open-label study with long-term follow-up. Ann Clin Transl Neurol. (2016) 3:552–5. doi: 10.1002/acn3.314
42. Clifford, KM, Hobson-Webb, LD, Benatar, M, Burns, TM, Barnett, C, Silvestri, NJ, et al. Thymectomy may not be associated with clinical improvement in MuSK myasthenia gravis. Muscle Nerve. (2019) 59:404–10. doi: 10.1002/mus.26404
43. Zisimopoulou, P, Evangelakou, P, Tzartos, J, Lazaridis, K, Zouvelou, V, Mantegazza, R, et al. A comprehensive analysis of the epidemiology and clinical characteristics of anti-LRP4 in myasthenia gravis. J Autoimmun. (2014) 52:139–45. doi: 10.1016/j.jaut.2013.12.004
44. Tzartos, JS, Zisimopoulou, P, Rentzos, M, Karandreas, N, Zouvelou, V, Evangelakou, P, et al. LRP4 antibodies in serum and CSF from amyotrophic lateral sclerosis patients. Ann Clin Transl Neurol. (2014) 1:80–7. doi: 10.1002/acn3.26
45. Huijbers, MG, Niks, EH, Klooster, R, de Visser, M, Kuks, JB, Veldink, JH, et al. Myasthenia gravis with muscle specific kinase antibodies mimicking amyotrophic lateral sclerosis. Neuromuscul Disord. (2016) 26:350–3. doi: 10.1016/j.nmd.2016.04.004
46. Romi, F, Skeie, GO, Aarli, JA, and Gilhus, NE. Muscle autoantibodies in subgroups of myasthenia gravis patients. J Neurol. (2000) 247:369–75. doi: 10.1007/s004150050604
47. Yamamoto, AM, Gajdos, P, Eymard, B, Tranchant, C, Warter, JM, Gomez, L, et al. Anti-titin antibodies in myasthenia gravis. Arch Neurol. (2001) 58:885–90. doi: 10.1001/archneur.58.6.885
48. Punga, AR, Maddison, P, Heckmann, JM, Guptill, JT, and Evoli, A. Epidemiology, diagnostics, and biomarkers of autoimmune neuromuscular junction disorders. Lancet Neurol. (2022) 21:176–88. doi: 10.1016/S1474-4422(21)00297-0
49. Romi, F, Skeie, GO, Aarli, JA, and Gilhus, NE. The severity of myasthenia gravis correlates with the serum concentration of titin and ryanodine receptor antibodies. Arch Neurol. (2000) 57:1596–600. doi: 10.1001/archneur.57.11.1596
50. Evoli, A, Antonini, G, Antozzi, C, DiMuzio, A, Habetswallner, F, Iani, C, et al. Italian recommendations for the diagnosis and treatment of myasthenia gravis. Neurol Sci. (2019) 40:1111–24. doi: 10.1007/s10072-019-03746-1
51. Chan, KH, Lachance, DH, Harper, CM, and Lennon, VA. Frequency of seronegativity in adult-acquired generalized myasthenia gravis. Muscle Nerve. (2007) 36:651–8. doi: 10.1002/mus.20854
52. Titulaer, MJ, Maddison, P, Sont, JK, Wirtz, PW, Hilton-Jones, D, Klooster, R, et al. Clinical Dutch-English Lambert-Eaton Myasthenic syndrome (LEMS) tumor association prediction score accurately predicts small-cell lung cancer in the LEMS. J Clin Oncol. (2011) 29:902–8. doi: 10.1200/JCO.2010.32.0440
53. Lennon, VA, Kryzer, TJ, Griesmann, GE, O'Suilleabhain, PE, Windebank, AJ, Woppmann, A, et al. Calcium-channel antibodies in the Lambert-Eaton syndrome and other paraneoplastic syndromes. N Engl J Med. (1995) 332:1467–75. doi: 10.1056/NEJM199506013322203
54. Motomura, M, Lang, B, Johnston, I, Palace, J, Vincent, A, and Newsom-Davis, J. Incidence of serum anti-P/O-type and anti-N-type calcium channel autoantibodies in the Lambert-Eaton myasthenic syndrome. J Neurol Sci. (1997) 147:35–42. doi: 10.1016/S0022-510X(96)05303-8
55. Benatar, M, Blaes, F, Johnston, I, Wilson, K, Vincent, A, Beeson, D, et al. Presynaptic neuronal antigens expressed by a small cell lung carcinoma cell line. J Neuroimmunol. (2001) 113:153–62. doi: 10.1016/S0165-5728(00)00431-8
56. Graus, F, Lang, B, Pozo-Rosich, P, Saiz, A, Casamitjana, R, and Vincent, A. P/Q type calcium-channel antibodies in paraneoplastic cerebellar degeneration with lung cancer. Neurology. (2002) 59:764–6. doi: 10.1212/WNL.59.5.764
57. Zalewski, NL, Lennon, VA, Lachance, DH, Klein, CJ, Pittock, SJ, and Mckeon, A. P/Q- and N-type calcium-channel antibodies: oncological, neurological, and serological accompaniments. Muscle Nerve. (2016) 54:220–7. doi: 10.1002/mus.25027
58. Majed, M, Zekeridou, A, Lennon, V, Pittock, S, McKeon, A, Klein, C, et al. Testing for N-type voltage gated calcium channel antibody has limited utility in evaluating patients with suspected Lambert-Eaton Myasthenic syndrome. Neurology. (2022) 99:S31–2. doi: 10.1212/01.wnl.0000903272.62514.70
59. Güre, AO, Stockert, E, Scanlan, MJ, Keresztes, RS, Jäger, D, Altorki, NK, et al. Serological identification of embryonic neural proteins as highly immunogenic tumor antigens in small cell lung cancer. Proc Natl Acad Sci USA. (2000) 97:4198–203. doi: 10.1073/pnas.97.8.4198
60. Titulaer, MJ, Klooster, R, Potman, M, Sabater, L, Graus, F, Hegeman, IM, et al. SOX antibodies in small-cell lung cancer and Lambert-Eaton myasthenic syndrome: frequency and relation with survival. J Clin Oncol. (2009) 27:4260–7. doi: 10.1200/JCO.2008.20.6169
61. Sabater, L, Titulaer, M, Saiz, A, Verschuuren, J, Güre, AO, and Graus, F. SOX1 antibodies are markers of paraneoplastic Lambert-Eaton myasthenic syndrome. Neurology. (2008) 70:924–8. doi: 10.1212/01.wnl.0000281663.81079.24
62. Lipka, AF, Verschuuren, JJ, and Titulaer, MJ. SOX1 antibodies in Lambert-Eaton myasthenic syndrome and screening for small cell lung carcinoma. Ann N Y Acad Sci. (2012) 1275:70–7. doi: 10.1111/j.1749-6632.2012.06772.x
63. Tschernatsch, M, Gross, O, Kneifel, N, Kaps, M, and Blaes, F. SOX-1 autoantibodies in patients with paraneoplastic neurological syndromes. Autoimmun Rev. (2009) 8:549–51. doi: 10.1016/j.autrev.2009.01.015
64. Ruiz-García, R, Martínez-Hernández, E, García-Ormaechea, M, Español-Rego, M, Sabater, L, Querol, L, et al. Caveats and pitfalls of SOX1 autoantibody testing with a commercial line blot assay in paraneoplastic neurological investigations. Front Immunol. (2019) 10:769. doi: 10.3389/fimmu.2019.00769
65. Kolter, T. Ganglioside biochemistry. Int Scholarly Res Notices. (2012) 2012:506160:1–36. doi: 10.5402/2012/506160
66. O'Hanlon, GM, Paterson, GJ, Veitch, J, Wilson, G, and Willison, HJ. Mapping immunoreactive epitopes in the human peripheral nervous system using human monoclonal anti-GM1 ganglioside antibodies. Acta Neuropathol. (1998) 95:605–16. doi: 10.1007/s004010050847
67. Ho, TW, Willison, HJ, Nachamkin, I, Li, CY, Veitch, J, Ung, H, et al. Anti-GD1a antibody is associated with axonal but not demyelinating forms of Guillain-Barre syndrome. Ann Neurol. (1999) 45:168–73. doi: 10.1002/1531-8249(199902)45:2<168:AID-ANA6>3.0.CO;2-6
68. Koga, M, Yoshino, H, Morimatsu, M, and Yuki, N. Anti-GT1a IgG in Guillain-Barre syndrome. J Neurol Neurosurg Psychiatry. (2002) 72:767–71. doi: 10.1136/jnnp.72.6.767
69. Sawaya, R. Anti-GQ1b Spectrum disorders. J Clin Neuromuscul Dis. (2019) 21:52–3. doi: 10.1097/CND.0000000000000241
70. Kaida, K, Kusunoki, S, Kamakura, K, Motoyoshi, K, and Kanazawa, I. GalNAc-GD1a in human peripheral nerve. Neurology. (2003) 61:465–70. doi: 10.1212/01.WNL.0000081229.88862.C6
71. Lleixà, C, Martín-Aguilar, L, Pascual-Goñi, E, Franco, T, Caballero, M, de Luna, N, et al. Autoantibody screening in Guillain-Barré syndrome. J Neuroinflammation. (2021) 18:2301. doi: 10.1186/s12974-021-02301-0
72. Yako, K, Kusunoki, S, and Kanazawa, I. Serum antibody against a peripheral nerve myelin ganglioside, LM1, in Guillain-Barré syndrome. J Neurol Sci. (1999) 168:85–9. doi: 10.1016/S0022-510X(99)00174-4
73. Kusunoki, S, Shiina, M, and Kanazawa, I. Anti-gal-C antibodies in GBS subsequent to mycoplasma infection: evidence of molecular mimicry. Neurology. (2001) 57:736–8. doi: 10.1212/WNL.57.4.736
74. Miyazaki, T, Kusunoki, S, Kaida, K, Shiina, M, and Kanazawa, I. Guillain–Barré syndrome associated with IgG monospecific to ganglioside GD1b. Neurology. (2001) 56:1227–9. doi: 10.1212/WNL.56.9.1227
75. Devaux, JJ, Odaka, M, and Yuki, N. Nodal proteins are target antigens in Guillain-Barré syndrome. J Peripher Nerv Syst. (2012) 17:62–71. doi: 10.1111/j.1529-8027.2012.00372.x
76. Sekiguchi, Y, Uncini, A, Yuki, N, Misawa, S, Notturno, F, Nasu, S, et al. Antiganglioside antibodies are associated with axonal Guillain–Barré syndrome: A Japanese–Italian collaborative study. J Neurol Neurosurg Psychiatry. (2012) 83:23–8. doi: 10.1136/jnnp-2011-300309
77. Hafer-Macko, C, Hsieh, ST, Ho, TW, Sheikh, K, Cornblath, DR, Li, CY, et al. Acute motor axonal neuropathy: An antibody-mediated attack on axolemma. Ann Neurol. (1996) 40:635644:635–44. doi: 10.1002/ana.410400414
78. Zautner, AE, Johann, C, Strubel, A, Busse, C, Tareen, AM, Masanta, WO, et al. Seroprevalence of campylobacteriosis and relevant post-infectious sequelae. Eur J Clin Microbiol Infect Dis. (2014) 33:1019–27. doi: 10.1007/s10096-013-2040-4
79. Sheikh, KA, Deerinck, TJ, Ellisman, MH, and Griffin, JW. The distribution of ganglioside-like moieties in peripheral nerves. Brain. (1999) 122:449660:449–60. doi: 10.1093/brain/122.3.449
80. Chiba, A, Kusunoki, S, Shimizu, T, and Kanazawa, I. Serum IgG antibody to ganglioside GQ1b is a possible marker of Miller fisher syndrome. Ann Neurol. (1992) 31:677–9. doi: 10.1002/ana.410310619
81. Odaka, M, Yuki, N, and Hirata, K. Anti-GQ1b IgG antibody syndrome: clinical and immunological range. J Neurol Neurosurg Psychiatry. (2001) 70:50–5. doi: 10.1136/jnnp.70.1.50
82. Ito, M, Kuwabara, S, Odaka, M, Misawa, S, Koga, M, Hirata, K, et al. Bickerstaff’s brainstem encephalitis and fisher syndrome form a continuous spectrum: clinical analysis of 581 cases. J Neurol. (2008) 255:674–82. doi: 10.1007/s00415-008-0775-0
83. Jacobs, BC, Endtz, H, van der Meché, F, Hazenberg, MP, Achtereekte, HA, and van Doorn, P. Serum anti-GQ1b IgG antibodies recognize surface epitopes on Campylobacter jejuni from patients with Miller fisher syndrome. Ann Neurol. (1995) 37:260–4. doi: 10.1002/ana.410370218
84. Rojas-García, R, Querol, L, Gallardo, E, de Luna Salva, N, Juarez, C, Garces, M, et al. Clinical and serological features of acute sensory ataxic neuropathy with antiganglioside antibodies. J Peripher Nerv Syst. (2012) 17:158–68. doi: 10.1111/j.1529-8027.2012.00407.x
85. Wakerley, BR, and Yuki, N. Pharyngeal-cervical-brachial variant of Guillain-Barre syndrome. J Neurol Neurosurg Psychiatry. (2014) 85:339–44. doi: 10.1136/jnnp-2013-305397
86. Fehmi, J, Davies, AJ, Walters, J, Lavin, T, Keh, R, Rossor, AM, et al. IgG1pan-neurofascin antibodies identify a severe yet treatable neuropathy with a high mortality. J Neurol Neurosurg Psychiatry. (2021) 92:1089–95. doi: 10.1136/jnnp-2021-326343
87. Quarles, RH. Myelin-associated glycoprotein (MAG): past, present and beyond. J Neurochem. (2007) 100:1431–48. doi: 10.1111/j.1471-4159.2006.04319.x
88. Dalakas, MC. Advances in the diagnosis, immunopathogenesis and therapies of IgM-anti-MAG antibody-mediated neuropathies. Ther Adv Neurol Disord. (2018) 11:1756285617746640. doi: 10.1177/1756285617746640
89. Kuijf, ML, Eurelings, M, Tio-Gillen, AP, van Doorn, PA, van den Berg, LH, Hooijkaas, H, et al. Detection of anti-MAG antibodies in polyneuropathy associated with IgM monoclonal gammopathy. Neurology. (2009) 73:688–95. doi: 10.1212/WNL.0b013e3181b59a80
90. Liberatore, G, Giannotta, C, Sajeev, BP, Morenghi, E, Terenghi, F, Gallia, F, et al. Sensitivity and specificity of a commercial ELISA test for anti-MAG antibodies in patients with neuropathy. J Neuroimmunol. (2020) 345:577288. doi: 10.1016/j.jneuroim.2020.577288
91. Hänggi, P, Aliu, B, Martin, K, Herrendorff, R, and Steck, AJ. Decrease in serum anti-MAG autoantibodies is associated with therapy response in patients with anti-MAG neuropathy: retrospective study. Neurol Neuroimmunol Neuroinflamm. (2021) 9:e1109. doi: 10.1212/NXI.0000000000001109
92. Stino, AM, Elsheikh, B, and Allen, JA. Anti-myelin-associated glycoprotein neuropathy: where do we stand? Muscle Nerve. (2023) 68:823–32. doi: 10.1002/mus.27954
93. Delmont, E, Attarian, S, Antoine, JC, Paul, S, Camdessanché, JP, Grapperon, AM, et al. Relevance of anti-HNK1 antibodies in the management of anti-MAG neuropathies. J Neurol. (2019) 266:1973–9. doi: 10.1007/s00415-019-09367-0
94. van den Bergh, P, van Doorn, PA, Hadden, RDM, Avau, B, Vankrunkelsven, P, Allen, JA, et al. European academy of neurology/peripheral nerve society guideline on diagnosis and treatment of chronic inflammatory demyelinating polyradiculoneuropathy: report of a joint task force-second revision. Eur J Neurol. (2021) 28:3556–83. doi: 10.1111/ene.14959
95. le Cann, M, Bouhour, F, Viala, K, Simon, L, Tard, C, Rossi, C, et al. CANOMAD: a neurological monoclonal gammopathy of clinical significance that benefits from B-cell-targeted therapies. Blood. (2020) 136:2428–36. doi: 10.1182/blood.2020007092
96. Yuki, N, and Uncini, A. Acute and chronic ataxic neuropathies with disialosyl antibodies: a continuous clinical spectrum and a common pathophysiological mechanism. Muscle Nerve. (2014) 49:629–35. doi: 10.1002/mus.24192
97. Willison, HJ, O'Leary, CP, Veitch, J, Blumhardt, LD, Busby, M, Donaghy, M, et al. The clinical and laboratory features of chronic sensory ataxic neuropathy with anti-disialosyl IgM antibodies. Brain. (2001) 124:1968–77. doi: 10.1093/brain/124.10.1968
98. Garcia-Santibanez, R, Zaidman, CM, Sommerville, RB, Lopate, G, Weihl, CC, Pestronk, A, et al. CANOMAD and other chronic ataxic neuropathies with disialosyl antibodies (CANDA). J Neurol. (2018) 265:1402–9. doi: 10.1007/s00415-018-8853-4
99. Peillet, C, Adams, D, Attarian, S, Bouhour, F, Cauquil, C, Cassereau, J, et al. Anti-disialosyl-immunoglobulin M chronic autoimmune neuropathies: a nationwide multicenter retrospective study. Eur J Neurol. (2022) 29:3547–55. doi: 10.1111/ene.15523
100. Roth, G, Rohr, J, Magistris, MR, and Ochsner, F. Motor neuropathy with proximal multifocal persistent conduction block, fasciculations and myokymia. Psychiatr Neurol. (2004) 25:416–23. doi: 10.1159/000116045
101. Pestronk, A, Cornblath, DR, Ilyas, AA, Baba, H, Quarles, RH, Griffin, JW, et al. A treatable multifocal motor neuropathy with antibodies to GM1 ganglioside. Ann Neurol. (1988) 24:73–8. doi: 10.1002/ana.410240113
102. Yeh, WZ, Dyck, PJ, van den Berg, LH, Kiernan, MC, and Taylor, BV. Multifocal motor neuropathy: controversies and priorities. J Neurol Neurosurg Psychiatry. (2020) 91:140–8. doi: 10.1136/jnnp-2019-321532
103. Vlam, L, van der Pol, WL, Cats, EA, Straver, DC, Piepers, S, Franssen, H, et al. Multifocal motor neuropathy: diagnosis, pathogenesis and treatment strategies. Nat Rev Neurol. (2011) 8:48–58. doi: 10.1038/nrneurol.2011.175
104. Taylor, BV, Gross, L, and Windebank, AJ. The sensitivity and specificity of anti-GM1 antibody testing. Neurology. (1996) 47:951–5. doi: 10.1212/WNL.47.4.951
105. Kollewe, K, Wurster, U, Sinzenich, T, Körner, S, Dengler, R, Mohammadi, B, et al. Anti-ganglioside antibodies in amyotrophic lateral sclerosis revisited. PLoS One. (2015) 10:e0125339. doi: 10.1371/journal.pone.0125339
106. Dalakas, MC, and Engel, WK. Immunoglobulin and complement deposits in nerves of patients with chronic relapsing polyneuropathy. Arch Neurol. (1980) 37:637640
107. Dubey, D. Autoimmune neuromuscular disorders associated with neural antibodies. Continuum. (2024) 30:1136–59. doi: 10.1212/CON.0000000000001461
108. Dubey, D, Lennon, VA, Gadoth, A, Pittock, SJ, Flanagan, EP, Schmeling, JE, et al. Autoimmune CRMP5 neuropathy phenotype and outcome defined from 105 cases. Neurology. (2018) 90:e103–10. doi: 10.1212/WNL.0000000000004803
109. Dubey, D, Jitprapaikulsan, J, Bi, H, do Campo, RV, McKeon, A, Pittock, SJ, et al. Amphiphysin-IgG autoimmune neuropathy: a recognizable clinicopathologic syndrome. Neurology. (2019) 93:e1873–80. doi: 10.1212/WNL.0000000000008472
110. Camdessanché, JP, Ferraud, K, Boutahar, N, Lassabliére, F, Mutter, M, Touret, M, et al. The collapsin response mediator protein 5 onconeural protein is expressed in Schwann cells under axonal signals and regulates axon-Schwann cell interactions. J Neuropathol Exp Neurol. (2012) 71:298–311. doi: 10.1097/NEN.0b013e31824d1df2
111. Antoine, JC, and Camdessanché, JP. Paraneoplastic neuropathies. Curr Opin Neurol. (2017) 30:513–20. doi: 10.1097/WCO.0000000000000475
112. Graus, F, Vogrig, A, Muñiz-Castrillo, S, Antoine, JCG, Desestret, V, Dubey, D, et al. Updated diagnostic criteria for paraneoplastic neurologic syndromes. Neurol Neuroimmunol Neuroinflamm. (2021) 8:e1014. doi: 10.1212/NXI.0000000000001014
113. Willison, HJ, Veitch, J, Swan, AV, Baumann, N, Comi, G, Gregson, NA, et al. Inter-laboratory validation of an ELISA for the determination of serum anti-ganglioside antibodies. Eur J Neurol. (1999) 6:71–7. doi: 10.1046/j.1468-1331.1999.610071.x
114. Collet, R, Caballero-Ávila, M, and Querol, L. Clinical and pathophysiological implications of autoantibodies in autoimmune neuropathies. Rev Neurol. (2023) 179:831–43. doi: 10.1016/j.neurol.2023.02.064
115. van Doorn, PA, van den Bergh, PYK, Hadden, RDM, Avau, B, Vankrunkelsven, P, Attarian, S, et al. European academy of neurology/peripheral nerve society guideline on diagnosis and treatment of Guillain-Barré syndrome. Eur J Neurol. (2023) 30:3646–74. doi: 10.1111/ene.16073
116. Joint Task Force of the EFNS and the PNS. European Federation of Neurological Societies/peripheral nerve society guideline on management of multifocal motor neuropathy. Report of a joint task force of the European Federation of Neurological Societies and the peripheral nerve society – first revision. J Peripher Nerv Syst. (2010) 15:295–301. doi: 10.1111/j.1529-8027.2010.00290.x
117. Martín-Aguilar, L, Lleixà, C, and Pascual-Goñi, E. Autoimmune nodopathies, an emerging diagnostic category. Curr Opin Neurol. (2022) 35:579–85. doi: 10.1097/WCO.0000000000001107
118. Querol, L, Nogales-Gadea, G, Rojas-Garcia, R, Martinez-Hernandez, E, Diaz-Manera, J, Suárez-Calvet, X, et al. Antibodies to contactin-1 in chronic inflammatory demyelinating polyneuropathy. Ann Neurol. (2013) 73:370–80. doi: 10.1002/ana.23794
119. Doppler, K, Appeltshauser, L, Villmann, C, Martin, C, Peles, E, Krämer, HH, et al. Auto-antibodies to contactin-associated protein 1 (Caspr) in two patients with painful inflammatory neuropathy. Brain. (2016) 139:2617–30. doi: 10.1093/brain/aww189
120. Querol, L, Nogales-Gadea, G, Rojas-Garcia, R, Diaz-Manera, J, Pardo, J, Ortega-Moreno, A, et al. Neurofascin IgG4 antibodies in CIDP associate with disabling tremor and poor response to IVIg. Neurology. (2014) 82:879–86. doi: 10.1212/WNL.0000000000000205
121. Delmont, E, Manso, C, Querol, L, Cortese, A, Berardinelli, A, Lozza, A, et al. Autoantibodies to nodal isoforms of neurofascin in chronic inflammatory demyelinating polyneuropathy. Brain. (2017) 140:1851–8. doi: 10.1093/brain/awx124
122. Vallat, JM, Yuki, N, Sekiguchi, K, Kokubun, N, Oka, N, Mathis, S, et al. Paranodal lesions in chronic inflammatory demyelinating polyneuropathy associated with antiNeurofascin 155 antibodies. Neuromuscul Disord. (2017) 27:290–3. doi: 10.1016/j.nmd.2016.10.008
123. Martín-Aguilar, L, Lleixà, C, Pascual-Goñi, E, Caballero-Ávila, M, Martínez-Martínez, L, Díaz-Manera, J, et al. Clinical and laboratory features in anti-NF155 autoimmune Nodopathy. Neurol Neuroimmunol Neuroinflamm. (2022) 9:e1098. doi: 10.1212/NXI.0000000000001129
124. Kira, JI. Anti-Neurofascin 155 antibody-positive chronic inflammatory demyelinating polyneuropathy/combined central and peripheral demyelination: strategies for diagnosis and treatment based on the disease mechanism. Front Neurol. (2021) 12:665136. doi: 10.3389/fneur.2021.665136
125. Dubey, D, Honorat, JA, Shelly, S, Klein, CJ, Komorowski, L, Mills, JR, et al. Contactin-1 autoimmunity: serologic, neurologic, and pathologic correlates. Neurol Neuroimmunol Neuroinflamm. (2020) 7:e771. doi: 10.1212/NXI.0000000000000771
126. Taieb, G, le Quintrec, M, Pialot, A, Szwarc, I, Perrochia, H, Labauge, P, et al. “Neuro‐renal syndrome” related to anti‐contactin‐1 antibodies. Muscle Nerve. (2019) 59:E19621. doi: 10.1002/mus.26392
127. Appeltshauser, L, Weishaupt, A, Sommer, C, and Doppler, K. Complement deposition induced by binding of anti-contactin-1 auto-antibodies is modified by immunoglobulins. Exp Neurol. (2017) 287:84–90. doi: 10.1016/j.expneurol.2016.10.006
128. Pascual-Goñi, E, Fehmi, J, Lleixà, C, Martín-Aguilar, L, Devaux, J, Höftberger, R, et al. Antibodies to the Caspr1/contactin-1 complex in chronic inflammatory demyelinating polyradiculoneuropathy. Brain. (2021) 144:1183–96. doi: 10.1093/brain/awab014
129. Liu, B, Zhou, L, Sun, C, Wang, L, Zheng, Y, Hu, B, et al. Clinical profile of autoimmune nodopathy with anti-neurofascin 186 antibody. Ann Clin Transl Neurol. (2023) 10:944–52. doi: 10.1002/acn3.51775
130. Uncini, A. Autoimmune nodo-paranodopathies 10 years later: clinical features, pathophysiology and treatment. J Peripher Nerv Syst. (2023) 28 Suppl 3:S23–35. doi: 10.1111/jns.12569
131. Stengel, H, Vural, A, Brunder, AM, Heinius, A, Appeltshauser, L, Fiebig, B, et al. Anti-pan-neurofascin IgG3 as a marker of fulminant autoimmune neuropathy. Neurol Neuroimmunol Neuroinflamm. (2019) 6:e603. doi: 10.1212/NXI.0000000000000603
132. Zhang, X, Kira, JI, Ogata, H, Imamura, T, Mitsuishi, M, Fujii, T, et al. Anti-LGI4 antibody is a novel Juxtaparanodal autoantibody for chronic inflammatory demyelinating polyneuropathy. Neurol Neuroimmunol Neuroinflamm. (2023) 10:e200081. doi: 10.1212/NXI.0000000000200081
133. Vernino, S, Low, PA, Fealey, RD, Stewart, JD, Farrugia, G, and Lennon, VA. Autoantibodies to ganglionic acetylcholine receptors in autoimmune autonomic neuropathies. N Engl J Med. (2000) 343:847–55. doi: 10.1056/NEJM200009213431204
134. Vernino, S, Lindstrom, J, Hopkins, S, Wang, Z, and Low, PAMuscle Study Group. Characterization of ganglionic acetylcholine receptor autoantibodies. J Neuroimmunol. (2008) 197:63–9. doi: 10.1016/j.jneuroim.2008.03.017
135. Li, Y, Jammoul, A, Mente, K, Li, J, Shields, RW, Vernino, S, et al. Clinical experience of seropositive ganglionic acetylcholine receptor antibody in a tertiary neurology referral center. Muscle Nerve. (2015) 52:386–91. doi: 10.1002/mus.24559
136. Gibbons, CH, and Freeman, R. Antibody titers predict clinical features of autoimmune autonomic ganglionopathy. Auton Neurosci. (2009) 146:8–12. doi: 10.1016/j.autneu.2008.11.013
137. Cutsforth-Gregory, JK, McKeon, A, Coon, EA, Sletten, DM, Suarez, M, Sandroni, P, et al. Ganglionic antibody level as a predictor of severity of autonomic failure. Mayo Clin Proc. (2018) 93:1440–7. doi: 10.1016/j.mayocp.2018.05.033
138. Sandroni, P, and Low, PA. Other autonomic neuropathies associated with ganglionic antibody. Auton Neurosci. (2009) 146:13–7. doi: 10.1016/j.autneu.2008.10.022
139. Urriola, N, and Adelstein, S. Autoimmune autonomic ganglionopathy: ganglionic acetylcholine receptor autoantibodies. Autoimmun Rev. (2022) 21:102988. doi: 10.1016/j.autrev.2021.102988
140. Karagiorgou, K, Dandoulaki, M, Mantegazza, R, Andreetta, F, Furlan, R, Lindstrom, J, et al. Novel cell-based assay for Alpha-3 nicotinic receptor antibodies detects antibodies exclusively in autoimmune autonomic Ganglionopathy. Neurol Neuroimmunol Neuroinflamm. (2022) 9:e1162. doi: 10.1212/NXI.0000000000001162
141. Camdessanche, JP, Jousserand, G, Ferraud, K, Vial, C, Petiot, P, Honnorat, J, et al. The pattern and diagnostic criteria of sensory neuronopathy: a case-control study. Brain. (2009) 132:1723–33. doi: 10.1093/brain/awp136
142. Mori, K, Iijima, M, Koike, H, Hattori, N, Tanaka, F, Watanabe, H, et al. The wide spectrum of clinical manifestations in Sjögren’s syndrome-associated neuropathy. Brain. (2005) 128:2518–34. doi: 10.1093/brain/awh605
143. Antoine, J-C, Boutahar, N, Lassabliere, F, Reynaud, E, Ferraud, K, Rogemond, V, et al. Antifibroblast growth factor receptor 3 antibodies identify a subgroup of patients with sensory neuropathy. J Neurol Neurosurg Psychiatry. (2015) 86:1347–55. doi: 10.1136/jnnp-2014-309730
144. Tholance, Y, Moritz, CP, Rosier, C, Ferraud, K, Lassablière, F, Reynaud-Federspiel, E, et al. Clinical characterisation of sensory neuropathy with anti-FGFR3 autoantibodies. J Neurol Neurosurg Psychiatry. (2020) 91:49–57. doi: 10.1136/jnnp-2019-321849
145. Kovvuru, S, Cardenas, YC, Huttner, A, Nowak, RJ, and Roy, B. Clinical characteristics of fibroblast growth factor receptor 3 antibody-related polyneuropathy: a retrospective study. Eur J Neurol. (2020) 27:1310–8. doi: 10.1111/ene.14180
146. Jungnickel, J, Klutzny, A, Guhr, S, Meyer, K, and Grothe, C. Regulation of neuronal death and calcitonin gene-related peptide by fibroblast growth factor-2 and FGFR3 after peripheral nerve injury: evidence from mouse mutants. Neuroscience. (2005) 134:1343–50. doi: 10.1016/j.neuroscience.2005.04.066
147. do, LD, Moritz, CP, Muñiz-Castrillo, S, Pinto, AL, Tholance, Y, Brugiere, S, et al. Argonaute autoantibodies as biomarkers in autoimmune neurologic diseases. Neurol Neuroimmunol Neuroinflamm. (2021) 8:e1032. doi: 10.1212/NXI.0000000000001032
148. Moritz, CP, Tholance, Y, Vallayer, PB, do, LD, Muñiz-Castrillo, S, Rogemond, V, et al. Anti-AGO1 antibodies identify a subset of autoimmune sensory Neuronopathy. Neurol Neuroimmunol Neuroinflamm. (2023) 10:e200105. doi: 10.1212/NXI.0000000000200105
149. Sagar, H, and Read, D. Subacute sensory neuropathy with remission: an association with lymphoma. J Neurol Neurosurg Psychiatry. (1982) 45:83–5. doi: 10.1136/jnnp.45.1.83
150. Denny-Brown, D. Primary sensory neuropathy with muscular changes associated with carcinoma. J Neurol Neurosurg Psychiatry. (1948) 11:73–87. doi: 10.1136/jnnp.11.2.73
151. Giometto, B, Grisold, W, Vitaliani, R, Graus, F, Honnorat, J, Bertolini, G, et al. Paraneoplastic neurologic syndrome in the PNS Euronetwork database: a European study from 20 centers. Arch Neurol. (2010) 67:330–5. doi: 10.1001/archneurol.2009.341
152. Camdessanché, J, Antoine, J, Honnorat, J, Vial, C, Petiot, P, Convers, P, et al. Paraneoplastic peripheral neuropathy associated with anti‐Hu antibodies. Brain. (2002) 125:166–75. doi: 10.1093/brain/awf006
153. Szabo, A, Dalmau, J, Manley, G, Rosenfeld, M, Wong, E, Henson, J, et al. HuD, a paraneoplastic encephalomyelitis antigen, contains RNA-binding domains and is homologous to Elav and sex-lethal. Cell. (1991) 67:325–33. doi: 10.1016/0092-8674(91)90184-Z
154. Schwenkenbecher, P, Chacko, LP, Wurster, U, Pars, K, Pul, R, Sühs, KW, et al. Intrathecal synthesis of anti-Hu antibodies distinguishes patients with paraneoplastic peripheral neuropathy and encephalitis. BMC Neurol. (2016) 16:136. doi: 10.1186/s12883-016-0657-5
155. Molinuevo, JL, Graus, F, Serrano, C, Reñe, R, Guerrero, A, and Illa, I. Utility of anti-Hu antibodies in the diagnosis of paraneoplastic sensory neuropathy. Ann Neurol. (1998) 44:976–80. doi: 10.1002/ana.410440620
156. Lladó, A, Mannucci, P, Carpentier, AF, Paris, S, Blanco, Y, Saiz, A, et al. Value of Hu antibody determinations in the follow-up of paraneoplastic neurologic syndromes. Neurology. (2004) 63:1947–9. doi: 10.1212/01.WNL.0000144340.03364.BF
157. Sabater, L, Gaig, C, Gelpi, E, Bataller, L, Lewerenz, J, Torres-Vega, E, et al. A novel non-rapid-eye movement and rapid-eye-movement parasomnia with sleep breathing disorder associated with antibodies to IgLON5: a case series, characterisation of the antigen, and post-mortem study. Lancet Neurol. (2014) 13:575–86. doi: 10.1016/S1474-4422(14)70051-1
158. Werner, J, Jelcic, I, Schwarz, EI, Probst-Müller, E, Nilsson, J, Schwizer, B, et al. Anti-IgLON5 disease: A new bulbar-onset motor neuron mimic syndrome. Neurol Neuroimmunol Neuroinflamm. (2021) 8:e962. doi: 10.1212/NXI.0000000000000962
159. Sista, SR, Crum, B, Aboseif, A, Devine, MF, Zekeridou, A, Hammami, MB, et al. Motor-neuron-disease-like phenotype associated with IgLON5 disease. J Neurol. (2022) 269:6139–44. doi: 10.1007/s00415-022-11262-0
160. Grüter, T, Möllers, FE, Tietz, A, Dargvainiene, J, Melzer, N, Heidbreder, A, et al. Clinical, serological and genetic predictors of response to immunotherapy in anti-IgLON5 disease. Brain. (2023) 146:600–11. doi: 10.1093/brain/awac090
161. Vogrig, A, Joubert, B, Maureille, A, Thomas, L, Bernard, E, Streichenberger, N, et al. Motor neuron involvement in anti-Ma2-associated paraneoplastic neurological syndrome. J Neurol. (2019) 266:398–410. doi: 10.1007/s00415-018-9143-x
162. Diard-Detoeuf, C, Dangoumau, A, Limousin, N, Biberon, J, Vourc'h, P, Andres, CR, et al. Association of a paraneoplastic motor neuron disease with anti-Ri antibodies and a novel SOD1 I18del mutation. J Neurol Sci. (2014) 337:212–4. doi: 10.1016/j.jns.2013.11.025
163. Dubey, D, Kryzer, T, Guo, Y, Clarkson, B, Cheville, JC, Costello, BA, et al. Leucine zipper 4 autoantibody: A novel germ cell tumor and paraneoplastic biomarker. Ann Neurol. (2021) 89:1001–10. doi: 10.1002/ana.26050
164. Terkelsen, AJ, Karlsson, P, Lauria, G, Freeman, R, Finnerup, NB, and Jensen, TS. The diagnostic challenge of small fibre neuropathy: clinical presentations, evaluations, and causes. Neurology. (2017) 16:934–44. doi: 10.1016/S1474-4422(17)30329-0
165. de Greef, BTA, Hoeijmakers, JGJ, Gorissen-Brouwers, CML, Geerts, M, Faber, CG, and Merkies, ISJ. Associated conditions in small fiber neuropathy - a large cohort study and review of the literature. Eur J Neurol. (2018) 25:348–55. doi: 10.1111/ene.13508
166. Pestronk, A, Choksi, R, Logigian, E, and Al-Lozi, MT. Sensory neuropathy with monoclonal IgM binding to a trisulfated heparin disaccharide. Muscle Nerve. (2003) 27:188–95. doi: 10.1002/mus.10301
167. Pestronk, A, Schmidt, RE, Choksi, RM, Sommerville, RB, and Al-Lozi, MT. Clinical and laboratory features of neuropathies with serum IgM binding to TS-HDS. Muscle Nerve. (2012) 45:866–72. doi: 10.1002/mus.23256
168. Levine, TD, Kafaie, J, Zeidman, LA, Saperstein, DS, Massaquoi, R, Bland, RJ, et al. Cryptogenic small-fiber neuropathies: serum autoantibody binding to trisulfated heparan disaccharide and fibroblast growth factor receptor-3. Muscle Nerve. (2020) 61:512–5. doi: 10.1002/mus.26748
169. Chompoopong, P, Rezk, M, Mirman, I, Berini, SE, Dyck, PJB, Mauermann, M, et al. TS-HDS autoantibody: clinical characterization and utility from real-world tertiary care center experience. J Neurol. (2023) 270:4523–8. doi: 10.1007/s00415-023-11798-9
170. Zeidman, LA, and Kubicki, K. Clinical features and treatment response in immune-mediated small Fiber neuropathy with Trisulfated heparin disaccharide or fibroblast growth factor receptor 3 antibodies. J Clin Neuromuscul Dis. (2021) 22:192–9. doi: 10.1097/CND.0000000000000355
171. Olsen, GM, Tormey, CA, Tseng, B, Hendrickson, JE, and Sostin, N. Therapeutic plasma exchange for peripheral neuropathy associated with trisulfated heparan disaccharide IgM antibodies: A case series of 17 patients. J Clin Apher. (2022) 37:13–8. doi: 10.1002/jca.21944
172. Gibbons, CH, Rajan, S, Senechal, K, Hendry, E, McCallister, B, and Levine, TD. A double-blind placebo-controlled pilot study of immunoglobulin for small fiber neuropathy associated with TS-HDS and FGFR-3 autoantibodies. Muscle Nerve. (2023) 67:363–70. doi: 10.1002/mus.27745
173. Fujii, T, Yamasaki, R, Iinuma, K, Tsuchimoto, D, Hayashi, Y, Saitoh, BY, et al. A novel autoantibody against Plexin D1 in patients with neuropathic pain. Ann Neurol. (2018) 84:208–24. doi: 10.1002/ana.25279
174. Fujii, T, Yamasaki, R, Miyachi, Y, Iinuma, K, Hashimoto, Y, Isobe, N, et al. Painful trigeminal neuropathy associated with anti-Plexin D1 antibody. Neurol Neuroimmunol Neuroinflamm. (2020) 7:e819. doi: 10.1212/NXI.0000000000000819
175. Fujii, T, Lee, EJ, Miyachi, Y, Yamasaki, R, Lim, YM, Iinuma, K, et al. Antiplexin D1 antibodies relate to small Fiber neuropathy and induce neuropathic pain in animals. Neurol Neuroimmunol Neuroinflamm. (2021) 8:e1028. doi: 10.1212/NXI.0000000000001028
176. Chan, ACY, Wong, HY, Chong, YF, Lai, PS, Teoh, HL, Ng, AYY, et al. Novel autoantibodies in idiopathic small Fiber neuropathy. Ann Neurol. (2022) 91:66–77. doi: 10.1002/ana.26268
177. De Wel, B, and Claeys, KG. Neuromuscular hyperexcitability syndromes. Curr Opin Neurol. (2021) 34:714–20. doi: 10.1097/WCO.0000000000000963
178. Comperat, L, Pegat, A, Honnorat, J, and Joubert, B. Autoimmune neuromyotonia. Curr Opin Neurol. (2022) 35:597–603. doi: 10.1097/WCO.0000000000001104
179. Binks, SNM, Klein, CJ, Waters, P, Pittock, SJ, and Irani, SR. LGI1, CASPR2 and related antibodies: a molecular evolution of the phenotypes. J Neurol Neurosurg Psychiatry. (2018) 89:526–34. doi: 10.1136/jnnp-2017-315720
180. Dawes, JM, Weir, GA, Middleton, SJ, Patel, R, Chisholm, KI, Pettingill, P, et al. Immune or genetic-mediated disruption of CASPR2 causes pain hypersensitivity due to enhanced primary afferent excitability. Neuron. (2018) 97:806–822.e10. doi: 10.1016/j.neuron.2018.01.033
181. Gastaldi, M, de Rosa, A, Maestri, M, Zardini, E, Scaranzin, S, Guida, M, et al. Acquired neuromyotonia in thymoma-associated myasthenia gravis: a clinical and serological study. Eur J Neurol. (2019) 26:992–9. doi: 10.1111/ene.13922
182. Fels, E, Muñiz-Castrillo, S, Vogrig, A, Joubert, B, Honnorat, J, and Pascual, O. Role of LGI1 protein in synaptic transmission: from physiology to pathology. Neurobiol Dis. (2021) 160:105537. doi: 10.1016/j.nbd.2021.105537
183. Farah, A, Patel, R, Poplawski, P, Wastie, BJ, Tseng, M, Barry, AM, et al. A role for leucine-rich, glioma inactivated 1 in regulating pain sensitivity. Brain. (2024):awae302. doi: 10.1093/brain/awae302
184. Lamb, GD. Rippling muscle disease may be caused by "silent" action potentials in the tubular system of skeletal muscle fibers. Muscle Nerve. (2005) 31:652–8. doi: 10.1002/mus.20307
185. Betz, RC, Schoser, BG, Kasper, D, Ricker, K, Ramírez, A, Stein, V, et al. Mutations in CAV3 cause mechanical hyperirritability of skeletal muscle in rippling muscle disease. Nat Genet. (2001) 28:218–9. doi: 10.1038/90050
186. Dubey, D, Beecher, G, Hammami, MB, Knight, AM, Liewluck, T, Triplett, J, et al. Identification of Caveolae-associated protein 4 autoantibodies as a biomarker of immune-mediated rippling muscle disease in adults. JAMA Neurol. (2022) 79:808–16. doi: 10.1001/jamaneurol.2022.1357
187. Svahn, J, Coudert, L, Streichenberger, N, Kraut, A, Gravier-Dumonceau-Mazelier, A, Rotard, L, et al. Immune-mediated rippling muscle disease associated with thymoma and anti-MURC/ cavin-4 autoantibodies. Neurol Neuroimmunol Neuroinflamm. (2023) 10:e200068. doi: 10.1212/NXI.0000000000200068
188. Selva-O'Callaghan, A, Pinal-Fernandez, I, Trallero-Araguás, E, Milisenda, JC, Grau-Junyent, JM, and Mammen, AL. Classification and management of adult inflammatory myopathies. Lancet Neurol. (2018) 17:816–28. doi: 10.1016/S1474-4422(18)30254-0
189. Lundberg, IE, Fujimoto, M, Vencovsky, J, Aggarwal, R, Holmqvist, M, Christopher-Stine, L, et al. Idiopathic inflammatory myopathies. Nat Rev Dis Primers. (2021) 7:86. doi: 10.1038/s41572-021-00321-x
190. Mariampillai, K, Granger, B, Amelin, D, Guiguet, M, Hachulla, E, Maurier, F, et al. Development of a new classification system for idiopathic inflammatory myopathies based on clinical manifestations and myositis-specific autoantibodies. JAMA Neurol. (2018) 75:1528–37. doi: 10.1001/jamaneurol.2018.2598
191. Lundberg, IE, Tjärnlund, A, Bottai, M, Werth, VP, Pilkington, C, Visser, M, et al. 2017 European league against rheumatism/American College of Rheumatology classification criteria for adult and juvenile idiopathic inflammatory myopathies and their major subgroups. Ann Rheum Dis. (2017) 76:1955–64. doi: 10.1136/annrheumdis-2017-211468
192. Betteridge, Z, and McHugh, N. Myositis-specific autoantibodies: an important tool to support diagnosis of myositis. J Intern Med. (2016) 280:8–23. doi: 10.1111/joim.12451
193. Pinal-Fernandez, I, Casal-Dominguez, M, and Mammen, AL. Immune-mediated necrotizing myopathy. Curr Rheumatol Rep. (2018) 20:21. doi: 10.1007/s11926-018-0732-6
194. Allenbach, Y, Benveniste, O, Stenzel, W, and Boyer, O. Immune-mediated necrotizing myopathy: clinical features and pathogenesis. Nat Rev Rheumatol. (2020) 16:689–701. doi: 10.1038/s41584-020-00515-9
195. Kaji, K, Fertig, N, Medsger, TA Jr, Satoh, T, Hoshino, K, Hamaguchi, Y, et al. Autoantibodies to RuvBL1 and RuvBL2: a novel systemic sclerosis-related antibody associated with diffuse cutaneous and skeletal muscle involvement. Arthritis Care Res. (2014) 66:575–84. doi: 10.1002/acr.22163
196. Lilleker, JB, Naddaf, E, Saris, CGJ, Schmidt, J, de Visser, M, Weihl, CC, et al. 272nd ENMC international workshop: 10 years of progress - revision of the ENMC 2013 diagnostic criteria for inclusion body myositis and clinical trial readiness. 16-18 June 2023, Hoofddorp, The Netherlands. Neuromuscul Disord. (2024) 37:36–51. doi: 10.1016/j.nmd.2024.03.001
197. Benjamin Larman, H, Salajegheh, M, Nazareno, R, Lam, T, Sauld, J, Steen, H, et al. Cytosolic 5′-nucleotidase 1A autoimmunity in sporadic inclusion body myositis. Ann Neurol. (2013) 73:408–18. doi: 10.1002/ana.23840
198. Lloyd, TE, Christopher-Stine, L, Pinal-Fernandez, I, Tiniakou, E, Petri, M, Baer, A, et al. Cytosolic 5′-nucleotidase 1A as a target of circulating autoantibodies in autoimmune diseases. Arthritis Care Res. (2016) 68:66–71. doi: 10.1002/acr.22600
199. Amlani, A, Choi, MY, Tarnopolsky, M, Brady, L, Clarke, AE, Garcia-de la Torre, I, et al. Anti-NT5c1A autoantibodies as biomarkers in inclusion body myositis. Front Immunol. (2019) 10:745. doi: 10.3389/fimmu.2019.00745
200. Lee, SA, Lee, HJ, Suh, BC, Shin, HY, Kim, SW, Yoon, BA, et al. Clinical significance of anti-NT5c1A autoantibody in Korean patients with inflammatory myopathies. PLoS One. (2023) 18:e0284409. doi: 10.1371/journal.pone.0284409
201. Tansley, SL, Li, D, Betteridge, ZE, and McHugh, NJ. The reliability of immunoassays to detect autoantibodies in patients with myositis is dependent on autoantibody specificity. Rheumatology. (2020) 59:2109–14. doi: 10.1093/rheumatology/keaa021
202. Halilu, F, and Christopher-Stine, L. Myositis-specific antibodies: overview and clinical utilization. Immunol Res. (2022) 3:1–10. doi: 10.2478/rir-2022-0001
203. Ghirardello, A, Rampudda, M, Ekholm, L, Bassi, N, Tarricone, E, Zampieri, S, et al. Diagnostic performance and validation of autoantibody testing in myositis by a commercial line blot assay. Rheumatology. (2010) 49:2370–4. doi: 10.1093/rheumatology/keq281
204. Damoiseaux, J, Mammen, AL, Piette, Y, Benveniste, O, Allenbach, Y, Allenbach, Y, et al. 256th ENMC international workshop: myositis specific and associated autoantibodies (MSA-ab): Amsterdam, The Netherlands, 8-10 October 2021. Neuromuscul Disord. (2022) 32:594–608. doi: 10.1016/j.nmd.2022.05.011
Keywords: autoantibodies, neuromuscular diseases, detection method, clinical practice, review
Citation: Loser V, Vicino A and Théaudin M (2024) Autoantibodies in neuromuscular disorders: a review of their utility in clinical practice. Front. Neurol. 15:1495205. doi: 10.3389/fneur.2024.1495205
Edited by:
Jian-Quan Shi, Nanjing Medical University, ChinaReviewed by:
Wladimir Bocca Vieira De Rezende Pinto, Federal University of São Paulo, BrazilLuca Leonardi, Sapienza University of Rome, Italy
Copyright © 2024 Loser, Vicino and Théaudin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Valentin Loser, dmFsZW50aW4ubG9zZXJAY2h1di5jaA==