 Shu-Yan Feng1,2†
Shu-Yan Feng1,2†- 1Department of Neurophysiology, Henan Provincial People's Hospital, Zhengzhou, China
- 2Zhengzhou University People's Hospital, Zhengzhou, China
- 3Department of Neurology, Fujian Medical University Union Hospital, Fuzhou, China
- 4Institute of Clinical Neurology, Fujian Medical University, Fuzhou, China
Mutations in the valosin-containing protein (VCP) gene have been linked to amyotrophic lateral sclerosis (ALS) in the Caucasian populations. However, the phenotype of VCP mutations in Chinese patients with (ALS) remains unclear. Targeted next-generation sequencing covered 28 ALS-related genes including the VCP gene was undertaken to screen in a Chinese cohort of 275 sporadic ALS cases and 15 familial ALS pedigrees. An extensive literature review was performed to identify all patients with ALS carrying VCP mutations previously reported. The clinical characteristics and genetic features of ALS patients with VCP mutations were reviewed. One known p.R155C mutation in the VCP gene was detected in two siblings from a familial ALS pedigree and two sporadic individuals. In addition, the same VCP p.R155C mutation was detected in an additional patient with ALS referred in 2021. Three patients with VCP p.R155C mutation presented with muscular weakness starting from proximal extremities to distal extremities. The other patient developed a phenotype of Paget's disease of bone in addition to the progressive muscular atrophy. We reported the first VCP mutation carrier manifesting ALS with Paget's disease of bone in the Chinese population. Our findings expand the phenotypic spectrum of the VCP mutations in Chinese patients with ALS and suggest that ALS patients with VCP p.R155C mutations tend to present with relatively young onset, symmetrical involvement of proximal muscles weakness of arms or legs, and then progressed to distal muscles of limbs.
Introduction
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disorder characterized by motor dysfunction in limbs (muscle weakness, atrophy, and spasticity) and bulbar palsy, such as dysarthria and dysphagia. Cognitive decline or behavioral impairment occurs in some cases. Approximate two-thirds of patients with ALS had a limb onset. The typical disease course of ALS cases is aggressive, ending in death due to respiratory failure within 3–5 years (1).
Approximately 5–10% of ALS cases present in a familial pattern, while the others have no family history. The genetic background of ALS is complicated, correlated with a growing spectrum of genes, such as C9orf72, SOD1, FUS, and TDP-43 (1). Valosin-containing protein (VCP) gene codes a highly conserved triple A-adenosine triphosphatase (triple A-ATPase), which operates as a regulatory factor in the procedure of endoplasmic reticulum-associated degradation (2). When the triple A-ATPase has functional deficits, the ubiquitin-dependent recycling or degradation by the proteasome will be disrupted. Additionally, the process of membrane fusion, transcriptional activation, and apoptosis heavily relies on the VCP-coded ATPase (3). Mutations in the VCP gene have been determined as a causative gene of the syndrome, inclusion body myopathy (IBM) with Paget's disease of bone (PDB), and frontotemporal dementia (FTD) (IBMPFD) since 2001 (4). The clinical spectrum of VCP gene-related diseases was expanded to ALS when a four-generation Italian ALS pedigree with VCP mutation was detected by whole-exome sequencing in 2010 (5). Subsequent studies in Caucasian populations detected VCP mutations in both familial and sporadic patients with ALS (5, 6). Mutations in the VCP gene in patients with ALS of Chinese origin have been rarely reported (7). Here, we reported the phenotype of four patients with ALS carrying VCP mutation of Chinese origin.
Materials and Methods
Subjects
In total, a cohort of 275 sporadic ALS cases and 15 familial ALS pedigrees was recruited at Fujian Medical University Union Hospital and Henan Provincial People's Hospital between January 2017 and December 2018. Another sporadic ALS case referred to Fujian Medical University Union Hospital in 2021 was included. Diagnosis of ALS was made according to the revised El Escorial criteria (8). Familial ALS was diagnosed if one or more first- or second-degree relatives developed ALS. The study was approved by the Ethics Committee of Fujian Medical University Union Hospital and Henan Provincial People's Hospital. All subjects involved in this research have offered written consent.
Genetic Studies
Genomic DNA extracted from venous peripheral blood lymphocytes of both sporadic cases and the proband of the family were subjected to the targeted next-generation sequencing on Illumina Hiseq sequencer (Illumina Inc., San Diego, CA, USA). An ALS-specific gene panel which included 28 genes (SOD1, FUS, TARDBP, VCP, VAPB, SPG11, OPTN, PFN1, ANG, ALS2, DAO, UBQLN2, SIGMAR1, SETX, FIG4, DCTN1, TUBA4A, TBK1, SQSTM1, CHCHD10, MATR3, HNRNPA1, HNRNPA2B1, KIF5A, ANXA11, TIA1, CCNF, and NEK1) was designed. The targeted regions were designed to include all exons and flanking regions of the 29 genes which contained the VCP gene (NM_007126.5). The GGGGCC expansions in C9orf72 were screened as previously described (9).
As a result of sequencing, the mean on-target coverage was 880 × with an average percentage of targets covered greater or equal to 100 × of 100%. Variant filtering process has been described (10). The identified variants were subsequently validated by Sanger sequencing. Bioinformatic analysis of the variants was performed as previously described (10).
Literature Review
We conducted a literature search in Medline to identify previous studies that screened for VCP mutations in patients with ALS. The following keywords were used: “Valosin-containing protein” OR “VCP”, in combination with “amyotrophic lateral sclerosis” OR “ALS” OR “motor neuron(e) disease” OR “MND”. Only English language literature was included in the review. For each eligible publication, the following information was extracted: name of the first author, publication year, population, the sample size of familial ALS (FALS), and/or sporadic ALS (SALS), numbers of VCP mutation carriers in FALS and/or SALS. For patients with ALS carrying VCP mutations, the following information was extracted from the relevant papers: first author, year of study, population, sex, age, family history, clinical features, and genetic characteristics.
Statistical Analysis
In each study, the mutation frequencies of the VCP gene were reported as the number of the mutation carriers among all cases of FALS or SALS screened. Single case reports, such as the additional VCP p.R155C mutated ALS referred to our center in 2021 were not included in the meta-analysis. The mutation frequencies in different populations were combined using a fixed-effects model. A statistical analysis was carried out using the Meta function of R (R version 3.64) (https://www.r-project.org/).
Results
Clinical Features of the ALS Cohort
Between January 2017 and December 2018, 275 sporadic cases and 15 familial pedigrees meeting the diagnostic criteria of ALS were enrolled in our study. There were 173 men and 117 women with mean onset age of 55.3 years (SD, 11.6). Of the total cases, 19.3% of patients reported a bulbar onset, 79.0% had a limb onset, and 1.7% had a respiratory onset.
Genetic Analysis
One known heterozygous missense mutation in the VCP gene, c.463C>T (p.R155C) (Figure 1A) was identified in one familial ALS proband (III-6) and his affected sister (III-5) (Figure 1B), as well as another sporadic patient in the cohort of 275 sporadic ALS cases and 15 familial ALS pedigrees. The same VCP p.R155C mutation was detected in the additional patient referred to Fujian Medical University Union Hospital in 2021. No parental DNA samples of patient 3 and patient 4 were available for sequencing. No variants in other ALS-related genes were identified in these patients.
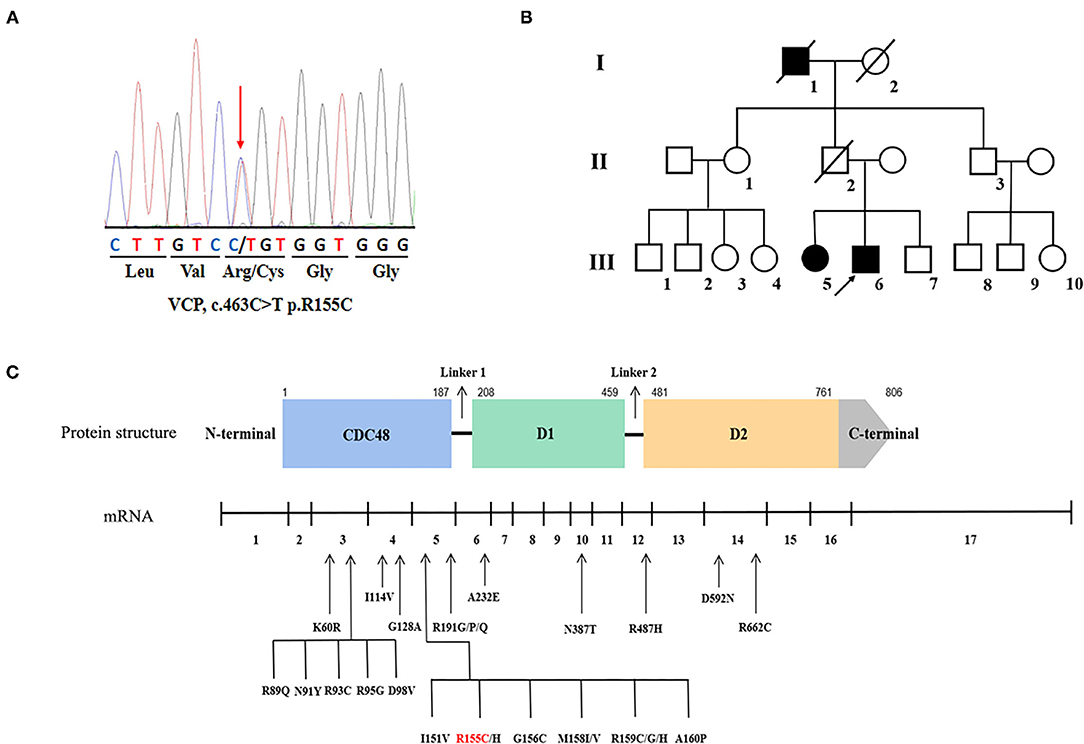
Figure 1. Genetic information of the valosin-containing protein (VCP) mutation in our study and overview of the VCP mutations associated with amyotrophic lateral sclerosis (ALS). (A) Sequencing chromatograms of the VCP p.R155C mutation. (B) The pedigree of the familial ALS with VCP p.R155C mutation. Arrow indicates the index patient. (C) Schematic graph of the VCP protein and overview of the VCP mutations linked to ALS. The locations of mutations are depicted in the mRNA structure where exons are numbered 1–17.
Clinical Features of Patients With VCP p.R155C Mutation
Clinical features of the four patients carrying VCP p.R155C mutations in this study are summarized in Table 1.
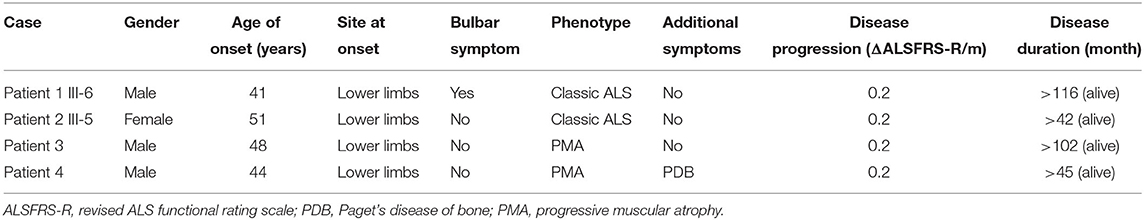
Table 1. Clinical features of the amyotrophic lateral sclerosis (ALS) patients with valosin-containing protein (VCP) p.R155C mutation in this study.
The familial ALS proband (III-6, Patient 1) was a 51-year-old male with progressive weakness in four limbs. At the age of 42 years, he complained of weakness in the lower limbs. He felt clumsy when climbing upstairs and unusual fatigue when walking a long distance. He began to have difficulties in uplifting his arms 4 years later. In the following years, he developed weakness of hands. No cognitive deficits or behavioral changes were noted. The neurologic examination at 7 years after onset revealed obvious weakness and atrophy of four limbs (MRC 4/5), more severe in the proximal muscles of limbs, with diffused reduction of deep tendon reflexes. Babinski's sign was elicited bilaterally. The sensory system and cognition were not affected. Serum creatine kinase (CK) level was normal. Electromyography (EMG) demonstrated fibrillations and positive sharp waves in muscles of four limbs and left thoracic paravertebral muscles, with muscle unit action potentials of increased amplitude, prolonged duration, and reduced recruitment in muscles of limbs. The ALS Functional Rating Scale-Revised (ALSFRS-R) score was 39/48. The score of Montreal Cognitive Assessment (MoCA) testing was 27/30. He developed exertional dyspnea 9 years after onset and began to use the non-invasive positive-pressure ventilation. At the last follow-up at 116 months after onset, he could still walk slowly with help and do daily life activities without assistance. He had neither dysarthria nor dysphagia. A repeated ALSFRS-R score was 30 with an estimated progression rate of 0.2 score/month since symptom onset. The sister of proband (III-5, Patient 2) had paraparesis in the legs since she was 51 years. She only had some trouble running and climbing upstairs. Neurological examination at 2 years after onset revealed mild atrophy and weakness of lower limbs (MRC 4/5). The deep tendon reflexes were brisk in four limbs. Hoffmann's sign and Babinski's sign were elicited bilaterally, with no sensory and bulbar involvement. Serum CK level was normal. EMG revealed acute and chronic neurogenic changes in all limbs and thoracic muscles. The ALSFRS-R score was 46/48. The score of MoCA testing was 28/30. At the last follow-up at 42 months after onset, she could still walk and go upstairs slowly with a cane, without the involvement of upper limbs. The ALSFRS-R score was 41 with an estimated progression rate of 0.2 score/month since symptom onset. Their father (II-2) died of acute myocardial infarction at the age of 72 years without evident symptoms of limb weakness, and their unaffected mother was still alive. The grandfather (I-1) of the proband exhibited signs of weakness in lower limbs and stayed bedridden for over 10 years before he died in his 70s. However, he was not formally diagnosed with ALS.
Patient 3 was a 57-year-old male who noticed weakness in the legs at the age of 48 years. Initially, he only had some difficulties in climbing stairs and standing up from a squatting position. In the following years, the weakness progressed distally to the feet and he began to have troubles in running and long-distance walking. Four years later, the patient was referred to physicians for his difficulty in lifting his arms and aggravated trouble of walking. There were no behavioral symptoms. Neurological examination 5 years after onset revealed obvious atrophy of muscles of arms and lower limbs with fasciculations. Deep tendon reflexes were decreased in all limbs. Palm-chin reflexes and Babinski's sign was not elicited. EMG indicated the pattern of neurogenic changes in muscles of four limbs, such as abnormal spontaneous potentials, motor unit potentials of prolonged duration, increased amplitude, and reduced recruitment. Serum CK level was 298 IU/L (normal value 22–270 IU/L). The biopsy sample of the biceps muscle demonstrated grouped atrophic fibers with both type I and type II fibers. He scored 42/48 on ALSFRS-R and 28/30 on MoCA testing. He began to use wheelchair 7 years after onset. At the last follow-up visit at 102 months after onset, he was bedridden, but still independent for daily life activities, such as writing, dressing, and eating meals, without bulbar involvement. The ALSFRS-R score was 31/48 with an estimated progression rate of 0.2 score/month since symptom onset. His parents were in their 80s and healthy.
Patient 4 presented with weakness of the left leg at the age of 44 years and felt clumsy when climbing upstairs. After 2 years, he developed weakness of the right leg. Muscle atrophy with fasciculation in lower limbs was noticed. Serum CK level was normal. Serum alkaline phosphatase level was 1,532 U/L (normal value 50–135 IU/L). Shoulder, pelvic, and lumbar spine radiographs showed osteolysis, osteosclerosis, and cortical thickening. An isotope bone scan showed increased tracer uptake in the affected bones. He did not suffer from bone pain. He was diagnosed with PDB and treated with zoledronic acid injection, calcitriol, and calcium carbonate. The serum alkaline phosphatase level decreased obviously to about 200 U/L. He gradually developed weakness of both arms and had difficulties lifting arms. Neurological examination at 33 months after onset revealed obvious muscle atrophy of arms and legs with fasciculations. Muscle strength was decreased in the upper limbs (MRC 4/5 in distal limbs and MRC 3/5 in proximal limbs) and the lower limbs (MRC 3/5 in distal limbs and MRC 4/5 in proximal limbs). Deep tendon reflexes were decreased in all limbs. Babinski and Hoffman's signs were not elicited. He was cognitively normal, with a score of 28/30 on MoCA testing. He scored 42/48 on ALSFRS-R. EMG demonstrated chronic and active denervation of the upper and lower limbs, rectus abdominis, and sternocleidomastoid muscles. At the last phone follow-up at 45 months after onset, he could still walk independently for 1 km and do daily life activities, such as writing, dressing, eating meals, and driving, without bulbar involvement. The ALSFRS-R score was 39/48 with an estimated progression rate of 0.2 score/month since symptom onset. Both parents of patient 4 were healthy in their 70s.
Prevalence of VCP Mutations in Patients With ALS in Different Populations
We identified 44 studies screened VCP mutations in patients with FALS and/or SALS (Table 2). The frequency of VCP mutations is 0.08% (95% CI 0.00–1.26%) in patients with FALS and 0.02% (95% CI 0.00–0.15%) in patients with SALS in Chinese. The frequency of VCP mutations is 0.37% (95% CI 0.00–1.43%) in patients with FALS and 0.09% (95% CI 0.00–0.38%) in patients with SALS in Japanese. The frequency of VCP mutations is 0.28% (95% CI 0.12–0.52%) in patients with FALS and 0.08% (95% CI 0.03–0.15%) in patients with SALS in Caucasian populations. In pooled analysis, the frequency of VCP mutations is 0.28% (95% CI 0.12–0.50%) in patients with FALS and 0.06% (95% CI 0.02–0.12%) in patients with SALS (Table 2).
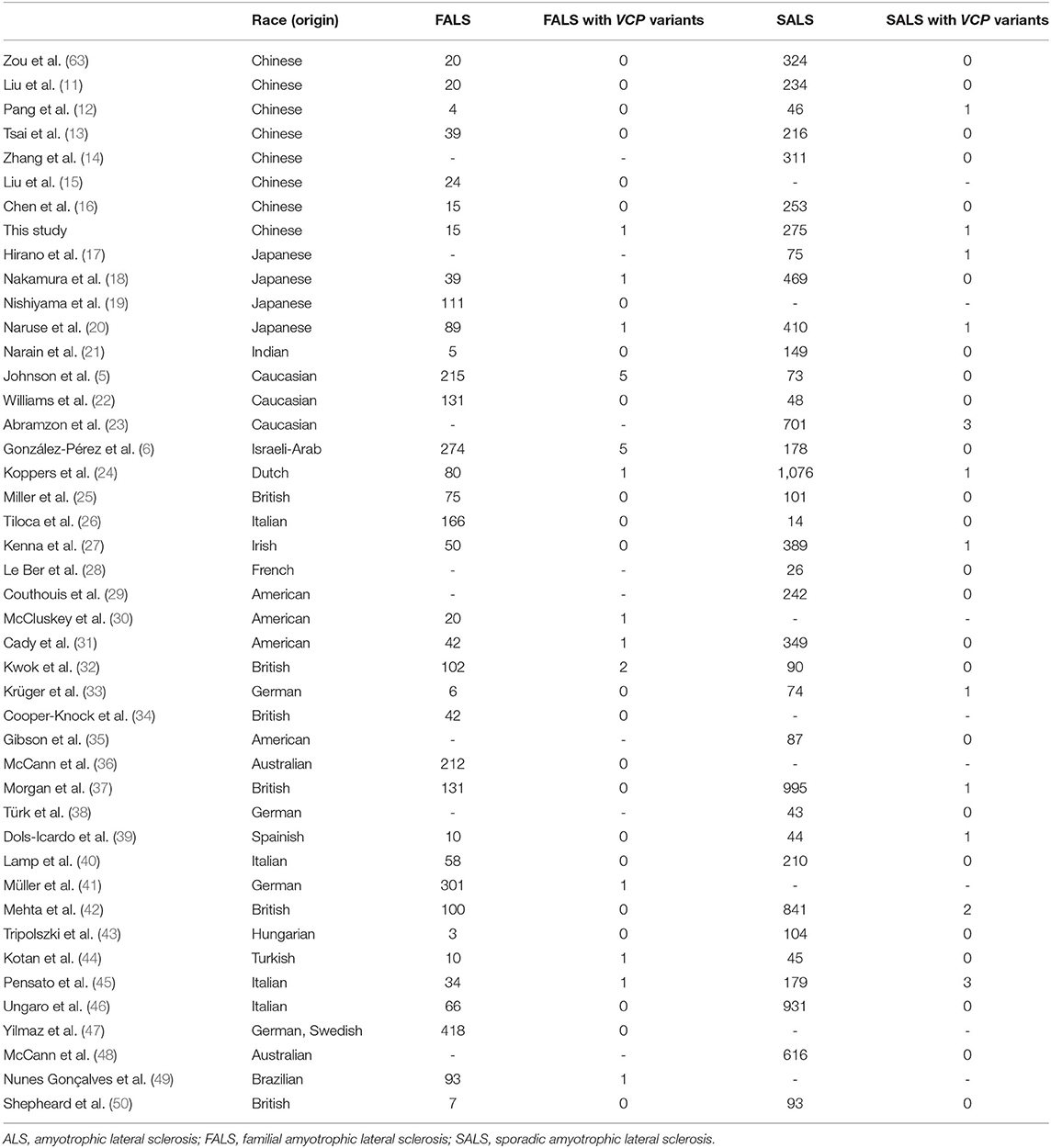
Table 2. Summary of studies screened VCP variants in patients with ALS.
Literature Review of the Phenotype of ALS Patients With VCP Mutations
In addition, 46 VCP mutated ALS patients with detailed clinical features in previous research were identified. The clinical characteristics of these patients are summarized in Table 3. The mean age at onset was 50.29 ± 10.55 years, ranging from 24 to 68 years. Most of the VCP-related patients with ALS were Caucasian (76.1%, 35/46), only 9 cases were of Asian origin (19.6%, 9/46). Thirty-seven patients (80.4%) had a family history of ALS, FTD, dementia, parkinsonism, or PDB, including 31 patients (67.4%) who had a family history of ALS. Thirty-two (69.6%) patients claimed that the weakness started from limbs, and only six 13.0% patients developed a bulbar-onset. Co-occurrence of FTD, PDB, parkinsonism, myopathy, or psychiatric diseases was reported in 18 (39.1%, 18/46) ALS cases carrying VCP mutations.
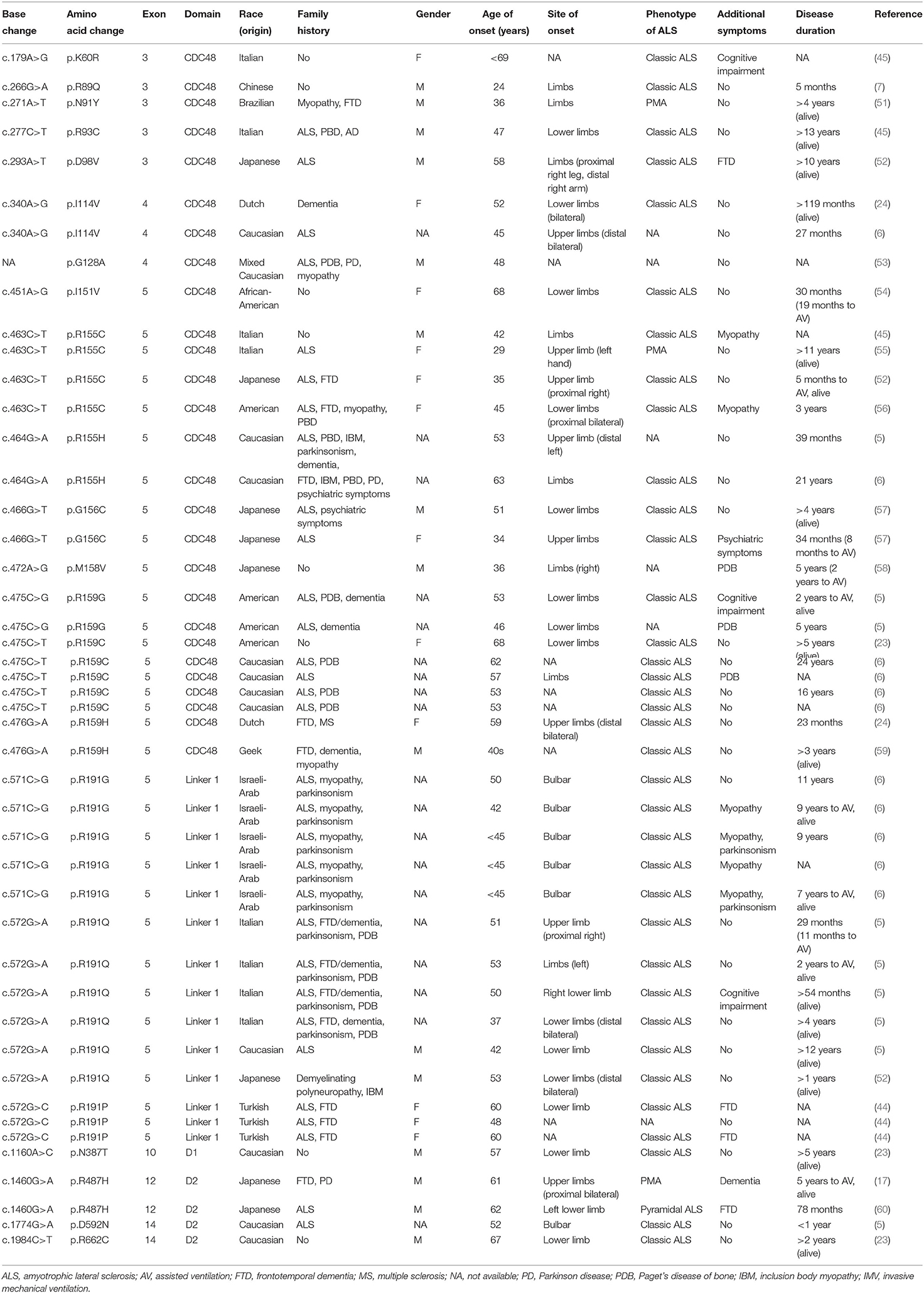
Table 3. Summary of previously published VCP-causing ALS cases with detailed records.
Discussion
In the present study, we reported the phenotype of four patients carrying the known VCP p.R155C mutation, such as two siblings from a familial ALS pedigree and a sporadic individual from a Chinese ALS cohort of 275 sporadic and 15 familial ALS pedigrees.
Mutations in the VCP gene have previously been identified in Caucasian patients with ALS (5, 6, 24, 27, 45, 61, 62). Our meta-analysis showed that the presence of VCP mutations was 0.28% (95% CI 0.12–0.52%) in FALS and 0.08% (95% CI 0.03–0.15%) in patients with SALS in the Caucasian populations. The prevalence of VCP mutations in the Asian populations has not been well determined since the published ALS-VCP cases were mostly of Japanese origin (17, 52, 57, 58, 60). A VCP p.R487H mutation was identified in one out of 75 Japanese patients with SALS (1.3%) (17), and a p.R155C mutation was detected in one out of 39 Japanese FALS pedigrees (2.6%) but no VCP mutations were found in 469 SALS individuals (18). Our meta-analysis showed that the presence of VCP mutations was 0.37% (95% CI 0.00–1.43%) in FALS and 0.09% (95% CI 0.00–0.38%) in patients with SALS in Japanese (17–20). No VCP mutations have been discovered among Chinese patients with ALS (11, 13–16, 63) except Pang SY et al. reported a p.G157R mutation in one out of 46 SALS (12) and a recent study identified a p.R89Q mutation in one SALS case in a cohort of 27 unrelated young-onset patients with ALS (7). The higher frequency of VCP mutations in FALS (6.7%, 1/15) and SALS (0.4%, 1/275) in our study may be due to the small sample. Our meta-analysis showed that the frequency of VCP mutations is 0.08% (95% CI 0.00–1.26%) in FALS and 0.02% (95% CI 0.00–0.15%) in SALS in Chinese, which is lower than Japanese and Caucasian populations. The overall pooled mutation frequency of VCP mutations is 0.28% (95% CI 0.12–0.50%) in patients with FALS and 0.06% (95% CI 0.02–0.12%) in patients with SALS.
Valosin-containing protein p.R155C mutation was first associated with ALS in 2012 (6), before which it was found in patients with IBMPFD (64). The mutation was subsequently observed in two ALS cases without FTD or PBD from a large cohort study of 190 individuals carrying VCP variants (61) and another survey on 36 families with diverse VCP mutants (62). The onset age of the VCP p.R155C mutated patients in our study ranged from 42 to 51 years, consistent with the onset age of four VCP p.R155C-mutated ALS cases reported previously (29, 35, 42, 45, respectively) (Table 3). It seems that patients carrying VCP p.R155C mutations tend to have a young onset. The three patients carrying VCP p.R155C mutation in our study all had a limb-onset. Our system review revealed that 69.6% of VCP-related ALS had limb-onset ALS, and only patients carrying VCP p.R191G and p.D592N mutation had bulbar onset (Table 3). Patient 1 and 2 in our study demonstrated a phenotype of ALS while patient 3 had a phenotype of progressive muscular atrophy (PMA). They all presented with rather symmetrical proximal muscle weakness in the lower limbs at onset and subsequently progressed to distal lower limbs and upper limbs. Patient 4 manifested weakness in the left leg and developed a phenotype of PMA. Interestingly, two VCP p.R155C mutated patients with ALS reported previously also presented with symmetrical involvement of proximal muscles weakness of arms or legs (52, 56). The system review of previous studies showed that patients carrying VCP p.R487H and p.R159G mutation demonstrated the same phenotype (5, 17). The four p.R155C mutation carriers in our study demonstrated a slow progression (ΔALSFRS-R/month = 0.2 score/month) and two had a long survival duration of more than 102 months and 116 months, respectively. The grandfather (I-1) of the proband also exhibited signs of weakness in lower limbs and stayed bedridden for over 10 years before he died in his 70s. An Italian ALS patient with VCP p.R155C mutation reported by Battistini et al. also had a survival of more than 11 years (55). However, a 35-year-old female with p.R155C mutation progressed rapidly and received tracheotomy positive-pressure ventilation within 5 months after onset (52). It is interesting that in our study the father of proband who was supposed to carry the VCP p.R155C mutation had no symptom of muscle weakness and atrophy before he died of acute myocardial infarction at the age of 72 years, indicated incomplete penetrance of VCP p.R155C mutation. Phenotypic variability in VCP p.R155C mutated ALS pedigree has been reported. In an Italian family, the proband with harboring VCP p.R155C mutation developed young-onset ALS with diffuse severe weakness and wasting in the limbs and rimmed vacuoles in muscle biopsy, while his mother and maternal aunt suffered from mild symptoms limited to lower limbs (45). Our system review showed that ALS patients with some VCP mutations (p.R93C, p.D98V, p.I114V, p.R155C, p.R155H, p.R159C, p.R191G, and p.R191Q) had a relatively slow progression and survival of more than 10 years (Table 3), which is consistent with the phenotype of our patients. However, some ALS patients with VCP mutations (p.R89Q, p.I114V, p.I151V, p.R155C, p.G156C, p.R159H, p.R191Q, and p.D592N) developed a rapid progression and had a survival of fewer than 3 years (Table 3). Phenotype variability associated with different or the same VCP mutation suggests the possible role of modifying genes and/or environmental factors. Extensive genetic studies in different populations to identify more ALS patients with VCP mutations may provide more insight into the genotype-phenotype correlations and the diversity of clinical phenotypes of VCP mutations.
Patient 4 carrying VCP p.R155C mutation developed a phenotype of PDB in addition to PMA. A VCP p.G97E mutation was reported in a Chinese family with IBMPFD without ALS (65). Patient 4 in our study was the first VCP mutation carrier manifesting PDB in addition to ALS in the Chinese population. The co-occurrence of FTD, PDB, parkinsonism, myopathy, or psychiatric diseases was commonly seen in the ALS cases carrying VCP mutations (Table 3). The co-existence of FTD or cognitive impairment was found in patients with seven VCP mutations (p.K60R, p.D98V, p.R155C, p.R159G, p.R191Q, p.R191P, and p.R487H) (5, 17, 44, 45, 52, 56, 60), while PDB was diagnosed in ALS patients with six VCP mutations (p.R93C, p.G128A, p.R155C, p.M158V, p.R159G, and p.R159C) (5, 6, 45, 53, 56, 58). Co-occurrence of myopathy was reported in ALS patients with VCP p.R155C and p.R191G mutations (6, 45, 56), and psychiatric disorders were found in patients with p.G156C and p.R159G mutations (5, 57). Parkinsonism is presented in familial ALS cases with VCP p.R191G mutation (6). Increasing evidence has shown that VCP-related disease may be a multisystem proteinopathy that has a wide clinical spectrum of IBM, FTD, PDB, and parkinsonism apart from ALS (66–68). The phenotypic diversity of the same VCP mutation may indicate the possible role of modifying genes and/or environmental factors.
Except for the hexanucleotide expansions and single base-pair substitutions in the 5' UTR or 3' UTR region of VCP predicted as pathogenic without definite experimental evidence (32), 26 mutations in 17 exons of the VCP gene have been identified in patients with ALS (Figure 1C). All mutations are heterozygous missense mutations. Residues of R155, R159, and R191 are three hot spots of VCP mutations of patients with ALS. VCP is divided into 4 domains, such as one N-terminal CDC48 region, two AAA ATPase domains (D1 and D2), and one C-terminal domain. VCP mutations are predominantly located within the N-terminal ubiquitin-binding domain (69.2%, 18/26), indicating that the malfunction of poly-ubiquitinated protein degradation may be the major pathogenesis of ALS (2). Within the ring-shaped VCP hexamer, the N-terminal domain serves as an indispensable binding region for interaction with target co-factor molecules (2). When pathogenic mutations occur in the N-domain of VCP protein, such as p.R155C, p.R159H, p.R95G, p.G97E, and p.A232E, the stress response is impaired resulting in the incorrect translocation of this hexameric ATPase and assembly disorder (69). Besides the aberrant aggregation of transactive response DNA-binding protein of 43 kDa (TDP-43) (5, 70), the nuclear-to-cytoplasmic mislocalization of fused in sarcoma (FUS) protein in motor neurons has been identified as another pathogenic feature of VCP-mutated ALS, which was ascribed to the increased intron retention (IR) in splicing factor proline and glutamine rich (SFPQ) transcripts (71, 72). Using ALS patient-specific induced pluripotent stem cell (iPSC) models, Patani et al. further unveiled that the four abnormal accumulated sequence-specific intron retention transcripts (IRTs) in VCP mutations included SFPQ, OGT, TUSC3, and DDX39, and their binding affinity to RNA binding proteins (RBPs) could be the key attributes for RBP localization (73, 74). In future, further experiments on pathomechanism of VCP-mutated ALS should be conducted on transgenic animal models as well as patient-specific iPSC models.
In summary, we reported the first VCP mutation carrier manifesting ALS with PDB of ALS in the Chinese population. Our findings expand the phenotypic spectrum of VCP mutations in Chinese patients with ALS and suggest that ALS patients with VCP p.R155C mutation tend to present with relatively young onset, symmetrical involvement of proximal muscles weakness of arms or legs, and then progressed to distal muscles of limbs.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/, sra/PRJNA791140.
Ethics Statement
The studies involving human participants were reviewed and approved by Ethics Committee of Fujian Medical Union Hospital and Henan Provincial People's Hospital. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
Z-YZ and S-YF designed and conceived the study. Z-YZ, S-YF, and HL performed the analysis of mutations in all the patients. HL wrote the manuscript. Z-YZ critically revised the manuscript. All remaining authors participated in the analysis of data, discussion of the final manuscript.
Funding
This study was supported by grants from the National Natural Science Foundation of China (81671271 and 81974199) and Joint Funds for the Innovation of Science and Technology, Fujian province (2017Y9002).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank the patients and their families for their cooperation. Dr. Ton-Hai Dou from Shanghai Amplicongene Bioscience Co., Ltd., Shanghai, China, for kind help in the analysis of the sequencing data.
References
1. Brown RH, Al-Chalabi A. Amyotrophic lateral sclerosis. N Engl J Med. (2017) 377:162–72. doi: 10.1056/NEJMra1603471
2. Bodnar N, Rapoport T. Toward an understanding of the Cdc48/p97 ATPase. F1000Res. (2017) 6:1318. doi: 10.12688/f1000research.11683.1
3. Nishimura N, Radwan MO, Amano M, Endo S, Fujii E, Hayashi H, et al. Novel p97/VCP inhibitor induces endoplasmic reticulum stress and apoptosis in both bortezomib-sensitive and -resistant multiple myeloma cells. Cancer Sci. (2019) 110:3275–87. doi: 10.1111/cas.14154
4. Kovach MJ, Waggoner B, Leal SM, Gelber D, Khardori R, Levenstien MA, et al. Clinical delineation and localization to chromosome 9p13.3-p12 of a unique dominant disorder in four families: hereditary inclusion body myopathy. Paget disease of bone, and frontotemporal dementia. Mol Genet Metab. (2001) 74:458–75. doi: 10.1006/mgme.2001.3256
5. Johnson JO, Mandrioli J, Benatar M, Abramzon Y, Van Deerlin VM, Trojanowski JQ, et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron. (2010) 68:857–64. doi: 10.1016/j.neuron.2010.11.036
6. Gonzalez-Perez P, Cirulli ET, Drory VE, Dabby R, Nisipeanu P, Carasso RL, et al. Novel mutation in VCP gene causes atypical amyotrophic lateral sclerosis. Neurology. (2012) 79:2201–8. doi: 10.1212/WNL.0b013e318275963b
7. Deng J, Wu W, Xie Z, Gang Q, Yu M, Liu J, et al. Novel and recurrent mutations in a cohort of chinese patients with young-onset amyotrophic lateral sclerosis. Front Neurosci. (2019) 13:1289. doi: 10.3389/fnins.2019.01289
8. Brooks BR, Miller RG, Swash M, Munsat TL. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. (2000) 1:293–9. doi: 10.1080/146608200300079536
9. Zou ZY, Li XG, Liu MS, Cui LY. Screening for C9orf72 repeat expansions in Chinese amyotrophic lateral sclerosis patients. Neurobiol Aging. (2013) 34:1710.e1715–1716. doi: 10.1016/j.neurobiolaging.2012.11.018
10. Feng SM, Che CH, Feng SY, Liu CY, Li LY, Li YX, et al. Novel mutation in optineurin causing aggressive ALS+/-frontotemporal dementia. Ann Clin Transl Neurol. (2019) 6:2377–83. doi: 10.1002/acn3.50928
11. Liu Q, Liu F, Cui B, Lu CX, Guo XN, Wang RR, et al. Mutation spectrum of Chinese patients with familial and sporadic amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. (2016) 87:1272–4. doi: 10.1136/jnnp-2016-313337
12. Pang SY, Hsu JS, Teo KC, Li Y, Kung MHW, Cheah KSE, et al. Burden of rare variants in ALS genes influences survival in familial and sporadic ALS. Neurobiol Aging. (2017) 58:238.e239–e215. doi: 10.1016/j.neurobiolaging.2017.06.007
13. Tsai PC, Liao YC, Chen PL, Guo YC, Chen YH, Jih KY, et al. Investigating CCNF mutations in a Taiwanese cohort with amyotrophic lateral sclerosis. Neurobiol Aging. (2018) 62:243.e241–6. doi: 10.1016/j.neurobiolaging.2017.09.031
14. Zhang H, Cai W, Chen S, Liang J, Wang Z, Ren Y, et al. Screening for possible oligogenic pathogenesis in Chinese sporadic ALS patients. Amyotroph Lateral Scler Frontotemporal Degener. (2018) 19:419–25. doi: 10.1080/21678421.2018.1432659
15. Liu ZJ, Lin HX, Wei Q, Zhang QJ, Chen CX, Tao QQ, et al. Genetic spectrum and variability in chinese patients with amyotrophic lateral sclerosis. Aging Dis. (2019) 10:1199–206. doi: 10.14336/AD.2019.0215
16. Chen W, Xie Y, Zheng M, Lin J, Huang P, Pei Z, et al. Clinical and genetic features of patients with amyotrophic lateral sclerosis in southern China. Eur J Neurol. (2020) 27:1017–22. doi: 10.1111/ene.14213
17. Hirano M, Nakamura Y, Saigoh K, Sakamoto H, Ueno S, Isono C, et al. VCP gene analyses in Japanese patients with sporadic amyotrophic lateral sclerosis identify a new mutation. Neurobiol Aging. (2015) 36:1604.e1601–6. doi: 10.1016/j.neurobiolaging.2014.10.012
18. Nakamura R, Sone J, Atsuta N, Tohnai G, Watanabe H, Yokoi D, et al. Next-generation sequencing of 28 ALS-related genes in a Japanese ALS cohort. Neurobiol Aging. (2016) 39:219.e211–8. doi: 10.1016/j.neurobiolaging.2015.11.030
19. Nishiyama A, Niihori T, Warita H, Izumi R, Akiyama T, Kato M, et al. Comprehensive targeted next-generation sequencing in Japanese familial amyotrophic lateral sclerosis. Neurobiol Aging. (2017) 53:194.e1–8. doi: 10.1016/j.neurobiolaging.2017.01.004
20. Naruse H, Ishiura H, Mitsui J, Takahashi Y, Matsukawa T, Tanaka M, et al. Burden of rare variants in causative genes for amyotrophic lateral sclerosis (ALS) accelerates age at onset of ALS. J Neurol Neurosurg Psychiatry. (2019) 90:537–42. doi: 10.1136/jnnp-2018-318568
21. Narain P, Pandey A, Gupta S, Gomes J, Bhatia R, Vivekanandan P. Targeted next-generation sequencing reveals novel and rare variants in Indian patients with amyotrophic lateral sclerosis. Neurobiol Aging. (2018) 71:265.e9–14. doi: 10.1016/j.neurobiolaging.2018.05.012
22. Williams KL, Solski JA, Nicholson GA, Blair IP. Mutation analysis of VCP in familial and sporadic amyotrophic lateral sclerosis. Neurobiol Aging. (2012) 33:1488.e15–e16. doi: 10.1016/j.neurobiolaging.2011.11.022
23. Abramzon Y, Johnson JO, Scholz SW, Taylor JP, Brunetti M, Calvo A, et al. Valosin-containing protein (VCP) mutations in sporadic amyotrophic lateral sclerosis. Neurobiol Aging. (2012) 33:2231.e1–6. doi: 10.1016/j.neurobiolaging.2012.04.005
24. Koppers M, van Blitterswijk MM, Vlam L, Rowicka PA, van Vught PW, Groen EJ VCP mutations in familial and sporadic amyotrophic lateral sclerosis. Neurobiol Aging. (2012) 33:837.e837–e813. doi: 10.1016/j.neurobiolaging.2011.10.006
25. Miller JW, Smith BN, Topp SD, Al-Chalabi A, Shaw CE, Vance C. Mutation analysis of VCP in British familial and sporadic amyotrophic lateral sclerosis patients. Neurobiol Aging. (2012) 33:2721.e1–2. doi: 10.1016/j.neurobiolaging.2012.06.003
26. Tiloca C, Ratti A, Pensato V, Castucci A, Sorar ù G, Del Bo R, et al. Mutational analysis of VCP gene in familial amyotrophic lateral sclerosis. Neurobiol Aging. (2012) 33:630.e1–2. doi: 10.1016/j.neurobiolaging.2011.10.025
27. Kenna KP, McLaughlin RL, Byrne S, Elamin M, Heverin M, Kenny EM, et al. Delineating the genetic heterogeneity of ALS using targeted high-throughput sequencing. J Med Genet. (2013) 50:776–83. doi: 10.1136/jmedgenet-2013-101795
28. Le Ber I, Van Bortel I, Nicolas G, Bouya-Ahmed K, Camuzat A, Wallon D, et al. hnRNPA2B1 and hnRNPA1 mutations are rare in patients with “multisystem proteinopathy” and frontotemporal lobar degeneration phenotypes. Neurobiol Aging. (2014) 35:934.e5–6. doi: 10.1016/j.neurobiolaging.2013.09.016
29. Couthouis J, Raphael AR, Daneshjou R, Gitler AD. Targeted exon capture and sequencing in sporadic amyotrophic lateral sclerosis. PLoS Genet. (2014) 10:e1004704. doi: 10.1371/journal.pgen.1004704
30. Mccluskey L, Vandriel S, Elman L, Van Deerlin VM, Powers J, Boller A, et al. ALS-Plus syndrome: non-pyramidal features in a large ALS cohort. J Neurol Sci. (2014) 345:118–24. doi: 10.1016/j.jns.2014.07.022
31. Cady J, Allred P, Bali T, Pestronk A, Goate A, Miller TM, et al. Amyotrophic lateral sclerosis onset is influenced by the burden of rare variants in known amyotrophic lateral sclerosis genes. Ann Neurol. (2015) 77:100–13. doi: 10.1002/ana.24306
32. Kwok CT, Wang HY, Morris AG, Smith B, Shaw C, de Belleroche J. VCP mutations are not a major cause of familial amyotrophic lateral sclerosis in the UK. J Neurol Sci. (2015) 349:209–13. doi: 10.1016/j.jns.2015.01.021
33. Krüger S, Battke F, Sprecher A, Munz M, Synofzik M, Schöls L, et al. Rare variants in neurodegeneration associated genes revealed by targeted panel sequencing in a german ALS cohort. Front Mol Neurosci. (2016) 9:92. doi: 10.3389/fnmol.2016.00092
34. Cooper-Knock J, Robins H, Niedermoser I, Wyles M, Heath PR, Higginbottom A, et al. Targeted genetic screen in amyotrophic lateral sclerosis reveals novel genetic variants with synergistic effect on clinical phenotype. Front Mol Neurosci. (2017) 10:370. doi: 10.3389/fnmol.2017.00370
35. Gibson SB, Downie JM, Tsetsou S, Feusier JE, Figueroa KP, Bromberg MB, et al. The evolving genetic risk for sporadic ALS. Neurology. (2017) 89:226–33. doi: 10.1212/WNL.0000000000004109
36. Mccann EP, Williams KL, Fifita JA, Tarr IS, O'connor J, Rowe DB, et al. The genotype-phenotype landscape of familial amyotrophic lateral sclerosis in Australia. Clin Genet. (2017) 92:259–66. doi: 10.1111/cge.12973
37. Morgan S, Shatunov A, Sproviero W, Jones AR, Shoai M, Hughes D, et al. A comprehensive analysis of rare genetic variation in amyotrophic lateral sclerosis in the UK. Brain. (2017) 140:1611–8. doi: 10.1093/brain/awx082
38. Türk M, Schröder R, Khuller K, Hofmann A, Berwanger C, Ludolph AC, et al. Genetic analysis of VCP and WASH complex genes in a German cohort of sporadic ALS-FTD patients. Neurobiol Aging. (2017) 56:213.e1–5. doi: 10.1016/j.neurobiolaging.2017.04.023
39. Dols-Icardo O, García-Redondo A, Rojas-García R, Borrego-Hernández D, Illán-Gala I, Muñoz-Blanco JL, et al. Analysis of known amyotrophic lateral sclerosis and frontotemporal dementia genes reveals a substantial genetic burden in patients manifesting both diseases not carrying the C9orf72 expansion mutation. J Neurol Neurosurg Psychiatry. (2018) 89:162–8. doi: 10.1136/jnnp-2017-316820
40. Lamp M, Origone P, Geroldi A, Verdiani S, Gotta F, Caponnetto C, et al. Twenty years of molecular analyses in amyotrophic lateral sclerosis: genetic landscape of Italian patients. Neurobiol Aging. (2018) 66:179.e5–16. doi: 10.1016/j.neurobiolaging.2018.01.013
41. Müller K, Brenner D, Weydt P, Meyer T, Grehl T, Petri S, et al. Comprehensive analysis of the mutation spectrum in 301 German ALS families. J Neurol Neurosurg Psychiatry. (2018) 89:817–27. doi: 10.1136/jnnp-2017-317611
42. Mehta PR, Jones AR, Opie-Martin S, Shatunov A, Iacoangeli A, Al Khleifat A, et al. Younger age of onset in familial amyotrophic lateral sclerosis is a result of pathogenic gene variants, rather than ascertainment bias. J Neurol Neurosurg Psychiatry. (2019) 90:268–71. doi: 10.1136/jnnp-2018-319089
43. Tripolszki K, Gampawar P, Schmidt H, Nagy ZF, Nagy D, Klivényi P, et al. Comprehensive genetic analysis of a hungarian amyotrophic lateral sclerosis cohort. Front Genet. (2019) 10:732. doi: 10.3389/fgene.2019.00732
44. Kotan D, Ozozen Ayas Z, Tunca C, Gungen BD, Akcimen F, Basak AN. Phenotypic and genotypic features of patients diagnosed with ALS in the city of Sakarya, Turkey. Acta Neurol Belg. (2020) 120:1411–8. doi: 10.1007/s13760-020-01441-z
45. Pensato V, Magri S, Bella ED, Tannorella P, Bersano E, Soraru G, et al. Sorting rare ALS genetic variants by targeted re-sequencing panel in Italian patients: OPTN, VCP, and SQSTM1 variants account for 3% of rare genetic forms. J Clin Med. (2020) 9:412. doi: 10.3390/jcm9020412
46. Ungaro C, Sprovieri T, Morello G, Perrone B, Spampinato AG, Simone IL, et al. Genetic investigation of amyotrophic lateral sclerosis patients in south Italy: a two-decade analysis. Neurobiol Aging. (2021) 99:99.e7–14. doi: 10.1016/j.neurobiolaging.2020.08.017
47. Yilmaz R, Müller K, Brenner D, Volk AE, Borck G, Hermann A, et al. SQSTM1/p62 variants in 486 patients with familial ALS from Germany and Sweden. Neurobiol Aging. (2020) 87:139.e9–15. doi: 10.1016/j.neurobiolaging.2019.10.018
48. Mccann EP, Henden L, Fifita JA, Zhang KY, Grima N, Bauer DC, et al. Evidence for polygenic and oligogenic basis of Australian sporadic amyotrophic lateral sclerosis. J Med Genet. (2020). doi: 10.1136/jmedgenet-2020-106866 [Epub ahead of print].
49. Nunes Gonçalves JP, Leoni TB, Martins MP, Peluzzo TM, Dourado MET Jr, Saute J, et al. Genetic epidemiology of familial ALS in Brazil. Neurobiol Aging. (2021) 102: 227.e1–4. doi: 10.1016/j.neurobiolaging.2021.01.007
50. Shepheard SR, Parker MD, Cooper-Knock J, Verber NS, Tuddenham L, Heath P, et al. Value of systematic genetic screening of patients with amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. (2021) 92:510–8. doi: 10.1136/jnnp-2020-325014
51. Abrahao A, Abath Neto O, Kok F, Zanoteli E, Santos B, Pinto WB, et al. One family, one gene and three phenotypes: a novel VCP (valosin-containing protein) mutation associated with myopathy with rimmed vacuoles, amyotrophic lateral sclerosis and frontotemporal dementia. J Neurol Sci. (2016) 368:352–8. doi: 10.1016/j.jns.2016.07.048
52. Ando T, Nakamura R, Kuru S, Yokoi D, Katsuno M. The wide-ranging clinical and genetic features in Japanese families with valosin-containing protein proteinopathy. Neurobiol Aging. (2020) 100:120.e1–6. doi: 10.1016/j.neurobiolaging.2020.10.028
53. Al-Tahan S, Al-Obeidi E, Yoshioka H, Lakatos A, Weiss L, Grafe M, et al. Novel valosin-containing protein mutations associated with multisystem proteinopathy. Neuromuscul Disord. (2018) 28:491–501. doi: 10.1016/j.nmd.2018.04.007
54. Dejesus-Hernandez M, Desaro P, Johnston A, Ross OA, Wszolek ZK, Ertekin-Taner N, et al. Novel p.Ile151Val mutation in VCP in a patient of African American descent with sporadic ALS. Neurology. (2011) 77:1102–3. doi: 10.1212/WNL.0b013e31822e563c
55. Battistini S, Ricci C, Benigni M, Casali S, Giannini F. Phenotypic variability associated with the R155C VCP gene mutation. In: International Symposium on Amyotrophic Lateral Sclerosis and Motor Neurone Disease 2013, Milan.
56. Surampalli A, Angèle N, Donkervoort S, Manaswitha K, Annabel W, Rudolph C, et al. A clinicopathologic case report of a female with valosin-containing protein (VCP) gene mutation related disease. Int J Neurodegener Dis. (2018) 1:006. doi: 10.23937/ijnd-2017/1710006
57. Segawa M, Hoshi A, Naruse H, Kuroda M, Bujo H, Ugawa Y. A patient with familial amyotrophic lateral sclerosis associated with a new valosin-containing protein (VCP) gene mutation. Rinsho Shinkeigaku. (2015) 55:914–20. doi: 10.5692/clinicalneurol.cn-000765
58. Ayaki T, Ito H, Fukushima H, Inoue T, Kondo T, Ikemoto A, et al. Immunoreactivity of valosin-containing protein in sporadic amyotrophic lateral sclerosis and in a case of its novel mutant. Acta Neuropathol Commun. (2014) 2:172. doi: 10.1186/s40478-014-0172-0
59. Papadimas GK, Paraskevas GP, Zambelis T, Karagiaouris C, Bourbouli M, Bougea A, et al. The multifaceted clinical presentation of VCP-proteinopathy in a Greek family. Acta Myol. (2017) 36:203–6.
60. Matsubara T, Izumi Y, Oda M, Takahashi M, Maruyama H, Miyamoto R, et al. An autopsy report of a familial amyotrophic lateral sclerosis case carrying VCP Arg487His mutation with a unique TDP43 proteinopathy. Neuropathology. (2021) 41:118–26. doi: 10.1111/neup.12710
61. Mehta SG, Khare M, Ramani R, Watts GD, Simon M, Osann KE, et al. Genotype-phenotype studies of VCP-associated inclusion body myopathy with Paget disease of bone and/or frontotemporal dementia. Clin Genet. (2013) 83:422–31. doi: 10.1111/cge.12000
62. Al-Obeidi E, Al-Tahan S, Surampalli A, Goyal N, Wang AK, Hermann A, et al. Genotype-phenotype study in patients with valosin-containing protein mutations associated with multisystem proteinopathy. Clin Genet. (2018) 93:119–25. doi: 10.1111/cge.13095
63. Zou ZY, Liu MS, Li XG, Cui LY. Screening of VCP mutations in Chinese amyotrophic lateral sclerosis patients. Neurobiol Aging. (2013) 34:1519.e1513–4. doi: 10.1016/j.neurobiolaging.2012.10.002
64. Watts GD, Wymer J, Kovach MJ, Mehta SG, Mumm S, Darvish D, et al. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet. (2004) 36:377–81. doi: 10.1038/ng1332
65. Gu JM, Ke YH, Yue H, Liu YJ, Zhang Z, Zhang H, et al. A novel VCP mutation as the cause of atypical IBMPFD in a Chinese family. Bone. (2013) 52:9–16. doi: 10.1016/j.bone.2012.09.012
66. Taylor JP. Multisystem proteinopathy: intersecting genetics in muscle, bone, and brain degeneration. Neurology. (2015) 85:658–60. doi: 10.1212/WNL.0000000000001862
67. Evangelista T, Weihl CC, Kimonis V, Lochmüller H. 215th ENMC International Workshop VCP-related multi-system proteinopathy (IBMPFD) 13-15 November 2015, Heemskerk, The Netherlands. Neuromuscul Disord. (2016) 26:535–47. doi: 10.1016/j.nmd.2016.05.017
68. Regensburger M, Türk M, Pagenstecher A, Schröder R, Winkler J. VCP-related multisystem proteinopathy presenting as early-onset Parkinson disease. Neurology. (2017) 89:746–8. doi: 10.1212/WNL.0000000000004240
69. Wang T, Xu W, Qin M, Yang Y, Bao P, Shen F, et al. Pathogenic Mutations in the Valosin-containing Protein/p97(VCP) N-domain Inhibit the SUMOylation of VCP and Lead to Impaired Stress Response. J Biol Chem. (2016) 291:14373–84. doi: 10.1074/jbc.M116.729343
70. Hall CE, Yao Z, Choi M, Tyzack GE, Serio A, Luisier R, et al. Progressive Motor Neuron Pathology and the Role of Astrocytes in a Human Stem Cell Model of VCP-Related ALS. Cell Rep. (2017) 19:1739–49. doi: 10.1016/j.celrep.2017.05.024
71. Tyzack GE, Luisier R, Taha DM, Neeves J, Modic M, Mitchell JS, et al. Widespread FUS mislocalization is a molecular hallmark of amyotrophic lateral sclerosis. Brain. (2019) 142:2572–80. doi: 10.1093/brain/awz217
72. Harley J, Hagemann C, Serio A, Patani R. FUS is lost from nuclei and gained in neurites of motor neurons in a human stem cell model of VCP-related ALS. Brain. (2020) 143:e103. doi: 10.1093/brain/awaa339
73. Luisier R, Tyzack GE, Hall CE, Mitchell JS, Devine H, Taha DM, et al. Intron retention and nuclear loss of SFPQ are molecular hallmarks of ALS. Nat Commun. (2018) 9:2010. doi: 10.1038/s41467-018-04373-8
Keywords: amyotrophic lateral sclerosis, Paget's disease of bone, valosin-containing protein, R155C, phenotype
Citation: Feng S-Y, Lin H, Che C-H, Huang H-P, Liu C-Y and Zou Z-Y (2022) Phenotype of VCP Mutations in Chinese Amyotrophic Lateral Sclerosis Patients. Front. Neurol. 13:790082. doi: 10.3389/fneur.2022.790082
Received: 06 October 2021; Accepted: 06 January 2022;
Published: 07 February 2022.
Edited by:
Kewei Chen, Banner Alzheimer's Institute, United StatesReviewed by:
Nicola Ticozzi, University of Milan, ItalyCaroline Vance, King's College London, United Kingdom
Copyright © 2022 Feng, Lin, Che, Huang, Liu and Zou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhang-Yu Zou, fmuzzy@fjmu.edu.cn
†These authors have contributed equally to this work and share first authorship